

Abstract
The site-selective functionalization of proteins has broad application in chemical biology, but can be limited when mixtures result from incomplete conversion or the formation of protein containing side products. It is shown here that when proteins are covalently tagged with pyridyl-tetrazines, the nickel-iminodiacetate (Ni-IDA) resins commonly used for His-tags can be directly used for protein affinity purification. These Affinity Bioorthogonal Chemistry (ABC) tags serve a dual role by enabling affinity-based protein purification while maintaining rapid kinetics in bioorthogonal reactions. ABC-tagging works with a range of site-selective bioconjugation methods with proteins tagged at the C-terminus, N-terminus or at internal positions. ABC-tagged proteins can also be purified from complex mixtures including cell lysate. The combination of site-selective conjugation and clean-up with ABC-tagged proteins also allows for facile on-resin reactions to provide protein-protein conjugates.
Keywords: bioorthogonal, affinity chromatography, protein purification, tetrazine, protein-protein conjugation
Graphical Abstract

Affinity Bioorthogonal Chemistry tags (ABC-tags) based on 2-pyridyltetrazines serve a dual role by enabling protein purification through metal chelation and still maintain their rapid kinetics in bioorthogonal reactions. ABC-tagging is successful for proteins tagged at the C- or N-terminus or at internal positions, is successful in complex mixtures including cell lysate, and can be carried out on-resin to generate multidomain proteins.
Introduction
The site-selective modification of proteins through chemical or enzymatic reactions is important to diverse fields including cellular and in vivo imaging,[1] proteomics,[2] drug-release[3] and material science.[4] Increasingly, these methods are used to incorporate tags that can engage in bioorthogonal chemistry,[5] which in turn dramatically increases the range of molecules that can be conjugated to the protein with site-selectivity. Chemical and enzyme-catalyzed site-selective labeling methods with amino acid specificity are actively being developed to meet this need.[6] Several chemical methods exploit the lowered pKa of a protein’s N-terminus for direct labeling or work in cohort with terminal amino acid side chains.[7] Naturally occurring enzymes have been engineered to accept new substrates for N-terminal,[8] C-terminal,[9] and internal[10] labeling of proteins bearing an appropriate amino-acid recognition sequence. These methods have been transformative for labeling proteins site-specifically, but differences in substrate specificity, reaction kinetics, or even reagent stability may result in incomplete labeling and a mixture of inseparable labeled and unlabeled proteins. The use of genetically encoded affinity tags for purification of complex mixtures such as lysates[11] or enzyme-catalyzed labeling reactions are limited to removal of a single protein species from a mixture.[12] Proteins labeled with small molecule affinity handles can also be selectively purified by affinity chromatography using binding partner interactions such as biotin to avidin[13] and lithocholic acid or chromophores to β-cyclodextrin.[14] Kuan, Weil and co-workers have elegantly demonstrated that boronic acid tags can be site-selectively introduced to proteins using a disulfide rebridging strategy.[15] Here, the boronic acid group is both used both for protein purification and subsequent protein conjugation via dynamic-covalent chemistry. While bioorthogonal chemistry has also been used to covalently attach affinity tags to proteins[2] or to covalently capture proteins on resin,[16] this approach ‘spends’ the bioorthogonal group on protein purification rather than on the conjugation of functional molecules to the protein target. A wide range of site-selective protein conjugation methods could benefit from a small molecule, dual-purpose tag where a single functional group can both facilitate protein purification while serving as a handle for subsequent rapid and quantitative bioorthogonal labeling.
Tetrazines represent a particularly attractive handle because of their ability to undergo inverse electron demand Diels-Alder reactions; the most rapid bioorthogonal reaction known, with reaction rates as fast as k2 104-106 M−1s−1 when combined with trans-cyclooctene (TCO) dienophiles.[17] Recent improvements to tetrazine synthesis have also increased access to smaller and more diverse substrates.[18] Tetrazine ligation has found widespread use in the field of chemical biology for cellular labeling,[19] in vivo imaging,[20] drug-delivery,[21] and proteomics.[22] Site-specific incorporation of tetrazine substrates onto proteins has been successfully adopted for chemical,[23] enzyme-catalyzed,[9c, 19a, 19b, 24] and genetic code expansion techniques.[25] These methods have been instrumental for multi-step preparation of protein-protein[26] or protein-DNA[27] conjugates utilizing the tetrazine ligation and standard protein purification methods. In addition to their popularity as bioorthogonal chemistry reagents, tetrazines have served as ligands in coordination chemistry. 2-Pyridyltetrazines bind to metals in a manner analogous to 2,2’-bipyridine– a group which has been incorporated into peptides and proteins[28] for metal binding. Symmetric, bidentate 3,6-di-(2-pyridyl)-tetrazine ligands are known to coordinate metal centers such as Ni2+, Cu2+, Fe2+ and Ag+ in organic solvents.[29] More recently, 3,6-di-(2-pyridyl)-tetrazine ligands have been used in aqueous buffered systems for supramolecular gelation with Fe2+ and Ni2+ salts[30] and to enhance reactivity of vinylboronic acids dienophiles mediated by boronic acid coordination.[31] Similarly, 6-methyl-3-(2-pyridyl)tetrazine iridium(III) complexes were constructed as luminogenic probes for live cell labeling.[32] In addition to their capacity for metal binding, the 2-pyridyl substituent also provides a favorable balance of reactivity and stability to tetrazine reagents.[33]
Here, we describe Affinity Bioorthogonal Chemistry tags (ABC-tags) for the dual role of facilitating protein purification as well as serving as a handle for subsequent rapid and quantitative bioorthogonal labeling (Fig 1A). Derivatives of 3-methyl-6-(2-pyridyl)tetrazine were designed to chelate to immobilized metal-ion affinity chromatography (IMAC)-resins commonly used for protein purification (Fig 1B). These small, dual-purpose tags serve as tools that can work in conjunction with site-selective protein conjugation methods at the C-terminus, N-terminus or internal residues. Because the dihydropyridazine products of tetrazine ligation also maintain affinity for nickel iminoacetate (Ni-IDA) resin, protein-protein conjugation reactions can be carried out ‘on-resin’ to provide dimeric and heterotrimeric protein conjugates without additional purification steps.
Figure 1.

(A) ABC-tags for Ni(IDA) purification of tagged proteins and bioorthogonal reactions (B) Pyridyl-tetrazine tagged proteins chelated to Ni(IDA) resin (PDB_ID 3KZY).
Results and Discussion
ABC-tag Screening and Optimization with Cysteine-tagged Proteins
A series of potential ABC tags were designed and evaluated for their ability to bind Ni-IDA agarose resin when attached to a model protein (Figure 2). Green-fluorescent protein (GFP) was site-specifically labeled with each of the tags (confirmed by ESI-MS) and separated by gel-filtration into pH 6.0 adjusted 2-(N-morpholino)ethanesulfonic acid (MES) buffer (Figure S1–9). MES buffer was selected as a non-coordinating buffer[34] and the pH was adjusted to 6.0 to alleviate non-specific protein interactions with Ni-IDA resin.[35] A 300 μL solution containing 100 μM labeled protein was added by gravity flow to 100 μL of Ni-IDA resin. The saturated resin was washed with 5 column volumes MES buffer, followed by 5 column volumes MES buffer with 10 mM imidazole to remove non-specifically bound protein. Finally, protein was eluted with 400 mM imidazole in phosphate buffer and the total bound protein was determined by UV-vis spectroscopy (Figure 2). Initial findings show the importance of the pyridyl substituent as compounds 1a-1c did not demonstrate any binding (Figure 2), whereas pyridyl-Tz 1d had the highest binding capacity of 3.4 mg/mL. This is within an order of magnitude of the binding capacity for His-tagged proteins on GoldBio Ni(IDA) resin, which is stated by the manufacturer to be greater than 25 mg/ml for a 60 kDa protein. Attachment of a bis-pyridyl-Tz tag (1g) increased the binding capacity to 9 mg/mL, a 2.6-fold increase with respect to 1d, demonstrating that approaches using multivalent pyridyl-Tz attachments can be used for increasing binding capacities. Pyridyl-dihydropyridazine derivative 1f, the product of the reaction between 1d and syn-1,2-dihydroxy-5-trans-cyclooctene, also bound to Ni(IDA) resin but with a weakened binding capacity of 1 mg/mL (Figure 2). Pyridyl-triazole (1e), the product of a copper(I) catalyzed azide-alkyne cycloaddition between an alkyne and pyridyl-azide, also displayed a similar binding capacity to 1f (Figure 2). ABC tags containing a pyridyl-Tz core displayed the best binding and were chosen as the lead compounds for testing selective purification of site-specifically labeled proteins from chemoenzymatic ligation mixtures.
Figure 2.
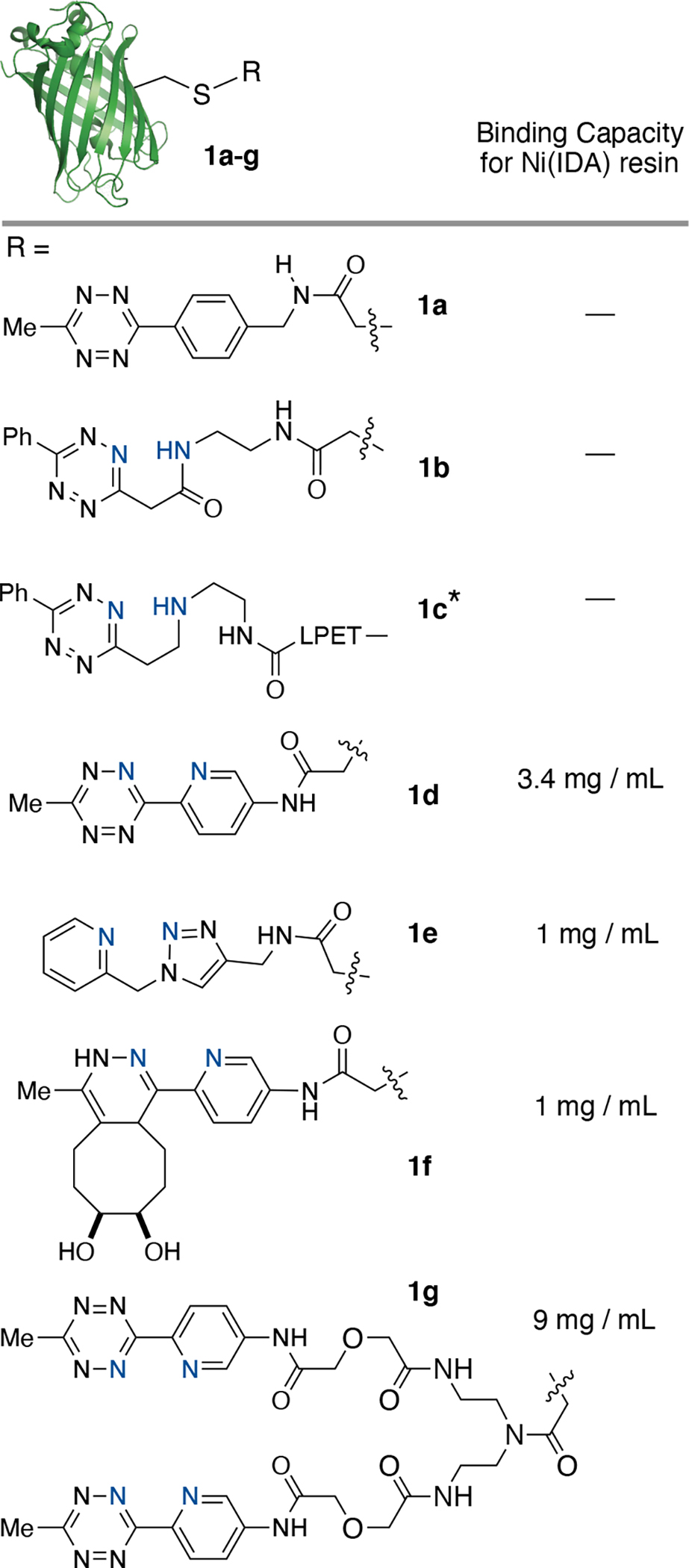
GFP (PDB_ID 2B3P) was site-specifically modified at cysteine with a panel of tetrazine compounds. Conjugates were used for measuring binding capacity to Ni(IDA) resin. *For 1c, GFP was modified at the C-terminus using sortase ligation.
Tetrazines and their TCO-Diels-Alder conjugates display differences in their UV-vis absorption spectra that can be used to monitor conjugation efficiency. The UV-vis spectra for a pyridyltetrazine (λmax 305 nm) and its 5-hydroxy-trans-cyclooctene Diels-Alder adduct (λmax 276 nm) are displayed in the SI as Figure S59. Absorption at 305 nm can also be used to monitor conjugation of GFP-pyridyltetrazine 1d with syn-dihydroxy-trans-cyclooctene to give Diels-Alder conjugate 1f (Figure S60). Using fluorescence spectroscopy, the rate of the Diels-Alder reaction between GFP-pyridyltetrazine (1d, 200 nM) and syn-1,2,-dihydroxy-5-trans-cyclooctene (10–30 μM) to form GFP-dihydropyridizine product 1f was measured to be 2260 (± 220) M−1s−1 in 97:3 PBS:DMSO (Fig S-56). Product 1f is more fluorescent than 1d due to the quenching of GFP fluorescence of 1d by the attached tetrazine.[25a, 36] We also carried the same reaction out in the presence of 1 μM NiCl2 to model the effect of Ni-coordination in resin-bound tetrazines. While Re- and B-coordination to 2-pyridyltetrazines has been shown to activate the rate of tetrazine ligation,[31, 37] with NiCl2 a slight reduction in rate k2 1780 (± 60) M−1s−1 was actually observed (Fig S-57). Nonetheless, protein conjugations are rapid, and reaction rates can be further accelerated by using s-TCO derivatives (vide infra) which are 160 times[17b] more reactive than regular TCO derivatives.
C-Terminal Tagging and Purification
Enzyme-catalyzed bioconjugations have emerged as a useful and simple method for site-specific incorporation of bioorthogonal handles to proteins. The transpeptidation reaction catalyzed by sortase A from Staphylococcus aureus is popular for labeling the N- or C-terminus of proteins because of its diverse substrate scope, continued improvement of sortase A enzymes, and simple reaction conditions.[38] For direct C-terminal labeling, an LPXTG sequence is encoded on the protein of interest (POI), this unique sequence is recognized by sortase A and facilitates amide bond formation with the N-terminus of an aminoglycine substrate. Recently, the SrtA7M mutant of sortase A was demonstrated by Cochran and coworkers to accept a variety of small molecule amine compounds.[24c] A minimal SrtA7M substrate 2 was synthesized and used to demonstrate the effectiveness of ABC tags for enzyme-catalyzed bioconjugations and purification. A diverse panel of eight proteins bearing a C-terminal LPETGG recognition sequence were tested for labeling and purification with ABC tag 2 using SrtA7M mediated bioconjugation. Labeling was conducted using 200 μM POI-LPETGG, 10 μM SrtA7M, and 2 mM 2 at room temperature for up to 2 hours. Gel-filtration of the reaction mixtures was used to stop the reaction by removing ABC tag 2. The percentage of labeled POI was determined by ESI-MS (Table S1). Consistent with previous reports for SrtA7M bioconjugations, the desired conjugates (POI-LPET-pyTz) were obtained in good yields for each of the protein reaction mixtures; for all proteins except GFP and Her2-affibody, the percentage of labeled protein was greater than 50% by ESI-MS analysis (Table S1). Also present in the reaction mixtures were impurities consistent with protein starting material (POI-LPETGG), SrtA7M enzyme, and/or hydrolyzed protein (POI-LPET) (Figure 3). Selective capture and purification of POI-LPET-pyTz conjugates were achieved on small-scale using 1 mL of Ni-IDA resin and following the procedure described previously. To pre-equilibrated Ni-IDA resin in MES buffer, the desalted sortase reaction mixture was added at 20% of the resin’s binding capacity. The resin was subsequently washed with MES buffer to remove non-specifically bound protein. The flow-through from the loading and washing steps were combined, collected and analyzed by ESI-MS. The only observable mass peaks were those consistent with protein impurities: no mass peaks containing POI-LPET-pyTz were observed, suggesting high retention to the Ni-IDA resin. Following column washing, bound protein was eluted with 5 column-volumes of 400 mM imidazole in phosphate buffer. Protein recovery was determined by UV-Vis, and purity assessed by ESI-MS. For each elution, only POI-LPET-pyTz conjugates were observed, with recoveries ranging from 72–95% (Figure 3, Table S-1). Automated purifications using 12.5 mL Ni-IDA with an AKTA fast-protein liquid chromatography (FPLC) were also tested with GFP, SnapTag, 5F7 nanobody, and NanoLuc luciferase. FPLC based purification methods offer a distinct advantage in terms of gradient profiling, in-line UV-Vis monitoring, column choice, and automated fraction collection. For each of the protein’s purified by FPLC, recoveries were high (≥ 85%) and contained pure product by ESI-MS (Figure S39–46).
Figure 3.
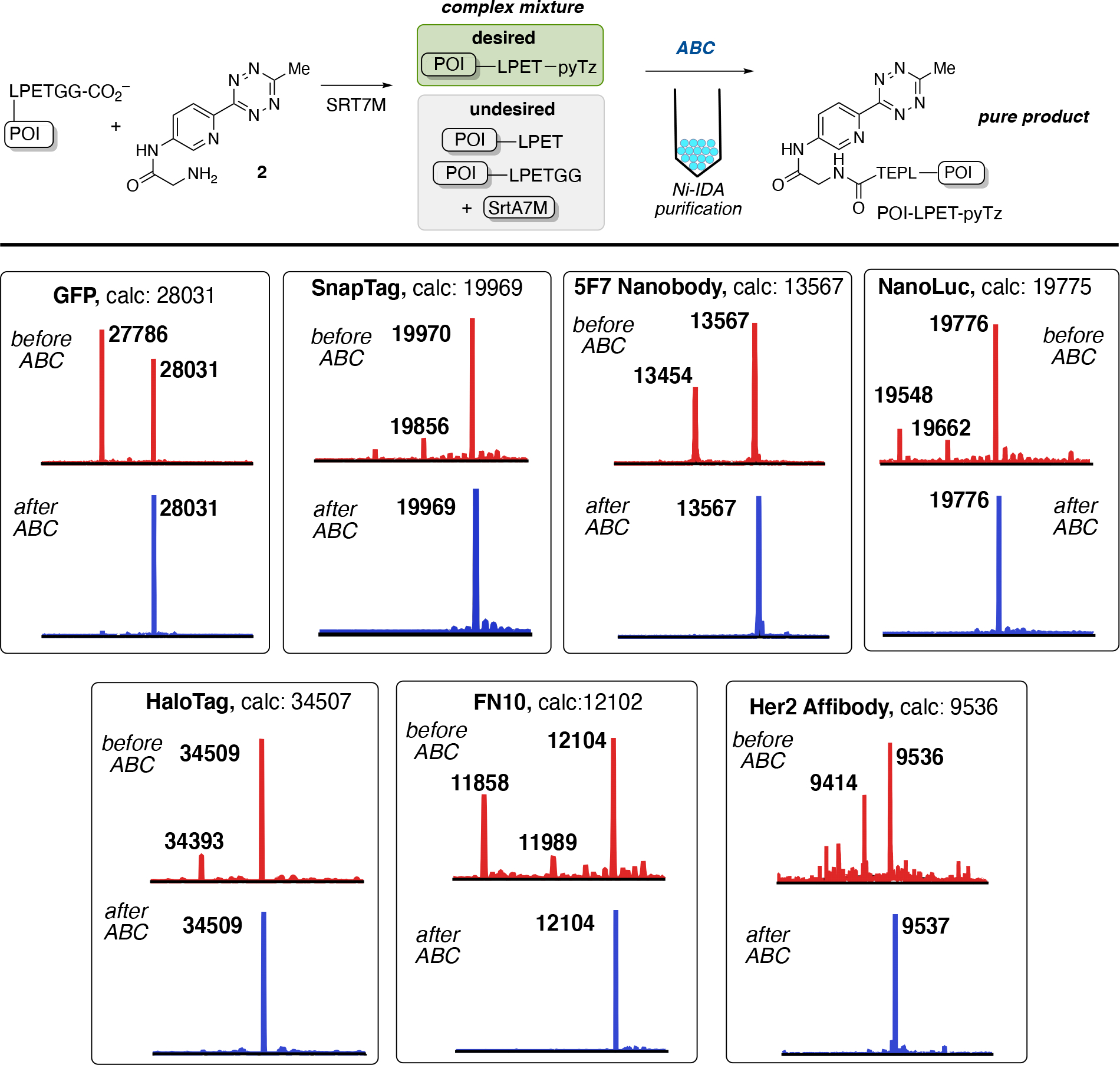
Purification from complex mixtures: C-terminal Sortase ligation. POI-LPETGG (200 μM) was treated with 2 (2 mM) and SrtA7M (10 μM) for 2 hours at room temperature resulting in a mixture of desired POI-LPET-pyTz and undesired POI-LPET, POI-LPETGG, and SrtA7M. The mixture was ‘ABC purified’ to obtain pure POI-LPET-pyTz. ESI-MS of the sortase reaction mixture (before ABC) and pure POI-LPET-pyTz (after ABC) are shown for GFP, SnapTag, 5F7 nanobody, NanoLuc, HaloTag, FN10, and Her2 affibody.
N-Terminal Tagging and Purification
Direct N-terminal labeling of proteins is another important method for site-specific protein labeling which often employs chemical methodology.[7a, 39] In 2009, cyanobenzothiazole (CBT) reagents were shown to react specifically with proteins bearing an N-terminal cysteine under reducing conditions.[7f] An ABC-tagged CBT ligand (3) was synthesized for demonstrating direct N-terminal labeling to monomeric streptavidin 2 (mSA2). Following established protocols, each protein was labeled under reducing conditions (2 mM TCEP) and 1 mM 3 for 2 hours at room temperature. Labeling was quenched by gel-filtration removal of 3 and the percentage of labeled protein was determined by ESI-MS (Figure 4B). In addition to the desired conjugate, multiple impurity peaks were observed in the ESI-MS spectrum; impurities are consistent with oxidized starting material (Figure S-37). ABC tagged protein was separated from these impurities by Ni-IDA purification and the eluted proteins was free from lower molecular weight impurities. The ESI-MS of the purified protein contained a peak corresponding to the mSA2-tetrazine conjugate and a minor peak due to a single oxidation. The recovery of the mSA2-tetrazine conjugate was 88% (Figure 4).
Figure 4.

Purification of N-terminal modified mSA2 using CBT ligation. (A) mSA2 was treated with 3 (1 mM) under reducing conditions with TCEP (2 mM) and then ABC purified. (B) ESI-MS for the N-terminal ligation mixture (before ABC) and purified mSA2 (after ABC)
ABC-tags Enable Purification from Complex Mixtures
Additional experiments were carried out to demonstrate that ABC-tagging is a successful purification strategy even when the pyridyltetrazine-tagged POI is only a minor component in a mixture of proteins. Enzyme-catalyzed or chemical conjugation methods are preferably performed in vitro under user-defined conditions as minor impurities can be anticipated for a given reaction. The presence of unknown impurities is always of concern, especially as some labeling methods require purification from cell lysates where the POI is of low abundance. Purification of low abundant proteins was initially tested using the model proteins GFP and thioredoxin (Trx). Each protein was labeled site-specifically by pyTz (15 μM, 1 eq) and mixed with unlabeled protein (135 μM, 9 eq) to produce a 9:1 mixture of unlabeled to labeled protein (Figure 5B). GFP-pyTz does not elute with 10 mM imidazole in pH 6.0 MES, and GFP-pyTz can be cleanly separated from unmodified GFP as determined by MS analysis before and after purification (Fig 5C). As shown in Fig 5D, the same procedure could be used to purify unmodified thioredoxin (Trx) from protein that was site-selectively tagged by a single pyridyl tetrazine. We then tested ABC purification from a complex mixture of unknown proteins, a defined amount of GFP-pyTz was doped at low abundance to a bacterial cell lysate mixture (Figure 5E). The bacterial lysate was purified by Ni-IDA and pure GFP-pyTz (>90%, gel) was recovered in 83% yield.
Figure 5.

(A,B) ABC purification of a pyridyltetrazine-tagged POI is only a minor component (<10%) in a mixture of proteins. (C,D) Separation of (C) GFP-pyTz and (D) Trx-pyTz from a mixture with excess, non-tetrazine tagged protein. (E) GFP-pyTz (Lane A) was added to bacterial cell lysate (Lane B) and the combined mixture (Lane C) was ABC purified to recover pure GFP-pyTz (Lane D); samples were treated with TCO-TAMRA and analyzed by SDS-PAGE to identify tetrazine labeled protein by in-gel fluorescence and total protein by coomassie stain.
On-resin Protein-Protein Conjugation
The ability for pyTz-proteins to bind Ni-IDA resin in situ for purification purposes led us to consider if the resin-bound protein could facilitate reactions with a free sTCO-protein counterpart for efficient protein coupling. Leading methods for preparing site-specific protein-protein conjugates are typically performed in solution with reaction times requiring hours for high-conversion. To obtain the desired conjugate, removal of excess or unlabeled proteins involves an additional protein chromatography step.[26c, 26e] With ABC-tagged proteins, successful conjugates would be formed in minutes, remain resin-bound through a dihydropyridazine linker (as observed with 1f, Table 1) and impurities would be washed away. This new methodology would enable efficient protein-protein conjugation and clean-up in a single step. C-terminal SnapTag conjugates bearing an sTCO (5) or pyTz (6) were prepared via sortase ligation as described above (Figure S-47,48). Resin-bound SnapTag-pyTz (6) was prepared by loading the sortase reaction mixture (100 μL, 40 μM Tz, 1 eq) onto 200 μL Ni-IDA resin and washing away unbound impurities. A solution of SnapTag-sTCO (5) (100 μL, 120 μM sTCO, 3 equiv.) sortase mixture was prepared, and without purification directly added to the top of the column bed containing 6 for on bead coupling. The solution was allowed to flow by gravity through the column over the course of 2 minutes, and unreacted protein was simply washed away. The successful, C-terminally linked SnapTag conjugates (7) remained bound to the resin through a dihydropyridazine linker and was cleanly eluted as determined by ESI-MS and SDS-PAGE (Figure 6A). The stability of pyridyl-tetrazine when bound to the resin was assessed by incubating resin-bound 1d for at room temperature in MES buffer. After 60 minutes, 1d was eluted and reacted with syn-1,2-dihydroxy-5-trans-cyclooctene to form 1f with complete conversion (Figure S58). The use of synthetic linkers cannot only enable protein-dimer conjugations but can expand access to hetero-trimeric.
Figure 6.

On-resin protein coupling reactions. (A) Reaction mixture of SnapTag C-terminally modified with sTCO 4 by sortase ligation (5) was added in excess to Ni(IDA) resnin bound SnapTag C-terminally modified with tetrazine 2 by sortase ligation (6). Purified SnapTag dimer 7 (5 + 6) was analyzed by ESI-MS and SDS-PAGE (coomassie). (B) GFP site-specifically modified with bis-pyTz (1g) was preloaded onto Ni(IDA) resin (8) and reacted with an excess of sTCO modified Trx (9). Purified protein trimer GFP-(Trx)2 (10) was analyzed by ESI-MS and SDS-PAGE (coomassie).
The use of synthetic linkers cannot only enable direct protein-protein conjugations but can expand access to give protein heterotrimers. To demonstrate this, GFP-(pyTz)2 (1g) (100 μL, 40 μM protein, 2 equiv. pyTz) was preloaded onto 100 μL Ni-IDA resin (8) and a thioredoxin-sTCO conjugate (9) (338 μL, 71 μM, 6 eq sTCO) was added in excess with respect to tetrazine (3 equiv. sTCO per Tz). The reaction was complete by flow and unreacted 9 was washed away, yielding a heterotrimeric GFP-(Trx)2 conjugate (10) that was cleanly eluted as observed by ESI-MS and SDS-PAGE (Figure 6B).
Conclusion
In summary, 2-pyridyl-tetrazine serves as an Affinity Bioorthogonal Chemistry (ABC) tag for the dual purpose of enabling protein purification via in situ coordination to Ni(IDA) resin and as a handle for bioorthogonal chemistry reactions. This method has been successful for obtaining pure, site-specifically modified proteins using C-terminal sortase ligation, N-terminal CBT ligation, and cysteine alkylation methods on 9 different proteins. On-resin reactions between ABC-tagged proteins and complementary sTCO-tagged proteins, that remain bound through a dihydropyridazine linker, has enabled the construction of dimeric and heterotrimeric protein conjugates in minutes with defined geometries in a single step with high purity. We anticipate ABC-tags will find general use for applications requiring pure bioconjugates and rapid in vitro protein assembly.
Supplementary Material
Acknowledgements
This work was supported by NIH (R01GM132460) and Pfizer. Instrumentation was supported by NIH awards P20GM104316, P20GM103446, S10OD025185, S10OD026951, S10OD016267, S10 OD016361, and S10 OD30321. Facilities and instrumentation were also supported by NSF through the University of Delaware Materials Research Science and Engineering Center, DMR-2011824.
References
- [1].Schneider AFL, Hackenberger CPR, Curr. Opin. Biotechnol. 2017, 48, 61–68. [DOI] [PubMed] [Google Scholar]
- [2].Parker CG, Pratt MR, Cell 2020, 180, 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Agarwal P, Bertozzi CR, Bioconjug. Chem. 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spicer CD, Pashuck ET, Stevens MM, Chem. Rev. 2018, 118, 7702–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scinto SL, Bilodeau DA, Hincapie R, Lee W, Nguyen SS, Xu M, am Ende CW, Finn MG, Lang K, Lin Q, Pezacki JP, Prescher JA, Robillard MS, Fox JM, Nat Rev Methods Primers 2021, 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Spicer CD, Davis BG, Nat. Commun. 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- [7].a) Rosen CB, Francis MB, Nat. Chem. Biol. 2017, 13, 697–705; [DOI] [PubMed] [Google Scholar]; b) Dawson PE, Muir TW, Clark-Lewis I, Kent SB, Science 1994, 266, 776–779; [DOI] [PubMed] [Google Scholar]; c) Blanco-Canosa JB, Dawson PE, Angew. Chem. Int. Ed. 2008, 47, 6851–6855; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Muir TW, Sondhi D, Cole PA, Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6705–6710; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mitchell NJ, Malins LR, Liu X, Thompson RE, Chan B, Radom L, Payne RJ, J. Am. Chem. Soc. 2015, 137, 14011–14014; [DOI] [PubMed] [Google Scholar]; f) Ren H, Xiao F, Zhan K, Kim YP, Xie H, Xia Z, Rao J, Angew. Chem. Int. Ed. 2009, 48, 9658–9662; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) MacDonald JI, Munch HK, Moore T, Francis MB, Nat. Chem. Biol. 2015, 11, 326–331. [DOI] [PubMed] [Google Scholar]
- [8].a) Theile CS, Witte MD, Blom AE, Kundrat L, Ploegh HL, Guimaraes CP, Nat. Protoc. 2013, 8, 1800–1807; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weeks AM, Wells JA, Nat. Chem. Biol. 2018, 14, 50–57; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nguyen GK, Cao Y, Wang W, Liu CF, Tam JP, Angew. Chem. Int. Ed. 2015, 54, 15694–15698; [DOI] [PubMed] [Google Scholar]; d) Kulkarni C, Kinzer-Ursem TL, Tirrell DA, ChemBioChem 2013, 14, 1958–1962; [DOI] [PubMed] [Google Scholar]; e) Witus LS, Moore T, Thuronyi BW, Esser-Kahn AP, Scheck RA, Iavarone AT, Francis MB, J. Am. Chem. Soc. 2010, 132, 16812–16817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Schumacher D, Helma J, Mann FA, Pichler G, Natale F, Krause E, Cardoso MC, Hackenberger CP, Leonhardt H, Angew. Chem. Int. Ed. 2015, 54, 13787–13791; [DOI] [PubMed] [Google Scholar]; b) Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, Ploegh HL, Nat. Protoc. 2013, 8, 1787–1799; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rehm FBH, Harmand TJ, Yap K, Durek T, Craik DJ, Ploegh HL, J. Am. Chem. Soc. 2019, 141, 17388–17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Fernandez-Suarez M, Baruah H, Martinez-Hernandez L, Xie KT, Baskin JM, Bertozzi CR, Ting AY, Nat. Biotechnol. 2007, 25, 1483–1487; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Boer E, Rodriguez P, Bonte E, Krijgsveld J, Katsantoni E, Heck A, Grosveld F, Strouboulis J, Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 7480–7485; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lopez Aguilar A, Briard JG, Yang L, Ovryn B, Macauley MS, Wu P, ACS Chem. Biol. 2017, 12, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Young CL, Britton ZT, Robinson AS, Biotechnol. J. 2012, 7, 620–634. [DOI] [PubMed] [Google Scholar]
- [12].Policarpo RL, Kang H, Liao X, Rabideau AE, Simon MD, Pentelute BL, Angew. Chem. Int. Ed. 2014, 53, 9203–9208. [DOI] [PubMed] [Google Scholar]
- [13].Cronan JE Jr., J. Biol. Chem. 1990, 265, 10327–10333. [PubMed] [Google Scholar]
- [14].a) Rosen CB, Kwant RL, MacDonald JI, Rao M, Francis MB, Angew. Chem. Int. Ed. 2016, 55, 8585–8589; [DOI] [PubMed] [Google Scholar]; b) Nguyen T, Joshi NS, Francis MB, Bioconjug. Chem. 2006, 17, 869–872. [DOI] [PubMed] [Google Scholar]
- [15].Zegota MM, Wang T, Seidler C, Wah Ng DY, Kuan SL, Weil T, Bioconj. Chem. 2018, 29, 2665–2670. [DOI] [PubMed] [Google Scholar]
- [16].a) Nessen MA, Kramer G, Back J, Baskin JM, Smeenk LE, de Koning LJ, van Maarseveen JH, de Jong L, Bertozzi CR, Hiemstra H, de Koster CG, Proteome Res J 2009, 8, 3702–3711; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Biedka S, Schmidt BF, Frey NM, Boothman SM, Minden JS, Wilson AL, Proteome Res J 2021, 20, 4787–4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Blackman ML, Royzen M, Fox JM, J. Am. Chem. Soc. 2008, 130, 13518–13519; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Taylor MT, Blackman ML, Dmitrenko O, Fox JM, J. Am. Chem. Soc. 2011, 133, 9646–9649; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Darko A, Wallace S, Dmitrenko O, Machovina MM, Mehl RA, Chin JW, Fox JM, Chem. Sci. 2014, 5, 3770–3776; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lambert WD, Scinto SL, Dmitrenko O, Boyd SJ, Magboo R, Mehl RA, Chin JW, Fox JM, Wallace S, Org. Biomol. Chem. 2017, 15, 6640–6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Lambert WD, Fang Y, Mahapatra S, Huang Z, am Ende CW, Fox JM, J. Am. Chem. Soc. 2019, 141, 17068–17074; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xie Y, Fang Y, Huang Z, Tallon AM, am Ende CW, Fox JM, Angew. Chem. Int. Ed. 2020, 59, 16967–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Murrey HE, Judkins JC, am Ende CW, Ballard TE, Fang Y, Riccardi K, Di L, Guilmette ER, Schwartz JW, Fox JM, Johnson DS, J. Am. Chem. Soc. 2015, 137, 11461–11475; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baalmann M, Ziegler MJ, Werther P, Wilhelm J, Wombacher R, Bioconjug. Chem. 2019, 30, 1405–1414; [DOI] [PubMed] [Google Scholar]; c) Beliu G, Kurz AJ, Kuhlemann AC, Behringer-Pliess L, Meub M, Wolf N, Seibel J, Shi ZD, Schnermann M, Grimm JB, Lavis LD, Doose S, Sauer M, Commun. Biol. 2019, 2, 261; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jemas A, Xie Y, Pigga JE, Caplan JL, am Ende CW, Fox JM, J. Am. Chem. Soc. 2022, 144, 1647–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Rossin R, Verkerk PR, van den Bosch SM, Vulders RC, Verel I, Lub J, Robillard MS, Angew. Chem. Int. Ed. 2010, 49, 3375–3378; [DOI] [PubMed] [Google Scholar]; b) Zeglis BM, Sevak KK, Reiner T, Mohindra P, Carlin SD, Zanzonico P, Weissleder R, Lewis JS, J. Nucl. Med. 2013, 54, 1389–1396; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang M, Vannam R, Lambert WD, Xie Y, Wang H, Giglio B, Ma X, Wu Z, Fox J, Li Z, Chem. Commun. 2019, 55, 2485–2488; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang C, Zhang H, Zhang T, Zou X, Wang H, Rosenberger JE, Vannam R, Trout WS, Grimm JB, Lavis LD, Thorpe C, Jia X, Li Z, Fox JM, J. Am. Chem. Soc. 2021, 143, 10793–10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Versteegen RM, Rossin R, ten Hoeve W, Janssen HM, Robillard MS, Angew. Chem. Int. Ed. 2013, 52, 14112–14116; [DOI] [PubMed] [Google Scholar]; b) van Onzen A, Versteegen RM, Hoeben FJM, Filot IAW, Rossin R, Zhu T, Wu J, Hudson PJ, Janssen HM, Ten Hoeve W, Robillard MS, J. Am. Chem. Soc. 2020, 142, 10955–10963. [DOI] [PubMed] [Google Scholar]
- [22].a) Kang K, Park J, Kim E, Proteome Sci 2016, 15, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qian L, Pan S, Lee JS, Ge J, Li L, Yao SQ, Chem. Commun. 2019, 55, 1092–1095. [DOI] [PubMed] [Google Scholar]
- [23].a) Gamache RF, Zettlitz KA, Tsai WK, Collins J, Wu AM, Murphy JM, Chem. Sci. 2020, 11, 1832–1838; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu L, Raabe M, Zegota MM, Nogueira JCF, Chudasama V, Kuan SL, Weil T, Org. Biomol. Chem. 2020, 18, 1140–1147. [DOI] [PubMed] [Google Scholar]
- [24].a) Macias-Contreras M, He H, Little KN, Lee JP, Campbell RP, Royzen M, Zhu L, Bioconjug. Chem. 2020, 31, 1370–1381; [DOI] [PubMed] [Google Scholar]; b) Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, Cragnolini J, Swee LK, Victora GD, Weissleder R, Ploegh HL, Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 6146–6151; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Glasgow JE, Salit ML, Cochran JR, J. Am. Chem. Soc. 2016, 138, 7496–7499. [DOI] [PubMed] [Google Scholar]
- [25].a) Blizzard RJ, Backus DR, Brown W, Bazewicz CG, Li Y, Mehl RA, J. Am. Chem. Soc. 2015, 137, 10044–10047; [DOI] [PubMed] [Google Scholar]; b) Jang HS, Jana S, Blizzard RJ, Meeuwsen JC, Mehl RA, J. Am. Chem. Soc. 2020, 142, 7245–7249; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, Chin JW, J. Am. Chem. Soc. 2012, 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Lorenz DA, Garner AL, Chem. Commun. 2016, 52, 8267–8270; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lim SI, Cho J, Kwon I, Chem. Commun. 2015, 51, 13607–13610; [DOI] [PubMed] [Google Scholar]; c) Baalmann M, Neises L, Bitsch S, Schneider H, Deweid L, Werther P, Ilkenhans N, Wolfring M, Ziegler MJ, Wilhelm J, Kolmar H, Wombacher R, Angew. Chem. Int. Ed. 2020, 59, 12885–12893; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rutkowska A, Plass T, Hoffmann JE, Yushchenko DA, Feng S, Schultz C, ChemBioChem 2014, 15, 1765–1768; [DOI] [PubMed] [Google Scholar]; e) Van Fossen EM, Bednar RM, Jana S, Franklin R, Beckman J, Karplus PA, Mehl RA, Sci. Adv. 2022, 8, eabm6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) van Buggenum JAG, Gerlach JP, Tanis SEJ, Hogeweg M, Jansen P, Middelwijk J, van der Steen R, Vermeulen M, Stunnenberg HG, Albers CA, Mulder KW, Nat. Commun. 2018, 9, 2384; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Agasti SS, Wang Y, Schueder F, Sukumar A, Jungmann R, Yin P, Chem. Sci. 2017, 8, 3080–3091; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) van Buggenum JA, Gerlach JP, Eising S, Schoonen L, van Eijl RA, Tanis SE, Hogeweg M, Hubner NC, van Hest JC, Bonger KM, Mulder KW, Sci. Rep. 2016, 6, 22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Yang M, Song WJ, Nat. Commun. 2019, 10, 5545; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mills JH, Sheffler W, Ener ME, Almhjell PJ, Oberdorfer G, Pereira JH, Parmeggiani F, Sankaran B, Zwart PH, Baker D, Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 15012–15017; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bersellini M, Roelfes G, Org. Biomol. Chem. 2017, 15, 3069–3073; [DOI] [PubMed] [Google Scholar]; d) Kang M, Light K, Ai HW, Shen W, Kim CH, Chen PR, Lee HS, Solomon EI, Schultz PG, ChemBioChem 2014, 15, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Stetsiuk O, Abherve A, Avarvari N, Dalton Trans. 2020, 49, 5759–5777; [DOI] [PubMed] [Google Scholar]; b) Kaim W, Coord. Chem. Rev. 2002, 230, 127–139. [Google Scholar]
- [30].Kawamoto K, Grindy SC, Liu J, Holten-Andersen N, Johnson JA, ACS Macro Lett. 2015, 4, 458–461. [DOI] [PubMed] [Google Scholar]
- [31].Eising S, Engwerda AHJ, Riedijk X, Bickelhaupt FM, Bonger KM, Bioconjug. Chem. 2018, 29, 3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tang TS, Liu HW, Lo KK, Chem. Commun. 2017, 53, 3299–3302. [DOI] [PubMed] [Google Scholar]
- [33].a) Maggi A, Ruivo E, Fissers J, Vangestel C, Chatterjee S, Joossens J, Sobott F, Staelens S, Stroobants S, Van Der Veken P, Wyffels L, Augustyns K, Org. Biomol. Chem. 2016, 14, 7544–7551; [DOI] [PubMed] [Google Scholar]; b) Svatunek D, Wilkovitsch M, Hartmann L, Houk KN, Mikula H, J. Am. Chem. Soc. 2022, 144, 8171–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Good NE, Winget GD, Winter W, Connolly TN, Izawa S, Singh RM, Biochemistry 1966, 5, 467–477. [DOI] [PubMed] [Google Scholar]
- [35].Bornhorst JA, Falke JJ, Methods Enzymol. 2000, 326, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tang L, Bednar RM, Rozanov ND, Hemshorn ML, Mehl RA, Fang C, Natural Sciences, n/a, e20220028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schnierle M, Blickle S, Filippou V, Ringenberg MR, Chem. Commun. 2020, 56, 12033–12036. [DOI] [PubMed] [Google Scholar]
- [38].Pishesha N, Ingram JR, Ploegh HL, Annu. Rev. Cell Dev. Biol. 2018, 34, 163–188. [DOI] [PubMed] [Google Scholar]
- [39].Agouridas V, El Mahdi O, Diemer V, Cargoet M, Monbaliu JM, Melnyk O, Chem. Rev. 2019, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.