


Abstract
The major human AP-endonuclease 1 (APE1) is a multifunctional protein that plays a central role in the repair of damaged DNA by acting as a dual-function nuclease in the base excision repair pathway. This enzyme was also independently identified as a redox activator of AP-1 DNA-binding activity and has subsequently been shown to activate a variety of transcription factors via a redox mechanism. In a third distinct role, APE1 was identified as a component of a trans-acting complex that acts as a repressor by binding to the negative calcium responsive elements (nCaRE)-A and nCaRE-B, which were first discovered in the promoter of the human parathyroid gene and later in the APE1 promoter itself. Here we show that the nuclear protein complex which binds to the nCaRE-B2 of the hAPE1 gene contains APE1 itself and the heterogeneous nuclear ribonucleoprotein L (hnRNP-L). The interaction between the APE1 and hnRNP-L proteins does not require the presence of nCaRE-B2. Our results support the possibility that the APE1 gene is down-regulated by its own product, which would be the first such example of the regulation of a DNA repair enzyme, and identify a novel function of hnRNP-L in transcriptional regulation.
INTRODUCTION
The major human apurinic/apyrimidinic (AP) endonuclease, APE1, is a multifunctional protein. APE1 (also named APEX, HAP-1 and Ref-1) has been extensively characterized in the repair of damaged DNA, specifically for its role in the base excision repair (BER) pathway (reviewed in 1–3). In the BER process, APE1 acts as a dual-function nuclease (4,5). As an endonuclease, it cleaves the DNA strand 5′ to AP sites generated as a result of excision of abnormal bases by monofunctional DNA glycosylases. APE1 also acts as a 3′-phosphodiesterase to remove 3′ blocking groups generated directly in DNA by reactive oxygen species or after removal of oxidized base lesions by complex DNA glycosylase/AP lyases (6,7).
APE1 was independently identified as a redox activator of AP-1 DNA-binding activity and named Ref-1 (8). In vitro studies showed that APE1/Ref-1 converts the oxidized and inactive form of c-Jun into a reduced, active form, presumably via a thiol exchange reaction between oxidized Cys272 of c-Jun and Cys65 in APE1 (9,10). Furthermore, oxidized APE1 was shown to be reduced by thioredoxin, which interacts with the N-terminal domain of APE1 (11,12). APE1 has also been shown to activate p53, by both redox-dependent and -independent mechanisms, resulting in its nuclear translocation and increased DNA-binding activity (13). A link between p53 and APE1 in tumor induction was provided by genetic studies (14). Finally, APE1 was recently shown to mediate activation of several other transcription factors, including Pax 5, Pax 8 and basic helix–loop–helix transcription factors (reviewed in 15).
A third and apparently distinct function of APE1 was discovered during investigation of trans-acting factors that act as Ca2+-dependent repressors of the parathyroid hormone (PTH) gene by binding to the negative Ca2+ response elements (nCaRE) in its promoter. Expression of the PTH gene is down-regulated in response to increases in extracellular calcium (16), which is mediated by nCaRE-A and nCaRE-B (17,18). Subsequent experiments revealed that APE1 (Ref-1) is a component of the nuclear protein complexes that bind to nCaRE-A and nCaRE-B elements; however, APE1 was incapable of binding alone (19). It was suggested, therefore, that APE1 requires an additional factor(s) to form a competent DNA-binding complex. Use of DNA affinity chromatography led to the identification of Ku antigen p70 and p86 as partners of APE1 in binding to nCaRE-A, but not to nCaRE-B, of the PTH promoter (20).
We have previously characterized the promoter region of the human APE1 gene and identified a negative regulatory region ∼2 kb upstream of transcriptional initiation, a distance comparable to that found in the PTH gene (21). Sequencing of this region revealed the presence of a single nCaRE-A-type and two nCaRE-B-type sequences. Further deletion and mutational analysis revealed that this repression was eliminated with the removal of one of the nCaRE-B elements, nCaRE-B2 (21). Additionally, we showed that HeLa cell nuclear extract formed a discrete binding complex with the nCaRE-B2 oligonucleotide and formation of this binding complex could be inhibited by preincubation with anti-APE1 antibodies (21). However, recombinant APE1 alone, purified from Escherichia coli, was incapable of binding to this element, consistent with previous reports indicating that APE1 protein requires additional factors to form competent binding complexes with nCaRE elements (19,20). In this report we show that the binding complex for the nCaRE-B2 element in the APE1 promoter contains APE1 and heterogeneous nuclear ribonucleoprotein L (hnRNP-L).
MATERIALS AND METHODS
Purification of nCaRE-B2-binding protein by affinity chromatography
The nCaRE-B2 duplex oligo was generated by annealing synthetic oligonucleotides (Gibco Life Technologies) with 5′-phosphate: top strand, 5′-GATCCTTTTTGAGACAGAGTTTCACTCTTG-3′ (nCaRE-B palindromic sequence underlined; 18), corresponding to nucleotides –1719 to –1690 in the APE1 promoter; bottom strand, 3′-AAAAACTCTGTCTCAAAGTGAACCTAGG-5′, which contains an additional 5 nt at the 5′-end to facilitate concatemer formation. Following annealing of complementary strands, 75 µg duplex oligo was treated overnight at room temperature with T4 DNA ligase (Promega) to generate concatemers that contained, an average, trimers of the oligo and up to 8mers, as judged by agarose gel electrophoresis (data not shown). The ligation mixture was phenol extracted, ethanol precipitated, resuspended in HEPES buffer (30 mM HEPES, pH 7.9, 100 mM NaCl) and then coupled to 3 ml CNBr-activated Sephadex 4B (Amersham Pharmacia) according to the manufacturer’s directions. Coupling efficiency was calculated to be ∼20% based on a small-scale coupling experiment using 32P-end-labeled nCaRE-B2 concatemers.
A 5 ml column containing nCaRE-B2–agarose was equilibrated with 10 column vol binding buffer (20 mM HEPES, pH 7.8, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT and 10% v/v glycerol). Then, 75 mg HeLa S3 nuclear extract in 15 ml of this buffer were preincubated with sheared salmon sperm DNA (50 µg/ml) for 1 h at 8°C with gentle rocking and then passed through the affinity column. The flow-through was recycled three times and then the column was washed with 10 column vol binding buffer. Finally, the bound proteins were eluted with binding buffer containing 0.5 M NaCl. Fractions (500 µl) were collected and 20 µl aliquots were assayed for binding activity (in 50 µl aliquots) by electrophoretic mobility shift assay (EMSA). Fractions showing binding activity were pooled and concentrated (Microcon 10 and Centricon 10 cartridges; Amicon) to ∼25 µl, analyzed on a 4–20% SDS–PAGE gradient gel (NOVEX) and finally transferred to a PVDF membrane (Immobilon P; Millipore). The membrane was longitudinally cut in half; half was stained with Coomassie brilliant blue R250 and the other half was used for western analysis using anti-APE1 antibodies.
Amino acid microsequencing
The Coomassie brilliant blue stained 66 kDa band was subjected to automated amino acid sequencing. Initial attempts at Edman degradation for N-terminal sequencing failed to yield any sequence data, presumably due to a blocked N-terminus. Subsequent samples were treated by mild trypsin digestion followed by microcapillary HPLC and N-terminal sequencing of three peptide products. These procedures were performed by the University of Texas Medical Branch (UTMB) Protein Chemistry Core Facility.
Tissue culture and preparation of nuclear extracts
HeLa S3 cells were initially grown as adherent cells in Eagle’s medium supplemented with 10% fetal calf serum (FCS) (Gibco BRL), streptomycin and penicillin (Sigma) and then adapted to grow in suspension in Spinner flasks in successively larger volumes. The cells were maintained at 1–5 × 105 cells/ml in an air incubator at 37°C; then ∼1 × 109 cells were harvested from 8 l of culture and the nuclear extract was isolated by scaling up a published procedure (22), after addition of the protease inhibitors (1 µg/ml each) Antipain, Leupeptin, Chymostatin and Pepstatin A (Sigma). The nuclear extracts were aliquoted, quick frozen and stored at –85°C; protein concentration was measured by Bradford assay (Bio-Rad).
Transient transfection of APE1 and hnRNP-L expression vectors
Full-length cDNAs for human APE1 and hnRNP-L (a generous gift from Dr Gideon Dreyfuss) were cloned in-frame into the pcDNA4HisMax expression vector (Invitrogen) and the correct reading frame was confirmed by DNA sequencing at the UTMB Protein Chemistry Core Facility. Vector-directed expression results in N-terminal X-Press epitope-tagged fusion proteins. For transient expression analysis, COS-1 cells were plated in triplicate on 60 mm dishes in DMEM (Gibco BRL) supplemented with 10% FCS at a density of 4–5 × 105 cells/dish and then transfected with 0.8 µg/dish empty vector, APE1–pcDNA4HisMax, hnRNP-L–pcDNA4HisMax or combined APE1– and hnRNP-L–pcDNA4HisMax constructs, using a liposome-mediated transfection reagent (Effectene; Qiagen). Nuclear and cytosolic extracts were isolated 48 h post-transfection using the procedure described above.
Generation of anti-hnRNP-L antibodies
A peptide, STPEQAAKNRIQHPSNVLHC, corresponding to amino acid residues 455–473 of human hnRNP-L, was synthesized and purified by HPLC at the UTMB Protein Chemistry Core Facility and was used for production of polyclonal antibodies in New Zealand White rabbits (Alpha Diagnostic). The rabbit antisera were enriched for IgG by ammonium sulfate precipitation and chromatography on DEAE (23). The generation of polyclonal antibodies against recombinant APE1 protein was described earlier (21).
Western analysis
Nuclear extracts were fractionated on 4–20% gradient polyacrylamide SDS–PAGE gels (Novex) and electrophoretically transferred to PVDF membranes (Millipore). After blocking the membranes with 5% (w/v) Blocker (Bio-Rad) in Tris-buffered saline, pH 7.4, containing 0.5% Tween-20 (TBS-T) for 1 h with gentle rocking at room temperature, the membranes were incubated with antibodies at 1:1000–1:2000 dilution in TBS-T for 1 h and the protein bands were visualized using ECL (Supersignal WestPico; Pierce) according to the manufacturer’s directions.
Electrophoretic mobility shift assays
The following oligonucleotides in duplex form were used: APE1 nCaRE-B2, 5′-TTTTTGAGACAGAGTTTCACTTTG-3′; mutant nCaRE-B2, 5′-TTTTTGCTAAAGAGCGTCACTTTG-3′; PTH nCaRE-A, 5′-CCATTTGTGTATGCACAA-3′. The nCaRE-B palindromic sequence is underlined and divergent sequences in the mutant nCaRE-B2 sequence are in bold. The conditions for EMSA, described earlier (21), were modified as follows: 20 µg nuclear extract was used in each reaction, unless otherwise indicated, with the addition of 1 µg sheared salmon sperm DNA per reaction to prevent non-specific binding of the labeled oligo. Reconstitution of EMSA using nCaRE-B2 was carried out under the reaction conditions described above with the addition of 2–3 nmol purified baculovirus-expressed hnRNP-L and bacterially expressed APE1 protein (24) and 0.1 µg sheared salmon sperm DNA per reaction.
Generation of recombinant hnRNP-L protein
Full-length human cDNA for hnRNP-L was cloned in-frame into the baculovirus transfer vector pAcGHLT-B (Pharmingen) by PCR-based cloning, which resulted in an N-terminal GST–hnRNP-L fusion protein. The sequence was verified by the UTMB Molecular Biology Core Facility. The GST domain was removed by cleavage with thrombin at an engineered thrombin site 5′ of the hnRNP-L coding sequence. Recombinant baculoviruses were generated by co-transfecting the hnRNP-L–pAcGHLT construct with linear BaculoGold DNA into Sf9 cells according to the protocol of Pharmingen. The GST–hnRNP-L fusion protein was purified from the extract of recombinant virus-infected Sf9 cells by affinity chromatography on glutathione–Sephadex (Sigma). After treatment with thrombin (Biotinylated Thrombin; Novagen), uncleaved fusion protein, GST and thrombin were removed by the addition of glutathione–Sephadex and streptavidin–agarose, respectively, using a Thrombin Cleavage Capture Kit (Novagen). Relative purity and concentration of recombinant hnRNP-L were estimated by SDS–PAGE analysis and Coomassie brilliant blue staining.
GST pull-down assay
Approximately 200 ng purified GST–hnRNP-L fusion protein was incubated with 80 µg COS-1 cell nuclear extract in 100 µl EMSA reaction buffer with or without 100 ng duplex nCaRE-B2 oligo for 30 min at 8°C on a rocking platform. Subsequently, 50 µl of reduced glutathione–agarose beads (30 mg/ml), prewashed in cold TBS, pH 7.4, was added and the mixture incubated for an additional 30 min at 8°C. The beads containing bound proteins were then collected by centrifugation (500 g for 2 min at 8°C), washed three times with 0.5 ml of cold TBS-T, resuspended in 20 µl of 10 mM reduced glutathione and incubated for 10 min on a rocking platform at 8°C. After removal of the beads by centrifugation, the supernatant was used for western analysis.
RESULTS
Purification of the nCaRE-B2-binding nuclear protein complex
In an effort to identify the proteins that bind to nCaRE-B2 in the APE1 promoter, we utilized DNA affinity chromatography and eluted proteins that were tightly bound to the nCaRE-B2 oligo (Fig. 1A). SDS–PAGE analysis of fractions with strong binding activity (Fig. 1A, lanes 5 and 6) showed the presence of two major proteins, at 66 and 36 kDa (Fig. 1B, left, lane 2). The 36 kDa band was identified as APE1, based on its immunoreactivity to anti-APE1 antibodies (Fig. 1B, right, lane 3). The 66 kDa band appeared to be the binding partner of APE1. After failing to identify the N-terminal sequence of this protein, we were able to sequence three internal peptides after trypsin digestion (Fig. 1C). A BLASTP search showed that these peptides corresponded exactly to sequences in hnRNP-L. The hnRNP-L protein, previously characterized in HeLa cell extracts, was shown to have a molecular mass of 64–68 kDa (25). Therefore, we conclude that the 66 kDa band was indeed hnRNP-L.
Figure 1.

Purification of nCaRE-B2-binding proteins. (A) EMSA of fractions (1–14) eluted from a nCaRE-B2–Sepharose column with 0.5 NaCl. Lanes with probe alone (free) and control (con) unfractionated HeLa nuclear extracts are indicated. (B) (Left) Coomassie brilliant blue stained PVDF membrane. Lane 1, marker proteins; lane 2, eluted fraction 5. Bands at 66 and 36 kDa are indicated by arrows. (Right) Western analysis of eluted fraction 5 (lane 3) and control HeLa nuclear extract (lane 4) using anti-APE1 antibody. (C) Sequences of three internal peptides from the 66 kDa species and the numbers indicate the positions of the terminal residues in the hnRNP-L polypeptide.
EMSA with nCaRE-B2
After co-purification of APE1 and hnRNP-L from the nCaRE-B2 affinity column, we investigated the interaction between these proteins and further defined the nature of the complex utilizing several experimental approaches. In an effort to show supershift of the nCaRE-B2 complex after treatment with anti-hnRNP-L mAb clone 4D11 (a generous gift from Dr Dreyfuss), we observed an ∼3.5-fold reduction in the amount of complex (Fig. 2, lanes 2 and 5). A similar treatment with anti-APE1 antibody resulted in a 2-fold reduction in nCaRE-B2-binding activity (Fig. 2, lane 4). The addition of preimmune serum had no effect on binding complex formation (data not shown). The sequence specificity of this binding complex is also shown in Figure 2. Addition of a 20-fold molar excess of unlabeled nCaRE-B2 oligonucleotides resulted in almost complete elimination of binding complex formation (compare lanes 2 and 3). On the other hand, addition of a 20-fold molar excess of unlabeled mutant nCaRE-B2 oligo, which lacks the consensus binding motif, did not compete with the nCaRE-B2-binding complex to a significant extent (compare lanes 7 and 8), demonstrating that an intact palindrome is required for interaction with the proteins. The addition of a 20-fold molar excess of unlabeled PTH nCaRE-A caused a slight reduction in nCaRE-B2-binding complex formation (lane 9).
Figure 2.
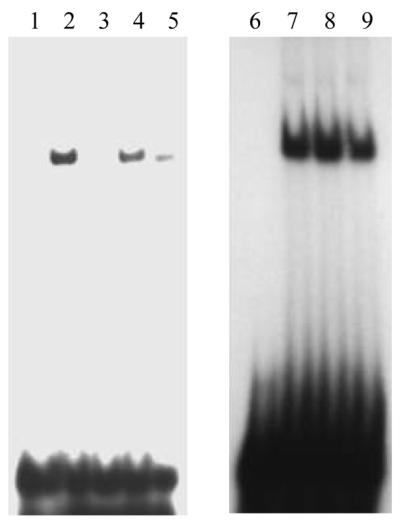
Competitive EMSA using mutant nCaRE-B2 sequence. Sequence-specific binding of HeLa nuclear extract to nCaRE-B2. Lanes 1 and 6, free probe; lanes 2–5 and 7–9, 20 µg HeLa extract with 20-fold molar excess each of unlabeled nCaRE-B2 (lane 3), mutant nCaRE-B2 (lane 8), nCaRE-A probe (lane 9) or after preincubation of extract with 4 µl of anti-APE1 (lane 4) or anti-hnRNP-L antibody (lane 5). Lanes 2 and 7, HeLa nuclear extract alone.
Transient transfection of APE1 and hnRNP-L expression vectors
In order to confirm the initial observation that APE1 and hnRNP-L comprise the nCaRE-B2-binding complex, APE1 and hnRNP-L cDNAs were cloned into the mammalian expression vector pcDNA4HisMax and transiently expressed in COS-1 cells. These plasmid-encoded proteins contain N-terminal epitope tags (X-Press epitope) that allow discrimination between endogenous and ectopically expressed APE1 and hnRNP-L. Representative western analyses of ectopically expressed APE1, hnRNP-L and APE1 + hnRNP-L nuclear protein levels are shown in Figure 3B. EMSA was performed using nuclear extracts from COS-1 cells transiently transfected with APE1 and hnRNP-L expression vectors. With a limiting amount (3.5 µg) of extract, we did not observe a mobility shift with nuclear extracts from untransfected COS-1 cells (Fig. 3C, lanes 2 and 3). Ectopic expression of APE1 and/or hnRNP-L, however, significantly increased nCaRE-B2-binding activity with the same amount of COS-1 nuclear extract (Fig. 3C and D). The increased binding activity observed with APE1 and hnRNP-L transfected alone suggests that these ectopically expressed proteins are capable of recruiting endogenous binding partners to participate in the nCaRE-B2-binding complex. Interestingly, while co-transfection of both APE1 and hnRNP-L resulted in increased binding activity, it was not synergistic or additive compared to the activity observed with APE1 or hnRNP-L transfected alone (Fig. 3C, lanes 4–6). We interpret these observations as suggesting that covalent modification of APE1 and/or hnRNP-L, such as acetylation or phosphorylation, rather than simple mass action, may be required for the observed nCaRE-B2-binding activities.
Figure 3.

EMSA with extracts of transiently transfected COS-1 cells. (A) Formation of the nCaRE-B2-binding complex with increasing amounts of nuclear extract of untransfected COS-1 cells. (B) Western analysis of transfected cell extracts. Lane 1, control (C); lane 2, cells transfected with empty vector (EV); lane 3, APE1 vector (A); lane 4, hnRNP-L vector (L); lane 5, both APE1 and hnRNP-L expression vectors (A + L). The protein bands were detected with anti-Xpress antibody. (C) nCaRE-B2 binding with 3.5 µg nuclear extract. Lane 1, free probe; lane 2, control cells; lane 3, cells transfected with empty vector; lane 4, APE1 cDNA; lane 5, hnRNP-L cDNA; lane 6, both APE1 and hnRNP-L cDNAs. The spots observed in control lanes 1 and 2 in the same position as the gel shifted band are artifacts. (D) Densitometric analysis (arbitrary scale) of nCaRE-B2-binding complexes shown in (C); nd, not detected. Error bars represent standard deviation (six samples).
In vitro reconstitution of the nCaRE-B2-binding complex
In order to determine whether the APE1 and hnRNP-L proteins together are sufficient to form the nCaRE-B2-binding complex and to test whether recombinant proteins are as efficient as the endogenous proteins in complex formation, we reconstituted this complex using recombinant APE1 and hnRNP-L proteins (Fig. 4). Recombinant human hnRNP-L purified from insect cells and bacterially expressed human APE1 were used in nCaRE-B2 EMSAs (Fig. 4C). Binding reactions contained 2 nmol recombinant APE1 (lane 4), 3 nmol hnRNP-L (lane 5) or APE1 and hnRNP-L together (lane 3) under nCaRE-B2 EMSA binding conditions and were compared to a control with HeLa S3 nuclear extract (lane 2). These results clearly demonstrate the formation of one major nCaRE-B2-binding complex in reactions containing both recombinant APE1 and hnRNP-L that migrates to the same position as seen with the HeLa S3 control extract. Recombinant APE1 alone failed to form a detectable complex, consistent with previous reports (19–21). However, recombinant hnRNP-L alone displayed weak binding activity (lane 5). It is surprising that the mobility of the complex with hnRNP-L alone was not significantly different from that containing both hnRNP-L and APE1 in lanes 2 and 3. The mobility of complexes in EMSA should depend on their molecular masses, which are obviously different in the case of hnRNP-L versus hnRNP-L together with APE1. However, we have often observed a lack of correlation between the relative mobility of a complex and the total molecular mass of proteins in the complex in these assays. In any event, these results suggest that hnRNP-L possesses an ability for nCaRE-B recognition and that its initial binding is strengthened or stabilized by APE1. There is an alternative possibility that the weak binding observed with hnRNP-L alone was due to interaction with an APE1-like protein present in Sf9 cells that co-purified with hnRNP-L as a minor contaminant.
Figure 4.

Reconstitution of the nCaRE-B2-binding complex and peptide interference assay. (A) Domain organization of the GST–hnRNP-L fusion protein. (B) Purification of recombinant hnRNP-L protein from Sf9 cells. (Left) Coomassie brilliant blue staining: lanes 1 and 4, protein standards; lane 2, uninfected Sf9 extract; lane 3, Sf9 extract expressing GST–hnRNP-L; lanes 5 and 6, purified GST–hnRNP-L before (lane 5) and after (lane 6) cleavage with thrombin. (Right) Western analysis of the proteins in lanes 5 and 6 with anti-hnRNP-L antibody. (C) In vitro reconstitution of nCaRE-B2-binding activity by EMSA. Lane 1, no protein; lane 2, HeLa nuclear extract; lane 3, APE1 and hnRNP-L; lane 4, APE1 alone; lane 5, hnRNP-L alone.
GST pull-down assay
We next examined whether the interaction between APE1 and hnRNP-L required the presence of nCaRE-B2 sequences. Repeated attempts at co-immunoprecipitation experiments using our anti-APE1 and anti-hnRNP-L polyclonal antibodies failed, probably due to the characteristics of our antisera. However, the GST–hnRNP-L fusion protein enabled its effective isolation, with associated factors, via interaction of GST with immobilized glutathione. As shown in Figure 5, 200 ng GST–hnRNP-L incubated with 80 µg COS-1 nuclear extracts with or without nCaRE-B2 oligonucleotides, followed by the addition of reduced glutathione–Sephadex and three washes of the bound GST–hnRNP-L and then western analysis, demonstrated that the association between APE1 and hnRNP-L is not dependent on the presence of cognate DNA elements. These results demonstrate that APE1 and hnRNP-L associate in vitro and may be complexed in vivo in the absence of the nCaRE-B2 oligo.
Figure 5.

Interaction between hnRNP-L and APE1. HeLa nuclear extract (NE) (100 µg) was incubated with recombinant GST–hnRNP-L in the presence or absence of nCaRE-B2 oligo, mixed with glutathione–agarose resin and the bound proteins eluted with glutathione for western analysis. Lane 1, HeLa extract alone; lane 2, with GST–hnRNP-L fusion protein; lane 3, both GST–hnRNP-L and nCaRE-B2 oligo.
DISCUSSION
The nCaRE-B2-binding complex consists of APE1 and hnRNP-L
We have demonstrated that the nCaRE-B2-binding complex contains both APE1 and hnRNP-L polypeptides. Identification of APE1 in this complex confirms earlier results reported by us and others and was thus predictable (19–21). By analogy with the situation of the nCaRE-A-binding complex, which was shown to include APE1 and Ku70, we expected the presence of a second protein in the nCaRE-B2-binding complex. Identification of this second protein as hnRNP-L was, however, completely unexpected (Fig. 1). Its presence in this binding complex was verified by EMSA (Fig. 2) and by reconstitution of the nCaRE-B2-binding activity (Fig. 4). Furthermore, the interaction between APE1 and hnRNP-L was shown using a GST pull-down assay (Fig. 5), which also indicated that the cognate cis element is not required for stable interaction between these two proteins. While our results demonstrate that the APE1 and hnRNP-L proteins are components of the binding complex, we cannot exclude the possibility that additional factors contribute to the formation of this binding complex and that covalent modifications of these proteins may be required for optimum binding to the nCaRE-B sequence.
A novel function of hnRNP-L
Pre-mRNAs, termed heterogeneous nuclear RNAs (hnRNAs), are synthesized in the nucleus by RNA polymerase II and require extensive post-transcriptional processing to generate mature mRNAs. These RNAs are complexed with a group of proteins named heterogeneous nuclear ribonucleoproteins (hnRNPs) (25–26). While the primary in vivo role of the hnRNP family, which consists of about 20 members with molecular masses ranging between 34 and 120 kDa, is in the packaging of nascent hnRNAs (27), recent studies suggest broader and unrelated functions for these proteins, e.g. in transcriptional regulation of gene expression and other cellular processes (28,29). For example, while hnRNP-L was first identified and characterized as a component of hnRNP complexes in HeLa cell nuclear extracts, immunohistochemical analysis showing significant amounts of hnRNP-L in the free form suggests that it has a distinct activity in the uncomplexed state (25). Several diverse functions for hnRNP-L have indeed been reported, including its binding to a cis-acting RNA sequence that facilitates nuclear export and expression of a chimeric, intronless gene (30). Furthermore, hnRNP-L was shown to interact with the polypyrimidine tract-binding protein (hnRNP-I) located at the 3′-border of the internal ribosome entry site of hepatitis C virus, suggesting a role in viral translational control (31,32). More recently, hnRNP-L has been shown to regulate the stability of human endothelial growth factor mRNA in response to hypoxia (33). To date, our studies are the first to identify a novel role of hnRNP-L in the regulation of gene expression. Interestingly, several other hnRNP family members have been reported to have similar roles in transcriptional regulation. For example, hnRNP-K was shown to bind to the CT element of the c-myc promoter to activate transcription (34,35). hnRNP-F has been shown to possess a transactivation function, via which it binds to DNA and interacts with RNA polymerase II (36). Nucleolin and an isoform of hnRNP-D have been shown to comprise the B cell- and sequence-specific DNA-binding complex LR1 (37). More recent studies suggest a role for hnRNP-D in telomere maintenance (38). It is thus becoming increasingly apparent that the hnRNP family members have a much broader role in vivo than in mRNA biogenesis alone.
Potential model for hnRNP-L–APE1 interaction
The hnRNP-L polypeptide contains a stretch of basic amino acids similar to those found in the transcription factors HLF, c-Fos and c-Jun (Fig. 6). The N-terminal redox domain of APE1 has been shown to be required for reductive activation of these transcription factors at specific, conserved cysteine residues in these proteins (9,39). While we do not propose a similar reductive activation of hnRNP-L, as the conserved cysteine is replaced by arginine in hnRNP-L and EMSA experiments were unaffected by DTT (data not shown), we propose that this region may function as an interaction domain that mediates the coupling of hnRNP-L and APE1. This would involve an interaction of the N-terminal region of APE1 with a region near the C-terminus of hnRNP-L. This model is consistent with the interaction between APE1 and Ku70/Ku86, which has been shown by Chung et al. (20) to require the N-terminal region of APE1, i.e. the region encompassing the redox domain. Furthermore, the authors identified a sequence, AALCR, corresponding to residues 395–399 in Ku70, which is involved in this interaction. This is consistent with our proposal that this region may function as a common protein–protein interaction domain for APE1.
Figure 6.

Partial sequence alignment of basic regions in hnRNP-L, HLF, c-Fos and c-Jun. The sequence containing the Cys residue involved in redox regulation is boxed. The numbers indicate residue numbers of these proteins.
Our observation that HeLa nuclear extract showed a much higher efficiency of binding to nCaRE-B2 than a mixture of recombinant APE1 and hnRNP-L (Fig. 4C) suggests that some other component is required for optimum complex formation. One possibility is that a third protein or small molecule is present in the shifted complex which does not affect the position of the band. It is more likely, however, that one or both of these proteins are post-translationally modified in vivo and that the modified APE1 and hnRNP-L interact more strongly with each other and with nCaRE-B2. This may also explain how moderately abundant proteins like APE1 and hnRNP-L can carry out such specialized functions in specific DNA sequence recognition, in addition to carrying out their predominant activities not related to transcriptional regulation.
Potential for negative autoregulation of the APE1 gene
Identification of APE1 as a component of nCaRE-B2-binding complexes and the role of nCaRE-B elements as functional negative regulatory elements (19,20) suggest that the APE1 gene may be down-regulated by its own product, as proposed earlier (21). This is the first such example of autoregulation of a eukaryotic DNA repair enzyme. The present studies show that APE1, with the ability to recognize AP sites in DNA, and hnRNP-L, with a primary function other than DNA binding, interact to form a functional binding complex for a unique DNA sequence. How the cell ‘directs’ the activities of these two multifunctional proteins to this unique function remains to be elucidated.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr G. Dreyfuss for providing the hnRNP-L cDNA clone and anti-hnRNP-L antibody. We thank Ms Wanda Smith for secretarial assistance. This research was supported by United States Health Service grant R01 ES08457 (S.M.) and an AG10514 Program Project Grant from the National Institute on Aging. Publication no. 115 of the J. Papaconstantinou laboratory.
REFERENCES
- 1.Seeberg E., Eide,L. and Bjoras,M. (1995) The base excision repair pathway. Trends Biochem. Sci., 20, 391–397. [DOI] [PubMed] [Google Scholar]
- 2.Mitra S., Izumi,T., Boldogh,I., Ramana,C.V., Hsieh,C.C., Saito,H., Lock,J. and Papaconstantinou,J. (1999) Repair of oxidative DNA damage and aging: central role of AP-endonuclease. In Karakaya,D.A. (ed.) Advances in DNA Damage and Repair. Plenum, New York, NY.
- 3.Mol C.D., Hosfield,D.J. and Tainer,J.A. (2000) Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res., 460, 211–229. [DOI] [PubMed] [Google Scholar]
- 4.Mitra S., Hazra,T.K., Roy,R., Ikeda,S., Biswas,T., Lock,J., Boldogh,I. and Izumi,T. (1997) Complexities of DNA base excision repair in mammalian cells. Mol. Cell, 7, 305–312. [PubMed] [Google Scholar]
- 5.Suh D., Wilson,D.M. and Povirk,L.F. (1997) 3′-Phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res., 25, 2495–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nash H.M., Bruner,S.D., Scharer,O.D., Kawate,T., Addona,T.A., Spooner,E., Lane,W.S. and Verdine,G.L. (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol., 6, 968–980. [DOI] [PubMed] [Google Scholar]
- 7.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 8.Xanthoudakis S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xanthoudakis S., Miao,G.G. and Curran,T. (1994) The redox and DNA repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl Acad. Sci. USA, 91, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirota K., Matsui,M., Iwata,S., Nishiyama,A., Mori,K. and Yodoi,J. (1997) AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl Acad. Sci. USA, 94, 3633–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin J., Clore,G.M., Kennedy,W.P., Kuszewski,J. and Gronenborn,A.M. (1996) The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure, 4, 613–620. [DOI] [PubMed] [Google Scholar]
- 13.Jayaraman L., Murthy,K.G., Zhu,C., Curran,T., Xanthoudakis,S. and Prives,C. (1997) Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev., 11, 558–570. [DOI] [PubMed] [Google Scholar]
- 14.Meira L.B., Cheo,D.L., Hammer,R.E., Burns,D.K., Reis,A. and Friedberg,E.C. (1997) Genetic interaction between HAP1/Ref-1 and p53. Nature, 17, 145. [DOI] [PubMed] [Google Scholar]
- 15.Evans A.R., Limp-Foster,M. and Kelley,M.R. (2000) Going APE over Ref-1. Mutat. Res., 461, 83–108. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto M., Igarashi,T., Muramatsu,M., Fukagawa,M., Motokura,T. and Ogata,E. (1989) Hypocalcemia increases and hypercalcemia decreases the steady-state level of parathyroid hormone messenger RNA in the rat. J. Clin. Invest., 83, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okazaki T., Zajac,J.D., Igarashi,T., Ogata,E. and Kronenberg,H.M. (1991) Negative regulatory elements in the human parathyroid hormone gene. J. Biol. Chem., 266, 21903–21910. [PubMed] [Google Scholar]
- 18.Okazaki T., Ando,K., Igarashi,T., Ogata,E. and Fujita,T. (1992) Conserved mechanisms of negative gene regulation by extracellular calcium. J. Clin. Invest., 89, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazaki T., Chung,U., Nishishita,T., Ebisu,S., Usuda,S., Mishiro,S., Xanthoudakis,S., Igarashi,T. and Ogata,E. (1994) A redox factor protein, ref-1, is involved in negative gene regulation by extracellular calcium. J. Biol. Chem., 269, 27855–27862. [PubMed] [Google Scholar]
- 20.Chung U., Igarashi,T., Nishishita,T., Iwanari,H., Iwamatsu,A., Suwa,A., Mimori,T., Hata,K., Ebisu,S., Ogata,E., Fujita,T. and Okazaki,T. (1996) The interaction between ku antigen and ref-1 protein mediates negative gene regulation by extracellular calcium. J. Biol. Chem., 271, 8593–8598. [DOI] [PubMed] [Google Scholar]
- 21.Izumi T., Henner,W.D. and Mitra,S. (1996) Negative regulation of the major human ap-endonuclease, a multifunctional protein. Biochemistry, 35, 14679–14683. [DOI] [PubMed] [Google Scholar]
- 22.Schreiber E., Matthias,P., Muller,M.M. and Schaffner,W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 24.Izumi T., Malecki,J., Chaudhry,M.A., Weinfeld,M., Hill,J.H., Lee,J.C. and Mitra,S. (1999) Intragenic suppression of an active site mutation in the human apurinic/apyrimidinic endonuclease. J. Mol. Biol., 287, 47–57. [DOI] [PubMed] [Google Scholar]
- 25.Pinol-Roma1 S., Swanson,M.S., Gall,J.G. and Dreyfuss,G. (1989) A novel heterogeneous nuclear rnp protein with a unique distribution on nascent transcripts. J. Cell Biol., 109, 2575–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dreyfuss G., Matunis,M.J., Pinol-Roma,S. and Burd,C.G. (1993) hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 27.Dreyfuss G., Swanson,M.S. and Pinol-Roma,S. (1988) Heterogeneous nuclear ribonucleoprotein particles and the pathway of mRNA formation. Trends Biochem. Sci., 13, 86–91. [DOI] [PubMed] [Google Scholar]
- 28.Weighardt F., Biamonti,G. and Riva,S. (1996) The roles of heterogeneous nuclear ribonucleoproteins (hnRNP) in RNA metabolism. Bioessays, 18, 747–756. [DOI] [PubMed] [Google Scholar]
- 29.Ladomery M. (1997) Multifunctional proteins suggest connections between transcription and post-transcriptional processes. Bioessays, 19, 903–909. [DOI] [PubMed] [Google Scholar]
- 30.Liu X. and Mertz,J.E. (1995) hnRNP L binds a cis-acting RNA sequence element that enables intron-independent gene expression. Genes Dev., 9, 1766–1780. [DOI] [PubMed] [Google Scholar]
- 31.Hahm B., Cho,O.H., Kim,J.E., Kim,Y.K., Kim,J.H., Oh,Y.L. and Jang,S.K. (1998) Polypyrimidine tract-binding protein interacts with hnRNP L. FEBS Lett., 425, 401–406. [DOI] [PubMed] [Google Scholar]
- 32.Hahm B., Kim,Y.K., Kim,J.H., Kim,T.Y. and Jang,S.K. (1998) Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitus C virus. J. Virol., 72, 8782–8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shih S.C. and Claffey,K.P. (1999) Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J. Biol. Chem., 274, 1359–1365. [DOI] [PubMed] [Google Scholar]
- 34.Michelotti E.F., Michelotti,G.A., Aronsohn,A.I. and Levens,D. (1996) Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol. Cell. Biol., 16, 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bomsztyk K., Van Seuningen,I., Suzuki,H., Denisenko,O. and Ostrowski,J. (1997) Diverse molecular interactions of the hnRNP K protein. FEBS Lett., 403, 113–115. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida T., Kokura,K., Makino,Y., Ossipow,V. and Tamura,T. (1999) Heterogeneous nuclear RNA-ribonucleoprotein F binds to DNA via an oligo (dG)-motif and is associated with RNA polymerase II. Genes Cells, 4, 707–719. [DOI] [PubMed] [Google Scholar]
- 37.Dempsey L.A., Hanakahi,L.A. and Maizels,N. (1998) A specific isoform of hnRNP D interacts with DNA in the LR1 heterodimer: canonical RNA binding motifs in a sequence-specific duplex DNA binding protein. J. Biol. Chem., 273, 29224–29229. [DOI] [PubMed] [Google Scholar]
- 38.Eversole A. and Maizels,N. (2000) In vitro properties of the conserved mammalian protein hnRNP D suggest a role in telomere maintenance. Mol. Cell. Biol., 20, 5425–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lando D., Pongratz,I., Poellinger,L. and Whitelaw,M.L. (2000) A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1α and the HIP-like factor. J. Biol. Chem., 275, 4618–4627. [DOI] [PubMed] [Google Scholar]