

Abstract
Genomic regulatory elements active in the developing human brain are notably enriched in genetic risk for neuropsychiatric disorders, including autism spectrum disorder (ASD), schizophrenia, and bipolar disorder. However, prioritizing the specific risk genes and candidate molecular mechanisms underlying these genetic enrichments has been hindered by the lack of a single unified large-scale gene regulatory atlas of human brain development. Here, we uniformly process and systematically characterize gene, isoform, and splicing quantitative trait loci (xQTLs) in 672 fetal brain samples from unique subjects across multiple ancestral populations. We identify 15,752 genes harboring a significant xQTL and map 3,739 eQTLs to a specific cellular context. We observe a striking drop in gene expression and splicing heritability as the human brain develops. Isoform-level regulation, particularly in the second trimester, mediates the greatest proportion of heritability across multiple psychiatric GWAS, compared with eQTLs. Via colocalization and TWAS, we prioritize biological mechanisms for ~60% of GWAS loci across five neuropsychiatric disorders, nearly two-fold that observed in the adult brain. Finally, we build a comprehensive set of developmentally regulated gene and isoform co-expression networks capturing unique genetic enrichments across disorders. Together, this work provides a comprehensive view of genetic regulation across human brain development as well as the stage-and cell type-informed mechanistic underpinnings of neuropsychiatric disorders.
Graphical Abstract:
Thousands of genetic risk loci have now been robustly associated with neurodevelopmental and psychiatric disorders by large-scale GWAS (1, 2). However, as most associated GWAS variants reside within non-coding regions of the human genome, often in large linkage disequilibrium (LD) blocks, the true underlying causal variant(s) and their target gene(s) remain largely unknown. Consequently, the critical defining obstacle of the post-GWAS era is to pinpoint the specific, locus-level molecular impact of GWAS variants at scale (3). As risk loci are enriched in known regulatory regions of the human genome (4–6), one major approach to address this challenge has been to connect risk variants with tissue-specific reference panels of expression quantitative trait loci (eQTLs), through statistical colocalization, transcriptome-wide association (TWAS), and related approaches (7–9). This has prompted several large-scale efforts (10–16) to generate comprehensive functional genomic compendia connecting population-level allelic variation with gene expression profiles in the human brain. Yet, while these resources have provided biologically interpretable and meaningful annotations for dozens of neuropsychiatric risk loci, the majority remain mechanistically unannotated (15, 17).
Gene regulation is known to be highly dependent on the specific underlying developmental stage, tissue, and cellular context (7, 17–20), and increasing evidence implicates the developing human brain in the genetic risk for neuropsychiatric disorders, including autism spectrum disorder (ASD) and schizophrenia (SCZ) (21–23). Consequently, several efforts have begun to characterize the genetic control of gene expression during human brain development, finding that gene regulation is tightly controlled during brain development and enriched for neuropsychiatric risk (11, 12, 24–28). However, as these studies are individually small in sample size, the power to pinpoint the developmental timing or elucidate the full extent of gene regulation in the developing human brain has been limited.
The regulation of transcript-isoform structure and diversity, through alternative local splicing and differences in transcriptional start and termination sites, is also known to be a mechanism critical for human brain development that is implicated in disease pathogenesis (14, 29–32). Indeed, relative to other tissues and species, the landscape of alternative splicing is particularly extensive in the human brain, and its regulation is notably distinct from that of gene expression (16, 33). While individual studies have begun to investigate splicing and isoform regulation (e.g., sQTL and isoQTLs) in the developing brain (24, 25), a lack of uniform data processing has precluded a systematic characterization of these critical mechanisms and their potential relationships with genetic risk factors for neuropsychiatric disorders.
To address these critical gaps, here we present the most comprehensive investigation into the genetic regulation of the transcriptome during human brain development by uniformly processing data from 672 distinct samples spanning 4-39 post-conception weeks (PCW), most of which are within the first and second trimesters. The full integrated dataset is highly diverse, comprising several major continental ancestries including European (EUR; 45%), Hispanic/Latino (AMR; 25%), African (AFR; 22%), and East Asian/Southeast Asian (EA/SEA; 8%). This well-powered, cross-population resource provides an unprecedented view of the developmental timing and regulatory landscape of human brain development at gene, isoform, and splicing levels. We observe a substantial drop in the heritability of gene expression and local splicing with development, particularly from 10-18 PCW. Yet, psychiatric GWAS signals appear more concentrated within the second trimester. We find that isoform-level regulation mediates substantially more psychiatric GWAS heritability than gene or local splicing regulation. We are able to connect ~60% of GWAS loci of five neuropsychiatric disorders with specific molecular mechanisms through colocalization and TWAS. Finally, we contextualize these risk mechanisms within a comprehensive set of gene and isoform-level co-expression networks across fetal brain development.
Large-Scale Functional Genomic Interrogation of Human Brain Development
Here, we present a comprehensive investigation of gene expression, splicing, and isoform regulation during human brain development by integrating data across five individual cohorts (12, 24–27) (Fig. S1) comprising 672 distinct donors. Following uniform data processing, including imputation into the multi-ancestry TOPMed reference panel (34), and strict quality control (Fig. S2; Methods), we retained 654 samples with matched genotype and cortical RNA-seq data spanning 4-39 PCW, the majority falling within the first two trimesters (Tri1, n=216; Tri2, n=433; Table S1). Altogether, our large sample size enabled detection of 10,094 genes with at least one cis-eQTL at FDR < 0.05 (termed “eGenes”), following permutation-based and FDR correction for multiple comparisons while accounting for local LD structure (Fig. 1A; Fig. S3; Table S1; Methods).
Fig. 1. The landscape of gene, splicing, and isoform regulation in the developing human brain.
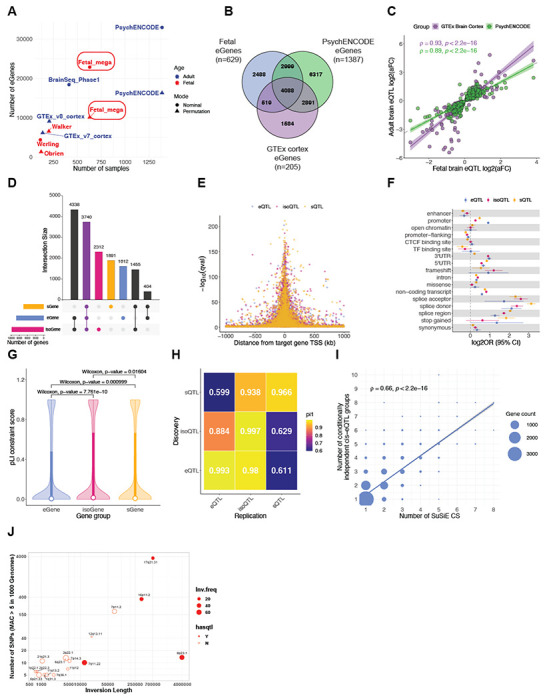
(A) The number of eGenes discovered here versus sample size, compared with other human brain studies. (B) Overlap of eGenes between fetal brain (n=629), GTEx v8 Brain Cortex (n=205), and PsychENCODE (n=1,387). (C) Correlation of eQTL effect size, measured by allelic fold change (aFC), between fetal and adult brain. Each dot is a shared pair of eGene-primary eQTL between fetal brain and GTEx (247 pairs) or PsychENCODE (253 pairs). (D) Overlap among eGenes, isoGenes, and sGenes. (E) Distance between the transcription start site (TSS) of each target gene of cis-eQTL, cis-isoQTL, and cis-sQTL SNPs. (F) Enrichment of cis-eQTL, cis-isoQTL, and cis-sQTLs within functional regions of the genome. (G) Loss-of-function mutation intolerance, as measured by pLI score, of eGenes, isoGenes, and sGenes. (H) Storey’s pi1 statistic of the proportion of true associations in the discovery group of QTL (y-axis) that are also true associations in the replication group of QTL (x-axis). (I) Number of fine-mapping credible sets versus number of conditionally independent eQTLs discovered. The size of the dots is scaled to the number of genes. (J) Common recurrent Inversion-QTLs in the developing brain. Inversions are displayed according to their length and the number of overlapping SNPs. Inversions with significantly associated eGenes have filled circles. The size of the circle indicates the population frequency of the inversions.
Compared with adult brain eQTL datasets from PsychENCODE (13, 14) and GTEx (16), we identified 2,488 unique “fetal-specific” eGenes in this dataset (Fig. 1B; Table S1). Among conserved eQTL-eGene pairs between the developing and adult human brain cohorts, we observe substantial consistency in effect size as measured by allelic fold change (GTEx: Spearman rho=0.93, p<2.2e-16; PsychENCODE: Spearman rho=0.89, p<2.2e-16; Fig. 1C; Fig. S3). Notably, these 2,488 fetal-specific eGenes were significantly more intolerant to loss-of-function mutations, as measured by the pLI score (35), compared with eGenes shared between fetal and adult timepoints (Wilcoxon p<2e-16; Fig. S3), suggesting that these context-specific annotations may be more relevant for interpretation of neuropsychiatric GWAS signals, which are known to be under negative selection. These fetal-specific eGenes were enriched for spliceosomal pathways and cell-type-specific marker genes (36) for a subtype of ventricular radial glia (vRG-0; OR 3.3, FDR-corrected P=0.054, Fisher’s exact test; Fig. S3; Table S1).
Dysregulation of RNA splicing has been strongly implicated in complex disease risk (29) as well as alterations in brain development (24). To systematically investigate RNA splicing regulation in the developing human brain, we used two complementary approaches. First, guided by existing GENCODE reference transcriptome annotations (37), we imputed isoform expression from short-read RNA-seq using Salmon (38). Second, we profiled local alternative splicing events via LeafCutter (39), an annotation-free method that quantifies intron excision ratios. Next, we identified genetic variants that are significantly associated with these isoform- and splicing-level quantitative traits (Fig. S4; Fig. S5; Methods), identifying 11,845 and 7,490 genes with a significant cis isoQTL or sQTL (termed “isoGenes” and “sGenes”; Table S1), respectively. Among the identified xQTL containing-genes, 3,740 are shared, and 2,312/1,891 only have significant genetic regulation detected at isoform/local splicing levels (Fig. 1D). As expected, cis-xQTL are strongly clustered near their target genes’ transcription start site (TSS) (Fig. 1E) and are correspondingly enriched in functional regions of the genome (Fig. 1F; Table S1). In addition, we observed that isoGenes and sGenes are significantly more intolerant to loss-of-function mutations than eGenes (Fig. 1G). To investigate the variant-level overlap among cis-e/iso/sQTLs, we calculated Storey’s pi1 statistic (40) and found that genetic variants directly associated with RNA splicing (especially isoQTL) capture independent and additional signals from eQTLs (Fig. 1H; Methods). Altogether, these results highlight the importance of studying regulation at the level of isoforms and splicing.
We next sought to characterize the extent of allelic heterogeneity in the developing human brain, in which multiple causal variants regulate gene expression at a given locus. Through stepwise conditional QTL mapping (41) (Fig. S3; Table S1; Methods), we identified multiple independent regulatory signals for 3,570 eGenes in the fetal brain, with some exhibiting up to 10 groups of conditionally independent cis-eQTL signals. As expected, the primary cis-eQTL signal (median - 0.0340 kb) was closer to the corresponding target gene’s TSS than was the secondary cis-eQTL signal (median 0.4820 kb; P<2.2e-16, Wilcoxon rank sum). The same was also found to be true for tertiary and higher-rank cis-eQTL signals. As an orthogonal approach, we also performed statistical fine-mapping with SuSiE (42), which estimates credible sets (CS) of causal variants. The number of SuSiE-estimated CS was highly concordant with the number of conditionally independent QTL signals for each eGene (Spearman correlation 0.66, p<2.2e-16; Fig. 1I), indicating that fine-mapping is a complementary approach to conditional QTL mapping for detecting independent regulatory signals. Each CS prioritized a median of 5 SNPs, with 2,423 containing exactly one SNP, which are strong candidates for functional validation (Data S1).
Compared to SNPs and indels, the impact of other classes of genetic variation (e.g., structural variants) on downstream gene expression remains underexplored. Nevertheless, complex structural variants such as large recurrent inversions have known associations with brain-relevant traits and can impact gene expression extensively (25, 43). To address this, we imputed the genotypes of 17 common (MAF > 0.05) inversions into our uniformly processed fetal brain data and quantified their effects on gene expression across the transcriptome. Longer inversions are more likely to significantly affect downstream gene expression. We found 49 inversion-associated expression quantitative loci (Inv-eQTLs) both in cis and in trans, for four of the inversions located at 17q21.31, 16p11.2, 7q11.22 and 8p23.1 (Fig. 1J; Fig. S6; Table S1; Methods).
Cross-Ancestry Gene Regulation and Fine-mapping
Differences in genetic variation (e.g., allele frequency and LD) across ancestries (44) has the potential to increase power for statistical fine-mapping (45, 46). After filtering and imputing the genotype data (Fig. S2; Methods), we inferred genetic ancestries in the fetal brain samples in reference to the 1000 Genomes (44). Of the 654 samples, 292 (44.6%) were labeled as “European” (EUR), 164 (25.1%) as “Hispanic/Latino” (AMR), 145 (22.2%) as “African” (AFR), 29 (4.4%) as “Southeast Asian” (SEA), and 24 (3.7%) as “East Asian” (EA) (Fig. 2A; Fig. S1; Table S1). Following the multi-ancestry QTL mapping pipeline, we independently mapped xQTLs in the three largest ancestry groupings: EUR, AMR, and AFR. 986 eGenes were shared between the multi-ancestry dataset and the three ancestry groupings (Fig. 2B; Table S2). Among the shared eGene-eQTL pairs between ancestries, we observe a highly consistent effect size measured by allelic fold change (47) (AMR-EUR Spearman correlation=0.97, p<2.2e-16; AFR-EUR Spearman correlation=0.97, p<2.2e-16; Fig. 2C). We then performed statistical fine-mapping separately in the ancestry groupings (42), and show that AFR, despite of the smallest sample size, has on average the smallest credible sets (CS) of causal variants (Fig. 2D; Data S2, S3, S4). Next, we found that the multi-ancestry dataset substantially reduced the CS size. Taking gene MTFR1 as an example (Fig. 2E), we show that the gain in fine-mapping resolution is not solely due to larger sample size, but the potential of multiancestry data to leverage distinct patterns of LD. MTFR1 has complex LD structure in EUR and AMR, reflected in its large CS (EUR n=49, AMR n=57), whereas in AFR, with simpler LD, the CS is downsized to 9 variants. CS sizes are further reduced in multi-ancestry fine-mapping (n=5), and in a trans-ancestry fine-mapping framework that leverages functional annotation (48, 49) (n=3). With these results, we highlight the promise of multi-ancestry data in refining statistical fine-mapping.
Fig. 2. Cross-population gene regulation and fine-mapping.
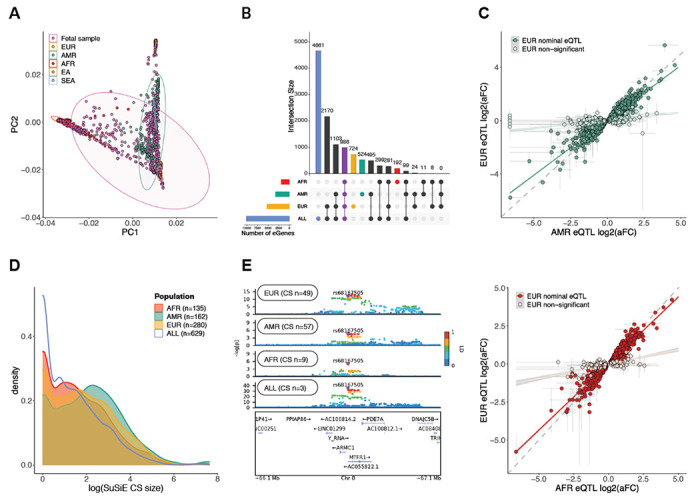
(A) Genotype PCA of the fetal brain samples. Population structure was inferred by merging imputed genotypes with 1000 Genomes. (B) Comparison of eGenes discovered in the full multi-population dataset (“ALL”, n=629) and in the separate sub-populations, EUR (n=280), AMR (n=162), and AFR (n=135). (C) Correlation of eQTL effect sizes between AMR/AFR (top/bottom) and EUR, as measured by allelic fold change. Each dot is an AMR/AFR eGene-primary eQTL pair and colored by its nominal significance in EUR. Grey lines denote the lower and upper bounds of aFC. (D) Comparison of fine-mapping credible set sizes between the populations. (E) Cis associations for the gene MTFR1 in the cross-population, EUR, AMR, and AFR datasets.
Trimester-Specific Genetic Regulation
Previous work has implicated the mid-fetal period of human brain development as a critical window convergently impacted by multiple distinct rare genetic risk factors for ASD and SCZ (21–23). Although common variant-mediated gene regulation is also known to be dependent on developmental context, this has not been systematically evaluated in the developing human brain. To address this critical gap, we conducted trimester-specific analyses to investigate the specificity of genetic regulation during distinct periods of brain development. We called QTLs after first separating samples into equally powered trimester 1 (4-13 PCW; EUR, n=143) and trimester 2 (14-26 PCW; EUR, n=145) windows, although there were too few samples in trimester 3 (n=4) to conduct a similar analysis. Surprisingly, we identified almost two-fold more trimester 1 eGenes (‘Tri1’; n=4,211) than Tri2 (n=2,220), with 1,261 eGenes shared between the two trimesters (Fig. 3A–B; Fig. S7; Table S3). To further explore this unexpected result, we next calculated the cis-window SNP-based heritability (cis-h2SNP) for gene expression -- a related measure that is more robust to sample size variation -- in Tri1 and Tri2 samples, as well as in adult samples from PsychENCODE (n=1,387). Mirroring eQTL results, we found that cis heritability drops significantly from Tri1 to Tri2 (Fig. 3C; Wilcoxon p < 2.2e-16; Table S3; Data S5, S6). Remarkably, we further observed significantly higher cis-h2SNP in both Tri1 and Tri2 compared with adult brain samples (Tri1 vs adult Wilcoxon p = 7.8e-8; Tri2 vs adult Wilcoxon p < 2.2e-16) (Fig. 3C).
Fig. 3. Trimester-specific patterns of gene expression and splicing regulation.
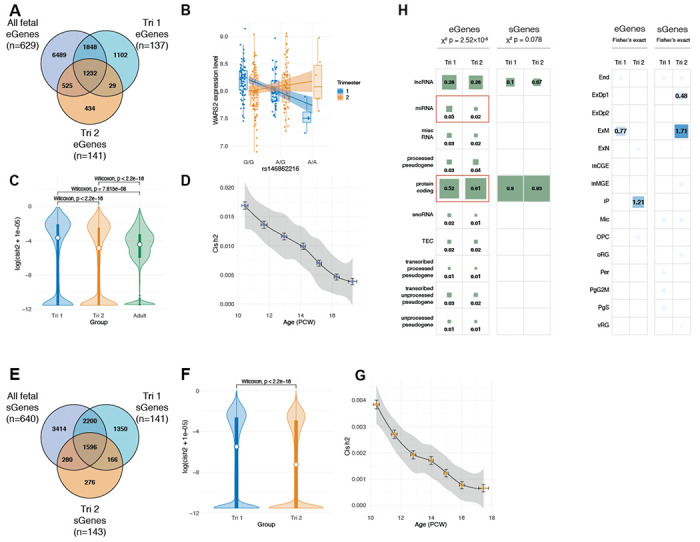
(A) Comparison of eGenes discovered in Tri1, Tri2, and the full dataset. Notably, we identify many more eGenes in Tri1 than Tri2 despite similar sample sizes. (B) Trimester-specific eQTLs for WARS2, where rs146862216 (G>A) is an eQTL in Tri1 (beta=−0.89, FDR=3.88e-13) but not in Tri2 (beta=−0.03, P-value=0.71). (C) cis-heritability of gene expression drops from Tri1 to Tri2 timepoints, and between fetal and adult (PsychENCODE) samples. (D) cis-heritability of gene expression across 10-18 post-conception weeks (PCW) in fetal brain EUR samples. For D and G, each dot represents a sliding set of temporally ordered samples (n=150), with mean age (+/−SD) on the x-axis, and median cis-h2SNP (+/−SD) on the y-axis. (E) Comparison of sGenes discovered in Tri1, Tri2, and the full dataset. (F) cis heritability of intron excision ratios in Tri1 vs Tri2. (G) cis heritability of local splining in fetal brain EUR samples over development, as in (D). (H) Comparison of gene and cell type enrichment of Tri1-only and Tri2-only e/sGenes.
We next sought to determine whether a particular developmental inflection point could contextualize this drop in heritability. To address this, we rank ordered EUR samples by PCW, and performed a sliding window analysis of cis-h2SNP across development in equally powered batches (n=150; Fig. 3D). We again observed a striking linear drop in cis-h2SNP across the entire range from 10-20 weeks. There were no detectable differences in the number of conditionally independent eQTLs (Fig. S7), gene expression levels, or phenotypic variance across Tri1 and Tri2 that could be driving these results (Fig. S8). To investigate other potential biological explanations for this drop, we next assessed whether Tri1 or Tri2 eGenes were differentially enriched among specific gene bio-types, cell-types, and/or pathways. Notably, from Tri1 to Tri2, we found that eGenes exhibited a significant shift from non-coding to protein-coding biotypes (chi-square=37.1, df=9, p=2.5e-5). Furthermore, Tri2 eGenes were significantly more enriched for deep layer excitatory neurons, a pattern not observed in Tri1 (Fig. 3H). Finally, to determine whether these observations extended beyond gene expression, we conducted similar analyses using sQTLs, as local splicing event quantifications are highly distinct from expression. Here again, we observed a striking drop from Tri1 to Tri2 in the number of genes harboring sQTLs, as well as in their cis-h2SNP from 10-18 weeks (Fig. 3E–G; Fig. S7; Table S3; Data S7, S8). This pattern also appeared to be biologically driven by a shift from non-coding to protein-coding biotypes, with proportional changes in sGenes approaching nominal significance (chi-square=3.1, df=1, p=0.08) (Fig. 3H). Altogether, these results underscore the temporally dynamic, context-specific nature of gene regulation in the human brain.
Temporal and Molecular Specificity of Neuropsychiatric GWAS
To prioritize the potential underlying biological context(s) through which genetic risk is conferred, we next sought to integrate our expanded set of functional genomic annotations with neuropsychiatric GWAS results. Using the well-powered SCZ GWAS as an example, we observed substantial enrichment of test statistics when subsetting to fetal brain cis e-, iso-, and sQTLs compared with background variants (Fig. 4A). Using stratified LD-score regression (S-LDSC (6, 50)), we next sought to estimate the degree to which SNP-based heritability was enriched among these xQTLs. We generated continuous annotations for genetic variants based on fine-mapping posterior probabilities (51) and found SCZ GWAS signals to be highly enriched among fetal brain regulatory elements -- more so than in the adult brain (Fig. 4B; Table S4; Methods). When jointly modeling fetal brain e-, iso-, and sQTL, we observed that splicing and isoform QTLs captured greater enrichment than eQTLs (Fig. S9; Table S4; Methods). These observations were consistent when extending across multiple neuropsychiatric GWAS as well as within trimester-specific annotations (Fig. S9; Table S4). Overall, these analyses highlight the critical importance of the fetal brain context -- as well as splicing and isoform regulation -- for interpreting potential mechanisms underlying psychiatric GWAS signals.
Fig. 4. Integrative analysis of fetal xQTLs with neuropsychiatric GWAS.
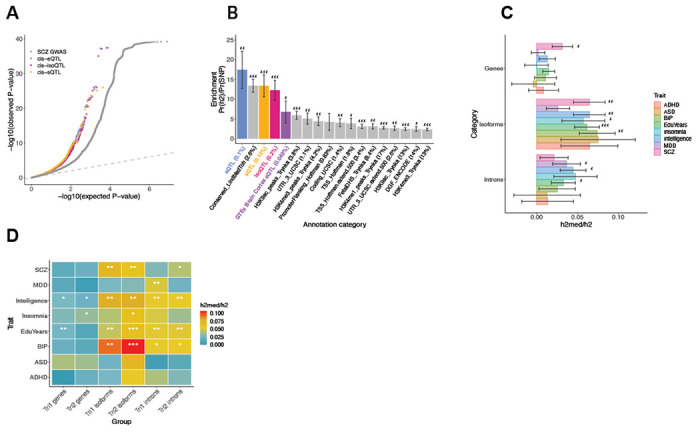
(A) Quantile-quantile plot of SCZ GWAS p-values, subsetted by top cis-eQTLs, cis-isoQTLs, and cis-sQTLs in comparison to all background GWAS SNPs. (B) s-LDSC enrichment of SCZ GWAS heritability within fetal brain xQTLx, adult brain cortex eQTLs (GTEx v8), compared with background functional annotations. The proportional genomic coverage of SNPs within each annotation are shown in parentheses. For B, C, D: *** FDR<0.001, ** FDR<0.01, * FDR<0.05. (C) Estimated proportion (+/−SE) of GWAS h2SNP mediated by the cis genetic component of gene, isoform, and intron (splicing) regulation. Isoform-level QTLs mediate the greatest degree of heritability for multiple neuropsychiatric traits in the developing brain, compared with e/sQTLs. (D) Estimated proportion of GWAS h2SNP mediated by the cis genetic component of trimester-stratified gene-, isoform-, and intron (splicing)-regulation.
Although the S-LDSC results clearly demonstrate greater enrichment of SNP-heritability among prenatal (relative to postnatal) gene regulatory variants, and splicing/isoform regulation (relative to total gene expression), these analyses are based on small genomic windows centered around top xQTLs. It can be difficult to compare results across annotations that differ substantially in genomic coverage, and it has been hypothesized that the top (e.g., most heritable) xQTLs may not overlap strongly with complex traits under negative selection (52, 53). To address these issues, we next leveraged the mediated expression score regression (MESC) framework (53) to estimate the proportion of heritability that is mediated by the cis-genetic component of assayed genes, isoforms, and introns (h2med/h2g). Notably, while S-LDSC is restricted to subset of significant xQTLs, MESC estimates enrichments genome-wide, including assayed features with low expression heritability (53). We chose five GWAS of brain-relevant neuropsychiatric traits SCZ, ASD, bipolar disorder (BIP), attention deficit/hyperactivity disorder (ADHD), and major depression disorder (MDD) (1, 2, 54–56). Across these traits, we consistently observed greater heritability mediated by isoform regulation (isoform h2med/h2g 6.45±1.92%) than that of gene expression (3.15±1.25%) or splicing (2.13±1.74%), although the overall extent of mediation remains small (Fig. 4C; Table S4; Methods). Extending these analyses across trimester-specific annotations, although less well powered, pointed to isoform-regulation in Tri2 as being particularly important, especially for BIP (Fig. 4D; Table S4). Altogether, our results highlight the importance of fetal isoform expression regulation as a critical mediator of psychiatric GWAS heritability, and the second trimester as the time frame during which genetic risks converge in the developing human brain.
Colocalization and Isoform-level Transcriptome-wide Association Study (isoTWAS)
To move from broad GWAS enrichments to locus-specific risk genes and molecular mechanisms, we next conducted a systematic colocalization analysis using eCAVIAR (9), which performs joint probabilistic fine-mapping and estimates the colocalization posterior probability (CLPP) that a given variant is “causal” in both GWAS and xQTL datasets. Across the combined 485 psychiatric GWAS loci, we are able to prioritize 292 (60.2%) with CLPP above the established cutoff of 0.01 (Fig. 5A, B; Fig. S10; Table S5; Methods). For context, restricting our results to eQTLs in SCZ and BIP, we identify 80 loci with prioritized eQTLs, while a recent, much larger adult brain eQTL study with an effective sample size of 3,154 identified 20 significant colocalizations (CLPP>0.01) for the two disorders (15).
Fig. 5. Neuropsychiatric risk gene prioritization through colocalization and isoTWAS.
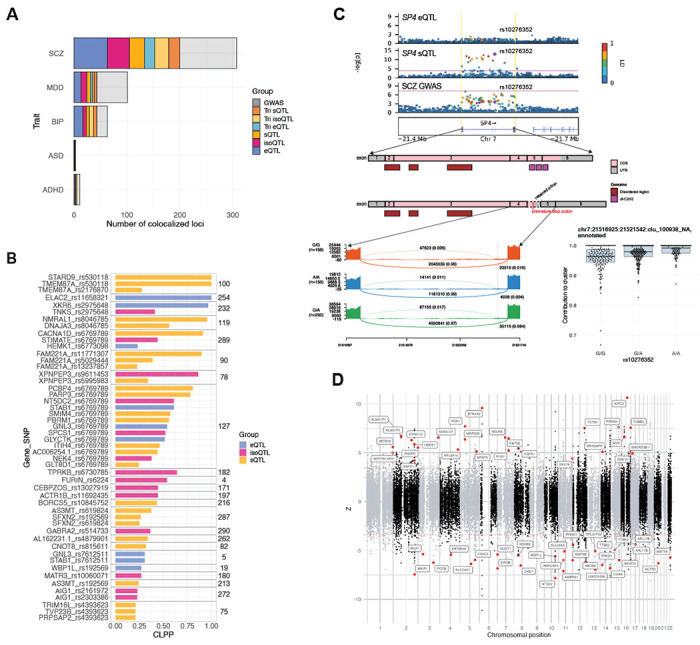
(A) Total number of GWAS loci exhibiting significant colocalization (CLPP>0.01) with specific fetal brain xQTL annotations. From left to right, bars represent the cumulative number of GWAS loci colocalized with each additional annotation. Shown in grey are GWAS loci that are not colocalized with any annotation. (B) Colocalization between SCZ GWAS and fetal brain xQTLs, ranked by CLPP and grouped by GWAS locus, as indicated by the number to the right. (C) Top: locus plots of SCZ GWAS with SP4 e- and sQTLs. A significant colocalization is observed for SCZ GWAS with a cryptic splicing event in SP4. Notably, SP4 does not have a detectable eQTL in the fetal brain. Middle: Gene structure of SP4 with and without cryptic exon inclusion, likely resulting in nonsense mediated decay. Bottom left: sashimi plot shows the density of exon and junction read mapping for intron cluster chr7:21516925-21521542, stratified by the colocalized sQTL. Bottom right: sQTL rs10276352 (G>A) increases the contribution to cluster of annotated intron chr:21516925-21521542. The SCZ risk allele increases cryptic exon inclusion. (D) Fetal brain isoTWAS associations with SCZ GWAS. Each dot represents an isoform and genes with significant, prioritized isoforms are colored in red.
Consistent with heritability enrichment patterns, we observed many more GWAS loci harboring colocalized iso-QTLs and sQTLs, compared to standard eQTLs (Fig. 5A,B; Fig. S10; Table S5). As a validation, we recover the well-established neuropsychiatric risk gene FURIN, with an isoQTL colocalization observed across both SCZ and BIP (Fig. 5B; Fig. S10) (10). The colocalized variant rs6224 (located at intron 13) is in moderately strong LD (R2=0.7) with rs4702 (located at the 3’-UTR of FURIN) that has been CRISPR-validated as an eQTL (57, 58). Further, we uncover a common-variant mechanism underlying the SCZ GWAS association at SP4, a transcription factor with a high-confidence rare variant implicated SCZ risk gene (1, 59). While SP4 was known to be the target gene in this GWAS locus, variant-to-gene mapping was limited by the lack of an observable SP4 eQTL signal in adult or fetal brain. Here, we identify a novel cryptic splicing event in SP4 colocalizing with both SCZ and BIP GWAS (Fig. 5C; Fig. S10). The risk variant rs10276352 (G>A) was identified as an sQTL associated with increased inclusion of a 181 bp cryptic, unannotated exon (chr7:21521120-21521300). The inclusion of this cryptic exon is predicted to introduce a frameshift and premature stop codon between canonical exons 4 and 5, likely resulting in nonsense-mediated decay. The resulting truncated protein, if any, would be missing the zinc finger domain that is critical for SP4’s DNA-binding activity (Fig. 5C).
Other novel disease mechanisms prioritized by these analyses include: downregulation of the GABAA-receptor subunit GABRA2 in SCZ (eQTL: rs514733, CLPP=0.32; Fig. 5B; Table S5), providing genetic evidence for GABAergic dysfunction hypothesized to contribute to disease pathophysiology (60); splicing dysregulation of ADCY2 in BIP (top colocalization sQTL: rs11750832, CLPP=0.97; Fig. S10; Table S5) which encodes adenylate cyclase, a cell membrane-bound enzyme regulating cAMP signaling and a target of the mood stabilizer lithium (61); splicing dysregulation in BIP of SCN2A (sQTL: rs17183814, CLPP=0.70; Fig. S10; Table S5), a voltage gated sodium channel subunit with rare variant associations in ASD and epilepsy (62, 63); and in MDD, the dysregulated splicing of CBLL1 (CLPP=0.43; Fig. S10; Table S5), a highly constrained E3 ubiquitin-protein ligase and regulator of N6-methyladenosine RNA modification involved in neural-immune activation (64).
We next conducted an isoform-centric transcriptome-wide association study (isoTWAS) (65), to identify genes and their isoforms whose cis regulated expression is associated with SCZ risk. isoTWAS identified 536 isoforms across 271 unique genes with significant isoTWAS associations (Bonferroni-corrected P < 0.05 across genes, FWER-corrected P < 0.05 across transcripts of the same gene, permutation P < 0.05; Methods). To leverage local LD and to account for SNP weight correlations, we performed fine-mapping on isoTWAS associations that passed permutation testing (Fig. 5D; Table S5). This analysis resulted in 129 putatively causal isoforms from 107 unique genes that fall in 90% credible sets, with 22 of these genes with pLI score > 0.8. Of these, 69 isoforms from 57 unique genes are within 500 kb of a GWAS-significant risk SNP. In addition, among LD-independent blocks that have SCZ GWAS significant SNPs, we identified 41 independent GWAS SNPs within 500 kb of an isoTWAS-prioritized isoform. Comparing isoforms prioritized through isoTWAS and/or isoQTL colocalization revealed 14 isoforms that were prioritized with both methods, including SLC9C2, KMT2E, and ABCB9. Of note, the relatively low overlap could be due to the low power of probabilistic colocalization methods (66). With its increased power, isoTWAS also captured additional notable associations, including genetically mediated upregulation of HCN1-201, the dominant isoform of the hyperpolarization-activated cation channel HCN1. An additional advantage of isoTWAS over colocalization analyses is the ability to map isoform-level associations outside of GWAS-significant loci, additionally identifying putatively causal 60 isoforms across 50 unique genes that were found outside a GWAS-significant locus.
When examining results from gene prioritization analyses, we noticed several instances in which a single variant was associated with multiple distinct QTLs and significantly overlapped with disease GWAS signal. For example, rs6769789 was identified as an xQTL for multiple genes on chromosome 3 that colocalized with SCZ GWAS (Fig. 5B). Due to pleiotropy, it can be particularly difficult to distinguish whether one (of many potential) molecular effects is the true mediator of the SNP-trait effect. In attempt to address this, we employed MRLocus (67) which leverages a Mendelian randomization framework for loci with allelic heterogeneity, using conditionally-independent QTLs for these mediators to further support (or refute) observed gene-to-trait effects. Among 14 TWAS-identified genes with >3 conditionally independent eQTLs, the MRLocus framework provided additional support for 7 genes with FDR<0.2 (Table S5; Methods). In the pleiotropic rs6769789 locus described above, for example, there was an association between eQTL and GWAS effect sizes among conditionally independent eQTLs for NT5DC2 (FDR=0.125), providing additional evidence in support of this gene-trait association (Fig. S11).
Network-level Contextualization of Developmental Gene Regulation
To identify developmentally relevant transcriptional networks, interrogate them for common and rare genetic risk, and connect that risk with key biological and cellular processes, we performed robust weighted gene co-expression network analysis (rWGCNA) on our dataset, an unsupervised method that clusters genes into modules based on shared patterns of expression (22, 68–70). We constructed networks using gene and isoform-level quantifications across all samples (n=642), as well as within samples filtered by trimester (Tri1, Tri2) or chromosomal sex (XX, XY). In total, we identified 124 gene and isoform co-expression modules demonstrating enrichment for all major developmental cell types (36) and recapitulating early biochemical processes and developmental pathways (Fig. 6A; Methods). These networks expand on previous work (12, 18, 19, 21, 22, 24) by incorporating an order of magnitude greater number of samples, generating isoform-level modules for over 120,000 transcripts, and contextualizing development-wide findings within trimester- and sex-specific contexts.
Fig. 6. Systems-level integration of risk variation with developmental gene and isoform co-expression.
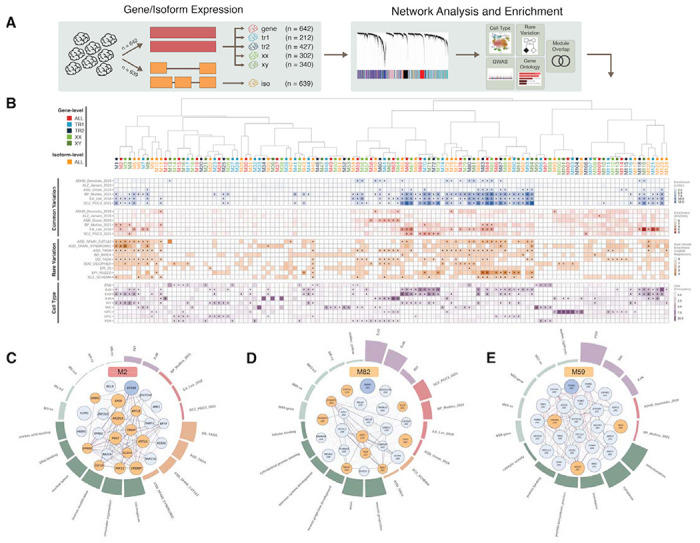
(A) Workflow for construction of gene and isoform-level co-expression networks, followed by cell-type, pathway, and disease gene enrichment analyses. Separate gene co-expression networks were built to capture trimester and sex-specific effects. (B) Top: hierarchical clustering of modules from gene, isoform, trimester and sex-stratified networks through bi-weight mid-correlation of eigengenes. Middle: heatmaps depict module-level enrichment for neuropsychiatric GWAS signal (−log10Penrich from s-LDSC and MAGMA) and odds ratios (OR) for rare variation and cell type enrichment (truncated at 10). Triangles indicate FDR-corrected P<0.1 significance. (C) Annotations for M2, a development-wide disease-associated chromatin regulation module. Center: top module (‘hub’) genes with circle size reflecting module membership (kME) and orange shading indicating genes with associated high-confidence neuropsychiatric disorder associated rare variants. Thin edges represent topologic overlap, solid edges indicate protein-protein interactions from the STRING database. Surrounding: circular bar plot highlighting module enrichment for cell types (purple), common (red) and rare (red-orange) variation, gene ontology terms (dark green), and module overlap (light green). (D) Annotations for M82, a SCZ/BIP-associated deep layer neuron-projection isoform module. (E) Annotations for M59, an ADHD-associated mitochondrial/proteasome isoform module.
Overlaying enrichment for common and rare variation within the hierarchical structure defined by co-expression pinpointed groups of modules that harbor most of neurodevelopmental disease risk (Fig. 6B; Table S6; Methods). Broadly, we find that isoform modules are more likely to capture cell-type marker enrichment (two-sided Fisher’s exact test on FDR<0.1 hits, p=8e-5, OR=2.37), demonstrating the importance of incorporating splicing and isoform expression in a network context. Neuropsychiatric GWAS signals were found to localize within modules enriched for deep layer excitatory, maturing excitatory, and interneuron cell populations, while exhibiting depletion for neural progenitor modules. Rare variation was more likely to be enriched in deep layer excitatory, maturing excitatory, and oligodendrocyte precursor modules, suggesting that the majority of disease-associated variation perturbs the maturation of neuronal cell types (Fig. S12).
At the gene level, a group of modules (M1, M2, M3) enriched for chromatin remodeling and histone modification pathways, with hub genes including EP300, EP400, ARID1A, KMT2E and POGZ, converged with excitatory and inhibitory neuron marker genes and showed strong enrichment for rare variation associated with ASD and developmental delay (DD) (Fig. 6C). These modules highly overlapped with the DD- and ASD-risk yellow module described in Walker et al. (24), building upon the previously described excitatory neuron enrichment for these disorders. Remarkably, our analysis partitions the hub genes of the yellow module into another group of excitatory-enriched synaptic projection modules (M83, M84, M85; Fig. S12), which exhibits strong enrichment for cross-disorder common variation and rare variation risk for ASD, epilepsy, and SCZ. Among the strongest genetic risk for SCZ was observed in two groups of interrelated synaptic modules (group 1: M93, M94; group 2: M86, M87, M88; Fig. S12), which strongly enriched for synaptic gene ontologies and excitatory neuron marker genes. A closely related isoform module, M82, uniquely captured this SCZ signal and harbored hub transcripts with known risk, like the canonical transcripts of TRIO and SYNGAP1, as well as multiple transcripts of ANK3 (Fig. 6D). Using module preservation analysis, we identified M17 as a first trimester-specific module that shows unique enrichment for neurodevelopmental disorders (NDD) risk genes and ontologies for neurogenesis and DNA binding, with hub genes such as GREB1L, LRP2, and CASZ1 (Fig. S12).
At the isoform level, M59 stood out for its consistent and unique association with ADHD common variant risk and GO term enrichment for mitochondrial and bioenergetic pathways. Hub genes for this module included multiple proteasome subunit genes (PSMD family), and cell type analysis revealed association with excitatory, microglial, and pericyte populations (Fig. 6E). Furthermore, a group of neuronally-enriched modules (M65, M66, M67; Fig. S12) displayed robust BIP, education attainment (EA), and SCZ enrichment while harboring high risk hub genes like MEF2C and SATB2 – similar to the prenatally-enriched M37 module described in Li et al (18). Of note, distinct transcripts of known ASD risk gene SOX5 acted as hub genes for two isoform modules, M120 and M122, but M120 alone showed a strong and specific signal for ASD rare variation (Fig. S12).
Cell-type Specificity through Module-interaction QTLs
Finally, as gene regulation is often known to occur within cell-type-specific contexts, we leveraged two orthogonal approaches to integrate cellular specificity into our gene regulatory results in the developing human brain. First, we applied the network-based framework implemented in CellWalker to integrate fetal single-cell chromatin accessibility with eQTL results (71). CellWalker mapped 21.7% of eQTL-containing genes to a specific cellular context in the developing human brain, corresponding to 3,739 of the bulk-derived eQTLs (Fig. 7A; Table S7; Methods). Second, we leveraged the fact that co-expression modules are known to capture cell-type-specific processes (Fig. 6A) to identify module-interacting eQTL (ieQTLs). To identify such ieQTLs, in which SNP-gene associations are modulated by the levels of a given module, we tested the interaction effect between genotype and module eigengenes on gene and isoform expression (Fig. 7B; Methods). Across all gene and isoform modules, 8,008 ieQTLs were identified after the permutation test (BH-corrected FDR<0.05; Methods), in which 3,960 ieQTLs were specific to a single module (Fig. 7C; Data S9). To validate the cell-type specificity of module ieQTLs, we examined the sharing between module ieQTLs with eQTLs identified separately within cultured neurons and progenitors (72). Using the Storey’s pi1 statistic (40), we observed substantial concordance between module interacting SNP-gene pairs that are true associations in external neuron/progenitor eQTL datasets (Fig. 7C). 40 modules have a pi1 difference greater than 0.2 between neuron and progenitor eQTLs, suggesting cell-type-specific genetic regulation (Fig. 7C). Finally, integrating disease GWAS, we identified significant colocalizations (CLPP>0.01) between ieQTLs and 22 SCZ GWAS loci, 13 of which are not found with bulk cA-eQTLs (Table S5). For example, we identified a SCZ colocalization with an ieQTL of BRINP2 and the Tri1 gene module M93 (rs17659437, CLPP = 0.01653) (Fig. 7D). M93 is enriched for deep layer excitatory neuron markers (ExD), cross-disorder psychiatric GWAS signal, and synapse-related pathways (Fig. 7E), with eigengene expression increasing across development (Fig. 7F). Concordantly, this module shows greater pi1 concordance with neuronal compared to progenitor eQTLs (Fig. 7C). BRINP2 is involved in BMP and retinoic acid signaling pathways, both of which play critical roles in brain development and neuropsychiatric disease. In sum, these analyses demonstrate how publicly available data can be used to annotate bulk QTLs to specific cell types, adding further context for detecting gene regulatory variation relevant to neuropsychiatric disease mechanisms in the developing human brain.
Fig. 7. Module interacting eQTLs and context-specific GWAS colocalization.
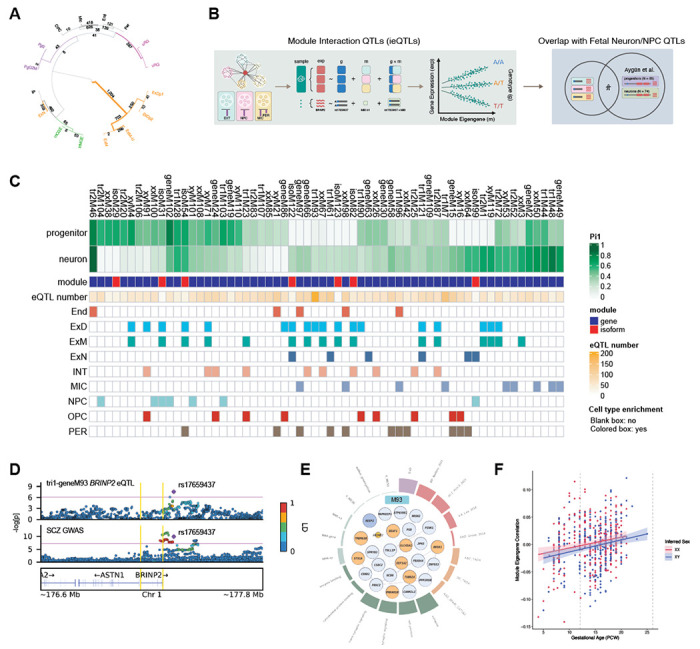
(A) Hierarchical clustering of cis-eQTL enrichments among specific fetal brain cell-types as mapped by CellWalker. Outermost numbers denote results from single cell type label analysis. Inner numbers denote results from a broader, multi-level label analysis. See Methods for details. (B) A schematic of module interaction ieQTL mapping and validation in cultured neurons and progenitors. (C) Results from module ieQTL mapping. From top to bottom, pi1 statistics depicting ieQTL overlap with eQTLs from cultured neurons or neural progenitor cells (NPCs), molecular feature (gene/isoform), number of ieQTLs, and cell type enrichment. 62 modules with pi1>0.2 in either neurons/NPCs are shown. (D) Colocalization between SCZ GWAS and BRINP2 ieQTL rs17659437 in M93 (CLPP=0.02). (E) Annotation for M93, a SCZ/BIP enriched deep-layer neuronal/synaptic module. (F) Trajectory of M93 eigengene expression across brain development, colored by biological sex.
Discussion
Here, we present an unprecedented view of the landscape of gene, isoform, and splicing regulation in the developing human brain, across more than 650 distinct donors. We further provide xQTL maps specific to the first and second trimesters of human brain development, as well as within 3 genetic ancestries, by leveraging the resulting allelic diversity through trans-ancestry fine-mapping to narrow in on underlying causal genetic variants. With this work, we are able to prioritize 2-fold more risk genes and molecular mechanisms underlying neurodevelopmental and psychiatric disorder GWAS loci compared with much larger adult brain reference panels, highlighting the importance of developmental context when interpreting genetic risk variation. Finally, we build gene and isoform-level co-expression networks to place this polygenic risk within an unsupervised, systems-level developmental and cell-type-specific contextualization.
Our results have several notable implications for future investigations of the functional genomic underpinnings of brainrelevant complex traits and disorders. First, several findings highlight the substantial impact of developmental stage on gene regulation, including a striking drop from 10-18 PCW in cis-heritability of gene expression and splicing which was not attributable to differences in gene expression magnitude or variance over time. As eQTL discovery and cis heritability are interrelated, this likely explains why we are able to identify eGenes on par with larger adult brain studies. Furthermore, while disease genes tend to be under negative selection, which is inversely related to gene expression heritability (52, 53), our data suggest that this relationship is likely moderated by developmental stage. Indeed, the fetal brain -- and Tri2, in particular -- harbors greater enrichment for neuropsychiatric GWAS signal than at postnatal timepoints, consistent with results from rare variant enrichment analyses (21–23).
Second, we observe striking differences in the ability of distinct molecular features to capture neuropsychiatric risk mechanisms. Specifically, iso form-level regulation mediated substantially more heritability than eQTLs, across several distinct disease GWAS. Further, iso- and sQTL colocalizations explained many more GWAS loci than standard eQTLs from the fetal or adult brain. Given that isoform quantification requires accurate imputation from short-read RNA-seq, guided by genomic reference annotations which are notably incomplete with respect to the human brain (73), we expect these differences to only become further accentuated as our understanding of the landscape of alternative splicing and isoform complexity grows, through the continued adoption of emerging long-read RNA-sequencing technologies.
Third, with the ever-increasing number of available functional genomic reference panels, including from this study, it is becoming increasingly clear that there will soon be multiple potential, prioritized (e.g., colocalized) functional genomic mechanisms at a given GWAS locus. This can occur when a GWAS SNP tags a haplotype containing an underlying structural variant (e.g., inversion), with multiple pleiotropic effects on cis gene regulation. Mendelian randomization-based approaches can help address this challenge, but will require larger reference panels capable of capturing the full extent of allelic heterogeneity across the transcriptome. Further, while single-cell QTL studies have been heralded of late, they will only exacerbate this issue while lacking the ability to profile splicing and isoform-level regulation which we show to be most high-yield for mechanistic inference.
Finally, given our finding that isoform-level co-expression networks in particular are able to recapitulate cell-type-specific biological processes and genetic regulation, future work should continue to build larger bulk-tissue reference panels of the developing human brain guided by more complete, matching isoform-level annotations from long-read sequencing. Greater visibility is needed, especially in the third trimester of brain development, which is critically underrepresented in our sample.
Supplementary Material
Acknowledgements:
We are grateful to the brain tissue donors and their families, without whom this work would not have been possible. This work was supported by the Simons Foundation (SFARI Bridge to Independence Award to M.J.G.), NIMH (R01-MH121521, R01-MH123922 to M.J.G.). Data were generated as part of the PsychENCODE Consortium, supported by: U01DA048279, U01MH103339, U01MH103340, U01MH103346, U01MH103365, U01MH103392, U01MH116438,U01MH116441, U01MH116442, U01MH116488, U01MH116489, U01MH116492, U01MH122590, U01MH122591, U01MH122592, U01MH122849, U01MH122678, U01MH122681, U01MH116487, U01MH122509, R01MH094714, R01MH105472, R01MH105898, R01MH109677, R01MH109715, R01MH110905, R01MH110920, R01MH110921, R01MH110926, R01MH110927, R01MH110928, R01MH111721, R01MH117291, R01MH117292, R01MH117293, R21MH102791, R21MH103877, R21MH105853, R21MH105881, R21MH109956, R56MH114899, R56MH114901, R56MH114911, R01MH125516, R01MH126459, R01MH129301, R01MH126393, R01MH121521, R01MH116529, R01MH129817, R01MH117406, and P50MH106934 awarded to: Alexej Abyzov, Nadav Ahituv, Schahram Akbarian, Kristin Brennand, Andrew Chess, Gregory Cooper, Gregory Crawford, Stella Dracheva, Peggy Farnham, Michael Gandal, Mark Gerstein, Daniel Geschwind, Fernando Goes, Joachim F. Hallmayer, Vahram Haroutunian, Thomas M. Hyde, Andrew Jaffe, Peng Jin, Manolis Kellis, Joel Kleinman, James A. Knowles, Arnold Kriegstein, Chunyu Liu, Christopher E. Mason, Keri Martinowich, Eran Mukamel, Richard Myers, Charles Nemeroff, Mette Peters, Dalila Pinto, Katherine Pollard, Kerry Ressler, Panos Roussos, Stephan Sanders, Nenad Sestan, Pamela Sklar, Michael P. Snyder, Matthew State, Jason Stein, Patrick Sullivan, Alexander E. Urban, Flora Vaccarino, Stephen Warren, Daniel Weinberger, Sherman Weissman, Zhiping Weng, Kevin White, A. Jeremy Willsey, Hyejung Won, and Peter Zandi. Author Contributions: This study was conceived and designed by MJG and CW with input from CL and KSP. Data was provided by DHG, NS, NJB, DRW, AEJ, JEK, TMH, RLW. Analyses were performed by CW, MM, RD, PZ, PFP, DDV, AB, NM, CJ, MK, ET, CH, NA, MIL, supervised by MJG, with contributions from CC, DC, HP, MG, NPD, ZW, KR, AG, BP, MAP, JLS, MIL, KSP, CL. CW and MJG wrote the manuscript, with major contributions from MM, AB and critical input from all authors.
Competing Interests:
M.J.G. and D.H.G. receive grant funding from Mitsubishi Tanabe Pharma America.
Footnotes
Code Availability:
Analysis code can be found on GitHub at https://github.com/gandallab/Fetal_metaQTL
Data Availability:
Supplemental data can be found on Synapse at https://www.synapse.org/#!Synapse:syn50897018.3/datasets/
References and Notes
- 1.Trubetskoy V., Pardiñas A. F., Qi T., Panagiotaropoulou G., Awasthi S., Bigdeli T. B., Bryois J., Chen C.-Y., Dennison C. A., Hall L. S., Lam M., Watanabe K., Frei O., Ge T., Harwood J. C., Koopmans F., Magnusson S., Richards A. L., Sidorenko J., Wu Y., Zeng J., Grove J., Kim M., Li Z., Voloudakis G., Zhang W., Adams M., Agartz I., Atkinson E. G., Agerbo E., Al Eissa M., Albus M., Alexander M., Alizadeh B. Z., Alptekin K., Als T. D., Amin F., Arolt V., Arrojo M., Athanasiu L., Azevedo M. H., Bacanu S. A., Bass N. J., Begemann M., Belliveau R. A., Bene J., Benyamin B., Bergen S. E., Blasi G., Bobes J., Bonassi S., Braun A., Bressan R. A., Bromet E. J., Bruggeman R., Buckley P. F., Buckner R. L., Bybjerg-Grauholm J., Cahn W., Cairns M. J., Calkins M. E., Carr V. J., Castle D., Catts S. V., Chambert K. D., Chan R. C. K., Chaumette B., Cheng W., Cheung E. F. C., Chong S. A., Cohen D., Consoli A., Cordeiro Q., Costas J., Curtis C., Davidson M., Davis K. L., de Haan L., Degenhardt F., DeLisi L. E., Demontis D., Dickerson F., Dikeos D., Dinan T., Djurovic S., Duan J., Ducci G., Dudbridge F., Eriksson J. G., Fañanás L., Faraone S. V., Fiorentino A., Forstner A., Frank J., Freimer N. B., Fromer M., Frustaci A., Gadelha A., Genovese G., Gershon E. S., Giannitelli M., Giegling I., Giusti-Rodríguez P., Godard S., Goldstein J. I., González Peñas J., González-Pinto A., Gopal S., Gratten J., Green M. F., Greenwood T. A., Guillin O., Gülöksüz S., Gur R. E., Gur R. C., Gutiérrez B., Hahn E., Hakonarson H., Haroutunian V., Hartmann A. M., Harvey C., Hayward C., Henskens F. A., Herms S., Hoffmann P., Howrigan D. P., Ikeda M., Iyegbe C., Joa I., Julià A., Kähler A. K., Kam-Thong T., Kamatani Y., Karachanak-Yankova S., Kebir O., Keller M. C., Kelly B. J., Khrunin A., Kim S.-W., Klovins J., Kondratiev N., Konte B., Kraft J., Kubo M., Kučinskas V., Kučinskiene Z. A., Kusumawardhani A., Kuzelova-Ptackova H., Landi S., Lazzeroni L. C., Lee P. H., Legge S. E., Lehrer D. S., Lencer R., Lerer B., Li M., Lieberman J., Light G. A., Limborska S., Liu C.-M., Lönnqvist J., Loughland C. M., Lubinski J., Luykx J. J., Lynham A., Macek M. Jr, Mackinnon A., Magnusson P. K. E., Maher B. S., Maier W., Malaspina D., Mallet J., Marder S. R., Marsal S., Martin A. R., Martorell L., Mattheisen M., McCarley R. W., McDonald C., McGrath J. J., Medeiros H., Meier S., Melegh B., Melle I., Mesholam-Gately R. I., Metspalu A., Michie P. T., Milani L., Milanova V., Mitjans M., Molden E., Molina E., Molto M. D., Mondelli V., Moreno C., Morley C. P., Muntané G., Murphy K. C., Myin-Germeys I., Nenadić I., Nestadt G., Nikitina-Zake L., Noto C., Nuechterlein K. H., O’Brien N. L., O’Neill F. A., Oh S.-Y., Olincy A., Ota V. K., Pantelis C., Papadimitriou G. N., Parellada M., Paunio T., Pellegrino R., Periyasamy S., Perkins D. O., Pfuhlmann B., Pietiläinen O., Pimm J., Porteous D., Powell J., Quattrone D., Quested D., Radant A. D., Rampino A., Rapaport M. H., Rautanen A., Reichenberg A., Roe C., Roffman J. L., Roth J., Rothermundt M., Rutten B. P. F., Saker-Delye S., Salomaa V., Sanjuan J., Santoro M. L., Savitz A., Schall U., Scott R. J., Seidman L. J., Sharp S. I., Shi J., Siever L. J., Sigurdsson E., Sim K., Skarabis N., Slominsky P., So H.-C., Sobell J. L., Söderman E., Stain H. J., Steen N. E., Steixner-Kumar A. A., Stögmann E., Stone W. S., Straub R. E., Streit F., Strengman E., Stroup T. S., Subramaniam M., Sugar C. A., Suvisaari J., Svrakic D. M., Swerdlow N. R., Szatkiewicz J. P., Ta T. M. T., Takahashi A., Terao C., Thibaut F., Toncheva D., Tooney P. A., Torretta S., Tosato S., Tura G. B., Turetsky B. I., Üçok A., Vaaler A., van Amelsvoort T., van Winkel R., Veijola J., Waddington J., Walter H., Waterreus A., Webb B. T., Weiser M., Williams N. M., Witt S. H., Wormley B. K., Wu J. Q., Xu Z., Yolken R., Zai C. C., Zhou W., Zhu F., Zimprich F., Atbaşoğlu E. C., Ayub M., Benner C., Bertolino A., Black D. W., Bray N. J., Breen G., Buccola N. G., Byerley W. F., Chen W. J., Cloninger C. R., Crespo-Facorro B., Donohoe G., Freedman R., Galletly C., Gandal M. J., Gennarelli M., Hougaard D. M., Hwu H.-G., Jablensky A. V., McCarroll S. A., Moran J. L., Mors O., Mortensen P. B., Müller-Myhsok B., Neil A. L., Nordentoft M., Pato M. T., Petryshen T. L., Pirinen M., Pulver A. E., Schulze T. G., Silverman J. M., Smoller J. W., Stahl E. A., Tsuang D. W., Vilella E., Wang S.-H., Xu S., Indonesia Schizophrenia Consortium, PsychENCODE, Psychosis Endophenotypes International Consortium, SynGO Consortium, Adolfsson R., Arango C., Baune B. T., Belangero S. I., Børglum A. D., Braff D., Bramon E., Buxbaum J. D., Campion D., Cervilla J. A., Cichon S., Collier D. A., Corvin A., Curtis D., Forti M. D., Domenici E., Ehrenreich H., Escott-Price V., Esko T., Fanous A. H., Gareeva A., Gawlik M., Gejman P. V., Gill M., Glatt S. J., Golimbet V., Hong K. S., Hultman C. M., Hyman S. E., Iwata N., Jönsson E. G., Kahn R. S., Kennedy J. L., Khusnutdinova E., Kirov G., Knowles J. A., Krebs M.-O., Laurent-Levinson C., Lee J., Lencz T., Levinson D. F., Li Q. S., Liu J., Malhotra A. K., Malhotra D., McIntosh A., McQuillin A., Menezes P. R., Morgan V. A., Morris D. W., Mowry B. J., Murray R. M., Nimgaonkar V., Nöthen M. M., Ophoff R. A., Paciga S. A., Palotie A., Pato C. N., Qin S., Rietschel M., Riley B. P., Rivera M., Rujescu D., Saka M. C., Sanders A. R., Schwab S. G., Serretti A., Sham P. C., Shi Y., St Clair D., Stefánsson H., Stefansson K., Tsuang M. T., van Os J., Vawter M. P., Weinberger D. R., Werge T., Wildenauer D. B., Yu X., Yue W., Holmans P. A., Pocklington A. J., Roussos P., Vassos E., Verhage M., Visscher P. M., Yang J., Posthuma D., Andreassen O. A., Kendler K. S., Owen M. J., Wray N. R., Daly M. J., Huang H., Neale B. M., Sullivan P. F., Ripke S., Walters J. T. R., O’Donovan M. C., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature (2022), doi: 10.1038/s41586-022-04434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grove J., Ripke S., Als T. D., Mattheisen M., Walters R. K., Won H., Pallesen J., Agerbo E., Andreassen O. A., Anney R., Awashti S., Belliveau R., Bettella F., Buxbaum J. D., Bybjerg-Grauholm J., Bækvad-Hansen M., Cerrato F., Chambert K., Christensen J. H., Churchhouse C., Dellenvall K., Demontis D., De Rubeis S., Devlin B., Djurovic S., Dumont A. L., Goldstein J. I., Hansen C. S., Hauberg M. E., Hollegaard M. V., Hope S., Howrigan D. P., Huang H., Hultman C. M., Klei L., Maller J., Martin J., Martin A. R., Moran J. L., Nyegaard M., Nærland T., Palmer D. S., Palotie A., Pedersen C. B., Pedersen M. G., dPoterba T., Poulsen J. B., Pourcain B. S., Qvist P., Rehnström K., Reichenberg A., Reichert J., Robinson E. B., Roeder K., Roussos P., Saemundsen E., Sandin S., Satterstrom F. K., Davey Smith G., Stefansson H., Steinberg S., Stevens C. R., Sullivan P. F., Turley P., Walters G. B., Xu X., Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium, BUPGEN, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, 23andMe Research Team, Stefansson K., Geschwind D. H., Nordentoft M., Hougaard D. M., Werge T., Mors O., Mortensen P. B., Neale B. M., Daly M. J., Børglum A. D., Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gandal M. J., Leppa V., Won H., Parikshak N. N., Geschwind D. H., The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci. 19, 1397–1407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward L. D., Kellis M., Evidence of abundant purifying selection in humans for recently acquired regulatory functions. Science. 337, 1675–1678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., Johnson A. K., Canfield T. K., Giste E., Diegel M., Bates D., Hansen R. S., Neph S., Sabo P. J., Heimfeld S., Raubitschek A., Ziegler S., Cotsapas C., Sotoodehnia N., Glass I., Sunyaev S. R., Kaul R., Stamatoyannopoulos J. A., Systematic localization of common disease-associated variation in regulatory DNA. Science. 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finucane H. K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K., Ripke S., Day F. R., ReproGen Consortium, Schizophrenia Working Group of the Psychiatric Genomics Consortium, RACI Consortium, Purcell S., Stahl E., Lindstrom S., Perry J. R. B., Okada Y., Raychaudhuri S., Daly M. J., Patterson N., Neale B. M., Price A. L., Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez L. M., Kim M., Hoftman G. D., Haney J. R., de la Torre-Ubieta L., Pasaniuc B., Gandal M. J., Transcriptomic Insight Into the Polygenic Mechanisms Underlying Psychiatric Disorders. Biol. Psychiatry. 89, 54–64 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B. W. J. H., Jansen R., de Geus E. J. C., Boomsma D. I., Wright F. A., Sullivan P. F., Nikkola E., Alvarez M., Civelek M., Lusis A. J., Lehtimäki T., Raitoharju E., Kähönen M., Seppälä I., Raitakari O. T., Kuusisto J., Laakso M., Price A. L., Pajukanta P., Pasaniuc B., Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hormozdiari F., van de Bunt M., Segrè A. V., Li X., Joo J. W. J., Bilow M., Sul J. H., Sankararaman S., Pasaniuc B., Eskin E., Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 99, 1245–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fromer M., Roussos P., Sieberts S. K., Johnson J. S., Kavanagh D. H., Perumal T. M., Ruderfer D. M., Oh E. C., Topol A., Shah H. R., Klei L. L., Kramer R., Pinto D., Gümüş Z. H., Cicek A. E., Dang K. K., Browne A., Lu C., Xie L., Readhead B., Stahl E. A., Xiao J., Parvizi M., Hamamsy T., Fullard J. F., Wang Y.-C., Mahajan M. C., Derry J. M. J., Dudley J. T., Hemby S. E., Logsdon B. A., Talbot K., Raj T., Bennett D. A., De Jager P. L., Zhu J., Zhang B., Sullivan P. F., Chess A., Purcell S. M., Shinobu L. A., Mangravite L. M., Toyoshiba H., Gur R. E., Hahn C.-G., Lewis D. A., Haroutunian V., Peters M. A., Lipska B. K., Buxbaum J. D., Schadt E. E., Hirai K., Roeder K., Brennand K. J., Katsanis N., Domenici E., Devlin B., Sklar P., Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaffe A. E., Straub R. E., Shin J. H., Tao R., Gao Y., Collado-Torres L., Kam-Thong T., Xi H. S., Quan J., Chen Q., Colantuoni C., Ulrich W. S., Maher B. J., Deep-Soboslay A., BrainSeq Consortium, Cross A. J., Brandon N. J., Leek J. T., Hyde T. M., Kleinman J. E., Weinberger D. R., Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci. 21, 1117–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werling D. M., Pochareddy S., Choi J., An J.-Y., Sheppard B., Peng M., Li Z., Dastmalchi C., Santpere G., Sousa A. M. M., Tebbenkamp A. T. N., Kaur N., Gulden F. O., Breen M. S., Liang L., Gilson M. C., Zhao X., Dong S., Klei L., Cicek A. E., Buxbaum J. D., Adle-Biassette H., Thomas J.-L., Aldinger K. A., O’Day D. R., Glass I. A., Zaitlen N. A., Talkowski M. E., Roeder K., State M. W., Devlin B., Sanders S. J., Sestan N., Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 31, 107489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F. C. P., Clarke D., Gu M., Emani P., Yang Y. T., Xu M., Gandal M. J., Lou S., Zhang J., Park J. J., Yan C., Rhie S. K., Manakongtreecheep K., Zhou H., Nathan A., Peters M., Mattei E., Fitzgerald D., Brunetti T., Moore J., Jiang Y., Girdhar K., Hoffman G. E., Kalayci S., Gümüş Z. H., Crawford G. E., PsychENCODE Consortium, Roussos P., Akbarian S., Jaffe A. E., White K. P., Weng Z., Sestan N., Geschwind D. H., Knowles J. A., Gerstein M. B., Comprehensive functional genomic resource and integrative model for the human brain. Science. 362 (2018), doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandal M. J., Zhang P., Hadjimichael E., Walker R. L., Chen C., Liu S., Won H., van Bakel H., Varghese M., Wang Y., Shieh A. W., Haney J., Parhami S., Belmont J., Kim M., Moran Losada P., Khan Z., Mleczko J., Xia Y., Dai R., Wang D., Yang Y. T., Xu M., Fish K., Hof P. R., Warrell J., Fitzgerald D., White K., Jaffe A. E., PsychENCODE Consortium, Peters M. A., Gerstein M., Liu C., Iakoucheva L. M., Pinto D., Geschwind D. H., Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 362 (2018), doi: 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeng B., Bendl J., Kosoy R., Fullard J. F., Hoffman G. E., Roussos P., Multi-ancestry eQTL meta-analysis of human brain identifies candidate causal variants for brain-related traits. Nat. Genet., 1–9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umans B. D., Battle A., Gilad Y., Where Are the Disease-Associated eQTLs? Trends Genet. 37, 109–124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O. V., Gulden F. O., Pochareddy S., Sunkin S. M., Li Z., Shin Y., Zhu Y., Sousa A. M. M., Werling D. M., Kitchen R. R., Kang H. J., Pletikos M., Choi J., Muchnik S., Xu X., Wang D., Lorente-Galdos B., Liu S., Giusti-Rodríguez P., Won H., de Leeuw C. A., Pardiñas A. F., BrainSpan Consortium, PsychENCODE Consortium, PsychENCODE Developmental Subgroup, Hu M., Jin F., Li Y., Owen M. J., O’Donovan M. C., Walters J. T. R., Posthuma D., Reimers M. A., Levitt P., Weinberger D. R., Hyde T. M., Kleinman J. E., Geschwind D. H., Hawrylycz M. J., State M. W., Sanders S. J., Sullivan P. F., Gerstein M. B., Lein E. S., Knowles J. A., Sestan N., Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 362 (2018), doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang H. J., Kawasawa Y. I., Cheng F., Zhu Y., Xu X., Li M., Sousa A. M. M., Pletikos M., Meyer K. A., Sedmak G., Guennel T., Shin Y., Johnson M. B., Krsnik Z., Mayer S., Fertuzinhos S., Umlauf S., Lisgo S. N., Vortmeyer A., Weinberger D. R., Mane S., Hyde T. M., Huttner A., Reimers M., Kleinman J. E., Sestan N., Spatiotemporal transcriptome of the human brain. Nature. 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colantuoni C., Lipska B. K., Ye T., Hyde T. M., Tao R., Leek J. T., Colantuoni E. A., Elkahloun A. G., Herman M. M., Weinberger D. R., Kleinman J. E., Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 478, 519–523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willsey A. J., Sanders S. J., Li M., Dong S., Tebbenkamp A. T., Muhle R. A., Reilly S. K., Lin L., Fertuzinhos S., Miller J. A., Murtha M. T., Bichsel C., Niu W., Cotney J., Ercan-Sencicek A. G., Gockley J., Gupta A. R., Han W., He X., Hoffman E. J., Klei L., Lei J., Liu W., Liu L., Lu C., Xu X., Zhu Y., Mane S. M., Lein E. S., Wei L., Noonan J. P., Roeder K., Devlin B., Sestan N., State M. W., Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parikshak N. N., Luo R., Zhang A., Won H., Lowe J. K., Chandran V., Horvath S., Geschwind D. H., Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulsuner S., Walsh T., Watts A. C., Lee M. K., Thornton A. M., Casadei S., Rippey C., Shahin H., Consortium on the Genetics of Schizophrenia (COGS), PAARTNERS Study Group, Nimgaonkar V. L., Go R. C. P., Savage R. M., Swerdlow N. R., Gur R. E., Braff D. L., King M.-C., McClellan J. M., Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 154, 518–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker R. L., Ramaswami G., Hartl C., Mancuso N., Gandal M. J., de la Torre-Ubieta L., Pasaniuc B., Stein J. L., Geschwind D. H., Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell. 179, 750–771.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien H. E., Hannon E., Hill M. J., Toste C. C., Robertson M. J., Morgan J. E., McLaughlin G., Lewis C. M., Schalkwyk L. C., Hall L. S., Pardiñas A. F., Owen M. J., O’Donovan M. C., Mill J., Bray N. J., Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 19, 194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaffe A. E., Shin J., Collado-Torres L., Leek J. T., Tao R., Li C., Gao Y., Jia Y., Maher B. J., Hyde T. M., Kleinman J. E., Weinberger D. R., Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat. Neurosci. 18, 154–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindsay S. J., Xu Y., Lisgo S. N., Harkin L. F., Copp A. J., Gerrelli D., Clowry G. J., Talbot A., Keogh M. J., Coxhead J., Santibanez-Koref M., Chinnery P. F., HDBR Expression: A Unique Resource for Global and Individual Gene Expression Studies during Early Human Brain Development. Front. Neuroanat. 10, 86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birnbaum R., Jaffe A. E., Hyde T. M., Kleinman J. E., Weinberger D. R., Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am. J. Psychiatry. 171, 758–767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y. I., van de Geijn B., Raj A., Knowles D. A., Petti A. A., Golan D., Gilad Y., Pritchard J. K., RNA splicing is a primary link between genetic variation and disease. Science. 352, 600–604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raj B., Blencowe B. J., Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron. 87, 14–27 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Zhang X., Chen M. H., Wu X., Kodani A., Fan J., Doan R., Ozawa M., Ma J., Yoshida N., Reiter J. F., Black D. L., Kharchenko P. V., Sharp P. A., Walsh C. A., Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell. 166, 1147–1162.e15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weyn-Vanhentenryck S. M., Feng H., Ustianenko D., Duffié R., Yan Q., Jacko M., Martinez J. C., Goodwin M., Zhang X., Hengst U., Lomvardas S., Swanson M. S., Zhang C., Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 9 (2018), doi: 10.1038/s41467-018-04559-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garrido-Martín D., Borsari B., Calvo M., Reverter F., Guigó R., Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 12, 727 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taliun D., Harris D. N., Kessler M. D., Carlson J., Szpiech Z. A., Torres R., Taliun S. A. G., Corvelo A., Gogarten S. M., Kang H. M., Pitsillides A. N., LeFaive J., Lee S.-B., Tian X., Browning B. L., Das S., Emde A.-K., Clarke W. E., Loesch D. P., Shetty A. C., Blackwell T. W., Smith A. V., Wong Q., Liu X., Conomos M. P., Bobo D. M., Aguet F., Albert C., Alonso A., Ardlie K. G., Arking D. E., Aslibekyan S., Auer P. L., Barnard J., Barr R. G., Barwick L., Becker L. C., Beer R. L., Benjamin E. J., Bielak L. F., Blangero J., Boehnke M., Bowden D. W., Brody J. A., Burchard E. G., Cade B. E., Casella J. F., Chalazan B., Chasman D. I., Chen Y.-D. I., Cho M. H., Choi S. H., Chung M. K., Clish C. B., Correa A., Curran J. E., Custer B., Darbar D., Daya M., de Andrade M., DeMeo D. L., Dutcher S. K., Ellinor P. T., Emery L. S., Eng C., Fatkin D., Fingerlin T., Forer L., Fornage M., Franceschini N., Fuchsberger C., Fullerton S. M., Germer S., Gladwin M. T., Gottlieb D. J., Guo X., Hall M. E., He J., Heard-Costa N. L., Heckbert S. R., Irvin M. R., Johnsen J. M., Johnson A. D., Kaplan R., Kardia S. L. R., Kelly T., Kelly S., Kenny E. E., Kiel D. P., Klemmer R., Konkle B. A., Kooperberg C., Köttgen A., Lange L. A., Lasky-Su J., Levy D., Lin X., Lin K.-H., Liu C., Loos R. J. F., Garman L., Gerszten R., Lubitz S. A., Lunetta K. L., Mak A. C. Y., Manichaikul A., Manning A. K., Mathias R. A., McManus D. D., McGarvey S. T., Meigs J. B., Meyers D. A., Mikulla J. L., Minear M. A., Mitchell B. D., Mohanty S., Montasser M. E., Montgomery C., Morrison A. C., Murabito J. M., Natale A., Natarajan P., Nelson S. C., North K. E., O’Connell J. R., Palmer N. D., Pankratz N., Peloso G. M., Peyser P. A., Pleiness J., Post W. S., Psaty B. M., Rao D. C., Redline S., Reiner A. P., Roden D., Rotter J. I., Ruczinski I., Sarnowski C., Schoenherr S., Schwartz D. A., Seo J.-S., Seshadri S., Sheehan V. A., Sheu W. H., Shoemaker M. B., Smith N. L., Smith J. A., Sotoodehnia N., Stilp A. M., Tang W., Taylor K. D., Telen M., Thornton T. A., Tracy R. P., Van Den Berg D. J., Vasan R. S., Viaud-Martinez K. A., Vrieze S., Weeks D. E., Weir B. S., Weiss S. T., Weng L.-C., Willer C. J., Zhang Y., Zhao X., Arnett D. K., Ashley-Koch A. E., Barnes K. C., Boerwinkle E., Gabriel S., Gibbs R., Rice K. M., Rich S. S., Silverman E. K., Qasba P., Gan W., NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Papanicolaou G. J., Nickerson D. A., Browning S. R., Zody M. C., Zöllner S., Wilson J. G., Cupples L. A., Laurie C. C., Jaquish C. E., Hernandez R. D., O’Connor T. D., Abecasis G. R., Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., O’Donnell-Luria A. H., Ware J. S., Hill A. J., Cummings B. B., Tukiainen T., Birnbaum D. P., Kosmicki J. A., Duncan L. E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D. N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M. I., Moonshine A. L., Natarajan P., Orozco L., Peloso G. M., Poplin R., Rivas M. A., Ruano-Rubio V., Rose S. A., Ruderfer D. M., Shakir K., Stenson P. D., Stevens C., Thomas B. P., Tiao G., Tusie-Luna M. T., Weisburd B., Won H.-H., Yu D., Altshuler D. M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J. C., Gabriel S. B., Getz G., Glatt S. J., Hultman C. M., Kathiresan S., Laakso M., McCarroll S., McCarthy M. I., McGovern D., McPherson R., Neale B. M., Palotie A., Purcell S. M., Saleheen D., Scharf J. M., Sklar P., Sullivan P. F., Tuomilehto J., Tsuang M. T., Watkins H. C., Wilson J. G., Daly M. J., MacArthur D. G., Exome Aggregation Consortium, Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polioudakis D., de la Torre-Ubieta L., Langerman J., Elkins A. G., Shi X., Stein J. L., Vuong C. K., Nichterwitz S., Gevorgian M., Opland C. K., Lu D., Connell W., Ruzzo E. K., Lowe J. K., Hadzic T., Hinz F. I., Sabri S., Lowry W. E., Gerstein M. B., Plath K., Geschwind D. H., A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron. 103, 785–801.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J. M., Sisu C., Wright J., Armstrong J., Barnes I., Berry A., Bignell A., Carbonell Sala S., Chrast J., Cunningham F., Di Domenico T., Donaldson S., Fiddes I. T., García Girón C., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Hunt T., Izuogu O. G., Lagarde J., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Xu J., Yates A., Zerbino D., Zhang Y., Aken B., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Reymond A., Tress M. L., Flicek P., GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y. I., Knowles D. A., Humphrey J., Barbeira A. N., Dickinson S. P., Im H. K., Pritchard J. K., Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 50, 151–158 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storey J. D., Tibshirani R., Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delaneau O., Ongen H., Brown A. A., Fort A., Panousis N. I., Dermitzakis E. T., A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 8, 15452 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang G., Sarkar A., Carbonetto P., Stephens M., A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol. 82, 1273–1300 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikram M. A., Fornage M., Smith A. V., Seshadri S., Schmidt R., Debette S., Vrooman H. A., Sigurdsson S., Ropele S., Taal H. R., Mook-Kanamori D. O., Coker L. H., Longstreth W. T. Jr, Niessen W. J., DeStefano A. L., Beiser A., Zijdenbos A. P., Struchalin M., Jack C. R. Jr, Rivadeneira F., Uitterlinden A. G., Knopman D. S., Hartikainen A.-L., Pennell C. E., Thiering E., Steegers E. A. P., Hakonarson H., Heinrich J., Palmer L. J., Jarvelin M.-R., McCarthy M. I., Grant S. F. A., St Pourcain B., Timpson N. J., Smith G. D., Sovio U., Early Growth Genetics Consortium, Nalls M. A., Au R., Hofman A., Gudnason H., van der Lugt A., Harris T. B., Meeks W. M., Vernooij M. W., van Buchem M. A., Catellier D., Jaddoe V. W. V., Gudnason V., Windham B. G., Wolf P. A., van Duijn C. M., Mosley T. H. Jr, Schmidt H., Launer L. J., Breteler M. M. B., DeCarli C., Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium, Common variants at 6q22 and 17q21 are associated with intracranial volume. Nat. Genet. 44, 539–544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The 1000 Genomes Project Consortium, Auton A., Abecasis G. R., Altshuler D. M. (Co-Chair), Durbin R. M. (Co-Chair), Abecasis G. R., Bentley D. R., Chakravarti A., Clark A. G., Donnelly P., Eichler E. E., Flicek P., Gabriel S. B., Gibbs R. A., Green E. D., Hurles M. E., Knoppers B. M., Korbel J. O., Lander E. S., Lee C., Lehrach H., Mardis E. R., Marth G. T., McVean G. A., Nickerson D. A., Schmidt J. P., Sherry S. T., Wang J., Wilson R. K., Gibbs R. A. (Principal, Boerwinkle E., Doddapaneni H., Han Y., Korchina V., Kovar C., Lee S., Muzny D., Reid J. G., Zhu Y., Wang J. (Principal, Chang Y., Feng Q., Fang X., Guo X., Jian M., Jiang H., Jin X., Lan T., Li G., Li J., Li Y., Liu S., Liu X., Lu Y., Ma X., Tang M., Wang B., Wang G., Wu H., Wu R., Xu X., Yin Y., Zhang D., Zhang W., Zhao J., Zhao M., Zheng X., Lander E. S. (Principal, Altshuler D. M., Gabriel S. B. (Co-Chair), Gupta N., Gharani N., Toji L. H., Gerry N. P., Resch A. M., Flicek P. (Principal, Barker J., Clarke L., Gil L., Hunt S. E., Kelman G., Kulesha E., Leinonen R., McLaren W. M., Radhakrishnan R., Roa A., Smirnov D., Smith R. E., Streeter I., Thormann A., Toneva I., Vaughan B., Zheng-Bradley X., Bentley D. R. (Principal, Grocock R., Humphray S., James T., Kingsbury Z., Lehrach H. (Principal, Sudbrak R. (Project L, Albrecht M. W., Amstislavskiy V. S., Borodina T. A., Lienhard M., Mertes F., Sultan M., Timmermann B., Yaspo M.-L., Mardis E. R. (Co-Princi R. K. Wilson (Co-Princi L. Fulton, Fulton R., Sherry S. T. (Principal, Ananiev V., Belaia Z., Beloslyudtsev D., Bouk N., Chen C., Church D., Cohen R., Cook C., Garner J., Hefferon T., Kimelman M., Liu C., Lopez J., Meric P., O’Sullivan C., Ostapchuk Y., Phan L., Ponomarov S., Schneider V., Shekhtman E., Sirotkin K., Slotta D., Zhang H., McVean G. A. (Principal, Durbin R. M. (Principal, Balasubramaniam S., Burton J., Danecek P., Keane T. M., Kolb-Kokocinski A., McCarthy S., Stalker J., Quail M., Schmidt J. P. (Principal, Davies C. J., Gollub J., Webster T., Wong B., Zhan Y., Auton A. (Principal, Campbell C. L., Kong Y., Marcketta A., Gibbs R. A. (Principal, Yu F. (Project L, Antunes L., Bainbridge M., Muzny D., Sabo A., Huang Z., Wang J. (Principal, Coin L. J. M., Fang L., Guo X., Jin X., Li G., Li Q., Li Y., Li Z., Lin H., Liu B., Luo R., Shao H., Xie Y., Ye C., Yu C., Zhang F., Zheng H., Zhu H., Alkan C., Dal E., Kahveci F., Marth G. T. (Principal, Garrison E. P. (Project L, Kural D., Lee W.-P., Fung Leong W., Stromberg M., Ward A. N., Wu J., Zhang M., Daly M. J. (Principal, DePristo M. A. (Project L, Handsaker R. E. (Project L, Altshuler D. M., Banks E., Bhatia G., del Angel G., Gabriel S. B., Genovese G., Gupta N., Li H., Kashin S., Lander E. S., McCarroll S. A., Nemesh J. C., Poplin R. E., Yoon S. C. (Principal, Lihm J., Makarov V., Clark A. G. (Principal, Gottipati S., Keinan A., Rodriguez-Flores J. L., Korbel J. O. (Principal, Rausch T. (Project L, Fritz M. H., Stütz A. M., Flicek P. (Principal, Beal K., Clarke L., Datta A., Herrero J., McLaren W. M., Ritchie G. R. S., Smith R. E., Zerbino D., Zheng-Bradley X., Sabeti P. C. (Principal, Shlyakhter I., Schaffner S. F., Vitti J., Cooper D. N. (Principal, Ball E. V., Stenson P. D., Bentley D. R. (Principal, Barnes B., Bauer M., Keira Cheetham R., Cox A., Eberle M., Humphray S., Kahn S., Murray L., Peden J., Shaw R., Kenny E. E. (Principal, Batzer M. A. (Principal, Konkel M. K., Walker J. A., MacArthur D. G. (Principal, Lek M., Sudbrak R. (Project L, Amstislavskiy V. S., Herwig R., Mardis E. R. (Co-Princi, Ding L., Koboldt D. C., Larson D., Ye K., Gravel S., Swaroop A., Chew E., Lappalainen T. (Principal, Erlich Y. (Principal, Gymrek M., Frederick Willems T., Simpson J. T., Shriver M. D. (Principal, Rosenfeld J. A. (Principal, Bustamante C. D. (Principal, Montgomery S. B. (Principal, De La Vega F. M. (Principal, Byrnes J. K., Carroll A. W., DeGorter M. K., Lacroute P., Maples B. K., Martin A. R., Moreno-Estrada A., Shringarpure S. S., Zakharia F., Halperin E. (Principal, Baran Y., Lee C. (Principal, Cerveira E., Hwang J., Malhotra A. (Co-Projec, Plewczynski D., Radew K., Romanovitch M., Zhang C. (Co-Projec, Hyland F. C. L., Craig D. W. (Principal, Christoforides A., Homer N., Izatt T., Kurdoglu A. A., Sinari S. A., Squire K., Sherry S. T. (Principal, Xiao C., Sebat J. (Principal, Antaki D., Gujral M., Noor A., Ye K., Burchard E. G. (Principal, Hernandez R. D. (Principal, Gignoux C. R., Haussler D. (Principal, Katzman S. J., James Kent W., Howie B., Ruiz-Linares A. (Principal, Dermitzakis E. T. (Principal, Devine S. E. (Principal, Abecasis G. R. (Principal, Min Kang H. (Project L, Kidd J. M. (Principal, Blackwell T., Caron S., Chen W., Emery S., Fritsche L., Fuchsberger C., Jun G., Li B., Lyons R., Scheller C., Sidore C., Song S., Sliwerska E., Taliun D., Tan A., Welch R., Kate Wing M., Zhan X., Awadalla P. (Principal, Hodgkinson A., Li Y., Shi X. (Principal, Quitadamo A., Lunter G. (Principal, McVean G. A. (Principal, Marchini J. L. (Principal, Myers S. (Principal, Churchhouse C., Delaneau O., Gupta-Hinch A., Kretzschmar W., Iqbal Z., Mathieson I., Menelaou A., Rimmer A., Xifara D. K., Oleksyk T. K. (Principal, Fu Y. (Principal, Liu X., Xiong M., Jorde L. (Principal, Witherspoon D., Xing J., Eichler E. E. (Principal, Browning B. L. (Principal, Browning S. R. (Principal, Hormozdiari F., Sudmant P. H., Khurana E. (Principal, Durbin R. M. (Principal, Hurles M. E. (Principal, Tyler-Smith C. (Principal, Albers C. A., Ayub Q., Balasubramaniam S., Chen Y., Colonna V., Danecek P., Jostins L., Keane T. M., McCarthy S., Walter K., Xue Y., Gerstein M. B. (Principal, Abyzov A., Balasubramanian S., Chen J., Clarke D., Fu Y., Harmanci A. O., Jin M., Lee D., Liu J., Jasmine Mu X., Zhang J., Zhang Y., Li Y., Luo R., Zhu H., Alkan C., Dal E., Kahveci F., Marth G. T. (Principal, Garrison E. P., Kural D., Lee W.-P., Ward A. N., Wu J., Zhang M., McCarroll S. A. (Principal, Handsaker R. E. (Project L, Altshuler D. M., Banks E., del Angel G., Genovese G., Hartl C., Li H., Kashin S., Nemesh J. C., Shakir K., Yoon S. C. (Principal, Lihm J., Makarov V., Degenhardt J., Korbel J. O. (Principal, Fritz M. H., Meiers S., Raeder B., Rausch T., Stütz A. M., Flicek P. (Principal, Paolo Casale F., Clarke L., Smith R. E., Stegle O., Zheng-Bradley X., Bentley D. R. (Principal, Barnes B., Keira Cheetham R., Eberle M., Humphray S., Kahn S., Murray L., Shaw R., Lameijer E.-W., Batzer M. A. (Principal, Konkel M. K., Walker J. A., Ding L. (Principal, Hall I., Ye K., Lacroute P., Lee C. (Principal, Cerveira E., Malhotra A., Hwang J., Plewczynski D., Radew K., Romanovitch M., Zhang C., Craig D. W. (Principal, Homer N., Church D., Xiao C., Sebat J. (Principal, Antaki D., Bafna V., Michaelson J., Ye K., Devine S. E. (Principal, Gardner E. J. (Project L, Abecasis G. R. (Principal, Kidd J. M. (Principal, Mills R. E. (Principal, Dayama G., Emery S., Jun G., Shi X. (Principal, Quitadamo A., Lunter G. (Principal, McVean G. A. (Principal, Chen K., Fan X., Chong Z., Chen T., Witherspoon D., Xing J., Eichler E. E. (Principal, Chaisson M. J., Hormozdiari F., Huddleston J., Malig M., Nelson B. J., Sudmant P. H., Parrish N. F., Khurana E. (Principal, Hurles M. E. (Principal, Blackburne B., Lindsay S. J., Ning Z., Walter K., Zhang Y., Gerstein M. B. (Principal, Abyzov A., Chen J., Clarke D., Lam H., Jasmine Mu X., Sisu C., Zhang J., Zhang Y., Gibbs R. A. (Principal, Yu F. (Project L, Bainbridge M., Challis D., Evani U. S., Kovar C., Lu J., Muzny D., Nagaswamy U., Reid J. G., Sabo A., Yu J., Guo X., Li W., Li Y., Wu R., Marth G. T. (Principal, Garrison E. P., Fung Leong W., Ward A. N., del Angel G., DePristo M. A., Gabriel S. B., Gupta N., Hartl C., Poplin R. E., Clark A. G. (Principal, Rodriguez-Flores J. L., Flicek P. (Principal, Clarke L. (Project L, Smith R. E., Zheng-Bradley X., MacArthur D. G. (Principal, Mardis E. R. (Principal, Fulton R., Koboldt D. C., Gravel S., Bustamante C. D. (Principal, Craig D. W. (Principal, Christoforides A., Homer N., Izatt T., Sherry S. T. (Principal, Xiao C., Dermitzakis E. T. (Principal, Abecasis G. R. (Principal, Min Kang H., McVean G. A. (Principal, Gerstein M. B. (Principal, Balasubramanian S., Habegger L., Yu H. (Principal, Flicek P. (Principal, Clarke L., Cunningham F., Dunham I., Zerbino D., Zheng-Bradley X., Lage K. (Principal, Berg Jespersen J., Horn H., Montgomery S. B. (Principal, DeGorter M. K., Khurana E. (Principal, Tyler-Smith C. (Principal, Chen Y., Colonna V., Xue Y., Gerstein M. B. (Principal, Balasubramanian S., Fu Y., Kim D., Auton A. (Principal, Marcketta A., Desalle R., Narechania A., Wilson Sayres M. A., Garrison E. P., Handsaker R. E., Kashin S., McCarroll S. A., Rodriguez-Flores J. L., Flicek P. (Principal, Clarke L., Zheng-Bradley X., Erlich Y., Gymrek M., Frederick Willems T., Bustamante C. D. (Principal, Mendez F. L., David Poznik G., Underhill P. A., Lee C., Cerveira E., Malhotra A., Romanovitch M., Zhang C., Abecasis G. R. (Principal, Coin L. (Principal, Shao H., Mittelman D., Tyler-Smith C. (Principal, Ayub Q., Banerjee R., Cerezo M., Chen Y., Fitzgerald T. W., Louzada S., Massaia A., McCarthy S., Ritchie G. R., Xue Y., Yang F., Gibbs R. A. (Principal, Kovar C., Kalra D., Hale W., Muzny D., Reid J. G., Wang J. (Principal, Dan X., Guo X., Li G., Li Y., Ye C., Zheng X., Altshuler D. M., Flicek P. (Principal, Clarke L., Zheng-Bradley X., Bentley D. R. (Principal, Cox A., Humphray S., Kahn S., Sudbrak R. (Project L, Albrecht M. W., Lienhard M., Larson D., Craig D. W. (Principal, Izatt T., Kurdoglu A. A., Sherry S. T. (Principal, Xiao C., Haussler D. (Principal, Abecasis G. R. (Principal, McVean G. A. (Principal, Durbin R. M. (Principal, Balasubramaniam S., Keane T. M., McCarthy S., Stalker J., Chakravarti A. (Co-Chair), Knoppers B. M. (Co-Chair), Abecasis G. R., Barnes K. C., Beiswanger C., Burchard E. G., Bustamante C. D., Cai H., Cao H., Durbin R. M., Gerry N. P., Gharani N., Gibbs R. A., Gignoux C. R., Gravel S., Henn B., Jones D., Jorde L., Kaye J. S., Keinan A., Kent A., Kerasidou A., Li Y., Mathias R., McVean G. A., Moreno-Estrada A., Ossorio P. N., Parker M., Resch A. M., Rotimi C. N., Royal Charmaine D., Sandoval K., Su Y., Sudbrak R., Tian Z., Tishkoff S., Toji L. H., Tyler-Smith C., Via M., Wang Y., Yang H., Yang L., Zhu J., Bodmer W., Bedoya G., Ruiz-Linares A., Cai Z., Gao Y., Chu J., Peltonen L., Garcia-Montero A., Orfao A., Dutil J., Martinez-Cruzado J. C., Oleksyk T. K., Barnes K. C., Mathias R. A., Hennis A., Watson H., McKenzie C., Qadri F., LaRocque R., Sabeti P. C., Zhu J., Deng X., Sabeti P. C., Asogun D., Folarin O., Happi C., Omoniwa O., Stremlau M., Tariyal R., Jallow M., Sisay Joof F., Corrah T., Rockett K., Kwiatkowski D., Kooner J., Tinh Hiêǹ T., Dunstan S. J., Thuy Hang N., Fonnie R., Garry R., Kanneh L., Moses L., Sabeti P. C., Schieffelin J., Grant D. S., Gallo C., Poletti G., Saleheen D., Rasheed A., Brooks L. D., Felsenfeld A. L., McEwen J. E., Vaydylevich Y., Green E. D., Duncanson A., Dunn M., Schloss J. A., Wang J., Yang H., Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Min Kang H., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., Corresponding authors, Steering committee, Production group, Baylor College of Medicine, BGI-Shenzhen, Broad Institute of MIT and Harvard, Coriell Institute for Medical Research, European Molecular Biology Laboratory, European Bioinformatics Institute, Illumina, Max Planck Institute for Molecular Genetics, McDonnell Genome Institute at Washington University, US National Institutes of Health, University of Oxford, Wellcome Trust Sanger Institute, Analysis group, Affymetrix, Albert Einstein College of Medicine, Baylor College of Medicine, BGI-Shenzhen, Bilkent University, Boston College, Broad Institute of MIT and Harvard, Cold Spring Harbor Laboratory, Cornell University, European Molecular Biology Laboratory, European Molecular Biology Laboratory, European Bioinformatics Institute, Harvard University, Human Gene Mutation Database, Illumina, Icahn School of Medicine at Mount Sinai, Louisiana State University, Massachusetts General Hospital, Max Planck Institute for Molecular Genetics, McDonnell Genome Institute at Washington University, McGill University, National Eye Institute, NIH, New York Genome Center, Ontario Institute for Cancer Research, Pennsylvania State University, Rutgers Cancer Institute of New Jersey, Stanford University, Tel-Aviv University, The Jackson Laboratory for Genomic Medicine, T. F. Scientific, Translational Genomics Research Institute, US National Institutes of Health, University of California, San Diego, University of California, San Francisco, University of California, Santa Cruz, University of Chicago, University College London, University of Geneva, University of Maryland School of Medicine, University of Michigan, University of Montréal, University of North Carolina at Chapel Hill, University of North Carolina at Charlotte, University of Oxford, University of Puerto Rico, University of Texas Health Sciences Center at Houston, University of Utah, University of Washington, Weill Cornell Medical College, Wellcome Trust Sanger Institute, Yale University, Structural variation group, BGI-Shenzhen, Bilkent University, Boston College, Broad Institute of MIT and Harvard, Cold Spring Harbor Laboratory, Cornell University, European Molecular Biology Laboratory, European Molecular Biology Laboratory, European Bioinformatics Institute, Illumina, Leiden University Medical Center, Louisiana State University, McDonnell Genome Institute at Washington University, Stanford University, The Jackson Laboratory for Genomic Medicine, Translational Genomics Research Institute, US National Institutes of Health, University of California, San Diego, University of Maryland School of Medicine, University of Michigan, University of North Carolina at Charlotte, University of Oxford, University of Texas MD Anderson Cancer Center, University of Utah, University of Washington, Vanderbilt University School of Medicine, Weill Cornell Medical College, Wellcome Trust Sanger Institute, Yale University, Exome group, Baylor College of Medicine, BGI-Shenzhen, Boston College, Broad Institute of MIT and Harvard, Cornell University, European Molecular Biology Laboratory, European Bioinformatics Institute, Massachusetts General Hospital, McDonnell Genome Institute at Washington University, McGill University, Stanford University, Translational Genomics Research Institute, US National Institutes of Health, University of Geneva, University of Michigan, University of Oxford, Yale University, Functional interpretation group, Cornell University, European Molecular Biology Laboratory, European Bioinformatics Institute, Harvard University, Stanford University, Weill Cornell Medical College, Wellcome Trust Sanger Institute, Yale University, Chromosome Y group, Albert Einstein College of Medicine, American Museum of Natural History, Arizona State University, Boston College, Broad Institute of MIT and Harvard, Cornell University, European Molecular Biology Laboratory, European Bioinformatics Institute, New York Genome Center, Stanford University, The Jackson Laboratory for Genomic Medicine, University of Michigan, University of Queensland, Virginia Bioinformatics Institute, Wellcome Trust Sanger Institute, Data coordination center group, Baylor College of Medicine, BGIShenzhen, Broad Institute of MIT and Harvard, European Molecular Biology Laboratory, European Bioinformatics Institute, Illumina, Max Planck Institute for Molecular Genetics, McDonnell Genome Institute at Washington University, Translational Genomics Research Institute, US National Institutes of Health, University of California, Santa Cruz, University of Michigan, University of Oxford, Wellcome Trust Sanger Institute, Samples and ELSI group, Sample collection, British from England and Scotland (GBR), Colombians in Medellín, Colombia (CLM), Han Chinese South (CHS), Finnish in Finland (FIN), Iberian Populations in Spain (IBS), Puerto Ricans in Puerto Rico (PUR), African Caribbean in Barbados (ACB), Bengali in Bangladesh (BEB), Chinese Dai in Xishuangbanna, China (CDX), Esan in Nigeria (ESN), Gambian in Western Division-Mandinka (GWD), Indian Telugu in the UK (ITU) and Sri Lankan Tamil in the UK (STU), Kinh in Ho Chi Minh City, Vietnam (KHV), Mende in Sierra Leone (MSL), Peruvian in Lima, Peru (PEL), Punjabi in Lahore, Pakistan (PJL), Scientific management, Writing group, A global reference for human genetic variation. Nature. 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaitlen N., Paşaniuc B., Gur T., Ziv E., Halperin E., Leveraging genetic variability across populations for the identification of causal variants. Am. J. Hum. Genet. 86, 23–33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahajan A., Go M. J., Zhang W., Below J. E., Gaulton K. J., Ferreira T., Horikoshi M., Johnson A. D., Ng M. C. Y., Prokopenko I., Saleheen D., Wang X., Zeggini E., Abecasis G. R., Adair L. S., Almgren P., Atalay M., Aung T., Baldassarre D., Balkau B., Bao Y., Barnett A. H., Barroso I., Basit A., Been L. F., Beilby J., Bell G. I., Benediktsson R., Bergman R. N., Boehm B. O., Boerwinkle E., Bonnycastle L. L., Burtt N., Cai Q., Campbell H., Carey J., Cauchi S., Caulfield M., Chan J. C. N., Chang L.-C., Chang T.-J., Chang Y.-C., Charpentier G., Chen C.-H., Chen H., Chen Y.-T., Chia K.-S., Chidambaram M., Chines P. S., Cho N. H., Cho Y. M., Chuang L.-M., Collins F. S., Cornelis M. C., Couper D. J., Crenshaw A. T., van Dam R. M., Danesh J., Das D., de Faire U., Dedoussis G., Deloukas P., Dimas A. S., Dina C., Doney A. S. F., Donnelly P. J., Dorkhan M., van Duijn C., Dupuis J., Edkins S., Elliott P., Emilsson V., Erbel R., Eriksson J. G., Escobedo J., Esko T., Eury E., Florez J. C., Fontanillas P., Forouhi N. G., Forsen T., Fox C., Fraser R. M., Frayling T. M., Froguel P., Frossard P., Gao Y., Gertow K., Gieger C., Gigante B., Grallert H., Grant G. B., Groop L. C., Groves C. J., Grundberg E., Guiducci C., Hamsten A., Han B.-G., Hara K., Hassanali N., Hattersley A. T., Hayward C., Hedman A. K., Herder C., Hofman A., Holmen O. L., Hovingh K., Hreidarsson A. B., Hu C., Hu F. B., Hui J., Humphries S. E., Hunt S. E., Hunter D. J., Hveem K., Hydrie Z. I., Ikegami H., Illig T., Ingelsson E., Islam M., Isomaa B., Jackson A. U., Jafar T., James A., Jia W., Jöckel K.-H., Jonsson A., Jowett J. B. M., Kadowaki T., Kang H. M., Kanoni S., Kao W. H. L., Kathiresan S., Kato N., Katulanda P., Keinanen-Kiukaanniemi S. M., Kelly A. M., Khan H., Khaw K.-T., Khor C.-C., Kim H.-L., Kim S., Kim Y. J., Kinnunen L., Klopp N., Kong A., Korpi-Hyövälti E., Kowlessur S., Kraft P., Kravic J., Kristensen M. M., Krithika S., Kumar A., Kumate J., Kuusisto J., Kwak S. H., Laakso M., Lagou V., Lakka T. A., Langenberg C., Langford C., Lawrence R., Leander K., Lee J.-M., Lee N. R., Li M., Li X., Li Y., Liang J., Liju S., Lim W.-Y., Lind L., Lindgren C. M., Lindholm E., Liu C.-T., Liu J. J., Lobbens S., Long J., Loos R. J. F., Lu W., ’an Luan J., Lyssenko V., Ma R. C. W., Maeda S., Mägi R., Männistö S., Matthews D. R., Meigs J. B., Melander O., Metspalu A., Meyer J., Mirza G., Mihailov E., Moebus S., Mohan V., Mohlke K. L., Morris A. D., Mühleisen T. W., Müller-Nurasyid M., Musk B., Nakamura J., Nakashima E., Navarro P., Ng P.-K., Nica A. C., Nilsson P. M., Njølstad I., Nöthen M. M., Ohnaka K., Ong T. H., Owen K. R., Palmer C. N. A., Pankow J. S., Park K. S., Parkin M., Pechlivanis S., Pedersen N. L., Peltonen L., Perry J. R. B., Peters A., Pinidiyapathirage J. M., Platou C. G. P., Potter S., Price J. F., Qi L., Radha V., Rallidis L., Rasheed A., Rathmann W., Rauramaa R., Raychaudhuri S., Rayner N. W., Rees S. D., Rehnberg E., Ripatti S., Robertson N., Roden M., Rossin E. J., Rudan I., Rybin D., Saaristo T. E., Salomaa V., Saltevo J., Samuel M., Sanghera D. K., Saramies J., Scott J., Scott L. J., Scott R. A., Segrè A. V., Sehmi J., Sennblad B., Shah N., Shah S., Shera A. S., Shu X. O., Shuldiner A. R., Sigurðsson G., Sijbrands E., Silveira A., Sim X., Sivapalaratnam S., Small K. S., So W. Y., Stančáková A., Stefansson K., Steinbach G., Steinthorsdottir V., Stirrups K., Strawbridge R. J., Stringham H. M., Sun Q., Suo C., Syvänen A.-C., Takayanagi R., Takeuchi F., Tay W. T., Teslovich T. M., Thorand B., Thorleifsson G., Thorsteinsdottir U., Tikkanen E., Trakalo J., Tremoli E., Trip M. D., Tsai F. J., Tuomi T., Tuomilehto J., Uitterlinden A. G., Valladares-Salgado A., Vedantam S., Veglia F., Voight B. F., Wang C., Wareham N. J., Wennauer R., Wickremasinghe A. R., Wilsgaard T., Wilson J. F., Wiltshire S., Winckler W., Wong T. Y., Wood A. R., Wu J.-Y., Wu Y., Yamamoto K., Yamauchi T., Yang M., Yengo L., Yokota M., Young R., Zabaneh D., Zhang F., Zhang R., Zheng W., Zimmet P. Z., Altshuler D., Bowden D. W., Cho Y. S., Cox N. J., Cruz M., Hanis C. L., Kooner J., Lee J.-Y., Seielstad M., Teo Y. Y., Boehnke M., Parra E. J., Chambers J. C., Tai E. S., McCarthy M. I., Morris A. P., Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet. 46, 234–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohammadi P., Castel S. E., Brown A. A., Lappalainen T., Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kichaev G., Yang W.-Y., Lindstrom S., Hormozdiari F., Eskin E., Price A. L., Kraft P., Pasaniuc B., Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 10, e1004722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kichaev G., Pasaniuc B., Leveraging Functional-Annotation Data in Trans-ethnic Fine-Mapping Studies. Am. J. Hum. Genet. 97, 260–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulik-Sullivan B. K., Loh P.-R., Finucane H. K., Ripke S., Yang J., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N., Daly M. J., Price A. L., Neale B. M., LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hormozdiari F., Gazal S., van de Geijn B., Finucane H. K., Ju C. J.-T., Loh P.-R., Schoech A., Reshef Y., Liu X., O’Connor L., Gusev A., Eskin E., Price A. L., Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat. Genet. 50, 1041–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mostafavi H., Spence J. P., Naqvi S., Pritchard J. K., Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv (2022), p. 2022.05.07.491045. [Google Scholar]
- 53.Yao D. W., O’Connor L. J., Price A. L., Gusev A., Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet. 52, 626–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demontis D., Walters R. K., Martin J., Mattheisen M., Als T. D., Agerbo E., Baldursson G., Belliveau R., Bybjerg-Grauholm J., Bækvad-Hansen M., Cerrato F., Chambert K., Churchhouse C., Dumont A., Eriksson N., Gandal M., Goldstein J. I., Grasby K. L., Grove J., Gudmundsson O. O., Hansen C. S., Hauberg M. E., Hollegaard M. V., Howrigan D. P., Huang H., Maller J. B., Martin A. R., Martin N. G., Moran J., Pallesen J., Palmer D. S., Pedersen C. B., Pedersen M. G., Poterba T., Poulsen J. B., Ripke S., Robinson E. B., Satterstrom F. K., Stefansson H., Stevens C., Turley P., Walters G. B., Won H., Wright M. J., ADHD Working Group of the Psychiatric Genomics Consortium (PGC), Early Lifecourse & Genetic Epidemiology (EAGLE) Consortium, 23andMe Research Team, Andreassen O. A., Asherson P., Burton C. L., Boomsma D. I., Cormand B., Dalsgaard S., Franke B., Gelernter J., Geschwind D., Hakonarson H., Haavik J., Kranzler H. R., Kuntsi J., Langley K., Lesch K.-P., Middeldorp C., Reif A., Rohde L. A., Roussos P., Schachar R., Sklar P., Sonuga-Barke E. J. S., Sullivan P. F., Thapar A., Tung J. Y., Waldman I. D., Medland S. E., Stefansson K., Nordentoft M., Hougaard D. M., Werge T., Mors O., Mortensen P. B., Daly M. J., Faraone S. V., Børglum A. D., Neale B. M., Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howard D. M., Adams M. J., Shirali M., Clarke T.-K., Marioni R. E., Davies G., Coleman J. R. I., Alloza C., Shen X., Barbu M. C., Wigmore E. M., Gibson J., 23andMe Research Team, Hagenaars S. P., Lewis C. M., Ward J., Smith D. J., Sullivan P. F., Haley C. S., Breen G., Deary I. J., McIntosh A. M., Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 9, 1470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mullins N., Forstner A. J., O’Connell K. S., Coombes B., Coleman J. R. I., Qiao Z., Als T. D., Bigdeli T. B., Børte S., Bryois J., Charney A. W., Drange O. K., Gandal M. J., Hagenaars S. P., Ikeda M., Kamitaki N., Kim M., Krebs K., Panagiotaropoulou G., Schilder B. M., Sloofman L. G., Steinberg S., Trubetskoy V., Winsvold B. S., Won H.-H., Abramova L., Adorjan K., Agerbo E., Al Eissa M., Albani D., Alliey-Rodriguez N., Anjorin A., Antilla V., Antoniou A., Awasthi S., Baek J. H., Bækvad-Hansen M., Bass N., Bauer M., Beins E. C., Bergen S. E., Birner A., Bøcker Pedersen C., Bøen E., Boks M. P., Bosch R., Brum M., Brumpton B. M., Brunkhorst-Kanaan N., Budde M., Bybjerg-Grauholm J., Byerley W., Cairns M., Casas M., Cervantes P., Clarke T.-K., Cruceanu C., Cuellar-Barboza A., Cunningham J., Curtis D., Czerski P. M., Dale A. M., Dalkner N., David F. S., Degenhardt F., Djurovic S., Dobbyn A. L., Douzenis A., Elvsåshagen T., Escott-Price V., Ferrier I. N., Fiorentino A., Foroud T. M., Forty L., Frank J., Frei O., Freimer N. B., Frisén L., Gade K., Garnham J., Gelernter J., Giørtz Pedersen M., Gizer I. R., Gordon S. D., Gordon-Smith K., Greenwood T. A., Grove J., Guzman-Parra J., Ha K., Haraldsson M., Hautzinger M., Heilbronner U., Hellgren D., Herms S., Hoffmann P., Holmans P. A., Huckins L., Jamain S., Johnson J. S., Kalman J. L., Kamatani Y., Kennedy J. L., Kittel-Schneider S., Knowles J. A., Kogevinas M., Koromina M., Kranz T. M., Kranzler H. R., Kubo M., Kupka R., Kushner S. A., Lavebratt C., Lawrence J., Leber M., Lee H.-J., Lee P. H., Levy S. E., Lewis C., Liao C., Lucae S., Lundberg M., MacIntyre D. J., Magnusson S. H., Maier W., Maihofer A., Malaspina D., Maratou E., Martinsson L., Mattheisen M., McCarroll S. A., McGregor N. W., McGuffin P., McKay J. D., Medeiros H., Medland S. E., Millischer V., Montgomery G. W., Moran J. L., Morris D. W., Mühleisen T. W., O’Brien N., O’Donovan C., Olde Loohuis L. M., Oruc L., Papiol S., Pardiñas A. F., Perry A., Pfennig A., Porichi E., Potash J. B., Quested D., Raj T., Rapaport M. H., DePaulo J. R., Regeer E. J., Rice J. P., Rivas F., Rivera M., Roth J., Roussos P., Ruderfer D. M., Sánchez-Mora C., Schulte E. C., Senner F., Sharp S., Shilling P. D., Sigurdsson E., Sirignano L., Slaney C., Smeland O. B., Smith D. J., Sobell J. L., Søholm Hansen C., Soler Artigas M., Spijker A. T., Stein D. J., Strauss J. S., Świątkowska B., Terao C., Thorgeirsson T. E., Toma C., Tooney P., Tsermpini E.-E., Vawter M. P., Vedder H., Walters J. T. R., Witt S. H., Xi S., Xu W., Yang J. M. K., Young A. H., Young H., Zandi P. P., Zhou H., Zillich L., HUNT All-In Psychiatry, Adolfsson R., Agartz I., Alda M., Alfredsson L., Babadjanova G., Backlund L., Baune B. T., Bellivier F., Bengesser S., Berrettini W. H., Blackwood D. H. R., Boehnke M., Børglum A. D., Breen G., Carr V. J., Catts S., Corvin A., Craddock N., Dannlowski U., Dikeos D., Esko T., Etain B., Ferentinos P., Frye M., Fullerton J. M., Gawlik M., Gershon E. S., Goes F. S., Green M. J., Grigoroiu-Serbanescu M., Hauser J., Henskens F., Hillert J., Hong K. S., Hougaard D. M., Hultman C. M., Hveem K., Iwata N., Jablensky A. V., Jones I., Jones L. A., Kahn R. S., Kelsoe J. R., Kirov G., Landén M., Leboyer M., Lewis C. M., Li Q. S., Lissowska J., Lochner C., Loughland C., Martin N. G., Mathews C. A., Mayoral F., McElroy S. L., McIntosh A. M., McMahon F. J., Melle I., Michie P., Milani L., Mitchell P. B., Morken G., Mors O., Mortensen P. B., Mowry B., Müller-Myhsok B., Myers R. M., Neale B. M., Nievergelt C. M., Nordentoft M., Nöthen M. M., O’Donovan M. C., Oedegaard K. J., Olsson T., Owen M. J., Paciga S. A., Pantelis C., Pato C., Pato M. T., Patrinos G. P., Perlis R. H., Posthuma D., Ramos-Quiroga J. A., Reif A., Reininghaus E. Z., Ribasés M., Rietschel M., Ripke S., Rouleau G. A., Saito T., Schall U., Schalling M., Schofield P. R., Schulze T. G., Scott L. J., Scott R. J., Serretti A., Shannon Weickert C., Smoller J. W., Stefansson H., Stefansson K., Stordal E., Streit F., Sullivan P. F., Turecki G., Vaaler A. E., Vieta E., Vincent J. B., Waldman I. D., Weickert T. W., Werge T., Wray N. R., Zwart J.-A., Biernacka J. M., Nurnberger J. I., Cichon S., Edenberg H. J., Stahl E. A., McQuillin A., Di Florio A., Ophoff R. A., Andreassen O. A., Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou Y., Liang W., Zhang J., Li Q., Ou H., Wang Z., Li S., Huang X., Zhao C., Schizophrenia-associated rs4702 G allele-specific downregulation of FURIN expression by miR-338-3p reduces BDNF production. Schizophr. Res. 199, 176–180 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Schrode N., Ho S.-M., Yamamuro K., Dobbyn A., Huckins L., Matos M. R., Cheng E., Deans P. J. M., Flaherty E., Barretto N., Topol A., Alganem K., Abadali S., Gregory J., Hoelzli E., Phatnani H., Singh V., Girish D., Aronow B., Mccullumsmith R., Hoffman G. E., Stahl E. A., Morishita H., Sklar P., Brennand K. J., Synergistic effects of common schizophrenia risk variants. Nat. Genet. 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu J. M., Satterstrom F. K., Peng M., Brand H., Collins R. L., Dong S., Wamsley B., Klei L., Wang L., Hao S. P., Stevens C. R., Cusick C., Babadi M., Banks E., Collins B., Dodge S., Gabriel S. B., Gauthier L., Lee S. K., Liang L., Ljungdahl A., Mahjani B., Sloofman L., Smirnov A. N., Barbosa M., Betancur C., Brusco A., Chung B. H. Y., Cook E. H., Cuccaro M. L., Domenici E., Ferrero G. B., Gargus J. J., Herman G. E., Hertz-Picciotto I., Maciel P., Manoach D. S., Passos-Bueno M. R., Persico A. M., Renieri A., Sutcliffe J. S., Tassone F., Trabetti E., Campos G., Cardaropoli S., Carli D., Chan M. C. Y., Fallerini C., Giorgio E., Girardi A. C., Hansen-Kiss E., Lee S. L., Lintas C., Ludena Y., Nguyen R., Pavinato L., Pericak-Vance M., Pessah I. N., Schmidt R. J., Smith M., Costa C. I. S., Trajkova S., Wang J. Y. T., Yu M. H. C., Autism Sequencing Consortium (ASC), Broad Institute Center for Common Disease Genomics (Broad-CCDG), iPSYCH-BROAD Consortium, Cutler D. J., De Rubeis S., Buxbaum J. D., Daly M. J., Devlin B., Roeder K., Sanders S. J., Talkowski M. E., Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. (2022), doi: 10.1038/s41588-022-01104-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewis D. A., Hashimoto T., Volk D. W., Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Meltzer H. L., Lithium mechanisms in bipolar illness and altered intracellular calcium functions. Biol. Psychiatry. 21, 492–510 (1986). [DOI] [PubMed] [Google Scholar]
- 62.Wolff M., Johannesen K. M., Hedrich U. B. S., Masnada S., Rubboli G., Gardella E., Lesca G., Ville D., Milh M., Villard L., Afenjar A., Chantot-Bastaraud S., Mignot C., Lardennois C., Nava C., Schwarz N., Gérard M., Perrin L., Doummar D., Auvin S., Miranda M. J., Hempel M., Brilstra E., Knoers N., Verbeek N., van Kempen M., Braun K. P., Mancini G., Biskup S., Hörtnagel K., Döcker M., Bast T., Loddenkemper T., Wong-Kisiel L., Baumeister F. M., Fazeli W., Striano P., Dilena R., Fontana E., Zara F., Kurlemann G., Klepper J., Thoene J. G., Arndt D. H., Deconinck N., Schmitt-Mechelke T., Maier O., Muhle H., Wical B., Finetti C., Brückner R., Pietz J., Golla G., Jillella D., Linnet K. M., Charles P., Moog U., Õiglane-Shlik E., Mantovani J. F., Park K., Deprez M., Lederer D., Mary S., Scalais E., Selim L., Van Coster R., Lagae L., Nikanorova M., Hjalgrim H., Korenke G. C., Trivisano M., Specchio N., Ceulemans B., Dorn T., Helbig K. L., Hardies K., Stamberger H., de Jonghe P., Weckhuysen S., Lemke J. R., Krägeloh-Mann I., Helbig I., Kluger G., Lerche H., Møller R. S., Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 140, 1316–1336 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Zandi P. P., Jaffe A. E., Goes F. S., Burke E. E., Collado-Torres L., Huuki-Myers L., Seyedian A., Lin Y., Seifuddin F., Pirooznia M., Ross C. A., Kleinman J. E., Weinberger D. R., Hyde T. M., Amygdala and anterior cingulate transcriptomes from individuals with bipolar disorder reveal downregulated neuroimmune and synaptic pathways. Nat. Neurosci. 25, 381–389 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cao M., Zheng H., Tan X., Xu W., Rui Y., Li L., Liu X., Xu G., Cui G., Xu J., Cao J., Ke K., Wu Q., Upregulation of CBLL1 in rat brain cortex after lipopolysaccharide treated. J. Mol. Histol. 44, 135–145 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Bhattacharya A., Jops C., Kim M., Wen C., Vo D. D., Hervoso J. L., Pasaniuc B., Gandal M. J., Isoform-level transcriptome-wide association uncovers extensive novel genetic risk mechanisms for neuropsychiatric disorders in the human brain. bioRxiv (2022), , doi: 10.1101/2022.08.23.22279134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hukku A., Pividori M., Luca F., Pique-Regi R., Im H. K., Wen X., Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am. J. Hum. Genet. 108, 25–35 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu A., Matoba N., Wilson E. P., Tapia A. L., Li Y., Ibrahim J. G., Stein J. L., Love M. I., MRLocus: Identifying causal genes mediating a trait through Bayesian estimation of allelic heterogeneity. PLoS Genet. 17, e1009455 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langfelder P., Horvath S., WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gandal M. J., Haney J. R., Parikshak N. N., Leppa V., Ramaswami G., Hartl C., Schork A. J., Appadurai V., Buil A., Werge T. M., Liu C., White K. P., CommonMind Consortium, PsychENCODE Consortium, iPSYCH-BROAD Working Group, Horvath S., Geschwind D. H., Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 359, 693–697 (2018).29439242 [Google Scholar]
- 70.Parikshak N. N., Gandal M. J., Geschwind D. H., Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Przytycki P. F., Pollard K. S., CellWalker integrates single-cell and bulk data to resolve regulatory elements across cell types in complex tissues. Genome Biol. 22, 61 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aygün N., Elwell A. L., Liang D., Lafferty M. J., Cheek K. E., Courtney K. P., Mory J., Hadden-Ford E., Krupa O., de la Torre-Ubieta L., Geschwind D. H., Love M. I., Stein J. L., Brain-trait-associated variants impact cell-typespecific gene regulation during neurogenesis. Am. J. Hum. Genet. (2021), doi: 10.1016/j.ajhg.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang D., Guelfi S., Garcia-Ruiz S., Costa B., Reynolds R. H., D’Sa K., Liu W., Courtin T., Peterson A., Jaffe A. E., Hardy J., Botía J. A., Collado-Torres L., Ryten M., Incomplete annotation has a disproportionate impact on our understanding of Mendelian and complex neurogenetic disorders. Science Advances. 6, eaay8299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Das S., Forer L., Schönherr S., Sidore C., Locke A. E., Kwong A., Vrieze S. I., Chew E. Y., Levy S., McGue M., Schlessinger D., Stambolian D., Loh P.-R., Iacono W. G., Swaroop A., Scott L. J., Cucca F., Kronenberg F., Boehnke M., Abecasis G. R., Fuchsberger C., Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao H., Sun Z., Wang J., Huang H., Kocher J.-P., Wang L., CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 30, 1006–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andrews S., Others, FastQC: a quality control tool for high throughput sequence data (2010). [Google Scholar]
- 78.Frankish A., Diekhans M., Jungreis I., Lagarde J., Loveland J. E., Mudge J. M., Sisu C., Wright J. C., Armstrong J., Barnes I., Berry A., Bignell A., Boix C., Carbonell Sala S., Cunningham F., Di Domenico T., Donaldson S., Fiddes I. T., García Girón C., Gonzalez J. M., Grego T., Hardy M., Hourlier T., Howe K. L., Hunt T., Izuogu O. G., Johnson R., Martin F. J., Martínez L., Mohanan S., Muir P., Navarro F. C. P., Parker A., Pei B., Pozo F., Riera F. C., Ruffier M., Schmitt B. M., Stapleton E., Suner M.-M., Sycheva I., Uszczynska-Ratajczak B., Wolf M. Y., Xu J., Yang Y. T., Yates A., Zerbino D., Zhang Y., Choudhary J. S., Gerstein M., Guigó R., Hubbard T. J. P., Kellis M., Paten B., Tress M. L., Flicek P., GENCODE 2021. Nucleic Acids Res. 49, D916–D923 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ewels P., Magnusson M., Lundin S., Käller M., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soneson C., Love M. I., Robinson M. D., Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leek J. T., Johnson W. E., Parker H. S., Jaffe A. E., Storey J. D., The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 28, 882–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van de Geijn B., McVicker G., Gilad Y., Pritchard J. K., WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 12, 1061–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mostafavi S., Battle A., Zhu X., Urban A. E., Levinson D., Montgomery S. B., Koller D., Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge. PLoS One. 8, e68141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ongen H., Buil A., Brown A. A., Dermitzakis E. T., Delaneau O., Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 32, 1479–1485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruiz-Arenas C., Cáceres A., López-Sánchez M., Tolosana I., Pérez-Jurado L., González J. R., scoreInvHap: Inversion genotyping for genome-wide association studies. PLoS Genet. 15, e1008203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zerbino D. R., Wilder S. P., Johnson N., Juettemann T., Flicek P. R., The ensembl regulatory build. Genome Biol. 16 (2015), doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McLaren W., Gil L., Hunt S. E., Riat H. S., Ritchie G. R. S., Thormann A., Flicek P., Cunningham F., The ensembl variant effect predictor. Genome Biol. 17 (2016), doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wen X., Effective QTL Discovery Incorporating Genomic Annotations. bioRxiv (2015), , doi: 10.1101/032003. [DOI] [Google Scholar]
- 92.Danecek P., Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., Whitwham A., Keane T., McCarthy S. A., Davies R. M., Li H., Twelve years of SAMtools and BCFtools. Gigascience. 10 (2021), doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim M., Vo D. D., Kumagai M. E., Jops C. T., Gandal M. J., GeneticsMakie.jl: A versatile and scalable toolkit for visualizing locus-level genetic and genomic data. Bioinformatics (2022), doi: 10.1093/bioinformatics/btac786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou H., Sinsheimer J. S., Bates D. M., Chu B. B., German C. A., Ji S. S., Keys K. L., Kim J., Ko S., Mosher G. D., Papp J. C., Sobel E. M., Zhai J., Zhou J. J., Lange K., OPENMENDEL: a cooperative programming project for statistical genetics. Hum. Genet.. 139, 61–71 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Howard D. M., Adams M. J., Clarke T.-K., Hafferty J. D., Gibson J., Shirali M., Coleman J. R. I., Hagenaars S. P., Ward J., Wigmore E. M., Alloza C., Shen X., Barbu M. C., Xu E. Y., Whalley H. C., Marioni R. E., Porteous D. J., Davies G., Deary I. J., Hemani G., Berger K., Teismann H., Rawal R., Arolt V., Baune B. T., Dannlowski U., Domschke K., Tian C., Hinds D. A., 23andMe Research Team, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, Trzaskowski M., Byrne E. M., Ripke S., Smith D. J., Sullivan P. F., Wray N. R., Breen G., Lewis C. M., McIntosh A. M., Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Y., Chen S., Li Z., Morrison A. C., Boerwinkle E., Lin X., ACAT: A Fast and Powerful p Value Combination Method for Rare-Variant Analysis in Sequencing Studies. Am. J. Hum. Genet. 104, 410–421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shaffer J. P., Modified Sequentially Rejective Multiple Test Procedures. J. Am. Stat. Assoc. 81, 826–831 (1986). [Google Scholar]
- 98.Mancuso N., Freund M. K., Johnson R., Shi H., Kichaev G., Gusev A., Pasaniuc B., Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet. 51, 675–682 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y., Zhou R., Wang Y., Sashimi.py: a flexible toolkit for combinatorial analysis of genomic data. bioRxiv (2022), , doi: 10.1101/2022.11.02.514803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karczewski K. J., Francioli L. C., Tiao G., Cummings B. B., Alföldi J., Wang Q., Collins R. L., Laricchia K. M., Ganna A., Birnbaum D. P., Gauthier L. D., Brand H., Solomonson M., Watts N. A., Rhodes D., Singer-Berk M., England E. M., Seaby E. G., Kosmicki J. A., Walters R. K., Tashman K., Farjoun Y., Banks E., Poterba T., Wang A., Seed C., Whiffin N., Chong J. X., Samocha K. E., Pierce-Hoffman E., Zappala Z., O’Donnell-Luria A. H., Minikel E. V., Weisburd B., Lek M., Ware J. S., Vittal C., Armean I. M., Bergelson L., Cibulskis K., Connolly K. M., Covarrubias M., Donnelly S., Ferriera S., Gabriel S., Gentry J., Gupta N., Jeandet T., Kaplan D., Llanwarne C., Munshi R., Novod S., Petrillo N., Roazen D., Ruano-Rubio V., Saltzman A., Schleicher M., Soto J., Tibbetts K., Tolonen C., Wade G., Talkowski M. E., Genome Aggregation Database Consortium, Neale B. M., Daly M. J., MacArthur D. G., The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson K. A., Krishnan A., Robust normalization and transformation techniques for constructing gene coexpression networks from RNA-seq data. Genome Biol. 23, 1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Leeuw C. A., Mooij J. M., Heskes T., Posthuma D., MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sey N. Y. A., Hu B., Mah W., Fauni H., McAfee J. C., Rajarajan P., Brennand K. J., Akbarian S., Won H., A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 23, 583–593 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abrahams B. S., Arking D. E., Campbell D. B., Mefford H. C., Morrow E. M., Weiss L. A., Menashe I., Wadkins T., Banerjee-Basu S., Packer A., SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism. 4, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Feng Y.-C. A., Howrigan D. P., Abbott L. E., Tashman K., Cerrato F., Singh T., Heyne H., Byrnes A., Churchhouse C., Watts N., Solomonson M., Lal D., Heinzen E. L., Dhindsa R. S., Stanley K. E., Cavalleri G. L., Hakonarson H., Helbig I., Krause R., May P., Weckhuysen S., Petrovski S., Kamalakaran S., Sisodiya S. M., Cossette P., Cotsapas C., De Jonghe P., Dixon-Salazar T., Guerrini R., Kwan P., Marson A. G., Stewart R., Depondt C., Dlugos D. J., Scheffer I. E., Striano P., Freyer C., McKenna K., Regan B. M., Bellows S. T., Leu C., Bennett C. A., Johns E. M. C., Macdonald A., Shilling H., Burgess R., Weckhuysen D., Bahlo M., O’Brien T. J., Todaro M., Stamberger H., Andrade D. M., Sadoway T. R., Mo K., Krestel H., Gallati S., Papacostas S. S., Kousiappa I., Tanteles G. A., Štěrbová K., Vlčková M., Sedláčková L., Laššuthová P., Klein K. M., Rosenow F., Reif P. S., Knake S., Kunz W. S., Zsurka G., Elger C. E., Bauer J., Rademacher M., Pendziwiat M., Muhle H., Rademacher A., van Baalen A., von Spiczak S., Stephani U., Afawi Z., Korczyn A. D., Kanaan M., Canavati C., Kurlemann G., Müller-Schlüter K., Kluger G., Häusler M., Blatt I., Lemke J. R., Krey I., Weber Y. G., Wolking S., Becker F., Hengsbach C., Rau S., Maisch A. F., Steinhoff B. J., Schulze-Bonhage A., Schubert-Bast S., Schreiber H., Borggräfe I., Schankin C. J., Mayer T., Korinthenberg R., Brockmann K., Kurlemann G., Dennig D., Madeleyn R., Kälviäinen R., Auvinen P., Saarela A., Linnankivi T., Lehesjoki A.-E., Rees M. I., Chung S.-K., Pickrell W. O., Powell R., Schneider N., Balestrini S., Zagaglia S., Braatz V., Johnson M. R., Auce P., Sills G. J., Baum L. W., Sham P. C., Cherny S. S., Lui C. H. T., Barišić N., Delanty N., Doherty C. P., Shukralla A., McCormack M., El-Naggar H., Canafoglia L., Franceschetti S., Castellotti B., Granata T., Zara F., Iacomino M., Madia F., Vari M. S., Mancardi M. M., Salpietro V., Bisulli F., Tinuper P., Licchetta L., Pippucci T., Stipa C., Minardi R., Gambardella A., Labate A., Annesi G., Manna L., Gagliardi M., Parrini E., Mei D., Vetro A., Bianchini C., Montomoli M., Doccini V., Marini C., Suzuki T., Inoue Y., Yamakawa K., Tumiene B., Sadleir L. G., King C., Mountier E., Caglayan S. H., Arslan M., Yapıcı Z., Yis U., Topaloglu P., Kara B., Turkdogan D., Gundogdu-Eken A., Bebek N., Uğur-İşeri S., Baykan B., Salman B., Haryanyan G., Yücesan E., Kesim Y., Özkara Ç., Poduri A., Shiedley B. R., Shain C., Buono R. J., Ferraro T. N., Sperling M. R., Lo W., Privitera M., French J. A., Schachter S., Kuzniecky R. I., Devinsky O., Hegde M., Khankhanian P., Helbig K. L., Ellis C. A., Spalletta G., Piras F., Piras F., Gili T., Ciullo V., Reif A., McQuillin A., Bass N., McIntosh A., Blackwood D., Johnstone M., Palotie A., Pato M. T., Pato C. N., Bromet E. J., Carvalho C. B., Achtyes E. D., Azevedo M. H., Kotov R., Lehrer D. S., Malaspina D., Marder S. R., Medeiros H., Morley C. P., Perkins D. O., Sobell J. L., Buckley P. F., Macciardi F., Rapaport M. H., Knowles J. A., Fanous A. H., McCarroll S. A., Gupta N., Gabriel S. B., Daly M. J., Lander E. S., Lowenstein D. H., Goldstein D. B., Lerche H., Berkovic S. F., Neale B. M., Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am. J. Hum. Genet. 105, 267–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Palmer D. S., Howrigan D. P., Chapman S. B., Adolfsson R., Bass N., Blackwood D., Boks M. P. M., Chen C.-Y., Churchhouse C., Corvin A. P., Craddock N., Curtis D., Di Florio A., Dickerson F., Freimer N. B., Goes F. S., Jia X., Jones I., Jones L., Jonsson L., Kahn R. S., Landén M., Locke A. E., McIntosh A. M., McQuillin A., Morris D. W., O’Donovan M. C., Ophoff R. A., Owen M. J., Pedersen N. L., Posthuma D., Reif A., Risch N., Schaefer C., Scott L., Singh T., Smoller J. W., Solomonson M., Clair D. S., Stahl E. A., Vreeker A., Walters J. T. R., Wang W., Watts N. A., Yolken R., Zandi P. P., Neale B. M., Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat. Genet. 54, 541–547 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaplanis J., Samocha K. E., Wiel L., Zhang Z., Arvai K. J., Eberhardt R. Y., Gallone G., Lelieveld S. H., Martin H. C., McRae J. F., Short P. J., Torene R. I., de Boer E., Danecek P., Gardner E. J., Huang N., Lord J., Martincorena I., Pfundt R., Reijnders M. R. F., Yeung A., Yntema H. G., Borras S., Clark C., Dean J., Miedzybrodzka Z., Ross A., Tennant S., Dabir T., Donnelly D., Humphreys M., Magee A., McConnell V., McKee S., McNerlan S., Morrison P. J., Rea G., Stewart F., Cole T., Cooper N., Cooper-Charles L., Cox H., Islam L., Jarvis J., Keelagher R., Lim D., McMullan D., Morton J., Naik S., O’Driscoll M., Ong K.-R., Osio D., Ragge N., Turton S., Vogt J., Williams D., Bodek S., Donaldson A., Hills A., Low K., Newbury-Ecob R., Norman A. M., Roberts E., Scurr I., Smithson S., Tooley M., Abbs S., Armstrong R., Dunn C., Holden S., Park S.-M., Paterson J., Raymond L., Reid E., Sandford R., Simonic I., Tischkowitz M., Woods G., Bradley L., Comerford J., Green A., Lynch S., McQuaid S., Mullaney B., Berg J., Goudie D., Mavrak E., McLean J., McWilliam C., Reavey E., Azam T., Cleary E., Jackson A., Lam W., Lampe A., Moore D., Porteous M., Baple E., Baptista J., Brewer C., Castle B., Kivuva E., Owens M., Rankin J., Shaw-Smith C., Turner C., Turnpenny P., Tysoe C., Bradley T., Davidson R., Gardiner C., Joss S., Kinning E., Longman C., McGowan R., Murday V., Pilz D., Tobias E., Whiteford M., Williams N., Barnicoat A., Clement E., Faravelli F., Hurst J., Jenkins L., Jones W., Kumar V. K. A., Lees M., Loughlin S., Male A., Morrogh D., Rosser E., Scott R., Wilson L., Beleza A., Deshpande C., Flinter F., Holder M., Irving M., Izatt L., Josifova D., Mohammed S., Molenda A., Robert L., Roworth W., Ruddy D., Ryten M., Yau S., Bennett C., Blyth M., Campbell J., Coates A., Dobbie A., Hewitt S., Hobson E., Jackson E., Jewell R., Kraus A., Prescott K., Sheridan E., Thomson J., Bradshaw K., Dixit A., Eason J., Haines R., Harrison R., Mutch S., Sarkar A., Searle C., Shannon N., Sharif A., Suri M., Vasudevan P., Canham N., Ellis I., Greenhalgh L., Howard E., Stinton V., Swale A., Weber A., Banka S., Breen C., Briggs T., Burkitt-Wright E., Chandler K., Clayton-Smith J., Donnai D., Douzgou S., Gaunt L., Jones E., Kerr B., Langley C., Metcalfe K., Smith A., Wright R., Bourn D., Burn J., Fisher R., Hellens S., Henderson A., Montgomery T., Splitt M., Straub V., Wright M., Zwolinski S., Allen Z., Bernhard B., Brady A., Brooks C., Busby L., Clowes V., Ghali N., Holder S., Ibitoye R., Wakeling E., Blair E., Carmichael J., Cilliers D., Clasper S., Gibbons R., Kini U., Lester T., Nemeth A., Poulton J., Price S., Shears D., Stewart H., Wilkie A., Albaba S., Baker D., Balasubramanian M., Johnson D., Parker M., Quarrell O., Stewart A., Willoughby J., Crosby C., Elmslie F., Homfray T., Jin H., Lahiri N., Mansour S., Marks K., McEntagart M., Saggar A., Tatton-Brown K., Butler R., Clarke A., Corrin S., Fry A., Kamath A., McCann E., Mugalaasi H., Pottinger C., Procter A., Sampson J., Sansbury F., Varghese V., Baralle D., Callaway A., Cassidy E. J., Daniels S., Douglas A., Foulds N., Hunt D., Kharbanda M., Lachlan K., Mercer C., Side L., Temple I. K., Wellesley D., Vissers L. E. L. M., Juusola J., Wright C. F., Brunner H. G., Firth H. V., FitzPatrick D. R., Barrett J. C., Hurles M. E., Gilissen C., Retterer K., Deciphering Developmental Disorders Study, Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature (2020), doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh T., Poterba T., Curtis D., Akil H., Al Eissa M., Barchas J. D., Bass N., Bigdeli T. B., Breen G., Bromet E. J., Buckley P. F., Bunney W. E., Bybjerg-Grauholm J., Byerley W. F., Chapman S. B., Chen W. J., Churchhouse C., Craddock N., Cusick C. M., DeLisi L., Dodge S., Escamilla M. A., Eskelinen S., Fanous A. H., Faraone S. V., Fiorentino A., Francioli L., Gabriel S. B., Gage D., Gagliano Taliun S. A., Ganna A., Genovese G., Glahn D. C., Grove J., Hall M.-H., Hämäläinen E., Heyne H. O., Holi M., Hougaard D. M., Howrigan D. P., Huang H., Hwu H.-G., Kahn R. S., Kang H. M., Karczewski K. J., Kirov G., Knowles J. A., Lee F. S., Lehrer D. S., Lescai F., Malaspina D., Marder S. R., McCarroll S. A., McIntosh A. M., Medeiros H., Milani L., Morley C. P., Morris D. W., Mortensen P. B., Myers R. M., Nordentoft M., O’Brien N. L., Olivares A. M., Ongur D., Ouwehand W. H., Palmer D. S., Paunio T., Quested D., Rapaport M. H., Rees E., Rollins B., Satterstrom F. K., Schatzberg A., Scolnick E., Scott L. J., Sharp S. I., Sklar P., Smoller J. W., Sobell J. L., Solomonson M., Stahl E. A., Stevens C. R., Suvisaari J., Tiao G., Watson S. J., Watts N. A., Blackwood D. H., Børglum A. D., Cohen B. M., Corvin A. P., Esko T., Freimer N. B., Glatt S. J., Hultman C. M., McQuillin A., Palotie A., Pato C. N., Pato M. T., Pulver A. E., St Clair D., Tsuang M. T., Vawter M. P., Walters J. T., Werge T. M., Ophoff R. A., Sullivan P. F., Owen M. J., Boehnke M., O’Donovan M. C., Neale B. M., Daly M. J., Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature (2022), doi: 10.1038/s41586-022-04556-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Taylor-Weiner A., Aguet F., Haradhvala N. J., Gosai S., Anand S., Kim J., Ardlie K., Van Allen E. M., Getz G., Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 20, 228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Przytycki P. F., Pollard K. S., CellWalkR: an R package for integrating and visualizing single-cell and bulk data to resolve regulatory elements. Bioinformatics. 38, 2621–2623 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ziffra R. S., Kim C. N., Ross J. M., Wilfert A., Turner T. N., Haeussler M., Casella A. M., Przytycki P. F., Keough K. C., Shin D., Bogdanoff D., Kreimer A., Pollard K. S., Ament S. A., Eichler E. E., Ahituv N., Nowakowski T. J., Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 598, 205–213 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supplemental data can be found on Synapse at https://www.synapse.org/#!Synapse:syn50897018.3/datasets/