

Abstract
Positively charged repeat peptides are emerging as key players in neurodegenerative diseases. These peptides can perturb diverse cellular pathways but a unifying framework for how such promiscuous toxicity arises has remained elusive. We used mass-spectrometry-based proteomics to define the protein targets of these neurotoxic peptides and found that they all share similar sequence features that drive their aberrant condensation with these positively charged peptides. We trained a machine learning algorithm to detect such sequence features and unexpectedly discovered that this mode of toxicity is not limited to human repeat expansion disorders but has evolved countless times across the tree of life in the form of cationic antimicrobial and venom peptides. We demonstrate that an excess in positive charge is necessary and sufficient for this killer activity, which we name ‘polycation poisoning’. These findings reveal an ancient and conserved mechanism and inform ways to leverage its design rules for new generations of bioactive peptides.
Keywords: Phase separation, cathelicidin, protein aggregation, membraneless organelles, frontotemporal dementia, amyotrophic lateral sclerosis, antimicrobial peptide, crotamine, RAN translation, LL-37
INTRODUCTION
Nucleotide repeat expansions are implicated in an expanding group of human diseases1,2. There are several different subclasses. Polyglutamine (polyQ) diseases, at least 9 different diseases each caused by the expansion of an exonic CAG repeat, are perhaps the most well-known3. These disease genes harboring the CAG repeat expansions get translated into proteins with an expanded polyQ tract, which makes them aggregation-prone, resulting in the formation of inclusion bodies as the hallmark pathology. Besides polyQ, expansions of exonic GCG and CGG repeats produce aggregation-prone polyalanine and polyglycine proteins, and cause a series of other diseases4,5. Repeat expansions in non-coding parts of genes can also cause disease. For example, in Fragile X-syndrome, large GC-rich expansions within promoter or 5’ untranslated regions (UTR) become hypermethylated, inhibiting transcription and resulting in haploinsufficiency6. When expanded nucleotide repeats are transcribed into RNA, the repeat RNAs themselves can be toxic–for example, CUG/CCUG intronic/3’UTR RNA repeats in myotonic dystrophies7. These repeat RNAs have a propensity to condense and can trap important RNA-binding proteins in the process, impairing their function. Even though, these distinctions between coding and non-coding repeats seemed simple at first, the picture has become more complicated. The repeat RNA of the exonic CAG repeats can be just as toxic as the intronic CUG ones8. Expanded repeats also can generate antisense transcripts, implicating additional types of repeat RNAs and proteins9. Moreover, repeat RNAs (even non-coding ones) can be aberrantly translated in an AUG-independent manner (referred to as RAN translation)10, greatly expanding the range of proteins that can be encoded from these nucleotide repeats.
An archetype of this complexity is the repeat expansion in the C9orf72 gene. A hexanucleotide GGGGCC expansion in the first intron of a previously undescribed gene was found to be the leading genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)11,12. Upon the discovery of this repeat expansion, an immediate question was one of mechanism—how does this expansion drive human disease? Evidence has emerged linking all of the aforementioned mechanisms to C9orf72 pathogenesis. Hypermethylation of the expanded repeat affects transcription of the gene13, which seems to be implicated in autophagy14, vesicle trafficking15 and immune function16. Repeat RNA foci (both sense and antisense versions) accumulate in patient cells12,17. RAN translation of the sense and antisense repeat generates so-called dipeptide repeats (DPRs) that are detected in the brain and spinal cord of patients harboring this mutation17–19. These are poly-GA, poly-GP, poly-PA, poly-GR and poly-PR (abbreviated as their amino acid codes from here on). GP and PA do not seem to be toxic, whereas GA has mixed effects based on the model being used. The two arginine-rich DPRs, GR and PR, are potently toxic in diverse model systems—ranging from yeast to flies and worms, to vertebrates and iPSC-derived motor neurons20–23.
Because GR and PR seem to be so toxic, there have been many different studies investigating the mechanisms of this toxicity. These studies have implicated disturbances in nucleocytoplasmic transport24–26, defects in stress granules27,28, mitochondrial defects29, perturbation of axonal transport30, proteasome deficits31, and inhibition of translation28,32. All of these mechanisms could help explain the etiology of the disease, which is undoubtedly complex, but we wondered how can two amino acids repeated head to tail cause such widespread cellular defects? In other words, why are PR and GR such promiscuous killers that seemingly hit many unrelated biochemical pathways at once?
Since their discovery a decade ago, the apparent “aspecificity” of PR and GR toxicity has confused the field. A unifying framework to explain the biological connection between their myriad effects and the observed target promiscuity has remained elusive. Here, we unexpectedly discover that the interactome of GR and PR is actually encoded in their biophysical behavior. We and others previously showed that PR and GR can undergo biomolecular condensation in vitro and target biomolecular condensates in cells22,27,28,33. Using new unbiased proteomics studies, we find that even the non-condensate effects of PR and GR can be explained by their “sticky” multivalency. Moreover, we find that nucleic acids are key modulators of their target space (i.e., the set of proteins that interact with GR and PR), explaining previously unresolved discrepancies between nuclear and cytoplasmic effects, and between disease models and patient material. Further, the recent identification of arginine-rich repeat peptides in at least three other degenerative conditions points at the emergence of a new class of repeat disorders centering around cationic peptides34–36, with potentially a shared pathophysiology.
Since our framework was able to connect pathological condensation events in a divergent set of biological pathways, we wondered whether we could harness it to predict novel physiological condensate proteins. Using machine learning approaches, we discovered that the promiscuous condensation-based toxicity of PR and GR is not limited to human disease but is an integral part of ancient natural anti-microbial defense systems and spider, scorpion, and rattlesnake venoms. Similar cationic peptides have evolved countless times independently throughout the tree of life, and all can drive aberrant condensation via electrostatic interactions. Finally, leveraging protein design, we show that a surplus of cationic charge is sufficient to turn an inert protein into a toxic killer peptide. Thus, a mechanism we name polycation poisoning is emerging as a powerful and unifying framework that guides our understanding of promiscuous killer peptides in health and disease, and may aid in the design of new generations of therapeutic bioactive peptides.
RESULTS
RNA concentration tunes the target space of arginine-rich dipeptides.
In addition to C9orf72 ALS/FTD, other diseases also exhibit the same or similar arginine-rich peptides (Fig. 1A). RAN translation of GGCCTG (NOP56) and AGAGGG (TAF1) repeats, linked to spinocerebellar ataxia type 36 (SCA36) and X-linked dystonia and parkinsonism (XDP), generate PR and GR DPRs, respectively34,35. QAGR tetrapeptide repeats are produced from the CCUG repeat in myotonic dystrophy type 2 (DM2)36. As we have shown previously for PR and GR, these QAGR peptides similarly condense in vitro when added to cell lysate (U2OS, Fig. 1B). We previously showed that PR and GR form liquid droplets in the test tube when combined with nucleic acids27, and these assemblies—or ones using other cationic peptides—have been since used as very simple in vitro models to study the biophysics of more complex RNA-centric liquid condensates in the cell37–40. Surprisingly, when we add these peptides to cell lysate, we observe the formation of irregular networked condensates (Fig. 1C, Fig. S1A)—more reminiscent of the pathological gel-like condensates found in neurodegenerative disease41,42. Adding additional RNA into the mixture (i.e., total yeast RNA), caused these assemblies to change morphology and acquire spherical shapes more similar to the liquid-like behavior of some physiological condensates (e.g., nuclear bodies like nucleoli43). Because the addition of RNA dramatically affected the appearance of these test tube assemblies, we wondered what this would mean for their protein composition. As expected, its negative charge allows RNA to compete with proteins in the cell lysate for binding to the positively charged PR peptide (Fig. 1D, Fig. S1B-D). Using unlabeled quantitative mass spectrometry, we dissected what this competition looks like at the level of individual proteins (Table S1). We calculated the relative solubility of proteins in the test tube after spinning down the PR-induced condensates—as a measure of condensate partitioning—and clustered proteins according to their respective solubility profile as a function of the added RNA concentration (Fig. 1E-F). Around 50% of proteins in the mixture never partitioned into PR condensates and remained soluble throughout the experiment (cluster 1; grey proteins). For those proteins that condense with PR, 25% (clusters 2–5; red proteins) were outcompeted by low concentrations of RNA; the other 25% (clusters 6–9; blue proteins) were only outcompeted by high RNA concentrations or partitioned more strongly into the pellet upon the addition of RNA. Of note, this blue behavior is strikingly similar to the RNA-dependent re-entrant condensation behavior that has been observed for several RNA-binding proteins38,44,45. Thus, these results demonstrate that PR-interacting proteins can have distinct RNA-dependent interaction profiles. Interestingly, these three clusters—simply based on the in vitro condensation behavior of proteins with PR and RNA—each specifically enriched different functional categories of proteins (Fig. 1G). The blue class of proteins included canonical members of physiological condensates (e.g., nuclear speckles, nucleoli) or cellular assemblies that have been proposed to form via similar biophysical principles (i.e., heterochromatin46,47) (Fig. S2). Red proteins consisted of proteins not typically associated with physiological condensates but that have been implicated in protein aggregation or its modulation (Fig. 1H, Fig. S1E-H). Consistent with these findings, several of these distinct biological pathways—both red and blue—have been directly implicated in the toxicity observed in C9orf72 disease models and/or patients. These include nuclear import receptors24,48, protein arginine methyl transferases24,49,50, microtubules30, the proteasome31, the translational machinery28,32,51–53, splicing factors28,54, nucleoli27,28,55,56, and heterochromatin57, among others. Thus, our simple test tube model recapitulates the target promiscuity of arginine-rich DPRs and provides a framework to understand how local RNA concentration modulates this target space, with important implications for pathophysiology (see Discussion).
Figure 1: RNA modulates protein composition of ex vivo PR30 condensates.

(A) Scheme indicating toxicity of cationic RAN peptides. (B) Cationic RAN peptides drive condensation of U2OS cell lysate. Heatmap showing quantification of RNA-dependent solubility for 1324 proteins. (C) Morphology or PR30 condensates is modulated by exogenous RNA addition (2 µg/µl RNA). (D) Silver stain indicating differential protein composition of PR30-induced condensates in response to RNA addition. (E) Heatmap showing quantification of RNA-dependent solubility for 1324 proteins. (F) Proteins cluster according to solubility responses as a function of RNA concentration. % indicates fraction of total proteins in the cluster. (G) Clusters differentially enrich for GO terms relating to subcellular compartments. Font size = −log(p-value). (H) Overview indicating that the morphology and composition of PR30 condensates are a function of the ratio of RNA concentration over basic protein concentration. All of the highlighted protein assemblies and factors have been directly implicated in C9orf72 ALS/FTD pathogenesis. 3D reconstructions of PR condensates were obtained via soft x-ray tomography. See also Fig. S1.
RNA-dependent behavior is encoded in sequence and follows an electrostatics framework.
PR and GR’s apparent target promiscuity has remained enigmatic; how are they able to target such a wide range of unrelated biological pathways? Because we now have in hand an in vitro model of this target space, we could ask whether this behavior and its modulation by RNA are encoded by a protein’s sequence. First, overlapping our data with a set of experimentally validated RNA-binding proteins58 showed that, as expected, blue proteins are enriched for RNA-binding activity compared to red and grey proteins (Fig. 2A). Second, based on amino acid composition, blue and red proteins separate based on their charge (Fig. 2A-B); blue proteins are on average more basic, red ones more acidic, and grey ones more neutral. We can even observe this effect when looking within protein families. 60S ribosomal proteins are on average more positively charged than the ones making up the 40S subunit, and this correlated with lower solubility profiles in our mass spectrometry experiment (Fig. 2C).
Figure 2: Electrostatic charge is a sequence feature modulating PR30 condensate partitioning.

(A) Blue proteins are enriched for experimentally validated RNA-binding proteins and cationic amino acids. Red proteins have higher acidic amino acid composition. Grey proteins are enriched for neither. (B) On average, red proteins are more acidic and blue proteins are more basic compared to grey proteins (left). Moreover, on average, blue proteins have more clusters of basic residues in their IDRs (middle), while red proteins have more clusters of acidic residues in their folded domains (right) compared to grey proteins Kruskal-Wallis. (C) 40S and 60S ribosomal subunits show differential solubility profiles that correlate with changes in average pI. Two-way ANOVA and Mann-Whitney. (D) Positive charge clustering on the IDRs of DDX helicases correlates with their partitioning response to RNA (change in solubility upon addition of 0.4 µg/µl RNA. Linear regression. (E) Two example folded proteins with mirrored convex-concave solubility curves show opposite surface charge clustering (NAID = negative average inverse distance) and pI. Diablo (PDB: 1FEW)108, SnrpD2 (PDB: 1B34)109. See also Fig. S3-4. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001, **** p-value < 0.0001.
While average charge can be informative, how charged residues are distributed across intrinsically disordered regions (IDRs) or on the surface of folded domains can substantially impact molecular interactions59–62. Therefore, we co-opted and extended our previously applied sequence clustering analysis63 to evaluate the position of acidic and basic residues in both IDRs and on the surface of neighboring fold domains (in linear space and three-dimensional space, respectively) (Fig. S3). By integrating proteome-wide structural predictions, this analysis revealed that—on average—blue proteins possess clusters of basic residues within IDRs, while red proteins possess clusters of acidic residues on the surface of folded domains (Fig. 2B, Fig. S4). We highlight two examples to further illustrate these observations. First, DEAD-box helicases (DDX) are a family of RNA chaperones that share the same type of folded domain typically flanked by IDRs. We identified red, blue, and grey DDX proteins in our assay, and, importantly, the positive charge clustering of their respective IDRs dictates their RNA-dependent condensation (Fig. 2D, Fig. S1G-H). Second, we identified two proteins that had virtually mirrored solubility profiles. SnrpD2 (a spliceosome component) shows very little acidic clustering on its folded domain but has a strongly basic pI, while Diablo (a mitochondrial protein) shows specific subregions with clusters of acidic residues (Fig. 2E). Despite having the same initial and final solubility in our mass spectrometry experiment, these proteins show opposite RNA-dependence, in line with their respective charge profiles. These analyses indicate that the differential protein-partitioning we observed in our mass spec experiment—on average—follows simple rules relating to the density and positioning of acidic and basic residues that tune protein-protein and protein-RNA interactions. We stress that these analyses report on average properties. Charge or the ability to bind RNA is predictive, but there are many examples of proteins that enrich differently. This argues that other—yet unresolved and probably more complex—sequence features and protein-protein interactions modulate protein partitioning in our assay.
Generating a machine learning algorithm to predict protein condensation.
Because the condensate partitioning behavior in our mass spectrometry experiment correlated with certain sequence features, we reasoned that we could in principle use our experimental data set to learn to predict such behavior de novo. We trained a machine-learning algorithm64 (see Material & Methods) to predict blue proteins from their red and grey counterparts (Fig. 3A). Next, we asked the algorithm to rank intracellular proteins based on their “color” (Fig. 3B, Table S2). As expected, top predicted blue proteins enriched for gene ontology terms associated with nuclear bodies, RNA and chromatin metabolism (Fig. 3C). Interestingly, the algorithm also enriched intermediate filament proteins, which have been shown previously to engage with arginine-rich DPRs via their disordered head domains33. None of these condensation-related gene ontology terms came up for the red and grey class of proteins nominated by the algorithm.
Figure 3: Machine learning predicts RNA-modulated phase separation of uncharacterized proteins.

(A) Scheme illustrating workflow to predict red (concave) vs blue (convex) proteins. (B) The intracellular proteome ranked according to normalized confidence scores (Z score). (C) GO categories associated with predicted red and blue proteins. (D) Scheme highlighting breakdown of top 10 predicted blue unknown proteins. (E) 9 out of 10 predicted blue proteins target membraneless organelles. 8 do so in a manner that is dependent on nuclear or cytoplasmic RNA availability (2 µg/ml actinomycin D inhibits all transcription, 0.5 mM arsenite induces polysome disassembly and increases free cytoplasmic RNA levels). White arrowhead indicates nucleolar stress cap targeting of C3orf49. C9orf85 targets nucleoli and stress granules upon arsenite treatment. C19orf47 demixes from nuclear speckles upon transcription inhibition. C4orf50 condenses into nuclear bodies upon arsenite treatment. C20orf96 forms cytoplasmic bodies unresponsive to changes in RNA availability. C4orf54 binds the actin cytoskeleton (see Fig. S5).
Given blue proteins appear to consistently be associated with intracellular condensates, we decided to directly test our algorithm’s ability to predict condensation behavior for previously uncharacterized proteins. We tested the top ten uncharacterized proteins from our predicted blue protein list and expressed them in cells (Fig. 3D, Table S2). Seven of these formed or targeted condensates under normal conditions: C3orf49, C11orf98, C16orf87, C18orf21, and C19orf53 targeted nucleoli, C19orf47 localized to nuclear speckles, and C20orf96 formed unknown cytoplasmic condensates. This result clearly demonstrates that our machine-learning model can predict cellular and biophysical behavior. Since our initial mass spectrometry experiment investigated condensation as a function of RNA concentration, we wondered how changing RNA availability would influence the behavior of our seven condensate-forming proteins. To do this, we decided to stress cells in two ways to change nuclear and cytoplasmic RNA availability–transcriptional shutdown and polysome disassembly. First, we shut down all transcription with a high dose of actinomycin D65. This made four out of five of the nucleolar proteins demix from the nucleolus into the nucleoplasm—a well-known response of several nucleolar proteins66,67. C3orf49 also showed localization to the nucleolar periphery in assemblies reminiscent of nucleolar stress caps68. The nuclear speckle protein C19orf47 remained in a condensed state upon transcription inhibition, but demixed from the nuclear speckle, showing that its condensation behavior was also modulated by RNA. Second, stressing the cells with arsenite—known to cause polysome disassembly in the cytoplasm and rewire transcription in the nucleus69—made C9orf85 partition into stress granules. Peculiarly, in addition to stress granule partitioning, C9orf85 also translocated into nucleoli in parallel. C4orf50, previously found in the nucleoplasm, now formed abundant small nuclear granules that were clearly distinct from nuclear speckles. Transcription inhibition or arsenite stress did not affect C20orf96 condensates. Finally, C4orf54 localized to the actin cytoskeleton under all tested conditions (Fig. S4).
Combined, our algorithm predicted nine out of ten proteins correctly as condensate proteins, of which eight did so in a manner that was modulated by RNA availability. The only “incorrect” prediction, turns out to be a novel actin-binding protein. Interestingly, we and others have previously found that PR and GR can interact with actin50, microtubules30, and intermediate filaments33—highlighting that there may be more generally shared sequence features between cytoskeleton-binding and condensate proteins. Future studies will be required to investigate the functions of these new proteins and the functional significance of their condensate formation. In all, these findings indicate that our algorithm can predict condensate formation properties of proteins with high accuracy, allowing us and others to start systematically interrogating diverse proteomes.
Condensation is a novel mode of action of bioactive killer peptides.
In the above analysis, we restricted our search for novel condensate proteins to intracellular proteins since biomolecular condensates have been almost exclusively studied within the confines of the plasma membrane. But given the accuracy of our prediction tool, we reasoned that we could explore the possible existence of condensation events outside of the cellular context. When we fed sequences of secreted proteins into the algorithm, we unexpectedly found that it predicted several antimicrobial peptides (AMPs) to condense (Fig. 4A, Table S2). Animals, including humans, have evolved a wide array of AMPs70. A well-studied example is LL-37 (Fig. 4B). Some cationic AMPs have been shown to kill bacteria by permeabilizing their membranes, but for LL-37 and several other AMPs this does not seem to be their main mode of action71. Because of their positive charge, such peptides are expected to interact with membranes, hence, potentially crossing them. Indeed, cationic peptides are often called cell-penetrant since they are spontaneously taken up by both prokaryote and eukaryote cells72. This raised the question: what would happen once a cationic peptide like LL-37 enters the bacterial cytoplasm? To answer this, we first performed a similar ex vivo lysate assay as described above. Adding LL-37, or PR as a positive control, to E. coli lysate was able to potently drive condensate formation (Fig. 4C). Second, we treated live bacteria with LL-37 at a concentration corresponding to its minimum inhibitory concentration against a range of pathogens73. We subsequently imaged these bacteria using soft x-ray tomography, allowing us to gain information on their subcellular organization (Fig. 4D, Fig. S5A). LL-37 treated bacteria showed compacted nucleoids and evidence for the formation of dense cytoplasmic structures (Fig. 4E). Because DNA and ribosomes are the main components of the bacterial nucleoid and cytoplasm, respectively, we tested whether LL-37 could condense these biomolecules in the test tube; it did (Fig. 4F). Thus, we suggest that the intracellular condensation caused by LL-37 is responsible for its bactericidal activity. Since LL-37 condenses with negatively charged biological structures, similar to PR, this condensation can be described according to the framework of so-called complex coacervation—originally conceptualized by the polymer physics field to describe the condensation of oppositely charged (bio)polymers74,75.
Figure 4: Killer peptides drive coacervation of cellular components and target biomolecular condensates.

(A) The intracellular + secreted proteome ranked according to normalized confidence scores (Z score). GO categories associated with predicted red and blue proteins. (B) Scheme illustrating LL-37 as an AMP. (C) PR20 and LL-37 induce condensation of E. coli lysate. (D) Soft x-ray tomography of sham and LL-37-treated E. coli cells. n = nucleoid, c = cytoplasm. (E) Quantification of nucleoid compaction and cytoplasmic condensation. Mann-Whitney. ** p-value < 0.01, **** p-value < 0.0001. See also Fig. S6. (F) LL-37 condenses both E. coli ribosomes and plasmid DNA. (G) Positive charge is a common feature among AMPs. (H-I) Examples of natural AMPs and venom peptides that induce cell lysate condensation. (J) Crotamine and PR30 (500 nM) target endogenous biomolecular condensates in human cells.
Is LL-37 a sole example of such a mechanism, or could this be more widespread? We analyzed thousands of curated AMPs76, revealing that positive charge is a common feature among them (Fig. 4G). We tested a set of cationic AMPs from various vertebrates and invertebrates and found that several induce condensation in our ex vivo assay (Fig. 4H). These results support our emerging hypothesis that positively charged peptides could offer broad-spectrum antibiotic functionality through aberrant condensation of essential cellular components.
While AMPs target microbes, nature has also evolved biochemical tools to target large multicellular organisms in the form of venoms. Although several well-known (paralytic) venom peptides have a clear mode of action—usually specifically binding and modulating ion channels—venoms are complex mixtures containing a range of other peptides. Among them, so-called necrotic peptides are cationic and seem mainly responsible for tissue damage and cell death77. Indeed, we found that cationic peptides from the venoms of scorpions, spiders, and rattlesnakes drive condensation in our ex vivo lysate assay (Fig. 4I). These results suggest that they have the biophysical capacity to condense, but do they exhibit this behavior in cells? Crotamine is a well-studied cell-penetrant rattlesnake peptide whose mode of action is incompletely resolved78. We added fluorescently-labeled crotamine to the medium of human cells (Fig. 4J). As a control, we added fluorescently labeled PR, which has been documented to traffic to nucleoli55,79. PR targeted the granular center of the nucleolus, consistent with other reports28,56, while crotamine targeted nucleolar subcompartments. PR did not target nuclear speckles but crotamine did. Both these potent killer peptides—one aberrantly produced from an expanded repeat in disease, one evolved in a venomous animal—similarly targeted RNA-rich nuclear bodies. Yet despite these similarities, distinct structural and sequence features determined condensate partitioning, providing evidence that condensate target specificity can be encoded in the sequences of even small peptides.
Cationic charge is necessary and sufficient for killer activity.
Both the arginine-rich repeat peptides in human disease and the natural killer peptides have one main feature in common: positive charge. Could it be that charge is necessary and sufficient for killer activity? To address the first question, we looked more deeply at the specific amino acid enrichments we had seen above. While AMPs are enriched for arginine- and lysine-rich sequences, we did not see any enrichment for histidine (Fig. 4G)76. This observation is not unexpected, given histidine is expected to be largely neutral at a pH of 7.0 – 7.4. Nonetheless, two of our top ten secreted predicted blue proteins belonged to the histatin family—a histidine-rich group of AMPs (Fig. S5B). When tested in the same assay as LL-37, histatin-3 (HTN3) did not induce condensation in our ex vivo assay. However, histatins are expressed specifically in saliva, which is slightly acidic (≈ pH 6.5). Switching assay conditions from pH 7.5 to pH 6.5—coinciding with a net charge gain of +3—drove the strong condensation of HTN3 in E. coli lysate. This finding provides evidence that cationic charge is required for condensation, and is consistent with the enhanced bactericidal activity of histatins under mildly acidic conditions first observed three decades ago80. Thus, evolution seems to have used histidine residues to generate pH-dependent condensation switches that tune the activity of AMPs to their respective biofluid of origin.
Our work thus far suggests a net positive charge is necessary for toxicity of numerous AMPs, but is it sufficient? In other words, can we make a non-toxic protein toxic simply by altering its charge? GFP is probably the best-studied protein, with predictable behavior and folding, and is generally non-toxic to cells. By mutating residues facing out from the β-barrel, one can generate GFP mutants that still fold properly (i.e., remain fluorescent) but have an altered surface charge (Fig. 5A)81. When we expressed a library of super folder GFP (sfGFP) variants—with net charges ranging from −24 to +24—in E. coli, we observed that cationic GFP variants caused bacterial growth arrest (Fig. 5B). Imaging these cells, we observed that the cationic GFP variants, but not the anionic ones, formed aberrant condensates with ribosomal proteins (Fig. 5C) and RNA82 at the bacterial cell pole. Similarly, when we combined recombinantly purified GFP variants with E. coli ribosomes in the test tube, only the cationic ones formed condensates (Fig. 5D). Thus, like we saw for LL-37, these GFP variants can drive charge-based coacervation of ribosomes—killing the cells.
Figure 5: Net positive charge is sufficient for protein toxicity.
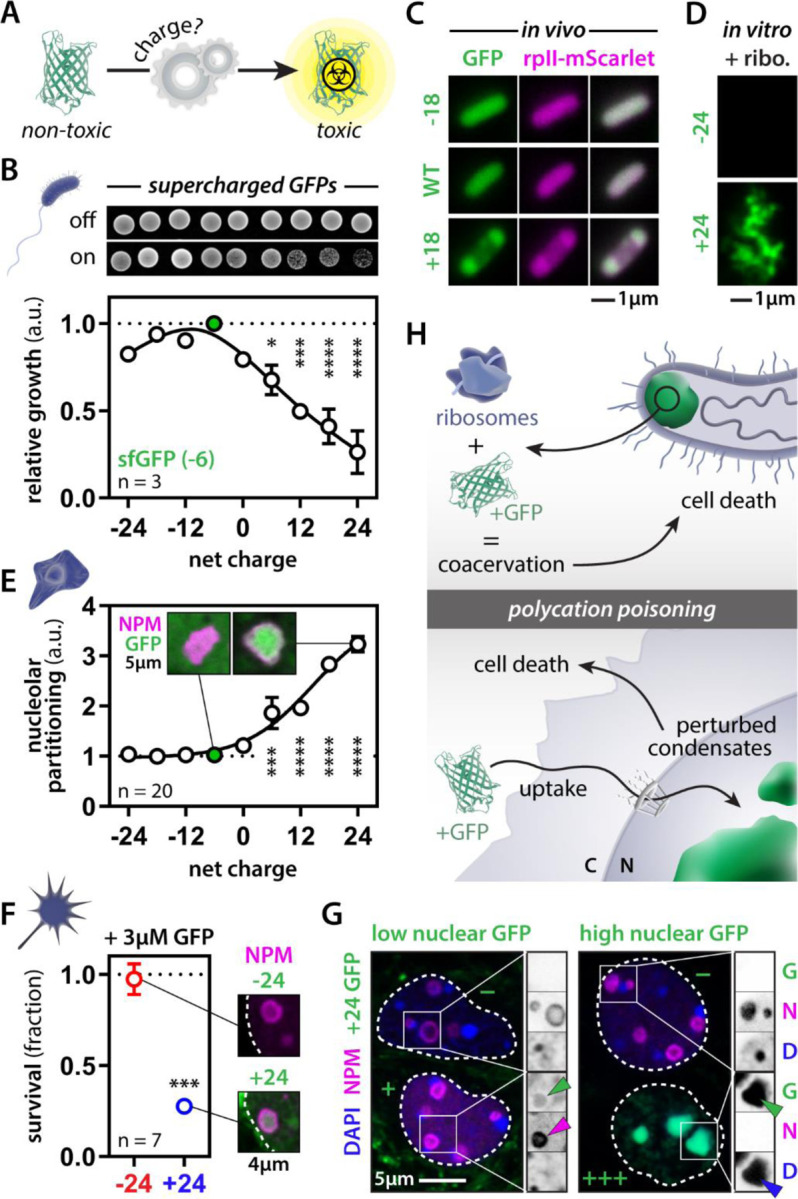
(A) Engineering supercharged GFP versions for toxicity assays. (B) Expression of cationic GFP variants drives toxicity in E. coli. One-way ANOVA. (C-D) Cationic GFPs condenses with ribosomal proteins in E. coli and the test tube. (E) GFP partitions into nucleoli of U2OS cells in a positive charge-dependent manner. One-way ANOVA. (F) Exogenous recombinant cationic GFP is taken up, targets nucleoli and kills mouse cortical neurons in the dish. Student’s t-test. (G) Cells with low levels of GFP (+) have a normal nuclear organization, whereas high levels (+++) perturb nucleoli and heterochromatin. Adjacent cells that did not take up GFP (−) are normal. (H) Scheme depicting polycation poisoning in pro- and eukaryote systems. * p-value < 0.05, *** p-value < 0.001, **** p-value < 0.0001.
We next tested the effect of these GFP variants in human cells. The cationic GFP variants spontaneously condensed in nucleoli and nuclear speckles in a charge-dependent fashion (Fig. 5E). Given cationic peptides often undergo spontaneous cellular entry, we wondered if our synthetic positively charged GFPs might also be sufficient for cellular uptake. To test this, we also added recombinant anionic and cationic GFP to the medium of cultured mouse cortical neurons. Virtually identical to our observations with PR and crotamine, cationic GFP was spontaneously taken up by cells and targeted the nucleolus, killing the neurons (Fig. 5F). Moreover, this cationic version of GFP did so at concentrations similar to or lower than the ones we and others have found GR to be toxic49,55,79. Importantly, this observation challenges that canonical expectation that cell-penetrant peptides must have evolved a specific and highly constrained mechanism for cellular uptake, instead implicating cellular entry as being driven in part if not entirely by a strong net positive charge. Finally, neuronal uptake of cationic GFP was not uniform; some cells rapidly accumulated large quantities of GFP, whereas in others the accumulation was slower or even absent. Note, this same heterogeneity has been observed for other cell-penetrant peptides as well83–85 and is linked to cellular tolerance against their toxic effects83. Cells with low levels of nuclear GFP showed nucleolar localization as expected (Fig. 5G). Surprisingly, cells with high levels of GFP had non-detectable nucleoli (based on NPM staining) and showed marked condensation of DNA. The oversaturation of the nuclear compartment with these charged molecules seems to upset the physiological balance of electrostatic interactions, dramatically perturbing nuclear organization and leading to the demise of the cell (Fig. 5H).
Taken together, our results suggest that positively charged peptides and proteins offer broad-scale cellular toxicity. In some cases, this toxicity is an evolved function, while in others, it may be an unfortunate mishap through spontaneous repeat expansions that underlie human disease. Our results demonstrate that while precise target selectivity can clearly be encoded in a killer peptide’s sequence (e.g., crotamine vs PR), we conclude that cationic charge is a unifying sequence feature that is sufficient to drive killer activity. This process, which we name polycation poisoning, follows the simple framework of complex coacervation and has independently evolved countless times across the tree of life.
DISCUSSION
For a decade, the observed target promiscuity of C9orf72 arginine-rich DPRs has puzzled the field. Compelling data has connected their toxicity to a bafflingly diverse array of biological pathways20–22. But a unifying framework has so far remained elusive. Were these studies monitoring the cause of cell death or simply the downstream consequences of dying cells? We and others had previously shown that these peptides can condense RNA and other biomolecules, explaining their effect on physiological condensates22,27,28,33. Here, we used proteomics to explore the target space of cationic dipeptide repeats and unexpectedly found that even their non-condensate targets fit into a simple electrostatics-based framework of complex coacervation. We propose that key actors in all these different biological pathways share the right sequence features that drive their aberrant interaction with these positively charged peptides.
Our framework also allows us to address the most common criticism against the pathological role for arginine-rich DPRs in patients. In every model system tested so far, PR—and to a lesser extent GR—targets the nucleolus, making it potently toxic. Yet, this has never been observed in any pathology report using patient post-mortem material. For reasons that are currently unresolved, but a topic of intense study, both PR and GR seem to get trapped in the cytoplasmic GA inclusions in patients, thus preventing them from entering the nucleus17–19. Our results unambiguously demonstrate that RNA tunes the target space of PR (Fig. 1). As such, which proteins PR (and, by extension GR) interact with within the cell will depend on the local RNA concentration around those DPRs. The concentration of RNA in the nucleus is estimated to be 36 times higher than the cytoplasm44, where most RNA is also trapped in polysomes. As such, our test tube experiments provide lysate mimics of either a nucleus-like (high RNA) or cytoplasm-like (low RNA) environment, enabling us to dissect which proteins are PR targets in these two compartments. Under high RNA conditions, we observe PR interacting with proteins associated with nuclear bodies and chromatin. This mirrors the subcellular localization observed in model systems, where PR interacts with nucleoli27,28,55,56 and heterochromatin57. Under low RNA concentrations, PR interacts with the translation initiation complex, ribosomes, axonal transport factors, and protein arginine methyl transferases. Analysis of patient post-mortem samples provides evidence that these kinds of proteins do associate with cytoplasmic GR/PR inclusions in patients24,30,49,52,53. Although defects in condensation seemed initially limited to disease models that failed to fully recapitulate human pathology, we now show that the same framework of electrostatic condensation is still applicable but is tuned by the local RNA concentration. To our knowledge, this is the first model that is able to explain a decade of disconnect between most C9orf72 disease model-based studies and human pathology reports.
Pathogenesis does not exist in a vacuum, but instead follows already-established biochemical principles. Can we use a framework designed to understand pathology to infer novel functional biology? We used our proteomics dataset to train a machine learning algorithm to detect similar electrostatics-based condensation behavior. When tested on the human proteome, we found already known condensate proteins but, importantly, also predicted this behavior in completely uncharacterized proteins. We experimentally validated this condensate behavior in nine out of the ten predicted proteins—showing that our approach allows for the discovery of new biology. The complete functional characterization of this set of novel condensate proteins is beyond the scope of this study, but we note that some of these proteins have substantial phenotypic effects in high-throughput knock-out studies86, arguing that their yet unknown function may carry cellular importance. It will be interesting to test if their condensation propensity is involved in their function, and whether they are implicated in human disease.
The (re)discovery of biomolecular condensation as an organizing principle of the cell besides membranes has revolutionized cell biology87,88. Ironically though, our exploitation of these insights has typically remained delimited by the plasma membrane. With our sequence-based condensate protein predictor in hand, we could now test for the condensation properties of extracellular proteins in a direct and unbiased way. To our initial surprise, we discovered that cationic killer peptides found in innate immune systems and venom cocktails are potent drivers of condensation, following the exact same set of rules we delineated for the human disease repeat peptides. Not impeded by GA inclusions like PR, venom peptides are able to gain entry to mammalian cells and readily target the nuclear centers of RNA production. AMPs condense both the nucleoid and ribosomes in bacteria. Our results demonstrate that two very different cellular systems (eukaryotes versus prokaryotes) with completely unrelated peptides follow the exact same framework of polycation poisoning. Indeed, using GFP as a scaffold we can engineer killer activity by simply making it positively charged, with its toxicity in neuronal culture rivaling that of the neurotoxic GR and PR peptides.
One question remains though: why positive and not negative charge? Although we do not rule out the existence of polyanion-based toxicity, there are some simple reasons why cationic peptides could be more disruptive. If we consider the cell as a collection of lipids, nucleic acids and proteins, a clear trend emerges. Lipids tend to have negatively charged head domains, nucleic acids are inherently anionic, but proteins can span the entire charge spectrum. Given all the negative charge on lipids, nucleic acids, and even the cytoskeleton (both actin filaments and microtubules have negatively charged surfaces), a cationic-biased proteome would promiscuously interact with all of these exposed biomolecular surfaces. Hence, it may come as no surprise that for organisms living at mesophilic pH and salt concentrations, proteomes tend to be—on average—more negatively charged89,90. Yet, we know many examples of highly cationic proteins in the cell, raising the question of how cells protect themselves from their own. By analyzing six model organisms, we indeed find no evidence for selection against positively charged proteins (Supplemental text, Fig. S7), despite our data suggesting that polycationic folded and disordered proteins drive cellular toxicity across the kingdoms of life. To better understand these seemingly contradictory observations, we performed an integrative bioinformatic analysis showing that while proteomes do possess many examples of proteins with a net positive charge, these are either expressed at low copy numbers, sequestered from the cellular environment via constitutive binding to anionic biomolecules, or neutralized through posttranslational modifications (Supplemental text, Fig. S8-S11, Table S3). Remarkably, we find that for essentially every single example of an abundant, positively charged protein, its charge is neutralized by a phosphate moiety—either in trans (nucleic acids or phospholipid headgroups) or in cis (through phosphorylation). We argue that this neutralization is essential to keep endogenous polycationic proteins in check to prevent them from poisoning the cell. Intriguingly, similar approaches are used by several human pathogens to neutralize cationic antimicrobial peptides and antibiotics91–93.
The above observations point at a seemingly universal proteome ‘design principle’. The net polarity for the large cellular surfaces ensures cellular proteomes do not aspecifically stick to lipids and nucleic acids. At the same time, it becomes relatively easy to evolve high-affinity domains that engage with these acidic moieties if needed. Cationic surface patches or disordered domains are pervasive interaction motifs in the proteome. For example, countless RNA binding proteins possess cationic disordered domains adjacent to their folded RNA binding domains27,94,95. Recent work has shown that even modest clusters of positively charged residues in IDRs from RNA binding proteins can have a profound impact on RNA binding affinity, mirroring our bioinformatic analyses96. Similarly, positively charged microtubule-binding proteins, such as tau, do so in part by interacting with the anionic tubulin tails that coat the microtubule lattice97. Membrane-binding proteins, like α-synuclein, do so via the exact same electrostatic interactions98. The use of cationic interaction and targeting motifs in a sea of anionic biomolecules is a powerful one but highlights the inherent weakness of cells against killer systems that corrupt this principle. It also shows why the cell has a variety of fail-safe mechanisms in place to prevent toxicity of its own cationic proteins. When we tested the cationic disordered domains of some human nucleic acid binding proteins in isolation, we found they act virtually identical to the killer peptides described in this study (Fig. S12). So how does the cell tame these sequences? Above we highlight the role that phosphorylation could have in regulating these proteins. Recent studies by us and others have also shown that these cationic domains are often paired with anionic or aromatic domains that can potently quench or regulate their activity99–103. Additionally, nuclear import factors that bind proteins translated in the cytoplasm via their nuclear localization sequence—usually cationic in nature—were recently shown to moonlight as bona fide protein chaperones104–107. Although philosophical, it is worth considering which function came first when transcription and translation became uncoupled during eukaryogenesis: chaperoning a condensation-prone protein in an RNA-poor environment (i.e., the cytosol) or translocating it to an RNA-rich environment (i.e., the nucleus)?
In this study, we provide a simple electrostatics-based framework that unifies the toxicity of cationic peptides in human disease and killer peptides in biology. McKnight and co-workers speculated that RAN translation of the C9orf72 repeat could be considered the “failed birth of a gene”55. Upon repeat expansion, a non-coding part of the genome all of a sudden becomes protein-coding. Yet, the gene product is toxic to the cell producing it. When venomous animals evolved their killer peptides, a requirement was that the venom did not kill its host. Unsurprisingly, venom peptide production is a tightly regulated process often involving extracellular processing and cleavage of quenching domains together with signal peptides correctly targeting the protein for secretion. Note that in the lumen of a secretory vesicle these peptides never “see” the cytoplasm of the cell they are produced in. With this in mind, one can only wonder whether we have witnessed the failed birth of a venom peptide that lacked its regulatory sequences.
Supplementary Material
ACKNOWLEDGEMENTS:
We thank all members of the Gitler lab, the Boeynaems lab, the Carnegie-Stanford Intrinsically Disordered Protein Scientific Interest Group (IDPSIG), IDPseminars, and FNZ for helpful discussion and suggestions. We are indebted to Dr. Bede Portz for providing invaluable feedback and insights that helped shape this manuscript. We thank the Stanford Neuroscience Microscopy Service for use of the core facility. Some of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. We thank members of the Water and Life Interface Institute (WALII), supported by NSF DBI grant # 2213983, for helpful discussions. We thank Dr. Richard Kriwacki and Dr. Aaron Phillips (St. Jude Children’s Research Hospital) for kindly providing us with recombinant NPM1.
FUNDING:
Work in the A.D.G. lab is supported by NIH (grant R35NS097263). A.D.G. is a Chan Zuckerberg Biohub Investigator. S.B. acknowledges an EMBO Long Term Fellowship. Work in the S.B. lab is supported by CPRIT (RR220094) and NSF (DBI # 2213983, WALII). The Stanford Neuroscience Microscopy Service is supported by NIH (grant NS069375). M.A.F.H. is supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo, 2022/00527–8; 2020/01107–7; 2019/13112–8), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – 001) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico No. 39337/2016–0). The National Center for X-ray Tomography is supported by NIH NIGMS (P41GM103445, P30GM138441) and the DOE’s Office of Biological and Environmental Research (DE-AC025CH11231). V.Y. and A.C.O. acknowledge an NSF CAREER award (DMR #1848388) for support. A.S.H. acknowledges support from the Longer Life Foundation, an RGA/Washington University collaboration.
Footnotes
DECLARATION OF INTERESTS:
A.D.G has served as a consultant for Aquinnah Pharmaceuticals, Prevail Therapeutics, and Third Rock Ventures and is a scientific founder of Maze Therapeutics. A.C.O. is a co-founder of Werewool. All other authors declare no competing interests. A.S.H. is a scientific consultant for Dewpoint Therapeutics and on the Scientific Advisory Board for Prose Foods.
REFERENCES
- 1.Malik I., Kelley C.P., Wang E.T., and Todd P.K. (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Depienne C., and Mandel J.-L. (2021). 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieberman A.P., Shakkottai V.G., and Albin R.L. (2019). Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol. 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boivin M., and Charlet-Berguerand N. (2022). Trinucleotide CGG repeat diseases: An expanding field of polyglycine proteins? Front. Genet. 13, 843014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes J.N., and Thomas P.Q. (2013). Molecular pathology of polyalanine expansion disorders: new perspectives from mouse models. Methods Mol. Biol. 1017, 135–151. [DOI] [PubMed] [Google Scholar]
- 6.Hagerman R.J., Berry-Kravis E., Hazlett H.C., Bailey D.B., Moine H., Kooy R.F., Tassone F., Gantois I., Sonenberg N., Mandel J.L., et al. (2017). Fragile X syndrome. Nature Reviews Disease Primers 3, 1–19. [DOI] [PubMed] [Google Scholar]
- 7.Lanni S., and Pearson C.E. (2019). Molecular genetics of congenital myotonic dystrophy. Neurobiol. Dis. 132, 104533. [DOI] [PubMed] [Google Scholar]
- 8.Li L.-B., Yu Z., Teng X., and Bonini N.M. (2008). RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro A.F., Loureiro J.R., Bessa J., and Silveira I. (2020). Antisense transcription across nucleotide repeat expansions in neurodegenerative and neuromuscular diseases: Progress and mysteries. Genes (Basel) 11, 1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cleary J.D., Pattamatta A., and Ranum L.P.W. (2018). Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 293, 16127–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu E.Y., Russ J., Wu K., Neal D., Suh E., McNally A.G., Irwin D.J., Van Deerlin V.M., and Lee E.B. (2014). C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 128, 525–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boivin M., Pfister V., Gaucherot A., Ruffenach F., Negroni L., Sellier C., and Charlet-Berguerand N. (2020). Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39, e100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Y., Lin S., Staats K.A., Li Y., Chang W.-H., Hung S.-T., Hendricks E., Linares G.R., Wang Y., Son E.Y., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burberry A., Suzuki N., Wang J.-Y., Moccia R., Mordes D.A., Stewart M.H., Suzuki-Uematsu S., Ghosh S., Singh A., Merkle F.T., et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8, 347ra93–347ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zu T., Liu Y., Bañez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T.M., Harms M.B., Falchook A.E., Subramony S.H., et al. (2013). RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U. S. A. 110, E4968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ash P.E.A., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.-L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W. 3rd, Rademakers R., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori K., Weng S.-M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz A., Pinheiro Marques J., Oertig I., Maharjan N., and Saxena S. (2021). Emerging perspectives on dipeptide repeat proteins in C9ORF72 ALS/FTD. Front. Cell. Neurosci. 15, 637548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freibaum B.D., and Taylor J.P. (2017). The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Odeh H.M., and Shorter J. (2020). Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg. Top. Life Sci. 4, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O.C., Pietrzyk J., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boeynaems S., Bogaert E., Michiels E., Gijselinck I., Sieben A., Jovičić A., De Baets G., Scheveneels W., Steyaert J., Cuijt I., et al. (2016). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6, 20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jovičić A., Mertens J., Boeynaems S., Bogaert E., Chai N., Yamada S.B., Paul J.W. 3rd, Sun S., Herdy J.R., Bieri G., et al. (2015). Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freibaum B.D., Lu Y., Lopez-Gonzalez R., Kim N.C., Almeida S., Lee K.-H., Badders N., Valentine M., Miller B.L., Wong P.C., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boeynaems S., Bogaert E., Kovacs D., Konijnenberg A., Timmerman E., Volkov A., Guharoy M., De Decker M., Jaspers T., Ryan V.H., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell 65, 1044–1055.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K.-H., Zhang P., Kim H.J., Mitrea D.M., Sarkar M., Freibaum B.D., Cika J., Coughlin M., Messing J., Molliex A., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Gonzalez R., Lu Y., Gendron T.F., Karydas A., Tran H., Yang D., Petrucelli L., Miller B.L., Almeida S., and Gao F.-B. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fumagalli L., Young F.L., Boeynaems S., De Decker M., Mehta A.R., Swijsen A., Fazal R., Guo W., Moisse M., Beckers J., et al. (2021). C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 7, eabg3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta R., Lan M., Mojsilovic-Petrovic J., Choi W.H., Safren N., Barmada S., Lee M.J., and Kalb R. (2017). The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro 4, ENEURO.0249–16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loveland A.B., Svidritskiy E., Susorov D., Lee S., Park A., Zvornicanin S., Demo G., Gao F.-B., and Korostelev A.A. (2022). Ribosome inhibition by C9ORF72-ALS/FTD-associated poly-PR and poly-GR proteins revealed by cryo-EM. Nat. Commun. 13, 2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin Y., Mori E., Kato M., Xiang S., Wu L., Kwon I., and McKnight S.L. (2016). Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789–802.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Todd T.W., McEachin Z.T., Chew J., Burch A.R., Jansen-West K., Tong J., Yue M., Song Y., Castanedes-Casey M., Kurti A., et al. (2020). Hexanucleotide repeat expansions in c9FTD/ALS and SCA36 confer selective patterns of neurodegeneration in vivo. Cell Rep. 31, 107616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyes C.J., Asano K., Todd P.K., Klein C., and Rakovic A. (2022). Repeat-associated non-AUG translation of AGAGGG repeats that cause X-linked dystonia-parkinsonism. Mov. Disord. 10.1002/mds.29183. [DOI] [PubMed]
- 36.Zu T., Cleary J.D., Liu Y., Bañez-Coronel M., Bubenik J.L., Ayhan F., Ashizawa T., Xia G., Clark H.B., Yachnis A.T., et al. (2017). RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 95, 1292–1305.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boeynaems S., Holehouse A.S., Weinhardt V., Kovacs D., Van Lindt J., Larabell C., Van Den Bosch L., Das R., Tompa P.S., Pappu R.V., et al. (2019). Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc. Natl. Acad. Sci. U. S. A. 116, 7889–7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banerjee P.R., Milin A.N., Moosa M.M., Onuchic P.L., and Deniz A.A. (2017). Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed Engl. 56, 11354–11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alshareedah I., Moosa M.M., and Raju M. (2020). Phase transition of RNA− protein complexes into ordered hollow condensates. Proceedings of the. [DOI] [PMC free article] [PubMed]
- 40.Fisher R.S., and Elbaum-Garfinkle S. (2020). Tunable multiphase dynamics of arginine and lysine liquid condensates. Nat. Commun. 11, 4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu S., Hu J., Arogundade O.A., Goginashvili A., Vazquez-Sanchez S., Diedrich J.K., Gu J., Blum J., Oung S., Ye Q., et al. (2022). Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 24, 1378–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mann J.R., Gleixner A.M., Mauna J.C., Gomes E., DeChellis-Marks M.R., Needham P.G., Copley K.E., Hurtle B., Portz B., Pyles N.J., et al. (2019). RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brangwynne C.P., Mitchison T.J., and Hyman A.A. (2011). Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. U. S. A. 108, 4334–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maharana S., Wang J., Papadopoulos D.K., Richter D., Pozniakovsky A., Poser I., Bickle M., Rizk S., Guillén-Boixet J., Franzmann T.M., et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henninger J.E., Oksuz O., Shrinivas K., Sagi I., LeRoy G., Zheng M.M., Andrews J.O., Zamudio A.V., Lazaris C., Hannett N.M., et al. (2021). RNA-mediated feedback control of transcriptional condensates. Cell 184, 207–225.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., and Karpen G.H. (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibson B.A., Doolittle L.K., Schneider M.W.G., Jensen L.E., Gamarra N., Henry L., Gerlich D.W., Redding S., and Rosen M.K. (2019). Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayes L.R., Duan L., Bowen K., Kalab P., and Rothstein J.D. (2020). C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife 9. 10.7554/eLife.51685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gittings L.M., Boeynaems S., Lightwood D., Clargo A., Topia S., Nakayama L., Troakes C., Mann D.M.A., Gitler A.D., Lashley T., et al. (2020). Symmetric dimethylation of poly-GR correlates with disease duration in C9orf72 FTLD and ALS and reduces poly-GR phase separation and toxicity. Acta Neuropathol. 139, 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radwan M., Ang C.-S., Ormsby A.R., Cox D., Daly J.C., Reid G.E., and Hatters D.M. (2020). Arginine in C9ORF72 dipolypeptides mediates promiscuous proteome binding and multiple modes of toxicity. Mol. Cell. Proteomics 19, 640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moens T.G., Niccoli T., Wilson K.M., Atilano M.L., Birsa N., Gittings L.M., Holbling B.V., Dyson M.C., Thoeng A., Neeves J., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 137, 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y.-J., Gendron T.F., Ebbert M.T.W., O’Raw A.D., Yue M., Jansen-West K., Zhang X., Prudencio M., Chew J., Cook C.N., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hartmann H., Hornburg D., Czuppa M., Bader J., Michaelsen M., Farny D., Arzberger T., Mann M., Meissner F., and Edbauer D. (2018). Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 1, e201800070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yin S., Lopez-Gonzalez R., Kunz R.C., Gangopadhyay J., Borufka C., Gygi S.P., Gao F.-B., and Reed R. (2017). Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep. 19, 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwon I., Xiang S., Kato M., Wu L., Theodoropoulos P., Wang T., Kim J., Yun J., Xie Y., and McKnight S.L. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White M.R., Mitrea D.M., Zhang P., Stanley C.B., Cassidy D.E., Nourse A., Phillips A.H., Tolbert M., Taylor J.P., and Kriwacki R.W. (2019). C9orf72 poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol. Cell 74, 713–728.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y.-J., Guo L., Gonzales P.K., Gendron T.F., Wu Y., Jansen-West K., O’Raw A.D., Pickles S.R., Prudencio M., Carlomagno Y., et al. (2019). Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 363, eaav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- 59.Das R.K., and Pappu R.V. (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U. S. A. 110, 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Greig J.A., Nguyen T.A., Lee M., Holehouse A.S., Posey A.E., Pappu R.V., and Jedd G. (2020). Arginine-enriched mixed-charge domains provide cohesion for nuclear speckle condensation. Mol. Cell 77, 1237–1250.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lyons H., Veettil R.T., Pradhan P., Fornero C., De La Cruz N., Ito K., Eppert M., Roeder R.G., and Sabari B.R. (2023). Functional partitioning of transcriptional regulators by patterned charge blocks. Cell 186, 327–345.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lasker K., Boeynaems S., Lam V., Scholl D., Stainton E., Briner A., Jacquemyn M., Daelemans D., Deniz A., Villa E., et al. (2022). The material properties of a bacterial-derived biomolecular condensate tune biological function in natural and synthetic systems. Nat. Commun. 13, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holehouse A.S., Ginell G.M., Griffith D., and Böke E. (2021). Clustering of aromatic residues in prion-like domains can tune the formation, state, and organization of biomolecular condensates. Biochemistry 60, 3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alley E.C., Khimulya G., Biswas S., AlQuraishi M., and Church G.M. (2019). Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 16, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity? [DOI] [PMC free article] [PubMed]
- 66.Yung B.Y., Bor A.M., and Chan P.K. (1990). Short exposure to actinomycin D induces “reversible” translocation of protein B23 as well as “reversible” inhibition of cell growth and RNA synthesis in HeLa cells. Cancer Res. 50, 5987–5991. [PubMed] [Google Scholar]
- 67.Schmidt H.B., Jaafar Z.A., Wulff B.E., Rodencal J.J., Hong K., Aziz-Zanjani M.O., Jackson P.K., Leonetti M.D., Dixon S.J., Rohatgi R., et al. (2022). Oxaliplatin disrupts nucleolar function through biophysical disintegration. Cell Rep. 41, 111629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lafita-Navarro M.C., and Conacci-Sorrell M. (2023). Nucleolar stress: From development to cancer. Semin. Cell Dev. Biol. 136, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bernstam L., and Nriagu J. (2000). Molecular aspects of arsenic stress. J. Toxicol. Environ. Health B Crit. Rev. 3, 293–322. [DOI] [PubMed] [Google Scholar]
- 70.Mookherjee N., Anderson M.A., Haagsman H.P., and Davidson D.J. (2020). Antimicrobial host defence peptides: functions and clinical potential. Nat. Rev. Drug Discov. 19, 311–332. [DOI] [PubMed] [Google Scholar]
- 71.Chongsiriwatana N.P., Lin J.S., Kapoor R., Wetzler M., Rea J.A.C., Didwania M.K., Contag C.H., and Barron A.E. (2017). Intracellular biomass flocculation as a key mechanism of rapid bacterial killing by cationic, amphipathic antimicrobial peptides and peptoids. Sci. Rep. 7. 10.1038/s41598-017-16180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Copolovici D.M., Langel K., Eriste E., and Langel Ü. (2014). Cell-penetrating peptides: design, synthesis, and applications. ACS Nano 8, 1972–1994. [DOI] [PubMed] [Google Scholar]
- 73.Dürr U.H.N., Sudheendra U.S., and Ramamoorthy A. (2006). LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758, 1408–1425. [DOI] [PubMed] [Google Scholar]
- 74.Sing C.E., and Perry S.L. (2020). Recent progress in the science of complex coacervation. Soft Matter 16, 2885–2914. [DOI] [PubMed] [Google Scholar]
- 75.Rumyantsev A.M., Jackson N.E., and de Pablo J.J. (2021). Polyelectrolyte complex coacervates: Recent developments and new frontiers. Annu. Rev. Condens. Matter Phys. 12, 155–176. [Google Scholar]
- 76.Pirtskhalava M., Amstrong A.A., Grigolava M., Chubinidze M., Alimbarashvili E., Vishnepolsky B., Gabrielian A., Rosenthal A., Hurt D.E., and Tartakovsky M. (2021). DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 49, D288–D297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhutia S.K., and Maiti T.K. (2008). Targeting tumors with peptides from natural sources. Trends Biotechnol. 26, 210–217. [DOI] [PubMed] [Google Scholar]
- 78.Rádis-Baptista G., and Kerkis I. (2011). Crotamine, a small basic polypeptide myotoxin from rattlesnake venom with cell-penetrating properties. Curr. Pharm. Des. 17, 4351–4361. [DOI] [PubMed] [Google Scholar]
- 79.Kramer N.J., Haney M.S., Morgens D.W., Jovičić A., Couthouis J., Li A., Ousey J., Ma R., Bieri G., Tsui C.K., et al. (2018). CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 50, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu T., Levitz S.M., Diamond R.D., and Oppenheim F.G. (1991). Anticandidal activity of major human salivary histatins. Infect. Immun. 59, 2549–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lawrence M.S., Phillips K.J., and Liu D.R. (2007). Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 129, 10110–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yeong V., Werth E.G., Brown L.M., and Obermeyer A.C. (2020). Formation of biomolecular condensates in bacteria by tuning protein electrostatics. ACS Cent. Sci. 6, 2301–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Snoussi M., Talledo J.P., Del Rosario N.-A., Mohammadi S., Ha B.-Y., Košmrlj A., and Taheri-Araghi S. (2018). Heterogeneous absorption of antimicrobial peptide LL37 in Escherichia coli cells enhances population survivability. Elife 7. 10.7554/eLife.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Safa N., Vaithiyanathan M., Sombolestani S., Charles S., and Melvin A.T. (2019). Population-based analysis of cell-penetrating peptide uptake using a microfluidic droplet trapping array. Anal. Bioanal. Chem. 411, 2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R., and Brock R. (2007). A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8, 848–866. [DOI] [PubMed] [Google Scholar]
- 86.Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S., et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L., et al. (2018). Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 28, 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shin Y., and Brangwynne C.P. (2017). Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- 89.Schavemaker P.E., Śmigiel W.M., and Poolman B. (2017). Ribosome surface properties may impose limits on the nature of the cytoplasmic proteome. Elife 6. 10.7554/elife.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Requião R.D., Fernandes L., de Souza H.J.A., Rossetto S., Domitrovic T., and Palhano F.L. (2017). Protein charge distribution in proteomes and its impact on translation. PLoS Comput. Biol. 13, e1005549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lewenza S. (2013). Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front. Microbiol. 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson L., Horsman S.R., Charron-Mazenod L., Turnbull A.L., Mulcahy H., Surette M.G., and Lewenza S. (2013). Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 13, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rao N.N., Gómez-García M.R., and Kornberg A. (2009). Inorganic polyphosphate: essential for growth and survival. Annu. Rev. Biochem. 78, 605–647. [DOI] [PubMed] [Google Scholar]
- 94.Thandapani P., O’Connor T.R., Bailey T.L., and Richard S. (2013). Defining the RGG/RG motif. Mol. Cell 50, 613–623. [DOI] [PubMed] [Google Scholar]
- 95.Castello A., Fischer B., Frese C.K., Horos R., Alleaume A.-M., Foehr S., Curk T., Krijgsveld J., and Hentze M.W. (2016). Comprehensive identification of RNA-binding domains in human cells. Mol. Cell 63, 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cubuk J., Alston J.J., Incicco J.J., Holehouse A.S., Hall K.B., Stuchell-Brereton M.D., and Soranno A. (2023). The disordered N-terminal tail of SARS CoV-2 Nucleocapsid protein forms a dynamic complex with RNA. bioRxiv. 10.1101/2023.02.10.527914. [DOI] [PMC free article] [PubMed]
- 97.Fung H.Y.J., McKibben K.M., Ramirez J., Gupta K., and Rhoades E. (2020). Structural characterization of tau in fuzzy tau:Tubulin complexes. Structure 28, 378–384.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davidson W.S., Jonas A., Clayton D.F., and George J.M. (1998). Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- 99.Boeynaems S., Dorone Y., Marian A., Shabardina V., Huang G., Kim G., Sanyal A., Şen N.-E., Docampo R., Ruiz-Trillo I., et al. (2021). Poly(A)-binding protein is an ataxin-2 chaperone that emulsifies biomolecular condensates. bioRxiv, 2021.08.23.457426. 10.1101/2021.08.23.457426. [DOI] [PMC free article] [PubMed]
- 100.Wang J., Choi J.-M., Holehouse A.S., Lee H.O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D., et al. (2018). A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang P., Mathieu C., Kolaitis R.-M., Zhang P., Messing J., Yurtsever U., Yang Z., Wu J., Li Y., Pan Q., et al. (2020). G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guillén-Boixet J., Kopach A., Holehouse A.S., Wittmann S., Jahnel M., Schlüßler R., Kim K., Trussina I.R.E.A., Wang J., Mateju D., et al. (2020). RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 181, 346–361.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bogaert E., Boeynaems S., Kato M., Guo L., Caulfield T.R., Steyaert J., Scheveneels W., Wilmans N., Haeck W., Hersmus N., et al. (2018). Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 24, 529–537.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshizawa T., Ali R., Jiou J., Fung H.Y.J., Burke K.A., Kim S.J., Lin Y., Peeples W.B., Saltzberg D., Soniat M., et al. (2018). Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo L., Kim H.J., Wang H., Monaghan J., Freyermuth F., Sung J.C., O’Donovan K., Fare C.M., Diaz Z., Singh N., et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.-D., Simons M., Niessing D., Madl T., et al. (2018). Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13. [DOI] [PubMed] [Google Scholar]
- 107.Qamar S., Wang G., Randle S.J., Ruggeri F.S., Varela J.A., Lin J.Q., Phillips E.C., Miyashita A., Williams D., Ströhl F., et al. (2018). FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 173, 720–734.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chai J., Du C., Wu J.W., Kyin S., Wang X., and Shi Y. (2000). Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862. [DOI] [PubMed] [Google Scholar]
- 109.Kambach C., Walke S., Young R., Avis J.M., de la Fortelle E., Raker V.A., Lührmann R., Li J., and Nagai K. (1999). Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96, 375–387. [DOI] [PubMed] [Google Scholar]
- 110.Cummings C.S., and Obermeyer A.C. (2018). Phase separation behavior of supercharged proteins and polyelectrolytes. Biochemistry 57, 314–323. [DOI] [PubMed] [Google Scholar]
- 111.Boeynaems S., De Decker M., Tompa P., and Van Den Bosch L. (2017). Arginine-rich peptides can actively mediate liquid-liquid phase separation. Bio Protoc. 7, e2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guse A., Fuller C.J., and Straight A.F. (2012). A cell-free system for functional centromere and kinetochore assembly. Nat. Protoc. 7, 1847–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Westhorpe F.G., Fuller C.J., and Straight A.F. (2015). A cell-free CENP-A assembly system defines the chromatin requirements for centromere maintenance. J. Cell Biol. 209, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.French B.T., Westhorpe F.G., Limouse C., and Straight A.F. (2017). Xenopus laevis M18BP1 Directly Binds Existing CENP-A Nucleosomes to Promote Centromeric Chromatin Assembly. Dev. Cell 42, 190–199.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lowary P.T., and Widom J. (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42. [DOI] [PubMed] [Google Scholar]
- 116.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chai Q., Singh B., Peisker K., Metzendorf N., Ge X., Dasgupta S., and Sanyal S. (2014). Organization of ribosomes and nucleoids in Escherichia coli cells during growth and in quiescence. J. Biol. Chem. 289, 11342–11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kuhlman T.E., and Cox E.C. (2010). Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 38, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chiva C., Olivella R., Borràs E., Espadas G., Pastor O., Solé A., and Sabidó E. (2018). QCloud: A cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS One 13, e0189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weinhardt V., Chen J.-H., Ekman A.A., Guo J., Remesh S.G., Hammel M., McDermott G., Chao W., Oh S., Le Gros M.A., et al. (2020). Switchable resolution in soft x-ray tomography of single cells. PLoS One 15, e0227601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen J.-H., Vanslembrouck B., Loconte V., Ekman A., Cortese M., Bartenschlager R., McDermott G., Larabell C.A., Le Gros M.A., and Weinhardt V. (2022). A protocol for full-rotation soft X-ray tomography of single cells. STAR Protoc. 3, 101176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Parkinson D.Y., Knoechel C., Yang C., Larabell C.A., and Le Gros M.A. (2012). Automatic alignment and reconstruction of images for soft X-ray tomography. J. Struct. Biol. 177, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang Z., Deng X., Liu Y., Gong W., and Li C. (2020). Analyses on clustering of the conserved residues at protein-RNA interfaces and its application in binding site identification. BMC Bioinformatics 21, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ginell G.M., Flynn A.J., and Holehouse A.S. (2022). SHEPHARD: a modular and extensible software architecture for analyzing and annotating large protein datasets. bioRxiv. 10.1101/2022.09.18.508433. [DOI] [PMC free article] [PubMed]
- 125.Emenecker R.J., Griffith D., and Holehouse A.S. (2021). Metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys. J. 120, 4312–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A., et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lalmansingh J.M., Keeley A.T., Ruff K.M., Pappu R.V., and Holehouse A.S. (2023). SOURSOP: A Python package for the analysis of simulations of intrinsically disordered proteins. bioRxivorg. 10.1101/2023.02.16.528879. [DOI] [PMC free article] [PubMed]
- 128.Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F., et al. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723. [DOI] [PubMed] [Google Scholar]
- 129.Smits A.H., Lindeboom R.G.H., Perino M., van Heeringen S.J., Veenstra G.J.C., and Vermeulen M. (2014). Global absolute quantification reveals tight regulation of protein expression in single Xenopus eggs. Nucleic Acids Res. 42, 9880–9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bassal M., Abukhalaf M., Majovsky P., Thieme D., Herr T., Ayash M., Tabassum N., Al Shweiki M.R., Proksch C., Hmedat A., et al. (2020). Reshaping of the Arabidopsis thaliana proteome landscape and co-regulation of proteins in development and immunity. Mol. Plant 13, 1709–1732. [DOI] [PubMed] [Google Scholar]
- 131.Wiśniewski J.R., and Rakus D. (2014). Quantitative analysis of the Escherichia coli proteome. Data Brief 1, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carpy A., Krug K., Graf S., Koch A., Popic S., Hauf S., and Macek B. (2014). Absolute proteome and phosphoproteome dynamics during the cell cycle of Schizosaccharomyces pombe (Fission Yeast). Mol. Cell. Proteomics 13, 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ghaemmaghami S., Huh W.-K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., and Weissman J.S. (2003). Global analysis of protein expression in yeast. Nature 425, 737–741. [DOI] [PubMed] [Google Scholar]
- 134.Matlock M.K., Holehouse A.S., and Naegle K.M. (2015). ProteomeScout: a repository and analysis resource for post-translational modifications and proteins. Nucleic Acids Res. 43, D521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., and Chang W. (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Holehouse A.S., Das R.K., Ahad J.N., Richardson M.O.G., and Pappu R.V. (2017). CIDER: Resources to analyze sequence-ensemble relationships of intrinsically disordered proteins. Biophys. J. 112, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mitrea D.M., Cika J.A., Guy C.S., Ban D., Banerjee P.R., Stanley C.B., Nourse A., Deniz A.A., and Kriwacki R.W. (2016). Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 5. 10.7554/eLife.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.