

Abstract
Background:
Homologous Recombination Deficiency (HRD) is a pan-cancer predictive biomarker that identifies patients who benefit from therapy with PARP inhibitors (PARPi). However, testing for HRD is highly complex. Here, we investigated whether Deep Learning can predict HRD status solely based on routine Hematoxylin & Eosin (H&E) histology images in ten cancer types.
Methods:
We developed a fully automated deep learning pipeline with attention-weighted multiple instance learning (attMIL) to predict HRD status from histology images. A combined genomic scar HRD score, which integrated loss of heterozygosity (LOH), telomeric allelic imbalance (TAI) and large-scale state transitions (LST) was calculated from whole genome sequencing data for n=4,565 patients from two independent cohorts. The primary statistical endpoint was the Area Under the Receiver Operating Characteristic curve (AUROC) for the prediction of genomic scar HRD with a clinically used cutoff value.
Results:
We found that HRD status is predictable in tumors of the endometrium, pancreas and lung, reaching cross-validated AUROCs of 0.79, 0.58 and 0.66. Predictions generalized well to an external cohort with AUROCs of 0.93, 0.81 and 0.73 respectively. Additionally, an HRD classifier trained on breast cancer yielded an AUROC of 0.78 in internal validation and was able to predict HRD in endometrial, prostate and pancreatic cancer with AUROCs of 0.87, 0.84 and 0.67 indicating a shared HRD-like phenotype is across tumor entities.
Conclusion:
In this study, we show that HRD is directly predictable from H&E slides using attMIL within and across ten different tumor types.
Keywords: Homologous Recombination Deficiency, Deep Learning, DNA repair mechanism, artificial intelligence, molecular pathology, pan cancer study
Background
Homologous recombination repair (HRR) is a DNA repair mechanism that ensures genomic integrity after DNA double-strand breaks (DSB), which occur regularly during the cell cycle (1). Homologous recombination deficiency (HRD) results in defective DNA break repair, increased somatic copy number alterations and genomic instability, driving malignant transformation and causing cancer (2). Poly(ADP-Ribose)-polymerase (PARP) plays pivotal roles in base excision repair of single strand DNA breaks (SSDBs), which is a compensatory DNA repair mechanism in the context of HRD. In the setting of homologous recombination (HR) proficiency PARP inhibition results in the accumulation of unrepaired SSDBs. These can eventually convert to DSBs, which can be repaired via HR thus maintaining genomic integrity and cell viability. However in the case of a HRD tumor, PARP inhibition-induced DSBs are no longer repaired, resulting in direct cytotoxicity.. This phenomenon of synthetic lethality is the reason why HRD is an important biomarker to select patients for PARP inhibitor (PARPi) treatment in several tumor types, especially in breast, ovarian, prostate and pancreatic cancer (3–6). Prevalences of HRD varies according to the genomic definition of HRD and among tumor types, ranging from 0% in thymoma or thyroid cancer up to 70% in ovarian cancer(7). The use of PARPi has led to improved disease-free survival in multiple clinical trials by increasing platinum sensitivity in ovarian (OV) and breast cancer (BRCA), and other tumor types (8,9).
The success of PARPi therapy is mainly limited by the challenge of diagnosing HRD. Many different test strategies are available. The most robust test for HRD are oncogenic mutations in the Breast Cancer genes 1 and 2 (BRCA1/2) (10,11). However, this approach excludes patients without BRCA1/2-related deficiencies in the HR pathway (12). Moreover, other mechanisms such as epigenetic modifications, germline and somatic mutations of genes related or non related to the HRR pathway may cause HRD (13). Unfortunately, non-BRCA HR mutations have not been reliably shown to predict HRD or PARPi benefit in the clinic. Certain patterns of mutations, like the single base substitution 3 (SBS3) are also associated with a defective HR and therefore a potential biomarker (14,15). Finally, another strategy for detecting HRD is to look for the consequence of HRD rather than the cause. This approach uses whole genome sequencing single nucleotide polymorphism (SNP) array data to identify loss of heterozygosity (LOH), telomeric allelic imbalance (TAI) and large-scale state transitions (LST), also defined as a genomic instability score (GIS). This combined score has been validated in randomized clinical trials as predictive of PARPi benefit (16–18). Biologically, this methods provides a more comprehensive assessment of genomic instability caused by HRD, rather than scores exclusively based on mutation or HRR genes (Figure 1A). However, the GIS is not yet implemented in routine diagnostics in clinical workflows (11,12,19). Combining the different components of HRD using algorithms (e.g. scarHRD, HRDetect, CHORD) may be the gold standard to determine the genomic “scar” associated with HRD (20–22). A non-DNA-based way to determine HRD is using a functional test such as the RAD51 focus formation assays (23,24). U.S. Food and Drug Administration (FDA)-approved genetic tests for HRD typically rely on a combination of alterations in BRCA1/2 genes and LOH (FoundationOne CDx, Foundation Medicine, Inc., Cambridge, MA) or GIS (myChoice CDx, Myriad Genetics Laboratories, Inc., Salt Lake City, UT) (10,11). However defining cut-off values for stratification between positive and negative cases is difficult (7,25). Taken together, the HRD testing landscape is highly complex. Many different tests coexist and they are not perfectly concordant. There is a high clinical need for a cheap, fast and standardized HRD test which captures a breadth of biological processes and not just alterations in individual genes. In this study, we hypothesized that the tumor phenotype as observed on histological whole slide images (WSI) of tumors reflects the HRD status and can be used to diagnose HRD.
Figure 1: Experimental Design and Study overview.
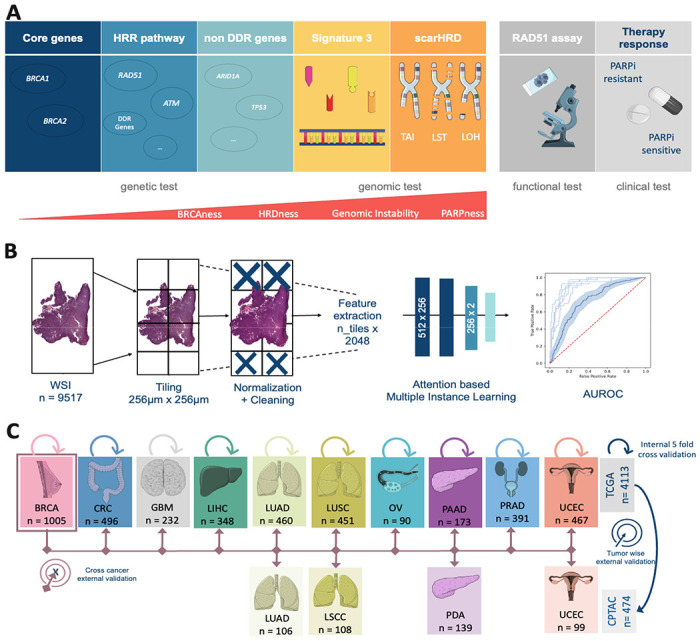
(A) Overview of the different Homologous Recombination Deficiency (HRD) scores, their content and assessment methods. (B) Workflow of our Deep Learning (DL) pipeline. A total of n=9517 Whole Slide Images (WSI) were processed and trained with an attention-based Multiple Instance Learning (attMIL) approach. The statistical endpoint was the Area under the receiving operating curve (AUROC). (C) Study design for the three main experiments (Internal 5-fold cross-validation, tumor-wise external validation and cross-cancer external validation) conducted and cohort overview for patients and tumor types included from The Cancer Genome Atlas (TCGA, n=4113 patients) and Clinical Proteomic Tumor Analysis Consortium (CPTAC, n=474 patients). Abbreviations: BRCA=breast cancer; CRC=colorectal cancer; GBM=glioblastoma; LIHC=liver cancer; LUAD=lung adenocarcinoma; LUSC/LSCC=lung squamous cell carcinoma; OV=ovarian cancer; PAAD/PDA=pancreatic adenocarcinoma; PRAD=prostate adenocarcinoma; UCEC=endometrial cancer; HRR=Homologous recombination repair. (This Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license)
Deep Learning (DL) is an artificial intelligence (AI)-based technology which has emerged as a powerful method to quantitatively mine data from histological WSI of tumors in the last five years. DL enables us to detect genetic alterations directly from histopathological image data (26–28). Specifically, DL has been shown to detect single mutations(29,30), as well as phenotypic manifestation of DNA instability mechanisms such as microsatellite instability (MSI), just by processing scanned WSI of tumor tissue stained with H&E (31,32). Today, several DL systems to predict genetic alterations and clinical outcomes have received regulatory approval and are available for routine diagnostic use in Europe and the USA (33,34). Some smaller pilot studies have shown encouraging data for DL-based prediction of HRD from H&E WSI (35,36). However, HRD is a pan-cancer biomarker and DL has not been systematically used to diagnose HRD across tumor types directly from routine H&E pathology slides.
Therefore, in the present study, we developed a DL system to predict HRD status directly from H&E pathology slides. We used the state-of-the-art technology “attention-based Multiple Instance Learning” (attMIL) in a weakly supervised experimental setup, using no spatial labels or manual annotations whatsoever (28)ruth to train the DL system, we used the calculated scarHRD, one of the most comprehensive HRD scores which integrates a variety of genomic changes (Figure 1B). We trained and evaluated the DL classifiers by cross-validation in a large cohort of n=4,113 patients from The Cancer Genome Atlas (TCGA), comprising 10 types of solid tumors. The models were then externally validated on four cancer types in an independent validation dataset (n=474) in a tumor-wise and cross-cancer experimental approach (Figure 1C). Taken together, our experimental results provide direct evidence that HRD is detectable from routine histology in different types of cancer with DL.
Methods
Data Acquisition
In total data from 5,155 patients of 10 tumor types from The Cancer Genome Atlas (TCGA) and 573 patients from five tumor types from the Clinical Proteomic Tumor Analysis Consortium (CPTAC, Figure 1C) were obtained from https://www.cbioportal.org/. Accordingly, the cancer types included in the present study were breast invasive carcinoma (TCGA-BRCA n=1,058), colorectal cancer (TCGA-CRC n=580), glioblastoma (TCGA-GBM n=420, CPTAC-GBM n=99), liver hepatocellular carcinoma (TCGA-LIHC n=364), lung adenocarcinoma (TCGA-LUAD n=536, CPTAC-LUAD n=111), lung squamous cell carcinoma (TCGA-LUSC n=497; CPTAC-LSCC n=109), ovarian cancer (TCGA-OV n=520), pancreatic adenocarcinoma (TCGA-PAAD n=177; CPTAC-PDA n=153), prostate adenocarcinoma (TCGA-PRAD n=488) and endometrial carcinoma (TCGA-UCEC n=515, CPTAC-UCEC n=101, Supplementary Figure 1A,B). Image data and clinical data were available in TCGA-BRCA for n=1005, TCGA-CRC for n=496, TCGA-GBM for n=232, CPTAC-GBM for n=99, TCGA-LIHC for n=348, TCGA-LUAD for n=460, CPTAC-LUAD for n=106, TCGA-LUSC for n=451, CPTAC-LSCC for n=108, TCGA-OV for n=90, TCGA-PAAD for n=173, CPTAC-PDA for n=139, TCGA-PRAD for n=391, TCGA-UCEC for n=467 and CPTAC-UCEC for n=99, therefore leaving us in total with n=4,565 patients for the analysis (Figure 1C, Supplementary Figure 1A,B). Moreover, some figures were created using https://www.cbioportal.org/ (37,38). For additional experiments on BRCA1/2 mutational status we retrieved data from Riaz et al previously published paper (39). Estrogen receptor data for the subgroup analysis was only available for n=661 patients in the TCGA-BRCA cohort.
Image Preprocessing
WSIs were downloaded for the above mentioned cohorts from the GDC Portal (https://portal.gdc.cancer.gov/) and The Cancer Imaging Archive (TCIA, https://www.cancerimagingarchive.net/). Initially, the images were tessellated into patches with an edge length of 256 μm and a resolution of 224x224 pixels. Secondly, the patches for each cohort were color normalized using the Macenko spectral matching technique(40) to enforce a standardized color distribution across cohorts. To train the prediction models, we used our in-house open-source DL pipeline “marugoto” (https://github.com/KatherLab/marugoto) consisting of a self-supervised learning (SSL) model using a pre-trained ResNet50 architecture with ImageNet weights, fine-tuned pan-cancer on approximately 32.000 WSI to extract a 2048-dimensional feature vector for each patch per patient (41). To obtain patient-level predictions, 512x2048 feature matrices (MIL bags) are constructed by concatenating 512 feature vectors selected at random per patient and fed into an attMIL framework with the following architecture: (512x256), (256x2) with a subsequent attention mechanism (Figure 1B). (42,43)
Calculation of HRD Scores
For the patient-wise calculation of HRD, single nucleotide polymorphism (SNP) data, generated by the Allele-Specific Copy number Analysis of Tumors (ASCAT) algorithm, was downloaded from the Genomic Data Commons (GDC) Portal: https://portal.gdc.cancer.gov/ (accessed 06/15/2022). In CPTAC, the respective data was only available for the CPTAC-3 cohort. The HRD score was calculated using the scarHRD (https://github.com/sztup/scarHRD), as described in previous studies (20,44). ScarHRD uses whole genome sequencing data in the form of SNP arrays to calculate the three subscores LOH, LST and TAI. The sum of these subscores makes up the patient-wise HRD score (Figure 1A). The cut-offs of the different subscores have been previously defined by Abkevich et al. for LOH, Popova et al. for LST and Birkbak et al. for TAI (16–18). Adding up the LOH, LST and TAI scores, patients can be divided into HRD high (HRD-H) and HRD low (HRD-L) at a cut-off of 42 (7). All patients in the CPTAC-GBM cohort were HRD-L. Hence, we excluded them from further analysis (Supplementary Figure 1A,B).
Experimental Design
In our study, we performed three main experiments (Figure 1B). To assess the baseline predictability of HRD from routine histology, we first trained an HRD classifier in a within-cohort approach using fivefold-cross-validation within each of 10 tumor entities mentioned above in the TCGA cohorts (internal validation). This was achieved by randomly splitting each cohort on the level of patients, creating non-overlapping training and test sets for model training. The ratio for splitting the training and testing set was 80:20 of the entire dataset, and the training and validation set was split 75:25 of the training set. Thus, the absolute split for training, internal validation and internal testing was 60, 20 and 20, respectively. Five different models were trained until each part was used as a test set once. Thus, no data leakage from the test set to the training set occurred. This process was repeated individually for each cancer type in the TCGA cohorts. A weighted cross-entropy loss function was used to assist the model with the imbalanced dataset. Secondly, we deployed the five models trained in the first experiments on the same tumor type from the CPTAC cohorts as an external validation. By utilizing this approach, we circumvent any potential claims of selecting the model with the highest AUROC in the external validation. Lastly, we trained an HRD classifier on the TCGA-BRCA cohort, which had the highest number of patients, and deployed it on all other TCGA cohorts (CRC, GBM, LIHC, LUAD, LUSC, PRAD, PAAD, OV, UCEC) as well as on all CPTAC cohorts (LUAD, LSCC, PDA, UCEC). In our study, we aimed to evaluate the performance of the models using the AUROC, which is commonly used for assessing the accuracy of binary classification tasks. Our primary statistical endpoint was the AUROC +/− 95%-confidence interval (CI) and Area under the precision recall curve (Supplementary Table 1). To further assess the performance of each model, we used a two-sided t-test to compare the patient-level prediction scores between the HRD-H and HRD-L patient groups as defined by the ground truth and report the p-values, assuming a significance level of 0.05 as statistically significant, without correction for multiple testing (Supplementary Table 1). As a final step to obtain a more profound understanding of the TCGA-BRCA cohort, we uploaded our custom HRD-H and HRD-L ground truth and predicted subgroups in cbioportal to examine the characteristics of these cases in the TCGA-BRCA PanCancer Atlas cohorts.
Explainability
To visualize the output of our model, we created high resolution heat maps that show the spatial distribution of our model’s attention and prediction scores on the WSI. Therefore, using RetCCL convolutional neural network image feature vectors for 32x32 pixel fields were extracted from the WSI. We then calculated attention and classification scores for each image region and normalized them across the distribution of scores within each patient cohort. Based on these scores, color heatmaps for each patient, with red indicating high attention or a positive classification and blue indicating low attention or a negative classification were generated. To ensure interpretability of the underlying morphology together with the attention and classification scores, we separately reconstructed the final attention and classification heatmaps by blending the raw color heatmaps with the image features. This approach allows us to interpret the output of our model in a way that is easy to understand and provides insight into the underlying morphology of the tumor.
Results
HRD is predictable from histology with attMIL
First, we investigated whether DL could predict HRD status from H&E types within 10 different types of cancer from the TCGA cohort. We used cross-validation on the level of patients to train and test an attMIL-based DL model within each cohort. In our dataset, the prevalence of HRD ranged from 3% in glioblastoma (GBM) up to 63% in OV (Supplements Figure 1C). We found that in five out of 10 cancer types, the mean prediction AUROC was above 0.6, and the 95% CI of the fold-wise HRD prediction AUROCs remained above the null hypothesis of 0.5. Among these, HRD prediction reached statistical significance with a p-value below 0.05 in three cancer types: endometrial cancer (UCEC, AUROC 0.79+/−0.04, p=0.0008), breast cancer (BRCA, AUROC 0.78+/−0.02, p<0.0001) and lung adenocarcinoma(LUAD, AUROC 0.66+/−0.05, p=0.02; Figure 2A). AUPRC values are reported in the Supplementary Table 1. Prediction of HRD was not possible in LUSC, LIHC, GBM, as their prediction AUROCs did not exceed the baseline (0.55+/−0.04 0.56+/−0.14, 0.58+/−0.38) with CIs above the null hypothesis or p-values below 0.05 (Supplementary Figure 2 A–J, Supplementary Table 1). For the tumor types PAAD, OV and PRAD, the AUROCs ranged from 0.58+/−0.22 to 0.6+/−0.09 to 0.76+/−0.22. Together, these data demonstrate that DL can predict HRD status from histology images alone in several tumor types.
Figure 2: Comparison of Area under the receiving operating curve (AUROC) for internal and tumor wise external validation experiment models.
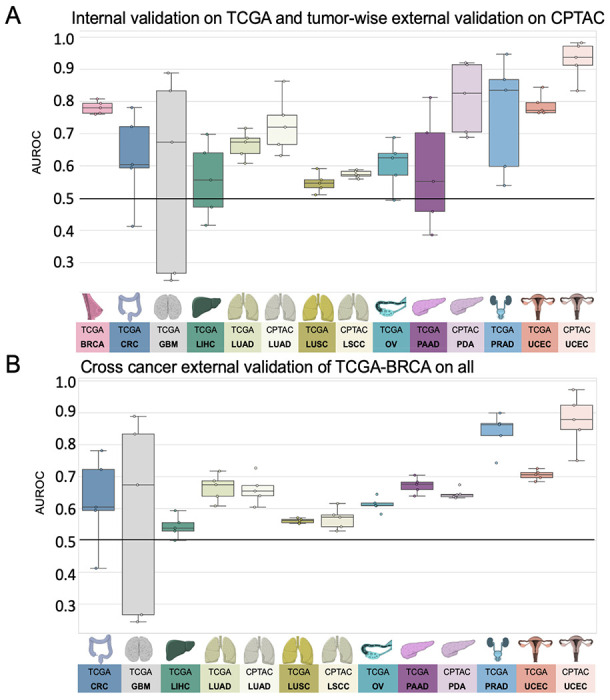
Boxplot displaying the distribution for the AUROC for (A) internal 5-fold cross-validation experiment of The Cancer Genome Atlas (TCGA) and tumor-wise external validation on the Clinical Proteomic Tumor Analysis Consortium (CPTAC); (B) AUROCs for the cross-cancer external validation experiment of the TCGA breast cancer cohort (TCGA-BRCA) on the TCGA and CPTAC cohort. The horizontal line indicates the median, whereas each box represents the interquartile range (IQR) between the first and third quartiles. The whiskers extend from the box to the minimum and maximum values, considering 1.5 times the IQR. Abbreviations: BRCA=breast cancer; CRC=colorectal cancer; GBM=glioblastoma; LIHC=liver cancer; LUAD=lung adenocarcinoma; LUSC/LSCC=lung squamous cell carcinoma; OV=ovarian cancer; PAAD/PDA=pancreatic adenocarcinoma; PRAD=prostate adenocarcinoma; UCEC=endometrial cancer
HRD is predictable from H&E histology with attMIL in an independent test set
A step that is germane to the successful development of DL models is external validation with WSIs from patient cohorts which are completely independent from the training set (45). Hence, for our external validation experiments, we deployed the classification models obtained from the cross-validation training on TCGA to analyze cohorts from the CPTAC dataset corresponding to the same cancer type. External validation cohorts in CPTAC were available for endometrial cancer (UCEC), pancreatic cancer (PDA), lung adenocarcinoma (LUAD), and lung squamous cell carcinoma (LSCC). In these external validation experiments, we noted that the prediction performance was highercompared to internal validation experiments. Once again, the best performance was obtained in UCEC, with an AUROC of 0.93+/−0.07, p=0.01. In LUAD, the performance increased in the external validation, yielding an AUROC of 0.73+/−0.11 and a significant p-value of 0.03. In the case of PAAD/PDA, where the internal validation was unsuccessful (internal validation AUROC 0.58+/−0.22), the external validation resulted in an improved AUROC reaching 0.81+/−0.14, albeit with a p-value of 0.07. Meanwhile, in LUSC/LSCC, no improvement in performance was observed in the external validation set compared to the internal training set (AUROC 0.57+/−0.01, p=0.23, Figure 2A, Supplementary Figure 2 K–N). Together, these data show that DL-based classifiers of HRD status generalize beyond the training cohort.
A HRD classifier trained on BRCA detects HRD across various types of cancer
Lastly, we aimed to investigate if HRD-related morphological features in one cancer type can help to predict HRD status in another cancer type. This would point to a shared set of morphological features across cancer types, potentially allowing us to develop a pan-cancer pathology-based prediction system for HRD status. To test this, we applied our trained HRD classifiers in a cross-cancer experimental design. We used the breast cancer cohort TCGA-BRCA to train the HRD classification model because this cohort had the highest number of patients. Subsequently, we deployed this model on all other cohorts obtained from the TCGA and CPTAC datasets. Surprisingly, the BRCA-based model was able to significantly predict HRD from non-BRCA tissue in UCEC, PRAD and PAAD. For those three cohorts, the external deployment of a BRCA-based model resulted in higher prediction AUROCs than the respective internal validation experiments, reaching AUROCs of 0.70+/−0.02, p<0.001 in TCGA-UCEC, 0.84+/−0.07 and p=0.004 in TCGA-PRAD 0.67+/−0.03, p=0.2 in TCGA-PAAD, 0.87+/−0.1 p=0.05 in CPTAC-UCEC and 0.65+/−0.02 p=0.26 in CPTAC-PDA, respectively (Figure 2B). In the tumor types LUAD and OV, the AUROCs remained with 0.62+/−0.03 for TCGA-LUAD, 0.66+/−0.06 for CPTAC-LUAD and 0.61+/−0.03 in TCGA-OV in a similar range to the internal validation results (Supplementary Figure 3A–M). Together, these data show that a classifier trained on breast cancer can predict HRD status from histology in other tumor types, indicating a shared “HRD morphology” between tumor types.
Molecular and histomorphological characterization of TCGA-BRCA HRD-H and HRD-L cases
Finally, we investigated which molecular and morphological patterns were associated with ground truth and DL-predicted HRD status. We used the TCGA-BRCA cohort to analyze this in detail, as this was the largest cohort. We observed that in the HRD-H subgroup, 45% were classified as basal-like breast cancers, 11% as HER2-enriched, 15% as Luminal A, and 26% as Luminal B. In contrast, only 7% of the cases in the HRD-L subgroup were basal-like, 7% were HER2-enriched, 64% were Luminal A, and 18% were Luminal B (Figure 3A) (46). Within our predicted groups, we observed a similar distribution among the BRCA subtypes (Figure 3B).
Figure 3: Molecular Characterization of The Cancer Genome Atlas breast cancer (TCGA-BRCA) cohort.
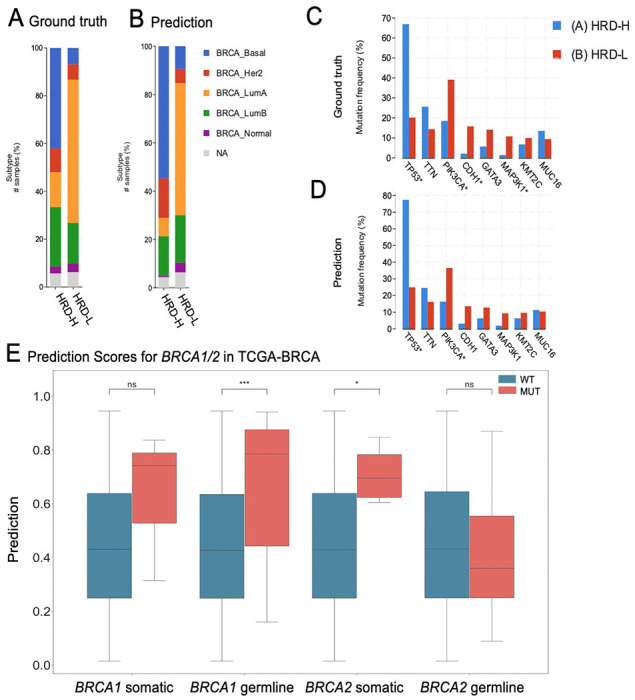
(A) Distribution of breast cancer subtypes for the Homologous Recombination deficiency high (HRD-H) and low (HRD-L) ground truth subgroups. (B) Distribution of the breast cancer subtypes for the HRD-H and HRD-L Deep Learning (DL) predicted subgroups. (C) Alteration Frequency for several genes of the HRD-H and HRD-L ground truth subgroups. (D) Alteration Frequency for several genes of the HRD-H and HRD-L within cohort internal results prediction subgroups. (E) Grouped Boxplots comparing the Homologous Recombination Deficiency high (HRD-H) prediction scores with the mutational status (mutated=MUT wildtype=WT) for the somatic and germline alterations of the BRCA1/2 genes. The central line represents the median value, while the box ranges between the first and third quartile (IQR) and the whiskers extend to the lowest and highest values within 1.5 times the IQR. The y-axis represents the Deep Learning (DL) HRD-H prediction values. An independent t–test was performed to calculate the p-values: ns: p <= 1.00e+00 *: 1.00e−02 < p <= 5.00e−02 **: 1.00e−03 < p <= 1.00e−02 ***: 1.00e−04 < p <= 1.00e−03
To reassure that our model predicts HRD detached from phenotypic differences of estrogen receptor negative (ER−) vs. ER-positive (ER+) breast cancers we calculated the receiving operating curve (ROC) and precision recall curve (PRC) for the subgroups: ER+/HER2+, ER+/HER2−, ER−/HER2+, ER−/HER2− , indicating that HRD was also predictable with AUROCs of 0.66+/−0.3, 0.8+/−0.09, 0.72+/−0.43 and 0.62+/−0.11 (Supplementary Figure 4A–H). Our analysis of the mutational landscape of both HRD-H and HRD-L ground truth revealed that TP53 had the highest alteration frequency with 67% in the HRD-H ground truth group, significantly higher than 20% in the HRD-L group, following alterations in the TTN (26% vs. 14%) gene. In contrast, the most enriched alterations in the HRD-L group were observed in the genes PIK3CA (39%) followed by CDH1 (16%), GATA3 (14%) and MAP3K1 (11%), whereas the prevalences in the HRD-H group of PIK3CA, CDH1, GATA3 and MAP3K1 were 19%, 2%, 6% and 1%, respectively (Figure 3C). For the HRD-H prediction subgroup alteration frequencies for TP53, were significantly higher at 77% (Figure 3D). Such divergences were not as noticeable in the HRD-L prediction group. These findings suggest that there are notable differences in alteration frequencies between the two subgroups, which are consistent across both the ground truth and prediction data. Moreover, we compared the HRD-H prediction score to the alteration status of somatic and germline mutations in the BRCA1/2 genes, whereupon we saw that there was a significant difference between the mutant and wild-type cases for BRCA1 germline and BRCA2 somatic mutations (Figure 3E). Methylation data indicated that the HRD-H group had most of its methylation alterations in the N-shore portion of the BRCA1 promoter region, whereas those in the HRD-L group were mainly located in the S-shore portion (Supplementary Figure 4I). Lastly, we proceeded to investigate the histomorphological patterns associated with the presence of HRD through whole slide prediction heatmaps in CPTAC-UCEC (Figure 4A–C). Our findings revealed that high grade, fibrosis, hemorrhage and lymphocytic infiltration are consistent features predictive of HRD across various tumor types, as shown in Figure 4 for BRCA and UCEC, particularly in the top predicted HRD-H tiles for the top three patients. Fibrosis was observed in HRD-positive cases, particularly in BRCA (Figure 4D). Moreover, hemorrhagic necrosis especially adjacent to tumor tissue and tumor stroma was consistently observed as highly predictive areas in the true HRD-H cases across various cancer types. (Supplementary Figure 5,6). In summary, these data show that known HRD morphology characteristics were found in our DL based top predicted HRD-H patients.
Figure 4: Visualization of predicted Homologous Recombination Deficiency high (HRD-H) tumor samples.
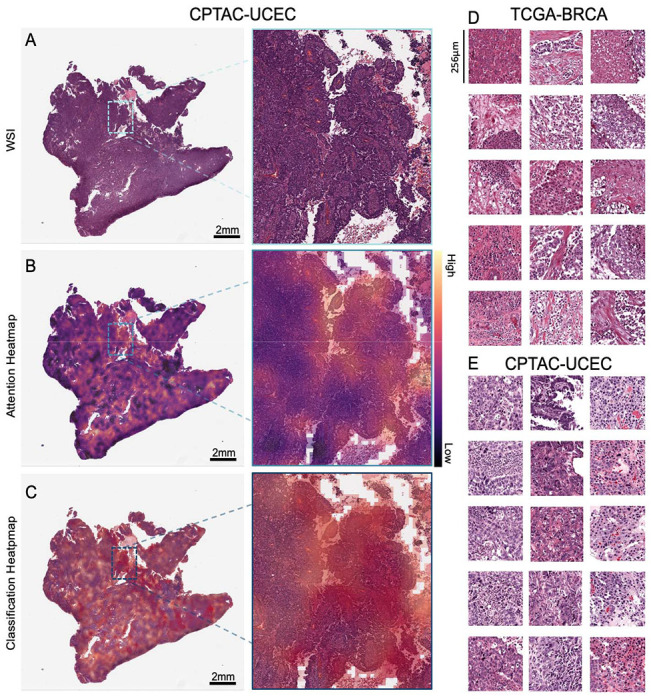
(A) Whole slide image (WSI) of an HRD-H predicted patient (ID: C3L-00358-21) from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) endometrial cancer (UCEC) cohort with magnification. (B) Attention heatmap for the same patient with magnification. (C) Classification Heatmap for the same patient with magnification. (D) Top predicted tiles for top three homologous recombination deficiency high (HRD-H) patients in The Cancer Genome Atlas (TCGA) breast cancer (BRCA). (E) Top predicted tiles for three HRD-H patients in the CPTAC-UCEC cohort.
Discussion
HRD has recently emerged as an important pan-cancer biomarker for targeted treatment in solid tumors (11,47). The assessment of HRD clinically, albeit indicated for all patients with gynecological tumors, remains challenging. This is due to the given availability of different methods with limited agreement and whose logistic complexities and inherent costs pose significant hurdles for their adoption. In this light, a pan-cancer test of HRD by DL-based image analysis on pathology slides could be a useful prescreening tool and reduce the load of genetic tests.
In this study, we demonstrated that DL can predict HRD status from H&E histology in different tumor types in both within-cohort and external validation experiments. Surprisingly, our findings revealed that a BRCA-based classifier could detect HRD from H&E slides across different tumor entities. As expected, the HRD prediction was significantly lower in tumors with a low prevalence of HRD. Our classifier has identified histomorphological characteristics such as hemorrhagic necrosis at tumor margins, lymphocyte infiltration, fibrosis, and high tumor cell density that are associated with HRD in BRCA(36). These findings validate the efficacy of our classifier. Moreover, despite having trained our classifier solely on BRCA, its consistent identification of HRD-associated morphological patterns across different tumor entities reiterates the value of our tool for broader applications. Compared to previous studies, we here show a pan-cancer DL-based prediction of a more comprehensive HRD score calculated from LOH, TAI, and LST as ground truth directly from H&E tumor slides. (35,36)
Our morphological analysis showed that UCEC or PAAD, achieved better predictive results compared to LUSC or LIHC, a trend previously observed in pan-cancer studies (30,48). In general, tumors with a complex structure, such as adenocarcinomas are morphologically susceptible to genetic alterations than solid tumors growing in rather syncytial patterns. HRD-positive tumors barely resemble glandular tissue anymore, which might be their main distinctive feature and therefore a potential explanation for this constellation. Additional studies with larger patient cohorts would be required to confirm this. A closer look at the TCGA-BRCA subgroups revealed that predicted HRD-H is more common in triple-negative breast cancer, which is known for its poor prognosis and resistance to conventional chemotherapy. In line with their ground truths, the majority of those patients were predicted to be HRD-positive by our classifier (Figure 3A,B) (46). Furthermore, clear molecular pathological differences were found in the two subgroups. Specifically, the HRD-H subgroup is characterized by TP53 alterations, while the HRD-L subgroup has a higher frequency of PIK3CA alterations, suggesting an interactive effect between the TP53 mutated cases and HRD-H patients (49,50). This is particularly true for BRCA1 mutated cancers, where HRD-H was predicted significantly better than in BRCA1 wildtype cases (Figure 3E) (51).
Recently, the EMA and FDA granted the first approval to use PARPi therapy for HRD positive ovarian cancer patients. Clinical trials with promising interim data are also underway for other tumor entities and further approvals are expected in the future. Despite the evident link between HRD and BRCA1/2 mutations, it is now well established that the total number of HRD-positive patients significantly exceeds the total number of BRCA-mutated patients in various cancer types (22,52). The patients who fall into this diagnostic gap can be identified with comprehensive HRD testing, as proposed in our study.41 HRD testing would thus complement BRCA1/2 testing as a biomarker test for PARPi use, such as with AI-based screening methods as applied here. Moving diagnostic routines towards phenotype-based instead of inconsistent, alteration-based HRD detection methods might extend our ability to identify patients who may benefit from PARPi and enroll them in clinical trials. Our study provides a proof of concept that there is indeed a pan-cancer preserved HRD morphology in histology slides which could potentially serve as an HRD marker. Prospective trials are needed to evaluate an AI-based HRD score as a biomarker to guide treatment decisions, potentially in a two-step approach leading to lower sequencing requirements and cost reduction.
Limitations
Our study has several limitations. Firstly, the sample sizes of our cohorts, particularly the CPTAC dataset, are relatively small. Moreover, the variation within the distribution of HRD prevalences between tumor types can result in class imbalances. Although the effect of imbalanced datasets on the accuracy of our classifiers was addressed via weighing techniques during the model training process, this could still have an effect on the statistical power of the results, as well as the generalisability of our models to a larger population. We observed higher AUROCs in the external validation cohort, which may be attributed to the smaller size and higher class imbalance in the test set. Further studies with larger patient cohorts are required to validate our findings. Furthermore, the quality of the data from the TCGA and CPTAC cohorts may vary, thus potentially impacting the accuracy of our predictions.
Conclusion
Our findings provide evidence that DL has the potential to not only contribute but improve diagnostic HRD testing, potentially saving time and costs as well as improving outcomes for patients by identifying subgroups who may benefit from targeted therapy. Current clinical practices face challenging factors such as high cost, time consumption, lack of availability, and inconsistency in HRD status screening methods. These logistic, analytic and financial challenges contribute to the partial identification of cancer patients who may benefit from PARPi therapy and to the limited genetic testing, which is further compounded by the panoply of HRD status assessment methods whose inter-assay concordance is limited. With the aid of AI, we have the opportunity to identify these subgroups and improve patient outcomes.
Supplementary Material
Funding
JNK is supported by the German Federal Ministry of Health (DEEP LIVER, ZMVI1-2520DAT111) and the Max-Eder-Programme of the German Cancer Aid (grant #70113864), the German Federal Ministry of Education and Research (PEARL, 01KD2104C), and the German Academic Exchange Service (SECAI, 57616814). This research was supported by the National Institute for Health and Care Research (NIHR, NIHR213331) Leeds Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. JSRF is funded in part by the Breast Cancer Research Foundation, a Susan G Komen Leadership Grant, the NIH/NCI P50 CA247749 01 grant and by the NIH/NCI Cancer Center Core Grant P30-CA008748.
List of Abbreviations
- AI
artificial intelligence
- ASCAT
Allele-Specific Copy number Analysis of Tumors
- attMIL
attention-weighted multiple instance learning
- AUROC
Area Under the Receiver Operating Characteristic curve
- BRCA
breast invasive carcinoma
- BRCA1/2
Breast Cancer genes 1 and 2
- CI
confidence interval
- CIOMS
Council for International Organizations of Medical Sciences
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- CRC
colorectal cancer
- DL
Deep Learning
- DSB
DNA double-strand breaks
- ER−
estrogen receptor negative
- ER+
estrogen receptor positive
- FDA
U.S. Food and Drug Administration
- GBM
glioblastoma
- GDC
Genomic Data Commons
- GIS
genomic instability score
- H&E
Hematoxylin & Eosin
- HR
Homologous recombination
- HRD-H
HRD high
- HRD-L
HRD low
- HRD
Homologous Recombination Deficiency
- HRR
Homologous recombination repair
- LIHC
liver hepatocellular carcinoma
- LOH
loss of heterozygosity
- LSCC
squamous cell carcinoma of the lung
- LST
large-scale state transitions
- LUAD
adenocarcinoma of the lung
- LUSC
squamous cell carcinoma of the lung
- OV
ovarian cancer (OV)
- PAAD
pancreatic adenocarcinoma
- PDA
pancreatic adenocarcinoma
- PARP
Poly(ADP-Ribose)-polymerase
- PARPi
Poly(ADP-Ribose)-polymerase inhibitor
- PRAD
prostate adenocarcinoma
- PRC
precision recall curve
- ROC
receiving operating curve
- SBS3
single base substitution 3
- SNP
single nucleotide polymorphism
- SSDBs
single strand DNA breaks
- SSL
self-supervised learning
- TAI
telomeric allelic imbalance
- TCGA
The Cancer Genome Atlas
- TRIPOD
Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis
- UCEC
endometrial carcinoma
- WSI
whole slide images
Footnotes
Ethics statement
The experiments in this study were carried out according to the Declaration of Helsinki and the International Ethical Guidelines for Biomedical Research Involving Human Subjects by the Council for International Organizations of Medical Sciences (CIOMS). The present study also adheres to the “Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis” (TRIPOD) statement.20. The Ethics Board at the Medical Faculty of Technical University Dresden (BO-EK-444102022) approved of the overall analysis in this study. The patient sample collection in each cohort was separately approved by the respective institutional ethics board.
Competing Interests
JNK reports consulting services for Owkin, France, Panakeia, UK and DoMore Diagnostics, Norway and has received honoraria for lectures by MSD, Eisai and Fresenius. JSRF reports a leadership (board of directors) role at Grupo Oncoclinicas, stock or other ownership interests at Repare Therapeutics and Paige.AI, and a consulting or Advisory Role at Genentech/Roche, Invicro, Ventana Medical Systems, Volition RX, Paige.AI, Goldman Sachs, Bain Capital, Novartis, Repare Therapeutics, Lilly, Saga Diagnostics, Swarm and Personalis. No other potential conflicts of interest are reported by any of the authors.
Data and Code availability
The WSI, molecular and clinical data for TCGA and CPTAC cohorts are publicly accessible at https://portal.gdc.cancer.gov/ and https://www.cbioportal.org/ (accessed, 08 March 2022). Script for calculating the HRD score is available under https://github.com/sztup/scarHRD (accessed 06 June 2022). All other source codes can be downloaded under https://github.com/KatherLab/marugoto. Our calculated HRD score is publicly available in Supplementary Table 2. Moreover, our custom TCGA-BRCA HRD-H and HRD-L group can be accessed for the PanCancer Atlas cohort at https://www.cbioportal.org/ (Supplementary Table 3).
References
- 1.Frey MK, Pothuri B. Homologous recombination deficiency (HRD) testing in ovarian cancer clinical practice: a review of the literature. Gynecol Oncol Res Pract. 2017. Feb 22;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001. May 17;411(6835):366–74. [DOI] [PubMed] [Google Scholar]
- 3.Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol. 2020. Sep 9;8:564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011. Apr 15;10(8):1192–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leary A, Auguste A, Mesnage S. DNA damage response as a therapeutic target in gynecological cancers. Curr Opin Oncol. 2016. Sep;28(5):404–11. [DOI] [PubMed] [Google Scholar]
- 6.Park W, Chen J, Chou JF, Varghese AM, Yu KH, Wong W, et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin Cancer Res. 2020. Jul 1;26(13):3239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takaya H, Nakai H, Takamatsu S, Mandai M, Matsumura N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci Rep. 2020. Feb 17;10(1):2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N Engl J Med. 2021. Jun 24;384(25):2394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016. Apr;27 Suppl 1:i40–4. [DOI] [PubMed] [Google Scholar]
- 10.Stewart MD, Merino Vega D, Arend RC, Baden JF, Barbash O, Beaubier N, et al. Homologous Recombination Deficiency: Concepts, Definitions, and Assays. Oncologist. 2022. Mar 11;27(3):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020. Dec;31(12):1606–22. [DOI] [PubMed] [Google Scholar]
- 12.Wagener-Ryczek S, Merkelbach-Bruse S, Siemanowski J. Biomarkers for Homologous Recombination Deficiency in Cancer. J Pers Med [Internet]. 2021. Jun 28;11(7). Available from: 10.3390/jpm11070612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuh K, Mullen M, Blachut B, Stover E, Konstantinopoulos P, Liu J, et al. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol Oncol. 2020. Dec;159(3):877–86. [DOI] [PubMed] [Google Scholar]
- 14.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020. Feb;578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulhan DC, Lee JJK, Melloni GEM, Cortés-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019. May;51(5):912–9. [DOI] [PubMed] [Google Scholar]
- 16.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012. Nov 6;107(10):1776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birkbak NJ, Wang ZC, Kim JY, Eklund AC, Li Q, Tian R, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012. Apr;2(4):366–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popova T, Manié E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012. Nov 1;72(21):5454–62. [DOI] [PubMed] [Google Scholar]
- 19.Westphalen CB, Fine AD, André F, Ganesan S, Heinemann V, Rouleau E, et al. Pan-cancer Analysis of Homologous Recombination Repair-associated Gene Alterations and Genome-wide Loss-of-Heterozygosity Score. Clin Cancer Res. 2022. Apr 1;28(7):1412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sztupinszki Z, Diossy M, Krzystanek M, Reiniger L, Csabai I, Favero F, et al. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer. 2018. Jul 2;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao EY, Shen Y, Pleasance E, Kasaian K, Leelakumari S, Jones M, et al. Homologous Recombination Deficiency and Platinum-Based Therapy Outcomes in Advanced Breast Cancer. Clin Cancer Res. 2017. Dec 15;23(24):7521–30. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen L, W M Martens J, Van Hoeck A, Cuppen E. Pan-cancer landscape of homologous recombination deficiency. Nat Commun. 2020. Nov 4;11(1):5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellegrino B, Herencia-Ropero A, Llop-Guevara A, Pedretti F, Moles-Fernandez A, Viaplana C, et al. Preclinical In Vivo Validation of the RAD51 Test for Identification of Homologous Recombination-Deficient Tumors and Patient Stratification. Cancer Res. 2022. Apr 15;82(8):1646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010. Dec 15;16(24):6159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.How JA, Jazaeri AA, Fellman B, Daniels MS, Penn S, Solimeno C, et al. Modification of Homologous Recombination Deficiency Score Threshold and Association with Long-Term Survival in Epithelial Ovarian Cancer. Cancers [Internet]. 2021. Feb 24;13(5). Available from: 10.3390/cancers13050946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmauch B, Romagnoni A, Pronier E, Saillard C, Maillé P, Calderaro J, et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat Commun. 2020. Aug 3;11(1):3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeffler CML, Ortiz Bruechle N, Jung M, Seillier L, Rose M, Laleh NG, et al. Artificial Intelligence-based Detection of FGFR3 Mutational Status Directly from Routine Histology in Bladder Cancer: A Possible Preselection for Molecular Testing? Eur Urol Focus. 2022. Mar;8(2):472–9. [DOI] [PubMed] [Google Scholar]
- 28.Shmatko A, Ghaffari Laleh N, Gerstung M, Kather JN. Artificial intelligence in histopathology: enhancing cancer research and clinical oncology. Nat Cancer. 2022. Sep;3(9):1026–38. [DOI] [PubMed] [Google Scholar]
- 29.Fu Y, Jung AW, Torne RV, Gonzalez S, Vöhringer H, Shmatko A, et al. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nature Cancer. 2020. Jul 27; 1—11. [DOI] [PubMed] [Google Scholar]
- 30.Kather JN, Heij LR, Grabsch HI, Loeffler C, Echle A, Muti HS, et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nature Cancer. 2020. Aug 1;1(8):789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kather JN, Pearson AT, Halama N, Jäger D, Krause J, Loosen SH, et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat Med. 2019. Jul;25(7):1054–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muti HS, Heij LR, Keller G, Kohlruss M, Langer R, Dislich B, et al. Development and validation of deep learning classifiers to detect Epstein-Barr virus and microsatellite instability status in gastric cancer: a retrospective multicentre cohort study [Internet]. The Lancet Digital Health. 2021. Available from: 10.1016/s2589-7500(21)00133-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saillard C, Dubois R, Tchita O, Loiseau N, Garcia T, Adriansen A, et al. Blind validation of MSIntuit, an AI-based pre-screening tool for MSI detection from histology slides of colorectal cancer [Internet]. bioRxiv. 2022. Available from: 10.1101/2022.11.17.22282460.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleppe A, Skrede OJ, De Raedt S, Hveem TS, Askautrud HA, Jacobsen JE, et al. A clinical decision support system optimising adjuvant chemotherapy for colorectal cancers by integrating deep learning and pathological staging markers: a development and validation study. Lancet Oncol. 2022. Sep;23(9):1221–32. [DOI] [PubMed] [Google Scholar]
- 35.Valieris R, Amaro L, Osorio CAB de T, Bueno AP, Rosales Mitrowsky RA, Carraro DM, et al. Deep Learning Predicts Underlying Features on Pathology Images with Therapeutic Relevance for Breast and Gastric Cancer. Cancers [Internet]. 2020. Dec 9; 12(12). Available from: 10.3390/cancers12123687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lazard T, Bataillon G, Naylor P, Popova T, Bidard FC, Stoppa-Lyonnet D, et al. Deep learning identifies morphological patterns of homologous recombination deficiency in luminal breast cancers from whole slide images. Cell Rep Med. 2022. Dec 20;3(12):100872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013. Apr 2;6(269):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012. May;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017. Oct 11;8(1):857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macenko M, Niethammer M, Marron JS, Borland D, Woosley JT, Guan X, et al. A method for normalizing histology slides for quantitative analysis. In: 2009 IEEE International Symposium on Biomedical Imaging: From Nano to Macro. 2009. p. 1107–10. [Google Scholar]
- 41.Wang X, Du Y, Yang S, Zhang J, Wang M, Zhang J, et al. RetCCL: Clustering-guided contrastive learning for whole-slide image retrieval. Med Image Anal. 2023. Jan 1;83:102645. [DOI] [PubMed] [Google Scholar]
- 42.Leiby JS, Hao J, Kang GH, Park JW, Kim D. Attention-based multiple instance learning with self-supervision to predict microsatellite instability in colorectal cancer from histology whole-slide images. Conf Proc IEEE Eng Med Biol Soc. 2022. Jul;2022:3068–71. [DOI] [PubMed] [Google Scholar]
- 43.Ilse M, Tomczak J, Welling M. Attention-based Deep Multiple Instance Learning. In: Dy J, Krause A, editors. Proceedings of the 35th International Conference on Machine Learning. PMLR; 10–15 Jul 2018. p. 2127–36. (Proceedings of Machine Learning Research; vol. 80). [Google Scholar]
- 44.Rempel E, Kluck K, Beck S, Ourailidis I, Kazdal D, Neumann O, et al. Pan-cancer analysis of genomic scar patterns caused by homologous repair deficiency (HRD). NPJ Precis Oncol. 2022. Jun 9;6(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleppe A, Skrede OJ, De Raedt S, Liestøl K, Kerr DJ, Danielsen HE. Designing deep learning studies in cancer diagnostics. Nat Rev Cancer. 2021. Mar;21(3):199–211. [DOI] [PubMed] [Google Scholar]
- 46.Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, et al. The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin Cancer Res. 2017. Jul 15;23(14):3859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ngoi NYL, Tan DSP. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: do we need it? ESMO Open. 2021. Jun;6(3):100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loeffler CML, Gaisa NT, Muti HS, van Treeck M. Predicting Mutational Status of Driver and Suppressor Genes Directly from Histopathology With Deep Learning: A Systematic Study Across 23 Solid Tumor …. Frontiers in [Internet]. 2021; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8889144/ [DOI] [PMC free article] [PubMed]
- 49.Takamatsu S, Brown JB, Yamaguchi K, Hamanishi J, Yamanoi K, Takaya H, et al. Utility of Homologous Recombination Deficiency Biomarkers Across Cancer Types. JCO Precis Oncol. 2022. May;6:e2200085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moukarzel LA, Ferrando L, Da Cruz Paula A, Brown DN, Geyer FC, Pareja F, et al. The genetic landscape of metaplastic breast cancers and uterine carcinosarcomas. Mol Oncol. 2021. Apr;15(4):1024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Na B, Yu X, Withers T, Gilleran J, Yao M, Foo TK, et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NPJ Breast Cancer. 2019. Apr 15;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai Z, Brosnan M, Sokol ES, Xie M, Dry JR, Harrington EA, et al. Landscape of homologous recombination deficiencies in solid tumours: analyses of two independent genomic datasets. BMC Cancer. 2022. Jan 3;22(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The WSI, molecular and clinical data for TCGA and CPTAC cohorts are publicly accessible at https://portal.gdc.cancer.gov/ and https://www.cbioportal.org/ (accessed, 08 March 2022). Script for calculating the HRD score is available under https://github.com/sztup/scarHRD (accessed 06 June 2022). All other source codes can be downloaded under https://github.com/KatherLab/marugoto. Our calculated HRD score is publicly available in Supplementary Table 2. Moreover, our custom TCGA-BRCA HRD-H and HRD-L group can be accessed for the PanCancer Atlas cohort at https://www.cbioportal.org/ (Supplementary Table 3).