

Abstract
Enzymatic processes, particularly those capable of performing redox reactions, have recently been of growing research interest. Substrate specificity, optimal activity at mild temperatures, high selectivity, and yield are among the desirable characteristics of these oxidoreductase catalyzed reactions. Nicotinamide adenine dinucleotide (phosphate) or NAD(P)H-dependent oxidoreductases have been extensively studied for their potential applications like biosynthesis of chiral organic compounds, construction of biosensors, and pollutant degradation. One of the main challenges associated with making these processes commercially viable is the regeneration of the expensive cofactors required by the enzymes. Numerous efforts have pursued enzymatic regeneration of NAD(P)H by coupling a substrate reduction with a complementary enzyme catalyzed oxidation of a co-substrate. While offering excellent selectivity and high total turnover numbers, such processes involve complicated downstream product separation of a primary product from the coproducts and impurities. Alternative methods comprising chemical, electrochemical, and photochemical regeneration have been developed with the goal of enhanced efficiency and operational simplicity compared to enzymatic regeneration. Despite the goal, however, the literature rarely offers a meaningful comparison of the total turnover numbers for various regeneration methodologies. This comprehensive Review systematically discusses various methods of NAD(P)H cofactor regeneration and quantitatively compares performance across the numerous methods. Further, fundamental barriers to enhanced cofactor regeneration in the various methods are identified, and future opportunities are highlighted for improving the efficiency and sustainability of commercially viable oxidoreductase processes for practical implementation.
Keywords: biocatalysis, cofactor regeneration, electrocatalysis, oxidoreductases, photocatalysis
Graphical Abstract

1. Introduction
A variety of enzymes produced by microorganisms has been gaining attention of researchers for extra-cellular production of various chemicals, including beverages, food additives, pharmaceutical products, and biofuels.[1] Enzymes may be beneficial over chemocatalysts owing to their ability of sustainable synthesis, high selectivity, substrate specificity, optimal activity at mild conditions, and high yield.[2] Enzymatic reactions have been studied for their extensive application in biomass conversion to commodity chemicals ranging from fuel hydrocarbons to aliphatic and aromatic alcohols, ketones, and carboxylic acids, as well as for production of chiral compounds.[3,4] Advances in molecular biology to produce more efficient recombinant enzymes and improvements in biocatalyst recovery and reuse by techniques like immobilization have further facilitated enzymes to become even more competitive to chemocatalysts.[5] Among all known enzymes, oxidoreductases constitute one quarter of all enzymes.[6] Although certain oxidoreductases possess prosthetic groups to facilitate redox reactions,[7] most of them are generally dependent on nonprotein cofactors like nicotinamide adenine dinucleotide (phosphate) [NAD(P)H] (Figure 1) to catalyze the transfer of electrons from an electron donor (reductant) to an electron acceptor (oxidant) molecule.[8] The cofactor thus works as an electron donor/acceptor in the redox reaction catalyzed by the enzyme while being exhausted in stoichiometric quantity. The exogenous addition of stoichiometric nicotinamide cofactors is commercially infeasible due to high costs,[9] hence continuous regeneration of the cofactor is required. To address this concern, precursor fermentation uses whole-cell biocatalysts to produce and regenerate cofactors for example, which are useful for one-pot enzymatic cascade systems.[10,11] Significant challenges are associated with whole-cell biocatalysis as it is difficult to control the amount of the different proteins produced by the organism, resulting in a mixture of byproducts, which leads to complicated downstream separation. In addition, metabolic demands of the organism might also reduce the overall efficiency of the biocatalytic process. In cell-free biocatalysis involving oxidoreductases, NAD(P)H regeneration is imperative for these processes to be commercially viable.
Figure 1.
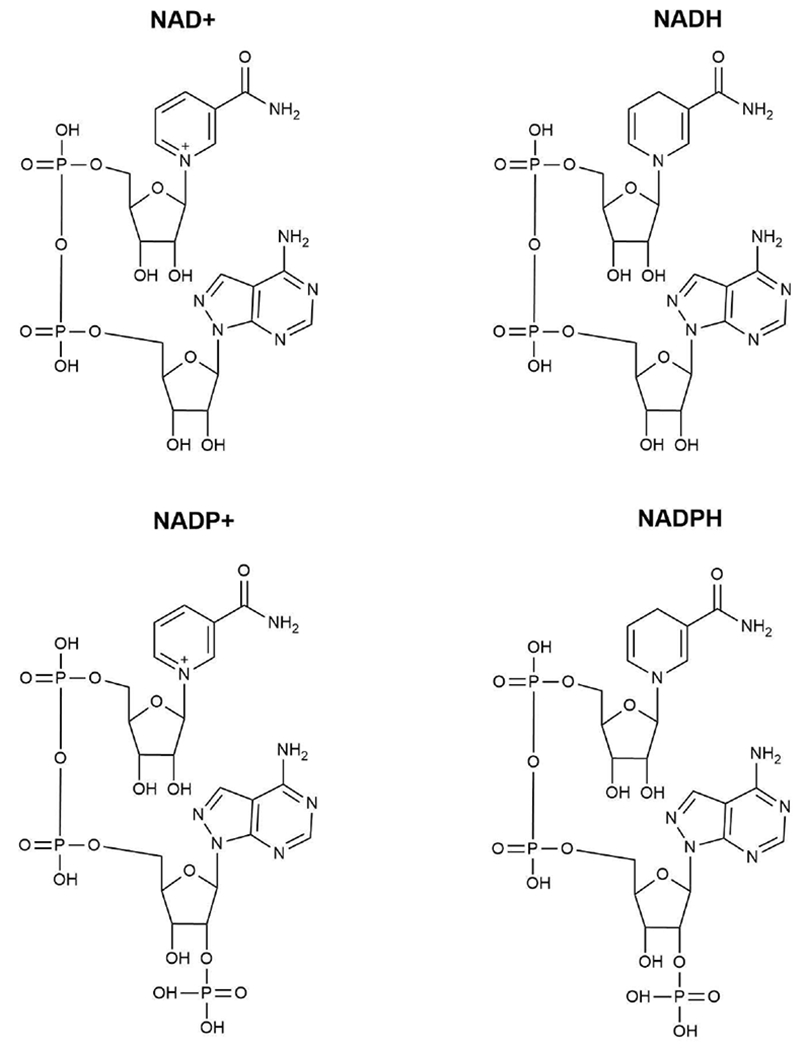
Molecular structures of nicotinamide adenine dinucleotide cofactors (non-phosphorylated and phosphorylated).
A quantitative measure of the cofactor regeneration can be estimated using the total turnover number (TTN), defined by Chenault et al. as the total number of moles of product formed per mole of cofactor during a complete reaction.[12] However, in several articles, the distinction between turnover number (TN) and TTN is ambiguous. For this Review, we have defined TN as the moles of product formed per mole of cofactor (TNcofactor) or per mole of enzyme (TNenzyme) or per mole of catalyst in general (TNcatalyst) used during a complete reaction.[12] Among all known regeneration methods, enzymatic regenerations have been reported to achieve the highest TNcofactor of >500000.[13] Enzymatic regenerations require coupled redox processes, in which a desired substrate oxidation is accompanied by a cosubstrate reduction. The NAD(P)+, thus, cycles between its reduced and oxidized forms, reducing the overall cost of using the cofactor.
In addition to enzymatic cofactor regeneration methods, other approaches can provide the required regeneration, including chemical, electrochemical, and photochemical regeneration, as shown in Figure 2. Chemical regeneration methods are somewhat similar to the enzymatic regeneration approach as the in-situ regeneration or direct electron transfer using hydride sources enables the cofactor to be used in catalytic amounts. Hydride acceptors such as pyridinium and flavin derivatives have been used in one of the earlier studies for regeneration of NAD+ ;[14] however, they were required in excess amount compared to the catalytic concentration of the oxidized cofactor. Pentamethylcyclopentadienyl rhodiumIII bipyridyl [Cp*Rh(bpy)] complexes have been efficiently used for formate-dependent NAD+ reduction[15] as well as pH-dependent reversible hydride exchange, which can facilitate regeneration of both reduced and oxidized cofactors.[16] Heterogeneous catalysts like platinum on alumina,[17] and pyrolytic graphite modified with a hydrogenase and a diaphorase subunit[18] have also been demonstrated to regenerate NADH using hydrogen as the hydride source.
Figure 2.

Schematic representation of different NAD(P)H regeneration strategies.
Electrochemical NAD(P)H regeneration uses electrons transferred between electrodes. This is an attractive method of regeneration as redox reactions and the corresponding cosubstrates or coproducts can be compartmentalized, simplifying downstream separation. Several research works in this area can be traced back to 1980s,[19–22] where authors regenerated the cofactors sometimes directly or via an electron-mediator like [Cp*Rh(bpy)(H2O)]2+.[23] Electrochemical regeneration mechanisms have been used extensively in biofuel cells and biosensors applications.[24,25]
Photochemical regeneration has also been used by exploiting light harvesting capability of artificial, biomimetic photosensitizers like cadmium sulfide (CdS) and titania (TiO2)[26–29] for the regeneration of NADH.[30] The small bandgap between the valence and conduction bands of these materials facilitates the excitation of electrons, which are then utilized in the electron transfer mechanism, responsible for the cofactor regeneration. However, the positive holes generated on the photosensitizer are required to be filled, which is commonly done by using a sacrificial donor like triethanolamine.[31,32]
A brief comparison of the different NAD(P)+ and NAD(P)H regeneration strategies (enzymatic, chemical, electrochemical, and photochemical) are listed in Table 1. Considering the large number of research articles published over the last two decades, multiple Reviews have also been published covering multienzymatic systems,[7,33,34] heterogeneous pathways-inspired regeneration,[9] organometallics-based approach for NAD(P)H regeneration,[35] and cofactor regeneration in oxidative biocatalytic processes,[36] but no comprehensive Review has appeared in nearly a decade, despite significant developments. Additionally, no Review has presented a uniform analysis comparing the productivity of various methods of regeneration. In this comprehensive Review, it is our goal to discuss and comment on different methods for nicotinamide cofactor regeneration and provide a quantitative comparison to highlight the effectiveness and utility of the tools or methodology used in these different modes of regeneration. TN based on cofactor and/or catalyst have been used to compare different strategies in enzymatic, chemical, electrochemical, and photochemical regeneration. For electrochemical regeneration methods, electrode potentials have also been used as a measure of comparison where TNs were not reported or could not be calculated. Most of the discussion comprises peer reviewed publications from the last decade; however, results from important studies published earlier have also been included based on their relevance to the context.
Table 1.
Comparison of the different cofactor regeneration strategies.
| Method | Advantages | Disadvantages |
|---|---|---|
| enzymatic | low impact on environment; high total turnover numbers; 100% selectivity; high enantioselectivity | enzyme denaturation; high cost of purified enzymes |
| chemical | moderate cost; hydrogen and oxygen could be used for reduced and oxidized cofactor regeneration, respectively | requires sacrificial donor; difficult downstream separation; mutual inactivation when used in enzymatic cascades; low total turnover numbers |
| electrochemical | renewable electricity sources can be used; enzymes can be immobilized on electrode surface; less complex downstream separtion; wide range of applications | low total turnover numbers; transition metal-based electron mediators required to enhance rate of regeneration and avoid inactive NAD2 dimer formation; high overpotentials required |
| photochemical | use of renewable energy (solar) for photoexcitation; broad applications | requires sacrificial donor; low total turnover numbers; requires transition metal-based electron mediators; quantum efficiency is low |
2. Enzymatic Regeneration
2.1. Types of enzymatic regeneration methods
Due to the growing emphasis on sustainable large-scale production of chemicals and pharmaceuticals, enzymatic processes, particularly those associated with redox enzymes, have gained traction over the years as reflected in several recent Review articles.[37–40] For NAD(P)-coupled redox enzymes, cofactor regeneration is essential for practical implementation. Two modes of enzymatic regeneration are (i) substrate-coupled and (ii) enzyme-coupled reactions. In a substrate-coupled reaction,[41] the same enzyme simultaneously oxidizes one substrate and reduces another to produce a product and coproduct (Figure 3a). The cofactor is regenerated toggling to and from its reduced to oxidized form. Alternatively, an enzyme-coupled reaction requires a separate regenerating enzyme (Figure 3b).[42] Multi-enzymatic cascade reactions use two or more enzymes with different substrates to facilitate cofactor regeneration and are classified into four types: (i) linear, (ii) orthogonal, (iii) parallel, and (iv) cyclic.[43] The orthogonal or parallel cascades are applicable for NAD(P)H-dependent dehydrogenase enzymes like the family of alcohol dehydrogenases (ADHs) to catalyze commercially important reactions like the production of enantiopure alcohols by stereoselective reduction of prochiral ketones.[44] Reduced nicotinamide cofactors are required in important organic syntheses like Bayer–Villiger oxidations (a parallel cascade) and dynamic kinetic resolution reactions (a cyclic cascade).[45] In the latter case, cofactor regeneration has been demonstrated in one-pot coupled enzymatic synthesis by parallel oxidation of racemic alcohols and reduction of the corresponding ketone to produce enantiopure secondary alcohols,[46,47] as a possible alternative to chemocatalytic dynamic kinetic resolution reactions.[48]
Figure 3.
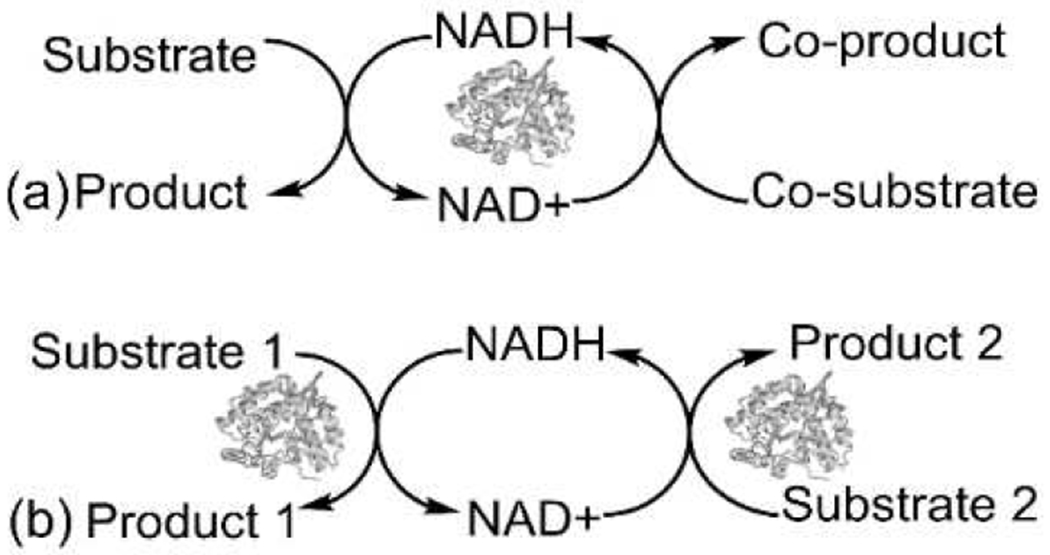
Schematic representation of cofactor regeneration using (a) substrate-coupled and (b) enzyme-coupled cascade reaction.
The importance of cofactor regeneration for applications in the syntheses of chiral drugs is readily apparent. Indeed, extracts from Rhodococcus ruber DSM 44541, which displayed different enantioselectivity for oxidation and reduction steps, were used by Voss et al. to demonstrate deracemization of pharmaceutically relevant secondary alcohols.[49] The system under investigation was a one-pot/two-step sequence and the enzymatic cofactor recycling system was switched back and forth between oxidation and reduction to achieve 100% pure enantiomers. Such strategies enable a continuous biotransformation process but also enhance the turnover number of both the enzyme and the cofactor several fold and make these processes more appealing towards industrial applications.
Another important reason for such enzymatic reactions gaining attraction is their capability of CO2 conversion to valuable chemicals or fuels like formate and methanol among others, in environmentally beneficial systems.[50–52] In one case, methanol yields as high as 95% were obtained from CO2 in a three-enzyme cascade.[53] One of the earliest enzymatic regenerations leveraging formate/CO2 interconversion with formate dehydrogenase (FDH) from Candida boidinii as the regenerating enzyme was demonstrated by Shaked and Whitesides[54] in the production of d-lactate from pyruvate. FDH has since become a common choice for a regenerating enzyme of either NAD+ or NADH. Partially entrapping these cofactors using nanofiltration membrane reactors can be of additional advantage as the soluble cofactors are retained within the reactor, which facilitates the design of a continuous reactor using similar coupled 2-enzyme regeneration approach.[55] Numerous NAD(P)H-dependent dehydrogenases have been utilized with in situ cofactor regeneration. Despite several advantages, challenges remain in these systems related to enzyme and cofactor retention and reuse, high cost, and difficult downstream separation.
2.2. Cofactor regeneration in whole-cell biocatalytic systems
Whole-cell biocatalysis with co-expression of two enzymes like leucine dehydrogenase (LeuDH) and FDH has been demonstrated to work in an NADH recycling cascade for the production of enantiopure drug precursors.[56] Further improvement of such two-enzyme cascades can be found in the work done by Jiang and Fang, who regulated the co-expression of individual enzymes by regulating the strength of ribosome binding sites to equilibrate the enzyme activities and achieve high yield of the product of interest.[57] However, the ratio of the NADH-dependent dehydrogenase enzymes is crucial in deciding the final product concentration in such cofactor regenerating cascade systems. For example, Qi et al. discovered that in a LeuDH/FDH-catalyzed enzyme-coupled cascade reaction for the production of l-norvaline, non-competitive inhibition by NADH at high concentration led to low equilibrium product concentration.[58] The authors maintained that optimizing the enzyme concentration ratio was important to maintain high productivity. Additionally, they also demonstrated a fed-batch reactor, which further enhanced the productivity of the cascade process, limiting the inhibition associated with high concentration of the d isomer. Some whole-cell biocatalytic processes have additional advantage as the cofactor is produced and recycled in vivo.[59] For instance, the drug precursor molecule (4R,6R)-actinol can be produced by enantioselective reduction of ketoisophorone by the action of two enzymes (alcohol dehydrogenase and enoate reductase), both of which are produced by Saccharomyces cerevisiae. Uzir and Najimudin demonstrated this important reaction by using whole-cell biotransformation using baker’s yeast where they maintained that the in-vivo produced cofactor was recycled within the cells by simultaneous oxidation of some arbitrary compound (Figure 4).[59] However, in other whole-cell biocatalysis systems with co-display of two enzymes, exogenous cofactor addition might be necessary, and in-vivo regeneration is not an option in such a case. Han et al. worked with such a system where glucoamylase (GA) and glucose dehydrogenase (GDH) were co-displayed on engineered bacterial surfaces for the production of glucono-δ-lactone using starch as the substrate.[60] They solved the issue of NAD+ regeneration by introducing a third enzyme, l-lactate dehydrogenase, which simultaneously reduced pyruvate to l-lactic acid. Similar tools like co-encapsulation and co-immobilization of redox enzymes, particularly those which are used for oxidation of alcohols, are frequently employed for efficient regeneration strategies.[38,61–65]
Figure 4.

Production of (4R,6R)-actinol from ketoisophorone using combination of two enzymes with cofactor recycling. ER = enoate reductase; ADH = alcohol dehydrogenase; Xred and Xox represent arbitrary species undergoing oxidation and reduction, respectively.
2.3. Cofactor regeneration in cell-free biocatalytic systems
2.3.1. Single enzyme cascade regeneration
Redox enzymes with a wide range of substrate specificity like that of yeast alcohol dehydrogenase (ADH) can regenerate the NAD(P)H cofactors by suitable coupling of substrates. Prediction of thermodynamic equilibria is particularly important to understand the feasibility of a redox reaction to proceed spontaneously.[66] For example, Jadhav et al. used a substrate-coupled strategy for the production of n-butanol from n-butyraldehyde with around 74% substrate conversion using ethanol as the cofactor regenerating substrate with a TNcofactor of 507.[41] Using single enzyme cascades for NAD(P)H regeneration by coupling suitable substrates is economically advantageous as it does not necessitate a second biocatalyst. Several ADHs, known for their catalytic promiscuity, like ADH2 from Haloferax volcanii[67] and ADH from Lactobacillus brevis,[68] have been used for enantioselective reduction of ketones with suitable co-substrates like ethanol or isopropanol. A wide range of substrate specificity enables these enzymes to carry out redox reactions without having to use a second enzyme for the cofactor regeneration, and the enantiopure products are typically expensive, which may be worth producing even at relatively low cofactor turnover numbers. Some other enzymes like glucose dehydrogenase do not generally accept substrates other than glucose, which necessitates the use of a second enzyme for the cofactor regeneration. However, in a recent impressive work, Yan et al. developed a rational design using saturation mutagenesis to create a mutant GDH enzyme with enhanced substrate specificity[69] and utilized this enzyme for the production of enantiopure (R)-2-chromandelic acid methyl ester, an important drug precursor with glucose as the co-substrate for NAD(P)H regeneration. However, not all alcohol dehydrogenases/ketoreductases can be used in single enzyme cascades for in-situ cofactor regeneration. In the case of organic retrosynthesis, where multiple substrates are required to be combined to produce a target molecule, it becomes necessary to develop multi-enzyme cascades, with cofactor regeneration as an important component of the holistic retrosynthetic strategy.[70,71]
2.3.2. Multienzymatic cascade regeneration
2.3.2.1. Multienzymatic cascade using formate dehydrogenase
Regeneration of NAD(P)H cofactors in cell-free biocatalysis, which is extremely important for the economic feasibility of cofactor-dependent redox enzyme-catalyzed processes, had been in use as early as 1980s, as reported by Kula and Wandrey,[72] who went a step further to modify these cofactors to demonstrate their efficient use in a continuous reactor for the production of l-leucine. Formate dehydrogenase from Candida boidinii has been a very popular choice as the regenerating enzyme for cofactor regeneration purpose along with other NAD(P)H-dependent oxidoreductases.[73–76] The interconversion of formate and CO2, both of which are benign materials, as well as the ease of downstream separation and compatibility of formate dehydrogenase to be coupled with other enzymes have driven this choice.[43,77] In fact, formate has been described as “a mediator between physicochemical and biological realms” by Yishai et al. in their Review, where they discussed several methods of formate production and use.[78] Coupling of enzymes in a multi-enzymatic cascade depends on multiple factors, like the optimum pH for individual enzymes, optimum temperature, and substrate or product inhibition effects. In certain studies, redox enzyme cascades were used for the sequential oxidation of alcohols to aldehydes to carboxylicacids, but cofactor regeneration was done electrochemically in alcohol-based biofuel cells.[79,80] Typically, in an enzymatic cofactor regeneration process, simultaneous oxidation and reduction of two substrates take place through transfer of electrons and H+ ions via intermediate enzyme-cofactor complexes. The stability of the regenerating enzyme is therefore of high importance as it is a cost-limiting factor in such processes.[81] Other factors like kinetics of enzyme-cofactor interaction as well as the kinetics of enzyme-cofactor complex interaction with the substrate might also affect the overall rate of a closed loop cascade reaction.[82] For instance, Wichmann et al. reported that NADH strongly inhibited FDH in an FDH/LEUDH (leucine dehydrogenase)-coupled cascade reaction. To achieve maximum substrate conversion, FDH required a higher fraction of total activity (55%) to overcome this inhibition.[83]
2.3.2.2. Multienzymatic cascade using other redox enzymes
While formate dehydrogenase is a common choice for enzyme-mediated cofactor regeneration, primarily due to the chemically benign nature of its substrate and product, other redox enzymes like glucose dehydrogenase (GDH), xylose dehydrogenase (XDH), NAD(P)H oxidase, and phosphite dehydrogenase (PDH) are some of the many NAD(P)H-dependent oxidoreductases that have been used for in-situ cofactor regeneration using one-pot cascade syntheses.[84–87] Enzymatic cofactor regeneration using one-pot cascade reactions allows multistep reactions without needing to isolate or purify intermediates, thus decreasing the energy and solvent requirements for intermediate separation processes, leading to higher economic feasibility.[88] So far, the highest TNcofactor among multienzymatic regeneration systems was achieved in the work reported by Angelastro et al., where the authors demonstrated NADP+ regeneration leveraging the chemistry of glutathione reductase (GR) combined with glutaredoxin (GRX).[13] In this novel strategy, NADP+ was regenerated from NADPH by GR with simultaneous reduction of glutathione disulfide (GSSG) to glutathione (GSH), which was complimented by GRX-catalyzed reduction of disulfide compounds like 2-hydroxyethyl disulfide (HED) or cystine. This regeneration strategy was used in the synthesis of 6-phosphogluconate from glucose-6-phosphate catalyzed by glucose-6-phosphate dehydrogenase, with a cofactor turnover number (TNcofactor) of 500000 (Figure 5). While offering astounding enzyme performance, the products are of somewhat limited commercial value. In a more recent study by Jia et al., NADP+ regeneration catalyzed by myoglobin was used for complimenting glucose dehydrogenase-catalyzed oxidation of d-glucose to gluconic acid.[89] Compared to the aforementioned study, while the TNcofactor was ten times lower (≈50000), the product is of greater commercial value, while also being able to regenerate the synthetic biomimetic cofactor 1-benzyl-1,4-dihydropyridine-3-carboxamide (BNA+) with higher catalytic efficiency compared to NADP+ and NAD+. Another noteworthy implementation of multienzyme one-pot cascade can be found in the work by Mutti et al., in which the authors set up a hydrogenborrowing cascade for the synthesis of chiral amines from alcohols.[90] The cascade method, which constituted an alcohol dehydrogenase and an amine dehydrogenase, demonstrated the ability to convert chiral secondary alcohols to amines with optical inversion, amination by retaining optical configuration of enantiomeric secondary alcohols, enantioselective conversion of racemic secondary alcohols to amines, and amination of primary alcohols.
Figure 5.

Glutathione reductase/glutaredoxin-based NADP + regeneration strategy for the conversion of glucose to 6-phosphogluconate using hexokinase and glucose-6-phosphate dehydrogenase.[13]
Although enzymatic synthesis is an efficient route for producing fine chemicals, the requirement of cofactor regeneration in NAD(P)H-dependent enzymes makes it difficult to use them for industrial purposes. For instance, an expensive and rare sugar, d-ribulose, which is also a useful precursor in asymmetric synthesis, can be produced by Enterobacter aerogenes ribitol dehydrogenase (EaRDH) using ribitol as the substrate.[91] Singh et al. used EaRDH in an enzyme coupled cascade with lipoamide dehydrogenase as the regenerating enzyme to produce d-ribulose with extremely efficient in-situ NAD regeneration.[92] The regeneration system, according to the authors, enabled them to increase d-ribulose yield from 0.6 to around 87%. In this case the oxidized cofactor was regenerated by simultaneous reduction of the dye 2,6-dichlorophenolindophenon catalyzed by lipoamide dehydrogenase. Another such example is the recent study by Su et al., whose objective was to produce l-tagatose from d-galactitol using galactitol dehydrogenase.[93] An efficient cofactor regeneration scheme using NADH oxidase as the regenerating enzyme enabled the authors to achieve high productivity using 3mm cofactor per 100 mm of substrate. Asymmetric transformations could benefit immensely from this kind of enzyme-catalyzed cascade reactions, using the right choice of regenerating enzyme with similar initial rate of substrate conversion using innocuous substrates that can also facilitate downstream separation. For example, tetrahydrofuran-3-one and tetrahydrothiofuran-3-one are extremely difficult to reduce to corresponding alcohols in enantiomeric excess, even with thermostable alcohol dehydrogenases.[94] These chiral alcohols are valuable in drug synthesis and can be produced by engineered thermostable alcohol dehydrogenase, as was done by Sun et al., who used triple-code saturation mutagenesis approach, a directed evolution, to modify the binding site of Thermoethanolicus brockii ADH to achieve the enantioselective reduction of such “difficult-to-reduce ketones”.[95] Although the authors’ intention was not to establish a NAD(P)H regenerating enzymatic cascade, but for enzymes with capabilities of catalyzing the production of such important drug precursors, it is needless to say that coupling with a suitable enzymatic cofactor regeneration system adds immense value to industrial application of these enzymes.
2.4. Enzymatic regeneration using immobilized enzyme/cofactors
Multienzymatic cascades are becoming increasingly popular as they provide access to several specialty chemicals under benign conditions of operation and especially because these cofactor regeneration schemes enable redox enzymes to a greater extent by extenuating the requirement of these expensive cofactors. Multienzyme cascade systems with cofactor regeneration are more advantageous with immobilized enzyme than free enzymes owing to higher enzyme stability, reusability, and ability to be used in continuous-flow biocatalysis. For example, in the study by Velasco-Lozano et al., co-immobilization of l-alanine dehydrogenase and FDH from Candida boidinii was done using polyethyleneimine as an anchoring layer to irreversibly immobilize the enzymes on agarose beads modified with aldehyde moieties.[65] There was progressive decrease in residual enzyme activity in each consecutive cycle, but the system retained 80% activity after 5 cycles. However, the turnover number was reported as 150 and the authors also cited diffusion resistance for the cofactors as a technical challenge. Another example worth mentioning is a study involving an expensive aromatic ester, cinnamyl cinnamate, which is extensively used in fragrance and flavoring applications and can be produced from condensation of cinnamic acid and cinnamyl alcohol catalyzed by lipase. By employing ADH in a cofactor regeneration cascade system with FDH as the regenerating enzyme, naturally occurring cinnamaldehyde was reduced to cinnamyl alcohol with an overall yield of about 54%.[64] To enhance the cofactor turnover number, researchers have also been able to tether or anchor the NAD cofactors alongside enzymes. Chen et al. featured a zeolitic imidazole framework-8 (ZIF-8) nanoreactor for co-encapsulation of ADH and LDH to demonstrate a self-sufficient cofactor recycling cascade system (Figure 6).[61] In addition to co-encapsulation of the redox enzymes, another major issue addressed by their work was encapsulation of the cofactor NAD+ by covalently tethering it to a phenylboronic acid conjugated poly(allylamine) polymer. This phenomenal study was able to demonstrate a roughly 5-fold increase in the rate of pyruvate reduction with the mentioned enzymatic cascade system compared to the homogeneous enzyme-cofactor assembly. Similar work by Hartley et al. used polyethylene glycol to modify NADH and used it in a modular cascade system leading to a cofactor turnover number > 10000.[96]
Figure 6.

NAD+-mediated two-enzyme biocatalytic cascade in ZIF-8 NMOF nanoreactor.[61] NMOF refers to ZIF-8 nano metal–organic framework; AlcDH = alcohol dehydrogenase, LacDH = lactate dehydrogenase. The red dots represent conjugated polymer-bound NAD+.
Advanced techniques like closed-loop cofactor regeneration strategy are possible, as demonstrated in a recent study by Baulmer et al., who used a substrate-coupled approach for NADH regeneration by setting up a modular system using an immobilized HaloTagged ADH from Lactobacillus brevis.[97] The authors demonstrated conversion of several prochiral ketones to corresponding alcohols with >99% enantioselectivity and turnover numbers up to 2023 molmol−1. The most noticeable innovation in this study was the use of a flow liquid–liquid extraction (FLLEX) system, which separated and recycled the cofactors back into the feed stream of this continuous reactor system.
From the discussion on enzymatic cofactor regeneration schemes, based on the references provided in this section, it can be noticed that the observed turnover numbers for the cofactor can vary to a great extent (Table 2). Using an immobilized enzyme or cofactor is one of the important strategies that may enhance the cofactor turnover number by several folds. The observed turnover numbers depend heavily on the design of the experiments and the reported values may be considered lower limits of the potential of any given system. For example, reports vary with respect to the number of cycles of batch reactions attempted and concentrations utilized. Additionally, it is not uniformly clear in reported studies if the enzymes or cofactors degrade chemically. Enzymatic cofactor regeneration strategies are clearly useful because of their simplicity in operation and the ability to bring in redox bioconversions of substrates that are otherwise extremely difficult. As enzyme stability in continuous operation is one of the major challenges, future research should be targeted towards addressing this issue, which can enhance the turnover numbers further and achieve greater industrial relevance.
Table 2.
Comparison of turnover numbers as observed in different studies using enzymatic regeneration methods.
| Enzyme | Substrate | TN [cofactor] | TN [enzyme] | Ref. |
|---|---|---|---|---|
| ADH | ammonia/ammonium | 5[b] | 236[b] | [90] |
| myoglobin | oxygen | 50000[a] | – | [89] |
| glutathione reductase/glutaredoxin | 2-hydroxyethyl disulfide | 500000[a] | – | [13] |
| FDH | formate | 6300[b] | – | [57] |
| XDH | xylose | 160[a] | 3500[b] | [85] |
| mutant phosphite dehydrogenase | sodium phosphite | 2630[a] | 90000[b] | [87] |
| diaphorase | 2,6-dichlorophenolindophenol | 174[b] | – | [92] |
| NADH oxidase | oxygen | 33[b] | 26000[b] | [93] |
| LbADH | 2-propanol | 12855[a] | – | [97] |
| GA-GDH | starch | 120[b] | – | [60] |
| NOx | oxygen | 11000[a] | 1843[a] | [96] |
| LbADH | (S)-5-nitrononane-2,8-dione | 14000[a] | – | [98] |
| ADH | ethanol | 507[b] | 30000[b] | [41] |
| ADH | ethanol | 370[b] | 12000[b] | [61] |
| FDH | formate | 112[b] | 37000[b] | [62] |
Used directly from reference.
Calculated from data provided in reference.
NOx = NADH oxidase, LbADH = Lactobacillus brevis alcohol dehydrogenase.
3. Chemical Regeneration Methods
Although enzymatic regeneration of cofactor has proven to be the most efficient technique based on turnover number,[99] complexities arise due to the requirement of additional substrate(s) and in some cases additional enzyme(s). For NAD-dependent enzymatic reactions focused on a single substrate, alternate methods of cofactor regeneration have been explored. Chemical regeneration primarily based on transition metal complexes like [Cp*Rh(bpy)]2+ (Figure 7) has long been employed in this regard to avoid addition of the expensive nicotinamide cofactors in stochiometric amounts.[9,17,100–102]
Figure 7.

Schematic representation of chemical regeneration of NADH.
3.1. Methods involving organometallic complexes in NAD(P)H regeneration
In the early 1980s, rhodium-based organometallic complexes gained huge attention owing to their excellent capability to function as electron relays in reduction of proton to hydrogen.[103] To exploit the redox property of such organometallic complexes, Steckhan and coworkers explored a pentamethyl bipyridyl rhodium(III) complex for direct chemical reduction of NAD+ using formate as the hydride source.[104] The intermediate rhodium-hydride complex was identified using 1H nuclear magnetic resonance (NMR) spectroscopy using 6,6′-dimethyl-2,2′-bipyridine as the ligand. Several challenges were identified associated with this mode of NADH regeneration, like degradation of NADH in tris buffer, replacement of the aquo ligand by hydroxo ligand at higher pH, and action of ammonia as a ligand in higher pH when ammonium formate was used as the hydride source. Some of these studies, which investigate the chemical mode of cofactor regeneration, have been done using NAD analogue molecules. For example, benzyl nicotina-midium (BNA+), an analogue of NAD+, was used as a model reductant in a study of enantioselective reduction of ketone catalyzed by alcohol dehydrogenase, which was subsequently demonstrated with NADH using ruthenium and rhodium complexes.[105] In-situ NADH regeneration was achieved under appropriate reaction conditions using [RuCl2(TPPTS)2]2 [TPPTS = tris(m-sulfonatophenyl)phosphine] and [Cp*(bpy)Rh(H2O)]Cl2 complexes and hydrogen (Figure 8). The simplicity of this process lies in the elimination of unwanted by-products as gaseous hydrogen acts as both the hydride and proton source for the enantioselective enzymatic reduction of ketones. Matsuo and Mayer characterized the mechanism of cofactor regeneration using these NADH analogues with cis-[RuIV(bpy)2(py)(O)]2+ as the catalyst for oxidation and found that the hydrogen atom transfer is rather the preferred kinetic mechanism than hydride transfer.[106]
Figure 8.

Schematic representation of Ru complex-based cofactor recycling with in-situ coupling with a reductase enzyme.
3.1.1. Mechanism of NAD+ reduction and regioselective nature of hydride transfer
One of the complexities in regenerating the reduced NAD(P)H cofactor lies in the regioselective reduction of the 1,4-pyridinium ring.[107] Reduction of NAD+ to NADH by transition metal complexes may give the preferred 1,4-addition product or the 1,6-addition product, which is not catalytically active. In an effort to unravel the mechanism of this regioselective reduction, Fish and co-workers used various pyridinium models of NAD+, with a variety of 3-substituents to investigate binding, steric, and electronic effects in their regioselective reductions conducted in 1:1 H2O/THF.[107] It was revealed that in presence of formate as the hydride donor, the aquo complex reacts with the formate, which further decomposes in a β-elimination process to produce CO2. The regioselective transfer of the hydride is facilitated by the amide functionality of the NAD+ and eventual replacement of the aqua complex occurs by displacement of the reduced 1,4-NADH from the coordination entity. To shed further light on the mechanism of hydride transfer, Miller and co-workers[108] isolated an intermediate pentamethylcyclopentadiene Rh complex, from fleeting rhodium hydride and studied the hydride transfer ability of the diene, including reduction of NAD+. The involvement of the pentamethylcyclopentadienyl ligand in hydride transfer was a novel finding for these Rh-complex mediated regenerations, and the proposed mechanism (Figure 9) was consistent with density functional theory (DFT) calculation results for several closely related Rh and Ir complexes. Based on studies on several RhIII and RuII complexes and comparing the corresponding turnover frequencies (TOF), it is now known that ligands in the coordination sphere have significant effect on the activity of the complexes,[109] and in general, RhIII complexes were found to be much more efficient than RuII complexes based on the TOF. One big advantage of such metal complexes is that they can be incorporated within enzymes like alcohol dehydrogenase from Thermonobacter brockii,[110] or protease enzymes like papain,[109,111] to synthesize a metalloenzyme, engineered as a biomimetic solution for NADH regeneration in enzymatic reactions.
Figure 9.

Proposed mechanism for the reduction of NAD+ through a [(Cp*H)Rh(bpy)]+ intermediate.
Analogous to enzymatic regeneration schemes with formate as the hydride donor, a study by Sadler and coworkers used [(η6-arene)Ru(en)Cl]PF6 (arene is hexamethylbenzene, p-cymene, indan; en is ethylenediamine) for reduction of NAD+ to 1,4-NADH with formate as the hydride donor.[112] The plot of TON vs. time was found to be a straight line, which indicated that the reduction of NAD+ was of zero order with respect to the NAD+ concentration. However, the turnover frequency vs. formate concentration plot showed Michaelis-type kinetics with respect to formate concentration, with a maximum TOF of 1.46 h−1 and Km = 58 mΜ (Figure 10).
Figure 10.

Plot of TOF against formate concentration. Plot constructed based on data provided in Ref. [112].
3.1.2. Effect of metal-ligand interaction
Metal–ligand interaction is a major factor that drives the reducing capability of organometallic complexes.[113,114] A recent study evaluated to role of substituent position on bipyridine ligand upon the catalytic efficiency of RhIII complexes for NADH regeneration. For the series of –CH2OH substituents, the 5,5’-substituted bipyridine RhIII complex had the lowest reduction potential of all positional isomers and could effectively regenerate NADH with a notably high turnover frequency of 1100 h−1.[115] Several studies have deemed Rh complexes superior over other transition metal complexes. While in a prior study by Macchioni and co-workers,[116] the TOF achieved with a novel Ir complex was about 143 h−1, a very recent study has achieved a TOF of 3731 h−1 using a pyridine-2-sulfonamidate-substituted iridium complex.[117] The enhanced acidity of the central metal atom induced by the electron-withdrawing –SO2–moiety and an amine was attributed to have such an effect. In several studies, like the one conducted by Sadler and co-workers,[118] it is evident that the transfer hydrogenation of NAD+ as cofactor can be made reversible by varying the pH. Additionally, the reduced nicotinamide cofactor can be used as a hydride source to reduce ketones in presence of the cyclopentadienyl bipyridyl derivatives of RuII and IrIII catalysts and an appropriate enzyme.
3.2. Other methods of chemical cofactor regeneration
Early studies like that conducted by Jones et al.[119] demonstrated sodium dithionite as a possible reagent for regioselective regeneration of 1,4-NADH and the maximum TNcofactor was 105 with low product yield. However, enzymes are likely to suffer deactivation by high concentration of sodium dithionite, hence they did not gain attraction for further studies. For enzyme/cofactor ratio, which gave high yield, the TNcofactor observed was as low as around 30 and the overall process of cyclohexanone reduction was only viable in a preparative scale. The laccase-mediator system (LMS) is based on a combination of 2,2’-azino-bis-3-ethylbenzthiazoline-6-sulfonic acid (ABTS)-catalyzed oxidation of NAD(P)H and laccase-catalyzed reduction of molecular oxygen as terminal oxidant. LMS systems are useful for coupling with ADH-catalyzed oxidation reactions, and in a recent study, chemical regeneration of NAD+ was achieved with turnover numbers of 300 and 16000 with respect to NADH and ABTS, respectively.[120] Natural flavin adenine mononucleotide (FMN), which is a metal-free organocatalyst capable of NAD+ regeneration,[14] suffers from slow kinetics of hydride transfer, making it difficult to achieve high turnover numbers. In an impressive recent work, Zhu et al. developed a synthetic, water-soluble bridged flavinium organocatalyst and demonstrated atom-economical NADP+ regeneration using oxygen as the regenerating substrate.[121] With a TNcofactor of around 50, the reaction rate tripled with the NADP+ regeneration system compared to natural FMN catalyst. Further, the regeneration system supported three different glucose dehydrogenase-catalyzed oxidations of monosaccharides like xylose, mannose, and glucose.
Although the use of transition metal-based pentamethyl cyclopentadienyl bipyridyl complexes has successfully demonstrated NAD(P)H cofactor regeneration capability by chemical means, major limitations remain due to the highly regioselective nature of the hydride transfer, requirement of an electron donor, and mutual inactivation in chemoenzymatic systems.[17,32,105,122] Table 3 features some of chemically (mostly transition metal complex) catalyzed cofactor regeneration methods discussed in this section, and it can be observed that low turnover number in chemical regeneration is common, making it less viable when compared to enzymatic regeneration methods. Another limitation of transition metal complex-based regeneration, which impedes its large-scale application, is the cost associated with expensive metals like Rh and Ru and the fact that it is a homogeneous catalyst. Accordingly, its separation, recovery, and recycling are prohibitive challenges. In recent studies, attempts to immobilize such catalysts on solid support, particularly on periodic mesoporous organosilica, have gained attention,[123–126] which might pave the way for a new generation of chemocatalytic regeneration with improved cost for large-scale application.
Table 3.
Comparison of turnover numbers as observed in different studies using chemical regeneration methods.
| Catalyst | Substrate | TN [cofactor] | TN [catalyst] | Ref. |
|---|---|---|---|---|
| (bis)phosphine Rh complex | H2 | 49[a] | 1470[a] | [102] |
| (Cp)*Rh(bpy) complex | formate | – | 43–170[a] | [109] |
| (Cp)*Rh(bpy) complex | formate | – | 550[b] | [115] |
| FMN derivative | O2 | 50[b] | – | [121] |
| ABTS | O2 | 300[a] | 16000[a] | [120] |
| (Cp)*Rh(bpy) complex | formate | 5.7[a] | 13[b] | [123] |
| (Cp)*Rh(bpy) complex | formate | 0.75[b] | 1500[a] | [127] |
| (Cp)*Rh(bpy) complex | formate | 20.5[b] | 41[b] | [128] |
| (Cp)*Rh(bpy) complex | formate | 10[b] | 50[b] | [129] |
| (Cp)*IrCl(phen) complex | formate | 2[b] | 1[b] | [130] |
Used directly from reference
Calculated from data provided in reference.
4. Electrochemical Regeneration Methods
Electrochemical methods are appealing for NADH regeneration processes as the electricity to drive the reaction can be sourced from renewable energy and the fixed electrode can facilitate easier product separation. In general, electrochemical regeneration can be broadly classified into direct and indirect electrochemical regeneration. Some of the of the earliest works on direct electrochemical regeneration of nicotinamide cofactors, both reduced (NADH) and oxidized (NAD+) forms, were done by Aizawa et al.[131,132] in attempts to demonstrate a cofactor regeneration strategy similar to those of chemical regeneration without having to use electron mediators. Although direct regeneration of enzymatically active cofactors has been successfully demonstrated, it has been proven to be difficult due to formation of inactive dimers, requirement of high cathodic overpotential for NAD+ reduction and slow kinetics of second electron transfer and protonation.[133–136] Aizawa et al. resolved the problem of inactive NAD2 dimer formation by covalently tethering the cofactor on to sodium alginate,[131] but also recognized that characterizing the cofactors during NADH regeneration was challenging due to their adsorption on the electrode surface.
While indirect regeneration can avoid issues associated with high overpotential and inactive dimer formation (in NAD+ reduction), the addition of electron shuttles can complicate downstream processing and purification. Here, we discuss the state of the art of direct and indirect electrochemical NAD(P)H and NAD(P)+ regeneration and provide yield and electrode potential as measures of comparison to other regeneration methods. Notably, the electrochemical regeneration literature rarely provides sufficient data to calculate TNenzyme and TNcofactor, complicating comparisons to other methods of cofactor regeneration.
4.1. Direct electrochemical regeneration of NADH
The electrochemical reduction of NAD+ for regeneration of 1,4-NADH is accomplished in two-step electron transfer[137] as shown below [Eqs. (1) and (2):
| (1) |
| (2) |
It is now recognized that direct electrochemical reduction of NAD+ additionally leads to the formation of enzymatically inactive dimers (Figure 11).[131,135,138–140] It was observed by Moiroux et al.[141] that the reduction of NAD+ to the free radical (NAD•) occurred at a cathodic potential of −0.858 V vs. standard hydrogen electrode (SHE), which rapidly dimerized and that the second electron transfer to NAD• occurred −1.358 V vs. SHE. It was concluded that the second electron transfer was kinetically less favorable than dimerization because of the adsorption of NAD• radical on the electrode surface. Several researchers have tested different electrode materials and modifications on them to overcome these problems.[135,137–140,142–145]
Figure 11.

Nicotinamide adenine dinucleotide in its oxidized form (NAD+), and its reduction to enzymatically active 1,4-NADH and enzymatically inactive dimer NAD2. R stands for adenosine diphosphoribose. Reproduced from Ref. [138]. Copyright (2006) with permission from Elsevier.
Ali et al. illustrated the effect of electrode potentials on NAD+ reduction using glassy carbon (GC) electrode and achieved yield as high as 96% of enzymatically active 1,4-NADH at high cathodic overpotentials.[139] It was observed that the reduction of NAD+ commenced at around −0.785 V vs. SHE but the slow rate of NADH production was evident from UV/Vis spectrophotometry. At high overpotential of around −1.685 V (vs. SHE), the absorbance plateau was reached at 3.5 h indicating a faster rate of NAD+ reduction to yield enzymatically active 1,4-NADH (more than 3 times higher). This trend was not observed when they tried electrochemical reduction of NAD+ on Au and Cu electrodes in an earlier work,[140] suggesting the importance of adsorption. On bare Au electrode, at low cathodic potential, the rate of NAD+ reduction was lower than that at higher overpotentials, but the yield of enzymatically active 1,4-NADH was inverse, varying from 75–28% at low versus high overpotentials. On bare Cu, a similar trend was observed as yield was found to vary from 71–52% at low versus high overpotentials. When the Au electrode was surface-modified with deposits of Pt, the yield of active reduced cofactor increased from 28 up to 63%. It was hypothesized that the Pt modification enhanced the kinetics of the second electron transfer and simultaneous hydrogenation of the NAD radical and minimized the dimerization of neighboring NAD radicals. In fact, it was shown in a contemporary study that changing the overpotential for NAD+ reduction from −0.273 to −0.573 V (vs. SHE) on a polycrystalline gold electrode, the ratio of 1,4-NADH to the inactive NAD2 dimer decreased from 0.78 to 0.28.[138] However, earlier studies suggest that the reduction peak at an electrode potential of −0.958 V vs. SHE on bare GC electrode corresponds to reduction of NAD+ to NAD radical in a single electron transfer step, but in case of a Ru-modified GC electrode at the same overpotential, a pronounced reduction peak appeared, corresponding to two-electron reduction of NAD+ to 1,4-NADH.[135,146] It was postulated that a submonolayer Ru modification on GC electrode can help avoid the formation of NAD2 dimer, thereby increasing the yield of NADH up to 96%. It was also evident from an electro-impedance study that the irreversible NAD+ reduction on Ru/GC was mass transfer-controlled below cathodic potentials of around −0.858 V vs. SHE. A possible reason for requirement of high overpotential for NAD+ reduction is the large electrontunneling distance between the electrode and the nicotinamide ring of the NAD+ molecule. Not only can Ru modification reduce the electron tunneling distance by reorientation of the adsorbed NAD species on the electrode surface, but also Ru sites act as proton providing sites.[135] It has also been reported that on GC electrodes, reduction of NAD+ is accompanied by hydrogen evolution.[135,142,144] This observation is further bolstered by relevant studies, which suggest that Ru-modified GC electrode is an excellent candidate for hydrogen evolution reaction.[147–149] In this context, a relatively recent study by Rahman et al. investigated two different types of Ru modification of glassy carbon electrodes (nanoparticle and film type) and studied the electrochemical response from each of these surface-modified electrodes with respect to NAD+ reduction and the role of hydrogen adsorption on the electrode surface during NAD+ reduction.[150] From cyclic voltametric studies, it was evident that Ru nanoparticle-modified GC electrode exhibited higher current density at the NAD+ reduction peak when compared to bare GC electrode but it could not clarify whether all of it was enzymatically active 1,4-NADH. In case of Ru-film-modified electrodes, a multifold increase in current density along with shift in the reduction peak voltage to around −0.678 V vs. SHE led the authors to conclude that hydrogen evolution reaction predominantly occurred in the Ru-film-modified electrode. However, it was finally concluded from UV absorption spectra at 340 nm that Ru-nanoparticle-modified GC electrode was more efficient with respect to NAD+ reduction. In a related work, nano-patterned Pt and Ni, electrochemically deposited on glassy carbon electrodes, were used to demonstrate direct reduction of NAD+ at lower electrode potentials compared to bare GC electrodes, and with higher recovery (up to 100%) of enzymatically active 1,4-NADH.[142] The nanoparticles on electrode surface provided adsorbed hydrogen at or adjacent to the NAD radical formation sites to promote faster radical protonation (Figure 12). Cyclic voltammograms showed characteristic peaks pertaining to hydrogen adsorption and desorption, which were not present on bare GC electrodes in the potential region investigated. Linear polarization voltammograms revealed that the current density on GC–Pt electrode was much higher than that on the bare GC electrode, which was due to the current generated due to hydrogen evolution reaction in addition to NAD+ reduction as observed by other researchers as well.[150] At an electrode potential of around −1.6 V, 100% recovery of 1,4-NADH was achieved with the nano-patterned GC–Pt electrode (Figure 13).[142]
Figure 12.

Representation of the bifunctional character of GC–Pt electrodes used for 1,4-NADH regeneration. Reproduced from Ref. [142]. Copyright (2012) with permission from Elsevier.
Figure 13.

Percentage recovery of enzymatically active 1,4-NADH at different electrode potential from reduction of 1 mm NAD+ in a batch electrochemical reactor using GC–Pt electrode. Reproduced from Ref. [142]. Copyright (2012) with permission from Elsevier.
Other carbon nanomaterials and transition metal-based electrodes have also been of great interest among researchers.[137,143–145,151] Ali et al. worked with bare Ti, Ni, Cd, and Co electrodes owing to their good hydrogen adsorption capacity.[144] The trend of regeneration of 1,4-NADH with increasing negative electrode potential was not uniform. The authors explained that Ti has a stronger affinity for H adsorption and a lower rate of hydrogen (H2) evolution reaction at a given potential than other metals. The larger concentration of surface adsorbed hydrogen facilitates desired NADH formation at a low electrode potential (96% yield at −0.185 V vs. SHE) compared to the undesired NAD dimerization. At higher potentials elevated rates of both NAD radical formation and H2 evolution explain the lower yield of NADH and the higher yield of NAD-dimer. By contrast, Ni, Cd, and Co electrodes exhibit lower M–Hads bond strength, higher H2 evolution rates, and lower yields of NADH. Despite its lower M–Hads bond strength than Pt, nickel nanoparticles were used to decorate multiwalled carbon nanotubes in a separate study of NADH regeneration.[137] A recovery of around 98% of 1,4-NADH was possible at an electrode potential of around −1.6 V. Although electrode modification for higher recovery of enzymatically active 1,4-NADH in direct electrochemical regeneration has been common, it suffers from limitations like higher expense of synthesis, loss of the modification layer, and possible denaturation of enzymes. Accordingly, their large-scale application has not been very widespread. To synthesize electrode material feasible for large-scale application, Barin et al. characterized pristine Cu foam, Ag- and Pt-modified Cu foam to study their efficacy for 1,4-NADH regeneration.[145] It was observed that foam deposition time affected the yield of NADH in all cases and pristine Cu foam could achieve higher yield of 1,4-NADH (up to 80%) than the bimetallic Cu foam electrodes. In subsequent study, the authors demonstrated coupled electroenzymatic CO2 reduction using the Cu foam electrodes in both batch and semi-continuous mode.[143,152] In the batch mode, up to 77% yield of active NADH was achieved at an electrode potential of −0.878 V vs. SHE. Although the authors did not comment on NADH regeneration efficiency in the semi-continuous mode, there was an increase in the final product (formate) yield by 42% compared to the batch mode. The increase in formate production could be attributed to the continuous removal of the product, which shifted the equilibrium towards product side and facilitated cofactor regeneration.
4.2. Indirect electrochemical regeneration of 1,4-NADH using electron mediators
Steckhan maintained that indirect electrochemical synthesis, via an electron transfer mediator, can offer a viable path for cofactor regeneration.[153] In these methods, the electron transfer mediator homogeneously reacts with the substrate (e. g., NAD+) leading to its reduction, after which the mediator is regenerated at the electrode surface. This approach avoids several problems associated with high overpotential requirements and inactive dimer formation. However, care must be exercised in selecting the mediator. Cp*RhIII(bpy) complexes are mostly used for this purpose, where the RhIII complex is reduced to RhI by the cathode, protonates to a Rh-hydride complex, which then transfers the hydride and the two electrons to the NAD(P)+ to regenerate the RhIII complex (Figure 14).[154] The principle was applied to regenerate NADH from NAD+ coupled with HLADH catalyzed ketone reduction using a bipyridyl rhodium complex as the electron mediator.[91] Although the method was effective, the intermediate Rh hydride was found to have been deposited on the electrode surface along with low observed rates of NAD+ reduction. Most of the studies thus far have not been successful in demonstrating direct electrochemical reduction of NAD+ to regenerate enzymatically active reduced cofactor with 100% recovery without surface modification of the electrode. As an alternative, indirect electrochemical regeneration of enzymatically active 1,4-NADH via electron mediators has been extensively studied to remediate the problem of NAD radical dimerization.[15] Following the footsteps of Steckhan and co-workers,[19] several researchers have demonstrated the use of electron mediators and discussed their efficiencies in regeneration of reduced nicotinamide cofactors while also limiting the formation of catalytically inactive NAD dimers.[126,154–162]
Figure 14.

Mechanism of [Cp*RhIII(bpy)Cl]Cl complex-mediated indirect electrochemical NADH regeneration method.
Among redox mediators, (2,2-bipyridyl)(pentamethyl cyclopentadienyl)rhodium [Cp*Rh(bpy)] is the most common owing to its less cathodic reduction potential to avoid direct NAD+ reduction and the capability of selective hydride transfer to yield the reduced form of the enzymatically active cofactor.[163] Hildebrand et al. functionalized the 2,2-bipyridine moiety to synthesize a series of substituted Rh complexes and investigated the possibility of faster overall rate to catch up to the rate of enzymatic reaction.[164] From a series of electrochemical measurements, it was clear that the electron transfer from the cathode to mediator was not influenced by change in the bipyridine moiety. Out of all the screened mediators, Cp*[Rh(4,4-dimethoxy-2-2’-bpy)] had a little higher reduction potential due to electron-donating effect but also showed significantly faster rate for cofactor regeneration leading to three times higher turnover frequency than the unsubstituted Cp*(bpy)Rh complex. Similar Rh complex like [Cp*Rh(bpy)Cl]+, which undergoes multistep redox transformation to enable efficient hydride transfer to regenerate 1,4-NADH, has been extensively studied in solution.[16,165,166] Free Rh complexes in solution might interact with the enzyme to decrease its activity, and it has also been observed that the pH of the medium can affect the proton transfer ability of Rh complex redox mediators, which can be attributed to loss of protons at higher pH.[155] Sivanesan and Yoon synthesized a bis(hydroxymethyl) pentamethyl bipyridyl Rh complex, which had a lower reduction peak at −0.549 V vs. SHE, about −0.17 V more anodic than the potential for NAD dimerization.[157] Reduction of NAD+ by this compound was achieved at −0.592 V vs. SHE, but the authors did not clarify the yield of enzymatically active 1,4-NADH. Walcarius et al. synthesized substituted derivatives of [Cp*Rh-(bpy)Cl]+ and studied the impact of immobilization of the mediator on to the SWCNT electrode surface. They observed mediator deactivation upon immobilization, which led to the suppression of catalytic reduction of NAD+.[154] The authors suggested that non-covalent immobilization methods, like π–π stacking, which has been previously applied to modify glassy carbon electrode with Rh complex-functionalized single-walled carbon nanotubes (SWCNT) can be used to immobilize the mediator of interest, [Cp*Rh(bpy)H2O]2+, to avoid its deactivation on the electrode surface and retain the catalytic activity.[167] Despite its limitations, covalent immobilization of Rh complexes on electrode surface can be a useful tool to facilitate reusability of electrodes and separation of the final reaction products in biocatalytic reactions. It is even more convenient to have the enzyme in close contact with these mediators as demonstrated in a relevant experimental work by Zhang et al. with a turnover frequency of 1.3 s−1 and faradaic efficiency of 83%.[160] Bucky paper electrode was chosen for excellent electrical properties and the bipyridyl ligand was grafted on to the surface by electroreduction. Subsequent treatment with [RhCp*Cl2]2 was done to generate the complex and the enzyme was immobilized on this electrode by overcoating on a glassy fiber layer. In a more recent study, carboxylic acid moieties were introduced into the bipyridyl ligand of a RhIII complex, used to transfer electrons from a fluorine-doped tin oxide (FTO) electrode coated with zirconia.[162] Owing to their excellent catalytic activity, Rh(bpy) complexes have been effectively featured in several research works of practical importance like CbFDH-catalyzed CO2 reduction[156] and quantitative detection of adenosine,[158] both of which require 1,4-NADH regeneration. In the case of photochemical cofactor regeneration, which is to be discussed later in this Review, the application of external electrical source is an alternative to sacrificial electron donors. Lee et al. have shown [Cp*Rh(bpy)Cl] can effectively transfer electrons from silicon nanowires, when used as photocathodes connected to an external bias in glucose dehydrogenase catalyzed production of l-glutamate from α-ketoglutarate (Figure 15).[168] Similar application of a Rh complex-mediated NADH regeneration is also featured in a water oxidation-driven photoelectrochemical cell (PEC) platform with simultaneous CO2 reduction by formate dehydrogenase.[169]
Figure 15.

Schematic illustration of the photo-electroenzymatic reaction using a silicon nanowire (SiNW) photocathode. Reproduced from Ref. [168]. Copyright (2014) with permission from Wiley-VCH GmbH & Co.
Other redox mediators like methyl viologen and ferredoxin can also be used as redox mediators in conjunction with NAD(P)H regenerating enzymes like diaphorase, ferredoxin-NAD(P) reductase, and other “VAPOR” (viologen accepting pyridine-nucleotide oxidoreductase) enzymes.[15] In conjunction with an oxidoreductase-catalyzed enzymatic reaction, lipoamide dehydrogenase can be used for 1,4-NADH regeneration via methyl viologen mediator, which transfers the electrons from the cathode. The regeneration typically takes place in a three-reaction sequence (Figure 16), as was shown in the work of Chen et al., where synthesis of lactate from pyruvate was achieved in a membrane packed-bed reactor.[170] Another instance of methyl viologen mediated electroreduction of NAD+ with lipoamide dehydrogenase (LDH), also known as diaphorase, occurs at low cathodic overpotential.[171] In-situ spectrophotometric study showed that the amount of NADH produced was incumbent on the LDH concentration while the rate of NADH regeneration depended on initial NAD+ concentration and followed Michaelis–Menten kinetics. Diaphoraseviologen conjugate formed by covalent attachment has been featured in a recent work by Dinh et al.,[172] which provides more flexible operational strategy as it can be separated from the reaction medium for further reuse. The authors also argued that the proximity of the enzyme-mediator would facilitate higher conversion of NAD+ based on their observation of different final conversion between viologen-diaphorase conjugate-catalyzed reaction versus native diaphorase-catalyzed reaction with exogenously added viologen.
Figure 16.

Three-reaction sequence of methyl viologen-lipoamide dehydrogenase-catalyzed NADH regeneration coupled to LDH-catalyzed lactate production. MV+/MV2+ = methyl viologen mediator; LipDH = lipoamide dehydrogenase; LDH = lactate dehydrogenase.
While it is evident that redox mediators can bypass high cathodic overpotential and dimer formation while regenerating enzymatically active 1,4-NADH during indirect electrochemical reduction, the process still requires downstream separation or immobilization of the cofactor on to the electrode surface. To overcome these problems, Yuan et al. demonstrated in their recent study that cobaltocene-modified poly-(allylamine), a redox polymer, can be potentially used to immobilize diaphorase, which can independently mediate the electroreduction of NAD+ in alcohol dehydrogenase-catalyzed reactions to produce methanol and propanol.[161] The attractive features of this regeneration system were relatively small applied overpotential and high current density, also considered to be an indicator of enhanced electron transfer kinetics. Table 4 enlists different direct and indirect electrochemical systems for NADH regeneration with electrode potentials and corresponding yield of 1,4-NADH.
Table 4.
Different electrode materials with corresponding yields, used in direct and indirect electrochemical regeneration methods.
| Electrode | Potential[a] [V vs. SHE] | Yield [%] | Type | Ref. |
|---|---|---|---|---|
| GC | −1.685 | 98 | direct | [139] |
| Cu | −0.585 | 52 | direct | [140] |
| Au | −0.585 | 28 | direct | [140] |
| Pt-modified Au | −0.485 | 63 | direct | [140] |
| GC-Pt | −0.985 | 100 | direct | [142] |
| GC-Ni | −0.885 | 100 | direct | [142] |
| Cu foam | −0.9 | 78 | direct | [143] |
| Ru-modified GC | −1.0 | 73 | direct | [150] |
| Cu | −0.8 | 91 | Rh mediator | [156] |
| GC or Pt | −0.8 | 36 | Rh mediator | [157] |
| Bucky paper | −0.575 | 9 | Rh mediator | [160] |
| FTO | −0.9 | 90 | Rh mediator | [162] |
Conversion of reference electrode potentials to potentials against SHE is done as outlined in Ref. [173].
4.3. Electrochemical regeneration of NAD+
The direct electrochemical regeneration of the oxidized cofactor, NAD+, reportedly occurs through a three-step process with the displacement of two electrons[136] shown as follows [Eqs. (3)–(5)]:
| (3) |
| (4) |
| (5) |
Similar to NAD(P)+ reduction, direct electrochemical oxidation of NAD(P)H on a bare electrode also takes place at a high overpotential.[17–178] Cyclic voltammograms from a carbon nanotube-modified GC electrode have been demonstrated to have a sharp, large anodic peak at −0.238 V vs. SHE, corresponding to direct oxidation of NAD(P)H, whereas on bare GC electrode, the anodic peak is observed at 0.961 V vs. SHE.[179] Although NAD radical dimerization does not seem to be a problem for direct electrochemical regeneration of the oxidized cofactor NAD(P), it is still necessary to modify the electrode surface or use redox mediators to overcome the high overpotential for electrochemical oxidation of NAD(P)H. Sosna et al. studied HMP (flavohaemoglobin)-modified graphite electrodes, but it was evident from CV studies that the electrode did not exhibit the characteristic redox peak from NADH oxidation.[180] However, when HMP was incorporated in an osmium-complex redox polymer matrix similar to that studied by Yuan et al.,[161] NADH oxidation was observed at reduced overpotentials. With an innovative advance by Megarity et al. in bioelectrocatalytic regeneration of NADP+, ferredoxin NADP reductase (FNR) enzyme was adsorbed in porous indium tin oxide (ITO) electrode, which was capable of regenerating the oxidized cofactor at a sufficiently low electrode potential of 0.08 V vs. SHE.[181,182] A second enzyme (alcohol dehydrogenase enzyme) was also co-immobilized in the ITO pores, which produced a nanoconfinement effect, leading to an increase in the local concentration of the NADPH, which facilitated the oxidative regeneration on the ITO electrode. This co-confined FNR/ADH enzyme on ITO electrode, also termed as “electrochemical leaf” by the authors, was used to explore the bidirectional nature of the NADP+/NADPH conversion by reversing the applied voltage to either direction to demonstrate ADH catalyzed deracemization of 4-phenyl-2-butanol to a single enantiomer.[183] This concept was extended to other studies as well, where multiple redox enzymes, including FNR, were nanoconfined in the electrode pores, used to demonstrate the production of aspartic acid,[184] and used to regenerate another biologically important coenzyme, adenosine triphosphate (ATP) from its monophosphate form (AMP).[185] A TNcofactor of 936 was achieved in the former study, while in the latter, TNcofactor was around 90. However, the nanoconfinement effect of the electrochemical leaf provides a unique way of combining multiple redox enzymes near high local concentrations of the cofactors and reaction intermediates, leading to high reaction rates and redox biotransformations at relatively low electrode potentials.
In recent times, the promising future prospect of biofuel cells has been driving the research for more efficient bioelectrodes, which requires easy and fast electron transfer from the active site of the biocatalyst to the electrode surface. Efforts to decrease the high overpotentials for NADH oxidation have led researchers to investigate certain redox mediators like Nile blue[186] and methylene green-modified multi-walled carbon nanotubes (MWCNT) electrodes[187] which have been employed for NAD(P)H oxidation in biofuel cell/biosensor applications. Integration of metallic nanoparticles like Au on MWCNT electrode surface displays excellent catalytic performance in terms of shifting the oxidation peak by 0.2 V.[188] Different types of surface-modified electrodes with their capabilities to decrease the NADH oxidation potential and immobilize enzymes and cofactors have been commonly used in biosensing applications.[189–192] Studies with SWCNT-coated Pt electrodes also yielded similar results in terms of steady-state current generation at fixed potential versus NADH concentration.[193] Linear amperometric response was observed with NADH concentration and the detection limit found to be as low as 0.17 μM.
Bioelectrocatalysis is an important field of interdisciplinary research, the aim of which is to achieve redox catalysis by synergistically coupling the advantages of biocatalysis like high selectivity and mild reaction conditions with those of electrocatalysis like possible utilization of renewable electricity.[194] The role of nicotinamide cofactors is indispensable as they facilitate the electron relay mechanism between the enzyme and substrate. Electrochemical regeneration has been so popular because they can eliminate additional enzymes and substrates required for cofactor regeneration in a cascade process and avoid complicated downstream separation processes to facilitate easier product recovery. Both direct and indirect electrochemical methods have been demonstrated to effectively reduce NAD+ to enzymatically active 1,4-NADH. However, most of these methods could not achieve TNs as high as enzymatic regeneration methods.[9,195] Table 5 lists different electrodemediator/catalyst combinations used for electrochemical NADH regeneration coupled to an enzymatic system and their corresponding turnover numbers. While TNcofactor are generally below 100, a single result from 2009 reached 18000 and incorporated methyl viologen mediator in a polymer modified electrode. This offers excitement to prompt reproduction and expansion of this technology.
Table 5.
Comparison of turnover numbers as observed in different studies using electrochemical regeneration methods.
| Electrode | Catalyst/mediator | Substrate | TNcofactor | Ref. |
|---|---|---|---|---|
| ZrO2-coated FTO | Cp*RhIII(bpy complex | CO2 | 79[a] | [162] |
| glassy carbon | cobaltocene modified PAA/Diaphorse | formaldehyde | 5.2[b] | [161] |
| Bucky paper | Cp*RhIII(bpy complex | d-fructose | 2.6[a] | [160] |
| carbon paper | ethyl carboxy viologen/diaphorase | pyruvate | <1[b] | [172] |
| ITO | ferredoxin NADP reductase | pyruvate | 936[a] | [184] |
| Cu | Cp*RhIII(bpy complex | CO2 | 6.4[b] | [156] |
| carbon cloth | cobaltocene modified PAA/Diaphorse | ethyl 4-chloroacetoacetate | 30[b] | [196] |
| graphite | MV2+/AMAPOR | 2-oxoglutarate/NH4+ | 1000[a] | [197] |
| carbon plate | MV2+/diaphorase | pyruvic acid/CO2 | 18000[a] | [198] |
| Au-Hg | MV2+/diaphorase | benzoylformate | 158[a] | [199] |
| carbon Felt | Cp*RhIII(bpy complex | acetophenone | 64[a] | [200] |
| glassy carbon | BPV-LPEI/diaphorase | acetoacetyl coenzyme A | 8[b] | [201] |
Used directly from reference
Calculated from data provided in reference.
In some cases, electrode modifications required to minimize overpotential and dimer formation (in case of NAD+ reduction) are expensive and sometimes complicated. Alternatively, redox mediators can be used, but they still need to be separated from the product solution or immobilized on electrode surface for repeated usage. Toxicity and sustainability of certain inorganic redox mediators must be evaluated and improved in indirect electrochemical methods. Nevertheless, there is yet a lot of scope in the field of electrochemical regeneration of nicotinamide cofactors with regards to electrode modification, development of environmentally benign redox mediators, development of immobilization techniques without affecting the kinetics of the redox process, and applicability of such methods in conjunction with enzyme-coupled process for a wide range of product syntheses.
5. Photocatalytic Regeneration Methods
The concept of photobiocatalysis has emerged from the natural photosynthetic process, which uses solar energy to catalyze biochemical reactions with very high efficiency, and thus, light-driven biocatalysis has emerged as a subject of great interest.[202–206] Solar energy being an abundant source of energy, photochemical routes for visible light-driven cofactor regeneration coupled with homogeneous enzymatic synthesis have become increasingly popular among researchers and a discussion topic for several Reviews.[206–212] Photocatalytic cofactor regeneration involves the coupling of nicotinamide cofactor reduction or oxidation with a photocatalyst, which is a semiconductor with a low bandgap for promotion of electrons under photoexcitation. These electrons are then transferred to nicotinamide cofactors during the enzyme-catalyzed redox reactions.[213] A terminal reductant is required in all cases to balance the electron loss from the photocatalyst when the reduced cofactor regeneration is required. Different types of materials have been studied based on their ability to generate electrons under photoexcitation and relay the electrons to be used in biocatalytic redox reactions. One of the bigger challenges while selecting an appropriate photocatalyst has been the issue of electron-hole recombination. In this section we discuss photoenzymatic systems in which a photocatalyst capable of regenerating the oxidized (NAD+) or reduced (NADH) cofactor is coupled with an enzymatic system acting with or without an electron mediator. Quantitative comparisons based on turnover number and yield have been made wherever possible.
5.1. Photoregeneration of the reduced cofactor 1,4-NADH
The electron transfer mechanism in photocatalytic NAD(P)H regeneration is similar to that of chemical regeneration, where a sacrificial electron donor like ethylene diamine tetraacetate (EDTA)[30] is exhausted to donate electrons to the photo-generated holes on a typical photocatalyst like TiO2[28,29,214] or g-C3N4[215] created by visible light excitation. The electrons eliminated from the photocatalyst surface are then available for NAD(P)+ reduction, which is generally coupled to a redox enzyme-mediated reaction requiring the reduced NAD(P)H cofactor.
One of the early works using a cadmium sulfide (CdS) semiconductor coupled with hydrogenase enzyme for NADH photoregeneration using formate as the electron source was done by Shumilin et al.[216] The study established a direct electron transfer mechanism facilitated by the CdS photocatalyst in presence of a NAD-dependent hydrogenase. The electrons available from the formate photooxidation were transferred directly to the hydrogenase electron transport chain via the CdS semiconductor. A high ratio of hydrogenase/photocatalyst enables the enzyme to utilize all available reducing equivalents in the system up to a certain maximum value. More recent studies like that by Park et al.[30] used a mediator for more efficient electron transfer to the cofactor (Figure 17). The redox mediator in this case is a Cp*Rh(bpy) complex, which effectively utilized the photoexcited electrons as well as improved the photonic efficiency.[16,28,217–219] The novelty of the work is the solid-state synthesis of the photocatalyst W2Fe4Ta2O17, which has been found to be superior to the benchmark photocatalyst TiO2 in terms of rate of NADH generation. Pristine TiO2 has been reported by Wang et al. to be used for electron mediated regeneration of NADH where EDTA was used as the sacrificial electron donor.[220] The study investigated the effects of mediator concentration, different electron donors, and pH on the yield of NADH. The photo-regeneration of 1,4-NADH achieved a high yield; however, trace amount of enzymatically inactive 1,6-NADH was reported by the authors.
Figure 17.

Schematic diagram of the photocatalyst–enzyme-coupled bioreactor functioning under visible light.
Owing to their low bandgap energy, zinc sulfide (ZnS) photocatalysts were studied for recycling of NADH by reducing NAD+ coupled to an enzymatic CO2 reduction to methanol.[221] ZnS–A (thioamide sulfur source) was observed to produce the highest concentration and was reported to have lower bandgap energy than 0.5% Ru-loaded ZnS–A and ZnS–C (commercial ZnS). Another interesting observation was the use of bioderived glycerol as the sacrificial electron donor. The authors observed higher rate of NADH regeneration when compared with isopropanol as the electron donor. In a subsequent work,[222] it was recognized that water could be used as an electron donor with a Cp*Rh(bpy) complex as an electron mediator and addition of glycerol improves the conversion yield and regeneration rate. Although the reduced thickness of the nanosheets and high surface area facilitate surface reactions, the authors were concerned that the use of sacrificial electron donor like triethanolamine (TEOA) may accumulate and deactivate the enzymes in the reaction medium. Although rhodium-based catalysts are excellent electron relaying agents, some of the challenges associated with Rh(bpy) complex mediated photoregeneration are the cost of these catalysts and their efficient separation from the aqueous reaction medium in an enzyme-coupled process. Other catalysts like cobaloxime have also been demonstrated by Kim et al.[223] in an effort to produce a cheaper catalyst compared to Pt catalysts or (Cp)*Rh(Bpy) complexes, which are also considered quite effective for photocatalytic NADH regeneration[28,224] A sacrificial electron donor is required in all cases, direct or indirect photoregeneration, to minimize electron–hole recombination on the photosensitizer surface. Pt nanoparticles, as demonstrated by Kim and co-workers,[224] could act as the photosensitizer as well as the photocatalyst at the same time, while the cobaloxime catalyst needed a photo-sensitizer as it acted merely as the electron relay agent to facilitate NAD+ reduction[223] through sequential redox cycles. Eosin Y, the most common form of eosin dye, was used as the photosensitizer and the system was effectively tested to carry out simultaneous enzymatic reduction of CO2 to formic acid catalyzed by NAD-dependent FDH. In fact, FDH-catalyzed CO2 reduction has been a significant target application in the area of biomimetic CO2 remediation by harvesting solar energy using photoenzymatic systems.[225] One of the most impressive recent works in this regard has been reported by Fard et al., where the authors demonstrated photoenzymatic CO2 reduction to formate using TEOA as the reductant.[226] The most ingenious aspect of this study was the spatial compartmentalization of immobilized FDH and an Rh complex on Janus type DNA nanosheet, which had two different DNA sequences on each side, enabling face-selective immobilization of the FDH and the Rh catalyst to avoid mutual inactivation. Based on the amount of formate produced, the TNcofactor reported was 1360, which, to the best of our knowledge, is the highest among photoenzymatic cofactor regeneration studies to date. This study clearly highlights the importance of spatial compartmentalization of the photocatalyst and the enzyme in regard to increasing the efficiency of photoenzymatic NAD(P)H regeneration systems. Another notable work using Rh complex was reported by Oppelt et al., where the authors synthesized a novel bipyridyl containing polymer ligand to generate a catalytically active rhodium complex, which could be used as a hydride transfer catalyst for the regeneration of NADH in chemoenzymatic and photoenzymatic systems.[227] Like most other photochemical regeneration systems, it requires a sacrificial electron donor for the reduction of NAD+, and TEOA was the choice in this case as well. It was also suggested that two different mechanisms of NAD+ reduction might exist, involving the possible photoexcitation of the Rh center or the polymeric chain in each case. The photochemical quantum yield was also comparable to the amount of NADH formed per unit area of the photocatalyst, per unit time. An additional advantage of this polymer-bound rhodium catalyst was that it could be coated on to glass beads and the authors demonstrated catalytic activity using the reaction scheme as depicted in (Figure 18).
Figure 18.

NAD+ regeneration scheme using formate as the reductant with polymer-bound Cp*Rh(bpy) catalyst on glass bead.
5.1.1. Metal-free photocatalysts for NADH regeneration
Bulk graphitic carbon nitride is an ideal example of metal-free photocatalyst but has issues from high rate of electron–hole recombination, which retards the rate of electron transfer.[215,228,229] Jones et al. suggested that it is difficult to get high yield of enzymatically active cofactor of the reduced form when no electron mediator is used.[230] Graphitic carbon nitride (g-C3N4) has been studied with and without electron mediator, and it was reported that in the direct case, the yield of 1,4-NADH regeneration was limited to around 50%, while the Rh complex-mediated regeneration yielded close to 100% without any inactive dimer formation.[219] Nanosheets of g-C3N4 and the hybrid g-C3N4/graphene films were studied by Jia et al.[228] to demonstrate NAD+ reduction with a simple Rh(bpy) complex mediated photocatalytic system as well as in photoelectrochemical NADH regeneration systems. Commercially available flavins and chemically synthesized flavins like deazariboflavin or dRf have been reported in the literature for photoregeneration of NADH;[231] however, the issue of enzymatically inactive complexes makes it relatively less attractive to use these photocatalysts for practical purposes. Other examples of polymeric photocatalysts for NADH regeneration include conjugate microporous polymers (CMPs) based on dibenzo-[b,d]thiophene polymer, which was able to demonstrate around 84% regeneration efficiency and excellent selectivity for the enzymatically active 1,4-NADH.[232]
5.1.2. Role of triethanolamine as electron donor
An interesting fact about TEOA as an electron donor is that the NAD+ photoreduction and oxidation of TEOA are not necessarily coupled.[233] In fact, it was noticed with several photocatalysts that glycolaldehyde formed from pre-irradiated TEOA induces NADH regeneration even in the absence of light or after removal of the photocatalyst. In other words, TEOA used an electron donor in photocatalytic NADH regeneration acts like a de facto precursor for the actual reducing agent, glycolaldehyde, which is presumably oxidized to glyoxal.
5.2. Photoregeneration of the oxidized cofactor NAD+
Oxidative photoregeneration of NAD+ has gained a lot of interest lately among researchers as the inverse reaction, photocatalytic NADH regeneration, has been studied more extensively in prior literature. The interaction between the photogenerated electrons and the cofactor is better understood than the interaction with the photogenerated holes. Zhang et al. studied this interaction using CdS as a model photocatalyst under anaerobic conditions to eliminate interferences from oxygen derived intermediates and explained that the photogenerated hole-induced NADH oxidation was not a one-step hydride transfer process.[234] Instead, it was hypothesized as a three-step process, where the photo-excited NADH underwent a single electron oxidation and turned into a NADH radical cation and subsequent deprotonation and additional single electron transfer could result in an enzymatically active NAD+ or fragmentation into ADP ribose radical and nicotinamide (Figure 19). Lin et al. used TiO2 photocatalyst and encapsulated alcohol dehydrogenase in a photocatalytic microcapsule assembly using TiO2 nanoparticles as the building block.[235] The enzyme encapsulated TiO2 microreactors were modified with a polydopamine layer and the microreactor system acted as an independent photo-enzymatic unit for the conversion of ethanol to acetaldehyde (Figure 20). Non-metallic photocatalysts like g-C3N4 are not traditionally used for NADH photooxidation due to their low efficacy. However, doping with iron increases its efficacy as reported by Zhang et al.[236] Iron-doped g-C3N4 indicated lower electron density than CN nanosheets due to coordination between the iron and pyridinic N species as observed from X-ray photoelectron spectroscopy (XPS) studies. The authors hypothesized that the enhanced activity with regards to NADH photooxidation could be ascribed to the synergistic effect of the Fe doping into the 2D CN nanosheets.
Figure 19.

NADH oxidation mechanism by photogenerated holes on CdS particles.
Figure 20.

Schematic illustration of the cycling of nicotinamide coenzymes NAD+/NADH catalyzed by the bioinspired ADH@TiO2 NP microreactors. Reproduced from Ref. [235]. Copyright (2018), with permission from MDPI.
5.3. Metal-free photocatalysts for NAD+ regeneration
Conjugated polymeric materials can facilitate visible light absorption by the virtue of their delocalized π-system and have thus found wide applications is photonics and photocatalysis.[237] Ma et al. developed a metal-free conjugated polymer-based module, which was able to regenerate the oxidized form of the cofactor, NAD+, in a photo-enzymatic system.[238] The conjugated microporous photocatalysts were encapsulated in polymer vesicles to produce autonomous microreactors, which catalyze mediator-free oxidation of NADH as demonstrated by evaluation of the cofactor’s biological activity using enzymatic oxidation of glycerol to dihydroxyacetone. A similar study based on conjugated microporous polymer nanoparticles was reported by Jo et al., where the researchers demonstrated a biomimetic cellular antioxidant defense system using these nanoparticles as photocatalysts, coupled with enzymatic glucose oxidation.[239] Oxygen scavenging enzymes were combined to protect the glucose dehydrogenase enzyme from reactive oxygen species and the photocatalytic properties of the conjugated polymer were used to regenerate NAD+ in cofactor recycling system. Other researchers reported similar polymer-based photocatalysts like a conjugate polymer hydrogel photocatalyst for NADH oxidation.[240]
Photochemical regeneration is a relatively new area of research, and it is emerging as an intriguing alternative for biomimetic cofactor regeneration. It synergistically combines photochemistry with biocatalytic redox chemistry to synthesize extremely beneficial compounds without the expense of multiple enzymes. Several different materials have been used as discussed earlier in this section, and Table 6 features some of those materials along with the corresponding turnover numbers. It is difficult to have an accurate estimate of the turnover numbers using photocatalysts because these are not soluble and, in most cases, published literature does not have the exact estimate of the active sites’ concentration. Despite being an environment-friendly strategy, it is contingent on sacrificial electron donors, which are needed in equimolar amount, as well requiring post-synthesis separation. This is a substantial limitation at this time but may be addressed by identifying electron donors, which yield valuable coproducts, analogous to enzyme regeneration cascade systems.
Table 6.
Comparison of turnover numbers as observed in different studies using photochemical regeneration methods.
| Catalyst | Substrate | Mediator | TNcofactor | Ref. |
|---|---|---|---|---|
| W2Fe4Ta2O17 | EDTA | Rh complex | 0.15[b] | [30] |
| porphyrin derivative on ZIF-8 | TEOA | Rh complex | 0.078[a] | [217] |
| Eosin Y | TEOA | Rh complex immobilized on DNA nanosheets | 1360[a] | [226] |
| Rh-coordinated polymer | TEOA | – | 0.236[a] | [227] |
| Eosin Y | TEOA | Co complex | 1.5[b] | [223] |
| ZIF-8/gC3N4 | TEOA | Rh complex | 0.3[a] | [218] |
| ZnS-A | glycerol | Ru complex | 9.6[a] | [221] |
| conjugate polymer | TEOA | Rh complex | 3.3[a] | [232] |
Used directly from reference
Calculated from data provided in reference.
6. Summary and Outlook
Enzymatic catalysis has sustained the biological world since its inception. Redox enzymes have served as the powerhouse of cellular catalysis and fueled the interests of the scientific community to invest research efforts towards sustainable manufacture of food, fuels, and pharmaceuticals. However, the intrinsic requirement of the nicotinamide cofactors is a bottle-neck to the commercial application of many redox enzymes.
We have discussed enzymatic, chemical, electrochemical, and photochemical routes of nicotinamide cofactor regeneration and critically evaluated the pioneering studies and the current state of the art. In making comparisons across families of technologies, this Review tabulates or calculates the efficiency of cofactor regeneration using the common metric of cofactor turnover number (TNcofactor). Present data confirm that enzymatic regeneration remains the benchmark technology, generally offering the highest TNcofactor (up to 500000) but does engender complications associated with scalability and co-product separations. Chemical, electrochemical, and photochemical regeneration technologies have notably advanced in efficiency in the last ten years, and opportunities for further improvement including utilization of inexpensive, environmentally benign co-products, reduced reliance upon noble metal components, and further enhancement of selectivity have been highlighted herein. Other challenges like the need for post-synthesis enzyme recovery in free-enzyme-catalyzed reactions, mutual inactivation of catalysts in case of organometallic complex-catalyzed regeneration coupled with enzymatic synthesis, formation of NAD2 dimer in direct electrochemical regeneration, and the need for equimolar amounts of a sacrificial donor in photochemical regeneration techniques have been described.
Different authors have tried addressing these challenges in their research using novel methodologies, with a general goal of increasing the turnover numbers. However, the most successful methods are still the ones involving enzymatic catalysis to regenerate the cofactors. Especially those research works that feature immobilized redox enzymes along with immobilized cofactors have achieved the highest turnover numbers, which reiterates the unmatched efficiency of enzymes. However, major developments have also been achieved in other non-conventional routes, like immobilization of redox enzymes on photocatalytic inorganic supports or electrode surfaces for cofactor regeneration using light and electrical energy, respectively.
While this Review has striven to provide a technical comparison based on performance metrics, including TN, equally important is the consideration of the different technologies’ sustainability. Common approaches to sustainability assessment for low technology readiness levels include technoeconomic assessment, life cycle assessment, and material flow analysis. Sustainability tools used in combination with research advancements is referred to as prospective assessments or quantitative sustainable design.[241] Combining these assessments can identify potential research advancements that maximize improvements to the technology’s sustainability.[242] In addition, only preliminary assessments have been performed to compare the relative environmental sustainability of different nicotinamide adenine dinucleotide (NAD+/NADH) regeneration technologies.[243] Several research improvements for NAD+/NADH regeneration have been studied and presented in this Review, including enzyme immobilization to increase stability and reusability and the use of Earth-abundant metal-based regeneration in place of rare metals. To ensure these improvements are impactful, researchers need to incorporate sustainability analysis, which will facilitate a complete technical comparison of NAD+/NADH regeneration mechanisms.
Acknowledgements
This work was supported in part by a research grant from the Kansas Corn Commission under award number 2205 and by National Institute of General Medical Sciences (NIGMS) under award number P20 GM113117 (JMH).
Biographies

Dr. Alan M. Allgeier is Associate Professor of Chemical Engineering and Associate Director of the Center for Environmentally Beneficial Catalysis at the University of Kansas (KU), where his research program explores sustainable manufacturing routes to chemicals and materials and seeks molecular-level insights into the performance of catalysts. He earned a Ph.D. in inorganic chemistry at Northwestern University and spent 21 years working in industrial research in the pharmaceutical and chemical industries before joining KU in 2017.

Dr. Justin M. Hutchison is currently an Assistant Professor in the department of Civil, Environmental, and Architectural Engineering at the University of Kansas. He holds B.A. degrees in Biology and Biochemistry from Augustana College and M.S. and Ph.D. degrees in Environmental Engineering from the University of Illinois at Urbana-Champaign. Dr. Hutchison’s interests center on enzyme (biocatalytic) based applications to promote sustainable water treatment.

Victor K. Sharma is currently a graduate student in the department of Chemical & Petroleum Engineering at the University of Kansas. He earned his Master and Bachelors’s degrees in Chemical Engineering from the Indian Institute of Technology Guwahati and West Bengal University of Technology, respectively. His research interests include sustainable production of chemicals and pharmaceuticals, biocatalysis, and novel composite materials synthesis.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Paul PEV, Sangeetha V, Deepika RG, in Recent Developments in Applied Microbiology and Biochemistry (Ed.: Buddolla V), Academic Press, 2019, pp. 107–125. [Google Scholar]
- [2].Robinson PK, Essays Biochem 2015, 59, 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Straathof AJJ, Chem. Rev 2014, 114, 1871–1908. [DOI] [PubMed] [Google Scholar]
- [4].Kataoka M, Miyakawa T, Shimizu S, Tanokura M, Appl. Microbiol. Biotechnol 2016, 100, 5747–5757. [DOI] [PubMed] [Google Scholar]
- [5].Sheldon RA, Brady D, ChemSusChem 2019, 12, 2859–2881. [DOI] [PubMed] [Google Scholar]
- [6].Sellés Vidal L, Kelly CL, Mordaka PM, Heap JT, Biochim. Biophys. Acta BBA-Proteins Proteomics 2018, 1866, 327–347. [DOI] [PubMed] [Google Scholar]
- [7].Liu W, Wang P, Biotechnol. Adv 2007, 25, 369–384. [DOI] [PubMed] [Google Scholar]
- [8].“Oxidoreductase Introduction – Creative Enzymes,” can be found under https://www.creative-enzymes.com/resource/oxidoreductase-introduction_19.html.
- [9].Wang X, Saba T, Yiu HHP, Howe RF, Anderson JA, Shi J, Chem 2017, 2, 621–654. [Google Scholar]
- [10].Sheldon RA, Brady D, Chem. Commun 2018, 54, 6088–6104. [DOI] [PubMed] [Google Scholar]
- [11].Garzón-Posse F, Becerra-Figueroa L, Hernández-Arias J, Gamba-Sánchez D, Molecules 2018, 23, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chenault HK, Simon ES, Whitesides GM, Biotechnol. Genet. Eng. Rev 1988, 6, 221–270. [DOI] [PubMed] [Google Scholar]
- [13].Angelastro A, Dawson WM, Luk LYP, Allemann RK, ACS Catal. 2017, 7, 1025–1029. [Google Scholar]
- [14].Bryan Jones J, Taylor KE, J. Chem. Soc. Chem. Commun 1973, 6, 205–206. [Google Scholar]
- [15].Weckbecker A, Gröger H, Hummel W, in Biosystems Engineering I: Creating Superior Biocatalysts (Eds.: Wittmann C, Krull R), Springer, Berlin, Heidelberg, 2010, pp. 195–242. [Google Scholar]
- [16].Hollmann F, Witholt B, Schmid A, J. Mol. Catal. B 2002, 19–20, 167–176. [Google Scholar]
- [17].Wang X, Yiu HHP, ACS Catal. 2016, 6, 1880–1886. [Google Scholar]
- [18].Reeve HA, Lauterbach L, Ash PA, Lenz O, Vincent KA, Chem. Commun 2012, 48, 1589–1591. [DOI] [PubMed] [Google Scholar]
- [19].Grammenudi S, Franke M, Vögtle F, Steckhan E, J. Inclusion Phenom 1987, 5,695–707. [Google Scholar]
- [20].Komoschinski J, Steckhan E, Tetrahedron Lett. 1988, 29, 3299–3300. [Google Scholar]
- [21].Wienkamp R, Steckhan E, Angew. Chem. Int. Ed. Engl 1982, 21, 782–783. [Google Scholar]
- [22].Shaked Z, Barber JJ, Whitesides GM, J. Org. Chem 1981, 46, 4100–4101. [Google Scholar]
- [23].Hollmann F, Arends IWCE, Buehler K, ChemCatChem 2010, 2, 762–782. [Google Scholar]
- [24].Campbell E, Meredith M, Minteer SD, Banta S, Chem. Commun 2012, 48, 1898–1900. [DOI] [PubMed] [Google Scholar]
- [25].Wang Z, Etienne M, Quilès F, Kohring G-W, Walcarius A, Biosens. Bioelectron 2012, 32, 111–117. [DOI] [PubMed] [Google Scholar]
- [26].Ryu J, Lee SH, Nam DH, Park CB, Adv. Mater 2011, 23, 1883–1888. [DOI] [PubMed] [Google Scholar]
- [27].Ha Lee S, Ryu J, Heon Nam D, Beum Park C, Chem. Commun 2011, 47, 4643–4645. [DOI] [PubMed] [Google Scholar]
- [28].Shi Q, Yang D, Jiang Z, Li J, J. Mol. Catal. B 2006, 43, 44–48. [Google Scholar]
- [29].Geng J, Yang D, Zhu J, Chen D, Jiang Z, Mater. Res. Bull 2009, 44, 146–150. [Google Scholar]
- [30].Beum Park C, Ha Lee S, Subramanian E, Kale BB, Mi Lee S, Baeg J-O, Chem. Commun 2008, 0, 5423–5425. [DOI] [PubMed] [Google Scholar]
- [31].Yang D, Zhang Y, Zhang S, Cheng Y, Wu Y, Cai Z, Wang X, Shi J, Jiang Z, ACS Catal. 2019, 9, 11492–11501. [Google Scholar]
- [32].Wang Y, Sun J, Zhang H, Zhao Z, Liu W, Catal. Sci. Technol 2018, 8, 2578–2587. [Google Scholar]
- [33].Sperl JM, Sieber V, ACS Catal. 2018, 8, 2385–2396. [Google Scholar]
- [34].Hummel W, Trends Biotechnol. 1999, 17, 487–492. [DOI] [PubMed] [Google Scholar]
- [35].Quinto T, Köhler V, Ward TR, Top. Catal 2014, 57, 321–331. [Google Scholar]
- [36].Rodriguez C, Lavandera I, Gotor V, Curr. Org. Chem 2012, 16, 2525–2541. [Google Scholar]
- [37].Aalbers FS, Fraaije MW, ChemBioChem 2019, 20, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Puetz H, Puch’ova E, Vrankova K, Hollmann F, Catalysts 2020, 10, 952. [Google Scholar]
- [39].de Gonzalo G, Alcantara AR, Catalysts 2021, 11, 605. [Google Scholar]
- [40].Wei X, Han P, You C, Chin. J. Chem. Eng 2020, 28, 2799–2809. [Google Scholar]
- [41].Jadhav SB, Harde S, Bankar SB, Granström T, Ojamo H, Singhal RS, Survase SA, RSC Adv. 2014, 4, 14597. [Google Scholar]
- [42].Parmentier S, Arnaut F, Soetaert W, Vandamme EJ, Biocatal. Biotransform 2005, 23, 1–7. [Google Scholar]
- [43].Ricca E, Brucher B, Schrittwieser JH, Adv. Synth. Catal 2011, 353, 2239–2262. [Google Scholar]
- [44].Voss CV, Gruber CC, Kroutil W, Synlett 2010, 2010, 991–998. [Google Scholar]
- [45].Rioz-Martínez A, Bisogno FR, Rodríguez C, de Gonzalo G, Lavandera I, Pazmiño DET, Fraaije MW, Gotor V, Org. Biomol. Chem 2010, 8, 1431–1437. [DOI] [PubMed] [Google Scholar]
- [46].Kroutil W, Mang H, Edegger K, Faber K, Curr. Opin. Chem. Biol 2004, 8, 120–126. [DOI] [PubMed] [Google Scholar]
- [47].Kroutil W, Mang H, Edegger K, Faber K, Adv. Synth. Catal 2004, 346, 125–142. [Google Scholar]
- [48].Pellissier H, Tetrahedron 2003, 59, 8291–8327. [Google Scholar]
- [49].Voss CV, Gruber CC, Kroutil W, Tetrahedron: Asymmetry 2007, 18, 276–281. [Google Scholar]
- [50].Schlager S, Dibenedetto A, Aresta M, Apaydin DH, Dumitru LM, Neugebauer H, Sariciftci NS, Energy Technol. 2017, 5, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sultana S, Sahoo PC, Martha S, Parida K, RSC Adv. 2016, 6, 44170–44194. [Google Scholar]
- [52].Gamba I, Bioinorg. Chem. Appl 2018, 2018, e2379141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xu S, Lu Y, Li J, Jiang Z, Wu H, Ind. Eng. Chem. Res 2006, 45, 4567–4573. [Google Scholar]
- [54].Shaked Z, Whitesides GM, J. Am. Chem. Soc 1980, 102, 7104–7105. [Google Scholar]
- [55].Lin S-S, Miyawaki O, Nakamura K, J. Biosci. Bioeng 1999, 87, 361–364. [DOI] [PubMed] [Google Scholar]
- [56].Liu W, Ma H, Luo J, Shen W, Xu X, Li S, Hu Y, Huang H, Biochem. Eng. J 2014, 91, 204–209. [Google Scholar]
- [57].Jiang W, Fang B, Sci. Rep 2016, 6, 30462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Qi Y, Yang T, Zhou J, Zheng J, Xu M, Zhang X, Rao Z, Yang S-T, Process Biochem. 2017, 55, 104–109. [Google Scholar]
- [59].Uzir MH, Najimudin N, J. Mol. Catal 2018, 447, 56–64. [Google Scholar]
- [60].Han L, Liang B, Song J, J. Ind. Microbiol. Biotechnol 2018, 45, 111–121. [DOI] [PubMed] [Google Scholar]
- [61].Chen W-H, Vázquez-González M, Zoabi A, Abu-Reziq R, Willner I, Nat. Catal 2018, 1, 689–695. [Google Scholar]
- [62].Peng F, Ou X-Y, Guo Z-W, Zeng Y-J, Zong M-H, Lou W-Y, Int. J. Biol. Macromol 2020, 162, 445–453. [DOI] [PubMed] [Google Scholar]
- [63].Nagy F, Gyujto I, Tasnadi G, Barna B, Balogh-Weiser D, Faber K, Poppe L, Hall M, J. Biotechnol 2020, 323, 246–253. [DOI] [PubMed] [Google Scholar]
- [64].Engelmann C, Johannsen J, Waluga T, Fieg G, Liese A, Bubenheim P, Catalysts 2020, 10, 1216. [Google Scholar]
- [65].Velasco-Lozano S, da Silva ES, Llop J, López-Gallego F, ChemBioChem 2018, 19, 395–403. [DOI] [PubMed] [Google Scholar]
- [66].Voges M, Fischer F, Neuhaus M, Sadowski G, Held C, Ind. Eng. Chem. Res 2017, 56, 5535–5546. [Google Scholar]
- [67].Alsafadi D, Alsalman S, Paradisi F, Org. Biomol. Chem 2017, 15, 9169–9175. [DOI] [PubMed] [Google Scholar]
- [68].Rodríguez C, Borzęcka W, Sattler JH, Kroutil W, Lavandera I, Gotor V, Org. Biomol. Chem 2013, 12, 673–681. [DOI] [PubMed] [Google Scholar]
- [69].Yan Q, Zhang X, Chen Y, Guo B, Zhou P, Chen B, Huang Q, Wang J, ACS Catal. 2022, 12, 6746–6755. [Google Scholar]
- [70].de Souza ROMA, Miranda LSM, Bornscheuer UT, Chem. Eur. J 2017, 23, 12040–12063. [DOI] [PubMed] [Google Scholar]
- [71].Luhavaya H, Sigrist R, Chekan JR, McKinnie SMK, Moore BS, Angew. Chem. Int. Ed 2019, 58, 8394–8399; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 8482–8487. [Google Scholar]
- [72].Kula M-R, Wandrey C, in Methods in Enzymology, Academic Press, 1987, pp. 9–21. [DOI] [PubMed] [Google Scholar]
- [73].Alpdagtas S, Binay B, Biocatal. Biotransform 2021, 39, 260–268. [Google Scholar]
- [74].Seelbach K, Riebel B, Hummel W, Kula M-R, Tishkov VI, Egorov AM, Wandrey C, Kragl U, Tetrahedron Lett. 1996, 37, 1377–1380. [Google Scholar]
- [75].Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K, Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- [76].Sivanathan S, Körber F, Tent JA, Werner S, Scherkenbeck J, J. Org. Chem 2015, 80, 2554–2561. [DOI] [PubMed] [Google Scholar]
- [77].Slatner M, Nagl G, Haltrich D, Kulbe KD, Nidetzky B, Biocatal. Biotransform 1998, 16, 351–363. [DOI] [PubMed] [Google Scholar]
- [78].Yishai O, Lindner SN, Gonzalez J Cruz de la, Tenenboim H, Bar-Even A, Curr. Opin. Chem. Biol 2016, 35, 1–9. [DOI] [PubMed] [Google Scholar]
- [79].Lau C, Moehlenbrock MJ, Arechederra RL, Falase A, Garcia K, Rincon R, Minteer SD, Banta S, Gupta G, Babanova S, Atanassov P, Int. J. Hydrogen Energy 2015, 40, 14661–14666. [Google Scholar]
- [80].Kim YH, Campbell E, Yu J, Minteer SD, Banta S, Angew. Chem. Int. Ed 2013, 52, 1437–1440; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 1477–1480. [Google Scholar]
- [81].Orlich B, Schomaecker R, Biotechnol. Bioeng. 1999, 65, 6. [DOI] [PubMed] [Google Scholar]
- [82].Slatner M, Nidetzky B, Kulbe KD, Biochemistry 1999, 38, 10489–10498. [DOI] [PubMed] [Google Scholar]
- [83].Wichmann R, Wandrey C, Bückmann AF, Kula M, Biotechnol. Bioeng 1981, 23,2789–2802. [DOI] [PubMed] [Google Scholar]
- [84].Wang Y, Li L, Ma C, Gao C, Tao F, Xu P, Sci. Rep. 2013, 3, 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Marpani F, Sárossy Z, Pinelo M, Meyer AS, Biotechnol. Bioeng 2017, 114, 2762–2770. [DOI] [PubMed] [Google Scholar]
- [86].Lim S, Yoo H, Sarak S, Kim B, Yun H, J. Ind. Eng. Chem 2021, 98, 358–365. [Google Scholar]
- [87].Johannes TW, Woodyer RD, Zhao H, Biotechnol. Bioeng 2007, 96, 18–26. [DOI] [PubMed] [Google Scholar]
- [88].Schrittwieser JH, Sattler J, Resch V, Mutti FG, Kroutil W, Curr. Opin. Chem. Biol 2011, 15, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jia H-Y, Zong M-H, Zheng G-W, Li N, ACS Catal. 2019, 9, 2196–2202. [Google Scholar]
- [90].Mutti FG, Knaus T, Scrutton NS, Breuer M, Turner NJ, Science 2015, 349, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Adachi O, Fujii Y, Ano Y, Moonmangmee D, Toyama H, Shinagawa E, Theeragool G, Lotong N, Matsushita K, Biosci. Biotechnol. Biochem 2001, 65, 115–125. [DOI] [PubMed] [Google Scholar]
- [92].Singh R, Singh RK, Kim S-Y, Sigdel S, Park J-H, Choi J-H, Kim I-W, Lee J-K, Biochem. Eng. J 2016, 109, 189–196. [Google Scholar]
- [93].Su W-B, Li F-L, Li X-Y, Fan X-M, Liu R-J, Zhang Y-W, New Biotechnol. 2021, 62, 18–25. [DOI] [PubMed] [Google Scholar]
- [94].Li G, Wang J, Reetz MT, Bioorg. Med. Chem 2018, 26, 1241–1251. [DOI] [PubMed] [Google Scholar]
- [95].Sun Z, Lonsdale R, Ilie A, Li G, Zhou J, Reetz MT, ACS Catal. 2016, 6, 1598–1605. [Google Scholar]
- [96].Hartley CJ, Williams CC, Scoble JA, Churches QI, North A, French NG, Nebl T, Coia G, Warden AC, Simpson G, Frazer AR, Jensen CN, Turner NJ, Scott C, Nat. Catal 2019, 2, 1006–1015. [Google Scholar]
- [97].Baumer B, Classen T, Pohl M, Pietruszka J, Adv. Synth. Catal 2020, 362, 2894–2901. [Google Scholar]
- [98].Peschke T, Bitterwolf P, Gallus S, Hu Y, Oelschlaeger C, Willenbacher N, Rabe KS, Niemeyer CM, Angew. Chem. Int. Ed 2018, 57,17028–17032; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 17274–17278. [Google Scholar]
- [99].Wu H, Tian C, Song X, Liu C, Yang D, Jiang Z, Green Chem. 2013, 15, 1773–1789. [Google Scholar]
- [100].Fukuzumi S, Lee Y-M, Nam W, J. Inorg. Biochem 2019, 199, 110777. [DOI] [PubMed] [Google Scholar]
- [101].Bardea A, Katz E, Buckmann A, Willner I, J. Am. Chem. Soc 1997, 119, 9114–9119. [Google Scholar]
- [102].Abril O, Whitesides GM, J. Am. Chem. Soc 1982, 104, 1552–1554. [Google Scholar]
- [103].Kölle U, Grützel M, Angew. Chem. Int. Ed 1987, 26, 567–570; [Google Scholar]; Angew. Chem 1987, 99, 572–574. [Google Scholar]
- [104].Steckhan E, Herrmann S, Ruppert R, Dietz E, Frede M, Spika E, Organometallics 1991, 10, 1568–1577. [Google Scholar]
- [105].Wagenknecht PS, Penney JM, Hembre RT, Organometallics 2003, 22, 1180–1182. [Google Scholar]
- [106].Matsuo T, Mayer JM, Inorg. Chem 2005, 44, 2150–2158. [DOI] [PubMed] [Google Scholar]
- [107].Lo HC, Buriez O, Kerr JB, Fish RH, Angew. Chem. Int. Ed 1999, 38, 1429–1432; [DOI] [PubMed] [Google Scholar]; Angew. Chem 1999, 111, 1524–1527. [Google Scholar]
- [108].Pitman CL, Finster ONL, Miller AJM, Chem. Commun 2016, 52, 9105–9108. [DOI] [PubMed] [Google Scholar]
- [109].Haquette P, Talbi B, Barilleau L, Madern N, Fosse C, Salmain M, Org. Biomol. Chem 2011, 9, 5720–5727. [DOI] [PubMed] [Google Scholar]
- [110].Morra S, Pordea A, Chem. Sci 2018, 9, 7447–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lachmanova SN, Pospisil L, Sebera J, Talbi B, Salmain M, Hromadova M, J. Electroanal. Chem 2020, 859, 113882. [Google Scholar]
- [112].Yan YK, Melchart M, Habtemariam A, Peacock AFA, Sadler PJ, JBICJ. Biol. Inorg. Chem 2006, 11, 483–488. [DOI] [PubMed] [Google Scholar]
- [113].Khusnutdinova JR, Milstein D, Angew. Chem. Int. Ed 2015, 54, 12236–12273; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 12406–12445. [Google Scholar]
- [114].Hopkins JA, Lionetti D, Day VW, Blakemore JD, Organometallics 2019, 38, 1300–1310. [Google Scholar]
- [115].Ganesan V, Sivanesan D, Yoon S, Inorg. Chem 2017, 56, 1366–1374. [DOI] [PubMed] [Google Scholar]
- [116].Bucci A, Dunn S, Bellachioma G, Menendez Rodriguez G, Zuccaccia C, Nervi C, Macchioni A, ACS Catal 2017, 7, 7788–7796. [Google Scholar]
- [117].Tensi L, Macchioni A, ACS Catal. 2020, 10, 7945–7949. [Google Scholar]
- [118].Betanzos-Lara S, Liu Z, Habtemariam A, Pizarro AM, Qamar B, Sadler PJ, Angew. Chem. Int. Ed 2012, 51, 3897–3900; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 3963–3966. [Google Scholar]
- [119].Jones JB, Sneddon DW, Higgins W, Lewis AJ, J. Chem. Soc. Chem. Commun 1972, 15, 856–857. [Google Scholar]
- [120].Aksu S, Arends IWCE, Hollmann F, Adv. Synth. Catal 2009, 351, 1211–1216. [Google Scholar]
- [121].Zhu C, Li Q, Pu L, Tan Z, Guo K, Ying H, Ouyang P, ACS Catal. 2016, 6, 4989–4994. [Google Scholar]
- [122].Poizat M, Arends IWCE, Hollmann F, J. Mol. Catal. B 2010, 63, 149–156. [Google Scholar]
- [123].Himiyama T, Waki M, Maegawa Y, Inagaki S, Angew. Chem. Int. Ed 2019, 58, 9150–9154; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 9248–9252. [Google Scholar]
- [124].Deng Y, Odziomek M, Sanchez C, Back O, Mougel V, Fontecave M, ChemCatChem 2020, 12,1236–1243. [Google Scholar]
- [125].Matsui K, Maegawa Y, Waki M, Inagaki S, Yamamoto Y, Catal. Sci. Technol 2018, 8, 534–539. [Google Scholar]
- [126].Zhang L, Vila N, Kohring G-W, Walcarius A, Etienne M, ACS Catal. 2017, 7,4386–4394. [Google Scholar]
- [127].Hollmann F, Kleeb A, Otto K, Schmid A, Tetrahedron: Asymmetry 2005, 16, 3512–3519. [Google Scholar]
- [128].Canivet J, Süss-Fink G, štěpnička P, Eur. J. Inorg. Chem 2007, 2007, 4736–4742. [Google Scholar]
- [129].Grau MM, Poizat M, Arends IWCE, Hollmann F, Appl. Organomet. Chem 2010, 24, 380–385. [Google Scholar]
- [130].Shen Y, Zhan Y, Li S, Ning F, Du Y, Huang Y, He T, Zhou X, Chem. Sci 2017, 8, 7498–7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Aizawa M, Coughlin RW, Charles M, Biotechnol. Bioeng 1976, 18, 209–215. [DOI] [PubMed] [Google Scholar]
- [132].Aizawa M, Coughlin RW, Charles M, Biochim. Biophys. Acta BBA – Gen. Subj 1975, 385, 362–370. [DOI] [PubMed] [Google Scholar]
- [133].Paxinos AS, Günther H, Schmedding DJM, Simon H, Bioelectrochem. Bioenerg 1991, 25, 425–436. [Google Scholar]
- [134].Deng C, Chen J, Chen X, Xiao C, Nie Z, Yao S, Electrochem. Commun 2008, 10, 907–909. [Google Scholar]
- [135].Azem A, Man F, Omanovic S, J. Mol. Catal. Chem 2004, 219, 283–299. [Google Scholar]
- [136].Immanuel S, Sivasubramanian R, Gul R, Dar MA, Chem. Asian J 2020, 15, 4256–4270. [DOI] [PubMed] [Google Scholar]
- [137].Ali I, Ullah N, McArthur MA, Coulombe S, Omanovic S, Can. J. Chem. Eng 2018, 96, 68–73. [Google Scholar]
- [138].Damian A, Omanovic S, J. Mol. Catal. Chem 2006, 253, 222–233. [Google Scholar]
- [139].Ali I, Soomro B, Omanovic S, Electrochem. Commun 2011, 13, 562–565. [Google Scholar]
- [140].Damian A, Maloo K, Omanovic S, Chem. Biochem. Eng. Q 2007, 21, 21–32. [Google Scholar]
- [141].Moiroux J, Deycard S, Malinski T, J. Electroanal. Chem. Interfacial Electrochem 1985, 194, 99–108. [Google Scholar]
- [142].Ali I, Gill A, Omanovic S, Chem. Eng. J. 2012, 188, 173–180. [Google Scholar]
- [143].Barin R, Biria D, Rashid-Nadimi S, Asadollahi MA, J. CO2 Util 2018, 28, 117–125. [Google Scholar]
- [144].Ali I, Khan T, Omanovic S, J. Mol. Catal. Chem 2014, 387, 86–91. [Google Scholar]
- [145].Barin R, Rashid-Nadimi S, Biria D, Asadollahi MA, Electrochim. Acta 2017, 247, 1095–1102. [Google Scholar]
- [146].Man F, Omanovic S, J. Electroanal. Chem 2004, 568, 301–313. [Google Scholar]
- [147].üosiewicz B, Martin M, Lebouin C, Lasia A, J. Electroanal. Chem 2010, 649, 198–205. [Google Scholar]
- [148].Magdić K, Kvastek K, Horvat-Radošević V, Electrochim. Acta 2015, 167, 455–469. [Google Scholar]
- [149].Breiter MW, J. Electroanal. Chem. Interfacial Electrochem 1984, 178, 53–59. [Google Scholar]
- [150].Rahman G, Mian SA, Shah A. ul H. A., Joo O-S, J. Appl. Electrochem. 2016, 46, 459–468. [Google Scholar]
- [151].Immanuel S, Sivasubramanian R, Mater. Sci. Eng. B 2020, 262, 114705. [Google Scholar]
- [152].Barin R, Biria D, Rashid-Nadimi S, Asadollahi MA, Chem. Eng. Process. Process Intensif 2019, 140, 78–84. [Google Scholar]
- [153].Steckhan E, Angew. Chem. Int. Ed. Engl 1986, 25, 683–701. [Google Scholar]
- [154].Walcarius A, Nasraoui R, Wang Z, Qu F, Urbanova V, Etienne M, Gollu M, Demir AS, Gajdzik J, Hempelmann R, Bioelectrochemistry 2011, 82, 46–54. [DOI] [PubMed] [Google Scholar]
- [155].Gajdzik J, Lenz J, Natter H, Walcarius A, Kohring GW, Giffhorn F, Gollu M, Demir AS, Hempelmann R, J. Electrochem. Soc 2012, 159, F10–F16. [Google Scholar]
- [156].Kim S, Kim MK, Lee SH, Yoon S, Jung K-D, J. Mol. Catal. B 2014, 102, 9–15. [Google Scholar]
- [157].Sivanesan D, Yoon S, Polyhedron 2013, 57, 52–56. [Google Scholar]
- [158].Han D, Kim H-M, Chand R, Kim G, Shin I-S, Kim Y-S, Appl. Biochem. Biotechnol 2015, 177, 812–820. [DOI] [PubMed] [Google Scholar]
- [159].Tan B, Hickey DP, Milton RD, Giroud F, Minteer SD, J. Electrochem. Soc 2015, 162, H102–H107. [Google Scholar]
- [160].Zhang L, Etienne M, Vila N, Le TXH, Kohring G-W, Walcarius A, ChemCatChem 2018, 10, 4067–4073. [Google Scholar]
- [161].Yuan M, Kummer MJ, Milton RD, Quah T, Minteer SD, ACS Catal. 2019, 9, 5486–5495. [Google Scholar]
- [162].Chen Y, Li P, Noh H, Kung C-W, Buru CT, Wang X, Zhang X, Farha OK, Angew. Chem. Int. Ed 2019, 58, 7682–7686; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 7764–7768. [Google Scholar]
- [163].Steckhan E, in Electrochemostry V (Ed.: Steckhan E), Springer, Berlin, Heidelberg, 1994, pp. 83–111. [Google Scholar]
- [164].Hildebrand F, Kohlmann C, Franz A, Lütz S, Adv. Synth. Catal 2008, 350,909–918. [Google Scholar]
- [165].Hollmann F, Schmid A, Steckhan E, Angew. Chem. Int. Ed 2001, 40, 169–171; [PubMed] [Google Scholar]; Angew. Chem 2001, 113, 190–193. [Google Scholar]
- [166].Vuorilehto K, Lütz S, Wandrey C, Bioelectrochemistry 2004, 65, 1–7. [DOI] [PubMed] [Google Scholar]
- [167].Salimi A, Izadi M, Hallaj R, Soltanian S, Hadadzadeh H, J. Solid State Electrochem 2009, 13, 485–496. [Google Scholar]
- [168].Lee SH, Ryu GM, Nam DH, Kim JH, Park CB, ChemSusChem 2014, 7, 3007–3011. [DOI] [PubMed] [Google Scholar]
- [169].Nam DH, Kuk SK, Choe H, Lee S, Ko JW, Son EJ, Choi E-G, Kim YH, Park CB, Green Chem. 2016, 18, 5989–5993. [Google Scholar]
- [170].Chen X, Fenton JM, Fisher RJ, Peattie RA, J. Electrochem. Soc 2004, 151, E56. [Google Scholar]
- [171].Tam TK, Chen B, Lei C, Liu J, Bioelectrochemistry 2012, 86, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Dinh TH, Lee SC, Hou CY, Won K, J. Electrochem. Soc 2016, 163, H440. [Google Scholar]
- [173].McCafferty E, in Introduction to Corrosion Science (Ed.: McCafferty E), Springer, New York, NY, 2010, pp. 33–56. [Google Scholar]
- [174].Bartlett PN, Simon E, J. Am. Chem. Soc 2003, 125,4014–4015. [DOI] [PubMed] [Google Scholar]
- [175].Catalin Popescu I, Dominguez E, Narváez A, Pavlov V, Katakis I, J. Electroanal. Chem 1999, 464, 208–214. [Google Scholar]
- [176].Pereira AR, de Souza JCP, Gonçalves AD, Pagnoncelli KC, Crespilho FN, Pereira AR, de Souza JCP, Gonçalves AD, Pagnoncelli KC, Crespilho FN, J. Braz. Chem. Soc 2017,28, 1698–1707. [Google Scholar]
- [177].Ma Y, Jin Z, Peng B, Ding J, Wang N, Zhou M, J. Electrochem. Soc 2015, 162, H317. [Google Scholar]
- [178].Kochius S, Magnusson AO, Hollmann F, Schrader J, Holtmann D, Appl. Microbiol. Biotechnol 2012, 93, 2251–2264. [DOI] [PubMed] [Google Scholar]
- [179].Chen J, Cai C-X, Chin. J. Chem 2004, 22, 167–171. [Google Scholar]
- [180].Sosna M, Bonamore A, Gorton L, Boffi A, Ferapontova EE, Biosens. Bioelectron 2013, 42, 219–224. [DOI] [PubMed] [Google Scholar]
- [181].Megarity CF, Siritanaratkul B, Heath RS, Wan L, Morello G, Patrick SRF, Booth RL, Sills AJ, Robertson AW, Warner JH, Turner NJ, Armstrong FA, Angew. Chem. Int. Ed 2019, 58, 4948–4952; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 5002–5006. [Google Scholar]
- [182].Megarity CF, Siritanaratkul B, Cheng B, Morello G, Wan L, Sills AJ, Heath RS, Turner NJ, Armstrong FA, ChemCatChem 2019, 11, 5662–5670. [Google Scholar]
- [183].Wan L, Heath RS, Megarity CF, Sills AJ, Herold RA, Turner NJ, Armstrong FA, ACS Catal. 2021, 11, 6526–6533. [Google Scholar]
- [184].Morello G, Megarity CF, Armstrong FA, Nat. Commun 2021, 12, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Megarity CF, Weald TRI, Heath RS, Turner NJ, Armstrong FA, ACS Catal. 2022, 12(15), 8811–8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Yan Y-M, Yehezkeli O, Willner I, Chem. Eur. J 2007, 13, 10168–10175. [DOI] [PubMed] [Google Scholar]
- [187].Aquino Neto S, Almeida TS, Meredith MT, Minteer SD, De Andrade AR, Electroanalysis 2013,25, 2394–2402. [Google Scholar]
- [188].Aquino Neto S, Almeida TS, Belnap DM, Minteer SD, De Andrade AR, Power Sources J 2015, 273, 1065–1072. [Google Scholar]
- [189].Teymourian H, Salimi A, Hallaj R, Talanta 2012, 90, 91–98. [DOI] [PubMed] [Google Scholar]
- [190].Haque A-MJ, Nandhakumar P, Yang H, Electroanalysis 2019, 31, 876–882. [Google Scholar]
- [191].Stolarczyk K, Rogalski J, Bilewicz R, Bioelectrochemistry 2020, 135, 107574. [DOI] [PubMed] [Google Scholar]
- [192].Narváez Villarrubia CW, Artyushkova K, Garcia SO, Atanassov P, J. Electrochem. Soc 2014, 161, H3020–H3028. [Google Scholar]
- [193].Feng L, Li H-P, Galatsis K, Monbouquette HG, J. Electroanal. Chem 2016, 773, 7–12. [Google Scholar]
- [194].Chen H, Simoska O, Lim K, Grattieri M, Yuan M, Dong F, Lee YS, Beaver K, Weliwatte S, Gaffney EM, Minteer SD, Chem. Rev 2020, 120, 12903–12993. [DOI] [PubMed] [Google Scholar]
- [195].Tassano E, Hall M, Chem. Soc. Rev 2019, 48, 5596–5615. [DOI] [PubMed] [Google Scholar]
- [196].Dong F, Chen H, Malapit CA, Prater MB, Li M, Yuan M, Lim K, Minteer SD, J. Am. Chem. Soc 2020, 142, 8374–8382. [DOI] [PubMed] [Google Scholar]
- [197].Schulz M, Leichmann H, Günther H, Simon H, Appl. Microbiol. Biotechnol 1995, 42, 916–922. [Google Scholar]
- [198].Zheng H, Ohno Y, Nakamori T, Suye S, J. Biosci. Bioeng 2009, 107, 16–20. [DOI] [PubMed] [Google Scholar]
- [199].Kim M-H, Yun S-E, Biotechnol. Lett 2004, 26, 21–26. [DOI] [PubMed] [Google Scholar]
- [200].Hildebrand F, Lütz S, Tetrahedron: Asymmetry 2007, 18, 1187–1193. [Google Scholar]
- [201].Alkotaini B, Abdellaoui S, Hasan K, Grattieri M, Quah T, Cai R, Yuan M, Minteer SD, ACS Sustainable Chem. Eng 2018, 6, 4909–4915. [Google Scholar]
- [202].Barber J, Tran PD, J. R. Soc. Interface 2013, 10, 20120984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Djurišić AB, He Y, Ng AMC, APL Mater. 2020, 8, 030903. [Google Scholar]
- [204].Concepcion JJ, House RL, Papanikolas JM, Meyer TJ, Proc. Natl. Acad. Sci. USA 2012, 109, 15560–15564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Scholes GD, Nat. Chem 2011, 3, 12.21160508 [Google Scholar]
- [206].Kim JH, Nam DH, Park CB, Curr. Opin. Biotechnol 2014, 28, 1–9. [DOI] [PubMed] [Google Scholar]
- [207].Lee SH, Choi DS, Kuk SK, Park CB, Angew. Chem. Int. Ed. 2018, 57, 7958–7985; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 8086–8116. [Google Scholar]
- [208].Kim J, Park CB, Curr. Opin. Chem. Biol 2019, 49, 122–129. [DOI] [PubMed] [Google Scholar]
- [209].Seel CJ, Gulder T, ChemBioChem 2019, 20, 1871–1897. [DOI] [PubMed] [Google Scholar]
- [210].Özgen FF, Runda ME, Schmidt S, ChemBioChem 2021, 22, 790–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Zhang Y, Zhao Y, Li R, Liu J, Solar RRL 2021, 5, 2000339. [Google Scholar]
- [212].Chen J, Guan Z, He Y-H, Asian J Org. Chem 2019, 8, 1775–1790. [Google Scholar]
- [213].Antonio Macia-Agullo J, Corma A, Garcia H, Chem. Eur. J 2015, 21, 10940–10959. [DOI] [PubMed] [Google Scholar]
- [214].Kment S, Riboni F, Pausova S, Wang L, Wang L, Han H, Hubicka Z, Krysa J, Schmuki P, Zboril R, Chem. Soc. Rev 2017, 46, 3716–3769. [DOI] [PubMed] [Google Scholar]
- [215].Ong W-J, Tan L-L, Ng YH, Yong S-T, Chai S-P, Chem. Rev 2016, 116, 7159–7329. [DOI] [PubMed] [Google Scholar]
- [216].Shumilin IA, Nikandrov VV, Popov VO, Krasnovsky AA, FEBS Lett. 1992, 306, 125–128. [DOI] [PubMed] [Google Scholar]
- [217].Zhou J, Yu S, Kang H, He R, Ning Y, Yu Y, Wang M, Chen B, Renewable Energy 2020, 156, 107–116. [Google Scholar]
- [218].Yu S, Lv P, Xue P, Wang K, Yang Q, Zhou J, Wang M, Wang L, Chen B, Tan T, Chem. Eng. J 2020, 420, 127649. [Google Scholar]
- [219].Liu J, Antonietti M, Energy Environ. Sci 2013, 6, 1486–1493. [Google Scholar]
- [220].Wang Y, Zhao Z, Zhou R, Liu W, J. Mol. Catal. B 2016, 133, S188–S193. [Google Scholar]
- [221].Dibenedetto A, Stufano P, Macyk W, Baran T, Fragale C, Costa M, Aresta M, ChemSusChem 2012, 5, 373–378. [DOI] [PubMed] [Google Scholar]
- [222].Aresta M, Dibenedetto A, Baran T, Angelini A, tabuz P, Macyk W, Beilstein J. Org. Chem 2014, 10, 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Kim JA, Kim S, Lee J, Baeg J-O, Kim J, Inorg. Chem 2012, 51, 8057–8063. [DOI] [PubMed] [Google Scholar]
- [224].Bhoware SS, Kim KY, Kim JA, Wu Q, Kim J, J. Phys. Chem. C 2011, 115, 2553–2557. [Google Scholar]
- [225].Tian Y, Zhou Y, Zong Y, Li J, Yang N, Zhang M, Guo Z, Song H, ACS Appl. Mater. Interfaces 2020, 12, 34795–34805. [DOI] [PubMed] [Google Scholar]
- [226].Fard PT, Albert SK, Ko J, Lee S, Park S-J, Kim J, ACS Catal. 2022, 9698–9705. [Google Scholar]
- [227].Oppelt KT, Gasiorowski J, Egbe DAM, Kollender JP, Himmelsbach M, Hassel AW, Sariciftci NS, Knor G, J. Am. Chem. Soc 2014, 136, 12721–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Jia C, Hu W, Zhang Y, Teng C, Chen Z, Liu J, Inorg. Chem. Front 2020, 7, 2434–2442. [Google Scholar]
- [229].Qi K, Liu S, Zada A, J. Inst. Chem 2020, 109,111–123. [Google Scholar]
- [230].Jones W, Burnett JWH, Shi J, Howe RF, Wang X, Joule 2020, 4, 2055–2059. [Google Scholar]
- [231].Hoefler GT, Fernandez-Fueyo E, Pesic M, Younes SH, Choi E-G, Kim YH, Urlacher VB, Arends IWCE, Hollmann F, ChemBioChem 2018, 19, 2344–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Lan F, Wang Q, Chen H, Chen Y, Zhang Y, Huang B, Liu H, Liu J, Li R, ACS Catal 2020, 10, 12976–12986. [Google Scholar]
- [233].Kinastowska K, Liu J, Tobin JM, Rakovich Y, Vilela F, Xu Z, Bartkowiak W, Grzelczak M, Appl. Catal. B 2019,243, 686–692. [Google Scholar]
- [234].Zhang S, Shi J, Chen Y, Huo Q, Li W, Wu Y, Sun Y, Zhang Y, Wang X, Jiang Z, ACS Catal. 2020, 10, 4967–4972. [Google Scholar]
- [235].Lin S, Sun S, Wang K, Shen K, Ma B, Ren Y, Fan X, Nanomaterials 2018, 8, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Zhang Y, Huang X, Li J, Lin G, Liu W, Chen Z, Liu J, Chem. Res. Chin. Univ 2020, 36, 1076–1082. [Google Scholar]
- [237].Zhang G, Lan Z-A, Wang X, Angew. Chem. Int. Ed 2016, 55, 15712–15727; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 15940–15956. [Google Scholar]
- [238].Ma BC, Caire da Silva L, Jo S-M, Wurm FR, Bannwarth MB, Zhang KAI, Sundmacher K, Landfester K, ChemBioChem 2019, 20, 2593–2596. [DOI] [PubMed] [Google Scholar]
- [239].Jo S-M, Zhang KAI, Wurm FR, Landfester K, ACS Appl. Mater. Interfaces 2020, 12, 25625–25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Byun J, Landfester K, Zhang KAI, Chem. Mater 2019, 31, 3381–3387. [Google Scholar]
- [241].Shi R, Guest JS, ACS Sustainable Chem. Eng. 2020, 8, 18903–18914. [Google Scholar]
- [242].Hutchison JM, Mayer BK, Vega M, Chacha WE, Zilles JL, Water Res. 2021, 12, 100112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Burnett JWH, Sun Z, Li J, Wang X, Wang X, Green Chem. 2021, 23, 7162–7169. [Google Scholar]