

Abstract
Monkeypox (MPXV) is a globally growing public health concern with 80,328 active cases and 53 deaths have been reported. No specific vaccine or drug is available for the treatment of MPXV. Hence, the current study also employed structure-based drug designing, molecular simulation, and free energy calculation methods to identify potential hit molecules against the TMPK of MPXV, which is a replicatory protein that helps the virus to replicate its DNA and increase the number of DNAs in the host cell. The 3D structure of TMPK was modeled with AlphaFold and screening of multiple natural products libraries (4,71,470 compounds) identified TCM26463, TCM2079, and TCM29893 from traditional Chinese medicines database (TCM), SANC00240, SANC00984, and SANC00986 South African natural compounds database (SANCDB), NPC474409, NPC278434 and NPC158847 from NPASS (natural product activity and species source database) while CNP0404204, CNP0262936, and CNP0289137 were shortlisted from coconut database (collection of open natural products) as the best hits. These compounds interact with the key active site residues through hydrogen bonds, salt bridges, and pie-pie interactions. The structural dynamics and binding free energy results further revealed that these compounds possess stable dynamics with excellent binding free energy scores. Moreover, the dissociation constant (KD) and bioactivity analysis revealed stronger activity of these compounds exhibit stronger biological activity against MPXV and may inhibit it in in vitro conditions. All the results demonstrated that the designed novel compounds possess stronger inhibitory activity than the control complex (TPD-TMPK) from the vaccinia virus. The current study is the first to design small molecule inhibitors for the replication protein of MPXV which may help in controlling the current epidemic and also overcome the challenge of vaccine evasion.
Keywords: Monkeypox, TMPK, Molecular screening, Drugs, Binding free energy, Bioactivity
1. Introduction
In the early 1970s in the Democratic Republic of Congo, a novel zoonotic virus was detected known as Monkeypox [1,2]. The virus was identified to cause smallpox in humans while its human-to-human remained efficient initially [3]. The majority of global Monkeypox epidemic transmissions have been linked to sex, and they have mostly only afflicted sexually active homosexual, bisexual, and other males who have sex with men (GBMSM) [4]. In high-income countries, sporadic cases of Monkeypox have also been reported, which are linked to the exotic pet trade and international travel. It is believed that the virus gains entry into the body through either the respiratory tract, broken skin, or the mucous membranes present in the eyes, nose, or mouth [5]. Since, May 2022, the active cases of Monkeypox are reported and in 50 countries the cases reached up to 78000 prompting WHO (world health organization) to declare it as “evolving threat of moderate public health concern” [[6], [7], [8]]. The common symptoms of Monkeypox virus disease include fever, muscle pain, swollen glands and other influenza like symptoms. Among the other potential complications that may arise include secondary infections, pneumonia, sepsis, encephalitis, and severe eye infections that can result in vision loss [9]. Infections with the human Monkeypox virus had a comparable prevalence in males and females in western and central Africa before 2017. However, a higher percentage of infections (65%) occurred in men during the Nigerian epidemic in 2017–18 [[10], [11], [12]]. Similarly, when describing the sexual target for the 2022 Monkeypox outbreak majority of the cases are male while few or no women case has been reported. The endemic is self-limited and usually, the case fatality is 1–10% only [13]. People living with HIV are at higher risk of getting Monkeypox virus disease as according to a study by the Centers for Disease Control and Prevention (CDC) revealed that among the total cases of mpox 41% are HIV-positive [14]. In contrast to normal patients, individuals exposed to MPXV and have HIV reported more severe symptoms than the normal patients. For instance, inflammation in the lining of the rectum and rectal perforation in the mpox infected HIV patients have been reported [13,15]. Interestingly, the current Monkeypox virus strain remained undetected in the non-endemic regions and mimic to be another type of STI (sexually transmitted disease) through evolutionary analysis. Consequently, the enduring worldwide epidemic of Monkeypox virus infection in humans’ advocates changes in the biologic features of the virus and changes in behavior, or both [16]. The smallpox vaccines i.e. ACAM2000, and the JYNNEOS can be used against Monkeypox but there is no real data available on how much protection is provided by these vaccines against Monkeypox [17]. Moreover, no other therapeutics such as drugs or other molecules to treat smallpox and Monkeypox. Even though the Monkeypox virus has circulated for decades in regions where it has traditionally been endemic, research into Monkeypox has been neglected and underfunded. Hence, this demands further research to develop further therapeutic choices before it becomes a global pandemic.
The proteome of the DNA containing the Monkeypox virus is comprised of 198 proteins that are involved in different cellular processes. Among these D10L, B4R, VITF3L, E8L, A42R, I1L, P28, PRO132, thymidylate kinase (TMPK) and G9R are the most essential proteins required for the pathogenesis, replication, and protection [18,19]. For instance, some studies have targeted these proteins using high-throughput molecular screening and computational approaches [20,21]. The Orthopoxviruses has this unique feature that it synthesizes its own TMPK which is required for efficient replication of the virus inside the host cell. TMPK synthesizes thymidine 5′-triphosphate (TTP) and regulates the DNA replication and different phases of the cell cycle [22]. Besides these, TMPK has been reported to directly interact with DNA to activate precursor nucleoside analogues and consequently affect the host genetic material replication too [[23], [24], [25]]. Furthermore, an alarming point is the emergence of the emergence of mutations in the replication complex of the Monkeypox virus is reported which may further aggravate the situation [26]. Considering the essential role of TMPK in viral replication, it serves as an essential therapeutic target for the treatment of Monkeypox and other Orthopoxviruses treatments. Therefore, quick efforts are needed to design small molecule therapeutics/drugs that could efficiently inhibit TMPK.
Natural products serves as a natural remedy for the treatment of different diseases since the first disease identification [27]. Plants-derived natural products are used against different diseases i.e. Alzheimer's disease (AD), Parkinson's Disease and the current pandemic agent SARS-CoV-2 [28]. For instance, Kaempferol has been reported to inhibit the main protease of SARS-CoV-2 identified from the Traditional Chinese Medicines (TCM) database [29]. Moreover, natural compounds for spike proteins, Mpro, PLpro, RdRp, and other targets have also been identified from the South African natural compounds database, NPASS, and Coconut natural compounds databases [[30], [31], [32], [33], [34]]. Identification of these molecules is fastened by the use of state-of-the-art computational methods which include molecular screening, simulation, and free energy calculation methods [35].
Keeping in view the pharmacological potential of natural products, the speed and accuracy of computational methods in discovering novel pharmacological molecules, the current study also uses similar approaches to target the TMPK of the Monkeypox virus. High throughput molecular screening TCM, SANCDB, NPASS, and Coconut databases were performed and potential lead molecules were identified. Using molecular simulation and free energy calculations these compounds were validated. From each database top three compounds were analyzed for binding modes. This study provides a structural basis for the inhibition of TMPK and consequently viral replication.
2. Materials and methods
2.1. Structural modelling of TMPK with AlphaFold
The crystallographic structure of TMPK of Monkeypox virus is not yet available therefore we used AlphaFold 2.0 to model the 3D structure of the target protein [36]. For this purpose, the TMPK structure from vaccinia virus with PDB ID: 2V54 was considered as a template for modeling [37]. The pLDDT score was considered as an evaluation criterion for the validation of the structure. Further validations such as stereochemical quality were performed by using SAVES web server to determine the structural accuracy and folding. Moreover, further validations were also performed using ProSA-web and PROCHECK to determine the residue distribution and bonding angle using by plotting the Ramachandran plot [38,39]. The constructed structure was validated and minimized before further processing. Active site residues were identified prior to a molecular screening of drug libraries. THYMIDINE-5′-DIPHOSPHATE (TDP) bound to TMPK from the vaccinia virus was considered as a control to compare all the experiments.
2.2. Molecular screening of natural products libraries
The drug libraries including traditional Chinese medicines (TCM) http://tcm.cmu.edu.tw/, NPASS (natural product activity and species source database) https://bidd.group/NPASS/, SANCDB (south African natural compounds database) http://african-compounds.org/about/afrodb/and coconut (collection of open natural products) https://coconut.naturalproducts.net/were downloaded and processed to generate the correct required format [[40], [41], [42], [43]]. These databases were screened for Lipinski's rule of five (R5) to filter out the violating and toxic compounds using FAF4drug online webserver. Moreover, the obtained R5-obeying compounds from each database were screened against the active site of TMPK using smina (Scoring and Minimization with AutoDock Vina) [44]. It supports scoring function development and high-performance energy minimization. The best hits from the smina screening were then re-evaluated by using ADFR which performed flexible docking of the compounds with increased accuracy by employing the improved version of AutoDock 4 scoring function [45]. Finally, the top three hits from each database were subjected to visual analysis using PyMOL and Schrodinger Maestro (free academic version for visualization) and molecular simulation for further validations [46,47].
2.3. Molecular simulation of top scoring hits
The top scoring hits and control drug based on the high docking scores from each database were subjected to molecular simulation and free energy calculation-based validation was performed using AMBER20 [48,49]. For simulation, the drug topologies were generated and processed using antechamber and parmchk2 [50]. Topology and coordinates files were used to minimize each complex in two stages: 1) the first round of minimization of 12000 steps and 2) the second round of minimization for 6000 steps was achieved. Each complex was sequentially heated and equilibrated for 50ns. In the production stage, 300ns simulation for each complex was performed. The simulation was accelerated by using the GPU version of PMEMD.cuda and trajectories were processed by using PTRAJ and CPPTRAJ [51,52].
2.4. Post-simulation analysis of the protein-ligand complexes
The stability was computed as root mean square deviation (RMSD), the flexibility index of each residue was estimated through root mean square flexibility (RMSF), the protein size was calculated as Rg (radius of gyration) while the hydrogen bonding in each computed through h-bonds function.
2.5. Binding free energy calculation using MM/GBSA approach
The end-point total binding free energy was estimated by using the molecular mechanics generalized Born surface area (MM/GBSA) method which is arguably the most extensively employed and accurate approach. Using the MMPBSA.py script the stabilized fractions from the simulation trajectories were subjected to free energy calculations [53]. As a part of the total binding free energy, vdW which stands for van der Waal energy, electrostatic energy and GB (generalized Born surface area) were estimated using the following equations.
In this equation, ΔG bind represents total free binding energy, while others show the free energy of the complex, the protein, and the ligand. Specific energy term contributes to the whole Free energy was calculated by the equation:
Gbond, Gelectrostatic, and GvdW specify interactions among bonded, electrostatic, and van der Waals states. In contrast, Gpolar and Gnon-polar represent the polar and non-polar interaction to the free energy presumed through precise GB (Generalized Born).
This free energy calculation method is widely used by different studies to understand the binding energy of different ligands [30,54].
2.6. Dissociation constant (KD) and bioactivity prediction
The dissociation constant (KD) and bioactivity for the top hits were computationally predicted by using PRODIGY-LIGAND (https://wenmr.science.uu.nl/prodigy/lig) and Molinspiration (https://www.molinspiration.com/cgi-bin/properties) webservers [55]. These servers are previously used to predict the KD and bioactivity of different molecules used against different diseases [56].
3. Results
The advanced computational modeling and simulation approaches have greatly accelerated the drug development cycle. Hence, the current study also employed structure-based drug designing, molecular simulation, and free energy calculation methods to identify potential hit molecules against the TMPK of Monkeypox virus. As mentioned earlier it is a replicatory protein that helps the virus to replicate its DNA and increase the number of DNAs in the host cell. Hence, targeting TMPK would greatly reduce the disease burden. We modeled the 3D structure of TMPK and presented in Fig. 1 a. Using AlphaFold the structure revealed a pLDDT score of >90% which shows good structure folding. This approach is currently the most reliable and accurate approach for predicting the correct folding of a protein. The structure of TMPK from MPXV contains right alpha-helices, five β–strands, and eight loops that connect each alpha helix and β–strand. Structural comparison analysis of TMPK from MPXV and Vaccinia virus (Vac-V) revealed an RMSD difference of 0.736 Å with a similar secondary structural distribution. The active site of the MPXV was identified based on the bound ligand in the active site of Vac-V. A superimposed structure of TMPK from MPXV and Vac-V is given in Fig. 1b. Moreover, the structure was validated further for proper and accurate folding using ERRAT, VERIFY-3D, WHATCHECK, PROCHECK, and ProSA-web tools. ERRAT revealed the overall quality factor of the structure above 90% while VERIFY-3D demonstrated that more than 80% of the residues have averaged 3D-1D score≥0.1 which shows the folding reliability of the TMPK. Moreover, ProSA-web was used to calculate the Z score which shows the x-ray and NMR similarity of the predicted structure compared with all the available structures in the protein databank. The more negative the Z score the best the structural folding. Herein, our modeled structure revealed a Z score of −6.54 which further validates the accuracy of the method. On the other hand, Ramachandran plot analysis is the best approach to validate the structure by demonstrating the bonding angle, torsion, and other stereochemical features. The results revealed that 91.3% of the total residues lie in the core region, 7.1% of the total residues are in the allowed regions, 1.1% of the total residues are in the general region while only 0.5% of the total residues lie in the disallowed region. This shows that the predicted structure has high-quality bonding angles, torsion, and folding. The visual results for ProSA-web and Ramachandran plots are shown in Fig. 1c and d.
Fig. 1.

Structural modeling and comparison of TMPK from MPXV. (a) Represent the modeled 3D structure of TMPK from MPXV where each secondary element i.e. alpha-helix, beta-strand, and loops are labeled. (b) Shows the superimposed structure of TMPK from MPXV (green) and Vac-V (magenta). An RMSD difference of 0.736 Å was calculated using PyMOL.
3.1. Binding modes of top hits from the Traditional Chinese Medicines database (TCM)
TCM medicines are always a great source of medication for the treatment of different diseases. The database containing ∼56000 compounds from traditional Chinese herbs were filtered for Lipinski's rule of five which revealed 38000 compounds as potential R5-obeying compounds while the rest were discarded. In the initial screening, approximately 380 compounds were reported to have docking scores greater than −5.0 kcal/mol. In the second round using the Induced-Fit docking (IFD) approach three compounds i.e., TCM26463 (3-Methoxytyramine-βxanthin), TCM2079 and TCM29893 (5-O-Feruloylquinic acid) were obtained to have the best docking scores than all the screened compounds. For TCM26463 the docking score was calculated to be −11.59 kcal/mol, for TCM2079-TMPK complex the docking score was reported to be −10.28 kcal/mol while for TCM29893-TMPK the docking score was reported to be −9.57 kcal/mol respectively. This compound establishes five hydrogen bonds with residues including Asp13, Lys14, Thr18, Asp92, Ser97 and Glu142 respectively. The compound also reported a single salt bridge with Asp13 and a single pie-pie stacking with Phe68. This shows a more robust interaction of 3-Methoxytyramine-βxanthin with TMPK thus blocking the key amino acids from the essential function. TCM2079 on the other hand interacted with TMPK by establishing four hydrogen bonds and a single salt bridge. Among the four hydrogen bonds, Lys14, Phe38, Arg93 and Glu145 are involved while the only salt-bridge was established by Lys17 residue. Lys14, Asp38, Arg93, and Ser97 were involved in hydrogen bonding while a single pie-pie bond was established by Phe68 residue of TMPK. It can be seen that these compounds uniformly block the same essential residues and exhibit anti-viral activities thus making it a potential pharmacological choice for the inhibition of the Monkeypox virus and the treatment of pox disease. The binding modes are visualized in Fig. 2 a–c.
Fig. 2.

Binding mode of top three hits from TCM database against TMPK. (a) Represent the binding mode of TCM26463 with TMPK. (b) Shows the binding mode of TCM2079 with TMPK. (c) The binding mode of TCM29893 with TMPK. The complexes were visualized for interactions in Schrodinger Maestro (free academic version for visualization only).
3.2. Binding modes of top hits from SANCDB
The South African natural compounds database is a rich source of remedies for a vast number of diseases. Molecular screening of SANCDB revealed SANC00240, SANC00984 and SANC00986 as the best hits among the 600 compounds. The docking score for this compound was reported to be −8.63 kcal/mol. In terms of interaction, SANC00240 established four hydrogen bonds and one pie-pie stacking interaction. Interesting the interacting residues align with the previous compounds reported in TCM database. The binding residues include Lys14, Arg41, Ser97 and Glu142 while the only pie-pie interaction was established by Phe68 residue. This shows consistent interactions of these small molecules with the essential residues of TMPK. It has a docking score of −8.12 kcal/mol with four hydrogen bonds involving Phe38, Arg41, Arg72 and Glu145 while the only pie-pie stacking was established by Phe68. Similarly, Dibenzo-1,4-dioxin-1,3,6,8-tetraol (SANC00986) isolated from the brown alga, Ecklonia maxima (Osbeck) Papenfuss demonstrated a docking score of −8.26 kcal/mol. The interaction pattern of SANC00986 revealed that this molecule interacts with TMPK through hydrogen bonds involving Asp13, Phe38, Arg93 and Ser97. Moreover, a single pie-pie interaction was established by Phe68. Over these compounds shows similar interaction pattern with the previous compounds thus showing potential pharmacological activity against TMPK. The interaction pattern of each compound is shown in Fig. 3 a–c.
Fig. 3.

Binding mode of top three hits from SANCDB database against TMPK. (a) Represent the binding mode of SANC00240 with TMPK. (b) Shows the binding mode of SANC00984 with TMPK. (c) The binding mode of SANC00986 with TMPK. The complexes were visualized for interactions in Schrodinger Maestro (free academic version for visualization only).
3.3. Binding modes of top hits from NPASS database
With over 30,926 natural compounds piled in the NPASS database serve as the best source for designing natural products based remedies for different diseases. Among the 30,926, only 21000 were successful in passing the R5 criteria. Molecular screening of these 21,000 compounds revealed NPC474409, NPC278434, and NPC158847 as the best hits from this database. NPC474409 demonstrated a docking score of −9.73 kcal/mol with only two hydrogen bonds including Phe38 and Ser97. The only pie-pie interaction was established by Phe68 aligned with the previous compounds. Moreover, NPC278434 is an Indole-3-Carboxylic Acid isolated from various plant species with anti-inflammatory activity and demonstrated a docking score of −8.60 kcal/mol [62]. Likewise, NPC474409 also reported three interactions including Phe38, Ser97 and Phe68. 1,3-Dimethyl-7,9-Dihydropurine-2,6,8-Trione, NPC158847, has anti-colorectal cancer and anti-inflammatory activities [63]. Here in this study, this compound reported a docking score of −7.13 kcal/mol against the TMPK protein from the Monkeypox virus. Among the key interactions Phe38, Arg72, Tyr94, Tyr101 and Phe68 are involved. The identified compounds from this database possess smaller molecular structures with potential pharmacological activity against the TMPK of MPXV. The interaction pattern of each compound is shown in Fig. 4 a–c.
Fig. 4.

Binding mode of top three hits from NPASS database against TMPK. (a) Represent the binding mode of NPC474409 with TMPK. (b) Shows the binding mode of NPC278434 with TMPK. (c) The binding mode of NPC158847 with TMPK. The complexes were visualized for interactions in Schrodinger Maestro (free academic version for visualization only).
3.4. Binding modes of top hits from coconut database
The coconut natural products (NPs) database exhibits 4, 06747 unique NPs, among which 2, 75233 only obey R5 rules. Screening of these compounds revealed 19,680 as the best hits while 11 compounds were found to be the best only. We selected three compounds i.e. CNP0404204, CNP0262936 and CNP0289137 as the best hits based on the docking scores. CNP0404204 is Cimicifugic Acid A isolated from Actaea dahurica and Actaea elata exhibit a docking score of −12.07 kcal/mol. This compound established eight hydrogen bonds, one pie-pie interaction, and one pie-cation interaction. Among the hydrogen bonds Asn37, Phe38, Arg41, Asp92, Arg93, Ser97 and Glu145 while Phe68 is involved in pie-pie interaction and Arg93 is involved in Pie-cation interaction. Overall, this compound exhibits a better docking score and has a higher number of interactions among all. This compound act as a pharmacological choice for the treatment of MPXV. 2,3,4-trihydroxy-5-{[3-(4-hydroxyphenyl) prop-2-enoyl] oxy} hexanedioic acid, CNP0262936, is Glucuronic acid derivatives exhibit a docking score of −11.39 kcal/mol. With six hydrogen bonds including Lys14, Thr18, Arg41, Arg93, Ser97 and Glu145 while Phe68 was reported to be involved in pie-pie stacking. The 4,4′-bis(2-methylpropanamido)-[1,1′-biphenyl]-3,3′-dicarboxylic acid on the other hand reported seven hydrogen bonds, one pie-cation and one pie-pie stacking. The docking score for this compound was reported to be −10.63 kcal/mol. Among the hydrogen bonding interactions Lys14, Phe38, Arg41, Arg93, Ser97 and Tyr144 are involved while Phe68 and Arg93 are involved in pie-pie stacking and pie-cation interaction. These shortlisted compounds exhibit the best docking scores and the interaction paradigm covers the whole active site blocking the key residues to enforce the pharmacological potential of these small molecules. The interaction pattern of each compound is shown in Fig. 5 a–c.
Fig. 5.

Binding mode of top three hits from Coconut database against TMPK. (a) Represent the binding mode of CNP0404204 with TMPK. (b) Shows the binding mode of CNP0262936 with TMPK. (c) The binding mode of CNP0289137 with TMPK. The complexes were visualized for interactions in Schrodinger Maestro (free academic version for visualization only).
3.5. Dynamic stability assessment of top-scoring compounds
Assessing the dynamic stability of a protein bound to a ligand is essential to demonstrate the pharmacological activity of that particular compound. For instance, the stable binding of a ligand to the active pocket of protein is associated with better pharmacological potential than the relatively unstable one. The stability of a simulation trajectory can be demonstrated by using the RMSD function integrated with simulation tools. Thus to understand whether these compounds remain stable or not during simulation we also calculated RMSD for these trajectories as a function of time. The RMSD of each complex is shown in Fig. 6 a-6l. TPD in complex with TMPK from vaccinia virus was used as a control to compare the simulation results. The control complex reported a stable RMSD with an average of 1.5 Å. No significant perturbation was experienced except minor deviations before 150ns and 225ns. Overall, the complex reported stable dynamics. The top three compounds from TCM database reported stable dynamic behaviour. The compound TCM26463 stabilized at 1.2 Å soon after reaching 5ns. No significant deviation was observed during the simulation except a minor increment in the RMSD graph at 45–50 and 170–175ns. However, the complex demonstrated stable dynamics with an average RMSD of 1.10 Å. In the case of TCM2079 in complex with TMPK the RMSD initially remained stable with no major deviation until 160ns, however, then two major deviations between 165 and 225ns were observed. Then the RMSD gradually decreased and increased following this pattern until the end of 300ns simulation. An average RMSD for this complex was estimated to be 1.5 Å. In the TCM complex, the best hit i.e. TCM29893 initially reported a minor increase in the RMSD value until 100ns, and then an abrupt decline in the RMSD graph was seen. After 125ns, the RMSD increased back and stabilized at 1.25 Å and continued to follow the same pattern until 205ns. Afterward a minor increment was observed but the complex then stabilized at the same point and remained the same until 300ns. In sum the three compounds from TCM database reported stable dynamics, however, a higher fold of stability was favoured in the TCM26463 complex. The RMSD graphs for the top three hits from TCM database in complex with TMPK are given in Fig. 6a–c.
Fig. 6.

Dynamic stability analysis as RMSD of each hit from four databases in complex with TMPK. (a–c) represent the RMSDs for the TPD-TMPK (control) and top hits from TCM database with TMPK. (d–f) represent the RMSDs for the TPD-TMPK (control) and top hits from the SANC database with TMPK. (g–i) represent the RMSDs for the TPD-TMPK (control) and top hits from the NPASS database with TMPK. (j–l) represent the RMSDs for the TPD-TMPK (control) and top hits from the coconut database with TMPK.
The top hits from SANCDB were also subjected to molecular simulation-based stability analysis which revealed stable dynamic behaviour. The first top hit from SANCDB i.e. SANC00240 demonstrated alike behaviour as TCM26463. The RMSD of the complex stabilized at 1.2 Å with only a minor deviation between 60 and 80ns was observed. The complex then demonstrated a uniform straight graph of RMSD which determines the stable behaviour of this complex during the simulation. An average RMSD for this complex was calculated to be 1.21 Å. On the other hand, the SANC00984 though demonstrated an overall stable behaviour however, minor deviations between 100-140ns and 260–270ns were experienced. No significant perturbation was recorded however a comparatively higher RMSD value was calculated for this complex. An average RMSD for this complex was computed to be 1.38 Å. For the SANC00986 despite the very stable dynamics initially, an increase in the RMSD was recorded until 25ns. Afterward the RMSD abruptly declined backed but minor gradual increase was observed until the end of the simulation. An average RMSD for this complex was estimated to be 1.44 Å. The dynamic stability assessment of these complexes shows stable pharmacological behaviour and thus may produce better pharmacological efficacy in in vitro setup. The RMSD graphs for the top three hits from SANCDB database in complex with TMPK are given in Fig. 6d–f.
The binding stability of the top hits from the NPASS database i.e. NPC15847, NPC278434 and NPC474409 was also demonstrated from the simulation trajectories. As given in Fig. 6g, the NPC158847 reported stable dynamic behaviour with no significant structural perturbation. Although the RMSD gradually increased over time reaching 1.56 Å a maximum but a uniform straight RMSD was demonstrated and thus behave stably during the simulation. In contrast, the NPC278434 reported significant structural perturbation after 75ns. The RMSD continues to increase with abrupt increase and decrease between 80 ns and 120 ns. The complex demonstrated a uniform RMSD pattern between 125 and 250ns with the gradual increasing trend but then significant perturbations were seen in the complex at the end of the simulation. On the other hand, with a single peak in the RMSD graph at 198ns the NPC474409 demonstrated alike stable uniform stability behaviour as NPC158847. An average RMSD for NPC474409 complex was estimated to be 1.48 Å. Similarly, these compounds also induce sable pharmacological properties against TMKP in the all-atoms simulation setup and thus could act as a potential choice for the inhibition of MPXV replication. The RMSD graphs for the top three hits from NPASS database in complex with TMPK are given in Fig. 6g–i.
The RMSDs for the top hits from Coconut NPs database were also computed to reveal the stability profiles of these molecules. Among the top hits such as CNP0292636, CNP0289137 and CNP0404204 were subjected to RMSD analysis. The CNP0292636 complex initially reported higher RMSD which then gradually decreased after 140ns and remained lower till the end of the simulation. Furthermore, the other two compounds i.e. CNP0289137 and CNP0404204 demonstrated alike behaviour with no significant perturbation during the simulation and stabilized at 1.45 Å each. The CNP0289137 demonstrated a minor deviation between 101 and 125 ns but no major deviation was recorded. The RMSD for the CNP0404204 increased at the end of the simulation after 150ns. Although a higher RMSD was recorded but overall no structural perturbation was observed. The RMSD graphs for the top three hits from the coconut database in complex with TMPK are given in Fig. 6j–l. It can be seen that the control complex though reported stable RMSD but all the complexes reported comparatively lower RMSD and thus show more stable dynamics by the novel compounds in complex with TMPK. Overall, these results show that these compounds are the potential therapeutic choices for the treatment of MPXV based on replication inhibition through direct interaction with the DNA-binding protein TMPK.
3.6. Structural packing and protein size estimation for the top complexes
Protein packing determination or protein size estimation demonstrates information regarding the essential events that occurred during the molecular simulation. It has been a widely applicable approach to understand variations in the protein size or binding and unbinding events. This approach has been previously used to understand how the interaction between different proteins/protein-ligand affects the dynamics of a protein. Hence, considering the wider applicability we estimated the structural compactness of each complex during simulation by calculating the radius of gyration (Rg) as a function of time. For instance, the control complex reported a uniform Rg with an average Rg 16.70 Å. Consistent with the RMSDs results, compounds from TCM database demonstrated a similar Rg pattern as the RMSD. The TCM26463 overall presented a uniform Rg pattern thus convey a stable binding of the ligand with no significantly increase or decrease in the protein size during simulation. An average Rg of 16.70 Å was calculated for the TCM26463-TMPK complex. On the other hand, the TCM2079 also presented a similar pattern of Rg as the RMSD. Initially, the Rg continues to increase and after 125ns–175ns abrupt increases in the Rg were seen however after 175ns the Rg decreased back and stabilized at 16.80 Å. This could be due to the binding and unbinding or loop movement which causes an increase and decrease in the protein size. An average Rg for TCM2079 was estimated to be 16.83 Å. Furthermore, with the initially trajectory increasing the Rg value an abrupt peak before 100ns was seen for the TCM29893-TMPK complex. The Rg value increased and decreased back between 200 and 250ns and then stabilized by demonstrating lower Rg values. An average Rg for TCM29893-TMPK complex was calculated to be 16.76 Å. Overall these results greatly align with the RMSD results showing the reliability and consistency of the findings. The Rg graphs for the TCM top hits are given in Fig. 7 a–c.
Fig. 7.

Structural compactness analysis as Rg of for the TPD-TMPK (control) and each hit from four databases in complex with TMPK. (a–c) represent the Rg(s) for the TPD-TMPK (control) and top hits from the TCM database with TMPK. (d–f) represent the Rg(s) for the TPD-TMPK (control) and top hits from the SANC database with TMPK. (g–i) represent the Rg(s) for the TPD-TMPK (control) and top hits from the NPASS database with TMPK. (j–l) represent the Rg(s) for the TPD-TMPK (control) and top hits from the coconut database with TMPK.
The structural compactness of each complex from SANCDB was also estimated to see the variations caused during simulation. Interestingly the Rg patterns for the top complexes from SANCDB strongly aligns with the RMSD results. It can be seen that the Rg of SANC00240 started to increase initially reaching to 16.78 Å and stabilized at 10 ns? The Rg then demonstrated a straight graph with no significant changes in the protein size was observed, however, an abrupt decline in the Rg was observed at 175 ns? This short decline in the Rg then increased back after a shorter period and continued to remain consistent until the end of the simulation. An average Rg for this complex was estimated to be 16.78 Å. Consistent with the SANC00984 RMSD results, the Rg pattern also reported a gradual increase in the Rg trajectory. The Rg started from 16.50 Å and reached to a maximum of 17.50 Å at 300 ns? This shows significant structural perturbation occurred during the simulation and thus causes binding and unbinding of the ligand in the pocket. An average Rg was calculated to 17.32 Å. Unlike the SANC00984, the SANC986-TMPK complex demonstrated a stable Rg graph as SANC00240. The graph initially fluctuated but after reaching 25ns the Rg decline back and continues to follow the same pattern till the end of the simulation. A continuous decrease in the Rg pattern was seen and the minimum Rg was observed during the last time of the simulation. An average Rg of 16.70 Å was calculated for this complex. The Rg graphs for the SANCDB top hits are given in Fig. 7d–f.
The NPASS database top hits though demonstrated a little higher but comparatively more stable Rg for all the complexes. The Rg for NPC158847-TMPK complex started to increase initially and reached 17.10 Å at 15ns and then equilibrated. A straight uniform Rg was seen until 100ns and then an abrupt decline was observed in the Rg pattern. The Rg then continues to remain lower and no deviation was observed until the end of the simulation. An average Rg of 17.0 Å was calculated for this complex. The Rg for NPC278434-TMPK demonstrated a floating behaviour during 1–75 ns but then stabilized at 16.70 Å and continues to follow this pattern. No significant deviations were observed till the end of the simulation. An average Rg for this complex was estimated to be 16.68 Å. On the other hand, the Rg for NPC474409-TMPK stabilized at 16.80 Å and continues to follow the same pattern till 125ns however then a continuous increase and decrease behaviour in the protein size was observed till the end of the simulation. An average Rg for this complex was estimated to be 17.0 Å. The Rg graphs for the NPASS database top hits are given in Fig. 7g–i.
Furthermore, the Rg pattern for the Coconut database best hits compounds also demonstrated alike behaviour as the RMSD. The Rg for the CNP0292636-TMPK complex demonstrated a continuous gradual increase in the Rg graph. The Rg started from 16.60 Å and reached to the maximum 16.90 Å at the end of the simulation. An average Rg was calculated to be 16.80 Å for this complex. Similarly, the Rg for the CNP0289137-TMPK complex started the Rg from 16.60 Å and demonstrated a floating behaviour till the end of the simulation. The Rg pattern for the CNO0404204-TMPK complex is more like the CNP0289137-TMPK complex. An average Rg for both of these complexes were calculated to be 16.92 Å and 16.96 Å respectively. The Rg graphs for the coconut database top hits are given in Fig. 7j–l. In conclusion, the current findings demonstrated stable protein compactness with similar average Rg values and minimal unbinding events determine the pharmacological potential of these hits that could potentially lead to the termination of replication of MPXV by targeting TMPK with these compounds.
3.7. Hydrogen bonding analysis of the top hits
Hydrogen bonds among intermolecular chemical interactions have a significant role in influencing the extent of binding. This analysis helps to comprehend the binding conformation stability of the interacting partners, which further suggests the likelihood of molecular binding and disease control. Herein, we calculated the intermolecular hydrogen bonding over the simulation time. We calculated the total number of hydrogen bonds in each complex subjected to molecular simulation. Using the simulation trajectories average number of hydrogen bonds were estimated and presented in Fig. 8 a-8l. For the TPD-TMPK from the vaccinia virus reported 100 average bonds. In the case of the TCM database average number of hydrogen bonds in each complex was calculated to be 106 in TCM26463, 105 in TCM2079, and 99 in TCM29893. Moreover, in SANCDB the average number of hydrogen bonds were calculated to be 101 in SANC00240, 100 in SANC00984 while 106 were estimated in SANC00986. Moreover, in NPC158848 the average hydrogen bonds counts were 106, 104 in NPC278434 and 101 in NPC474409. Interesting the average number of hydrogen bonds in the coconut database top hit compounds were also similar to the aforementioned compounds. In CNP0292636 average hydrogen bonds were estimated to be 102, in CNP0289137 average number of hydrogen bonds were 106 while 92 in CNP0404204. The bonding graphs given in Fig. 8a–l shows a significant number of variations in the hydrogen bonds which consequently produce differential inhibitory effects and reduces the pathological role of TMPK in MPXV infection.
Fig. 8.

Hydrogen bonding analysis of the TPD-TMPK (control) and each hit from four databases in complex with TMPK. (a–c) represent the hydrogen bonds of the TPD-TMPK (control) and for the top hits from the TCM database with TMPK. (d–f) represent the hydrogen bonds of the TPD-TMPK (control) and for the top hits from the SANC database with TMPK. (g–i) represent the hydrogen bonds of the TPD-TMPK (control) and for the top hits from the NPASS database with TMPK. (j–l) represent the hydrogen bonds of the TPD-TMPK (control) and for the top hits from the coconut database with TMPK.
3.8. Residues flexibility indexing
Furthermore, the residues flexibility which supplies essential information regarding the molecular patterns of interaction, inter-residues communication, protein coupling, inhibition potential, biocatalysis and enzyme engineering was calculated for each residue and presented in Fig. 9 a–d. The residues flexibility of the control complex was observed to be similar to the top hits’ complexes. In the case of TCM database, all the compounds presented an almost similar pattern of residues flexibility. The region between 100 and 125 presented higher flexibility in TCM2079 complex only while the regions between 140-150 and 165–175 demonstrated higher fluctuation in all the complexes. The region 100–125 is a loop which connects the H3 and H4 while the 140–150 and 165–175 are also loop regions that connect alpha-helices and beta-sheets. Moreover, in case of SANCDB compound SANC00984 demonstrated significantly higher residues flexibility in the regions 40–75 and 135–150. These regions also comprise of loops that connect alpha-helices and beta sheets with each other. The NPASS complexes demonstrated differential flexibility effects where higher flexibility was recorded in regions 10–60 and 125–150. These regions are corresponding to loops regions. Furthermore, the significant fluctuation was observed in coconut database in which almost all the regions comparatively demonstrated higher fluctuations except 75–125. The RMSF graphs for each complex are shown in Fig. 9a–d. Overall this shows that the binding of each ligand affects the internal dynamics differentially and the regions 125–150 cover the active site residues hence, imply that the movement of this loop helps the drug to optimize in the cavity by increasing the volume of the pocket.
Fig. 9.

Residues flexibility analysis of each hit from four databases in complex with TMPK. (a) Represent the RMSF for the top hits from the TCM database with TMPK. (b) Represent the RMSF for the top hits from the SANC database with TMPK. (c) Represent the RMSF for the top hits from the NPASS database with TMPK. (d) Represent the RMSF for the top hits from the coconut database with TMPK.
3.9. Binding free energy calculation
Calculation of the binding energy is an imperative assessment to re-evaluate the binding conformation and accuracy of the interacting partner. This method has the advantage of higher accuracy and is least expensive in contrast to other. It has been previously implicated to determine the binding energy for small molecules targeting the other receptors and druggable proteins of SARS-CoV-2. Considering the wider applicability of the method, we have also computed the binding free energy for the top hits from each database using MD trajectories. We calculated the total binding energy (ΔG) and presented in Table 1 . The TBE for TCM26463 was calculated to be −126.09 kcal/mol, for TCM2079 the TBE was −79.72 kcal/mol while for TCM29893 the TBE was estimated to be −61.37 kcal/mol. On the other hand, the SANCDB best hits reported the TBE as: SANC00240–70.69 kcal/mol, for SANC00984 the TBE was −68.52 kcal/mol while for SANC00986 the TBE was −53.61 kcal/mol. Moreover, the TBE for NPASS best hits was estimated to be −44.03 kcal/mol for NPC474409, for NPC278434 the TBE was −38.69 kcal/mol while for NPC158847 the TBE was calculated to be −36.56 kcal/mol. Finally, the total binding energy for each complex from coconut database was estimated to be −69.21 kcal/mol for CNP0404204, for CNP0262936 the TBE was −74.41 kcal/mol while CNP0289137 demonstrated a TBE of −66.14 kcal/mol. On the other hand, the control complex (TPD-TMPK) reported the TBE of −50.63 kcal/mol. Overall the results reports revealed that these compounds exhibit stronger binding energy for the active site residues of TMPK and may play significant role in the inhibition of replication of MPXV. The TBE results are presented in Table 1.
Table 1.
3D structures, Database ID, KD, bioactivity and total binding free energy for the top hits from each database are given.
| S. No | 2D structure | Database ID | KD (ΔG) | Bioactivity | ΔG (kcal/mol) |
|---|---|---|---|---|---|
| 1. | 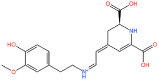 |
TCM26463 | −12.44 | 0.16 | −126.09 |
| 2. | TCM2079 | −11.63 | 0.35 | −79.72 | |
| 3. | 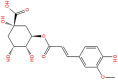 |
TCM29893 | −9.87 | 0.28 | −61.37 |
| 4. | 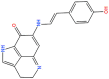 |
SANC00240 | −10.25 | 0.41 | −70.69 |
| 5. | 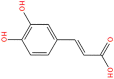 |
SANC00984 | −10.14 | 0.33 | −68.52 |
| 6. | 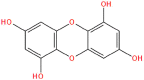 |
SANC00986 | −8.97 | 0.27 | −53.61 |
| 7. | 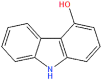 |
NPC474409 | −7.56 | −0.21 | −44.03 |
| 8. | 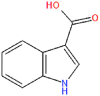 |
NPC278434 | −7.24 | −0.26 | −38.69 |
| 9. | 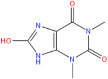 |
NPC158847 | −6.68 | −0.33 | −36.56 |
| 10. |
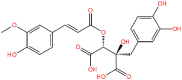 0 0 |
CNP0404204 | −12.13 | 0.31 | −69.21 |
| 11. | 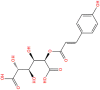 |
CNP0262936 | −12.62 | 0.24 | −74.41 |
| 12. | 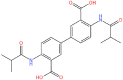 |
CNP0289137 | −11.98 | 0.33 | −66.14 |
| 13. | 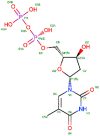 |
TPD (control) | −7.43 | 0.46 | −50.63 |
3.10. Dissociation constant and bioactivity analysis
PRODIGY-LIG generates the KD results as ΔG upon the submission of the complex. For the top hits the KD values were calculated to be TCM26463 (−12.44), TCM2079 (−11.63), TCM29893 (−9.87), SANC00240 (−10.25), SANC00984 (−10.14), SANC00986 (−8.97), NPC474409 (−7.56), NPC278434 (−7.24), NPC158847 (−6.68), CNP0404204 (−12.13), CNP0262936 (−12.62) and CNP0289137 (−11.98) respectively. The dissociation constant for the control complex was −7.30. This shows the stronger activity of these compounds against the TMPK. On the other hand, the bioactivity is calculated as −0.5 to 0.5. Interpretation of the results is such that a molecule having bioactivity score of more than 0.00 is most likely to exhibit considerable biological activities, while values 0.50 to 0.00 are expected to be moderately active and scores less than 0.50 is presumed to be inactive. The bioactivity for each of the top hit molecules was estimated to be TCM26463(0.16), TCM2079(0.35), TCM29893(0.28), SANC00240(0.41), SANC00984(0.33), SANC00986(0.27), NPC474409(−0.21), NPC278434(−0.26), NPC158847(−0.33), CNP0404204(0.31), CNP0262936(0.24) and CNP0289137(0.33) respectively. For the control complex (TPD-TMPK) the bioactivity was predicted to be 0.46. This shows that these compounds exhibit stronger biological activity against MPXV and may inhibit it in in vitro conditions. The KD and bioactivity results are given in Table 1.
4. Discussion
The current outbreak of MPXV has created an alarming situation with the potential to spread to many localities globally. Humans who get MPXV succumb to a serious febrile disease that may be fatal. Monkeypox virus infections do not yet have a particular antiviral therapy, and the only authorized vaccine is not advised for use in people with compromised immune systems [64]. Hence, designing effective small molecule therapy for the inhibition of the MPXV is highly recommended. Until now potential drug candidate's cidofovir and tecovirimat, ST-246 and CMX001 have been reported to have promising activity against MPXV in in vitro and in animal models [[65], [66], [67]]. The quest for a magical drug is still continue and therefore the current study also uses structure-based drug designing methods to identify potential hits for the inhibition of TMPK, a replicatory protein, of MPXV.
The current study uses structural modeling and simulation approaches to identify potential inhibitors for the inhibition of TMPK of MPXV to disintegrate the replication machinery of MPXV. Since the 3D structure of TMPK is not yet resolved experimentally, we used the most accurate and highly employed method alphaFold to model the structure of TMPK. For instance, this approach has been used to generate the 3D coordinates of the druggable proteins from MPXV. We used a molecular drug screening approach in various steps which revealed twelve potential hits from four different databases as the best inhibitors of TMPK. A similar approach has been used by other studies to identify or repurpose drugs against MPXV. Among the top hits many compounds are previously characterized to have anti-viral, anti-inflammatory, anti-cancerous or anti-bacterial activities. For instance, 3-Methoxytyramine-βxanthin is a phenethylamine alkaloid isolated from Polygonum cuspidatu has been previously characterized to exhibit anti-influenza activity against the oseltamivir-resistant H1 protein [57]. Moreover, 5-O-Feruloylquinic acid available in both Coffea arabica and Angelica taiwaniana possess antiviral activity against the SARS-CoV-2 [58]. The hits from the south afican database i.e. SANC00240, Makaluvamine M, is quinolone derivative from Strongylodesma aliwaliensis, a south African marine microorganism, possess anti-cancerous activity [59]. Similarly, SANC00984 which is a Caffeic acid isolated from different species of Salvia family from Africa [60]. NPC474409 which is 9H-Carbazol-4-Ol isolated from Murraya koenigii possess anti-oxidant activity [61]. In addition, NPC278434 is an Indole-3-Carboxylic Acid isolated from various plant species with anti-inflammatory activity and demonstrated a docking score of −8.60 kcal/mol [62]. Likewise, NPC474409 also reported three interactions including Phe38, Ser97 and Phe68. 1,3-Dimethyl-7,9-Dihydropurine-2,6,8-Trione, NPC158847, has anti-colorectal cancer and anti-inflammatory activities [63]. Characterization of the activity of these compounds by targeting wide range of disorders and infections demonstrates the promising potential of the shortlisted compounds against MPXV. Interestingly, brincidofovir, tecovirimat and auranofin have been previously identified to inhibit the replication of MPXV using computer simulation and in vitro experiments [68,69]. Similarly by combining high-throughput computational molecular screening and experimental methods also reported promising drugs that target A27 protein from MPXV. The models from these studies can be used to experimentally validate our hits and may be taken for further clinical consideration [70]. The structural dynamic features of our hits further demonstrated the robust pharmacological potential of our compounds in inhibiting the replication of MPXV. Moreover, binding free energy reports revealed that these compounds exhibit stronger binding energy for the active site residues of TMPK and may play a significant role in the replication of MPXV. In sum, these results hold promising clinical candidates for the treatment of mpox subjected to further in vitro and in vivo validations.
5. Conclusions
With the growing cases of monkeypox, the global health system is further pressurized that is already shaken by the recent COVID-19 pandemic. Hence, the current study employed structure-based drug designing, molecular simulation, and free energy calculation methods to identify potential hit molecules against the TMPK of Monkeypox virus. We identified TCM26463, TCM2079, TCM29893, SANC00240, SANC00984, SANC00986, NPC474409, NPC278434, NPC158847, CNP0404204, CNP0262936 and CNP0289137 from different database. The docking score, the simulation results, the binding free energy results, the dissociation constant and the bioactivity analysis confirmed the pharmacological activity of these compounds against MPXV by inhibiting the replicating protein TMPK. Further in vitro and in vivo tests are required to validate the activity of these compounds for potential clinical usage.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Funding
Dong-Qing Wei is supported by grants from the Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the National Science Foundation of China (Grant No. 32070662, 61832019, 32030063), the Science and Technology Commission of Shanghai Municipality (Grant No: 19430750600), as well as SJTU JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2021ZD02).
Availability of data and material
All the data is available on RCSB, UniProt and any simulation data would be provided on reasonable demand. The accession numbers to access this data are given in the manuscript.
Authors contribution
Conceptualization, Abbas Khan and Hafiza Qudsia; Data curation, Fahad M Alshabrmi; Formal analysis, Abbas Khan, Hafiza Qudsia and Yasir Waheed; Funding acquisition, Dongqing Wei; Investigation, Shoaib Adil and Yasir Waheed; Methodology, Shoaib Adil and Fahad M Alshabrmi; Resources, Yasir Waheed; Supervision, Dongqing Wei; Validation, Abbas Khan, Shoaib Adil and Hafiza Qudsia; Visualization, Fahad M Alshabrmi; Writing – original draft, Abbas Khan; Writing – review & editing, Abbas Khan and Dongqing Wei.
Declaration of competing interest
Declared none.
Acknowledgements
The computations were partially performed at the PengCheng Lab. and the Center for High-Performance Computing, Shanghai Jiao Tong University.
References
- 1.Ladnyj I., Ziegler P., Kima E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull. World Health Organ. 1972;46:593. [PMC free article] [PubMed] [Google Scholar]
- 2.Aplogan A., Szczeniowski M. Human monkeypox--kasai oriental, democratic republic of. MMWR (Morb. Mortal. Wkly. Rep.): Morb. Mortal. Wkly. Rep. 1997;46:1168–1171. [PubMed] [Google Scholar]
- 3.Durski K.N., McCollum A.M., Nakazawa Y., Petersen B.W., Reynolds M.G., Briand S., Djingarey M.H., Olson V., Damon I.K., Khalakdina A. Emergence of monkeypox—west and central Africa. Morb. Mortal. Wkly. Rep. 2018;67:306. doi: 10.15585/mmwr.mm6710a5. 1970–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girometti N., Byrne R., Bracchi M., Heskin J., McOwan A., Tittle V., Gedela K., Scott C., Patel S., Gohil J. Demographic and clinical characteristics of confirmed human monkeypox virus cases in individuals attending a sexual health centre in London, UK: an observational analysis. Lancet Infect. Dis. 2022;22:1321–1328. doi: 10.1016/S1473-3099(22)00411-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris E. What to know about monkeypox. JAMA. 2022;327:2278–2279. doi: 10.1001/jama.2022.9499. [DOI] [PubMed] [Google Scholar]
- 6.Bunge E.M., Hoet B., Chen L., Lienert F., Weidenthaler H., Baer L.R., Steffen R. The changing epidemiology of human monkeypox—a potential threat? A systematic review. PLoS Neglected Trop. Dis. 2022;16 doi: 10.1371/journal.pntd.0010141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guarner J., Del Rio C., Malani P.N. Monkeypox in 2022—what clinicians need to know. JAMA. 2022;328:139–140. doi: 10.1001/jama.2022.10802. [DOI] [PubMed] [Google Scholar]
- 8.Thornhill J.P., Barkati S., Walmsley S., Rockstroh J., Antinori A., Harrison L.B., Palich R., Nori A., Reeves I., Habibi M.S. Monkeypox virus infection in humans across 16 countries—april–June 2022. N. Engl. J. Med. 2022;387:679–691. doi: 10.1056/NEJMoa2207323. [DOI] [PubMed] [Google Scholar]
- 9.Ola P. 2022. The Origin of the Mysterious Multi-Country Monkeypox Outbreak in Non-endemic Countries. [Google Scholar]
- 10.Doshi R.H., Guagliardo S.A.J., Doty J.B., Babeaux A.D., Matheny A., Burgado J., Townsend M.B., Morgan C.N., Satheshkumar P.S., Ndakala N. Epidemiologic and ecologic investigations of monkeypox, likouala department, republic of the Congo. Emerg. Infect. Dis. 2019;25:273. doi: 10.3201/eid2502.181222. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yinka-Ogunleye A., Aruna O., Dalhat M., Ogoina D., McCollum A., Disu Y., Mamadu I., Akinpelu A., Ahmad A., Burga J. Outbreak of human monkeypox in Nigeria in 2017–18: a clinical and epidemiological report. Lancet Infect. Dis. 2019;19:872–879. doi: 10.1016/S1473-3099(19)30294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogoina D., Iroezindu M., James H.I., Oladokun R., Yinka-Ogunleye A., Wakama P., Otike-Odibi B., Usman L.M., Obazee E., Aruna O. Clinical course and outcome of human monkeypox in Nigeria. Clin. Infect. Dis. 2020;71:e210–e214. doi: 10.1093/cid/ciaa143. [DOI] [PubMed] [Google Scholar]
- 13.Thornhill J.P., Barkati S., Walmsley S., Rockstroh J., Antinori A., Harrison L.B., Palich R., Nori A., Reeves I., Habibi M.S., Apea V., Boesecke C., Vandekerckhove L., Yakubovsky M., Sendagorta E., Blanco J.L., Florence E., Moschese D., Maltez F.M., Goorhuis A., Pourcher V., Migaud P., Noe S., Pintado C., Maggi F., Hansen A.-B.E., Hoffmann C., Lezama J.I., Mussini C., Cattelan A., Makofane K., Tan D., Nozza S., Nemeth J., Klein M.B., Orkin C.M. Monkeypox virus infection in humans across 16 countries — april–june 2022. N. Engl. J. Med. 2022;387:679–691. doi: 10.1056/NEJMoa2207323. [DOI] [PubMed] [Google Scholar]
- 14.Boesecke C., Monin M.B., van Bremen K., Schlabe S., Hoffmann C. Severe monkeypox-virus infection in undiagnosed advanced HIV infection. Infection. 2022:1–2. doi: 10.1007/s15010-022-01901-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel A., Bilinska J., Tam J.C.H., Da Silva Fontoura D., Mason C.Y., Daunt A., Snell L.B., Murphy J., Potter J., Tuudah C., Sundramoorthi R., Abeywickrema M., Pley C., Naidu V., Nebbia G., Aarons E., Botgros A., Douthwaite S.T., van Nispen tot Pannerden C., Winslow H., Brown A., Chilton D., Nori A. Clinical features and novel presentations of human monkeypox in a central London centre during the 2022 outbreak: descriptive case series. BMJ. 2022;378 doi: 10.1136/bmj-2022-072410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Toole A., Rambaut A. ARTIC Network; 2022. Initial Observations about Putative APOBEC3 Deaminase Editing Driving Short-Term Evolution of MPXV since 2017. [Google Scholar]
- 17.Philpott D. vol. 71. Morbidity and Mortality Weekly Report; 2022. (Epidemiologic and Clinical Characteristics of Monkeypox cases—United States, May 17–July 22, 2022, MMWR). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soheili M., Nasseri S., Afraie M., Khateri S., Moradi Y., Mortazavi S.M.M., Gilzad-Kohan H. Monkeypox: virology, pathophysiology, clinical characteristics, epidemiology, vaccines, diagnosis, and treatments. J. Pharm. Pharmaceut. Sci. 2022;25:297–322. doi: 10.18433/jpps33138. [DOI] [PubMed] [Google Scholar]
- 19.Wang L., Shang J., Weng S., Aliyari S.R., Ji C., Cheng G., Wu A. Genomic annotation and molecular evolution of monkeypox virus outbreak in 2022. J. Med. Virol. 2022;95:1–7. doi: 10.1002/jmv.28036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng L., Meng J., Lin M., Lv R., Cheng H., Zou L., Sun J., Li L.X., Ren R., Wang S. Acta Materia Medica; 2022. Structure Prediction of the Entire Proteome of Monkeypox Variants. [Google Scholar]
- 21.Yang Q., Xia D., Syed A.A.S., Wang Z., Shi Y. Highly accurate protein structure prediction and drug screen of monkeypox virus proteome. J. Infect. 2022;86:66–117. doi: 10.1016/j.jinf.2022.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caillat C., Topalis D., Agrofoglio L.A., Pochet S., Balzarini J., Deville-Bonne D., Meyer P. Crystal structure of poxvirus thymidylate kinase: an unexpected dimerization has implications for antiviral therapy. Proc. Natl. Acad. Sci. USA. 2008;105:16900–16905. doi: 10.1073/pnas.0804525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia D.R., Souza F.R., Guimarães A.P., Valis M., Pavelek Z., Kuca K., Ramalho T.C., França T.C.C. 2021. Silico Studies of Potential Selective Inhibitors of Thymidylate Kinase from Variola Virus; p. 14. Pharmaceuticals (Basel, Switzerland) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Omari K., Solaroli N., Karlsson A., Balzarini J., Stammers D.K. Structure of vaccinia virus thymidine kinase in complex with dTTP: insights for drug design. BMC Struct. Biol. 2006;6:1–9. doi: 10.1186/1472-6807-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solaroli N., Johansson M., Persoons L., Balzarini J., Karlsson A. Substrate specificity of feline and canine herpesvirus thymidine kinase. Antivir. Res. 2008;79:128–132. doi: 10.1016/j.antiviral.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Kannan S.R., Sachdev S., Reddy A.S., Kandasamy S.L., Byrareddy S.N., Lorson C.L., Singh K. Mutations in the monkeypox virus replication complex: potential contributing factors to the 2022 outbreak. J. Autoimmun. 2022;133 doi: 10.1016/j.jaut.2022.102928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chevallier A. 2016. Encyclopedia of Herbal Medicine: 550 Herbs and Remedies for Common Ailments. Penguin. [Google Scholar]
- 28.Fan H.-M., Gu R.-X., Wang Y.-J., Pi Y.-L., Zhang Y.-H., Xu Q., Wei D.-Q. Destabilization of Alzheimer's Aβ42 protofibrils with a novel drug candidate wgx-50 by molecular dynamics simulations. J. Phys. Chem. B. 2015;119:11196–11202. doi: 10.1021/acs.jpcb.5b03116. [DOI] [PubMed] [Google Scholar]
- 29.A. Khan, W. Heng, Y. Wang, J. Qiu, X. Wei, S. Peng, S. Saleem, M. Khan, S.S. Ali, D.-Q. Wei, In Silico and in Vitro Evaluation of Kaempferol as a Potential Inhibitor of the SARS-CoV-2 Main Protease (3CLpro), Phytotherapy research: PTR. [DOI] [PMC free article] [PubMed]
- 30.Khan A., Kaushik A.C., Ali S.S., Ahmad N., Wei D.-Q. Deep-learning-based target screening and similarity search for the predicted inhibitors of the pathways in Parkinson's disease. RSC Adv. 2019;9:10326–10339. doi: 10.1039/c9ra01007f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan A., Khan M., Saleem S., Babar Z., Ali A., Khan A.A., Sardar Z., Hamayun F., Ali S.S., Wei D.-Q. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdiscipl. Sci. Comput. Life Sci. 2020;12:335–348. doi: 10.1007/s12539-020-00381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan M.T., Ali A., Wang Q., Irfan M., Khan A., Zeb M.T., Zhang Y.-J., Chinnasamy S., Wei D.-Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2. A molecular dynamic study. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1769733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva R.C., Freitas H.F., Campos J.M., Kimani N.M., Silva C.H., Borges R.S., Pita S.S., Santos C.B. Natural products-based drug design against SARS-CoV-2 Mpro 3CLpro. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ibrahim M.A., Abdeljawaad K.A., Abdelrahman A.H., Hegazy M.-E.F. Natural-like products as potential SARS-CoV-2 Mpro inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn. 2021;39:5722–5734. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sliwoski G., Kothiwale S., Meiler J., Lowe E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A. Highly accurate protein structure prediction with AlphaFold. Nature. 2021:1. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caillat C., Topalis D., Agrofoglio L.A., Pochet S., Balzarini J., Deville-Bonne D., Meyer P. Crystal structure of poxvirus thymidylate kinase: an unexpected dimerization has implications for antiviral therapy. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16900–16905. doi: 10.1073/pnas.0804525105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 39.Gopalakrishnan K., Sowmiya G., Sheik S., Sekar K. Ramachandran plot on the web (2.0) Protein Pept. Lett. 2007;14:669–671. doi: 10.2174/092986607781483912. [DOI] [PubMed] [Google Scholar]
- 40.Chen C.Y.-C. TCM Database@ Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico. PLoS One. 2011;6 doi: 10.1371/journal.pone.0015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng X., Zhang P., He W., Qin C., Chen S., Tao L., Wang Y., Tan Y., Gao D., Wang B. NPASS: natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018;46:D1217–D1222. doi: 10.1093/nar/gkx1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ntie-Kang F., Zofou D., Babiaka S.B., Meudom R., Scharfe M., Lifongo L.L., Mbah J.A., Mbaze L.M.a., Sippl W., Efange S.M. AfroDb: a select highly potent and diverse natural product library from African medicinal plants. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sorokina M., Merseburger P., Rajan K., Yirik M.A., Steinbeck C. COCONUT online: collection of open natural products database. J. Cheminf. 2021;13:1–13. doi: 10.1186/s13321-020-00478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravindranath P.A., Forli S., Goodsell D.S., Olson A.J., Sanner M.F. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeLano W.L. Pymol: an open-source molecular graphics tool. CCP4 Newsletter on protein crystallography. 2002;40:82–92. [Google Scholar]
- 47.Bell J., Cao Y., Gunn J., Day T., Gallicchio E., Zhou Z., Levy R., Farid R. 2012. PrimeX and the Schrödinger Computational Chemistry Suite of Programs. [Google Scholar]
- 48.Case D.A., Cheatham T.E., III, Darden T., Gohlke H., Luo R., Merz K.M., Jr., Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pearlman D.A., Case D.A., Caldwell J.W., Ross W.S., Cheatham T.E., III, DeBolt S., Ferguson D., Seibel G., Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995;91:1–41. [Google Scholar]
- 50.Wang J., Wang W., Kollman P.A., Case D.A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001;222:U403. [Google Scholar]
- 51.Salomon-Ferrer R., Götz A.W., Poole D., Le Grand S., Walker R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theor. Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 52.Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theor. Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 53.Chen F., Liu H., Sun H., Pan P., Li Y., Li D., Hou T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys. Chem. Chem. Phys. 2016;18:22129–22139. doi: 10.1039/c6cp03670h. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y., Khan A., Chandra Kaushik A., Junaid M., Zhang X., Wei D.-Q. The systematic modeling studies and free energy calculations of the phenazine compounds as anti-tuberculosis agents. J. Biomol. Struct. Dyn. 2019;37:4051–4069. doi: 10.1080/07391102.2018.1537896. [DOI] [PubMed] [Google Scholar]
- 55.Vangone A., Schaarschmidt J., Koukos P., Geng C., Citro N., Trellet M.E., Xue L.C., Bonvin A.M. Large-scale prediction of binding affinity in protein–small ligand complexes: the PRODIGY-LIG web server. Bioinformatics. 2019;35:1585–1587. doi: 10.1093/bioinformatics/bty816. [DOI] [PubMed] [Google Scholar]
- 56.Khan A., Ali S.S., Khan M.T., Saleem S., Ali A., Suleman M., Babar Z., Shafiq A., Khan M., Wei D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro) J. Biomol. Struct. Dyn. 2021;39:4659–4670. doi: 10.1080/07391102.2020.1779128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang S.-S., Huang H.-J., Chen C.Y.-C. High performance screening, structural and molecular dynamics analysis to identify H1 inhibitors from TCM Database@Taiwan. Mol. Biosyst. 2011;7:3366–3374. doi: 10.1039/c1mb05320e. [DOI] [PubMed] [Google Scholar]
- 58.Lingwan M., Shagun S., Pahwa F., Kumar A., Verma D.K., Pant Y., Kamatam L.V.K., Kumari B., Nanda R.K., Sunil S., Masakapalli S.K. Phytochemical rich Himalayan Rhododendron arboreum petals inhibit SARS-CoV-2 infection in vitro. J. Biomol. Struct. Dyn. 2021:1–11. doi: 10.1080/07391102.2021.2021287. [DOI] [PubMed] [Google Scholar]
- 59.Whibley C.E., Keyzers R.A., Soper A.G., Davies-Coleman M.T., Samaai T., Hendricks D.T. Antiesophageal cancer activity from southern african marine organisms. Ann. N. Y. Acad. Sci. 2005;1056:405–412. doi: 10.1196/annals.1352.031. [DOI] [PubMed] [Google Scholar]
- 60.Kamatou G.P.P., Viljoen A.M., Steenkamp P. Antioxidant, antiinflammatory activities and HPLC analysis of South African Salvia species. Food Chem. 2010;119:684–688. [Google Scholar]
- 61.Akimov M.G., Fomina-Ageeva E.V., Dudina P.V., Andreeva L.A., Myasoyedov N.F., Bezuglov V.V. ACTH(6-9)PGP peptide protects SH-SY5Y cells from H(2)O(2), tert-butyl hydroperoxide, and cyanide cytotoxicity via stimulation of proliferation and induction of prosurvival-related genes. Molecules. 2021;26 doi: 10.3390/molecules26071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sandberg G. Biosynthesis and metabolism of indole-3-ethanol and indole-3-acetic acid by Pinus sylvestris L. needles. Planta. 1984;161:398–403. doi: 10.1007/BF00394569. [DOI] [PubMed] [Google Scholar]
- 63.Mazzafera P. Catabolism of caffeine in plants and microorganisms. Front. Biosci. : J. Vis. Literacy. 2004;9:1348–1359. doi: 10.2741/1339. [DOI] [PubMed] [Google Scholar]
- 64.Elsayed S., Bondy L., Hanage W.P. Monkeypox virus infections in humans. Clin. Microbiol. Rev. 2022;35 doi: 10.1128/cmr.00092-22. e00092-00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCarthy M.W. Therapeutic strategies to address monkeypox. Expert Rev. Anti-infect. Ther. 2022;20:1249–1252. doi: 10.1080/14787210.2022.2113058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hooper J.W., Ferro A.M., Golden J.W., Silvera P., Dudek J., Alterson K., Custer M., Rivers B., Morris J., Owens G., Smith J.F., Kamrud K.I. Molecular smallpox vaccine delivered by alphavirus replicons elicits protective immunity in mice and non-human primates. Vaccine. 2009;28:494–511. doi: 10.1016/j.vaccine.2009.09.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rizk J.G., Lippi G., Henry B.M., Forthal D.N., Rizk Y. Prevention and treatment of monkeypox. Drugs. 2022;82:957–963. doi: 10.1007/s40265-022-01742-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu X., An S., Chu J., Liang B., Liao Y., Jiang J., Lin Y., Ye L., Liang H. Potential inhibitors of monkeypox virus revealed by molecular modeling approach to viral DNA topoisomerase I. Molecules. 2023;28:1444. doi: 10.3390/molecules28031444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jha S.K., Islam M., Kumar R., Rana L., Saifi M.A. 2023. Evaluation of Vernonia amygdalina del. containing phyto constituents a medicinal plant compound as new potential inhibitors of Monkey pox virus using molecular docking analysis. [Google Scholar]
- 70.Lam T.-P., Tran V.-H., Mai T.T., Lai N.V.-T., Dang B.-T.N., Le M.-T., Tran T.-D., Trinh D.-T.T., Thai K.-M. Identification of diosmin and flavin adenine dinucleotide as repurposing treatments for monkeypox virus: a computational study. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms231911570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data is available on RCSB, UniProt and any simulation data would be provided on reasonable demand. The accession numbers to access this data are given in the manuscript.