

Abstract
Prior to the early 2000s, patients with advanced gastrointestinal stromal tumors (GIST) had very poor prognoses owing to a lack of effective therapies. The development of tyrosine kinase inhibitors at the turn of the century significantly improved the overall survival for patients with GIST. The resounding success of imatinib in the first clinical trial of a tyrosine kinase inhibitor to treat GIST led to its approval for first-line therapy for advanced GIST; this study was open to all comers and not restricted to any GIST subtype(s). The trials that led to the approvals of second-, third-, and fourth-line therapy for advanced GIST were also open to all patients with advanced/metastatic GIST. Only in retrospect do we realize the role that the molecular subtypes played in the results observed in these studies. In this review, we discuss the studies that led to the US Food and Drug Administration approval of imatinib (first line), sunitinib (second line), regorafenib (third line), and ripretinib (fourth line) for advanced KIT-mutant GIST. In addition, we review how information about GIST molecular subtypes has been used to accelerate the approval of other targeted therapies for non-KIT mutant GIST, leading to the approval of five additional drugs indicated for the treatment of specific GIST molecular subtypes. We also discuss how our understanding of the molecular subtypes will play a role in the next generation of therapeutic approaches for treating advanced GIST.
1. Introduction
Gastrointestinal stromal tumors (GIST), the most common soft-tissue sarcoma, are driven by a variety of oncogenic drivers. The understanding of GIST molecular drivers has resulted in the clinical testing and eventual approval of multiple targeted therapies, specifically tyrosine kinase inhibitors (TKIs), which have revolutionized the treatment of the majority of patients with GIST [1]. Since the implementation of effective targeted therapies, the overall survival (OS) of patients with advanced GIST has improved to the current estimated range of 6–8 years, compared with historical results of approximately 1.5 years when treated with chemotherapy or radiation (neither of which is effective in GIST), or < 15 months with surgery alone [1–5]. This improvement in survival is mostly confined to the population of patients with GIST with driver mutations in the receptor tyrosine kinase (RTK) KIT, which can be effectively targeted with KIT TKIs. The KIT TKIs imatinib (first line), sunitinib (second line), regorafenib (third line), and ripretinib (fourth line) were approved by health authorities, such as the US Food and Drug Administration, based on studies in which eligibility required only a diagnosis of advanced GIST and no requirements of any particular molecular driver [4, 6–8]. For the purposes of classification, we have labeled these registrational studies as examples of a mutation-agnostic/histology-specific study design. This approach was successful for the clinical development of KIT TKIs because the vast majority (70%) of patients with GIST have KIT mutations (Fig. 1A) that can be effectively targeted by these agents [9]. However, we believe that this approach is not viable for the development of future therapies. It has become apparent that mutation-agnostic clinical studies using KIT TKIs to treat patients of other molecular subtypes outside of KIT-mutant GIST do not provide the same degree of clinical benefit, therefore leaving these patients with advanced GIST with few to no options for effective therapy [10–15].
Fig. 1.
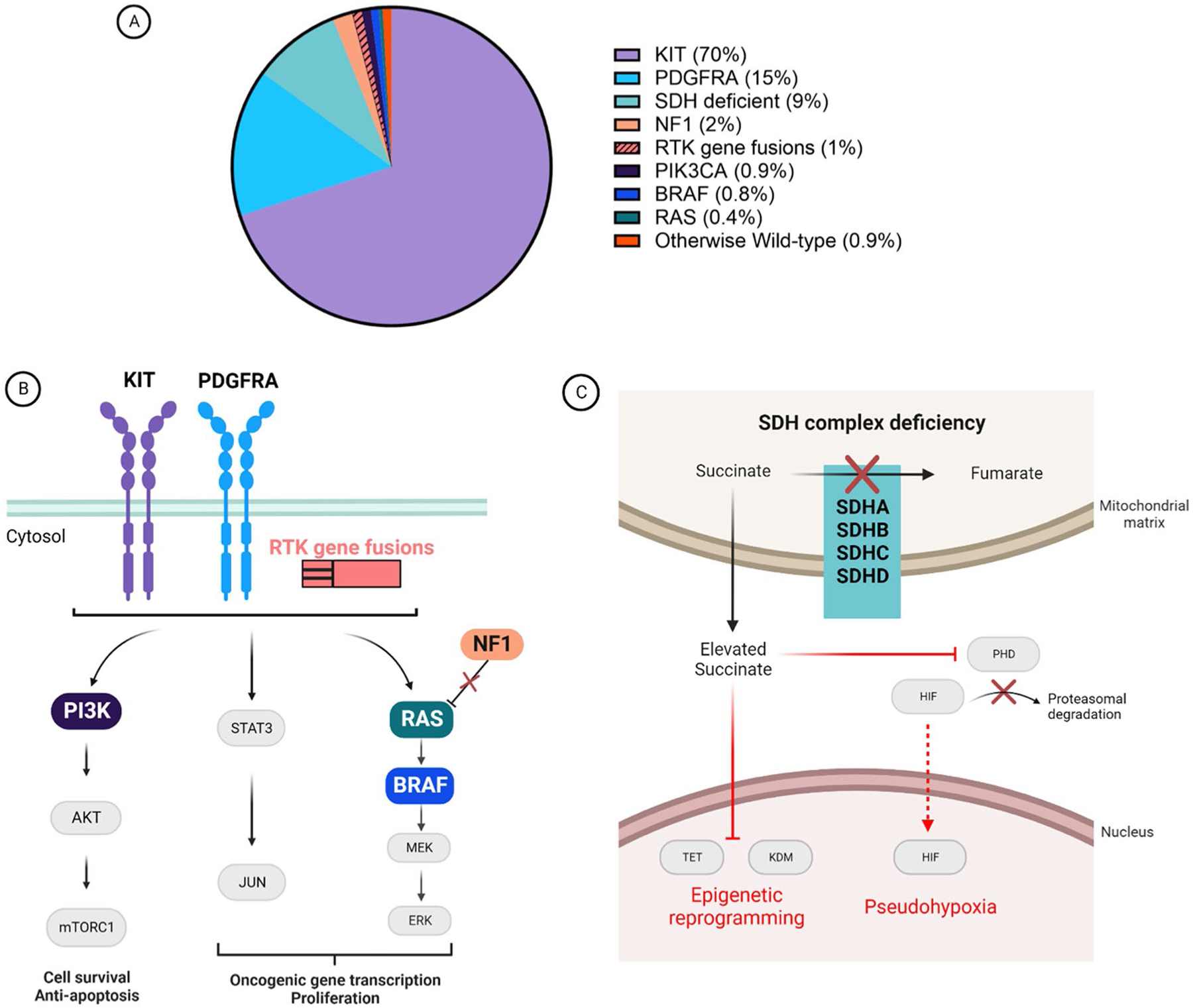
Molecular drivers of gastrointestinal stromal tumors (GIST) and their corresponding oncogenic signaling mechanisms. A Proportion of GIST cases based on oncogenic driver subtype. B The majority of genetic aberrations that drive GIST development result in the activation of downstream signaling pathways. The downstream pathways (JAK/STAT, PI3K/AKT/mTORC1, and RAS/RAF/MEK/ERK) activated are similar for GIST driven by KIT, platelet-derived growth factor receptor alpha (PDGFRA), or receptor tyrosine kinase (RTK) gene fusions (e.g., FGFR1, FGFR2, NTRK1, NTRK3, and ALK). Gastrointestinal stromal tumors driven by neurofibromatosis (type 1) (NF1) loss, RAS, BRAF, or PI3K activation also utilize similar pathways for oncogenesis. The colors for these genetic aberrations correspond to the pie chart in panel A. C A subset of GIST is driven by succinate dehydrogenase (SDH) complex deficiency. This deficiency results from an inactivating mutation of any one of the four SDH subunit genes (SDHA, SDHB, SDHC, or SDHD) or epimutation of the SDHC promoter leading to gene silencing. The SDH complex converts succinate to fumarate, but in SDH-deficient GIST the inactive complex results in the accumulation of succinate. This accumulated succinate leads to the inhibition of prolyl hydroxylase (PHD), which is involved in the proteasomal degradation of hypoxia-inducible factor (HIF). Elevated succinate levels also have an effect on epigenetic reprogramming, by inhibiting ten-eleven translocation methyl-cytosine dioxygenase (TET) and lysine-specific histone demethylase (KDM) family proteins. Image created using BioRender.com
Recently, rational drug design and biological approaches focused on these less common GIST molecular subtypes (remaining 30% of cases) have resulted in successful clinical studies testing novel therapeutics. These drugs include: avapritinib (a novel type I platelet-derived growth factor receptor alpha [PDGFRA]/KIT TKI), larotrectinib and entrectinib (potent NTRK TKIs for targeting NTRK fusions in GIST) [16–19], and the combination of a BRAF inhibitor (dabrafenib) and a MEK inhibitor (trametinib) for BRAF V600E-mutant GIST [20–22]. The recent approvals of these five treatments, each for a specific molecular subtype, were enabled by clinical studies with fundamentally different approaches for selecting eligible patients for participation than those used for the early KIT TKI trials. The inclusion criteria of avapritinib, larotrectinib or entrectinib, and the combination of dabrafenib and trametinib, phase I–II studies required the presence of a PDGFRA D842V mutation [23, 24], a NTRK family fusion, or a BRAF V600E mutation [16–19, 22], respectively. In this review, we refer to these studies as having a precision medicine study design (i.e., mutation-specific patient eligibility). This type of study design can be histology specific (e.g., GIST avapritinib approval) or histology agnostic (e.g., BRAF V600E-mutant solid tumors and approval of dabrafenib and trametinib combination [22]). We posit that precision medicine approaches, facilitated by diagnostic molecular testing and pre-clinical research, will be the most successful strategy for developing new GIST treatment strategies, especially for those less common, non-KIT mutant GIST subtypes.
2. GIST Drivers and Molecular Subtypes
Over the past 20 years, analysis of tens of thousands of cases of primary GIST have led to the identification of molecular drivers in almost all cases. Mutations in homologous RTKs, KIT and PDGFRA, account for 70 and 15% of cases, respectively (Fig. 1A) [1, 11, 25–28]. As is well known, most (RTKs), including mutant forms of KIT or PDGFRA, signal through the canonical JAK/STAT, PI3K/AKT/mTORC, and RAS/RAF/MEK/ERK pathways (Fig. 1B) [29–34]. Along this line, a minority of GIST cases have other RTKs in addition to KIT/PDGFRA as molecular drivers, including more recent descriptions of activating translocations of FGFR1, FGFR2, NTRK1, NTRK3, and ALK (Fig. 1A, B) [28, 35–38]. In addition, there are cases of GIST that arise because of activating mutations of PIK3CA, BRAF, or RAS family members (Fig. 1A, B) [39–42]. More recently, BRAF translocations have been reported as a molecular driver in GIST [43]. In addition, homozygous/hemizygous loss of neurofibromatosis (type 1) [NF1] is a well-established cause of some cases of GIST, likely due to dysregulation of the RAS/RAF/MEK/ERK pathway. Neurofibromatosis (type 1) loss giving rise to GIST can occur sporadically owing to a somatic mutation or in the setting of NF1 with a heterozygous germline NF1 mutation [44–48]. In total, cases of RTK translocation-mutant GIST or those with mutations of downstream signaling pathways account for approximately 5% of all cases of GIST (Fig. 1A) [9].
In addition to mutations that activate RTK downstream signaling pathways, a biologically and clinically distinct subtype of GIST is driven by succinate dehydrogenase (SDH) deficiency [49–51]. Gastrointestinal stromal tumors of this subtype result from inactivating hemizygous or compound heterozygous mutations of one the four SDH subunits (SDHA, SDHB, SDHC, or SDHD) or epimutation of the SDHC promoter (Fig. 1C) [49, 52–56]. In total, 9% of GIST are SDH deficient, comprising the third largest molecular subtype after KIT- and PDGFRA-mutant GIST. In contrast to GIST driven by activating mutations in RTKs or within the classical RTK-signaling pathways, these cases arise as a consequence of SDH loss of function. Succinate dehydrogenase loss of function results in increased intracellular succinate that inhibits a certain class of enzymes leading to activation of the VHL pathway (pseudohypoxia), as well as inhibition of ten-eleven translocation and KDM family enzymes with resultant genome-wide epigenetic reprogramming (Fig. 1C) [51, 53, 57, 58].
3. Traditional TKI Treatment Paradigm for Advanced GIST
Slightly more than two decades ago, the first clinical trial of a targeted therapy in GIST began [8, 59]. There are now nine targeted therapies approved for GIST, and while we now know that each drug is most effective for specific subtypes of GIST, four of these agents are still approved for the treatment of all forms of GIST because their registrational studies included all patients with advanced GIST (Table 1). The current treatment sequencing for KIT-mutant GIST remains in the order of original drug development and US Food and Drug Administration (FDA) approval: imatinib (first line), sunitinib (second line), regorafenib (third line), and ripretinib (fourth line). The trials that led to the approvals of imatinib, sunitinib, regorafenib, and ripretinib were open to all patients with advanced/metastatic GIST and only later was it fully appreciated the role molecular subtypes played in the results reported from those studies [4, 7, 8, 10–13, 60–65]. Figure 2 highlights the results from these studies and showcases that the traditional treatment paradigm was mostly successful for patients with KIT-mutant and certain non-exon 18 PDGFRA-mutant GIST and minimally effective for patients with other subtypes. In the following sections, we focus on the preclinical and clinical studies that led to the approval of imatinib, sunitinib, regorafenib, and ripretinib.
Table 1.
List of US FDA-approved therapies for the treatment of advanced/metastatic GIST with summary details on FDA-approved prescribing indication, study design leading to drug approval for this indication, and reported major clinical outcomes of interest
| Agent | Year of approval | Approval | Studies leading to approval | Inclusion criteria | ORR | PFS | OS | References |
|---|---|---|---|---|---|---|---|---|
| Imatinib (Gleevec) | 2002 | Patients with KIT (CD117) positive unresectable and/or metastatic malignant GIST | Phase I/II | Adults with a histologically confirmed, unresectable or metastatic GIST that expressed CD117 (a marker of KIT-receptor tyrosine kinase) |
Demetri [8] | |||
| Phase III | Patients with metastatic or unresectable GIST | 400 mg: 51%, 800 mg: 54% | 400 mg: 18.9 months; 800 mg: 23.2 months | 400 mg: 49.0 months, 800 mg: 48.7 months | Verweij [69] | |||
| Sunitinib (Sutent) | 2006 | GIST after disease progression on or intolerance to imatinib mesylate | Phase I/II | Patients with imatinib-resistant/intolerant GIST | 7% | 7.8 months | 19.0 months | Demetri [95] |
| Phase II (assess morning vs evening dosing) | Patients with imatinib-resistant/intolerant GIST | 34 weeks (7.8 months) | 107 weeks (24.6 months) | George [88] | ||||
| Phase III (NCT00075218) | Patients with unresectable GIST and progression following imatinib | 6.80% | 5.6 months | Demetri [7] | ||||
| Regorafenib (Stivarga) | 2012 | Locally advanced, unresectable or metastatic GIST who have been previously treated with imatinib mesylate and sunitinib malate | Phase II (NCT01068769) | Patients with advanced GIST after failure of at least imatinib and sunitinib | 18% | 13.2 months | 25.0 months | George [6] Ben-Ami [101] |
| Phase III (NCT01271712) | Patients with metastatic and/or unresectable GIST progressing after failure of at least imatinib and sunitinib | 4.50% | 4.8 months | NR | Demetri [61] | |||
| Ripretinib (Qinlock) | 2020 | Indicated for the treatment of adult patients with advanced GIST who have received prior treatment with 3 or more kinase inhibitors, including imatinib | Phase I (NCT02571036) | Patients with advanced GIST, intolerant to or experienced progression on 1st line of systemic therapy, and other advanced malignancies | 11.3% (> fourth line: 7.2%, second line: 19.4%) | 5.5 months (> fourth line), 10.7 months (second line) | NR | Janku [102] |
| Phase III (NCT03353753: INVICTUS) | Patients with previously treated, advanced GIST | 9.40% | 6.3 months | 15.1 months | Blay [4] | |||
| Phase III (NCT03673501: INTRIGUE) | Patients with advanced GIST after front-line imatinib failure vs sunitinib | NR | NR | NR | ||||
| Avapritinib (Ayvakit) | 2020 | Inhibitor indicated for the treatment of adults with unresectable or metastatic GIST harboring a PDGFRA exon 18 mutation, including PDGFRA D842V mutations | Phase I (NCT02508532) | Dose escalation: patients with unresectable GIST and progression following imatinib and > 1 other TKI or PDGFRA D842V mutation Dose expansion: patients with unresectable GIST |
Patients with PDGFRA D842V: 89%, patients with other PDGFRA ex 18 mutations: 84% | 29.5 months | NR | Heinrich [23] Jones [24] |
| Larotrectinib (Vitrakvi) | 2018 | Indicated for the treatment of adult and pediatric patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation, are metastatic or where surgical resection is likely to result in severe morbidity, and have no satisfactory alternative treatments or that have progressed following treatment | Phase I/II (NCT02122913) Phase I/II (NCT02637687) Phase I/II (NCT02576431) |
Pediatric and adult patients with unresectable or metastatic solid tumors with a NTRK gene fusion | 75% (patients with GIST [n = 3]: 100%) | NR specifically for GIST | NR specifically for GIST | Drilon [17] |
| Entrectinib (Rozyltrek) | 2019 | Indicated for the treatment of adult and pediatric patients 12 years of age and older with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation, whose disease is metastatic or where surgical resection is likely to result in severe morbidity, and who have progressed following treatment or have no satisfactory alternative therapy | Phase I (NCT02097810) Phase II (NCT02568267) |
Adult patients with unresectable or metastatic solid tumors with an NTRK fusion | NR specifically for GIST [n = 1] | NR specifically for GIST | NR specifically for GIST | Doebele [126] |
| Dabrafenib (Taflinar) given with Trametinib (Mekinist) | 2022 | Trametinib is indicated, in combination with dabrafenib, for the treatment of adult and pediatric patients 6 years of age and older with unresectable or metastatic solid tumors with a BRAF V600E mutation who have progressed following prior treatment and have no satisfactory alternative treatment options This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s) | Phase II (NCT02034110) NCT MATCH (NCT02465060) |
Adult patients with unresectable or metasttic solid tumors with a BRAF V600E mutation, excluding patients with melanoma, thyroid cancer, or colorectal cancer | 0% GIST [n = 1] | NR specifically for GIST | NR specifically for GIST | See prescribing information [20] |
FDA Food and Drug Administration, GIST gastrointestinal stromal tumors, NR not reported, NTRK neurotrophic tyrosine receptor kinase, ORR objective response rate, OS median overall survival, PDGFRA platelet-derived growth factor receptor alpha, PFS progression-free survival, REF primary study publication(s)
Fig. 2.
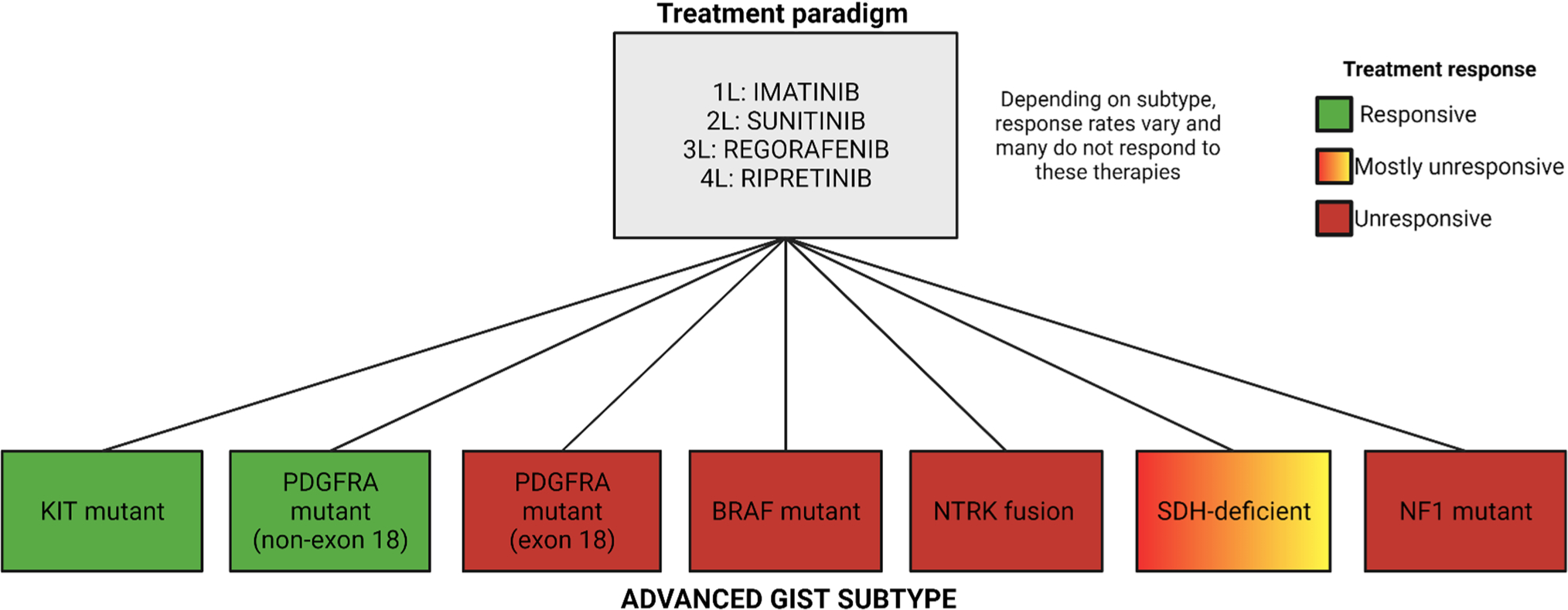
Traditional treatment paradigm for advanced gastrointestinal stromal tumors. The gray box shows the list of US Food and Drug Administration-approved drugs in the traditional treatment paradigm, and 1L, 2L, 3L, and 4L correspond to first-, second-, third-, and fourth-line therapy, respectively. The advanced gastrointestinal stromal tumor subtypes are listed in the colored boxes, and those colors correspond to patient treatment response to 1L, 2L, 3L, and 4L therapy. It was later discovered after clinical trials that based on the advanced gastrointestinal stromal tumor subtype, response rates vary amongst patients depending on the mutational status. NF1 neurofibromatosis (type 1), NTRK neurotrophic tyrosine receptor kinase, PDGFRA platelet-derived growth factor receptor alpha, SDH succinate dehydrogenase. Image created using BioRender.com
3.1. Imatinib
Imatinib, a novel agent originally designed to inhibit the aberrant enzymatic function of the oncogenic driver in chronic myelogenous leukemia, BCR::ABL1, was found to be similarly effective in inhibiting the enzymatic function of KIT, the main driver in the majority of GIST cases (Fig. 1) [66–68]. Despite being designed for targeted cancer therapy, the phase I, II, and III studies of imatinib that led to its approval for advanced GIST included patients of all GIST subtypes, the only criteria being “metastatic or advanced CD117+ GIST” (Table 1) [8, 59, 69, 70]. The response rate to imatinib in the initial studies was 50–80% [8, 11, 14, 70–73]. At that time, molecular analysis of GIST was in its infancy, thus the vast majority of patients were enrolled without any molecular testing results, but it was subsequently determined that almost all of the responders in this trial had driver mutations that were sensitive to imatinib (e.g., KIT exons 9, 11, or 13; PDGFRA exon 12) [Fig. 2] [13, 14, 70, 73]. Those with the third most common subtype, SDH-deficient GIST, had a < 5% objective response rate (ORR) to imatinib [51, 74]. With the appreciation of the molecular underpinnings of the success of imatinib as a targeted therapy in GIST, the search for additional targetable drivers accelerated. For example, analysis of a patient with a partial response to imatinib whose tumor lacked a KIT mutation led to investigations that identified an imatinib-sensitive PDGFRA mutation [11, 14]. Likewise, investigation of patients with “wild-type” KIT (meaning no KIT mutation) GIST who had rapid progression during imatinib therapy led to the identification of the imatinib-resistant primary PDGFRA D842V mutation [25, 75]. It is known that TKIs that target wild-type KIT also inhibit wild-type PDGFRA and the converse is true as well [66, 76–78]. Therefore, at the time, the treatment of PDGFRA mutant-GIST followed the same clinical development paradigm as the treatment of KIT-mutant GIST with the exception of patients with imatinib-resistant PDGFRA D842V, which we discuss in a later section.
After several years, the first reports about patients with GIST with delayed resistance to imatinib began to appear because of acquired secondary intra-allelic KIT mutations that disrupted drug binding [79–83]. We now appreciate that intra-allelic secondary kinase mutations are the most common mechanism of acquired resistance to imatinib in GIST [1, 9, 84–87]. This mechanism of resistance demonstrated the continued dependence of GIST on the enzymatic activity of the initial driver, and led to the development of additional targeted TKIs to overcome specific secondary KIT mutations in imatinib-resistant patients.
3.2. Sunitinib
The “next-generation” GIST TKIs arose from the rapid development of various small-molecule kinase inhibitors in the early 2000s. In most cases, KIT was not the primary target of the drug development program, but drugs designed to inhibit vascular endothelial growth factors and/or PDGFRs commonly had similar, or even greater, potency against KIT. Sunitinib, a type II multi-kinase inhibitor, entered phase I–II studies and then proceeded to be tested in a phase III clinical study in patients with imatinib-refractory GIST in 2003 [7, 88]. Again, these trials were conducted with a mutation-agnostic approach. “Imatinib-refractory” patients that enrolled in the registrational sunitinib phase study included both those with primary resistance (progression in < 6 months, e.g., PDGFRA D842V) and those with secondary resistance, meaning they initially responded to imatinib, but then their tumors progressed after months or even years of clinical response. No molecular testing was required for study eligibility (Table 1). Pre-clinically, sunitinib was shown to inhibit some imatinib-resistant KIT secondary mutations, including the most common imatinib-resistance mutation, V654A, and other secondary KIT mutations in exons 13 and 14, which encode the ATP binding pocket (ABP) [Fig. 3] [10, 12]. However, as sunitinib is a type II TKI like imatinib, it does not inhibit activation loop mutations that confer primary resistance (PDGFRA D842V and cases lacking a KIT/PDGFRA driver mutation [historically classified as “wild-type” cases]) nor those with secondary KIT activation loop (exons 17–18) mutations of the kinase domain [12, 89–94]. Notably, these studies were predictive of the results of the sunitinib clinical studies, specifically the greatly reduced response rate compared with the initial imatinib trials (6.8 vs ~ 51.4%, respectively), largely owing to the presence of heterogeneous tumor clones, some with sunitinib-sensitive mutations and others with resistant mutations [10, 12, 86, 87, 95]. It was also thought that a minority of SDH-deficient GIST cases may respond to sunitinib or regorafenib (discussed below), likely due to vascular endothelial growth factor receptor inhibitory activity against these agents [51, 62, 96, 97]. However, at that time, after progression on imatinib, patients had no other options, therefore the criteria for approval were low. Sunitinib was approved for second-line treatment of patients with GIST after progression on imatinib, although it is now clear that it mainly benefits only a select subset patients with the KIT mutation (those with secondary ABP or primary KIT exon 9 mutations) [98, 99]. Potentially, the results of these trials could have had a higher response rate if the eligibility criteria were limited to those with ABP mutations; however, there would still remain the issue of how to treat patients with heterogeneous tumor clones after progression on imatinib.
Fig. 3.
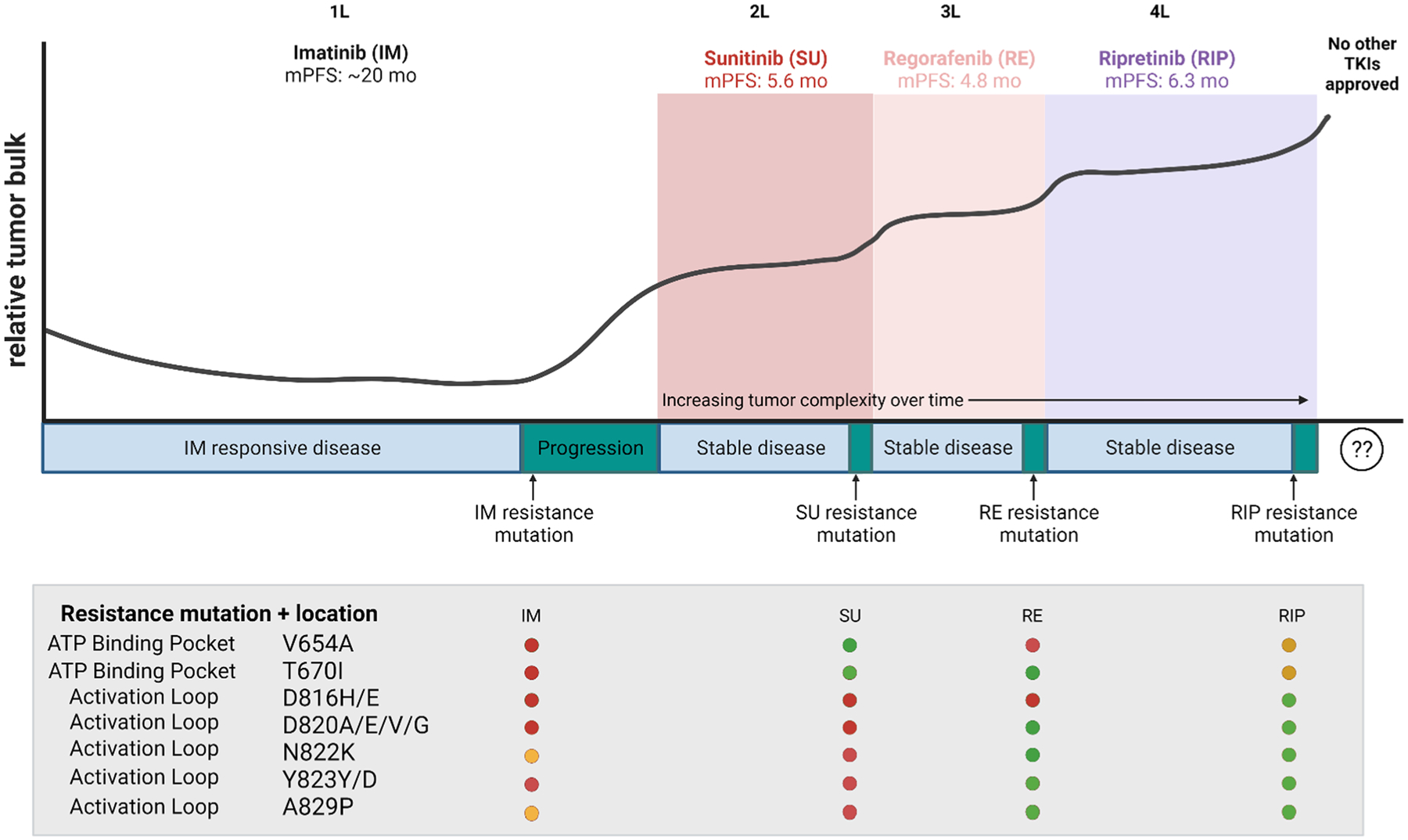
Representation of tyrosine kinase inhibitor (TKI) therapy and the emergence of secondary resistance in patient with hypothetical KIT-exon 11-mutant gastrointestinal stromal tumors. Initially KIT-exon 11-mutant gastrointestinal stromal tumor cells typically respond to first-line (1L) imatinib therapy, and correspond to a decrease in relative tumor bulk and lead to sustained stable disease. However, over time, residual tumor cells acquire imatinib-resistant mutations and lead to tumor progression. The table shows the various areas in which resistance mutations occur, either in the ATP binding pocket or activation loop of KIT. As tumor cells no longer respond to imatinib therapy, relative tumor bulk increases and a new therapy is required. Sunit common imatinib-resistant mutations. As a patient progresses through various therapies, tumor cell heterogeneity increases over time as additional mutations arise to ongoing therapy. Third- and fourth-line (3L and 4L, respectively) therapies can target previously acquired mutations, but not all of these are sensitive to a given drug. This is indicated in the graph as an overall increase in tumor bulk with intermittent stable disease. After ripretinib (4L), there are no other US Food and Drug Administration-approved therapies to target ripretinib-resistant tumor cells. mo months, mPFS median progression-free survival. Image created using BioRender.com
3.3. Regorafenib
Regorafenib, a more promiscuous type II TKI, entered clinical testing for all patients with GIST who had not responded to both imatinib and sunitinib (first- and second-line therapy) [100]. Pre-clinical testing indicated improved potency against activation loop mutations in exons 17 or 18 [100]. Unfortunately, potency against these mutations came at the expense of potency against secondary resistance mutations in KIT exons 13 and 14 (Fig. 3) [86]. Promising results were seen in a phase II study, with a partial response rate of 18% and a median progression-free survival (PFS) of 13 months (Table 1) [6, 61, 101]. This study led to a randomized, double-blind, placebo-controlled phase III study (NCT01271712) of regorafenib in patients with progression or intolerance to prior imatinib and sunitinib. This study was open to all mutational subgroups and in fact did not require any testing in order to be eligible for enrollment (mutation agnostic). In this study, the ORR was slightly lower than that seen in the phase III, second-line sunitinib study (4.5%) [61]. Despite this low response rate, regorafenib offered a statistically significant benefit in PFS compared with placebo and was FDA approved as a third-line GIST therapy in 2012 [6, 61, 101].
3.4. Ripretinib
A program to rationally design a TKI that could overcome secondary resistance mutations specifically led to the development of ripretinib (originally known as DCC-2618), a TKI that binds KIT in the “switch pocket” rather than the classical ABP, thereby preventing conformational change of the kinase to the active form [76]. Pre-clinical studies showed improved potency against many of the KIT secondary mutations when compared with approved KIT TKIs (Fig. 3) [76]. In the first in-human phase I trial (NCT02571036), promising results were seen in terms of ORR and PFS from patients treated with different numbers of prior lines of therapy (Table 1). Notably, the PFS for patients treated in the second, third, or fourth line or later was 10.7, 8.3, and 5.5 months, respectively [102]. Based on these results, a randomized, placebo-controlled, double-blind phase III trial (INVICTUS, NCT03353753) was conducted to study the efficacy of ripretinib in patients who had progressed on prior treatment with imatinib, sunitinib, and regorafenib or those who have documented intolerance to any of these treatments [102, 103]. The eligibility criteria specified prior lines of therapy but did not require molecular testing or exclude any molecular subtypes (similar to a mutation-agnostic study design). In this registrational study, treatment with ripretinib significantly improved PFS compared with placebo (6 months vs 1 month, hazard ratio = 0.15, p < 0.0001). These results led to FDA approval of ripretinib for treatment of patients with GIST who had not responded to at least imatinib, sunitinib, and regorafenib [4, 102]. Even though this study incorporated a cross-over from placebo to ripretinib at the time of progression, OS was improved for patients initially assigned to ripretinib treatment (15.1 months vs 6.6 months, hazard ratio = 0.36, no statistical testing because of the hierarchical study design). In addition, the ORR for ripretinib was numerically superior to that seen in the second-line sunitinib, or third/fourth-line regorafenib studies. These results suggest an improved ability of ripretinib to control complex heterogeneous TKI-resistant disease compared with sunitinib or regorafenib [104]. However, given that the PFS with ripretinib was only 6 months, the results also indicate that either ripretinib cannot durably control all secondary KIT resistance mutations and/or KIT-independent resistance mutations that arise in heavily pre-treated KIT-mutant GIST [105, 106]. In addition to the issue of KIT secondary resistance mutations, the study also enrolled patients lacking KIT or PDGFRA mutations (approximately 8%) or with tumors that had not been genotyped (approximately 15%), likely diluting some of the benefit from this therapy [4, 104].
These clinical results suggested that ripretinib might be able to control disease if used earlier in the treatment sequence, rather than as fourth-line or later therapy. A phase III clinical trial (INTRIGUE, NCT03673501) was conducted to compare the activity of ripretinib versus sunitinib for the treatment of patients with failure of prior imatinib therapy (progression or documented intolerance but no other TKI therapy). In a departure from previous studies, in order to be eligible for this study, a molecular pathology report was required, although no specific patients with subtypes of GIST were excluded. However, patients were stratified based on mutational status as well as a history of imatinib intolerance. The primary endpoint of the study was PFS, per the statistical design plan, this endpoint was analyzed first in the KIT exon 11 intention-to-treat patient population and then in the all-patient intention-to-treat population. Therefore, the overall study design was mutation agnostic, but the initial endpoint analyzed was for a mutation-specific patient population [103]. Despite the expectation that ripretinib would yield superior PFS results compared with sunitinib, the study failed to meet the endpoint of superior PFS with ripretinib compared with sunitinib. Specifically, the median PFS in the KIT exon 11 intention-to-treat population for ripretinib versus sunitinib was 8.3 and 7.0 months, respectively (hazard ratio 0.88, p = 0.36) [107, 108].
3.5. Disease Heterogeneity and Tumor Burden Increases with Successive Lines of Therapy in KIT-Mutant GIST
Figure 3 summarizes the overall tumor burden in a hypothetical KIT-exon 11-mutant patient with metastatic disease from initial imatinib therapy through the end of ripretinib treatment. With each line of therapy, there is a progressive increase in tumor burden, suggesting that it will not be possible to indefinitely add on sequential salvage therapies (e.g., fifth-, sixth-,…-, and nth-line) unless new therapies cause significant tumor regression. Eventually the increasing tumor burden will not be compatible with survival to the next line of therapy. This was suggested in the INVICTUS study, where the OS of patients initially randomized to placebo was inferior to patients initially assigned to ripretinib, despite the presence of a cross-over in the study design [4]. This is likely due to these placebo-assigned patients experiencing such clinical deterioration in which they were either too ill to cross-over or the tumor burden became so large that ripretinib could no longer provide sufficient clinical benefit and improve survival. Although the registrational studies for sunitinib, regorafenib, and ripretinib all included a blinded placebo arm, the results from INVICTUS suggest that further placebo-controlled studies in advanced GIST might violate the concept of equipoise [4, 7, 61, 109]. Notably, the PFS of placebo patients was very similar in all of these studies, averaging 4–6 weeks, suggesting that perhaps this duration could be used to conduct future single-arm studies of new agents in the fifth line or later.
As predicted by in vitro profiling, each of the approved KIT inhibitors has different liabilities against secondary resistance mutations, likely explaining the limited overall duration of PFS for agents used after imatinib (Fig. 3) [4, 7, 61, 62, 110, 111]. For example, sunitinib has minimal activity against secondary KIT activation loop mutations (e.g., D816H), but potently inhibits the common secondary ABP mutations such as V654A and T670I (Fig. 3) [10, 86]. Regorafenib has better activity than sunitinib against some but not all activation loop mutations, with inferior activity against V654A. Of the approved agents, ripretinib has the best activity across the entire spectrum of activation loop mutations, but may lack sufficient clinical activity against the ABP mutations [105, 106]. However, the mechanisms leading to ripretinib resistance are just beginning to be elucidated and may include both KIT-dependent (secondary mutations) and KIT-independent mechanisms that activate downstream signaling pathways (e.g., RAS mutations or NF1 loss [Fig. 1]) [85, 112]. Despite the data suggesting differential activity of sunitinib, regorafenib, and ripretinib against different secondary mutations, currently, treatment is prescribed based on the line of therapy, rather than any individualized biomarkers such as sequencing of tumor biopsy and/or circulating tumor DNA (ctDNA) samples. As we continue to understand resistance mechanisms and the molecular drivers of GIST, and with the development of new technologies and approaches, we posit that the use of precision medicine study designs may be more useful in creating new targeted therapies, especially for specific GIST subtypes.
4. Precision Medicine Approaches Accelerated FDA Approvals for Non-KIT Mutant GIST Therapies
While the mutation-agnostic approach to clinical drug development in GIST has undeniably benefited the majority of patients with GIST, increasing the median OS for patients with KIT-mutant/imatinib-sensitive GIST to the range of 6–8 years, there remained unmet clinical needs [1]. Outside of KIT-mutant GIST and certain patients with PDGFRA mutations outside of exon 18, the traditional mutation-agnostic approach and treatment paradigm provides limited benefit to other patients with GIST, leaving these patients subject to physical and financial toxicity (Fig. 2). Most notably was the challenge of primary TKI resistance associated with PDGFRA D842V-mutant GIST and the many historically designated patients with KIT/PDGFRA “wild-type” GIST. The traditional treatment paradigm highlighted above was unsuitable for addressing these rare, but not insignificant populations and therefore called for a different approach to clinical study designs. A shift to a mutation-focused treatment approach was facilitated by an increased understanding of the molecular subtypes of GIST and improved availability of molecular testing. This precision medicine study approach has demonstrated great success for patients with rare GIST subtypes that were not addressed in the original KIT TKI mutation-agnostic trials.
4.1. Avapritinib for PDGFRA-Exon 18-Mutant GIST
The most common PDGFRA driver mutation seen in GIST, exon 18 D842V (in the kinase activation loop), displays primary resistance to imatinib and other type II TKIs (e.g., sunitinib) [10, 25, 26, 73, 113]. It is now appreciated that certain PDGFRA activation loop mutations, such as PDGFRA D842V, lock the kinase in the active confirmation, which interferes with type II TKI binding [114]. Therefore, rational drug design was used to specifically develop a type I TKI to target activation loop mutation PDGFRA D842V, and the homologous mutation KIT D816V (seen in mastocytosis). Preclinical in vitro studies demonstrated that avapritinib (formally BLU-285) had potent biochemical activity against these KIT and PDGFRA activation loop mutations, which was further explored in clinical trials [115].
In 2015, the phase I NAVIGATOR (NCT02508532) study, a two-part, open-label, non-randomized trial, was initiated [23, 24]. NAVIGATOR represents a prototypical example of a precision medicine approach, which describes a mutation-specific, histology-specific study design. Patients with PDGFRA-D842V mutant GIST were a pre-specified subgroup within the overall safety population (20/46 in the dose-escalation phase and 36/204 for the dose-expansion phase). The other patients enrolled were those without PDGFRA D842V mutations treated with imatinib and one or more other TKIs (Table 1). At the time of data cut-off, the ORR for patients with PDGFRA D842V was 88%, with seven complete responses and 44 partial responses [24]. In contrast, the ORR for all other patients was 22% [23]. This highlights the importance of molecular testing and tailoring treatments based on mutational status, as avapritinib was initially developed to target PDGFRA activation loop mutations, and those patients without these mutations did not respond as well [116]. The success of avapritinib specifically in PDGFRA-D842V patients fast tracked its FDA approval in February 2020 for the treatment of all exon 18 PDGFRA-mutant GIST (Fig. 4) [117].
Fig. 4.
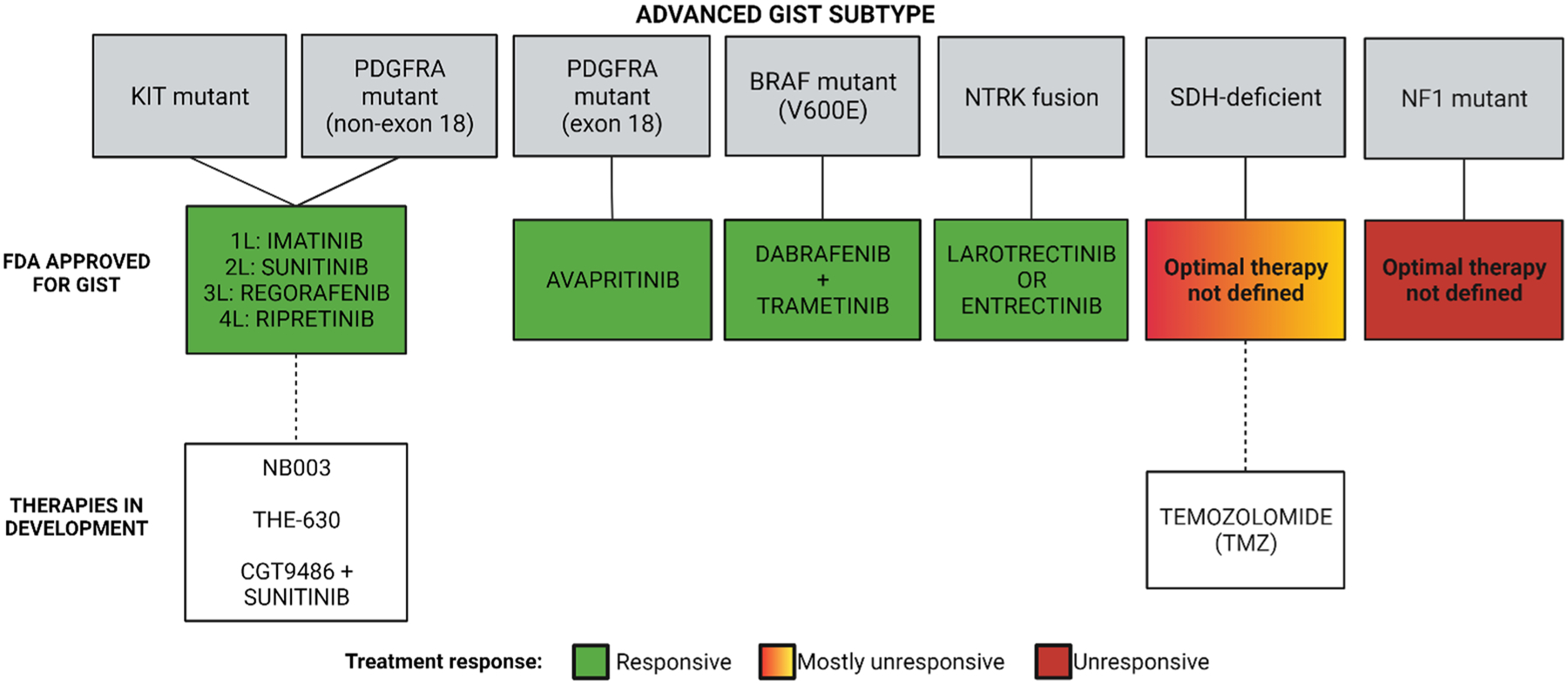
Using precision oncology approaches to treat advanced gastrointestinal stromal tumors (GIST) significantly improves patient outcomes and treatment options by improving the likelihood of clinical response and minimizing the treatment of patients who are unlikely to respond to a given therapy. Colors correspond to patient outcomes; white boxes indicate therapies that are in development. FDA Food and Drug Administration, NF1 neurofibromatosis (type 1), NTRK neurotrophic tyrosine receptor kinase, PDGFRA platelet-derived growth factor receptor alpha, SDH succinate dehydrogenase. Image created using BioRender.com
Despite a much lower response rate in non-PDGFRA D842V patients, clinical trials (specifically VOYAGER, NCT03465722) continued with avapritinib, primarily to compare its efficacy to regorafenib for third-line treatment in patients with GIST previously treated with imatinib and one or two other TKIs. This phase III study failed to show a significant improvement in median PFS (4.2 and 5.6 months for avapritinib and regorafenib, respectively) [116]. Based on this disappointing result, avapritinib is no longer being developed for the treatment of KIT-mutant GIST, although it is now FDA approved for the treatment of advanced systemic mastocytosis, another neoplasm driven by activating KIT mutations (most typically KIT D816V) [118]. These results highlight the contrast in success rates between mutation-specific (PDGFRA D842V) and mutation-agnostic (GIST treated with one to two other TKIs) studies for the same drug. In the case of avapritinib, this rationally designed drug did not fare well in a mutation-agnostic study with previously treated GIST, as mutational load increases in complexity as disease progresses and avapritinib could not overcome tumor heterogeneity any better than regorafenib.
4.2. Larotrectinib and Entrectinib for NTRK-Fused GIST
Gastrointestinal stromal tumors with unidentified drivers have been a continued area of ongoing research. Originally referred to as “wild type” because sequencing of the tumors failed to detect any of the known driver mutations, the proportion of these GIST cases has consistently shrunk as drivers have been identified [119–121]. The most recently identified and exceptionally rare drivers in GIST are RTK gene fusions (1% of GIST cases, Fig. 1A), including those involving NTRK family members [122, 123]. NTRK fusions as oncogenes had previously been discovered in other cancers, and as a result, NRTK TKIs were developed and clinically tested in these select populations [17, 19, 124]. The first NTRK-fusion genes were identified in GIST over 5 years ago, just when NTRK TKIs were entering clinical study. Shi et al. identified a fusion between NTRK3 and ETV6 in a patient with GIST who then was able to enter the phase I trial of larotrectinib (LOXO-101, NCT02122913) and responded well despite rapid progressive disease when previously treated with the KIT/PDGFRA TKIs imatinib (3 months), sunitinib (2 months), and sorafenib (2 months) [17, 19]. Since then, a handful of patients with GIST with NTRK fusions have been identified and successfully treated with larotrectinib. In a pooled analysis of three phase I/II trials, three patients with GIST were treated and all achieved complete responses (Table 1) [17, 19]. Because these studies defined eligibility using a molecular rather than a histological diagnosis, FDA approval was granted for this mutation-selected/histology-agnostic population, both for larotrectinib and subsequently for another NTRK TKI, entrectinib (Fig. 4) [16]. Because of the rarity of this type of GIST, four patients in total were included in the larotrectinib (n = 3, response rate 100%) and entrectinib (n = 1, response not reported) registrational studies (Table 1) [17–19, 124–126]. Therefore, it remains important that additional cases of response/non-response to these agents in patients with GIST be reported, thereby strengthening the rationale for physicians to identify and treat patients with NTRK-translocated GIST with these drugs.
4.3. Dabrafenib and Trametinib for BRAF-V600E Mutant GIST
BRAF V600E-mutant GIST accounts for approximately 0.8% of all cases of GIST (Fig. 1A). Because of the rarity, there are few reports describing this subtype, but one case report demonstrates the successful use of dabrafenib monotherapy in BRAF V600E-mutant GIST [127]. In June 2022, the FDA granted accelerated approval to the combination of dabrafenib and trametinib for the treatment of adult and pediatric BRAF V600E-mutant solid tumors after progression on prior treatment and for whom no satisfactory treatment options were available [21]. Previously, this combination had been approved for the treatment of BRAF-mutant melanoma (adjuvant and advanced disease), metastatic non-small cell lung cancer, and anaplastic thyroid cancer based on mutation-specific/histology-agnostic clinical studies, MFR1117019 (NCT02034110) and NCI-MATCH (NCT02465060) [20]. Between these two studies, only a single patient with GIST was treated, and this patient did not have an objective response endpoint reported. Despite this, the combination of dabrafenib and trametinib was still approved for BRAF V600E-mutant solid tumors, which would then include BRAF V600E-mutant GIST (Fig. 4). As noted above for the case of larotrectinib and entrectinib, post-marketing reporting of additional cases of treated patients with GIST will be helpful to understand the efficacy of this combination.
5. Future Directions in GIST Drug Development
5.1. Next Generation of KIT (and PDGFRA) Inhibitors
Currently, a number of new KIT inhibitors are entering phase I studies, including NB003 (formerly AZD3229, NCT04936178) and THE-630 (NCT05160168) [105, 106, 128]. Both of these agents have potency against a broader range of KIT TKI-resistant mutations, including both ABP and activation loop mutations, offering the possibility of controlling a broader range of TKI-resistant residual tumor cells and providing clinical activity even in late-line therapy. An alternative approach is being tested in the case of CGT-9486 (previously PLX-9486), where combination therapy using CGT-9486 (active against activation loop mutations) plus sunitinib (active against ABP mutations) is being tested in sunitinib-naïve patient to see if the combination is superior to standard single-agent sunitinib (NCT02401815) [129, 130].
Although registrational studies of these new agents could continue the historical practice of enrolling all patients with advanced GIST, we suggest that limiting enrollment to patients with KIT-mutant GIST as well a subset of PDGFRA-mutant GIST would result in superior ORRs and improved PFS compared with testing these agents against all patients with advanced GIST selected only based on the number and type of previous lines of therapy (Fig. 4). This type of design would be crucial to any attempt at regulatory approval based on a single-arm study in last-line therapy, where a sufficiently high ORR and duration of response would be required [131]. Eliminating GIST cases lacking KIT mutations from the denominator would improve the odds of success of such a study. Following approval as an “nth-line therapy”, future studies could test these new therapies against approved agents in a phase III study for earlier lines of therapy, but again the use of eligibility requirements that include a molecular definition for patient eligibility would be predicted to improve the odds of success. A limitation to this approach would be determining how patients with advanced heterogeneous disease (e.g., two or more resistance mutations) should be enrolled in these types of clinical studies. In order to change the current drug sequencing for KIT-mutant GIST, future studies could use ctDNA to select or exclude patients for studies that compare two approved agents [104, 132]. For example, despite the failure of ripretinib to be proven superior to sunitinib in a second-line phase III study [108], it remains possible that biomarker selection of patients might have yielded a different outcome. The ctDNA selection of patients could either enrich for patients expected to have a superior response to ripretinib versus sunitinib or exclude patients who would be predicted to have a better response to sunitinib versus ripretinib [133]. Currently, there are no published data that would allow an estimation of the likelihood of success of such a study design, but hopefully such data will be available in the future. One practical issue for the use of ctDNA for study eligibility is the observation that patients with GIST on average shed less tumor DNA than other types of solid tumors (e.g., lung cancer or melanoma), with 20–25% of patients with advanced GIST having undetectable levels of ctDNA [134–136]. In addition, the use of ctDNA as an eligibility criterion would require the development, validation, and regulatory approval of a companion diagnostic [137].
5.2. Targeting SDH-Deficient GIST
As mentioned previously, SDH-deficient GIST cases have a dismal ORR to imatinib (< 5%) and, at most, 20–30% of cases have a partial response to sunitinib or regorafenib [49, 51, 96, 101]. During the conduct of the mutation-agnostic registrational studies for these agents, these studies were open to patients who met eligibility criteria based on the number of lines of prior therapy and the drugs used for prior therapy. However, current treatment strategies are now focused on targeting unique characteristics in SDH-deficient GIST. Notably, compared with KIT and PDGFRA-mutant GIST, SDH-deficient GIST show global hyper-methylation [53, 138]. Succinate dehydrogenase deficiency in GIST leads to succinate accumulation and inhibition of histone lysine demethylases (KDM) and ten-eleven translocation enzymes, which leads to DNA and histone hyper-methylation (Fig. 1C) [139, 140]. A study by Ricci et al. reported that the epigenetic inactivation (i.e., methylation) of O6-methylguanine DNA methyl-transferase is higher in SDH-deficient GIST than compared with SDH-proficient GIST [141]. It has been reported that the inactivation of O6-methylguanine DNA methyl-transferase leads to an increased effectiveness of alkylating agents in several other cancers, such as gliomas, colorectal cancer, and large B-cell lymphoma [141, 142]. These results led to the hypothesis that DNA methylation could affect O6-methylguanine DNA methyl-transferase in SDH-deficient GIST, therefore inducing a favorable response to alkylating agents. This hypothesis was experimentally supported using patient-derived tumor models that were very sensitive to temozolomide (TMZ) treatment [143]. Notably, TMZ is an alkylating agent that is currently approved by the FDA for the treatment of glioblastoma multiforme and refractory anaplastic astrocytomas [144]. A 2014 study showed that in 15 patients with SDHB-mutant paraganglioma/pheochromocytoma, 50% had a partial response to TMZ [145]. These studies suggest that SDH deficiency could be a biomarker for TMZ sensitivity [74]. Another case study conducted by De Silva et al. shows the efficacy of using TMZ in one patient with SDH-deficient GIST, indicated by a durable partial response, ongoing through 18 cycles [146]. Based on these preliminary data, a two-arm phase II study of TMZ in patients with SDH-deficient GIST (NCT03556384) began in 2018 to determine the overall response rate of 6 months of TMZ therapy (Fig. 4). In an initial report, 2/5 patients had a partial response [143]. The primary results from this study are expected to be updated in the fall of 2022.
5.3. Other Potential Targetable Mutations
Other GIST subtypes that could be potentially treated with approved or emerging agents include ALK-translocated and FGFR-translocated GIST. To date, there have been very limited reports of where patients with GIST were treated with specific inhibitors of these molecular drivers, but presumably these GIST subtypes would respond to kinase inhibitors specifically targeting the underlying molecular driver [127]. In addition, NF1 mutant GIST remains without a known FDA-approved optimal therapy, and future studies should focus on new treatment strategies for these patients.
6. Conclusions
Although the historical pathway to GIST treatment approval has used a mutation-agnostic design, a precision medicine approach informed by the molecular underpinnings of GIST subtypes has shown incredible success for rare GIST subtypes. Advances in technology have facilitated the utility of this approach, both in GIST-specific studies (e.g., avapritinib) as well as in histology-agnostic (but mutation-specific) studies (e.g., larotrectinib and entrectinib). Using up-front molecular testing and precision oncology treatment of GIST results in superior clinical outcomes and minimizes the number of patients treated with biologically inactive therapies (Fig. 4). The use of a precision medicine approach necessitates the widespread use of comprehensive molecular profiling of all cases of advanced GIST requiring medical therapy. However, in certain countries including the USA, such molecular profiling occurs in less than 50% of patients, at least in certain countries including the USA [147, 148]. The push for molecular testing and profiling will definitely be needed for the less common forms of GIST for which there is no effective FDA-approved therapy (e.g., NF1-deficient or SDH-deficient GIST). We believe that future registration studies of KIT/PDGFRA inhibitors should also utilize a precision oncology approach and enroll only those patients with KIT/PDGFRA mutations that would be predicted to respond to these novel agents (Fig. 4).
A limitation to these precision medicine-driven clinical studies includes determining the criteria for those patients previously treated with other TKIs and who have highly heterogeneous disease. These patients differ from TKI-naïve patients, as their disease may not be effectively targeted by a single drug, as previously shown in Fig. 3. With the emergence of new technologies and models, we should focus on how to combat heterogeneous disease, and the optimal selection of patients for testing new targeted therapies. In recent years, mutation-agnostic studies that included all patients with advanced disease were successful for patients with a specific subtype of GIST. While precision medicine approaches narrow the inclusion criteria for a study, subjecting patients to new therapies in which they may not receive a clinical benefit unnecessarily exposes them to potential toxicities. To address this issue, correlative studies that compare tumor biopsy with ctDNA mutation profiling should be integrated into future studies, not only to identify those patients most likely to benefit from a new therapy, but also to identify mechanisms of acquired resistance to novel agents. Continuing to understand the mechanisms of acquired resistance and the molecular underpinnings of each subtype of GIST will aid in the development of the next generation of targeted therapies.
Key Points.
The discovery and use of tyrosine kinase inhibitors significantly improved the overall survival of patients with advanced/metastatic gastrointestinal stromal tumors (GIST), as surgery alone and traditional chemotherapy/radiation therapies provide little clinical benefit.
The studies that led to the approval of first-, second-, third-, and fourth-line therapies included all patients with advanced/metastatic GIST, regardless of mutational driver status, and only provided clinical benefit to a subset of patients.
Our understanding of the molecular mechanisms of various GIST subtypes has informed precision medicine-focused studies, and led to the development and approval of more successful targeted therapies for those patients with rarer GIST subtypes.
We propose that future drug development should focus on mutation-specific subsets of GIST, rather than simply selecting patients based only on the number of specific types of prior therapy.
Funding
The authors received no specific funding for this work. The general work of Michael C. Heinrich has been supported by grants from the Department of Veterans Affairs (1 I01 BX005358-01A1) and the National Institutes of Health National Cancer Institute (1 R21 CA263400-01), and by philanthropic donations from the GIST Cancer Research Fund and the Jonathan David Foundation.
Footnotes
Conflict of interest Homma M. Khosroyani and Lillian R. Klug have no conflicts of interest that are directly relevant to the content of this article. Michael C. Heinrich has the following conflicts of interest: honoraria: Novartis; consulting or advisory role: Novartis, Blueprint Medicines, Deciphera, Theseus Pharmaceuticals; and patents, royalties, other intellectual property: patent on treatment of GIST licensed to Novartis (Inst).
References
- 1.Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers. 2021;7(1):22. [DOI] [PubMed] [Google Scholar]
- 2.Edmonson JH, Marks RS, Buckner JC, Mahoney MR. Contrast of response to dacarbazine, mitomycin, doxorubicin, and cisplatin (DMAP) plus GM-CSF between patients with advanced malignant gastrointestinal stromal tumors and patients with other advanced leiomyosarcomas. Cancer Invest. 2002;20(5–6):605–12. [DOI] [PubMed] [Google Scholar]
- 3.Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2022;33(5):466–77. [DOI] [PubMed] [Google Scholar]
- 4.Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutton TL, Walker BS, Billingsley KG, Corless CL, Sheppard BC, Heinrich MC, et al. Ten-year survivorship in patients with metastatic gastrointestinal stromal tumors. Ann Surg Oncol. 2022. 10.1245/s10434-022-12063-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. [DOI] [PubMed] [Google Scholar]
- 8.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):47–80. [DOI] [PubMed] [Google Scholar]
- 9.Klug LR, Khosroyani HM, Kent JD, Heinrich MC. New treatment strategies for advanced-stage gastrointestinal stromal tumours. Nat Rev Clin Oncol. 2022. 10.1038/s41571-022-00606-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9. [DOI] [PubMed] [Google Scholar]
- 12.Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12(8):2622–7. [DOI] [PubMed] [Google Scholar]
- 13.Debiec-Rychter M, Sciot R, Le CA, Schlemmer M, Hohenberger P, Van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–103. [DOI] [PubMed] [Google Scholar]
- 14.Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40(5):689–95. [DOI] [PubMed] [Google Scholar]
- 15.von Mehren M, Heinrich MC, Shi H, Iannazzo S, Mankoski R, Dimitrijević S, et al. Clinical efficacy comparison of avapritinib with other tyrosine kinase inhibitors in gastrointestinal stromal tumors with PDGFRA D842V mutation: a retrospective analysis of clinical trial and real-world data. BMC Cancer. 2021;21(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thein KZ, Lemery SJ, Kummar S. Tissue-agnostic drug development: a new path to drug approval. Cancer Discov. 2021;11(9):2139–44. [DOI] [PubMed] [Google Scholar]
- 17.Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong DS, Bauer TM, Lee JJ, Dowlati A, Brose MS, Farago AF, et al. Larotrectinib in adult patients with solid tumours: a multicentre, open-label, phase I dose-escalation study. Ann Oncol. 2019;30(2):325–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trametinib. Prescribing information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/204114s024lbl.pdf. Accessed 6 Dec 2022.
- 21.Salama AKS, Li S, Macrae ER, Park JI, Mitchell EP, Zwiebel JA, et al. Dabrafenib and trametinib in patients with tumors with BRAF(V600E) mutations: results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol. 2020;38(33):3895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.US FDA. FDA grants accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinibunresectable-or-metastatic-solid. Accessed 6 Dec 2022.
- 23.Heinrich MC, Jones RL, von Mehren M, Schoffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGA-TOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21(7):935–46. [DOI] [PubMed] [Google Scholar]
- 24.Jones RL, Serrano C, von Mehren M, George S, Heinrich MC, Kang YK, et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer. 2021;145:132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–10. [DOI] [PubMed] [Google Scholar]
- 26.Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–64. [DOI] [PubMed] [Google Scholar]
- 27.Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. 2008;3:557–86. [DOI] [PubMed] [Google Scholar]
- 28.Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med. 2016;14(1):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23(22):3999–4006. [DOI] [PubMed] [Google Scholar]
- 30.Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26(54):7560–8. [DOI] [PubMed] [Google Scholar]
- 31.Paner GP, Silberman S, Hartman G, Micetich KC, Aranha GV, Alkan S. Analysis of signal transducer and activator of transcription 3 (STAT3) in gastrointestinal stromal tumors. Anticancer Res. 2003;23(3B):2253–60. [PubMed] [Google Scholar]
- 32.Gupta A, Ma S, Che K, Pobbati AV, Rubin BP. Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS ONE. 2021;16(7): e0252689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosbach B, Rossi F, Yozgat Y, Loo J, Zhang JQ, Berrozpe G, et al. Direct engagement of the PI3K pathway by mutant KIT dominates oncogenic signaling in gastrointestinal stromal tumor. Proc Natl Acad Sci USA. 2017;114(40):E8448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi Y, Zhao W, Wang Z, Xie Q, Cao J, Meng X. Cross regulation of signaling pathways in gastrointestinal stromal tumor. Oncol Lett. 2018;16(5):6770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W, Yuan W, Ren L, Xu C, Luo R, Zhou Y, et al. A novel fusion between CDC42BPB and ALK in a patient with quadruple wild-type gastrointestinal stromal tumor. Mol Genet Genomic Med. 2022;10(5):e1881. 10.1002/mgg3.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dermawan JK, Vanderbilt CM, Chang JC, Untch BR, Singer S, Chi P, et al. FGFR2::TACC2 fusion as a novel KIT-independent mechanism of targeted therapy failure in a multidrug-resistant gastrointestinal stromal tumor. Genes Chromosomes Cancer. 2022;61(7):412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’Alpino Peixoto R, Medeiros BA, Cronemberger EH. Resected high-risk rectal GIST harboring NTRK1 fusion: a case report and review of the literature. J Gastrointest Cancer. 2021;52(1):316–9. [DOI] [PubMed] [Google Scholar]
- 38.Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(6):1769–76. [DOI] [PubMed] [Google Scholar]
- 39.Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foerster A, Hartmann A, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol. 2009;62(7):613–6. [DOI] [PubMed] [Google Scholar]
- 40.Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2012;133(1):141–8. [DOI] [PubMed] [Google Scholar]
- 41.Lasota J, Felisiak-Golabek A, Wasag B, Kowalik A, Zięba S, Chłopek M, et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod Pathol. 2016;29(3):275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondo J, Huh WJ, Franklin JL, Heinrich MC, Rubin BP, Coffey RJ. A smooth muscle-derived, Braf-driven mouse model of gastrointestinal stromal tumor (GIST): evidence for an alternative GIST cell-of-origin. J Pathol. 2020;252(4):441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torrence D, Xie Z, Zhang L, Chi P, Antonescu CR. Gastrointestinal stromal tumors with BRAF gene fusions: a report of two cases showing low or absent KIT expression resulting in diagnostic pitfalls. Genes Chromosomes Cancer. 2021. 10.1002/gcc.22991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol. 2005;29(9):1170–6. [DOI] [PubMed] [Google Scholar]
- 45.Gasparotto D, Rossi S, Polano M, Tamborini E, Lorenzetto E, Sbaraglia M, et al. Quadruple-negative GIST is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin Cancer Res. 2017;23(1):273. [DOI] [PubMed] [Google Scholar]
- 46.Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013:398570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart DR, Corless CL, Rubin BP, Heinrich MC, Messiaen LM, Kessler LJ, et al. Mitotic recombination as evidence of alternative pathogenesis of gastrointestinal stromal tumours in neurofibromatosis type 1. J Med Genet. 2007;44(1): e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoller ME, Rembeck B, Oden A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79(11):2125–31. [PubMed] [Google Scholar]
- 49.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108(1):314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doyle LA, Nelson D, Heinrich MC, Corless CL, Hornick JL. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology. 2021;61(5):801–9. [DOI] [PubMed] [Google Scholar]
- 51.Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2(7):922–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44(4):285–92. [DOI] [PubMed] [Google Scholar]
- 53.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35(11):1712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011;103(12):983–7. [DOI] [PubMed] [Google Scholar]
- 56.Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney–Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79–88. [DOI] [PubMed] [Google Scholar]
- 57.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72(1):106–16. [DOI] [PubMed] [Google Scholar]
- 58.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato DP, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358(9291):1421–3. [DOI] [PubMed] [Google Scholar]
- 60.George S, von Mehren M, Heinrich MC, Wang Q, Corless CL, Butrynski JE, et al. , A multicenter phase II study of regorafenib in patients (pts) with advanced gastrointestinal stromal tumor (GIST), after therapy with imatinib (IM) and sunitinib (SU), ASCO Annual Meeting. J Clin Oncol. 2011;2011. [Google Scholar]
- 61.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.George S, Feng Y, Mehren MV, Choy E, Corless CL, Hornick JL, et al. Prolonged survival and disease control in the academic phase II trial of regorafenib in GIST: response based on genotype. J Clin Oncol. 2013;31(15_Suppl.):10511. [Google Scholar]
- 63.Rutkowski P, Nowecki ZI, Biec-Rychter M, Grzesiakowska U, Michej W, Wozniak A, et al. Predictive factors for long-term effects of imatinib therapy in patients with inoperable/metastatic CD117(+) gastrointestinal stromal tumors (GISTs). J Cancer Res Clin Oncol. 2007;133(9):589–97. [DOI] [PubMed] [Google Scholar]
- 64.Biron P, Cassier PA, Fumagalli E, Blesius M, Debiec-Rychter M, Adenis A, et al. Outcome of patients (pts) with PDGFRA D842V mutant gastrointestinal stromal tumor (GIST) treated with imatinib (IM) for advanced disease. J Clin Oncol. 2010;28:15s. [Google Scholar]
- 65.Cassier PA, Fumagalli E, Rutkowski P, Schoffski P, van Glabbeke M, Debiec-Rychter M, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18(16):4458–64. [DOI] [PubMed] [Google Scholar]
- 66.Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–45. [PubMed] [Google Scholar]
- 67.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96(3):925–32. [PubMed] [Google Scholar]
- 68.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–8. [DOI] [PubMed] [Google Scholar]
- 69.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–34. [DOI] [PubMed] [Google Scholar]
- 70.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32. [DOI] [PubMed] [Google Scholar]
- 71.Van Oosterom AT, Judson I, Verweij J, Di Paola E, van Glabbeke M, Dimitrijevic S, et al. STI571, an active drug in metastatic gastrointestinal stromal tumors (GIST) an EORTC phase I study. Proc Am Soc Clin Oncol. 2001;20:1a. [Google Scholar]
- 72.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard-versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–5. [DOI] [PubMed] [Google Scholar]
- 73.Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neppala P, Banerjee S, Fanta PT, Yerba M, Porras KA, Burgoyne AM, et al. Current management of succinate dehydrogenase–deficient gastrointestinal stromal tumors. Cancer Metastasis Rev. 2019;38(3):525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128(2):270–9. [DOI] [PubMed] [Google Scholar]
- 76.Smith BD, Kaufman MD, Lu WP, Gupta A, Leary CB, Wise SC, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35(5):738–51.e9. [DOI] [PubMed] [Google Scholar]
- 77.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–8. [PubMed] [Google Scholar]
- 78.Zaki K, Aslam S, Eisen T. Regorafenib (BAY 73–4506): stromal and oncogenic multikinase inhibitor with potential activity in renal cell carcinoma. Curr Oncol Rep. 2013;15(2):91–7. [DOI] [PubMed] [Google Scholar]
- 79.Weisberg E, Griffin JD. Resistance to imatinib (Glivec): update on clinical mechanisms. Drug Resist Updates. 2003;6(5):231–8. [DOI] [PubMed] [Google Scholar]
- 80.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–74. [DOI] [PubMed] [Google Scholar]
- 81.Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64(17):5913–9. [DOI] [PubMed] [Google Scholar]
- 82.Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–9. [DOI] [PubMed] [Google Scholar]
- 83.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–90. [DOI] [PubMed] [Google Scholar]
- 84.Barnett CM, Corless CL, Heinrich MC. Gastrointestinal stromal tumors: molecular markers and genetic subtypes. Hematol Oncol Clin North Am. 2013;27(5):871–88. [DOI] [PubMed] [Google Scholar]
- 85.Muhlenberg T, Ketzer J, Heinrich MC, Grunewald S, Marino-Enriquez A, Trautmann M, et al. KIT-dependent and KIT-independent genomic heterogeneity of resistance in gastrointestinal stromal tumors: TORC1/2 inhibition as salvage strategy. Mol Cancer Ther. 2019;18(11):1985–96. [DOI] [PubMed] [Google Scholar]
- 86.Serrano C, Marino-Enriquez A, Tao DL, Ketzer J, Eilers G, Zhu M, et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;20(6):612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.George S, Blay JY, Casali PG, Le CA, Stephenson P, DePrimo SE, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–68. [DOI] [PubMed] [Google Scholar]
- 89.Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA. 2009;106(5):1542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo T, Hajdu M, Agaram NP, Shinoda H, Veach D, Clarkson BD, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502–3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15(22):6862–70. 10.1158/1078-0432.CCR-09-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22(1):103–15. [DOI] [PubMed] [Google Scholar]
- 92.Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15(24):7510–8. [DOI] [PubMed] [Google Scholar]
- 93.Nishida T, Takahashi T, Nishitani A, Doi T, Shirao K, Komatsu Y, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol. 2009;14(2):143–9. [DOI] [PubMed] [Google Scholar]
- 94.Wang D, Zhang Q, Blanke CD, Demetri GD, Heinrich MC, Watson JC, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): long-term follow-up results of RTOG 0132, ASCO Annual Meeting. J Clin Oncol. 2011;29:10057. [Google Scholar]
- 95.Demetri GD, Heinrich MC, Fletcher JA, Fletcher CD, Van den Abbeele AD, Corless CL, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15(18):5902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Janeway KA, Albritton KH, Van Den Abbeele AD, D’Amato GZ, Pedrazzoli P, Siena S, et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer. 2009;52(7):767–71. [DOI] [PubMed] [Google Scholar]
- 97.Verschuur AC, Bajciova V, Mascarenhas L, Khosravan R, Lin X, Ingrosso A, et al. Sunitinib in pediatric patients with advanced gastrointestinal stromal tumor: results from a phase I/II trial. Cancer Chemother Pharmacol. 2019;84(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–96. [DOI] [PubMed] [Google Scholar]
- 99.Cabebe E, Wakelee H. Sunitinib: a newly approved small-molecule inhibitor of angiogenesis. Drugs Today (Barc). 2006;42(6):387–98. [DOI] [PubMed] [Google Scholar]
- 100.Overton LC, Heinrich MC. Regorafenib for treatment of advanced gastrointestinal stromal tumors. Expert Opin Pharmacother. 2014;15(4):549–58. [DOI] [PubMed] [Google Scholar]
- 101.Ben-Ami E, Barysauskas CM, von Mehren M, Heinrich MC, Corless CL, Butrynski JE, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27(9):1794–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Janku F, Abdul Razak AR, Chi P, Heinrich MC, von Mehren M, Jones RL, et al. Switch control inhibition of KIT and PDGFRA in patients with advanced gastrointestinal stromal tumor: a phase I study of ripretinib. J Clin Oncol. 2020;38(28):3294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nemunaitis J, Bauer S, Blay JY, Choucair K, Gelderblom H, George S, et al. Intrigue: phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib. Future Oncol. 2020;16(1):4251–64. [DOI] [PubMed] [Google Scholar]
- 104.Bauer S, Heinrich MC, George S, Zalcberg JR, Serrano C, Gelderblom H, et al. Clinical activity of ripretinib in patients with advanced gastrointestinal stromal tumor harboring heterogeneous KIT/PDGFRA mutations in the phase III INVICTUS study. Clin Cancer Res. 2021;27(23):6333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Banks E, Grondine M, Bhavsar D, Barry E, Kettle JG, Reddy VP, et al. Discovery and pharmacological characterization of AZD3229, a potent KIT/PDGFRalpha inhibitor for treatment of gastrointestinal stromal tumors. Sci Transl Med. 2020;12(541). 10.1126/scitranslmed.aaz2481. [DOI] [PubMed] [Google Scholar]
- 106.Rivera VM, Huang W-S, Lu M, Pritchard JR, Dalgarno D, Shakespeare WC. Preclinical characterization of THE-630, a next-generation inhibitor for KIT-mutant gastrointestinal stromal tumors (GIST) [abstract 1292]. Cancer Res. 2021;81(13 Suppl.):1292. [Google Scholar]
- 107.Heinrich MC, Jones RL, Gelderblom H, George S, Schöffski P, Mehren MV, et al. INTRIGUE: a phase III, randomized, open-label study to evaluate the efficacy and safety of ripretinib versus sunitinib in patients with advanced gastrointestinal stromal tumor previously treated with imatinib. J Clin Oncol. 2022;40(36_Suppl.):359881. [Google Scholar]
- 108.Bauer S, Jones RL, Blay JY, Gelderblom H, George S, Schoffski P, et al. Ripretinib versus sunitinib in patients with advanced gastrointestinal stromal tumor after treatment with imatinib (INTRIGUE): a randomized, open-label, phase III trial. J Clin Oncol. 2022. 10.1200/jco.22.00294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goss G, Merriam P, Manola J, Singer S, Fletcher C, Demetri G. Clinical and pathological characteristics of gastrointenstinal stromal tumors. Proc Am Soc Clin Oncol. 2001;19:559a. [Google Scholar]
- 110.Blanke CD, von Mehren M, Joensuu H, Roberts PJ, Esienberg B, Heinrich MC, et al. Evaluation of the safety and efficacy of an oral molecularly-targeted therapy, STI571, in patients with unresectable or metastatic gastrointestinal stromal tumors (GISTs) expressing c-kit (CD117). Proc Am Soc Clin Oncol. 2001:1a. [Google Scholar]
- 111.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri GD, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156(3):791–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Serrano C, Wang Y, Marino-Enriquez A, Lee JC, Ravegnini G, Morgan JA, et al. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-Kinase/AKT dysregulation. J Clin Oncol. 2015;33(22):e93–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125(3):660–7. [DOI] [PubMed] [Google Scholar]
- 114.Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “gatekeeper door”: exploiting the active kinase conformation. J Med Chem. 2010;53(7):2681–94. [DOI] [PubMed] [Google Scholar]
- 115.Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9(414). 10.1126/scitranslmed.aao1690 [DOI] [PubMed] [Google Scholar]
- 116.Kang YK, George S, Jones RL, Rutkowski P, Shen L, Mir O, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: a randomized, open-label phase III study. J Clin Oncol. 2021. 10.1200/jco.21.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dhillon S Avapritinib: first approval. Drugs. 2020;80(4):433–9. [DOI] [PubMed] [Google Scholar]
- 118.Gotlib J, Reiter A, DeAngelo DJ. Avapritinib for advanced systemic mastocytosis. Blood. 2022;140(15):1667–73. 10.1182/blood.2021014612. [DOI] [PubMed] [Google Scholar]
- 119.Alkhuziem M, Burgoyne AM, Fanta PT, Tang CM, Sicklick JK. The call of “the wild”-type GIST: it’s time for domestication. J Natl Compr Canc Netw. 2017;15(5):551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nannini M, Astolfi A, Urbini M, Indio V, Santini D, Heinrich MC, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST). BMC Cancer. 2014;14:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pantaleo MA, Nannini M, Corless CL, Heinrich MC. Quadruple wild-type (WT) GIST: defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015;4(1):101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi E, Chmielecki J, Tang C-M, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med. 2016. (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brenca M, Rossi S, Polano M, Gasparotto D, Zanatta L, Racanelli D, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol. 2016;238(4):543–9. [DOI] [PubMed] [Google Scholar]
- 124.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018;15(12):731–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res. 2020;152: 104609. [DOI] [PubMed] [Google Scholar]
- 126.Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21(2):271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Falchook GS, Trent JC, Heinrich MC, Beadling C, Patterson J, Bastida CC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4(2):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pilla Reddy V, Anjum R, Grondine M, Smith A, Bhavsar D, Barry E, et al. The pharmacokinetic-pharmacodynamic (PKPD) relationships of AZD3229, a novel and selective inhibitor of KIT, in a range of mouse xenograft models of GIST. Clin Cancer Res. 2020;26(14):3751–9. [DOI] [PubMed] [Google Scholar]
- 129.Gebreyohannes YK, Burton EA, Wozniak A, Matusow B, Habets G, Wellens J, et al. PLX9486 shows anti-tumor efficacy in patient-derived, tyrosine kinase inhibitor-resistant KIT-mutant xenograft models of gastrointestinal stromal tumors. Clin Exp Med. 2019;19(2):201–10. [DOI] [PubMed] [Google Scholar]
- 130.Wagner AJ, Severson PL, Shields AF, Patnaik A, Chugh R, Tinoco G, et al. Association of combination of conformation-specific KIT inhibitors with clinical benefit in patients with refractory gastrointestinal stromal tumors: a phase 1b/2a non-randomized clinical trial. JAMA Oncol. 2021. 10.1001/jamaoncol.2021.2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.US FDA. Clinical trial endpoints for the approval of cancer drugs and biologics: guidance for industry. Available from: https://www.fda.gov/media/71195/download. Accessed 6 Dec 2022.
- 132.Saluja H, Karapetis CS, Pedersen SK, Young GP, Symonds EL. The use of circulating tumor DNA for prognosis of gastrointestinal cancers. Front Oncol. 2018;8:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Mattos-Arruda L, Siravegna G. How to use liquid biopsies to treat patients with cancer. ESMO Open. 2021;6(2): 100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jeffers M, Kappeler C, Kuss I, Beckmann G, Mehnert DH, Fredebohm J, et al. Broad spectrum of regorafenib activity on mutant KIT and absence of clonal selection in gastrointestinal stromal tumor (GIST): correlative analysis from the GRID trial. Gastric Cancer. 2022;25(3):598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bauer S, Heinrich MC, George S, Zalcberg JR, Serrano C, Gelderblom H, et al. Clinical activity of ripretinib in patients with advanced gastrointestinal stromal tumor harboring heterogenous KIT/PDGFRA mutations in the phase 3 INVICTUS study. Clin Cancer Res. 2021. 10.1001/jamaoncol.2021.2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.George S, von Mehren M, Fletcher JA, Sun J, Zhang S, Pritchard JR, et al. Phase II study of ponatinib in advanced gastrointestinal stromal tumors: efficacy, safety, and impact of liquid biopsy and other biomarkers. Clin Cancer Res. 2022;28(7):1268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chan HT, Chin YM, Low SK. Circulating tumor DNA-based genomic profiling assays in adult solid tumors for precision oncology: recent advancements and Future challenges. Cancers (Basel). 2022;14(13):3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6(268):268ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Waterfall JJ, Killian JK, Meltzer PS. The role of mutation of metabolism-related genes in genomic hypermethylation. Biochem Biophys Res Commun. 2014;455(1–2):16–23. [DOI] [PubMed] [Google Scholar]
- 140.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–52. [DOI] [PubMed] [Google Scholar]
- 141.Ricci R, Martini M, Ravegnini G, Cenci T, Milione M, Lanza P, et al. Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents. Clin Epigenetics. 2019;11(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–4. [DOI] [PubMed] [Google Scholar]
- 143.Yebra M, Bhargava S, Kumar A, Burgoyne AM, Tang CM, Yoon H, et al. Establishment of patient-derived succinate dehydrogenase-deficient gastrointestinal stromal tumor models for predicting therapeutic response. Clin Cancer Res. 2022;28(1):187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ortiz R, Perazzoli G, Cabeza L, Jiménez-Luna C, Luque R, Prados J, et al. Temozolomide: an updated overview of resistance mechanisms, nanotechnology advances and clinical applications. Curr Neuropharmacol. 2021;19(4):513–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella C, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. 2014;135(11):2711–20. [DOI] [PubMed] [Google Scholar]
- 146.De Silva M, Rastogi S, Chan D, Angel C, Prall O, Gill A, et al. Succinate dehydrogenase-deficient gastrointestinal stromal tumor: from diagnostic dilemma to novel personalised therapy in 2 case reports. Transl Cancer Res. 2021;10(7):3588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Florindez J, Trent J. Low frequency of mutation testing in the United States: an analysis of 3866 GIST patients. Am J Clin Oncol. 2020;43(4):270–8. [DOI] [PubMed] [Google Scholar]
- 148.Barrios CH, Blackstein ME, Blay JY, Casali PG, Chacon M, Gu J, et al. The GOLD ReGISTry: a global, prospective, observational registry collecting longitudinal data on patients with advanced and localised gastrointestinal stromal tumours. Eur J Cancer. 2015;51(16):2423–33. [DOI] [PubMed] [Google Scholar]