

Abstract
The in vitro and in vivo anti-cancer activity of iodinated photosensitizers (PS) with and without erlotinib moiety was investigated in UMUC-3 [epidermal growth factor (EGFR) positive], T24 (EGFR low) cell lines, and tumored mice. Both the erlotinib conjugated PS 3 and 5 showed EGFR target specificity, but the position-3 erlotinib-PS conjugate 3 demonstrated lower photodynamic therapy (PDT) efficacy than the corresponding non-erlotinib analog 1, whereas the conjugate 5 containing an erlotinib moiety at position-17 of the PS gave higher tumor-uptake and long-term tumor cure (SCID mice bearing UMUC-3 tumors). PS-erlotinib conjugates in the absence of light were ineffective in vitro and in vivo, but robust apoptotic and necrotic cell death was observed in bladder cancer cells after exposing them with a laser light at 665 nm. In contrast to 18F-fluorodeoxyglucose (FDG), a positron emission tomography (PET) agent, the position-17 erlotinib conjugate (124I- analog 6) showed enhanced UMUC3 tumor contrast even at a low imaging dose of 15μCi/mouse.
Graphical Abstract
INTRODUCTION
Bladder cancer is the 4th most commonly diagnosed cancer in the US in men and the 10th in women1. Approximately two-thirds of newly diagnosed bladder cancers have not invaded the bladder smooth muscle and are considered non-muscle invasive bladder cancers (NMIBC)2. Despite a 5-year survival rate of 88%, up to 70% of NMIBCs recur after initial treatment3. For muscle-invasive bladder cancer (MIBC), the gold standard remains radical cystectomy (RC) and lymph node dissection (LND), with or without neoadjuvant chemotherapy. However, RC remains a major procedure with a high rate of complications. Minimally invasive RC has been incorporated to reduce the morbidity associated with RC4,5. Still, 5-year survival rates are only 50–70%, and complication rates exceed 606–10. Recently, trimodality (bladder preservation) therapy has emerged as an alternative to RC, with comparable survival rates in select patients. However, most guidelines do not recommend it as a standard treatment.
Treatment of bladder cancer is dependent on tumor grade and stage. Non-muscle invasive tumors are treated by transurethral resection with and without subsequent intra-vesical chemotherapy or immunotherapy. The high recurrence rate of NMIBC may be due to poor detection at diagnosis, or regrowth of residual tumor after incomplete resection. For MIBC, more interest has been recently spurred in bladder preservation strategies as a viable alternative to RC and LND in select patients. Therefore, the concept of having a dual functional agent (diagnostic and therapeutic) will have definite advantages.
Recently, epidermal growth factor receptor (EGFR) has gained much attention for the treatment of cancer11. EGFR is a member of the tyrosine kinase receptor family, and pathological expression of EGFR leads to uncontrolled cell proliferation12. It also results in promotion of angiogenesis and inhibition of apoptosis leading to malignant growth and progression. Overexpression of EGFR in bladder cancer has been widely reported, and several studies have shown EGFR is associated with high tumor stage, tumor progression, and poor clinical outcome13. Due to the central role of EGFR in several tumors, two main types of EGFR inhibitors have been reported: monoclonal antibodies14,15 and small molecule tyrosine kinase inhibitors16. Some of the small molecules (e.g., erlotinib and related analogs) are either clinically approved or are at various stages of clinical trials. These compounds alone or in combination with other chemotherapy agents have shown enhanced long-term patients’ survival. Thus, following this approach, PDT induced tumor destruction can be maintained and significantly improved by the administration of EGFR inhibitors. Bladder and prostate have a multitude of primary lymphatic landing sites, making the sentinel node sampling concept inapplicable. However, dual-imaging techniques could be helpful in guiding surgeons in the removal of suspicious nodes, avoiding excessive or suboptimal LND. Despite advances in various imaging techniques, their sensitivity and specificity are still limited, and histological examination of lymph nodes remains the standard for lymph node staging. Therefore, improved target-specific imaging agents (PET/fluorescence) with therapeutic potential (e.g. PDT or combination therapy) will have definite advantage(s) in treating these cancers.
There are only limited reports to address the choices of PS used for PDT on the basis of specificity or affinity for ABCG217–19. Indeed, many tumors do not appear to express large amounts of ABCG2, or if they do it is not necessarily present in the cell membrane where it functions as an effluxing pump, but in the cytoplasm or nucleus. However, it has become evident that a small subset of ABCG2-expressing cells known as side population (SP) is present in most types of tissues and in many tumors, and possesses stem cell-like characteristics. In many tumors, the SP has been demonstrated as enriched for cells with enhanced tumorigenic potential, known as tumor initiating cells (TIC), stem cell-like cancer cells (SCLCC) or cancer stem cells (CSC). The TIC and CSC have been postulated as the source and driving force of tumor growth and development. If the small number of SP cells present in tumors efflux ABCG2 substrate PS and evade PDT-mediated death, they could be responsible for repopulating tumors, causing recurrences and poor clinical outcome20.21. Recent demonstration of SP in bladder cancer necessitates further investigation of its potential role in characterizing CSCs. A recent report by Hepburn et al.22 confirmed that ABCG2 SP enriches for SCCs in human bladder cancer and provides evidence for the importance of the MAPK pathway as a suitable therapeutic target for the treatment. On the basis of published reports it could be advantageous to verify ABCG2 expression in tumors before selecting the most appropriate PS for PDT treatment, especially if the PS is a substrate and the ABCG2 positive SP cells are part of the tumor initiating population. If the non-SP or ABCG2 negative population include TIC, then the substrate specificity and affinity of the PS may be less important, since it will not be pumped out. An alternative method to ensure adequate PS levels in all tumor cells including bladder cancer would be to combine substrate PS with an ABCG2 inhibitor for enhanced PDT outcome.
Physicians at West Virginia University have evaluated Photofrin-PDT in 58 patients with refractory NMIBC and patients who could not receive local treatment with chemotherapy or BCG immunotherapy. With a single PDT treatment, 84% of patients with residual refractory papillary transitional cell carcinoma and 75% of patients with refractory carcinoma in situ demonstrated complete response. Now, over 4 years from treatment, 59% of responders are alive, and only 3 out of 34 patients experienced disease recurrence. PDT was safe and effective treatment for refractory carcinoma in situ or recurrent papillary transitional cell carcinoma. Clinical trials are ongoing to determine whether this form of treatment is superior to standard BCG immunotherapy or chemotherapy23. Unfortunately, Photofrin has several limitations, including skin photo-toxicity and limited tumor-specificity. Therefore, a multifunctional PS with high tumor-selectivity may help to detect the tumors by PET and improve the survival of bladder cancer patients by fluorescence-guided PDT alone, or in combination with erlotinib therapy.
Therefore, to make PDT more successful, our proposed study was aimed to develop a near-infrared (NIR) dual-function imaging agent (e.g.,124I-PET & fluorescence, which are complimentary to each other) to optimize tumor staging and treatment of bladder cancer. The use of NIR light source for fluorescence-image guided PDT24 will help to locate and precisely treat NMIBC, as well as MIBC as part of bladder preservation strategies. 124I-PET- imaging will improve lymph node and systemic staging, which will allow better control of tumor recurrence and long-term survival.
(A). RESULTS AND DISCUSSION
Chemistry:
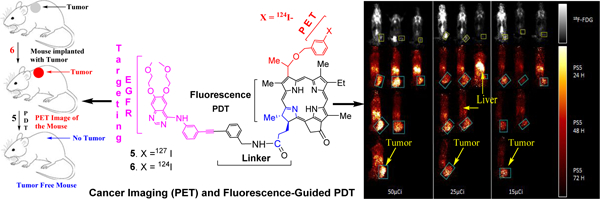
Our laboratory has been investigating the utility of porphyrin-based compounds in cancer-imaging and therapy for quite some time24. In our hands the structure-activity relationship (SAR) and quantitative structure activity relationship (QSAR) studies were extremely fruitful in selecting the most active agents in porphyrin and reduced porphyrin (chlorin and bacteriochlorin) systems26. We further extended this approach by preparing a series of carbohydrate (substrate for lectins) and cRGD (substrate for integrin) analogs. Some of the conjugates showed enhanced PDT efficacy, but the target-specificity following the inhibition approach could not be confirmed27. We also observed that on introducing the same functionality (including small molecules, e.g. carbohydrates to target galectins, known for their over expression in certain tumors) at various peripheral positions of the PS makes a significant difference in tumor-uptake and PDT efficacy28,29. Therefore, to investigate the impact of erlotinib conjugated PS for targeting EGFR known for its high expression in bladder cancer, the erlotinib moiety was introduced either at position-3 or at position-17 of PS 1, which has already been recognized for its PET and PDT potential in various tumor types30. The PS-erlotinib conjugates 3 and 5 bearing an erlotinib moiety at position-3 and -17 respectively were synthesized from methyl-3-[1’(m-iodobenzyloxy)ethyl]-3-devinylpyropheophorbide-a following two different synthetic strategies (PS 1 was used as a diasteriomeric mixture for the preparation of desired analogs because the respective R- and S- isomers showed similar in vitro and in vivo efficacy)31. In our first approach (Scheme 1B), PS 1 was conjugated with erlotinib 7 under Sonogashira cross coupling conditions32, and the intermediate 8 was isolated in 72% yield. The hydrolysis of the methyl ester analog under mild conditions gave the corresponding carboxylic acid 9 in >95% yield, which on subsequent reaction with m-iodobenzylamine afforded the desired non-radioactive iodinated PS-erlotinib conjugate 3 in 76% yield.
Scheme 1:
Structures of parent compounds (radioactive and non-radioactive) derived from chlorophyll-a, and the synthesis of the corresponding erlotinib conjugates.
In our second approach, as illustrated in Scheme 1C for the synthesis of conjugate 5, erlotinib was first coupled with m-iodobenzylamine. The resulting product 11 was reacted with the carboxylic acid 12, obtained by hydrolyzing the corresponding methyl ester analog 1, and the desired position-17 erlotinib analog was obtained in 71% yield.
For the preparation of PET-imaging agents with longer half-life, conjugates of the iodinated PS 3 and 5 were first converted to the corresponding trimethyl-tin analogs 10 and 13 respectively, which on subsequent reaction with 124I- sodium iodide (Na124I) afforded the desired PET imaging agents 4 and 6 by following the methodology reported for the preparation of 124I-agent 2 [30]. The purity of the radioactive PS was confirmed by HPLC analyses.
(B). Characterization of conjugates 2 and 3:
The structures of all the intermediate and final products (non-radioactive) were confirmed by NMR (1H-, 13C-, and 2D-) and mass spectrometry analyses. In addition to being generally consistent with the proposed structures, the NMR data also show some interesting differences between the spectra of these compounds. These spectral features suggest significant differences in molecular conformation, which are dependent on the location of the erlotinib conjugation to the PS.
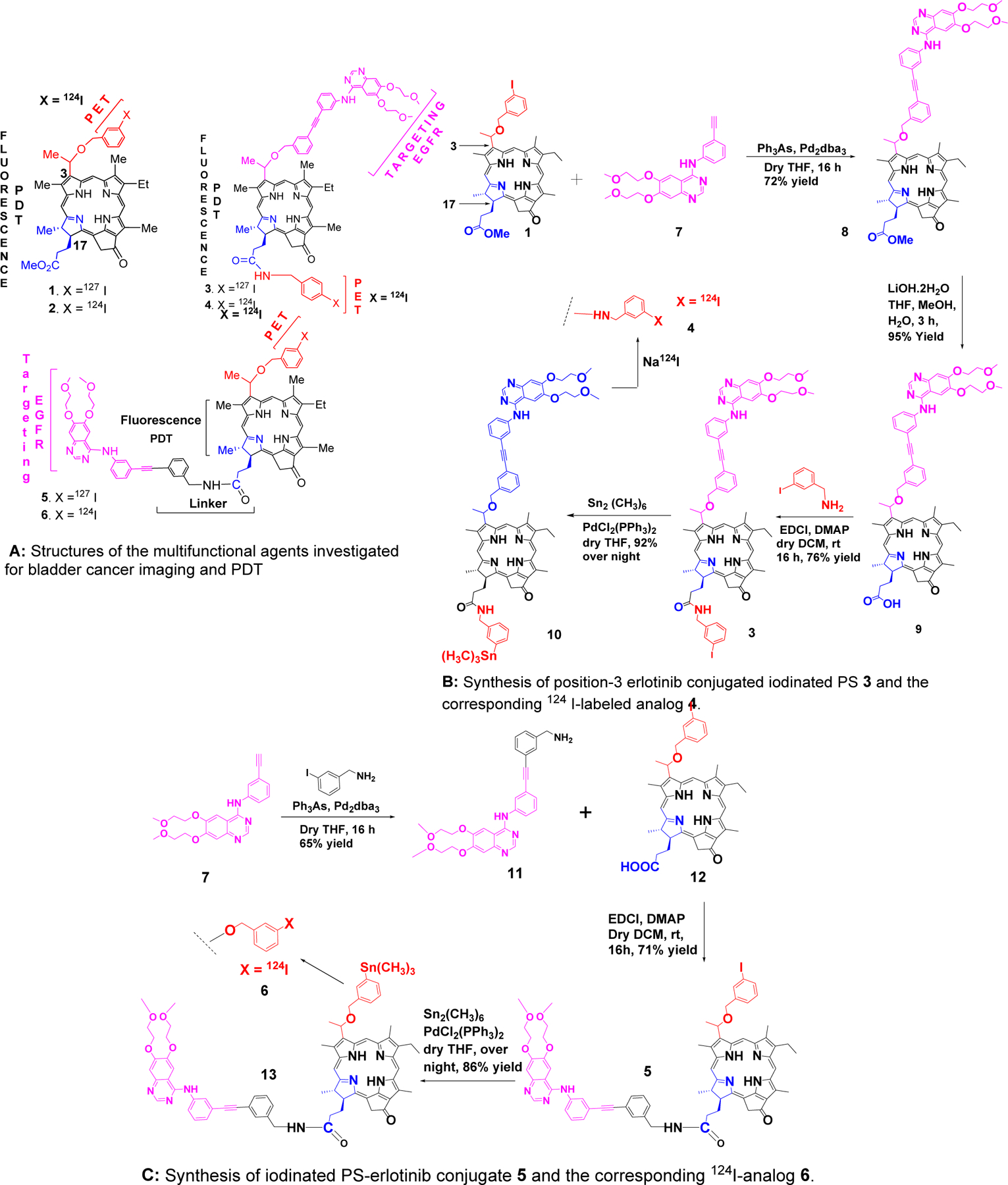
Three groups of photosensitizer protons – 10-H, 12-CH3 and one of the 132-CH2 group protons – exhibit substantial shielding in compound 5 relative to the chemical shifts observed in compounds 1 and 3 (Figure 1A). Each of these three groups is in close proximity to the macrocycle’s C-ring. The 10-H resonances of compounds 1 and 3 were observed at 9.54 and 9.34 ppm, respectively. In 5, however, 10-H appears at a much lower chemical shift (8.99 ppm). The 12-CH3 protons of 5 (2.33 ppm) are shielded by more than 1 ppm relative to both 1 (3.68 ppm) and 3 (3.43 ppm). In addition, one of the protons of the 132-CH2 group of 5 (4.56 ppm) exhibits a shielding of roughly 0.5 ppm relative to the 132-CH2 group protons of 1 (5.27/5.12 ppm) and 3 (5.20/5.05 ppm). These chemical shift data suggest that differences in molecular structure among these three compounds result in substantial changes in the electronic environment of specific protons, and that these changes are localized about the C-ring.
Figure 1.
A: 1H NMR spectral regions of 1, 3 and 5 show significant chemical shift differences for specific protons. In 5, protons in the vicinity of the photosensitizer moiety’s C-ring (10-H, 132-CHH, and 12-CH3) are significantly shielded with respect to the corresponding protons of 1 and 3. Also, 5-H of the erlotinib moiety of 3 exhibits a substantial shielding compared to the 5-H chemical shift observed in 5. These observations suggest structural differences that are dependent on the erlotinib orientation in the conjugates (See text for details).
B: 1H NMR spectra of 5 at 28 °C (bottom) and 55 °C (top). At elevated temperature, the chemical shifts of the 10-H (open triangle), 131-CHH (filled triangle), and 12-CH3 (arrow) proton signals exhibit a marked deshielding towards the positions observed for the corresponding protons of 1 and 3. This behavior is consistent with the breaking of pi-pi stacking interactions that may be responsible for the unusual shieldings observed for these protons in 5. (See figure in supporting information for additional spectra taken at other temperatures.)
The shieldings are observed only when the erlotinib moiety is linked through the 17-position of the photosensitizer macrocycle. Linkage through the 3-position does not yield similar shieldings. The relative flexibility of the linkers may play a role here. When linked through the 17-position (as in 5), the linker is longer and presumably more flexible than that of the 3-linked conjugate (PS 3). The greater flexibility may allow a conformation in which one or more of the aromatic rings of the erlotinib moiety interacts with the protons around the C-ring. Pi-pi stacking may render such a conformation more stable. Alternatively, the changes in chemical shifts might be due to differences in intermolecular stacking of macrocycle and/or erlotinib aromatic ring systems. ROESY data, however, showed no cross peaks indicating close proximity of these aromatic systems. This may be due to an unfavorable orientation of the stacked rings, which could place any potentially interacting protons too far from each other to generate observable NOEs.
Both stacking hypotheses agree with the 1H NMR spectra acquired on 5 at elevated temperature. These data (Figure 1B) exhibit substantial deshielding (relative to 1H deltas obtained at lower temperature) of the 10-H, 12-CH3 and 132-CHH protons as the temperature increases from 28 °C to 55 °C. So, all three groups of protons exhibiting shielding in 5, relative to the corresponding protons in 1 and 3, behave in a manner that is consistent with stacking of aromatic systems. A breaking of the pi-pi stacking is expected if energy is added to the system by increasing the temperature.
Chemical shift changes can arise from a variety of sources. These include differences in pH and analyte concentration, as well as temperature. In fact, 1H NMR spectra collected at 28°C using two different samples of 5 exhibit such changes (Figures 1A and 1B). These differences in chemical shifts, however, are relatively small compared to those obtained by changing temperature. The temperature dependence of the chemical shifts of the 10-H, 12-CH3 and 132-CHH protons is not only relatively large, it is also regular, and reversible (see supporting information, Figure 1, page S2).
A further chemical shift difference was observed for the 5-H resonance of the quinazoline ring system of the erlotinib moiety (Figure 1B). In 5, 5-H resonates at 7.82 ppm, similar to the chemical shift observed in unconjugated erlotinib analogs such as 11 (5-H δ = 7.65 ppm). In 3, however, this proton is shielded by more than 0.5 ppm, exhibiting a chemical shift of ~7.1 ppm. This chemical shift change may be due to differences in the orientation of the phenyl ring bound to the amino nitrogen at quinazoline C4. An orientation that places this ring near 5-H of erlotinib could result in the observed deshielding (in 5, relative to 3) due to ring current effects. On the other hand, rotation about the exocyclic C4–N bond can give a much different conformation, where the phenyl ring is nearer the quinazoline N3. In this arrangement, the phenyl group is far from 5-H and can impart no ring current induced deshielding at that site. This could account for the more shielded position of the 5-H resonance observed in 3.
Finally, these spectra differ in still another way. The frequency differences (Δν’s) between resonances of corresponding protons of the two diastereomeric forms of 3 are larger than those observed in 5 and 1. The 5-H (meso) proton of the photosensitizer moiety of 3 exhibits the largest Δν (60 Hz). This is much larger than that observed in either 5 (13 Hz) or 1 (0 Hz). The pyrimidine hydrogen (2-H of the quinazoline ring system) of 3 also displays a large Δν (45 Hz). In contrast, 5 shows no difference between the 2-H proton frequencies of its two diastereomers. The photosensitizer ring methyl proton resonances of the two diastereomers of 3 are also more widely separated (with Δν as large as 20 Hz) than those observed in either 5 or 1. Other protons show similar, though much smaller differences in Δν. In every case, the separation of the resonances of the two diastereomers is largest in 3, where the erlotinib is conjugated through the 3-position of the photosensitizer. The 3-position substituent also contains the stereogenic center responsible for the existence of diastereomers of these compounds. Conjugation of erlotinib at this point on the macrocycle places a large substituent with several (potentially interacting) functional groups in a location (i.e., on the chiral center) that allows different configurations, and these may generate large differences in conformation. This may be the source of the larger Δν’s seen in the NMR spectrum of 3. See the supporting material information (Figures S1-S15) for the NMR spectra of compounds 3,5, 8–11 and 13.
(C). Photophysical properties and singlet oxygen generation abilities of the photosensitizers with and without erlotinib moieties:
Photodynamic therapy is a three component system, which includes a photosensitizer (PS), light of an appropriate wavelength, and oxygen. In general, most PS are non-toxic, but upon light exposure the PS present in tumors is excited, and the excited state of PS on intersystem crossing releases the energy, which converts the oxygen present in the tumor to singlet oxygen which is responsible for tumor destruction, mainly by vascular shutdown. Therefore it is of utmost importance to determine the photophysical properties of the photosensitizer, including the singlet oxygen generation ability.
(C-1). Steady-state absorption and fluorescence spectra in toluene:
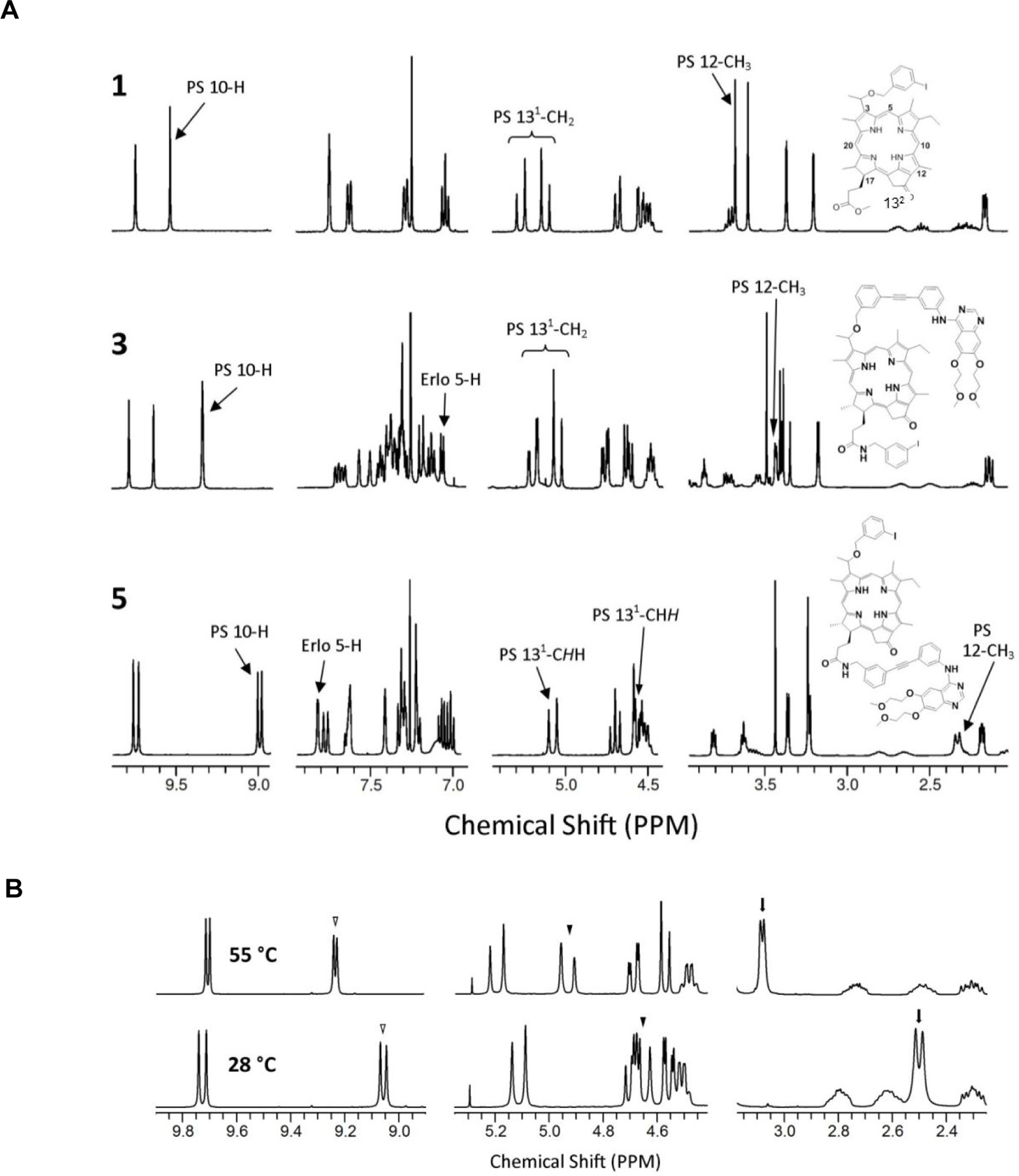
The absorption spectra of the investigated chlorin compounds are shown in Figure 2. As seen, the absorption spectrum of unsubstituted chlorin 1 exhibited a strong absorption for Q-band (at 665 nm) and Soret band at 411 nm. In addition, weak absorption bands were recorded at 317, 505, 535 and 607 nm. chlorins 3 and 5, showed spectral features similar to those of unsubstituted 1. The difference is the small red shift of the Q- and Soret bands (~2 nm) indicating weak ground state interaction between the ring and the two substituent moieties. In addition, weak absorption bands of the substituted moieties were recorded at 333 and 347 nm.
Figure 2:
Photophysical properties of photosensitizers with and without erlotinib moiety
The photochemical behavior of chlorin derivatives was investigated, first by using steady-state fluorescence measurements in toluene by using 610 nm excitation light. As shown in Figure 2, the emission spectrum of the unsubstituted chlorin 1 exhibited a strong emission band at 669 nm, and a shoulder at 713 nm. When turning to the erlotinib substituted PS 3 and 5, one can see similar spectra features with ~ 3 nm red shift compared to that of 1 indicating a considerable interaction between the ring and the substituted moieties. In addition, the spectrum of 5 showed a significant increase in the fluorescence intensity of the shoulder at 717 nm compared to 3. The fluorescence quantum yields in toluene were determined to be 0.176 (for 1), 0.183 (for 3), and 0.217 (for 5). Based on the fluorescence maxima, the energy of the singlet states was determined to be 1.85 eV (for 1) and 1.84 eV (for 3 and 5). As shown in inset Figure 2A (right), the fluorescence lifetime measurements in toluene were determined to be 6.96 ns (for 1), 7.24 ns (for 3), and 7.01 ns (for 5).
(C-2). Time-resolved transient absorption studies:
Femtosecond transient absorption spectroscopy was used to obtain further insight into the excited state of the examined chlorin compounds in deaerated toluene. Figure 2B (left) shows the differential absorption spectra of 1 upon excitation at 390 nm laser light. The strong bleaching of the band at ca. 670 nm revealed the instantaneous formation of the singlet excited chlorin, in addition to weak absorption bands at 485, 526, 553, 580, and 624 nm. By monitoring the decay of the 670 nm band with first-order kinetics, the rate of singlet decay and its lifetime were determined to be 1.0 × 109 s-1 and 1.0 ns, respectively. Similar spectral features were observed in the transient absorption spectra of the substituted chlorins 3 and 5 (Figures S16 & S17, supplemental information). By monitoring the decay of the 670 nm band, the rate constants and the lifetimes of the singlet-excited states were found to be 1.2 × 1010 s-1 and 83 ps (for 3) and 7.5 × 109 s-1 and 133 ps (for 5).
The complementary nanosecond transient measurements of chlorin derivative 1 in toluene with 420 nm laser excitation exhibited the absorption band in the visible region with a maximum at 450 nm (Figure 2B, right) that is assigned to the triplet-excited state of chlorin. The decay of the triplet excited state obeyed first-order kinetics and the decay rate constant of the triplet excited state of 3 in oxygen-free toluene solution was determined to be 6.24 x 104 s–1. The finding that the decay getting much faster in the presence of oxygen (1.96 x 106 s–1) suggests the energy transfer from the triplet state of 3 to oxygen (with a rate constant of 9.8 x 109 M–1 s–1) generating the singlet oxygen. When turning to the substituted derivatives 3 and 5, similar spectral features were observed (Figures S18 and S19). The triplet excited state decayed in Ar-saturated toluene solutions with the rate constants of 4.27 x 104 s–1 (for 3) and 9.8 x 104 s–1 (for 5). In oxygen-saturated solution, the triplet excited states decayed with the rate constants of 6.34 x 105 s–1 (for 3) and 2.07 x 105 s–1 (for 5). From these values, the rate constants of energy transfer from the triplet chlorins to oxygen were determined to be 3.17 x 109 M–1 s–1 (for 3) and 1.03 x 1010 M–1 s–1 (for 5).
As shown in Figures 2B and supporting information figures S19 and S20, the decay of the triplet excited states of chlorin derivatives in toluene was much accelerated on addition of oxygen because of the energy-transfer process from the triplet excited chlorins to oxygen generating the singlet oxygen. In order to determine the efficiency of singlet oxygen production of the examined chlorin derivatives, which is a necessary requirement to utilize PDT, two methods are generally used. The first method is to measure singlet oxygen generation such as photooxidation of the chemical quenchers for singlet oxygen, while the second method is to measure directly photoluminescence of singlet oxygen at 1274 nm33,34. The ability of chlorin derivatives to generate singlet oxygen was confirmed here by directly observing the characteristic emission band of generated singlet oxygen in the NIR region. As shown in Figure 2C, the photoluminescence (at 1274 nm) characteristic of singlet oxygen was observed in toluene. By comparison with fullerene C60 (the quantum yield of singlet oxygen generation in toluene = 0.76) used as a standard33–36, the quantum yield of the singlet oxygen generation in toluene was determined to be 0.60 (for 1), 0.59 (for 5), and 0.57 (for 3). These findings suggest relatively high efficiency of chlorin derivatives to generate singlet oxygen in less polar toluene. Slightly lower efficiency of 3 compared to other two derivatives coincides with the biological activities. From Figure 2 and the Supplemental Information Figures S17-20, the decay profiles of the singlet oxygen at 1274 nm were fitted with an exponential decay at a long time scale (up to 250 μs), from which the lifetime of the singlet oxygen was determined to be 29.06 μs (for 1), 29.13 μs (for 3), and 28.92 μs (for 5). In general, these values are close to the reported lifetime values of singlet oxygen in toluene.
(C-3). Impact of solvent on absorption and fluorescence intensities of PS 1 and erlotinib conjugates 3 & 5:
The PS with and without erlotinib moieties are water insoluble, and were formulated in 1% Tween80. The formulations of PS 1–3 were diluted to a concentration of 0.75 μM in methanol and in 17% FBS (v/v) DPBS (for mimicking physiological conditions). Before measuring the impact of solvents on fluorescence characteristics, absorption spectra at similar concentrations (0.75 μM) were obtained. The results illustrated in Figure 3 indicate that photosensitizers 1, 3 and 5 with and without erlotinib showed similar absorption bands (scale: 300–800 nm), but with reduced intensities in 17% FBS formulation. The fluorescence characteristics of the PS were determined using the same concentrations by exciting the absorption band at 415 nm, and recording the emission intensities in the 600–750 nm range. These are summarized in Figure 3. Among the compounds investigated, PS 1 gave the highest fluorescence emission intensity in both the solvents. However, it was reduced significantly for the erlotinib analogs 3 and 5 compared to the non-erlotinib compound 1, which could be due to differences in self-aggregation in aqueous FBS formulation.
Figure 3:
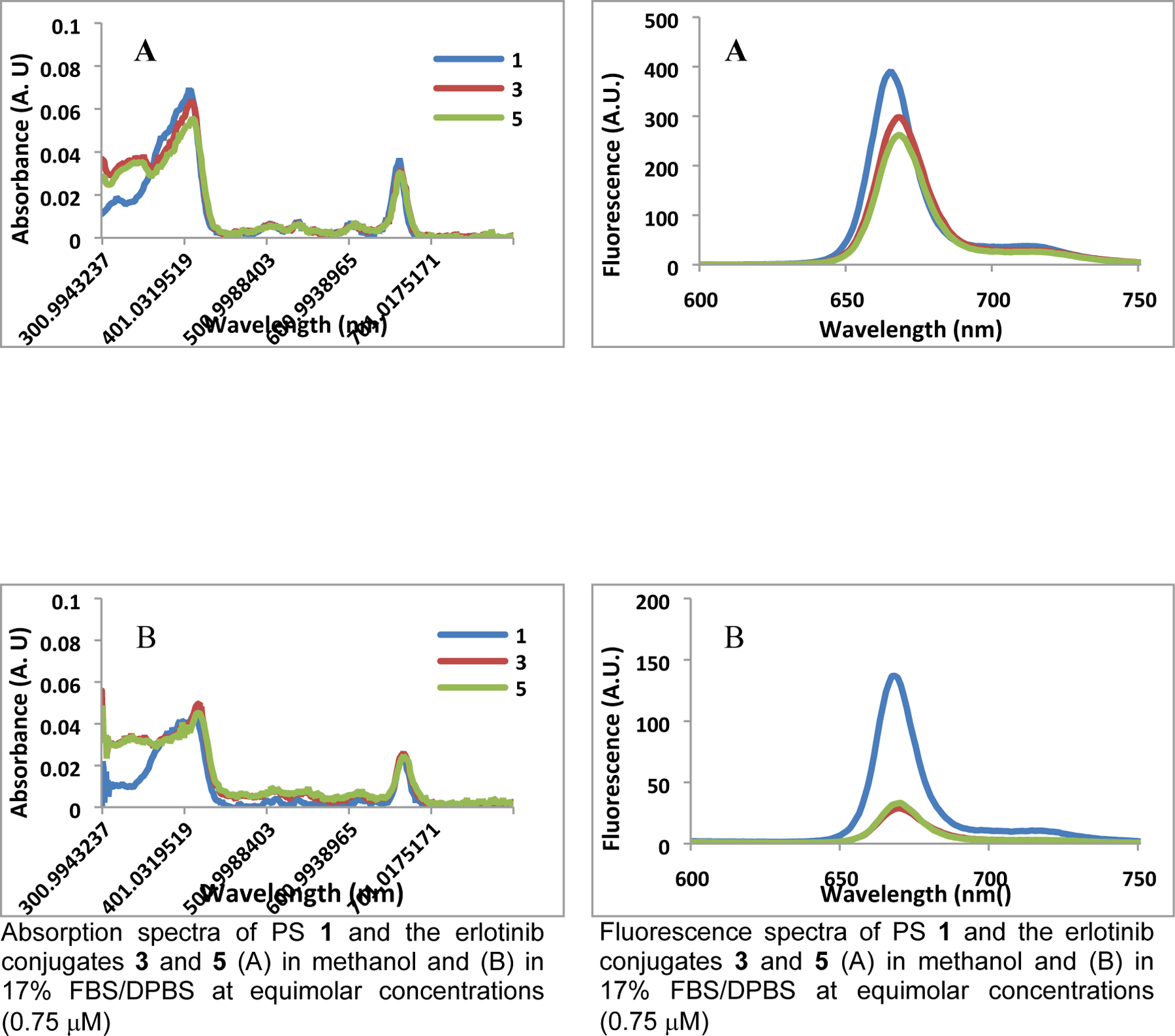
Impact of solvents in absorption and fluorescence spectra of photosensitizers with and without erlotinib moiety.
(D). Comparative biological activity of PS with and without erlotinib moiety
(D-1). Selection of formulation for the PS:
Most of the long-wavelength absorbing PS investigated in this study are derived from chlorophyll-a and are hydrophobic in nature, with limited solubility in water. Therefore, the selection of a suitable formulation(s) for in vitro/in vivo studies was extremely important. Formulation plays a critical role in the proper absorption of PS, its delivery, and release to the target. Increasing bioavailability within the system improves pharmacokinetics (PK). A proper PK profile can also reduce the side effects of the PS and can help in the reduction of overall toxicity.
Similar to most of the porphyrin-based compounds, PS 1–3 were water-insoluble and were formulated in 1%Tween80/5% dextrose solution and 2% Pluronic F-127/phosphate buffer37, and were evaluated for biological efficacy in both the formulations. with the aim to select the best formulation for the pharmacokinetic (PK) and toxicity studies of the desired PS, before moving it to Phase I human clinical trials. In both formulations, the PSs showed long-term stability (stored either at -4 °C or at -20 °C for two months) without any precipitation or change in pH.
(D-2). Comparative in vitro PDT efficacy of the PS 1 and the corresponding erlotinib analogs 3 & 5 in bladder cancer cell lines:
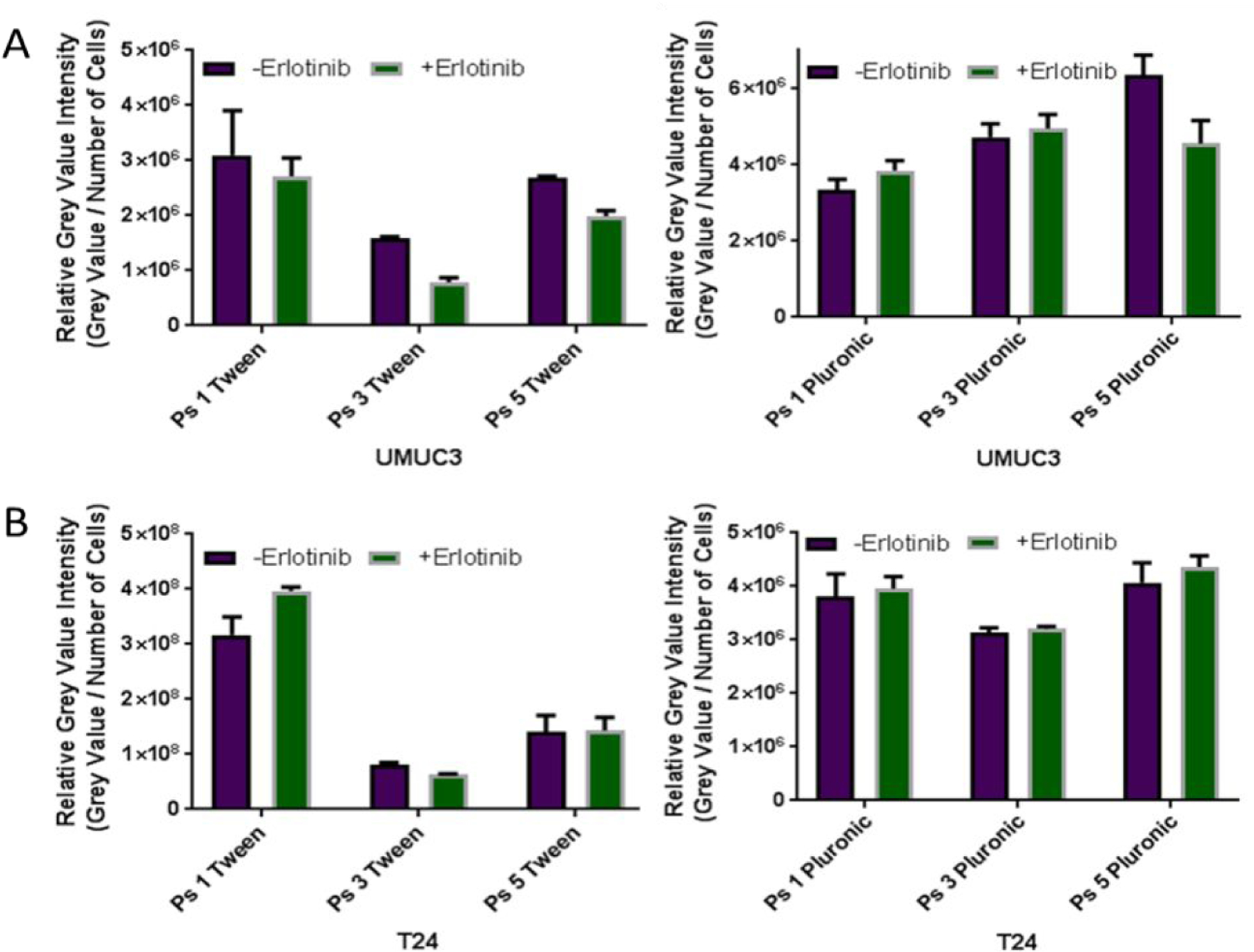
To compare cytotoxic potential of each compound, an MTT assay38,39 was conducted. In order to model EGFR target specificity, two cell lines were chosen: EGFR dependent cell line (UMUC3), and EGFR independent cell line (T24). The results are summarized in Table 2 and Figure 4. Briefly, cells were plated in 96 well plates at 1x104 cells per well and incubated with photosensitizer for 24 hours before being irradiated at peak absorbance (around 665 nm) for a total of 1 Joule. At 48 hours after light exposure, the cell viability was quantified. Interestingly, PS 1 (a non-erlotinib analog) showed similar photosensitizing activity in both the cell lines with low (T24) and high (UMUC3) EGFR expression (Figure 4A). However, under similar parameters, PS 3, in which erlotinib was introduced at the top half of the molecule, was less effective than 5, where it is conjugated at the lower half (Figure 4B). The Tween and Pluronic formulation of the PS investigated were effective in both cell lines with slight variations. Therefore, both formulations were investigated to determine their comparative tumor uptake in vivo.
Table 2:
Comparative in vitro PDT efficacy (IC50 values) of PS 1, 3 & 5
| Photosensitizer | IC50 µM | IC50 nM | ||
|---|---|---|---|---|
| UMUC3 | T24 | UMUC3 | T24 | |
| 1 (Tween) | 0.0952 | 0.0737 | 95.2 | 73.7 |
| 1 (Pluronic) | 0.1996 | 0.1775 | 199.6 | 177.5 |
| 3 (Tween) | 0.6083 | 1.026 | 608.3 | 1,026 |
| 3 (Pluronic) | 0.326 | 1.256 | 326.3 | 1,256 |
| 5 (Tween) | 0.0364 | 0.1369 | 36.4 | 136.9 |
| 5 (Pluronic) | 0.0584 | 0.3149 | 58.4 | 314.9 |
Figure 4:
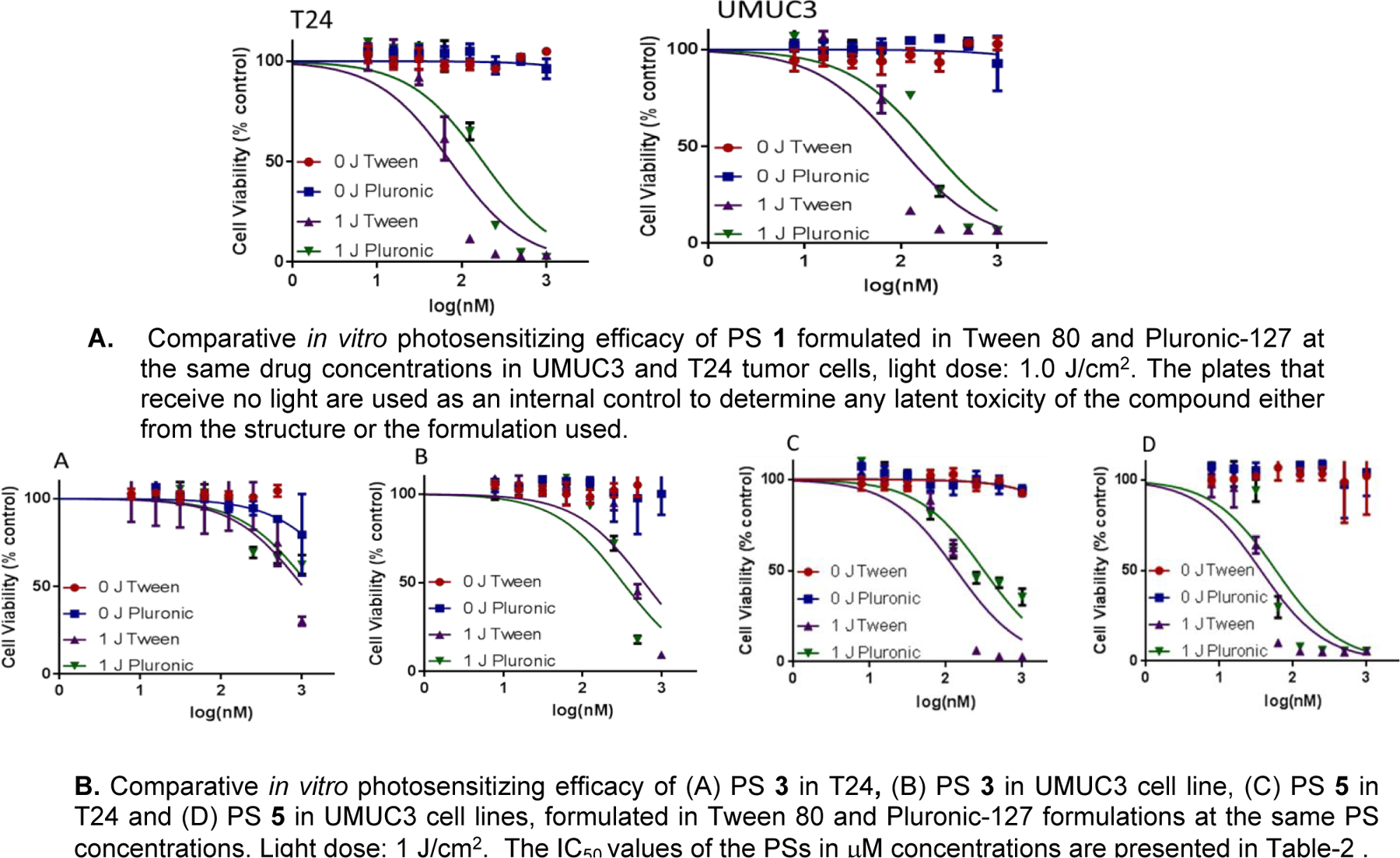
Comparative in vitro photosensitizing efficacy of the photosensitizers formulated in Tween80 and Pluronic formulations.
(D-3). Comparative cell-uptake and intracellular localization (mitochondria vs. lysosomes) of PS 1 and the erlotinib conjugates 3 & 5:
To investigate the impact of structural modifications on cell-uptake and intracellular localization ability of PS 1, 3 and 5 in UMUC 3 and T24 cell lines Mitotacker Red and FluoSphaeres were used to stain the mitochondria and the lysosome respectively40–42. The cell uptake of the PS in both formulations was determined by measuring the fluorescence intensity in tumor cells. Both PS 1 and the conjugate 3 showed similar uptake in UMUC3 and T24 cell lines, regardless of the delivery vehicle used. However, the PDT efficacy of 3 in UMUC3 (EGFR positive) was significantly higher than observed in T24 cells (EGFR low). This may be due to EGFR target-specificity of PS 3 as shown later in Figure 6. In contrast, PS 5 (erlotinib moiety introduced at position-17 of the PS) showed significantly more fluorescence in UMUC3 cells known for higher expression of EGFR than T24, indicating some degree of target specificity. Furthermore, the nature of formulation vehicles (Tween 80 vs. Pluronic) drastically produced a significant difference in the uptake of all of the compounds. Among the compounds investigated PS 5 in Pluronic formulation showed the highest uptake in UMUC3 cells.
Figure 6:
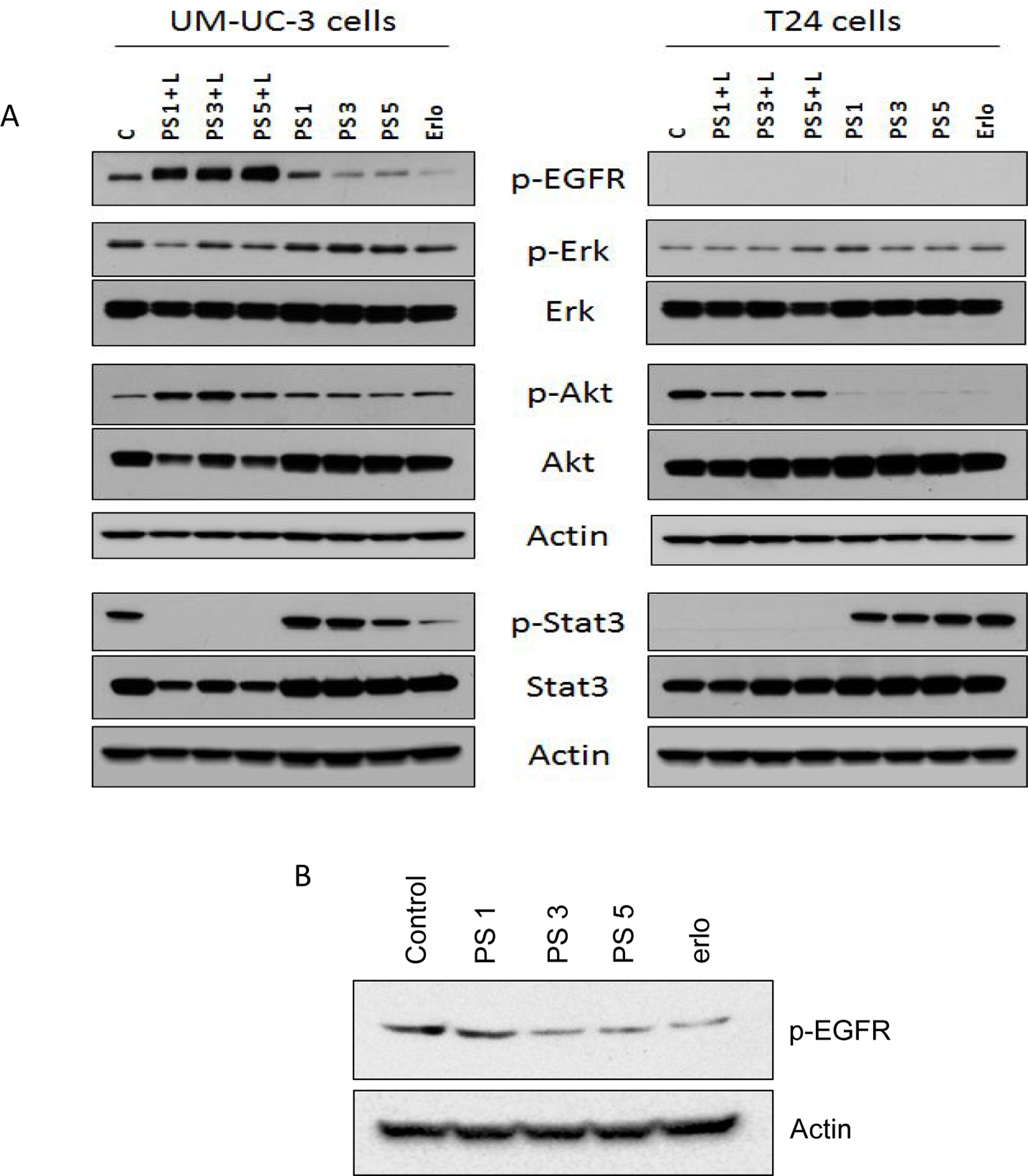
Comparative EGFR signal inhibition efficacy of erlotinib, PS 1 and the corresponding erlotinib conjugates 3 & 5 in bladder cancer cell lines UMUC-3 and T24 cell lines known for high and low expression of EGFR. (A) PS-erlotinib conjugates were treated at 1 µM concentration for 24 hours followed by PDT. (B) UM-UC-3 cells were treated with PS-erlotinib conjugates at 100 nM concentration for 24 hours.
To investigate the impact of erlotinib moiety in intracellular localization of the PSs, two different dyes that are known to localize in the mitochondria (Mitotracker Red) and the lysosome (Lysosphere green) respectively were used to confirm the site specificity. In both cell lines (UMUC3 and T24), the photosensitizers formulated in Tween80 and Pluronic showed localization in mitochondria and in lysosomes (Figure 5 C-F).
Figure 5:
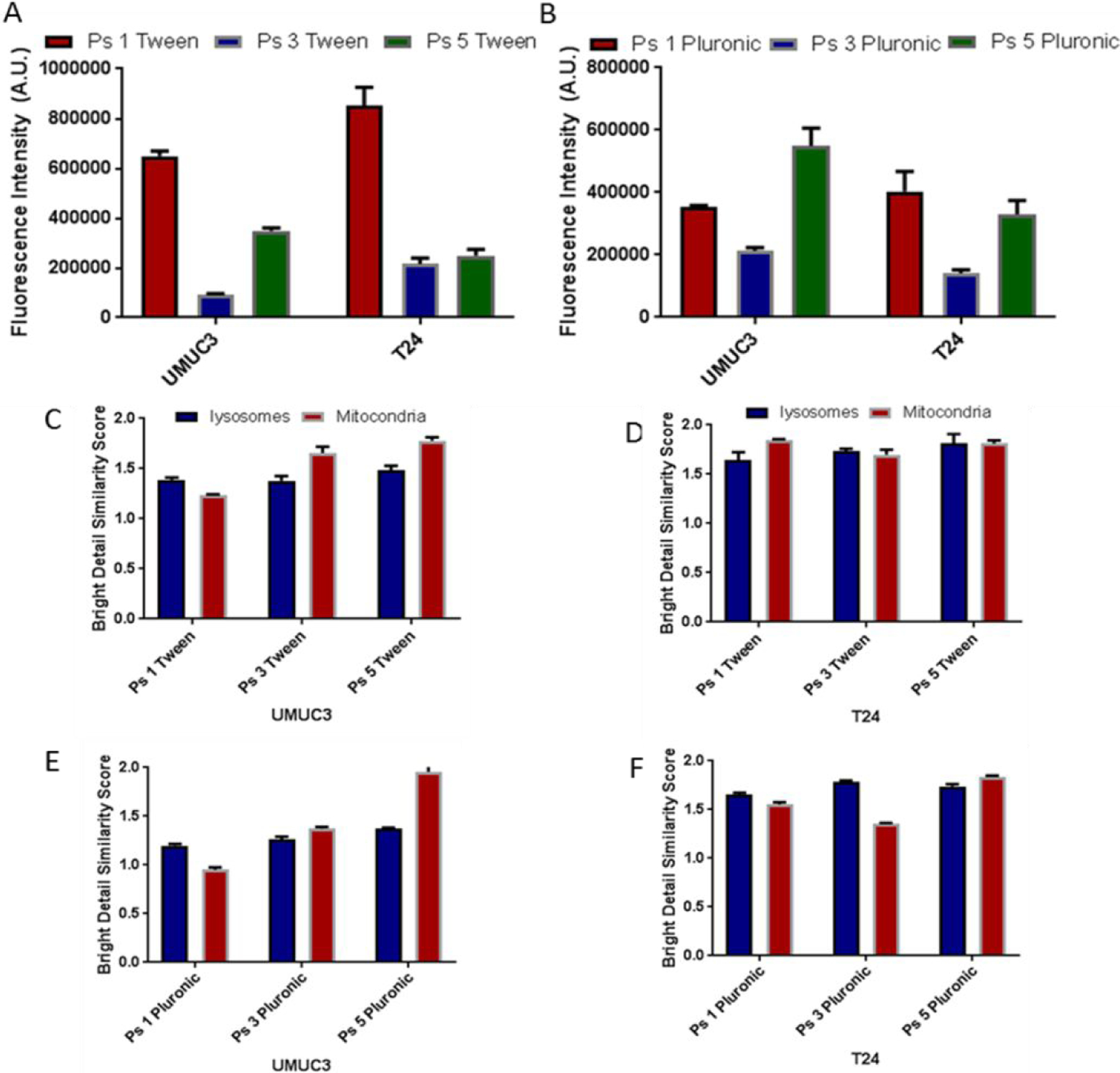
Comparative uptake and intracellular localization (mitochondria vs. lysosomes) of PSs 1, 3, 5 formulated either in Tween80 or Pluronic127F in UMUC3 and T24 cells. The uptake PSs in Tween (A) and Pluronic (B) after 24 h incubation, and the fluorescence was measured using a CARY fluorimeter. The degree of fluorescence in each cell was directly compared as a measure of uptake. The fluorescence of each cell was measured using an Image Stream. Simultaneously, cells were stained with lysosphere green and mitotracker red to determine the subcellular localization specificity the PSs: (C) PSs 1, 3 & 5 Tween80 formulation in UMUC3, (D) PS 1, 3 & 5 Tween formulation in T24, (E) PSs 1, 3 & 5 Pluronic formulation in UMUC3 and (F) PS 1, 3 & 5 Pluronic formulation in T 24 cell line. The higher bright detail similarity score to a particular site reflects stronger overlap of the PS fluorescence to that site, either lysosomes or the mitochondria.
(D-4). Compared to PS 1, EGFR conjugates PS 3 and 5 showed EGFR target specificity:
To demonstrate that the PS-erlotinib analogs are effective against tumor cells that express and rely upon EGFR activation, EGFR dependent bladder cancer cell line, UMUC3, was treated with PS 1, 3 and 5 for 30 hours and analyzed the levels of p-EGFR in whole cell lysates through Western blotting using phosphorylated-specific antibody. Our results showed that except compound 1 (non-Erlotinib conjugate) the erlotinib conjugates 3 and 5 were able to inhibit the auto-phosphorylation of EGFR at Ty1068 residue and modulated cellular signaling (Figure 6).
The overexpression of p-EGFR in presence of PS and light may be in response to robust cell death induction as shown in Figure 7. There are several reports available which suggest that EGFR phosphorylation in response to external stimuli can leads to cell death in several cell types43,44. We also observed inhibition in EGFR phosphorylation at Ty1068 residue following PS 3, PS 5 and erlotinib treatment at 100 nM dose to UM-UC-3 cells for 24 hours, suggesting that the PS-erlotinib conjugates are showing specificity towards EGFR.
Figure 7:
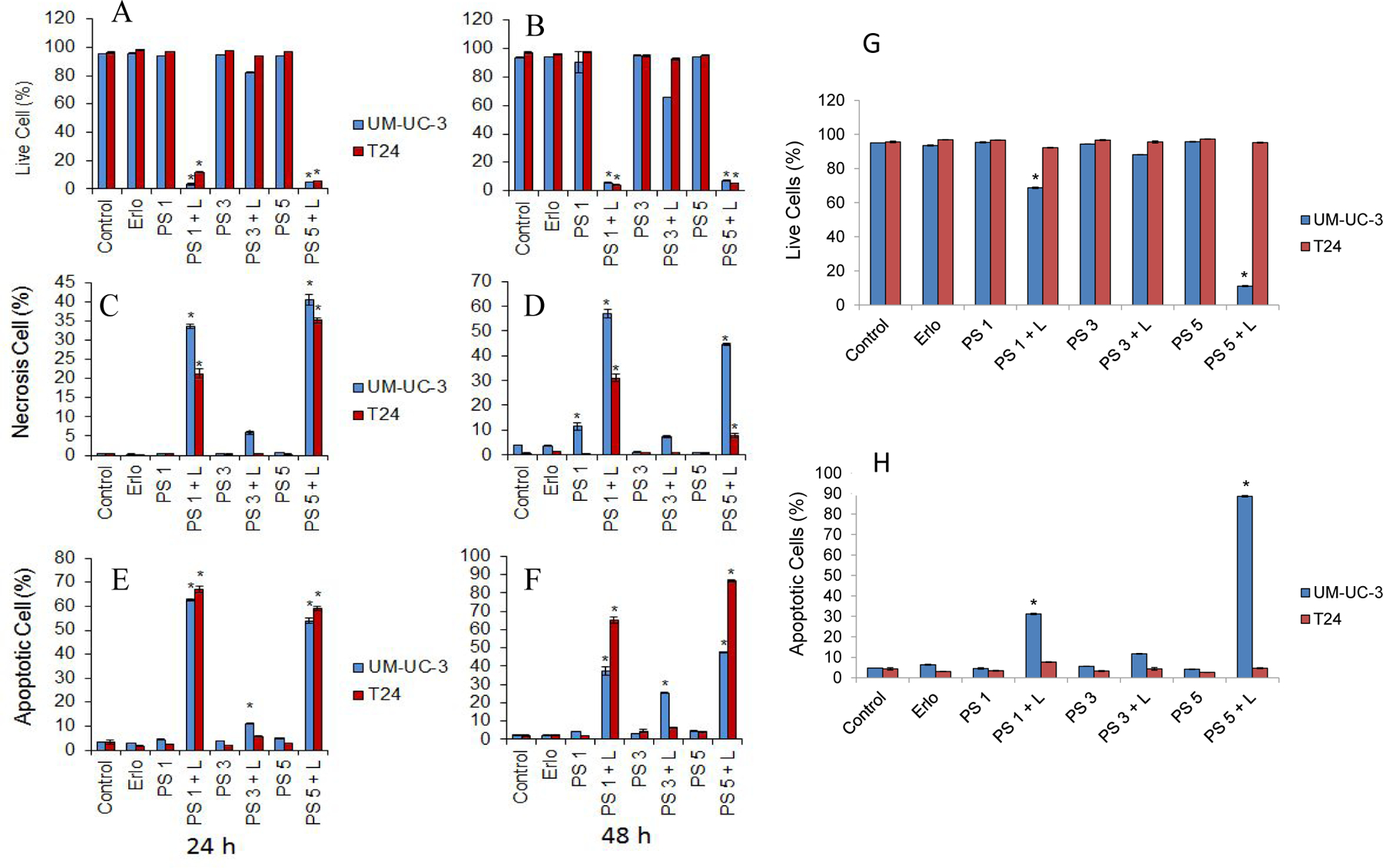
Erlotinib conjugates induce robust apoptotic and necrotic cell death upon PDT. UMUC-3 and T24 cells were treated with different erlotinib conjugates (1.0 μM) with and without PDT. Cell death was quantified using trypan blue assay (A and B). Necrotic (C and D) and Apoptotic (E and F) cell death was quantified using Annexin V/PI labeling. Live cells and Apoptotic cell death was quantified in UM-UC-3 and T24 cells treated with erlotinib conjugates (100 nM) with and without PDT (G and H).
To understand the molecular insights on cell death in response to PS-erlotinib conjugate in UMUC3 and T24 cells with or without PDT exposure, we quantified cell death using Annexin V/PI staining. In contrast to PS–erlotinib conjugate 3, PS 1 and the erlotinib conjugate 5 induced significant apoptotic and necrotic cell death upon PDT in UMUC3 and T24 cells. We also analyzed apoptotic cell death in these two cells following PS 1, PS 3 and PS 5 treatment at a lower concentration of 100 nM with or without PDT and we found that even at a lower dose of 100 nM, PS 1 and PS 5 produced significant induction in apoptotic cells and therefore cell death in only UMUC-3 cells with no effect on T24 cells upon light exposure (PDT). However, none of the PSs induced any cell death without exposing them to light of an appropriate wavelength, further confirming the potential therapeutic benefits and safety of these compounds.
(D-5). Mechanistic difference between PS1 vs PS 3 and PS 5: competitive uptake and ABCG2 efflux:
One major barrier of therapeutic efficacy for PDT is the photosensitizer’s efflux from the cell. Many cancer cells overexpress ABCG2 and certain chlorophyll-a derivatives, e. g., HPPH and PS 1 are reported as substrates for ABCG218,19. However, Erlotinib has been shown to not be effluxed by ABCG245. To investigate the influence of erlotinib in PSs (with and without conjugated with erlotinib moiety) as substrates for ABCG2 transport, tumor cells were co-incubated with or without Gleevec (5 µM), with photosensitizer 1, the erlotinib conjugates 3 and 5 (500 nM), and their relative cell-uptake was measured. Interestingly in UMUC3 cells, the addition of Gleevec significantly increased its uptake of PS 1 but no significant difference was observed with conjugates 3 and 5 (Figure 8). This observation is interesting as it could help in selecting an effective treatment option to cancer patients with and without expression of ABCG2 transporter. To further confirm the EGFR target-specificity of the erlotinib-PS conjugates, the tumor cells [UMUC3 (EGFR +) and T24 (EGFR -)] were first incubated with erlotinib for 24 h, before adding the PSs 1, 3 and 5 formulated in both Tween and Pluronic formulations, and the cell uptake was measured. The data summarized in Figure 9 indicate that both PSs 3 and 5 showed lower cell uptake than 1 in UMUC3 cells incubated with erlotinib. As expected, no such difference in cell uptake between the PSs with and without erlotinib moiety was observed in T24 cells, known for negative EGFR expression.
Figure 8:
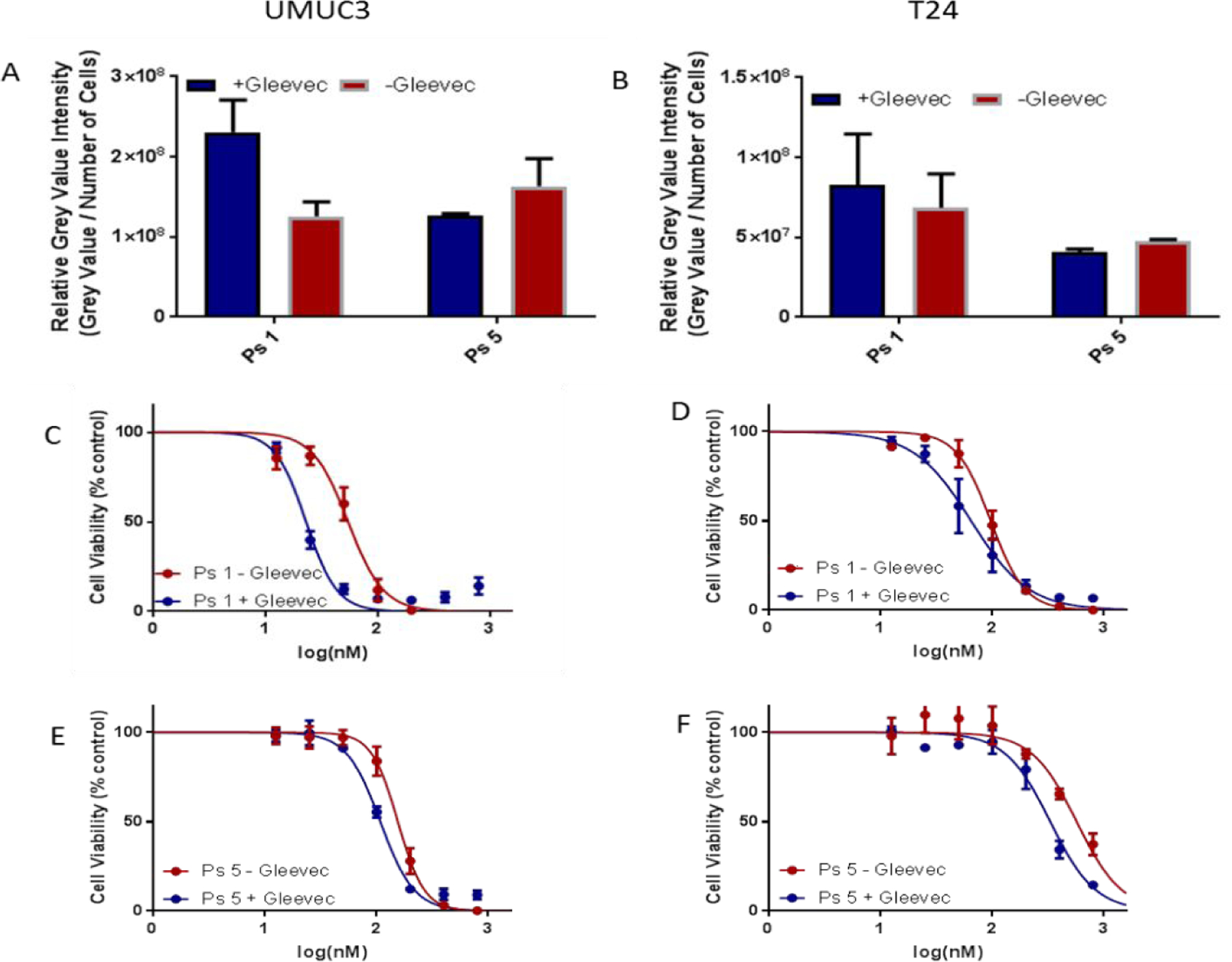
ABCG2 substrate specificity of PS with and without Erlotinib in UMUC3 and T24 cells. (A & B)) the uptake of PS 1 and 5 with and without the addition of Gleevec (small molecule inhibitor known to inhibit ABCG2 function) in UMUC3 and T24 cells. Cells were incubated with 500 nm of the Photosensitizers with or without the addition of Gleevec at 1uM concentration. (C-F) The effect of Gleevec on the toxicity of PS 1 and PS 5 at 48 h post incubation (light dose: I J).
Figure 9:
Erlotinib competition with PSs 1, 3, and 5 in UMUC3 and T24 cells. UMUC3 (A) or T24 (B) cells were incubated with photosensitizer at 1 uM or Erlotinib at 3 uM for 4 hours. The cells were washed with warmed PBS three times then imaged using a Zeis Fluorescence microscope.
In vivo tumor uptake, PDT efficacy and PET imaging of PS with and without erlotinib conjugates:
(E-1). Whole body in vivo fluorescence imaging and uptake of PS 1, 3 and 5 in SCID mice bearing T24 and UMUC-3 tumors:
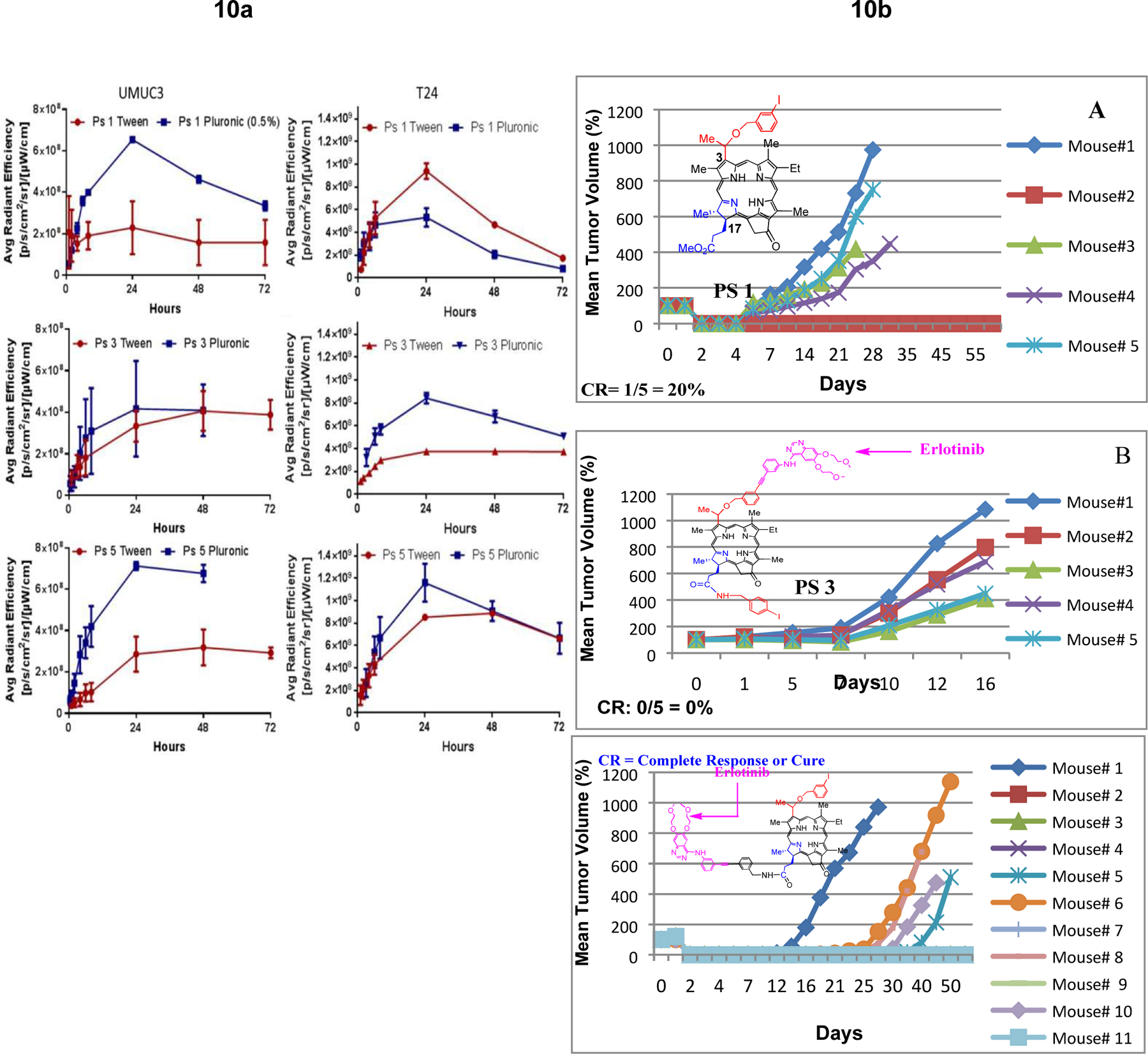
The main objective of this particular study was to investigate the impact of Tween 80 and Pluronic formulations in tumor uptake of PSs at various time points. The SCID mice (3 mice /group) bearing either UMUC3 or T24 tumors (4–5 mm size) were injected with the PS 1, and the corresponding erlotinib conjugates 3 and 5 (0.47 μmole/kg) in both Tween80 and Pluronic F-127 formulations. The tumor uptake was measured by IVIS system and interesting pharmacokinetic profiles of the PSs were observed (Figure 10a). For example all the PSs formulated in Pluronic showed higher tumor uptake in both UMUC3 tumor at 24 h post-injection, with most significant difference observed for the erlotinib conjugate 5 in which the erlotinib moiety was introduced at the lower half of the PS.
Figure 10.
a: Comparative uptake of PS 1, 3, 5 (formulated either in Tween or Pluronic) in tumor, liver and skin (SCID mice bearing UMUC3 and T24 tumors) at a dose of 0.47 μmol/kg)) at variable time points (λex: 675 nm, λem: 720), using the IVIS optical imaging system.
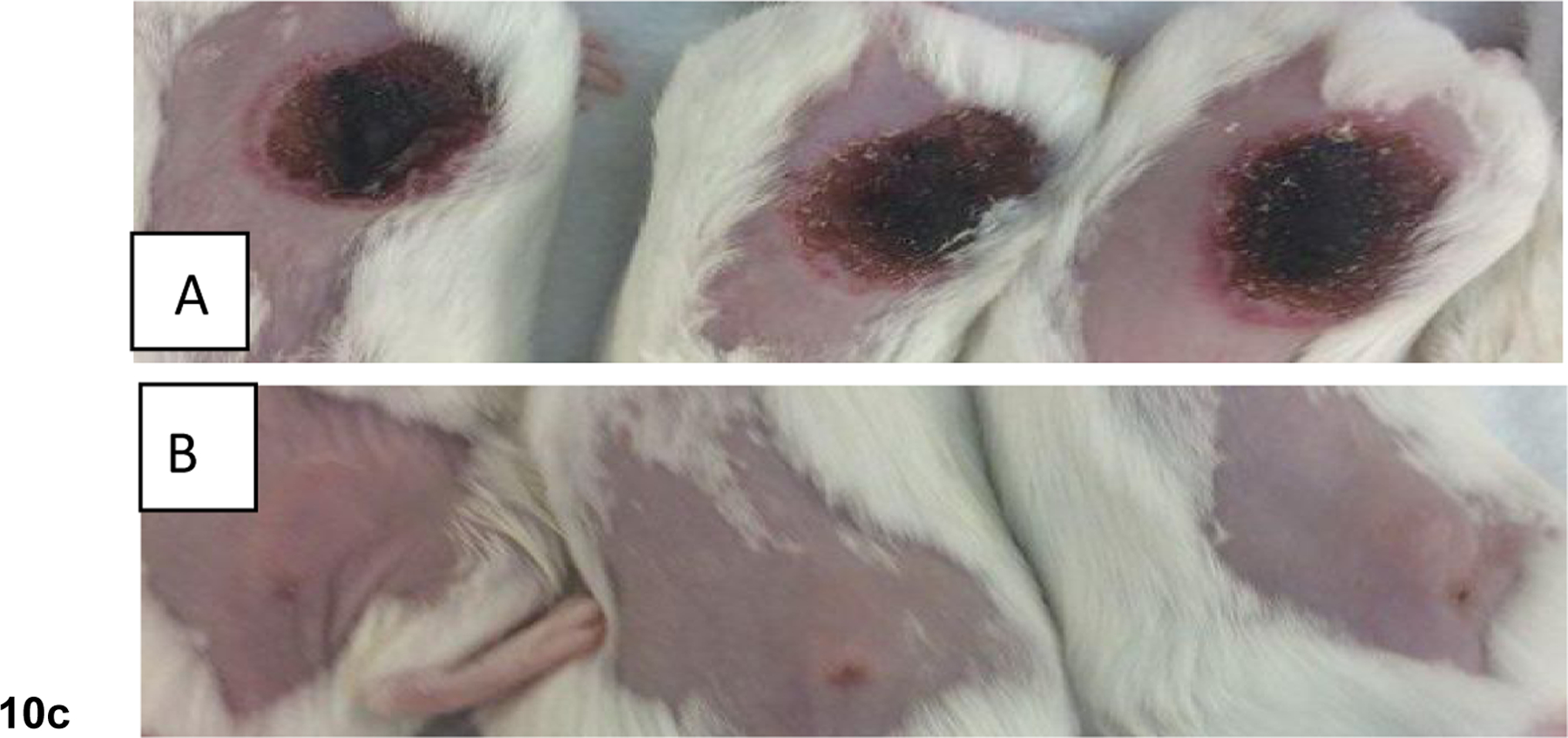
b: Comparative in vivo PDT efficacy (long-term antitumor activity) of PS with and without erlotinib conjugates. (A): PS 1 (non-erlotinib PS), (B): PS 3 erlotinib moiety is attached at position-3 of the PS (C): PS 5, erlotinib moiety is attached at position-17 of the PS. PS 1, 3, and 5 were individually injected (dose: 0.47 micromole/Kg) to SCID mice bearing UMUC-3 tumors at the flank. Mice were exposed to light (665 nm, 135 J/cm2, 75 mW/cm2) at 24 h post-injection (optimal uptake time) and tumor re-growth was monitored daily. These results suggest that position of the erlotinib moiety in the PS makes a remarkable difference in long-term tumor cure.
c: Comparative tumor necrosis of mice injected with (A) PS 3 (effective PDT agent), and (B) PS 5 that showed limited efficacy at 72h post-light exposure. Mice were treated under similar drug and light doses. Dug dose: 0.47 μmol/kg, light dose: 135 J/cm2, 75 mW/cm2 and the tumors were exposed to light (665 nm) at 24h post-injection of the PS.
(E-2). In vivo PDT efficacy of PSs 1, 3 and 5 in SCID mice bearing UMUC-3 tumors:
The focus of the study was to compare the in vivo efficacy of the PSs under similar drug/light treatment parameters. The treatment parameters were not optimized. The drug dose 0.47μmol/kg was selected on the basis of our previous study with PS 1 in other tumor types, where it showed moderate long-term anticancer activity at this dose. For our initial study UMUC-3 cells (1 x 109) SCID mice (5 mice/group) were implanted in the right flank of the mice, and when the tumor size was around 4–5 mm, the PS were intravenously injected. At 24h post-injection, the tumors were exposed to light, and tumor regrowth was measured daily. As per approved protocol, the mice with tumor >400 mm3 were euthanized. The initial results indicated that among the compounds investigated, PS 5 in which erlotinib moiety was present at the lower half of the PS showed the best efficacy. To confirm these results, PS 5 was again evaluated with a set of additional 6 tumored mice under similar treatment parameters, and the combined results from both sets of mice (6/11 mice were tumor free on day 60), are shown in Figure 10b.
(E-3): In contrast to PS 5, no tumor necrosis was observed with PS 3:
It has been shown by us and others that tumors containing effective PS on exposure with light show significant necrosis46,47 which is generally observed at 24h post-light exposure and lasts for a week or so. Interestingly, PS 5, which showed significant long-term PDT response (tumor cure) also produced tumor necrosis, whereas no such response was observed with PS 3 (Figure 10c), an ineffective PDT agent.
To understand the reasons for such a remarkable difference between the PDT efficacy of PS 3 and 5, the retention of the PS, singlet oxygen producing ability, overall hydrophobicity (HPLC retention time), intracellular localization and cell-efflux of the PS results were compared, but no significant difference was observed. The difference in antitumor activity (tumor cure) between the erlotinib conjugates 3 and 5 could not be explained only on the basis of the difference in their tumor uptake. Because, in spite of similar tumor uptake demonstrated by both PSs 1 and 5, the erlotinib conjugate 5 gave improved long-term tumor cure (20% vs. 55% cure on day 60). Therefore, besides higher tumor uptake of PS 5 over PS 3 in UMUC3 tumors its EGFR targeting ability could also be playing an important role in producing improved long-term tumor cure.
It is well-established that in PDT treatment the singlet oxygen, a reactive oxygen species (ROS) is a key cytotoxic agent for tumor destruction. Therefore, we measured the photophysical properties of the PSs in organic solvents (Table 1) and in aqueous formulation (with and without BSA, Figure 3), and it showed a significant difference in absorption and fluorescence characteristics. However, compared to the in vitro parameters used to study these PSs, the environment in living cells/tumors is quite different, which could have a significant impact in aggregation characteristics of the molecules, singlet oxygen production, and PDT efficacy. Several approaches to evaluate ROS production in vitro are currently being evaluated, and among these approaches, the use of carboxy-HDCFDA (2’, 7’-dichlorofluorescin diacetate), which is a non-fluorescent compound in the absent of ROS but it fluoresces in the presence of ROS, is of particular interest, and the fluorescence intensity directly correlates to the ROS production48. Therefore, we used this methodology to measure the ROS production of PSs 1, 3 and 5 in living cells.
Table 1:
Summary of the photophysical and photochemical data of PS 1, 3 and 5 in toluene
| 1 | 3 | 5 | |
|---|---|---|---|
| (nm) | 317, 411, 505, 535, 607, 665 | 301, 318, 333, 347, 413, 666 | 301, 333, 413, 507, 540, 609, 667 |
| (nm) | 669, 713 | 672, 717 | 672,718 |
| τf (ns) | 6.96 | 7.24 | 7.01 |
| Ф s | 0.176 | 0.182 | 0.217 |
| k (s-s) | 1.00 × 109 s−1 | 1.00 × 1010 s−1 | 7.50 × 109 s−1 |
| kT (s−1) | 6.24 × 104 s−1 | 4.27 × 104 s−1 | 9.80 × 104 s−1 |
| τT (μs) | 16 | 23 | 10 |
| Ф | 0.60 | 0.57 | 0.59 |
| τ ( 1 O 2 ) (μs) | 29.06 | 29.13 | 28.92 |
(E4). Comparative reactive oxygen species (ROS) of PSs 1, 3 and 5 in tumor cells;
To evaluate the ROS generation potential in vitro, UMUC3 cells were incubated with carboxy-H2DCFDA, and plated in a 96 well plate and incubated with 1 µM of each PS (1, 3 and 5). After 24 hours, 1 µM of DCFDA was incubated with the cells for 20 minutes. The fluorescence intensity of DCFDA significantly increased for both PS 1 and PS 5, and was about 2.5 and 4.5-fold higher from the background respectively, and it was only 0.9-fold higher for PS 3 (Figure 11). Such a dramatic change in DCFDA fluorescence intensity indicates that in a living system, the ROS production of 3 was relatively lower compared to PS 1 and 5, which directly correlates to their in vitro PDT efficacy.
Figure 11:
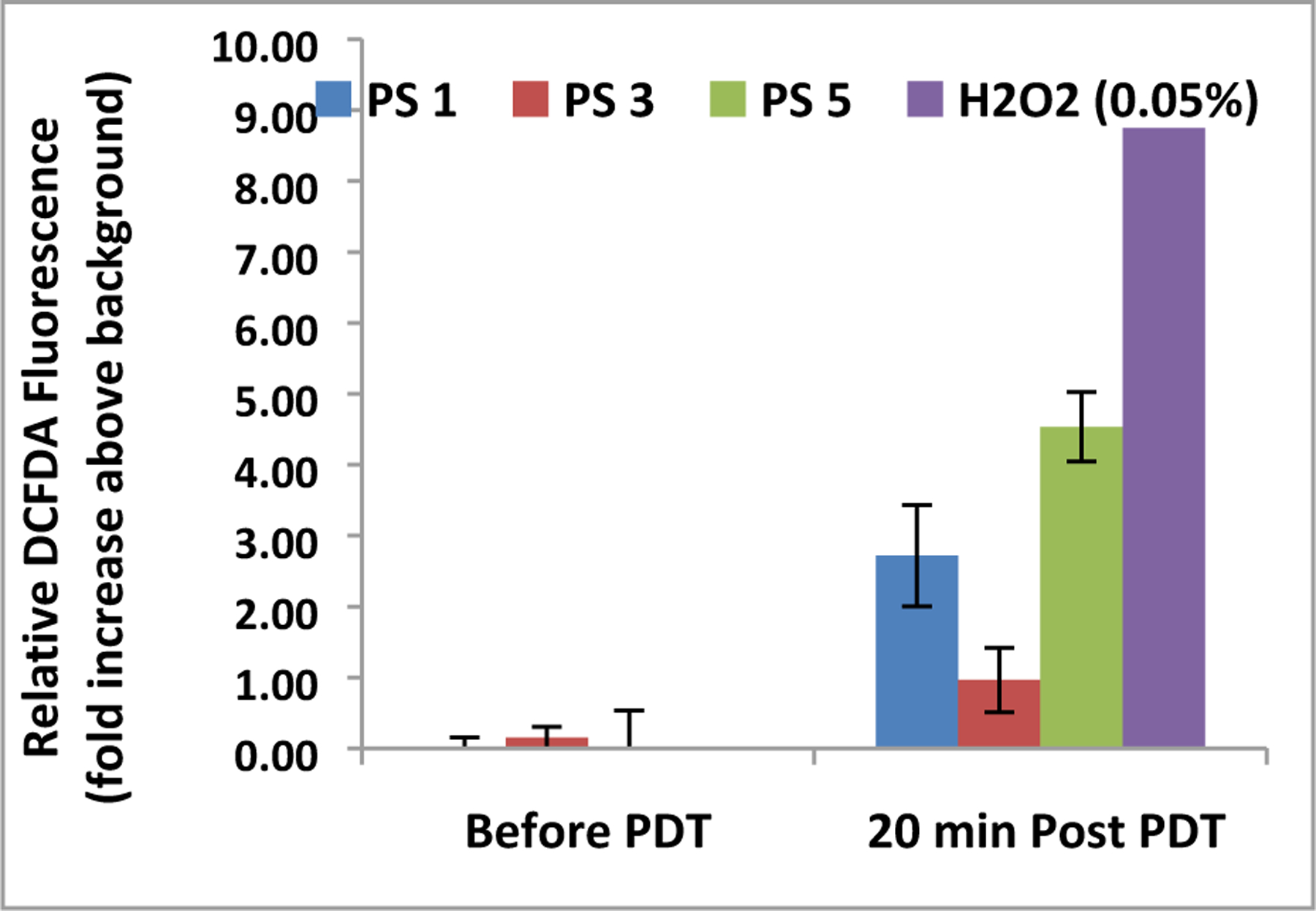
Comparative DCFDA fluorescence before and after exposure to 665 nm light. Plates were exposed to a total of 1 J of light (665 nm) at a rate of 3.2 mW/cm2.. Fluorescence value of cells with no DCFDA (background) was subtracted from before and after PDT. 0.05% H2O2 (30% stock was diluted to 0.18%, then 20 µL was added to each well) was added to 4 wells as a control before the plates were exposed to light. Photosensitizers were formulated in Tween80 formulation.
(E-5). Comparative bladder tumor imaging ability of erlotinib conjugate 124I-6 and 18F-FDG in SCID mice bearing UMUC-3 tumors:
Among the PET imaging agents, 18F-FDG is widely used to image a variety of cancers, but it shows limitations in imaging brain, kidney, prostate, pancreas and bladder cancers49. Due to short half-life (2 h) of 18F- radionuclide, its long-distance transportation is also a major issue. Thus it requires an expensive cyclotron facility at short distances for the production of the 18F-FDG. Therefore, efforts are underway in various laboratories, including ours to develop a PET imaging agent with high tumor avidity and longer half-life.
Due to high uptake/retention (24 to 72 h) of PS 5 in UMUC-3 tumors, and excellent long-term PDT efficacy in mice bearing UMUC-3 tumors, we converted it into the corresponding 124I-analog (half-life 4 days) and its PET imaging potential was investigated. In our initial approach, SCID mice bearing UMUC3 tumors (3 mice/group) were injected with 18F-FDG (50 μCi) and imaged at 110 min post-injection. At 24 h post-imaging, compound 6 (124I-analog of 5, 50 μCi) was injected (i. v.) and the mice were imaged at 24, 48 and 72h post-injection. The PET imaging experiments were repeated at lower doses (25 and 15 μCi) of both imaging agents. The results summarized in Figure 12 clearly demonstrate the superiority of PET-imaging agent 6 even at a dose of 15 μCi/mouse, and image acquisition recorded at 24, 48 and 72 h post injection. In contrast, 18F-FDG showed limited tumor imaging ability of UMUC3 tumors even at a dose of 50μCi/mouse.
Figure 12.
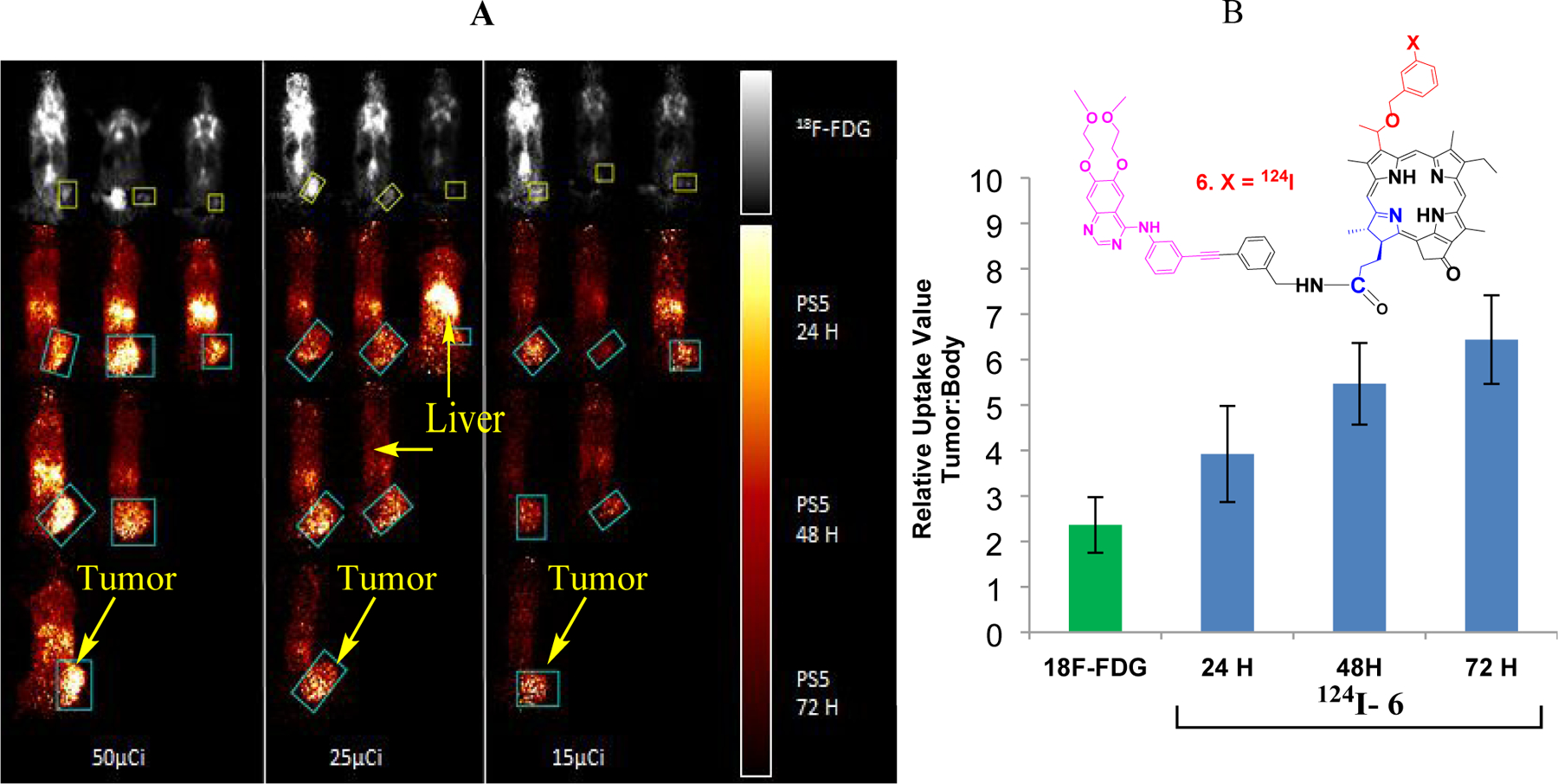
(A): PET images of SCID mice bearing UMUC3 tumors using 124I-6 and 18F-FDG. The images were analyzed using Amide Medical Imaging Software Boxes indicate Volume of Interest (VOI) in the coronal plane. Separate scale bars for 18F-FDG and 124 I-6 images are indicated on the right. The maximum threshold for color distribution was set to 10% in the 18F-FDG images and 50% in the 124I- 6 images. In each panel, the top row of mice show 18F-FDG uptake in the bladder, brain, chest and muscles, while tumor uptake is difficult to visualize. Tumors are well-visualized by 124I- 6 at alltime points, and uptake is primarily in the tumor and liver. 124I- 6 is retained in the tumor longer than in other parts of the body. (B): Relative Uptake Values were calculated as maximum tumor uptake divided by average whole body uptake. 18F- FDG (left) and 124I- 6 (right) RUVs indicate almost twice the specific uptake of 124I- 6 at 24 hours as compared to 18F-FDG. For II124 I- 6, the RUVs continue to rise at longer time points as it retains well in the tumor and less in the rest of the body.
The initial dose of radiation and the residual in the syringe were measured using a well gamma counter. Actual dose varied by no more than +/- 5% of intended dose. Mice were placed in the MicroPET at the same position for each round of imaging, with the bed set to the same height. All studies were acquired using the MicroPET Focus120™ (Concorde MicroSystems Inc.). Image analysis was performed using open source Amide Medical Imaging software (Sourceforge). The tumor was defined using an elliptical cylinder volume of interest (VOI) and the whole body VOI was defined using a 3D isocontour, as per the standardized Imaging SOP. The Relative Uptake Value (RUV) was calculated as maximum tumor value divided by average body value (Figure 12). The RUV enables comparison of specificity at earlier time points and retention at later time points.
Conclusion
The results presented in this manuscript show a remarkable difference in in vitro and in vivo PDT efficacy and photophysical properties of PSs with and without erlotininb moiety. The PDT efficacies of PSs 3 and 5 are significantly influenced by position of conjugation between the erlotinib and PS moieties. For example, PS 5, in which the erlotinib moiety was conjugated at position-17 of the PS, afforded far better efficacy than PS 3, in which the erlotinib moiety was attached at position-3 of the PS. Between the two PSs, no significant difference was observed in singlet oxygen yields (in organic solvent), EGFR targeting ability, cell uptake, intracellular localization, and mechanism of cell death (apoptosis vs. necrosis), but compared to 5, PS 3 showed significantly lower singlet oxygen production in biological environment. A significant difference in tumor-uptake, tumor necrosis after PDT, and long term PDT efficacy between the PS 3 and 5 were observed in vivo. The lower efficacy of PS 3, relative to 1 and 5, could also be due to its lower ROS production (singlet oxygen) in tumor cells. Further mechanistic studies to determine the factors responsible for such significant differences between PSs are currently underway.
Interestingly, the radioactive compound 6 (124I- analog of the most effective PDT agent 5) demonstrated excellent PET imaging ability of UMUC3 tumors implanted in SCID mice. In contrast to 18F-FDG (50μCi/mouse), the 124I-analog 6, even at a dose of 15μCi /mouse, showed excellent tumor contrast and imaging ability of UMUC3 tumors implanted in SCID mice at 24, 48 and 72 h post-injection (Figure 12). Due to a long half-life (4 days) of 124I- radionuclide, compound 6 in a single dose can also be used for monitoring the tumor response. In contrast to 18F-FDG (a current clinical standard, half-life 110 min), the 124I- analog can be shipped to long-distances, and could eliminate the necessity of having expensive cyclotrons at close distances of the imaging facilities. Besides, the requirements of 4 to 5- fold lower doses of 124I-analogs than the 18F-FDG in tumor imaging, the PET imaging with 124I- analog 6 should be more economical and widely acceptable. Due to the PS’s inherent fluorescence ability in the NIR region (abs: 665 nm, em: 715 nm), PS 5 (the non-radioactive analog of 6) showed excellent long-term tumor cure in SCID mice bearing UMUC3 tumors. Therefore PS 5 in combination with corresponding 124I-analog 6 provides an opportunity to image the bladder tumors by PET, and then treat them by fluorescence-guided PDT.
EXPERIMENTAL SECTION
Chemistry Methods:
All reactions were carried out in heat gun-dried glassware under an atmosphere of nitrogen and magnetic stirring. Thin layer chromatography (TLC) was done on pre-coated silica gel sheets (layer thickness 0.2m mm). Column chromatography was performed either using silica gel 60 (70–230 mesh) or neutral alumina grade III. In some cases preparative TLC was used for the purification of compounds. Purity of the compounds was ascertained by TLC and HPLC analysis. All compounds including the intermediates were >95% pure. UV-visible spectra were recorded on FT UV-visible spectrophotometer using methanol as a solvent. Mass spectrometry analyses were performed at the Mass Spectrometry Facility, University of Buffalo, NY.
NMR data were acquired at 28 °C (unless otherwise noted) on a Bruker Avance III HD NMR spectrometer equipped with a 9.4 T narrow-bore magnet, a 5-mm BBO Z-gradient probe, and Topspin 3.2 software. Observed frequencies for 1H and 13C observations were 400 MHz and 100 MHz, respectively. Chemical shifts were calibrated to the residual solvent resonance, and are reported relative to TMS at 0.00 ppm.
3-{1’-(3-[N-(3-ethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine]benzyloxy) ethyl} 17-(3-iodobenzyamine)-pyropheophorbide-a (3):
To a solution of compound 9 (50 mg, 0.0484 mmol) and 3-iodobenzylamine (16.90 mg, 0.0725 mmol) in 12 mL of dry dichloromethane, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydro chloride (EDCI, 18.53 mg, 0.0967 mmol) and 4-(dimethylamino) pyridine (DMAP, 11.81 mg, 0.0967 mmol) were added. The reaction mixture was stirred at room temperature under N2 atmosphere for overnight. It was then diluted with dichloromethane (40 mL), washed with water (3 x 50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 6% methanol in dichloromethane to obtain pure product 3 with 76% (45.90 mg) yield.1H NMR (400 MHz, CDCl3, δ ppm): 9.79/9.64 (1H, s, 5-H), 9.34 (1H, s, 10-H), 8.55/8.53 (1H, s, 20-H), 8.46/8.34 (1H, s, pyrimidine H), 7.71/7.67 (1H, m, phenyl H), 7.57/7.51 (1H, m, phenyl H), 7.45 (1H, m, phenyl H), 7.37–7.42 (2H, m, 2 x phenyl H), 7.34–7.37 (1H, m, phenyl H), 7.28–7.34 (3H, m, 3 x phenyl H), 7.21/7.18 (1H, s, erlotinib 8-H), 7.14/7.13 (1H, m, phenyl H), ~7.1–7.2 (1H, br s, amine H), 7.08/7.06 (1H, s, erlotinib 5-H), 6.85 (1H, m, phenyl H), 6.79/6.77 (1H, dd, J = 7.8, 7.8 Hz, phenyl H), 6.00/5.97 (1H, q, J = 6.7 Hz, 31-H), 5.72/5.66 (1H, t, J = 5.6 Hz, amide H), 5.20/5.19 (1H, d, J = 19.7 Hz, 132-CHH), 5.05 (1H, d, J = 19.7 Hz, 132-CHH), 4.76/4.75 (1H, d, J = 11.9 Hz, -OCHH-phenyl-), 4.63/4.61 (1H, d, J = 11.9 Hz, -OCHH-phenyl-), 4.49/4.47 (1H, dq, J ~ 7.2, 1.8 Hz, 18-H), 4.34 (1H, m, 17-H), 4.22–4.32 (2H, m, -OCH2CH2OCH3), 4.14 (2H, m, -OCH2CH2OCH3), 4.07–4.13 (1H, m, -NHCHH-phenyl-), 3.950/3.946 (1H, dd, J = 5.6, 15.0 Hz, -NHCHH-phenyl-), 3.87 (2H, m, -OCH2CH2OCH3), 3.69–3.76 (2H, m, -OCH2CH2OCH3), 3.55/3.54 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.49 (3H, s, - OCH2CH2OCH3), 3.44/3.43 (3H, s, 12-CH3), 3.41/3.39 (3H, s, -OCH2CH2OCH3), 3.40/3.35 (3H, s, 2-CH3), 3.18/3.17 (3H, s, 7-CH3), 2.68 (1H, m, 1H of 17-CH2CH2-), 2.50 (1H, m, 1H of 17-CH2CH2-), 2.24 (1H, m, 1H of 17-CH2CH2-), 2.15/2.13 (3H, d, J = 6.7 Hz, 31-CH3), 1.88 (1H, ddd, J = 5.1, 10.0, 14.6 Hz, 1H of 17-CH2CH2-), 1.79/1.77 (3H, d, J = 7.1 Hz, 18-CH3), 1.59/1.58 (3H, t, J = 7.6 Hz, 8-CH2CH3), 0.43 (1H, br s, core NH), -1.70/-1.72 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.0, 172.23/172.21, 171.70/171.69, 160.2/160.1, 156.0/155.9, 155.3/155.2, 154.81/154.78, 153.4/153.3, 151.0, 148.97/148.95, 148.9, 145.04/145.00, 141.3/141.2, 140.41/140.35, 138.8, 138.74/138.72, 138.48/138.44, 138.39, 137.7, 136.54/136.53, 136.4, 136.3, 135.5/135.3, 132.9/132.7, 131.91/131.86, 130.9/130.8, 130.4, 130.2, 128.93/128.89, 128.6/128.5, 128.27/128.17, 128.19, 127.03/127.02, 126.78/126.76, 124.14/124.06, 123.9, 123.3/123.2, 121.21/121.15, 109.1/109.0, 108.9, 106.13/106.11, 104.01/103.96, 102.7/102.6, 98.3/97.7, 94.3, 92.6, 89.37/89.35, 89.36, 71.7/71.3, 71.0/70.9, 70.74/70.60, 70.56, 69.5/69.4, 68.4, 59.31, 59.31/59.28, 51.64/51.62, 50.01/49.97, 48.06/48.05, 42.60/42.59, 32.60/32.56, 30.3, 24.6/24.3, 23.12/23.07, 19.4, 17.31/17.29, 11.89/11.87, 11.3, 11.2/11.1. UV-vis (CH3OH, λmax, nm (ε)): 663 (4.62 x 104), 606 (8.25 x 103), 537 (9.39 x 103), 506 (9.08 x 103), 409 (9.15 x 104), 349 (4.99 x 104). MS (ESI) m/z: 1249.43 (M + H+). HRMS (ESI): calcd for C69H70O7N8I (M + H+) 1249.4383; found, 1249.4371. Note: Small 1H impurity peaks were observed at 3.80 and 3.65 ppm. 1H impurity peaks at 1.60, 1.27, and 0.87 ppm (probably H2O, grease CH2 and CH3, respectively) are larger. 13C impurity peaks at 29.7 and 14.1 ppm are likely due to grease CH2 and CH3, respectively.
3-{1’-(3-iodobenzyloxy)ethyl}17-[N-(3-benzylamineethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine] pyropheophorbide-a (5):
To a solution of compound 12 (50 mg, 0.0661 mmol) and compound 11 (48.64 mg, 0.0976 mmol) in 12 mL of dry dichloromethane, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI, 25.30 mg, 0.132 mmol) and 4-(dimethylaminopyridine (DMAP, 16.13 mg, 0.132 mmol) were added. The reaction mixture was stirred at room temperature under N2 atmosphere for overnight. It was then diluted with dichloromethane (40 mL), washed with water (3 x 50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 6% methanol in dichloromethane to obtain pure product 5 with 71% (57.68 mg) yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.76/9.73 (1H, s, 5-H), 9.00/8.98 (1H s, 10-H), 8.87/8.86 (1H, s, amine H), 8.68 (1H, s, pyrimidine H), 8.53 (1H, s, 20-H), 8.20 (1H, d, J = 8.2 Hz, phenyl H), 7.823/7.818 (1H, s, erlotinib 5-H), 7.79/7.76 (1H, m, phenyl H), 7.61–7.66 (2H, m, 2 x phenyl H), 7.41 (1H, s, phenyl H), 7.27–7.34 (3H, m, 3 x phenyl H), 7.22 (1H, s, erlotinib 8-H), ~7.21 (1H, d, J ~ 8 Hz, phenyl H), ~7.10 (1H, br s, amide H), 7.07/7.05 (1H, t, J = 7.7 Hz, phenyl H), 7.01 (1H, t, J = 7.6 Hz, phenyl H), 6.91 (1H, d, J = 7.7 Hz, phenyl H), 5.98/5.97 (1H, q, J = 6.7 Hz, 31-H), 5.08 (1H, d, J = 19.8 Hz, 132-CHH), 4.72/4.69 (1H, d, J ~ 12 Hz, -OCHH-phenyl-), 4.57/4.56 (1H, d, J ~ 12 Hz, -OCHH-phenyl-), 4.56 (1H, d, J = 19.9 Hz, 132-CHH), 4.51 (1H, dq, J = 7.3, 1.4 Hz, 18-H), 4.38 (1H, dd, J = 6.6, 14.9 Hz, -NHCHH-phenyl-), 4.28 (1H, m, 17-H), 4.15–4.27 (4H, m, 2 x - OCH2CH2OCH3), 4.00 (1H, dd, J = 4.5, 14.7 Hz, -NHCHH-phenyl-), 3.81 (2H, m, - OCH2CH2OCH3), 3.63 (2H, m, -OCH2CH2OCH3), 3.59 (2H, m, 8-CH2CH3), 3.44 (3H, s, -OCH3), 3.37/3.35 (3H, s, 2-CH3), 3.239/3.238 (3H, s, -OCH3), 3.23/3.22 (3H, s, 7-CH3), 2.81 (1H, m, 1H of 17-CH2CH2-), 2.65 (1H, m, 1H of 17-CH2CH2-), 2.34/2.32 (3H, s, 12-CH3), ~2.31 (1H, m, 1H of 17-CH2CH2-), 2.19/2.18 (3H, d, J = 6.7 Hz, 31-CH3), 2.03 (1H, m, 1H of 17-CH2CH2-), 1.75 (3H, d, J = 7.3 Hz, 18-CH3), 1.63/1.62 (3H, t, J = 7.6 Hz, 8-CH2CH3), 0.56 (1H, br s, core NH), - 1.52 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.7, 173.15/173.14, 172.2, 160.20/160.19, 156.7, 155.55/155.53, 154.3, 153.6, 151.0, 149.0, 148.9, 147.4, 145.1, 141.5/141.4, 140.74/140.69, 139.5, 138.6, 138.5, 137.5, 136.89/136.86, 136.7, 136.40/136.39, 135.64/135.61, 133.1, 133.0, 130.77/130.75, 130.6, 130.14/130.12, 129.43/129.42, 128.9, 128.4, 127.82/127.81, 127.12/127.08, 126.7, 124.3, 123.7, 123.5, 122.1, 109.8, 108.6, 105.9, 103.9, 103.0, 97.9/97.8, 94.45/94.42, 92.8, 89.6, 88.9, 72.09/72.06, 70.7, 70.5, 70.28/70.25, 68.9, 68.3, 59.2, 59.1, 51.5, 49.8, 47.7, 43.2, 32.71/32.69, 31.0, 24.6/24.5, 22.89/22.88, 19.4, 17.4, 11.37/11.36, 11.09/11.07, 10.92/10.90. UV-vis (CH3OH, λmax, nm (ε)): 664 (4.76 x 104), 607 (8.76 x 103), 539 (8.96 x 103), 507 (9.06 x 103), 410 (8.74 x 104), 347 (5.17 x 104). MS (ESI) m/z: 1249.43 (M + H+). HRMS (ESI): calcd for C69H70O7N8I (M + H+) 1249.4376; found, 1249.4383. Note: 1H impurity peaks observed at 1.28 and 0.88 ppm are likely grease CH2 and CH3, respectively. 13C impurity peaks at 29.7 and 14.1 ppm are likely due to grease CH2 and CH3, respectively.
Synthesis of radioactive compound 6 (124I-labeled analog of PS 5):
Iodine-124 was purchased through Sofie (Richmond, VA). The radioactive solution was dried under flow of nitrogen gas at 110 °C. After cooling the tube, the dried radioactivity was dissolved in 100 µL of 5% acetic acid in methanol. Compound 13 (40 µg) was dissolved in 50 µL of 5% acetic acid in ethanol solutions and was transferred to the I-124 tube and mixed. 10 µL of N-chlorosuccinimide in methanol (1 mg/mL) was added. The reaction mixture was incubated at room temperature for 15 minutes, and the product was purified on a HPLC column (Waters Symmetry C18 5µm, 4.6 x 250 mm) and was eluted with a 97:3 mixture of methanol and water at a flow rate of 1 mL/minute. The output was monitored by UV (254 nm) and radioactivity detectors. The labeled product 6 was collected and dried under nitrogen gas flow at 90 °C. To the dried product tube 1mL of 2 mg/mL solution of Pluronic in ethanol was added and the activity was dissolved by vortexing the solution. The ethanol was evaporated under nitrogen flow at 90 °C. The labeled material was dissolved in one mL of 0.01 M PBS with vortexing. This final product solution was filtered through a 0.2 micron syringe filter with Tyffryn membrane (Pall, #28142–340). The labeled product was eluted at 13.5 min. The specific activity was found to be > 1 Ci/µmole.
3-{1’-(3-[N-(3-ethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine]benzyloxy) ethyl]pyropheophorbide-a methyl ester (8):
Triphenylarsine (18.76 mg, 0.06132 mmol) and Pd2dba3 (28.06 mg, 0.0306 mmol) were added to a stirred solution of compound 1 (60 mg, 0.0766) and Erlotinib 7 (45.22 mg, 0.1144) in dry THF (30 mL) and Et3N (6 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product. The remaining ¼ did not react even by increasing the catalyst quantity, temperature and duration of the reaction. The product 8 and the remaining starting material 1 were purified by preparative TLC plates with 72% yield (57.85 mg). 1H NMR (400 MHz, CDCl3, δ ppm): 9.76/9.73 (1H, s, 5-H), 9.48/9.47 (1H, s, 10-H), 8.64 (1H, s, pyrimidine H), 8.56 (1H, s, 20-H), 7.70–7.78 (2H, m, 2 x phenyl H), 7.54/7.52 (1H, br s, phenyl H), 7.46 (1H, m, phenyl H), 7.27–7.38 (4H, m, 3 x phenyl H & 1 x erlotinib NH), 7.26 (1H, s, aromatic H), 7.16 (1H, m, phenyl H), 7.14/7.11 (1H, br s, aromatic H), 6.01 (1H, q, J = 6.7 Hz, 31-H), 5.26/5.25 (1H, d, J = 19.8 Hz, 132-CHH), 5.112/5.109 (1H, d, J = 19.8 Hz, 132-CHH), 4.77 (1H, d, J = 11.8 Hz, -OCHH-phenyl-), 4.62/4.61 (1H, d, J = 11.9 Hz, - OCHH-phenyl-), 4.49 (1H, dq, J = 7.3, 1.8 Hz, 18-H), 4.26–4.33 (3H, m, 17-H & - OCH2CH2OCH3), 4.18 (2H, m, -OCH2CH2OCH3), 3.86 (2H, m, -OCH2CH2OCH3), 3.77 (2H, m, - OCH2CH2OCH3), 3.55–3.67 (8H, m, 8-CH2CH3 & ring CH3 & -COOCH3), 3.482/3.481 (3H, s, - OCH2CH2OCH3), 3.432/3.431 (3H, s, -OCH2CH2OCH3), 3.39 (3H, s, ring CH3), 3.20/3.19 (3H, s, ring CH3), 2.69 (1H, m, 1H of 17-CH2CH2-), 2.56 (1H, m, 1H of 17-CH2CH2-), 2.21–2.36 (2H, m, 2H of 17-CH2CH2-), 2.16 (3H, d, J = 6.7 Hz, 31-CH3), 1.82 (3H, d, J = 7.2 Hz, 18-CH3), 1.642/1.639 (3H, t, J = 7.7 Hz, 8-CH2CH3), 0.42 (1H, br s, core NH), -1.71 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.19/196.18, 173.5, 171.4, 160.32/160.31, 156.17/156.15, 155.2, 154.8, 153.3, 151.0, 149.1, 149.01/149.00, 145.0, 141.3/141.2, 138.7, 138.62, 138.58, 137.8, 136.5, 135.5/135.4, 132.8/132.7, 131.5/131.4, 130.9, 130.5, 129.0, 128.51/128.49, 128.4, 128.12/128.10, 127.27/127.25, 124.30/124.26, 123.93/123.92, 123.3, 121.50/121.48, 109.0, 108.6, 106.1, 104.1, 102.55/102.50, 97.94/97.87, 92.7, 89.52/89.51, 89.3/89.2, 71.8/71.7, 71.0, 70.71/70.66, 70.5, 69.4, 68.4, 59.32, 59.30, 51.68, 51.67, 50.0, 48.0, 30.9, 29.9, 24.52/24.50, 23.17/23.15, 19.4, 17.4, 12.0, 11.25/11.24, 11.2. MS (ESI) m/z: 1048.49 (M + H+). HRMS (ESI):calcd for C63H66N7O8 (M + H+) 1048.4928; found, 1048.4957. UV-vis (CH3OH, λmax, nm (ε)): 663 (4.58 x 104), 608 (8.19 x 103), 537 (9.1 x 103), 507 (8.99 x 103), 409 (8.8 x 104), 349 (4.93 x 104). Note: 1H impurity peaks at 1.27 ppm and 0.87 ppm are likely due to grease. One carbon peak is missing from this spectrum, presumably due to aggregation-induced line broadening. The 13C peak at 29.7 ppm is likely due to grease impurity.
3-{1’-(3-[N-(3-ethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine]benzyloxy) ethyl}pyropheophorbide-a carboxilic acid (9):
Aqueous LiOH (36.02 mg in 3 mL of H2O) was added to a solution of compound 8 (30 mg) in dry THF:MeOH (4.5:3 mL), and the reaction mixture was stirred under argon at room temperature for 3 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and washed with 2% AcOH in H2O (18 mL) and with H2O (3 x 50 mL), and the organic layer was dried over Na2SO4, concentrated, and purified over a preparative TLC plate using 8% MeOH in CH2Cl2 as eluent to yield 28.11 mg (95%) of product 9. 1H NMR (400 MHz, 90:10 CDCl3/CD3OD, δ ppm): 9.63 (1H, s, meso H), 9.30 (1H, br s, meso H), 8.49 (1H, s, meso H), 8.40 (1H, s, pyrimidine H), 7.68 (1H, d, J = 7.9 Hz, phenyl H), 7.64/7.60 (1H, s, phenyl H), 7.34–7.46 (3H, m, 3 x aromatic H), 7.23–7.32 (3H, m, 3 x aromatic H), 7.13 (1H, s, aromatic H), 7.10 (1H, d, J = 7.5 Hz, phenyl H), 5.95 (1H, q, J = 6.4 Hz, 31-H), 5.21 (1H, d, J = 19.9 Hz, 132-CHH), 5.01 (1H, d, J = 19.9 Hz, 132-CHH), 4.70 (1H, d, J = 11.8 Hz, -OCHH-phenyl-), 4.57 (1H, d, J = 11.7 Hz, -OCHH-phenyl-), 4.43 (1H, q, J ~ 6.5 Hz, 18-H), 4.28 (2H, br s, - OCH2CH2OCH3), ~4.19 (1H, m, 17-H), ~4.07 (2H, m, -OCH2CH2OCH3), 3.85 (2H, br s, - OCH2CH2OCH3), 3.73 (2H, br s, -OCH2CH2OCH3), 3.50 (2H, q, J = 7.5 Hz, 8-CH2CH3), 3.45 (3H, s, -OCH2CH2OCH3), ~3.44 (3H, br s, ring CH3), 3.41 (3H, s, -OCH2CH2OCH3), ~3.31 (3H, s, ring CH3), 3.11 (3H, s, ring CH3), 2.62 (1H, m, 1H of 17-CH2CH2-), 2.50 (1H, m, 1H of 17-CH2CH2-), 2.13–2.32 (2H, m, 2H of 17-CH2CH2-), 2.09 (3H, d, J = 6.3 Hz, 31-CH3), 1.73 (3H, d, J ~ 6.4 Hz, 18-CH3), 1.54 (3H, t, J = 7.3 Hz, 8-CH2CH3); 13C NMR (100 MHz, 90:10 CDCl3/CD3OD, δ ppm): 197.9, 178.7, 172.8, 161.7, 157.44/157.42, 156.08/156.06, 155.0, 153.4, 151.4, 149.4, 149.2, 146.9, 145.6, 141.8, 139.4, 138.9, 138.8, 137.8, 136.9, 136.0/135.9, 133.61/133.56, 132.0/131.9, 131.2, 130.0, 129.1, 128.9, 128.6, 128.5, 127.6/127.5, 125.7/125.6, 124.0, 123.7, 122.8/122.7, 109.9, 107.9, 106.1, 104.4, 103.7/103.6, 98.1/98.0, 93.4, 89.8, 89.5, 72.1/72.0, 71.13, 71.05/71.02, 70.9, 69.0, 68.7, 59.43, 59.40, 52.3, 50.5, 48.3, 33.0, 31.0, 24.5, 23.2, 19.6, 17.4, 11.9, 11.3, 11.2. MS (ESI) m/z: 1034.48 (M + H+). HRMS (ESI): calcd for C62H64N7O8 (M + H+) 1034.4814; found, 1034.4821. UV-vis (CH3OH, λmax, nm (ε)): 663 (4.58 x 104), 608 (8.19 x 103), 537 (9.1 x 103), 507 (8.99 x 103), 409 (8.8 x 104), 349 (4.93 x 104). Note: One carboxylic acid and three NH protons were not observed, presumably due to chemical exchange. Small 13C impurity peaks were observed at 180.2, 173.4, 29.6, and 14.2 (probably CH3 of grease) ppm. A somewhat larger impurity peak – likely CH2 of grease – was observed at 30.0 ppm.
3-{1’-(3-[N-(3-ethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine]benzyloxy) ethyl}-17-(3-trimethyltin-benzyamine)-pyropheophorbide-a (10):
To a solution of compound 3 (30 mg, 0.025 mmol), hexamethylditin (295.09 mg, 1.88 mmol) and PdCl2(PPh3)2 (8.43 mg, 0.0120 mmol) in 12 mL of dry THF. The reaction mixture was stirred at room temperature under N2 atmosphere overnight. It was then diluted with dichloromethane (40 mL), washed with water (3 x 50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 4% methanol in dichloromethane to obtain pure product 10 with 92% (28.42 mg) yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.77/9.68 (1H, s, 5-H), 9.37/9.36 (1H, s, 10-H), 8.55/8.53 (1H, s, 20-H), 8.55/8.48 (1H, s, pyrimidine H), 7.72 (1H, m, phenyl H), 7.65/7.61 (1H, m, phenyl H), 7.42–7.46 (2H, m, 2 x phenyl H), 7.27–7.36 (4H, m, amine H & 3 x phenyl H), 7.20–7.25 (2H, m, phenyl H & aromatic H), 7.12–7.17 (3H, m, aromatic H & 2 x phenyl H), 7.08/7.05 (1H, t, J = 7.4 Hz, phenyl H), 6.89/6.85 (1H, dt, J = 7.7, ~1.4 Hz, phenyl H), 6.00/5.97 (1H, q, J = 6.7 Hz, 31-H), 5.62/5.58 (1H, t, J = 5.6 Hz, amide H), 5.214/5.205 (1H, d, J = 19.8 Hz, 132-CHH), 5.06 (1H, d, J = 19.8 Hz, 132-CHH), 4.75 (1H, d, J = 11.8 Hz, -OCHH-phenyl-), 4.611/4.605 (1H, d, J = 11.8 Hz, -OCHH-phenyl-), 4.50/4.48 (1H, dq, J ~ 7.0, 1.8 Hz, 18-H), 4.34 (1H, m, 17-H), 4.28 (2H, m, -OCH2CH2OCH3), 4.23/4.22 (1H, dd, J = 5.6, 14.5 Hz, -NHCHH-phenyl-), 4.15 (2H, m, -OCH2CH2OCH3), 4.11 (1H, m, -NHCHH-phenyl), 3.86 (2H, m, -OCH2CH2OCH3), 3.73 (2H, m, -OCH2CH2OCH3), 3.57 (2H, q, J = 7.6 Hz, 8-CH2CH3), 3.48 (3H, s, -OCH2CH2OCH3), 3.47/3.46 (3H, s, ring CH3), 3.41/3.40 (3H, s, - OCH2CH2OCH3), 3.39/3.35 (3H, s, ring CH3), 3.19/3.18 (3H, s, ring CH3), 2.70 (1H, m, 1H of 17-CH2CH2-), 2.48 (1H, m, 1H of 17-CH2CH2-), 2.27 (1H, m, 1H of 17-CH2CH2-), 2.15/2.13 (3H, d, J = 6.7 Hz, 31-CH3), 1.92 (1H, m, 1H of 17-CH2CH2-), 1.79/1.77 (3H, d, J = 7.0 Hz, 18-CH3), 1.61/1.60 (3H, t, J = 7.6 Hz, 8-CH2CH3), 0.40 (1H, br s, core NH), 0.144/0.137 (9H, s, Sn(CH3)3), -1.70/-1.72 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.06, 172.04/172.01, 171.71/171.69, 160.32/160.26, 156.1/156.0, 155.23/155.21, 154.72/154.69, 153.6/153.5, 151.0, 148.93/148.91, 148.89, 147.64/147.61, 145.01/144.98, 142.8, 141.2, 138.9/138.8, 138.6/138.5, 138.4, 137.8, 137.4/137.3, 136.5, 135.4/135.3, 135.2, 134.9, 132.8/132.7, 131.73/131.71, 130.84/130.79, 130.4, 128.9, 128.50/128.48, 128.22/128.15, 128.15, 127.62/127.60, 127.0, 124.2/124.1, 123.8, 123.29/123.26, 121.34/121.30, 109.2/109.1, 108.9, 106.09/106.08, 104.00/103.97, 102.62/102.60, 98.1/97.8, 92.7, 89.4, 89.4/89.3, 71.7/71.5, 71.0/70.9, 70.7/70.6, 70.5, 69.4/69.3, 68.3, 59.29, 59.29/59.27, 51.7, 50.02/50.00, 48.0, 43.68/43.66, 32.8/32.7, 30.4, 24.5/24.4, 23.12/23.07, 19.4, 17.3, 11.90/11.88, 11.23, 11.20/11.10, -9.7. UV-vis (CH3OH, λmax, nm (abs)): 664 (0.663), 609 (0.120), 538 (0.136), 506 (0.131), 409 (1.315), 349 (0.719). MS (ESI) m/z: 1287.50 (M + H+). HRMS (ESI): calcd for C72H78O7N8Sn (M + H+) 1287.5000; found, 1287.5010. Note: 1H impurity peaks at 1.27 ppm and 0.88 ppm are likely due to grease CH2 and CH3, respectively. One carbon peak is missing, presumably due to line broadening caused by aggregation. The 13C peak at 29.7 ppm is likely due to grease (CH2) impurity.
[N-(3-(3’benzylamine)ethynylphenyl)-6,7-bis(2-methoxy-ethoxy)-4-quinazolinamine] (11):
Triphenylarsine (104.67 mg, 0.342 mmol) and Pd2dba3 (156.50 mg, 0.1709 mmol) were added to a stirred solution of compound 3-iodobenzylamine (100 mg, 0.427) and Erlotinib 7 (252.13 mg, 0.641) in dry THF (25 mL) and Et3N (5 mL). The reaction mixture was stirred at room temperature under an argon atmosphere overnight. As per TLC only ¾ amount of the starting material reacted to produce the desired product. The remaining ¼ did not react even by increasing the catalyst quantity, temperature and duration of the reaction. The product 11 was purified by column chromatography by using MeOH/DCM (2/8) with 69% yield (344 mg). 1H NMR (400 MHz, 80:20 CDCl3/CD3OD, δ ppm): 8.46 (1H, s, pyrimidine H), 7.88 (1H, br t, J ~ 1.7 Hz, phenyl H), 7.67 (1H, ddd, J ~ 8.0, 2.1, 1.1 Hz, phenyl H), 7.65 (1H, s, erlotinib 5-H), 7.46 (1H, br s, phenyl H), 7.40 (1H, dt, J ~ 7.3, 1.5 Hz, phenyl H), 7.34 (1H, t, J = 7.9 Hz, phenyl H), 7.31 (1H, t, J = 7.6 Hz, phenyl H), 7.24–7.29 (2H, m, 2 x phenyl H), 7.13 (1H, s, erlotinib 8-H), 4.30 (2H, m, -OCH2CH2OCH3), 4.27 (2H, m, -OCH2CH2OCH3), 3.84 (6H, m, 2 x - OCH2CH2OCH3 & -phenyl-CH2NH2), 3.47 (3H, s, -OCH2CH2OCH3), 3.45 (3H, s, - OCH2CH2OCH3); 13C NMR (100 MHz, 80:20 CDCl3/CD3OD, δ ppm): 157.5, 154.9, 153.4, 149.1, 147.0, 141.0, 139.3, 130.87, 130.85, 129.2, 129.1, 127.8, 127.7, 125.8, 123.93, 123.90, 123.1, 110.0, 107.9, 103.9, 89.7, 89.3, 71.1, 70.8, 69.2, 68.6, 59.42, 59.35, 45.4. Note: The three amino protons were not observed, presumably due to chemical exchange. 1H impurity peaks at 1.27 ppm and 0.84 ppm are likely due to grease CH2 and CH3, respectively. The 13C peak at 29.9 ppm is likely due to grease (CH2) impurity.
3-{1’-(3-trimethyltinbenzyloxy)ethyl}17-[N-(3-benzylamineethynylphenyl)-6,7-bis(2 methoxy-ethoxy)-4-quinazolinamine]pyropheophorbide-a (13):
To a solution of compound 5 (30 mg, 0.025 mmol), hexamethylditin (295.09 mg, 1.88 mmol) and PdCl2 (PPh3)2 (8.43 mg, 0.0120 mmol) in 12 mL of dry THF. The reaction mixture was stirred at room temperature under N2 atmosphere for overnight. It was then diluted with dichloromethane (40 mL), washed with water (3 x 50 mL), dried over anhydrous sodium sulfate and concentrated down to yield crude product which was purified by silica column by using 4% methanol in dichloromethane to obtain pure compound 13 with 86% (26.57 mg) yield. 1H NMR (400 MHz, CDCl3, δ ppm): 9.77/9.74 (1H, s, 5-H), 9.10/9.07 (1H, s, 10-H), 8.75 (1H, br s, amine H), 8.69 (1H, s, pyrimidine H), 8.52 (1H, s, 20-H), 8.21 (1H, d, J = 8.3 Hz, phenyl H), 7.79 (1H, s, aromatic H), 7.65 (1H, m, phenyl H), 7.41–7.49 (3H, m, 3 x phenyl H), 7.30–7.41 (4H, m, 4 x phenyl H), 7.24 (1H, s, aromatic H), 7.22 (1H, br d, J ~ 8 Hz, phenyl H), 7.06/7.04 (1H, dd, J = 1.6, 7.6 Hz, phenyl H), 6.93 (1H, br d, J = 7.4 Hz, phenyl H), 6.81 (1H, br s, amide H), 6.02/5.99 (1H, q, J = 6.8 Hz, 31-H), 5.13 (1H, d, J = 19.8 Hz, 132-CHH), 4.81/4.79 (1H, d, J = 11.6 Hz, -OCHH-phenyl-), 4.65/4.64 (1H, d, J = 19.8 Hz, 132-CHH), 4.64/4.62 (1H, d, J = 11.5 Hz, -OCHH-phenyl-), 4.53 (1H, q, J = 7.2 Hz, 18-H), 4.41/4.40 (1H, dd, J = 6.4, 14.8 Hz, -NHCHH-phenyl-), 4.31 (1H, m, 17-H), 4.27 (2H, m, - OCH2CH2OCH3), 4.23 (2H, m, -OCH2CH2OCH3), 4.04 (1H, dd, J = 4.2, 14.7 Hz, -NHCHH-phenyl-), 3.83 (2H, m, -OCH2CH2OCH3), 3.63 (2H, m, -OCH2CH2OCH3), ~3.61 (2H, m, 8-CH2CH3), 3.45 (3H, s, -OCH3), 3.37/3.34 (3H, s, ring-CH3), 3.234/3.230 (3H, s, -OCH3), 3.19/3.18 (3H, s, ring-CH3), 2.83 (1H, m, 1H of 17-CH2CH2-), 2.65 (1H, m, 1H of 17-CH2CH2-), 2.49 (3H, br s, 12-CH3), 2.32 (1H, m, 1H of 17-CH2CH2-), 2.162/2.156 (3H, d, J = 6.7 Hz, 31-CH3), 2.02 (1H, m, 1H of 17-CH2CH2-), 1.76 (3H, d, J = 7.2 Hz, 18-CH3), 1.65/1.63 (3H, t, J = 7.6 Hz, 8-CH2CH3), 0.62 (1H, br s, core NH), 0.22/0.20 (9H, s, Sn(CH3)3), -1.49 (1H, s, core NH); 13C NMR (100 MHz, CDCl3, δ ppm): 196.8, 173.0, 172.2, 160.2, 156.7, 155.7/155.6, 154.3, 153.6, 151.0, 149.1, 148.9, 147.5, 145.0, 142.6/142.5, 141.7/141.6, 139.5, 139.2/139.1, 138.5, 137.7/137.6, 137.5, 136.38/136.36, 135.90/135.87, 135.59/135.56, 135.3, 133.0/132.9, 130.7, 130.6, 129.5, 129.0, 128.5, 128.3/128.2, 128.1, 127.86/127.83, 127.81, 126.8, 124.3, 123.77/123.75, 123.5, 122.1, 109.8, 108.72/108.71, 105.8, 104.0, 103.0, 98.0/97.9, 92.7, 89.65/89.64, 89.0, 71.73/71.70, 71.5, 70.7, 70.5, 68.9, 68.3, 59.3, 59.1, 51.54/51.53, 49.8, 47.8, 43.2, 32.74/32.71, 31.03/31.00, 24.64/24.57, 22.9, 19.4, 17.4, 11.3, 11.13/11.09, 11.1, -9.60/-9.61. UV-vis (CH3OH, λmax, nm (abs)): 663 (0.622), 607 (0.119), 539 (0.122), 506 (0.119), 410 (1.314), 347 (0.664). MS (ESI) m/z: 1287.50 (M + H+). HRMS (ESI): calcd for C72H78O7N8Sn (M + H+) 1287.1876; found, 1287.1883. Note: 1H impurity peaks at 1.27 ppm and 0.89 ppm are likely due to grease CH2 and CH3, respectively. The 13C peak at 29.7 ppm is likely grease (CH2) impurity.
HPLC Analysis of iodo-analog 5 and the related trimethyl-tin derivative 13:
The solutions of 5 and 13 were prepared by dissolving 0.2 mg of individual compound in 25 μL THF and then 500 μL methanol was added and mixed. These solutions were then filtered through a 0.2 micron pore size regenerated cellulose 4 mm syringe filter. For analyzing the purity of these samples, a Waters 600 controller connected to a 996 Photodiode array detector, a Delta 600-Multisolvent delivery system, and Rheodyne injector (with a 20 uL loop) were used. The system was equipped with a Waters Symmetry C18 column of dimensions 4.6x250 mm and 5 micro particle size. The mobile phase was 97% methanol/ 3% water at a flow rate of 1.0 mL/min. Each of the two compounds were isolated as epimeric mixture, but the iodo analog 5 eluted at 14.69 min, whereas the trimethyl tin derivative 13 showed two peaks (partially resolved), with retention times 18.38 and 19.23 min. The purity of the radioactive analog 6 was also confirmed by the same HPLC parameters.
Cell lines:
UMUC-3 (CRL-1749) and T24 (HTB-4) cells were acquired from ATCC. The cells were grown in McCoy supplemented with 10% FBS and 1% Penicillin/Streptomycin, L-Glutamate, and HEPES buffer.
Cytotoxicity Assay:
Cells were plated in 96 well plates at a concentration of 10k cells per well in 100µl. After 24 hours incubation time, to allow the cells to adhere to the plate, photosensitizer was added to the plate in a two-fold dilution pattern where the top concentration is 1µM. The photosensitizer was allowed to incubate with the cells for 24 hours before the cells were exposed to the optimal wavelength for HPPH in an aqueous environment, 665 nm. Cells were exposed to light for a total of 1.0J/cm2 at a dose rate of 3.2mw/cm2. 48 hours after PDT, the cell viability was measured using an MTT assay. Briefly, 15 µl of MTT solution was added to each well and allowed to incubate for about 2 hours. The media was removed and the insoluble Formazan was suspended in DMSO. The plate is read at 570 nm. For the investigation on the impact of ABCG2 inhibition, cells were incubated with Ps as discussed above along with Gleevec at 5 µM concentration.
Mechanistic investigation utilizing fluorescence microscope:
100k cells were plated into 6 well plates and allowed to grow to 80% confluency. The cells were washed and fresh media was added. To investigate the impact of ABCG2 inhibition, 5 µM of Gleevec was added to the cells with 500 nM of each photosensitizer. The cells incubated with the drugs for 24 hours before being washed twice with PBS. Fresh media was added to the cells. Hoechst 33342 was added to the plates to better visualize the cells. the plates were visualized utilizing a Zies Fluorescence microscope. The images were analyzed in ImageJ where the grey value of each fluorescent field was normalized to the number of cells in the field. Each group was done in triplicate. Each well was imaged in triplicate. To investigate whether EGFR affinity of the erlotinib conjugated photosensitizer, free erlotinib was added at 3µM concentration. For this experiment the photosensitizer concentration was raised to 1µM. The fluorescence images were recorded, and analysis was performed by following the method described above.
Comparative ROS measurements in vitro:
10k UMUC3 cells were plated in 96 well plate. After 24 hours, the Ps 1, 3 or 5 was added at 1 µM. After 24 hour incubation, 1 µM of DCFDA was incubated with the cells for 20 minutes. The fluorescent intensity of DCFDA was measured utilizing a Spectra Max at 492 nm excitation and 517 emission. The plates were then exposed to 665 nm light for a total of 1 J. Additionally, 0.05% H2O2 was as a control to one well. The fluorescence was measured again using the same parameters as above.
Annexin V/PI staining:
UMUC3 and T24 cells were treated with erlotinib conjugates 1, 3 and 5 followed by PDT for indicated time points. Cells were trypsinized and washed with ice cold PBS once and suspended in 100 µl Annexin V binding buffer. 5 µl Annexin V-FITC and 1 µl PI (100 µg/ml) was added and incubated at room temperature. After 15 min incubation, 300 µl Annexin V binding buffer was added and analyzed using BD flow cytometer (LSRII B) at FITC and PE channels. Data were analyzed using BD Diva software. Annexin V- and PI- cells were considered live cells; Annexin V- and PI+ cells were considered necrosis cells; Annexin V+ cells were considered apoptotic cells.
Western blotting:
UMUC3 and T24 cells were treated with erlotinib conjugates 1, 3 and 5 followed by PDT for indicated time points. Cells were trypsinized and washed with ice cold PBS once and lysed in RIPA buffer supplemented with Protease inhibitor cocktail (Sigma Aldrich), 1 mM sodium orthovanadate, 5 mM sodium fluoride and 1 mM DTT. Protein content was quantified by micro BCA protein estimation kit (Thermo Scientific). Proteins samples were resolved on 4–20 % Criterion gels, transferred on nitrocellulose membranes (BioRad) and subjected to immunoblotting. Membranes were blocked in 5% fat free dry milk (Santa Cruz) prepared in PBS-T (Tween 20) and incubated overnight in indicated primary antibodies (1:1000 dilution) at 4°C with continuous shaking. After washing with PBS-T, membranes were incubated in HRP conjugated anti-mouse or anti-rabbit secondary antibody at room temperature for 1 hour. After washing with PBS-T, proteins were detected with Clarity chemiluminescent reagent (BioRad) and X-Ray films (ASI). Membranes were stripped with stripping buffer and probed with HRP conjugated beta-actin antibody to ensure equal loading of proteins.
In vivo studies:
The experimental details for in vivo studies are presented in the Results and Discussion part of the paper. All the animal studies presented in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Roswell Park Comprehensive Cancer Center, Buffalo, NY.
Instrumentation:
Measurement of fluorescence quantum yields were carried out on a Hamamatsu C9920−0X(PMA-12) U6039− 05 fluorescence spectrofluorometer. Femtosecond laser flash photolysis was conducted using a Clark-MXR 2010 laser system and an optical detection system provided by Ultrafast Systems (Helios). The source for the pump and probe pulses were derived from the fundamental output of Clark laser system (775 nm, 1 mJ pulse-1 and fwhm = 150 fs) at a repetition rate of 1 kHz. A second harmonic generator introduced in the path of the laser beam provided 412 nm laser pulses for excitation. A 95% of the fundamental output of the laser was used to generate the second harmonic, while 5% of the deflected output was used for white light generation. Prior to generating the probe continuum, the laser pulse was fed to a delay line that provided an experimental time window of 1.6 ns with a maximum step resolution of 7 fs. The pump beam was attenuated at 5 μJ pulse-1 with a spot size of 2 mm diameter at the sample cell where it was merged with the white probe pulse in a close angle (<10°). The probe beam after passing through the 2 mm sample cell was focused on a 200 μm fibre optic cable, which was connected to a CCD spectrograph (Ocean Optics, S2000-UV-vis for visible region and Horiba, CP-140 for NIR region) for recording the time-resolved spectra (450–800 and 800–1400 nm). Typically, 5000 excitation pulses were averaged to obtain the transient spectrum at a set delay time. The kinetic traces at appropriate wavelengths were assembled from the time-resolved spectral data. Nanosecond time-resolved transient absorption measurements were carried out using the laser system provided by UNISOKU Co., Ltd. Measurements of nanosecond transient absorption spectrum were performed according to the following procedure. A mixture solution in a quartz cell (1 cm × 1 cm) was excited by a Nd:YAG laser (Continuum SLII-10, 4–6 ns fwhm, λex = 355 nm, 80 mJ pulse–1, 10 Hz). The photodynamics were monitored by continuous exposure to a xenon lamp for visible region and halogen lamp for near-IR region as a probe light and a photomultiplier tube (Hamamatsu 2949) as a detector.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Mykhaylo Dukh of our research group for providing the starting material (iodinated photosensitizer 1) to synthesize a series of erlotinib analogs. The authors are highly thankful to Photolitec, LLC, and Roswell Park Alliance Foundation (RP and KG) for the financial support. A partial support from the shared resources of the RPCI support grant (P30CA16056) is also highly appreciated.
ABBREVIATIONS USED
- PS
photosensitizers
- PDT
photodynamic therapy
- EGFR
epidermal growth factor receptor
- PET
positron emission tomography
- CDFADA
2,7-dichlorofluorescein diacetate
- FDG
fluoro deoxyglucose
- NIR
near- infrared
- HPLC
high performance liquid chromatography
Footnotes
Ancillary Information
Supporting Information
The supporting information is available free of charge on the ACS Publication website at DOI: 10.1021/acschembio.7b00023.
Copies of the NMR (proton and C-13)spectra of certain compounds (Figures S2-S16), variable temperature NMR spectra of compound 5 (Figure S15), Comparative photophysical properties of compounds 1, 3 and 5 (Figures S6-20). Molecular formula strings file is attached.
REFERENCES
- (1).Siegel R; Ma J; Zou Z; Jemal A Cancer Statistics, 2014. CA Cancer J Clin 2014, 64, 9–29. [DOI] [PubMed] [Google Scholar]
- (2).Kaufman DS; Shipley WU; Feldman AS Bladder Cancer. Lancet 2009, 374, 239–249. [DOI] [PubMed] [Google Scholar]
- (3).American Cancer Society: Bladder Cancer; Survival rates for bladder cancer by stage, 2016. Available at http://www.cancer.org/cancer/bladdercancer/detailedguide/bladder-cancer-survival-rates (accessed December 10, 2014).
- (4).Grossman HB; Natale RB; Tangen CM; Speights VO; Vogelzang NJ; Trump DL; deVere White RW; Sarosdy MF; Wood DP; Raghavan D and Crawford ED, Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl. J. Med 2003, 349, 859–866 [DOI] [PubMed] [Google Scholar]
- (5).Hinata N; Hussein AA; George S; Trump DL; Levine EG; Omar K; Dasgupta P; Khan MS; Hosseini A; Wiklund P; Guru KA Impact of suboptimal neoadjuvant chemotherapy on peri-operative outcomes and survival after robot-assisted radical cystectomy: a multicentre multinational study. BJU Int 2017, 119(4), 605–611. [DOI] [PubMed] [Google Scholar]
- (6).Hussein AA; Dibaj S; Hinata N; Field E; O’Leary K; Kuvshinoff B; Mohler JL; Wilding G; Guru KA Development and validation of a quality assurance score for robot-assisted radical cystectomy: A 10-year Analysis. Urology 2016, 97, 124–129. [DOI] [PubMed] [Google Scholar]
- (7).Hussein AA; Hashmi Z; Dibaj S; Altartir T; Fiorica T; Wing J; Durrani M; Binkowski J; Boateng L; Wilding G; Guru KA Reoperations following robot-assisted radical cystectomy: A decade of experience. J Urol 2016, 195, 1368–1376. [DOI] [PubMed] [Google Scholar]
- (8).Novara G; Catto JW; Wilson T; Annerstedt M; Chan K; Murphy DG; Motttrie A; Peabody JO; Skinner EC; Wiklund PN; Guru KA; Yuh B Systematic review and cumulative analysis of perioperative outcomes and complications after robot-assisted radical cystectomy. Eur Urol 2015, 67, 376–401. [DOI] [PubMed] [Google Scholar]
- (9).Raza SJ; Wilson T; Peabody JO; Wiklund P; Scherr DS; Al-Daghmin A; Dibaj S; Khan MS; Dasgupta P; Mottrie A; Menon M; Yuh B; Richstone L; Saar M; Stoeckle M; Hosseini A; Kaouk J; Mohler JL; Rha KH; Wilding G; Guru KA Long-term oncologic outcomes following robot-assisted radical cystectomy: results from the International Robotic Cystectomy Consortium. Eur Urol 2015, 68, 721–728. [DOI] [PubMed] [Google Scholar]
- (10).Wilson TG; Guru K; Rosen RC; Wiklund P; Annerstedt M; Bochner BH; Chan KG; Montorsi F; Mottrie A; Murphy D; Novara G; Peabody JO; Palou Redorta J.; Skinner EC; Thalmann G; Stenzl A; Yuh B; Catto J; Pasadena Consensus Panel. Best practices in robot-assisted radical cystectomy and urinary reconstruction: recommendations of the Pasadena Consenus Panel. Eur Urol 2015, 67, 363–375. [DOI] [PubMed] [Google Scholar]
- (11).Tomas A; Futter CE; Eden ER EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 2014, 24, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yewale C; Baradia D; Vhora I; Patil S; Misra A Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [DOI] [PubMed] [Google Scholar]
- (13).Weintraub MD; Vourganti S; Li Q; Apolo AB; Metwalli AR; Agarwal PK Targeting the epidermal growth factor receptor in bladder cancer. J. Carcinogenesis & Mutagenesis 2013, 4 (2), 2157–2518. [Google Scholar]
- (14).Bianco R; Daniele G; Ciardiello F; Tortora G Monoclonal antibodies targeting the epidermal growth factor receptor. Curr Drug Targets 2005, 3, 275–287. [DOI] [PubMed] [Google Scholar]
- (15).Sartore-Bianchi A; Martini M; Molinari F; Veronese S; Nichelatti M; Artale S; Nicolantonio FD; Saletti P; Dosso SD; Mazzucchelli L; Frattini M; Siena S; Bardelli A PIK3CA mutations in colorectal cancer associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Research 2009, 69, 1853–1857. [DOI] [PubMed] [Google Scholar]
- (16).Janmaat ML; Giaccone G Small-molecule epidermal growth factor receptor tyrosine kinase inhibitors. The Oncologist 2003, 8, 576–586. [DOI] [PubMed] [Google Scholar]
- (17).Robey RW; Steadman K; Polgar O; Morisaki K; Blayney M; Mistry P; Bates SE Pheophorbide-a is a specific probe for ABCG2 function and inhibition. Cancer Res 2004, 64, 1242–1246. [DOI] [PubMed] [Google Scholar]
- (18).Liu W; Baer MR; Bowman MJ; Pera P; Zheng X; Morgan J; Pandey RK; Oseroff AR The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy in inhibiting ABCG2. Clin Cancer Res, 2007, 13(8), 2463–2470. [DOI] [PubMed] [Google Scholar]
- (19).Morgan J; Jackson JD; Zheng X; Pandey SK; Pandey RK Substrate affinity of photosensitizers derived from chlorophyll-a: The ABCG2 transporter affects the phototoxic response of side population stem cell-like cancer cells to photodynamic therapy. Mol. Pharm 2010, 7, 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Spring BQ; Rizvi I; Xu N; Hasan T The role of photodynamic therapy in overcoming cancer drug resistance. Photochem. Photobiol. Sci, 2015, 14, 1476–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hagiya Y; Fukuhara H; Matsumoto K; Endo Y; Nakajima M; Tanaka T; Okura I; Kurabayashi A; Furihata M; Inoue K; Shuin T; Ogura S Expression levels of PEPT1 and ABCG2 play key roles in 5-aminolevulenic acid (ALA)-induced tumor-specific protoporphyrin IX accumulation in bladder cancer. Photodiagnosis and Photodynamic Therapy 2013, 10, 288–295. [DOI] [PubMed] [Google Scholar]
- (22).Hepbum AC; Veeratterapillay R; Williamson SC; El-Sherif A; Sahay N; Thomas HD Side population in human non-muscle invasive bladder cancer enriches for cancer stem cells that are maintained by MAPK signaling. PLOS one 2012, 7(11): e50690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Brooks NA; O’Donnell M Treatment options in non-muscle invasive bladder cancer after BCG failure. Indian J Urology 2015, 3 (14), 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Azar FS; Intes X (Eds). Translational Multimodality Optical Imaging, Artech House, Boston, 2008. [Google Scholar]
- (25).Ethirajan M; Chen Y; Joshi P; Pandey RK The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev 2011, 40, 340–362. [DOI] [PubMed] [Google Scholar]
- (26).(a) Pandey RK; Sumlin AB; Constantine S; Aoudia M; Potter WR; Bellnier DA; Henderson BW; Rodgers MA; Smith KM; Dougherty TJ Alkyl ether analogs of chlorophyll-a derivatives: Part 1. Synthesis, photophysical properties and photodynamic therapy. Photochem. Photobiol 1996, 64, 194–204. [DOI] [PubMed] [Google Scholar]; (b) Henderson BW; Bellnier DA, Greco WR; Sharma A; Pandey RK; Vaughan LA; Weishaupt KR; Dougherty TJ An in vivo quantitative structure-activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res 1997, 57, 4000–4007. [PubMed] [Google Scholar]
- (27).Srivatsan A; Ethirajan E; Pandey SK; Dubey S; Zheng X; Liu T-H; Shibata M; Missert J; Morgan J and Pandey RK Conjugation of cRGD peptide to chlorophyll-a based photosensitizer (HPPH) alters its pharmacokinetics with enhanced tumor-imaging and photosensitizing (PDT) efficacy. Mol. Pharm 2011, 8(4), 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) Zheng X; Morgan J; Pandey SK; Bellnier D; Baumann H; Henderson BW; Oseroff A; Pandey RK Conjugation of HPPH to carbohydrates changes its subcellular biodistribution and enhances photodynamic activity in vivo. J. Med. Chem 2009, 52, 4306–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tracy EC; Bowman MJ; Pandey RK; Henderson BW; Baumann H Cell-type selective phototoxicity achieved with chlorophyll-a derived photosensitizers in a co-culture system of primary human tumor and normal lung cells. Photochemistry and Photobiology 2011, 1405–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pandey SK; Zheng X; Morgan J; Missert JR; Liu TH; Shibata M; Bellnier DA; Oseroff AR; Henderson BW; Dougherty TJ; Pandey RK Purpurinide carbohydrate conjugates: effect of the position of the carbohydrate moiety in photosensitizing efficacy. Mol. Pharm 2007, 4 (3), 448–464 [DOI] [PubMed] [Google Scholar]
- (30).Pandey SK; Gryshuk AL; Sajjad M; Zheng X; Chen Y; Abouzeid MM; Morgan J; Nabi HA; Oseroff A; Pandey RK Multimodality agents for tumor imaging (PET, Fluorescence) and photodynamic therapy, A possible “See and Treat” approach. J. Med. Chem 2005, 48 (20), 6286–6295. [DOI] [PubMed] [Google Scholar]
- (31).Srivatsan A; Pera P, Joshi P; Wang Y; Missert JR; Tracy EC; Tabaczynski WA; Yao R; Sajjad M; Baumann H; Pandey RK Effect of chirality on cellular uptake, imaging and photodynamic therapy of photosensitizers derived from chlorophyll-a. Bioorg. Med. Chem, 2015, 23, 3603–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chinchilla R and Najera C The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev, 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- (33).Yu C. Yu; Canteenwala T; El-Khouly ME; Araki Y; Pritzker K; Ito O; Wilson BC; Chiang LY Efficiency of singlet oxygen production from self-assembled nanospheres of molecular micelle-like photosensitizers FC4S. J. Mater. Chem 2005, 15, 1857–1864 [Google Scholar]
- (34).El-Khouly ME; Padmawar P; Araki Y; Verma S; Chiang LY; Ito O Photoinduced processes in a tricomponent molecule consisting of diphenylaminofluorene-dicyanoethylene-methano[60]fullerene. J. Phys. Chem 2006, 110, 884–891. [DOI] [PubMed] [Google Scholar]
- (35).Redmond RW; Gamlin JN Comparsion of singlet oxygen yields from relevant biological molecules. Photochem. Photobiol 1999, 70, 391–475. [PubMed] [Google Scholar]
- (36).Mathai S; Smith TA; Ghiggino KP Singlet oxygen quantum yields of potential porphyrin-based photosensitizers for photodynamic therapy. Photochem. Photobio. Sci 2007, 6, 995–1002. [DOI] [PubMed] [Google Scholar]
- (37).Barry AP; Barry NPE Pluronic block-copolymers in medicine: from chemical and biological versatility to rationalization and clinical advances. Royal Society of Chemistry 2014, 5, 3291–3297. [Google Scholar]
- (38).Riss TL; Moravec RA; Niles AL; Duellman S; Benink HA; Worzella TJ; Minor L Cell Viability Assay, In: Sittampalam GS; Coussens NP; Brimacombe K; Grossman A; Arkin M; Auld D; Austin C; Baell J; Bejcek B; Caaveiro JMM; Chung TDY; Dahlin JL; Devanaryan V; Foley TL; Glicksman M; Hall MD; Haas JV; Inglese J; Iversen PW; Kahl SD; Kales SC; Lal-Nag M; Li Z; McGee J; McManus O; Riss T; Trask OJ Jr.; Weidner JR; Wildey MJ; Xia M; Xu X editors. Assay Guidance Manual [Internet] Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004–2013 May 1 [updated 2016. Jul 1]. [Google Scholar]
- (39).Saenz C; Cheruku RR; Ohulchanskyy TY; Joshi P; Tabaczynski WA; Missert JR; Chen Y; Pera P; Tracy E; Marko A; Rohrbach D; Sunar U; Baumann H; Pandey RK Structural and epimeric isomers of HPPH: Effects on uptake and photodynamic therapy. ACS Chem. Biol 2017, 12 (4), 933–946. [DOI] [PubMed] [Google Scholar]
- (40).Kessel D; Luo Y; Deng Y; Chang CK The role of subcellular localization in initiation of apoptosis by photodynamic therapy., Photochem. Photobiol 1997, 65 (3), 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dutta S; Ongarora BG; Vicente G; Kolli BK; Chang KP Intracellular targetingspecificity of novel Phthalocyanines assessed in a host-parasite model for developing potential photodynamic medicine. PLOS one 2011, 6 (6), e20786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Castano AP; Demidova TN; Hamblin MR Mechanisms in photodynamic therapy: part one – photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn Ther 2004, 1 (4), 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Treda C; Popeda M; Ksiazkiewicz M; Grzela DP; Walczak MP; Banaszczyk M; Peciak J Stoczynska-Fidelus, F. Rieske, P. EGFR activation leads to cell death independent of P13K/ART/mTOR in an AD293 cell line. PloS one, 2016, 11 e0155230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jackson NM and Ceresa BP EGFR-mediated apoptosis via STAT3. Exp. Cell Res 2017, 356, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lainey E; Sebert M; Bouteloup C; Leroy C; Thepot S; Tailler M; Adres L; Gardin C; Kroemer G; Fenaux P; Boehrer S Erlotinib antagonizes efflux via ABC transporters and decreases P-Gp cell surface expression by inhibiting SRC kinase and mTOR pathways in acute myeloid leukemia (AML). Blood 2011, 118, 2564. [Google Scholar]
- (46).Mroz P; Yaroslasvsky A; Kharkwal CB; Hamblin MR Cell death pathways in photodynamic therapy of cancer. Cancers (Basel) 2011, 3, 2516–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).(a) Evans S; Mathews W; Perry R; Fraker D; Norton J; Pass HI Effect of photodynamic therapy on tumor necrosis factor production by murine macrophages. JNCI 1990, 82, 34–38. [DOI] [PubMed] [Google Scholar]; (b) Chen B; Pogue BW; Zhou XO; Hara JA; Solban N; Demidenko E; Hoopes P; J Hasan T Effect of tumor host microenvironment on photodynamic therapy in a rat prostate tumor model. Clin. Cancer Res 2005, 11, 720–727. [PubMed] [Google Scholar]
- (48).Wu D; Yotnda P Production and detection of the reactive oxygen species (ROS) in cancers. J. Vis. Exp 2011, 57, 3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Belhocine T; Spaepen K; Dusart M; Castaigne C; Muylle K; Bourgeois P; Bourgegis D; Dierickx L; Flamen P [18] FFDG PET in Oncology: The best and the worst (Review). International Journal of Oncology 2006, 28, 1249–1261. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.