

SUMMARY
Background:
Precision oncology is gradually advancing into mainstream clinical practice, demonstrating significant survival benefits. However, eligibility and response rates remain limited in many cases, calling for better predictive biomarkers.
Methods:
We present ENLIGHT, a transcriptomics-based computational approach that identifies clinically relevant genetic interactions and uses them to predict a patient’s response to a variety of therapies in multiple cancer types without training on previous treatment response data. We study ENLIGHT in two translationally oriented scenarios: personalized oncology (PO), aimed at prioritizing treatments for a single patient, and clinical trial design (CTD), selecting the most likely responders in a patient cohort.
Findings:
Evaluating ENLIGHT’s performance on 21 blinded clinical trial datasets in the PO setting, we show that it can effectively predict a patient’s treatment response across multiple therapies and cancer types. Its prediction accuracy is better than previously published transcriptomics-based signatures and is comparable with that of supervised predictors developed for specific indications and drugs. In combination with the interferon-γ signature, ENLIGHT achieves an odds ratio larger than 4 in predicting response to immune checkpoint therapy. In the CTD scenario, ENLIGHT can potentially enhance clinical trial success for immunotherapies and other monoclonal antibodies by excluding non-responders while overall achieving more than 90% of the response rate attainable under an optimal exclusion strategy.
Conclusions:
ENLIGHT demonstrably enhances the ability to predict therapeutic response across multiple cancer types from the bulk tumor transcriptome.
Funding:
This research was supported in part by the Intramural Research Program, NIH and by the Israeli Innovation Authority.
Graphical Abstract

Dinstag et al. describe ENLIGHT, a transcriptomics-based computational approach that identifies clinically relevant genetic interactions from the tumor transcriptome. ENLIGHT can predict treatment response across multiple therapies and cancer types better than published biomarkers, and it can potentially enhance clinical trial success by effectively excluding non-responders.
INTRODUCTION
The current paradigm of precision oncology (PO), rooted in the 1990s development of trastuzumab and imatinib, focuses on matching individual targets to molecularly derived therapies.1,2 In the past 20 years, cancer therapeutics, driven by treatments developed for specific oncogenes and by the advent of immunotherapy, has overwhelmingly dominated regulatory drug approvals.3 For patients with a qualifying biomarker and otherwise limited treatment options, this paradigm can demonstrate improvement in clinical outcomes,4–7 as in the I-PREDICT study,8 which uses DNA biomarkers to identify novel combination therapies for patients. However, large studies, such as NCI-MATCH, demonstrate the limitations of the variant-centered biomarker approach, with less than 20% of patients ultimately assigned to single-therapy treatment arms.9 Response rates in the setting of targeted therapy also show broad ranges, between 25% and 75% of patients.10 Although qualification for immunotherapy varies by cancer type, often only 20%–40% of patients respond to treatment.11,12
One strategy to broaden which patients qualify for treatment and to improve response rates is to leverage omics data beyond genomics, with the main focus of current research being on transcriptomics. Clinical programs, such as University of Michigan’s MIONCOSEQ, have sought to integrate DNA- and RNA-based sequencing to validate and support existing biomarkers.13,14 WINTHER was a prospective umbrella trial in which patients received personalized monotherapy or combination therapy based on genomic or transcriptomic data.15 This was the first clinical trial to examine the utility of RNA sequencing (RNA-seq) in a prospective clinical setting by targeting highly expressed cancer-associated genes, demonstrating that the tumor transcriptome could be used to provide complementary clinical information to DNA.
Other attempts to identify predictors of response from the transcriptome have only been studied in limited contexts to date.16–19 Notable examples include Sammut et al.,20 who demonstrated the ability of RNA-seq to improve prediction of pathologic complete response in early breast cancer (a space where RNA-seq has already demonstrated clinical utility21–23) beyond DNA-seq, pathology imaging, and clinical data; Jiang et al.16 and Cui et al.,24 who built predictors for response to immune checkpoint blockade (ICB) based on the tumor’s immune microenvironment, reflected by RNA data; and the OncoTarget/OncoTreat program at Columbia University that builds on using RNA-seq alone by mapping protein interaction networks derived from the transcriptome to prioritize cancer drivers for treatment and was evaluated in pancreatic neuroendocrine tumors.25 These important and novel approaches have, however, been limited to highly specific clinical contexts and treatments and each has different data requirements. Using such approaches in a scenario where multiple treatment options per patient exist is therefore a very challenging task.12,26–28
In a recent effort to overcome these limitations and develop a uniform systematic approach for stratifying patients to multiple therapies based on the tumor transcriptome, Lee et al.29 have demonstrated that synthetic lethality (SL) and synthetic rescue (SR) interactions can be leveraged to predict treatment response via the transcriptome. An SL interaction between two genes means that the simultaneous inactivation of both genes reduces the viability of the cell while the individual inactivation of each does not.30,31 An SR interaction is one in which the inactivation of one gene reduces cell viability, but an alteration of another gene’s activity, termed the rescuer, restores (rescues) viability.32,33 Their framework, SELECT, mines large-scale in vitro and TCGA patient data to computationally identify putative pairs of genetic interactions (GI) and uses these interactions to predict drug response and prioritize a variety of treatments. It showed that tumor expression could be analyzed systematically to predict patients’ response to a broad range of targeted therapies and immunotherapies in multiple cancer types with high accuracy. However, SELECT has a few notable limitations, which we have set to remedy.
To this end, here we substantially extend the work of Lee et al. and present ENLIGHT, a transcriptomics-based precision oncology pipeline based on the GI networks approach. First, our work has been motivated by a survey we have conducted, where we evaluated the ability of SELECT to identify GI networks for the genes that are inhibited by 105 FDA-approved targeted and immunotherapies, finding that it could build such GI networks for only 67% (70/105) of them, thus limiting its possible use in clinical settings. Addressing this challenge with a new GI-based scoring algorithm that considers different types of GIs concomitantly, ENLIGHT successfully extends the scope of SELECT to all these 105 therapies. Second, SELECT had focused on maximizing the area under the ROC curve (AUC)—a measure commonly used in data science but not directly applicable in clinical settings. In contrast, ENLIGHT’s design and parameter optimization is aimed at maximizing its performance on three key translational tasks: (1) drug coverage—determining the range of drugs for which predictions can be obtainedand extending the prior range of SELECT; (2) personalized oncology—evaluating and prioritizing multiple candidate treatment options for individual patients. Since a variety of treatment options are considered in this scenario, a good test should primarily have a high positive predictive value (PPV) (also known as precision) taking priority over sensitivity (recall), which can be moderate. (3) Improving clinical trial design (CTD) by excluding patients that are unlikely to respond to the treatment by employing a test that has a high negative predictive value (NPV).
To evaluate ENLIGHT’s performance, we analyzed 21 new cohorts not analyzed in the SELECT study, overall spanning 697 patients receiving 15 different drugs in 11 cancer indications, and, importantly, kept them blinded for evaluation purposes. We find that while SELECT is reassuringly predictive on the subset of those datasets for which it could produce results, ENLIGHT still performs better when evaluated using clinically relevant performance measures. We show that (1) ENLIGHT produces predictions for all the drugs used in these cohorts; (2) high ENLIGHT-matching scores (EMS) are associated with better response (odds ratio [OR] = 2.59; 95% confidence interval [CI] [1.85, 3.55]; p = 3.41e–−8). (3) ENLIGHT can be used to successfully exclude patients from clinical trials, achieving overall more than 90% of the response rate attainable under an optimal exclusion strategy for both immunotherapies and targeted monoclonal antibody (mAb) treatments. Finally, we find that ENLIGHT performs as well as supervised tests developed for specific indications and drugs and better than other known transcriptomics-based predictive and prognostic signatures. These results showcase the effectiveness of ENLIGHT as a tool to improve translational oncology research.
RESULTS
Tuning and evaluation datasets
To tune the parameters for ENLIGHT, including the GI network size and a decision threshold on the EMS, which is used for predicting a favorable response (see below), we selected eight tuning sets—six datasets34–39 already analyzed in Lee et al.29—along with two other datasets40,41 from the public domain. These were selected as they span a range of different treatments, therapeutic classes, response rates, and sample sizes, reflecting diverse real-world data, covering five targeted therapies and one ICB therapy.
To study the value of ENLIGHT on real-world data, which was not previously analyzed by either SELECT or ENLIGHT, we surveyed the public domain (GEO,42 ArrayExpress,43 CTRDB,44 and the broader literature) for available datasets of patients receiving targeted or immunotherapies, containing both pre-treatment transcriptomics and response information. In addition, we publish here a new dataset of breast cancer patients who received alpelisib or ribociclib that was obtained as part of a collaboration with Massachusetts General Hospital. Overall, we identified, obtained and set aside 21 datasets20,24,34,39,45–61—the evaluation sets—as unseen data for evaluation. See Table S1 and STAR Methods for full details.
Overview of the ENLIGHT pipeline
ENLIGHT’s drug response prediction pipeline generally follows the flow of SELECT,29 which comprises two steps (Figure 1A): (1) given a drug, the GI engine identifies the clinically relevant GI network of the drug’s target genes. As was done in SELECT, we start with a list of initial candidate SL/SR pairs by analyzing cancer cell line dependencies with RNAi, CRISPR-Cas9, or pharmacological inhibition, based on the principle that SL/SR interactions should decrease/increase tumor cell viability, respectively, when activated (e.g., in the SL case, when both genes are downregulated). Among these candidate pairs, we select those that are more likely to be clinically relevant by analyzing a database of tumor samples with associated transcriptomics and survival data, requiring a significant association between the joint inactivation of a putative SL gene pair and better patient survival, and analogously for SR interactions. Finally, among the candidate pairs that remain after these steps, we select those pairs that are supported by a phylogenetic profiling analysis (see STAR Methods for more details). When considering combination therapies, ENLIGHT computes a GI network based on the union of all the drug targets. (2) The drug-specific GI network is then used to predict a given patient’s response to the drug based on the gene expression profile of the patient’s tumor. The EMS, which evaluates the match between patient and treatment, is based on the overall activation state of the genes in the drug target’s GI network, reflecting the notion that a tumor would be more susceptible to a drug that induces more active SL interactions and fewer active SR interactions. An important prerequisite for a drug to be analyzed by this approach is that the gene targets are well defined. Thus, in this work we focus on targeted therapies and ICBs.
Figure 1. The ENLIGHT pipeline and its tuning for personalized oncology.
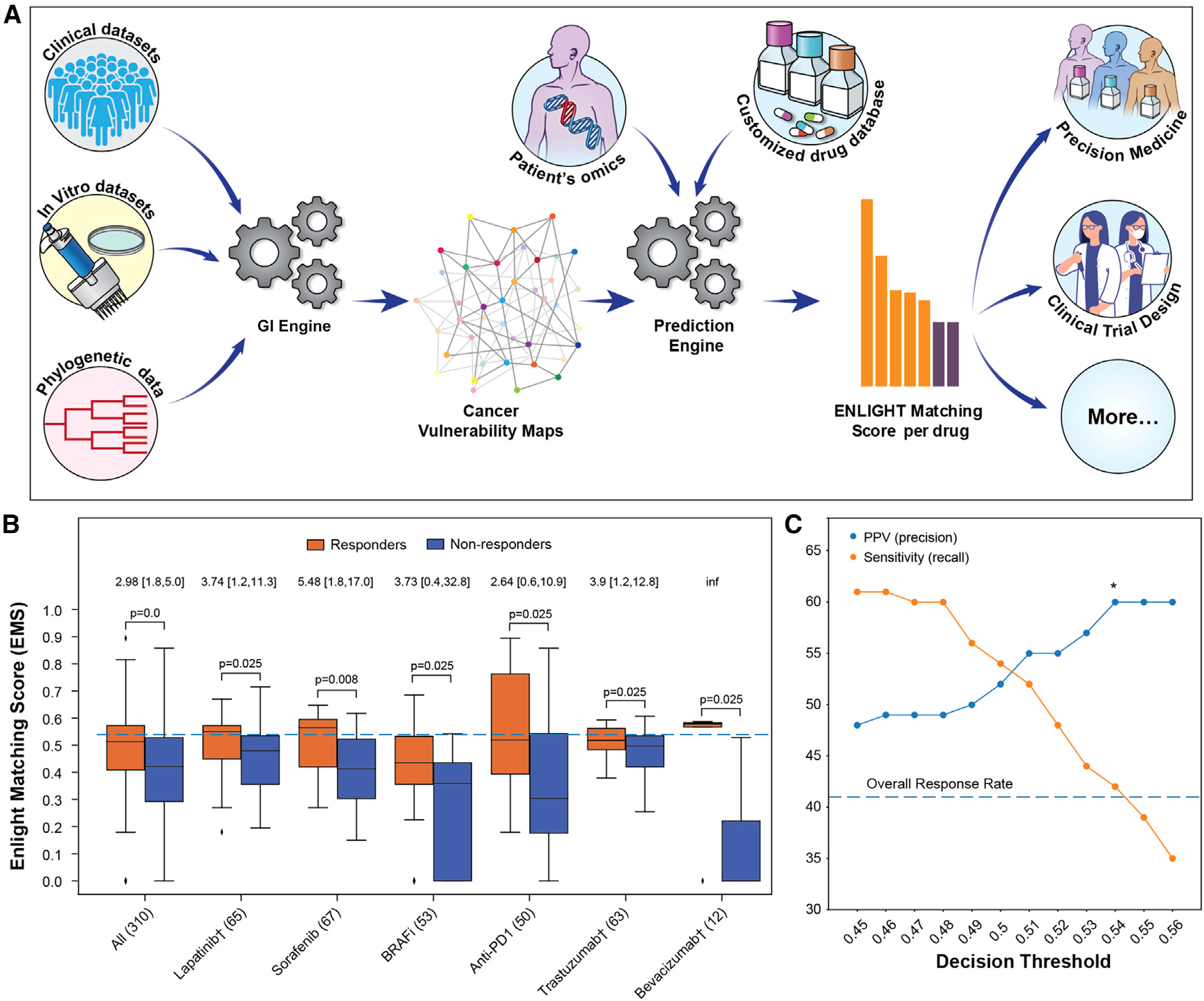
(A) ENLIGHT’s pipeline starts by generating the GI network of a given drug. The GI network and omics data from patients are then used to predict treatment response.
(B) ENLIGHT matching scores in responders (orange) are higher than in non-responders (blue) across the tuning cohorts. The three BRAFi cohorts are very small (n= 16, 17, and 20), so their results are presented in aggregate. The “All” column presents the results for all patients aggregated together. p values were calculated using a one-sided Mann-Whitney test. The horizontal line marks the EMS decision threshold. The odds ratio of ENLIGHT-matched cases appears on top of each pair of bars, with brackets indicating the 95% confidence interval. The number of patients in each cohort is provided in parentheses.
(C) The PPV (precision) and sensitivity (recall) on the aggregate tuning cohort (y axis) as a function of the decision threshold (x axis).
In developing ENLIGHT, we have extended and improved this basic framework, by introducing the following adaptations (see STAR Methods for more details): (1) ENLIGHT’s GI networks include both SL and SR interactions that are concomitantly identified for each drug, thereby considerably increasing drug coverage relative to SELECT, which utilizes only one type of interaction per network (for immunotherapy, where SELECT had no coverage limitation, we use the same SR interactions as in Lee et al., see Figure 5A in Lee et al.29 for details). (2) To improve the GI engine, we follow the idea presented in Lee et al.62 and Sahu et al.63 and add a depletion test (not present in SELECT), requiring that the joint inactivation of a candidate SL pair is underrepresented in tumors (and analogously for SR partners). (3) SELECT has used a Cox proportional hazard test on categorized expression data to identify candidate SL/SR pairs that confer favorable/unfavorable patient survival when the interaction is active. To increase robustness and statistical power, ENLIGHT applies a fully parametric test, based on an exponential survival model, on continuous expression data. (4) ENLIGHT GI networks are considerably larger than those of SELECT to reduce score variation across drugs and indications. (5) To improve the prediction engine, for immunotherapy and other mAbs, which are highly target specific, the EMS incorporates the expression of the target as an additional score component. The ENLIGHT pipeline is described in detail in STAR Methods, where we also describe a web service enabling researchers to apply ENLIGHT to RNA expression data.
We emphasize that the GI networks are generated without using any treatment response data. Notably, the parameters of ENLIGHT, namely the network size and thresholds for the GI engine tests, are optimized on the tuning datasets, but their values are then held fixed to predict all treatment outcomes across all 21 evaluation sets. These conservative procedures markedly mitigate the risk of deriving overfitted predictors that would not predict response in new datasets.
Setting the ENLIGHT decision threshold for personalized oncology based on the tuning cohorts
To define an ENLIGHT-based test for the personalized oncology scenario, we define a uniform decision threshold on the EMS, above which the probability of a patient to respond is high. The chosen decision threshold should maximize the proportion of true responders among those predicted to respond (the PPV), while the proportion of true responders identified by the test (termed its sensitivity, or recall) does not have to be very high since multiple treatment options are usually considered. In other words, in a real-life setting a physician needs to choose one of multiple treatment options, hence our objective is to maximize the PPV of the recommendations given that at least one match would be found for, ideally, each patient. Importantly, the decision threshold, set on the tuning cohorts, is fixed, and is used as is in evaluating ENLIGHT’s performance on all cohorts and treatments. The EMS distributions in the tuning cohorts show that the EMS are significantly higher among responders compared with non-responders (Figure 1B), testifying to its discriminatory power. Figure 1C shows that a decision threshold of 0.54, which will be used henceforth, maximizes the PPV while still maintaining a reasonable sensitivity on the tuning cohort (for the dataset-specific PPV and sensitivity curves see Figures S1 and S2). ENLIGHT’s performance is evaluated using the OR for response, which denotes the odds to respond when the treatment is recommended (ENLIGHT-matched, i.e., EMS above the decision threshold), divided by the odds when it is not. As shown in Figure 1B, ENLIGHT obtains an aggregate OR of 2.98 on the tuning cohorts (aggregation based on individual patients; 95% CI [1.8, 5]; p = 4e–05).
ENLIGHT successfully predicts patient treatment response in 21 different test datasets
We next turned to evaluate how ENLIGHT performs in identifying the true responders in the 21 unseen patient cohorts that we have collected, spanning ICBs, mAbs, and targeted small molecules. Notably, the response data for all evaluation cohorts was unblinded only after finalizing the ENLIGHT pipeline, including fixing the decision threshold and calculating EMS for all patients. Figure 2A shows that ENLIGHT-matched treatments are associated with better patient response (OR > 1) in all cohorts except for two (sorafenib2 and one ICB cohort), with an aggregate OR of 2.59 (95% CI, 1.89–3.55; p = 3.41e–8, n = 697). Correspondingly, Figure 2B shows that the overall PPV obtained for ENLIGHT-matched cases is markedly higher than the overall response rate (52% versus 38%, a 36.84% increase, p = 3.30e–13, one-sided proportion test, and see Table S2 for a more detailed account). Interestingly, ENLIGHT is more accurate in immunotherapies and other mAbs versus targeted small molecules, which aligns with its reliance on drugs that have accurate targets. More specifically, within the small-molecule class, ENLIGHT is only less predictive in drugs with many targets (sorafenib, a broad tyrosine kinase inhibitor and MK2206, a pan-AKT inhibitor). Notably, when a patient received a combination of targeted and chemotherapy agents (see cohorts marked with a cross), the EMS was calculated for the targeted agent alone; however, remarkably, the performance is still maintained.
Figure 2. ENLIGHT’s ability to stratify patients for therapy.
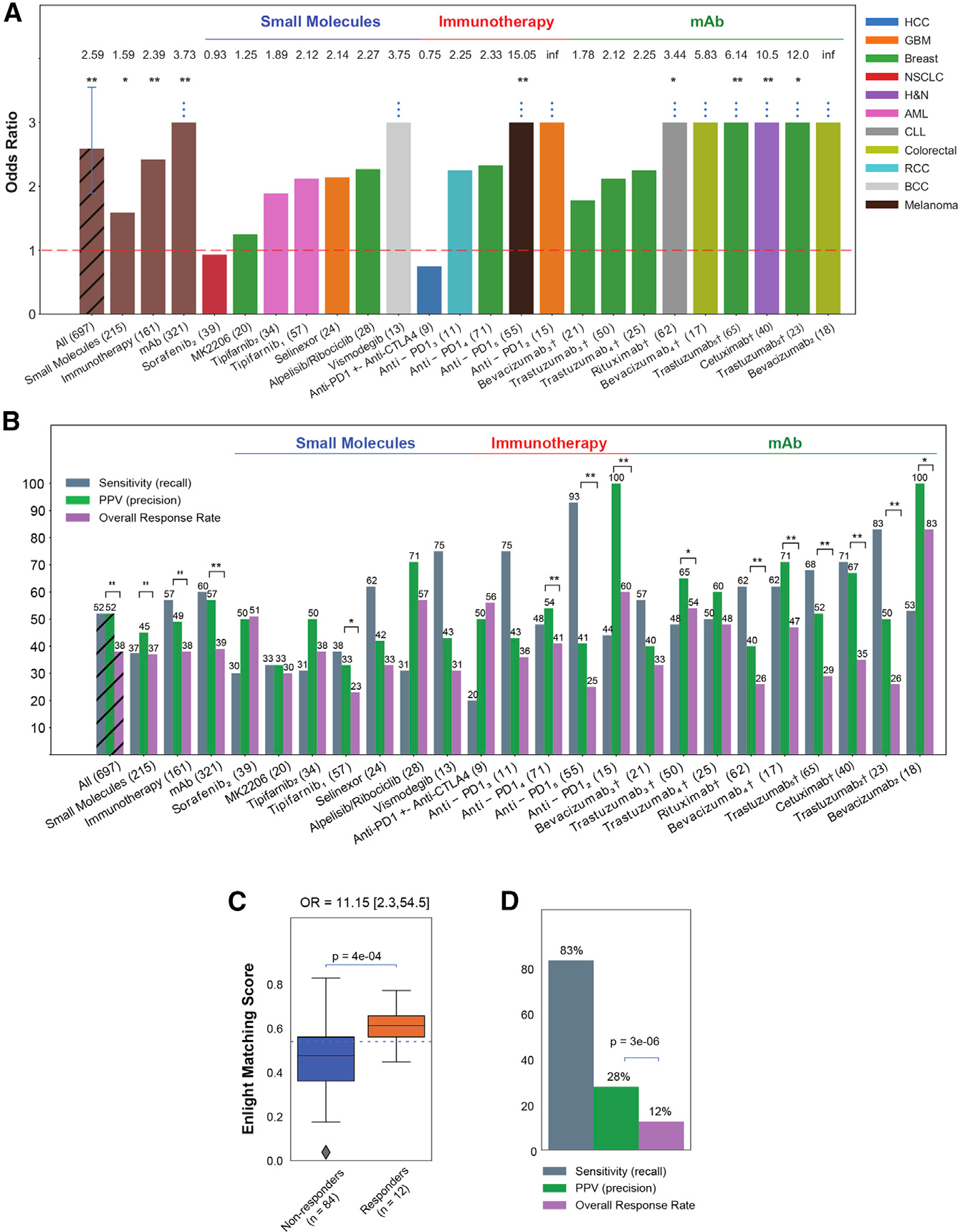
(A) The bar graphs show the OR for response of ENLIGHT-matched cases in the 21 evaluation cohorts (OR values appear on top of each bar; all eight patients predicted to respond in the bevacizumab2 cohort responded to the treatment, resulting in an infinite OR), along with the OR for the aggregation of all cohorts and aggregation based on therapeutic class. Sample sizes are denoted in parentheses. Cohorts for which OR is significantly larger than 1 according to Fisher’s exact test are denoted with asterisks. “Anti-PD1” encompasses three different drugs (nivolumab, pembrolizumab, and durvalumab) (see details in Table S1). Vertical error bars in the “All” bar denotes 95% confidence interval for the OR.
(B) Analogous to (A) but presenting the sensitivity and PPV of ENLIGHT-matched cases versus the overall response rate for the evaluation cohorts and their aggregations. Significant differences between PPV and response rate according to the one-sided proportion test are denoted with asterisks.
(C) In the WINTHER trial, responders (orange) have significantly higher EMS than non-responders (blue); the p value was calculated using a one-sided Mann-Whitney test. 95% confidence interval for the OR is denoted in brackets. The horizontal line marks the decision threshold (0.54).
(D) The sensitivity and PPV of ENLIGHT-matched cases versus overall response rate in the WINTHER trial. p value was calculated according to the one-sided proportion test. †Patients in these cohorts received a combination of targeted and chemotherapy; *p < 0.1, **p < 0.05.
In addition, we evaluated ENLIGHT as a personalized oncology tool in a multi-arm clinical trial setting, by analyzing data from the WINTHER trial, a large-scale prospective clinical trial that has incorporated genetic and transcriptomic data for cancer therapy decision making in adult patients with advanced solid tumors.15 ENLIGHT was able to provide predictions for all patients, except four (see STAR Methods). The EMS of the responders were significantly higher than those of non-responders (p = 4e–04, Figure 2C). The OR of ENLIGHT-matched treatments is 11.15 (p = 8e–04, Figure 2C), and the PPV is more than two times higher than the overall response rate (Figure 2D). Further analysis shows that responders had significantly higher EMS than non-responders also for the 24 patients treated with a combination of drugs (Figure S3A) and that ENLIGHT-matched treatments were associated with better response, without being hampered by the background of chemotherapy treatment (Table S3). Figure S3B depicts the landscape of different treatment alternatives with high EMS scores for each patient. We observe that 91/96 patients (94.8%) had at least one treatment with which they were ENLIGHT-matched, highlighting the potential coverage of ENLIGHT in real-world cases.
Except for tuning very few parameters on the tuning cohorts, as described above, ENLIGHT is essentially an unsupervised prediction method that relies on a series of biologically motivated statistical tests that underlie the identification of GIs and their utilization and is not trained on treatment outcome data. We next turned to compare its predictive power with that of transcriptomics-based biomarker signatures generated via supervised machine learning on specific training cohorts. To this end, we have studied four of the evaluation datasets, for which a supervised classifier was presented in the original publication and data enabling us to perform this comparison was provided (Figure 3A). In one of these datasets, Sammut et al.20 used supervised learning to predict response to chemotherapy with or without trastuzumab among HER2+ breast cancer patients. We focused on the subset of 56 patients who received trastuzumab and for which the model’s scores were published. ENLIGHT achieved a slightly higher AUC than that reported in the original study, without ever training on these data, nor on any other data of response to trastuzumab, and without using any clinical features. Similarly, Cui et al.24 developed a biomarker for response to anti-PD1 in melanoma (see anti-PD15 in Table S1) and Raponi et al.54 and Raponi et al.55 developed biomarkers for response to tipifarnib in AML to which we compared ENLIGHT’s performance. Overall, in three of the four datasets, ENLIGHT’s predictive performance is comparable with that of the biomarkers developed using a supervised approach for a specific treatment and indication. However, in one of these datasets (“Raponi et al. Blood”), while ENLIGHT has a slightly higher PPV, its sensitivity is much lower than that achieved by the supervised method. Taken together, these results demonstrate the remarkable predictive power of ENLIGHT as a pan-cancer and pan-treatment response predictor, which quite successfully competes with supervised models specifically trained for narrow predictive scenarios.
Figure 3. ENLIGHT’s predictive accuracy versus other biomarkers.
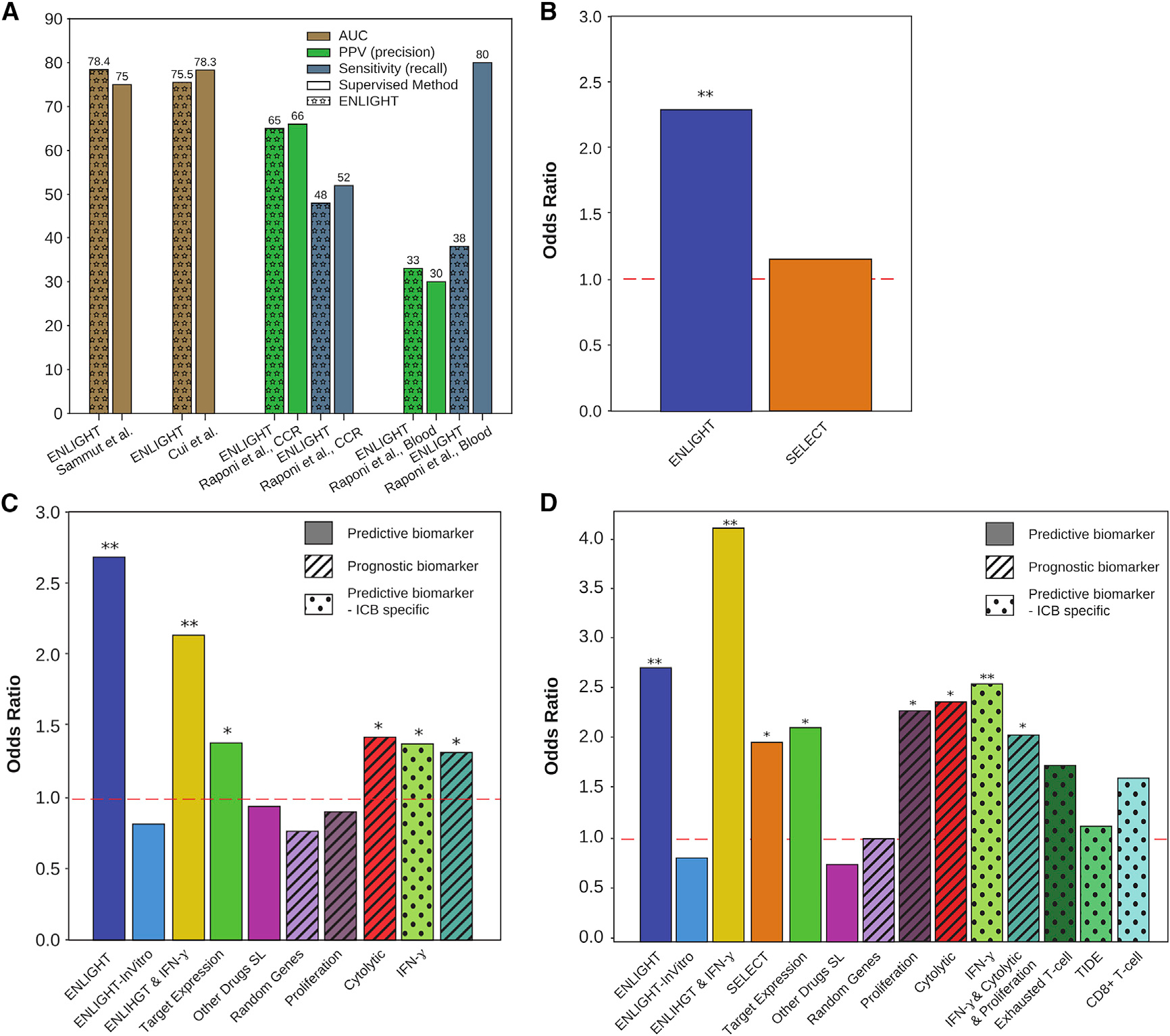
(A) Comparison of ENLIGHT’s performance to that of biomarkers developed using supervised methods. Comparison was performed on the performance measure presented in the respective publication.
(B) OR (y axis) of ENLIGHT (blue) and SELECT (orange) calculated for the 10 cohorts (n = 297) for which SELECT was able to generate a prediction.
(C) OR (y axis) of ENLIGHT (blue) and other transcriptomic and interaction-based biomarkers described in the text (see STAR Methods), calculated for all non-ICB targeted therapies (n= 512).
(D) OR (y axis) of ENLIGHT (blue) and the other biomarkers detailed in (C), as well as SELECT and three ICB-specific transcriptomic biomarkers, calculated for the ICB cohorts (n = 161). Biomarkers denoted by “X” and “Y” (e.g., “ENLIGHT & IFN-γ”) were calculated as the geometric mean of X and Y. “Predictive biomarker”: a biomarker that is drug specific, i.e., yields a different prediction per drug. “Prognostic biomarker”: a biomarker that is drug agnostic, i.e., it is predictive of patient survival/general inclination of response but cannot be used to guide therapy. “Predictive biomarker—ICB specific”: a biomarker that is used to predict general response to immunotherapy but without differentiation between treatments. Asterisks represent the corrected p value for a one-sided test of an OR greater than 1: *p < 0.1, **p < 0.05.
We next turned to compare ENLIGHT’s performance to (1) that of its predecessor, SELECT, (2) several known transcriptomics-deduced metrics (interferon [IFN]-γ signature,17 proliferation signature,64 cytolytic index,65 and the drug target expression levels), and, finally, (3) several GI-based scores. This comparison was done comprehensively on all 21 evaluation cohorts (see STAR Methods). Finally, (4) we added a comparison to three ICB specific biomarkers (exhausted T cell66 TIDE16 and CD8+ T cell abundance67) on the immunotherapy datasets. The performance of ENLIGHT could not be compared with that achieved by DNA-based markers, such as TMB or MSI, since all evaluation datasets available include only mRNA data. We note that several of the widely used biomarkers included in these comparisons do not provide differential scoring for different drugs, i.e., they cannot be used to guide therapy (e.g., proliferation signature), while others are specific to predicting response to ICB (e.g., exhausted T cell). Nevertheless, all are included for completeness.
As all biomarkers other than SELECT and TIDE do not suggest a threshold for determining favorable treatments, we set the decision threshold for those using the tuning sets to match ENLIGHT’s recall (see STAR Methods). For TIDE, we used the threshold of 0, as suggested in Jiang et al.,16 and for SELECT we used 0.44, as suggested in Lee et al.29 For 11 of the 21 evaluation cohorts (n = 400), the GI networks produced by SELECT were too small to produce robust predictions (having less than four GIs). Figure 3B compares the OR of ENLIGHT and SELECT on the remaining 10 cohorts (n = 297). While SELECT provides a beneficial discriminatory power on the evaluation cohorts to which it could be applied, especially for ICBs (Figure 3D), ENLIGHT can analyze a wider range of treatments than SELECT, with overall higher precision. Figures 3C and 3D compare ENLIGHT’s performance to all the biomarkers enumerated above. This is presented separately for non-ICB cohorts (n = 512, the selinexor dataset was excluded since IFN-γ, cytolytic index and target expression could not be calculated on this dataset due to missing data) and for the ICB cohorts (n = 152, the anti-PD1+-Anti-CTLA4 dataset was excluded since missing data prevented calculation of the proliferation signature), since three of the markers are ICB specific, and since SELECT could only be applied to a few of the non-ICB datasets, as described above. Table S4 contains the detailed statistics of all comparisons. In short, ENLIGHT has significantly higher OR than SELECT on the datasets for which SELECT could produce predictions (Figure 3B, p = 0.0487). In addition, ENLIGHT has higher OR than all other markers, with the improvement being statistically significant in all non-ICB datasets (Table S4).
In light of the high predictive power of several ICB-specific markers (namely, proliferation signature, cytolytic index, and IFN-γ signature), we studied if ENLIGHT’s combination with these biomarkers can further enhance the overall predictive power for response to ICB. To this end, we tested the predictive power of new combined biomarkers computed as the geometric mean of: (1) ENLIGHT and each of the three individual markers; (2) ENLIGHT and all three biomarkers; and (3) the three markers without ENLIGHT (for comparison). For each of these combined biomarkers, we optimized the decision threshold as explained in STAR Methods. We found that the combination of ENLIGHT with each of the three biomarkers is more predictive than either ENLIGHT or any of them on their own, with the combination of ENLIGHT and IFN-γ being superior to all other combinations and individual markers, achieving an OR of 4.07 in the ICB cohorts (Figures 3D and S4A). Notably, in the non-ICB cohorts ENLIGHT on its own is superior to the combined biomarkers (Figures 3C and S5).
Overall, these results provide strong support for the ability of ENLIGHT to provide clinical benefit in the precision medicine scenario with superior performance over known markers, spanning a variety of different treatments and cancer types.
ENLIGHT enables near optimal exclusion of non-responding patients in CTD
In the CTD scenario, we are interested in identifying a sub-population of non-responding patients who could be excluded from the trial a priori, thereby allowing smaller studies to achieve higher response rates with adequate statistical power. Figure 4 (top row) depicts the proportion of true non-responders among those predicted not to respond (NPV) as a function of the percent of patients excluded, where patients are excluded by order of increasing EMS. For both immunotherapy and other mAbs, ENLIGHT’s NPV curve is considerably higher than the NPV expected by chance, i.e., the percentage of non-responders, testifying to its benefit. For targeted small molecules, however, it is unable to reliably identify non-responders, an issue that should be further studied and improved upon in future work.
Figure 4. ENLIGHT can facilitate the exclusion of non-responding patients in clinical trials.

Each of the three columns depicts ENLIGHT’s results on the aggregate of all evaluation cohorts from a given therapeutic class. Panels on the top row display the NPV (percentage of true non-responders out of those predicted as non-responders) as a function of the percentage of patients excluded. The horizontal line denotes the actual percentage of non-responders in the corresponding aggregate cohort (i.e., the NPV expected by chance). Panels on the bottom row display the response rate among the remaining patients (y axis) after excluding a certain percentage of the patients (x axis). The horizontal line denotes the overall response rate in the aggregate cohort. The dotted-dashed line represents the upper bound on the response rate, achieved by the “all knowing” optimal classifier excluding only true non-responders.
The bottom row of Figure 4 depicts the response rate in the remaining cohort after excluding patients with EMS below the decision threshold. As evident, ENLIGHT-based exclusion considerably increases the response rate among the remaining patients (middle, solid line). The dotted-dashed line represents the limit performance of an optimal “all-knowing” classifier that excludes all non-responders, retaining only true responders (correspondingly, the x axes end when this optimal classifier excludes all true non-responders, achieving the optimal response rate of 100%). Focusing on a practical exclusion range of up to 25% of patients (shaded area), ENLIGHT-based exclusion achieves 87%–97% and 90%–99% of the optimal exclusion response rate, for both immunotherapy and other mAbs, respectively (see Table 1). It is important to acknowledge that the ENLIGHT-based exclusion strategy assumes knowledge of the EMS distribution in the trial, which may not be known a priori, but could be estimated using historical transcriptomics data from a reference population of the pertaining cancer indication and clinical characteristics.
Table 1.
The performance of ENLIGHT’s exclusion strategy in clinical trial design compared to the optimal upper bound
| % patients excluded | 5 | 10 | 15 | 20 | 25 | |
|---|---|---|---|---|---|---|
|
| ||||||
| % of optimal response rate | Immunotherapy (n = 161) | 97 | 97 | 93 | 90 | 87 |
| mAb (n = 321) | 99 | 98 | 96 | 94 | 90 | |
For each percent of patient exclusion (columns), the response rate among the remaining patients when excluding based on increasing EMS is given as a percentage of the upper bound response rate achieved by the “all knowing” optimal classifier that excludes only true non-responders.
DISCUSSION
Here, we present ENLIGHT, an algorithmic platform that leverages SL and rescue interactions to predict response to targeted therapies and immunotherapies. ENLIGHT is an unsupervised approach that, like its predecessor, SELECT, leverages large-scale data in cancer to generate GI networks associated with drug targets on a whole-genome scale, and then uses the activation patterns of the genes comprising these networks, as measured in the tumor, to generate a matching score for each possible treatment. Differing from its predecessor, ENLIGHT has been designed and evaluated with two real-world clinical scenarios in mind: personalized oncology, where one matches the best treatment to a patient based on a fixed decision threshold, and CTD, where the goal is to a priori exclude non-responding patients in the best possible manner. Testing ENLIGHT on 21 unseen clinical cohorts showed that patients whose treatments were recommended by ENLIGHT have markedly better odds of response than the others. ENLIGHT is a promising systematic approach that, in a comprehensive retrospective analysis, is shown to provide clinically relevant benefits across a broad array of treatments and indications. Despite this broad applicability and its general unsupervised nature, its performance is comparable with that of supervised classifiers trained and predictive on very narrow and specific treatment cohorts. This comparison is of much interest since, in theory, given sufficiently large datasets, such supervised methods are expected to yield higher performance on unseen datasets than unsupervised methods like ENLIGHT. However, if the training sets available are small (as is regrettably the case with the vast majority of the data currently available), practice may differ from theory and supervised methods may overfit and underperform.
ENLIGHT is also more predictive than its predecessor, SELECT, and other transcriptomics-based biomarkers. Some of the biomarkers in these comparisons are prognostic in nature, i.e., provide information about the patient’s overall cancer outcome, regardless of therapy (e.g., proliferation signature) or are specific to predicting response to ICB (e.g., T cell exhaustion). Unlike ENLIGHT, such markers cannot add information when one aims to choose between various treatments. Specifically, IFN-γ had an OR comparable with that of ENLIGHT on the ICB datasets, which are almost exclusively anti-PD1. However, unlike ENLIGHT, it will not be able to offer differential scoring as more ICB treatments enter the clinic, since its score is not treatment dependent. The combination of ENLIGHT and IFN-γ has increased predictive performance for ICB therapy, obtaining an impressive OR of 4, and offering differential scoring per treatment, due to ENLIGHT’s predictive nature. Finally, ENLIGHT can enhance CTD, by efficiently excluding non-responsive patients while attaining more than 90% of the maximum attainable response rate in targeted mAbs and ICB cohorts.
In the past several years, bulk RNA-seq has become increasingly available and reliable, opening the door for translational and clinical applications.68 The Pan-Cancer Analysis of whole genomes demonstrated how the majority of classical driver genes exhibit alterations that were potentially better characterized via RNA than DNA.69 Beyond developing drugs for new targets, we must consider ways to expand eligibility criteria for existing targeted therapies and immunotherapies. Data from ENLIGHT can provide a biologically informed basis to test strongly prioritized off-label therapies for specific patient populations through phase II trials. This may be especially helpful in rare cancer types or challenging clinical scenarios, where hypothesis generation will only require access to relevant RNA-seq samples. Second, building on the CTD capacity of ENLIGHT, the tool can offer simulations focused on a given therapeutic option and its performance across multiple types of patient populations—a computational version of the resource-challenging yet exciting basket trial design.
This study mainly establishes ENLIGHT’s power to predict monotherapies. However, in clinical practice, identifying combination therapy is often desirable. Notably in this context, ENLIGHT shows promising results when predicting response to targeted therapy on the background of chemotherapy, suggesting that a clinician could use ENLIGHT to identify favorable immune/targeted therapy and combine it with chemotherapy as deemed fit clinically.
While ENLIGHT’s performance is evaluated here on a broad array of unseen clinical trial datasets, its performance should obviously be further evaluated in prospective studies. Indeed, based on the results obtained so far by SELECT and ENLIGHT, the design of such a multi-arm study—SYNTHESIS—is now underway at the National Cancer Institute’s Center for Cancer Research. We hope that the results presented here will elicit additional prospective studies in other clinical centers across the globe and we are committed to facilitating such studies.
Limitations of the study
Like any response prediction approach, ENLIGHT has several limitations that should be acknowledged. First, as it operates on the drug target level, it has very limited utility in predicting response to chemotherapies and, more generally, its prediction accuracy depends on the accuracy with which the targets of a given drug have been identified. Second, most datasets included in this study are based on mRNA expression derived from microarrays, while the standard is shifting toward RNA-seq data. Third, current applications of ENLIGHT have focused on bulk tumor transcriptomics. Future work is needed to study its application for analyzing single-cell tumor transcriptomics, to build predictors that consider tumor heterogeneity and the important interplay between a tumor and its immune microenvironment. Fourth, while gene expression is at times strongly associated with protein levels, in other cases it is not, especially for lowly expressed genes. Notably, some of these limitations may point us to future directions for building even better GI-based predictive approaches. This includes approaches for identifying and harnessing cell-type specific interactions from single-cell and deconvolved expression data, approaches that chart the GI landscape at different regions of the tumor, and more. Finally, one may reasonably anticipate that the advent of much larger molecular and response datasets will on its own contribute to the accuracy of the methods already extant.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ranit Aharonov (ranit@pangeabiomed.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All transcriptomic and clinical information including treatment and outcome information of the datasets used in the paper are publicly available as individually described in Table S1. For ease of usage, all datasets analyzed in this manuscript can be found in https://github.com/PangeaResearch/enlight-data. See Key Resources Table for full details.
Any additional data required to reanalyze the data reported in this paper is available from the lead contact upon request.
A web service that generates EMS for the drugs described in this study as well as for any new data supplied by the user is available at https://ems.pangeabiomed.com/.
KEY RESOURCES TABLE.
METHOD DETAILS
Data collection
We surveyed the public domain for available cohorts of patients receiving targeted therapies or immunotherapies, containing both pre-treatment transcriptomics and response information (either RECIST or a binary classification of response). We identified a total of 23 real world datasets which were not previously analyzed by either SELECT or ENLIGHT, and can hence serve as unseen datasets: 22 datasets20,24,34,40,45–61 from GEO, ArrayExpress, CTRDB or the broader literature published by February 2022, and one dataset that was obtained as part of a collaboration with Massachusetts General Hospital (MGH) which we publish here for the first time. We selected six datasets34–39 already analyzed in Lee et al.29 along with two40,41 of these 23 unseen sets to serve as tuning sets. These eight tuning datasets were selected as they span a range of different treatments, therapeutic classes, response rates and sample sizes, reflecting diverse real-world data, covering five targeted therapies and one immune checkpoint blockade (ICB). These datasets were used to tune the parameters of ENLIGHT, including the GI network size and a decision threshold on the ENLIGHT Matching Score (EMS) that is used for predicting response (see below). We set aside the remaining 21 unseen datasets – the evaluation sets – as unseen data for evaluation.
Table S1 details the tuning and evaluation datasets used in this study. All datasets were coupled with response to treatment in the form of either: (i) RECIST criteria response evaluations or (ii) binary classifications of responders and non-responders that was not exclusively defined using RECIST and in several cases was not specified. In this study, we classify a patient as responder if he/she had a RECIST evaluation of CR/PR, or if he/she had a binary classification of responder. The rest were classified as non-responders. In each dataset we only analyzed patients for whom both pre-treatment transcriptomics and response data was available. N in the table denotes the number of patients that were analyzed. The Selinexor dataset encompasses data from a clinical trial containing several arms, each with a different treatment dosage which significantly affected the response rate. Here we only analyzed the largest arm of the trial (N = 24). In cohort Anti-PD14, which is a combination of immunotherapy and a targeted drug, we analyzed the score based on the immunotherapy only.
ENLIGHT improvements over SELECT
In developing ENLIGHT, we have extended and improved SELECT, by introducing the following adaptations:
ENLIGHT leverages a larger amount of in-vitro data that was updated since SELECT was published. This results in a more robust list of initial GI candidate pairs.
While SELECT uses 25 SL pairs in its targeted drug GI networks, ENLIGHT’s GI networks for targeted therapies include a combined list of 100 SL and/or SR interactions that are concomitantly identified for each drug. The fact that ENLIGHT utilizes both SL and SR interactions considerably increases the number of drugs for which it can generate a GI network, and hence produce predictions. In addition, using larger networks reduces the variance in score distributions across treatments and cancer types, allowing a uniform test for multiple drugs. For immunotherapies, SELECT had full coverage and hence ENLIGHT uses the same size 10 GI networks.
ENLIGHT follows Lee et al.62 and Sahu et al.63 in requiring SL/SR pairs to display a low joint disadvantageous/advantageous activation state in clinical samples, which is reflected by a depletion test, added as a step in the GI engine (not present in SELECT). In developing ENLIGHT, the depletion test went under complete revision. Instead of a hypergeometric test on categorized data to identify depletion for both SL and SR interactions, ENLIGHT’s depletion test differs between SL and SR: for the SL case, the depletion test is built on the fundamentals of the Gumbel copula, applied on continuous RNA expression data to identify pairs with low probability of being simultaneously inactive. The depletion test for SR requires that the activation state of a rescuer gene be conditioned on its partner being inactive.
SELECT uses cox proportional hazard test on categorized expression data to select candidate SL/SR pairs that confer favorable/unfavorable patient survival when the interaction is active. To increase robustness and statistical power, ENLIGHT applies a fully parametric test, based on an exponential survival model, on continuous expression data.
For treatments that are highly target specific, namely ICB and other mAbs, the ENLIGHT matching score incorporates the target expression, since the drug is expected to be more effective when the target expression is higher. Specifically, the EMS is a geometric mean of the network-based score and a logistic function of the target expression.
The ENLIGHT pipeline
As explained and detailed in the main text and above, ENLIGHT improves and extends upon SELECT, making important changes to the pipeline to improve and focus it on translational aspects. For ease of reading, we include here the entire ENLIGHT pipeline, including those parts which are taken from,29,62,63 and properly refer herein.
The ENLIGHT algorithm can be broadly divided into two steps (see Figure 1A for a visualization of the pipeline): (i) the GI engine and (ii) the prediction engine.
GI engine
The GI engine uses 4 statistical tests to identify interacting gene pairs:
1. in-vitro test
By definition, it is expected that gene A will be more essential when its SL partner gene B is inactive in a cancer cell line. Using a set of input genome-wide shRNA/siRNA/sgRNA screens mined from DepMap,70–72 ENLIGHT identifies pairs that show conditional essentiality: Gene A is defined as SL conditionally essential with gene B if its essentiality is significantly higher in cell-lines where gene B is inactive using Wilcoxon rank sum test. We use both mRNA expression and SCNA data to classify genes as over/under-active. Similarly, gene A is defined as SR-DU/SR-DD conditionally essential with gene B if its essentiality is significantly higher in the samples where gene B is underactive/overactive. An SR-DU/SR-DD interaction between gene A and gene B dictates that a cell can be rescued from cell death caused by the inhibition of gene A by the upregulation/downregulation of gene B respectively.
2. Depletion test
ENLIGHT follows the ideas of Lee et al.62 and Sahu et al.63 in requiring SL/SR pairs not to display joint activation patterns that are disadvantageous for SL/SR interactions in patient tumors. This requirement is implemented by a depletion test, added as a step to the GI engine. Conceptually, if an SL interaction exists between a pair of genes, we would expect not to observe tumors in which both genes are inactive, since this would have caused tumor cell lethality. Similarly, we would not expect patients with inactivation of a gene to have low/high activation of its SR-DU/SR-DD rescuer, since that would induce tumor cell death. To summarize, in both SL/SR cases we are looking for the statistical absence (or depletion) of a non-favorable joint activation pattern in observational cohorts to support a GI between gene pairs.
3. Survival test
This test identifies candidate SL/SR pairs that confer favorable/unfavorable patient survival when the interaction is active. When an SL pair is simultaneously inactive in a patient or a cell-line we term it active. Similarly, we term an SR-DU/SR-DD as active when gene A is inactive in conjunction with its SR-DU/SR-DD partner being underactive/overactive. The survival test used in ENLIGHT is a fully parametric test, based on an exponential survival model. Given the omics of a gene pair from a patient cohort, coupled with survival data, we first calculate a covariate value for each patient, reflecting its joint activation state of the gene pair. For a true SL/SR pair, patients in whom the joint activation of the pair is in a disadvantageous state in the tumor, are expected to have better survival, since this should lead to tumor cell death. Hence, the covariate value is positively associated with survival time in these cases. The statistical model of the test follows the common assumption that covariates have a log-linear effect on survival times, and the coefficient of the SL/SR covariate reflects whether a putative interaction confers a significant effect on patient survival. The model also controls for patient age, gender and stage as confounding factors. If data is missing for any of these three attributes in a patient, we set it to the mean of all other patients (or the majority in the case of gender). Altogether, the likelihood of observing a population with survival times t and covariates matrix X is given by:
where OBS are the set of deceased patients, S is the survival function, λ is the hazard function, xi are the covariates for patient i, ti is the time of death or censoring for patient i and β are the coefficients associated with the covariates. We solve this equation to identify the coefficient β associated with the interaction along with its 95% CI and p-value.
4. Phylogenetic test
The last test identifies SL/SR pairs with high similarity between their phylogenetic profiles, following observations that interacting genes were found to be conserved across different species73,74 and following the analysis of Lee et al.29,62 This is done by calculating the Euclidean distance between the genetic similarity profiles of two genes A and B across 86 species, while taking into account the baseline phylogenetic distance between the species (adopting the method of Tabach et al.75).
In order to build a GI network around specific drug target/s, we start by performing the above 4 tests for all putative SL/SR-DU/SR-DD pairs between the targets of the drug and all other genes in the genome. Then, we sequentially filter out non-significant pairs for each test, starting from all pairs and all interaction types, so that only pairs that pass all 4 tests are kept. We follow Lee et al. for setting the statistical significance thresholds. Finally, we rank the remaining interactions according to the survival test as it best reflects the clinical impact of the interactions, and use the top K interactions to build the GI network. In this study we tested K = 25, 50, 100, 150 and 200 on the tuning sets and selected K = 100 as it achieved the best PPV. For immunotherapies, ENLIGHT uses the same GI networks used in SELECT29 (K = 10).
Prediction engine
The prediction engine predicts the response to a given drug or a given combination of drugs with known drug targets based on quantitative RNA data (microarray or RNAseq). The prediction engine works as follows:
GI network generation
First, a GI network surrounding all drug targets is generated based on the GI engine.
Normalization
Next, the RNA data of the cohort is rank normalized to values in [0,1] twice: gene-wise and patient-wise. Gene-wise normalized values are used to identify gene activation states of SL/SR partners across comparable samples of the same tissue. Patient-wise normalized values are used to determine whether the drug targets are expressed to a minimal degree in a patient for the drug to have an effect.
Scoring
The ENLIGHT Matching Score (EMS) is defined as the fraction of SL/SR interactions that are in an advantageous predisposition for drug admission. That is, a drug that inhibits gene A is expected to work better in patients for whom an SL/SR-DU B of A is underexpressed, or for whom an SR-DD partner B of A is overexpressed. Thus, the fraction of under/over expressed partners in advantageous states (with respect to the interaction type) is expected to be positively associated with response. A gene is determined to be underexpressed if its normalized expression is equal to or below 1/3 (i.e. is in the bottom tertile across samples in the same dataset), or overexpressed if its normalized expression is equal to or above 2/3 (i.e. is in the top tertile across samples in the same dataset), similar to Lee et al.29 In addition, we zero the EMS of a patient if its mean patient-wise normalized expression of drug targets is below or equal to the 30th percentile across genes. This has been motivated by the notion that an antagonist drug will not be effective when its target genes are underexpressed. Finally, for treatments that are highly target specific, namely ICB and other mAbs, the EMS incorporates the target expression, since the drug is expected to be more effective when the target expression is higher.19,76 Specifically, the EMS is a geometric mean of the network-based score and a logistic function of the target expression. Drug target genes were mapped based on DrugBank (Law et al., 2014).
Analysis of the WINTHER trial
We analyzed 100 patients for whom both treatment outcome and transcriptomic data were available. Of these, 97 received at least one targeted or immunotherapy agent, 1 of which had missing values that deemed the case non-analyzable. Thus, in total, we calculated an ENLIGHT Matching Score for 96 patients. Among the remaining 96 patients, one patient had a complete response and 11 patients had a partial response. These 12 patients were considered responders, while the 15 patients with stable disease and the 69 patients with progressive disease, were considered non-responders. To generate a GI network for a regimen involving several drugs, we considered the union of the targets from all the drugs in the regimen. We did not consider the targets of chemotherapies or hormonal therapies that were part of the treatment.
Figure S3B shows the EMS of each patient for the prescribed regimen (the Winther row) as well as for all ENLIGHT-analyzable drugs in the WINTHER trial. We observe that ENLIGHT identified at least one favorable treatment for all but one patient, and thus, in theory, other drugs or drug combinations may have proven more beneficial for those patients who did not respond.
Biomarkers compared with ENLIGHT
In order to compare the OR for identifying beneficial treatments between different biomarkers, the focus and objective of this study, one has to set a clinical threshold for binary response classification. For two of the biomarkers compared in this study, such thresholds were given in the original publications. For SELECT, we used the threshold of 0.44 as described in Lee et al.29 and for TIDE we used the threshold of 0 as suggested by Jiang et al.16 For the remaining markers, we used the tuning sets to assign thresholds. For each biomarker, we calculated its scores in the tuning datasets, and searched for a threshold that matches ENLIGHT’s recall:
For comparing ENLIGHT to other biomarkers on non-ICB datasets (Figure 3C), we identified a threshold for each of the presented biomarkers based on all tuning datasets except Anti-PD1.
For comparing ENLIGHT to other biomarkers on ICB datasets (Figure 3D), we identified a threshold for each of the presented biomarkers based only on Anti-PD1.
The difference in threshold identification is intended to make an appropriate comparison to ENLIGHT’s recall which naturally varies between the two cases. The identified thresholds were then used to calculate OR based on the test datasets for Figure 3.
In order to avoid artifacts arising from varying mRNA quantification platform (i.e. RNA-seq vs. microarrays), for example the difference in the dynamic range of expression between platforms, all biomarkers that do not have explicit means for calculation (i.e. all markers except SELECT, TIDE and CD8+ abundance which have available code for calculation) were calculated based on gene-wise ranking of expression across samples, after applying proper normalizations to account for library size and gene lengths (if needed). The description of each marker is as follows:
Target expression
Mean expression of all drug targets in the treatment regimen.
ENLIGHT-InVitro
An EMS-like score that uses as the GI network, all interactions that passed the first test of ENLIGHT: the In-Vitro test.
Other drugs SL
An EMS-like score that uses the combined GI network of all drugs shown in Figure 2A except the given drug.
Random genes
An EMS-like score based on a GI network populated by a set of random genetic interactions. That is, we randomly choose 100 gene partners and an interaction type for each (SL/SR), and use them to score each sample in the same way the EMS is calculated.
IFN-γ
A score based on a signature that measures the expression of the Interferon-gamma pathway.17
Proliferation
A score based on the proliferation signature suggested by.64
Cytolytic
A score based on the Cytolytic index suggested by.65
Exhausted T-cell
A signature that measure T-cell exhaustion in the TME as suggested by.66
TIDE
a gene expression-based classifier that predicts response to ICB based on T-cell exclusion/exhaustion developed by.16
CD8+ T-cell
CD8+ T-cell abundance as calculated by CYBERSORT.67
QUANTIFICATION AND STATISTICAL ANALYSIS
Differences between distributions were analyzed using a one-sided Mann-Whitney test using the scipy.stats.mannwhitneyu function in python. Test of OR > 1 was done using Fisher’s exact test. Difference between OR was done using a one-sided test for difference in OR. Difference between PPV and overall response rates was done using a one-sided proportion test. Difference between OR of two methods was done using Chi square test. All statistical results were FDR corrected for multiple hypotheses throughout the paper. Throughout the paper, we denoted p < 0.1 with a single asterix (*) and p < 0.05 with two asterix (**). Throughout the paper, brackets denote 95% CI and N denotes the sample size of the dataset.
Supplementary Material
CONTEXT AND SIGNIFICANCE.
Improving cancer treatments requires both developing better treatments and finding new ways to best match them to individual patients. This precision oncology approach has been advancing into oncological practice, demonstrating its benefits but also calling for better ways to find the best tumor-drug match. Here, researchers at Pangea Biomed and the National Cancer Institute present ENLIGHT, a computational tool based on cancer gene expression profiles that aims at predicting the most appropriate drug and their dose for each patient, as well as improving clinical trial design by predicting patients unlikely to respond to the treatment. Their comprehensive results show that ENLIGHT can markedly enhance the ability to predict therapeutic response across multiple cancer types, laying a solid basis for testing it in clinical trials.
Highlights.
ENLIGHT enables clinical response prediction from the tumor transcriptome
ENLIGHT can effectively predict response across multiple therapies and cancer types
ENLIGHT does not require training on previous treatment response data
ENLIGHT successfully competes with known biomarkers and supervised predictors
ACKNOWLEDGMENTS
This research was supported in part by the Israeli Innovation Authority. This research was supported in part by the Intramural Research Program, National Institutes of Health, National Cancer Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
DECLARATION OF INTERESTS
G.D., E.D.S., E. Elis, D.S.B.-Z., O.T., E.M., E. Elalouf, B.T., P.V., T.B., and R.A. are employees of Pangea Biomed. I.M. is a paid consultant of Pangea Biomed. E.S. is the Chairman of the Board of Pangea Biomed. E.R. is a co-founder of MedAware, Metabomed, and Pangea Biomed (divested) and an unpaid member of Pangea Biomed’s scientific advisory board. Z.R. is a co-founder of Pangea Biomed and an unpaid member of its scientific advisory board. R.B. is a member of Pangea Biomed’s scientific advisory board.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2022.11.001.
REFERENCES
- 1.Schwartzberg L, Kim ES, Liu D, and Schrag D (2017). Precision oncology: who, how, what, when, and when not? Am. Soc. Clin. Oncol. Educ. Book. 37, 160–169. 10.1200/EDBK_174176. [DOI] [PubMed] [Google Scholar]
- 2.Doroshow DB, and Doroshow JH (2020). Genomics and the history of precision oncology. Surg. Oncol. Clin. N. Am. 29, 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olivier T, Haslam A, and Prasad V (2021). Anticancer drugs approved by the US food and drug administration from 2009 to 2020 according to their mechanism of action. JAMA Netw. Open 4, e2138793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn R, Patel R, Sison C, Singh A, and Zhu XH (2021). Impact of precision medicine on clinical outcomes: a single-institution retrospective study. Front. Oncol. 11, 659113. 10.3389/fonc.2021.659113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cutler DM (2020). Early returns from the era of precision medicine. JAMA 323, 109–110. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- 8.Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, De P, Krie A, Piccioni DE, Miller VA, et al. (2019). Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat. Med. 25, 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flaherty KT, Gray RJ, Chen AP, Li S, McShane LM, Patton D, Hamilton SR, Williams PM, Iafrate AJ, Sklar J, et al. (2020). Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: national cancer Institute molecular analysis for therapy choice (NCI-match). J. Clin. Oncol. 38, 3883–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gyawali B, D’Andrea E, Franklin JM, and Kesselheim AS (2020). Response rates and durations of response for biomarker-based cancer drugs in nonrandomized versus randomized trials. J. Natl. Compr. Canc. Netw. 18, 36–43. [DOI] [PubMed] [Google Scholar]
- 11.Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian SL, Taube JM, Anders RA, and Pardoll DM (2016). Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu Y-M, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. (2011). Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci. Transl. Med. 3, 111ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roychowdhury S, and Chinnaiyan AM (2016). Translating cancer genomes and transcriptomes for precision oncology. CA. Cancer J. Clin. 66, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodon J, Soria J-C, Berger R, Miller WH, Rubin E, Kugel A, Tsimberidou A, Saintigny P, Ackerstein A, Braña I, et al. (2019). Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat. Med. 25, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536. [DOI] [PubMed] [Google Scholar]
- 19.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792. 10.1056/nejm200103153441101. [DOI] [PubMed] [Google Scholar]
- 20.Sammut S-J, Crispin-Ortuzar M, Chin S-F, Provenzano E, Bardwell HA, Ma W, Cope W, Dariush A, Dawson S-J, Abraham JE, et al. (2022). Multi-omic machine learning predictor of breast cancer therapy response. Nature 601, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, Geyer CE Jr., Dees EC, Goetz MP, Olson JA Jr., et al. (2018). Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 379, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, Pierga J-Y, Brain E, Causeret S, DeLorenzi M, et al. (2016). 70-Gene signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 375, 717–729. [DOI] [PubMed] [Google Scholar]
- 23.National Comprehensive Cancer Network. Breast Cancer. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf.
- 24.Cui C, Xu C, Yang W, Chi Z, Sheng X, Si L, Xie Y, Yu J, Wang S, Yu R, et al. (2021). Ratio of the interferon-g signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genom. Med. 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 50, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsimberidou AM, Fountzilas E, Nikanjam M, and Kurzrock R (2020). Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 86, 102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malone ER, Oliva M, Sabatini PJB, Stockley TL, and Siu LL (2020). Molecular profiling for precision cancer therapies. Genome Med. 12, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng F, Shen B, Mou X, Li Y, and Li H (2021). Large-scale pharmacogenomic studies and drug response prediction for personalized cancer medicine. J. Genet. Genomics 48, 540–551. [DOI] [PubMed] [Google Scholar]
- 29.Lee JS, Nair NU, Dinstag G, Chapman L, Chung Y, Wang K, Sinha S, Cha H, Kim D, Schperberg AV, et al. (2021). Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell 184, 2487–2502.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen JP, and Ideker T (2018). Synthetic lethal networks for precision oncology: promises and pitfalls. J. Mol. Biol. 430, 2900–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lord CJ, and Ashworth A (2017). PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velimezi G, Robinson-Garcia L, Muñoz-Martínez F, Wiegant WW, Ferreira da Silva J, Owusu M, Moder M, Wiedner M, Rosenthal SB, Fisch KM, et al. (2018). Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48. Nat. Commun. 9, 2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eischen CM, Woo D, Roussel MF, and Cleveland JL (2001). Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell Biol. 21, 5063–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dieci MV, Prat A, Tagliafico E, Paré L, Ficarra G, Bisagni G, Piacentini F, Generali DG, Conte P, and Guarneri V (2016). Integrated evaluation of PAM50 subtypes and immune modulation of pCR in HER2-positive breast cancer patients treated with chemotherapy and HER2-targeted agents in the CherLOB trial. Ann. Oncol. 27, 1867–1873. [DOI] [PubMed] [Google Scholar]
- 35.Guarneri V, Dieci MV, Frassoldati A, Maiorana A, Ficarra G, Bettelli S, Tagliafico E, Bicciato S, Generali DG, Cagossi K, et al. (2015). Prospective biomarker analysis of the randomized CHER-LOB study evaluating the dual anti-HER2 treatment with trastuzumab and lapatinib plus chemotherapy as neoadjuvant therapy for HER2-positive breast cancer. Oncol. 20, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kakavand H, Rawson RV, Pupo GM, Yang JYH, Menzies AM, Carlino MS, Kefford RF, Howle JR, Saw RPM, Thompson JF, et al. (2017). PD-L1 expression and immune escape in melanoma resistance to MAPK inhibitors. Clin. Cancer Res. 23, 6054–6061. [DOI] [PubMed] [Google Scholar]
- 37.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. (2014). BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin. Cancer Res. 20, 1965–1977. [DOI] [PubMed] [Google Scholar]
- 38.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinyol R, Montal R, Bassaganyas L, Sia D, Takayama T, Chau G-Y, Mazzaferro V, Roayaie S, Lee HC, Kokudo N, et al. (2019). Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 68, 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Konishi T, Horie H, Ikeuchi H, et al. (2011). Gene expression signature and response to the use of leucovorin, fluorouracil and oxaliplatin in colorectal cancer patients. Clin. Transl. Oncol. 13, 419–425. [DOI] [PubMed] [Google Scholar]
- 41.Prat A, Bianchini G, Thomas M, Belousov A, Cheang MCU, Koehler A, Gómez P, Semiglazov V, Eiermann W, Tjulandin S, et al. (2014). Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin. Cancer Res. 20, 511–521. [DOI] [PubMed] [Google Scholar]
- 42.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Athar A, Füllgrabe A, George N, Iqbal H, Huerta L, Ali A, Snow C, Fonseca NA, Petryszak R, Papatheodorou I, et al. (2019). ArrayExpress update - from bulk to single-cell expression data. Nucleic Acids Res. 47, D711–D715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Liu J, Liu X, Wang X, Xie Q, Zhang X, Kong X, He M, Yang Y, Deng X, et al. (2022). CTR-DB, an omnibus for patient-derived gene expression signatures correlated with cancer drug response. Nucleic Acids Res. 50, D1184–D1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pentheroudakis G, Kotoula V, Fountzilas E, Kouvatseas G, Basdanis G, Xanthakis I, Makatsoris T, Charalambous E, Papamichael D, Samantas E, et al. (2014). A study of gene expression markers for predictive significance for bevacizumab benefit in patients with metastatic colon cancer: a translational research study of the Hellenic Cooperative Oncology Group (HeCOG). BMC Cancer 14, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Birkbak NJ, Li Y, Pathania S, Greene-Colozzi A, Dreze M, Bowman-Colin C, Sztupinszki Z, Krzystanek M, Diossy M, Tung N, et al. (2018). Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann. Oncol. 29, 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verstraete M, Debucquoy A, Dekervel J, van Pelt J, Verslype C, Devos E, Chiritescu G, Dumon K, D’Hoore A, Gevaert O, et al. (2015). Combining bevacizumab and chemoradiation in rectal cancer. Translational results of the AXEBeam trial. Br. J. Cancer 112, 1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. (2013). An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu JC, Voisin V, Bader GD, Deng T, Pusztai L, Symmans WF, Esteva FJ, Egan SE, and Zacksenhaus E (2012). Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERa-breast cancer. Proc. Natl. Acad. Sci. USA 109, 5832–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen K, Qi Y, Song N, Tian C, Rice SD, Gabrin MJ, Brower SL, Symmans WF, O’Shaughnessy JA, Holmes FA, et al. (2012). Cell line derived multi-gene predictor of pathologic response to neoadjuvant chemotherapy in breast cancer: a validation study on US Oncology 02–103 clinical trial. BMC Med. Genomics 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bossi P, Bergamini C, Siano M, Cossu Rocca M, Sponghini AP, Favales F, Giannoccaro M, Marchesi E, Cortelazzi B, Perrone F, et al. (2016). Functional genomics uncover the biology behind the responsiveness of head and neck squamous cell cancer patients to cetuximab. Clin. Cancer Res. 22, 3961–3970. [DOI] [PubMed] [Google Scholar]
- 52.Lassman AB, Wen PY, van den Bent MJ, Plotkin SR, Walenkamp AME, Green AL, Li K, Walker CJ, Chang H, Tamir S, et al. (2022). A phase II study of the efficacy and safety of oral selinexor in recurrent glioblastoma. Clin. Cancer Res. 28, 452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Magbanua MJM, Li W, Wolf DM, Yau C, Hirst GL, Swigart LB, Newitt DC, Gibbs J, Delson AL, Kalashnikova E, et al. (2021). Circulating tumor DNA and magnetic resonance imaging to predict neoadjuvant chemotherapy response and recurrence risk. NPJ Breast Cancer 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raponi M, Harousseau J-L, Lancet JE, Löwenberg B, Stone R, Zhang Y, Rackoff W, Wang Y, and Atkins D (2007). Identification of molecular predictors of response in a study of tipifarnib treatment in relapsed and refractory acute myelogenous leukemia. Clin. Cancer Res. 13, 2254–2260. [DOI] [PubMed] [Google Scholar]
- 55.Raponi M, Lancet JE, Fan H, Dossey L, Lee G, Gojo I, Feldman EJ, Gotlib J, Morris LE, Greenberg PL, et al. (2008). A 2-gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood 111, 2589–2596. [DOI] [PubMed] [Google Scholar]
- 56.Foà R, Del Giudice I, Cuneo A, Del Poeta G, Ciolli S, Di Raimondo F, Lauria F, Cencini E, Rigolin GM, Cortelezzi A, et al. (2014). Chlorambucil plus rituximab with or without maintenance rituximab as first-line treatment for elderly chronic lymphocytic leukemia patients. Am. J. Hematol. 89, 480–486. 10.1002/ajh.23668. [DOI] [PubMed] [Google Scholar]
- 57.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 25, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu C-L, Ou D-L, Bai L-Y, Chen C-W, Lin L, Huang S-F, Cheng A-L, Jeng Y-M, and Hsu C (2021). Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer 10, 346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ascierto ML, McMiller TL, Berger AE, Danilova L, Anders RA, Netto GJ, Xu H, Pritchard TS, Fan J, Cheadle C, et al. (2016). The intratumoral balance between metabolic and immunologic gene expression is associated with anti-PD-1 response in patients with renal cell carcinoma. Cancer Immunol. Res. 4, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, String-Reasor E, Boughey JC, Chien AJ, Elias AD, et al. (2021). Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang ALS, et al. (2015). Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee JS, Das A, Jerby-Arnon L, Arafeh R, Auslander N, Davidson M, McGarry L, James D, Amzallag A, Park SG, et al. (2018). Harnessing synthetic lethality to predict the response to cancer treatment. Nat. Commun. 9, 2546–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahu AD, S Lee J, Wang Z, Zhang G, Iglesias-Bartolome R, Tian T, Wei Z, Miao B, Nair NU, Ponomarova O, et al. (2019). Genome-wide prediction of synthetic rescue mediators of resistance to targeted and immunotherapy. Mol. Syst. Biol. 15, e8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitfield ML, George LK, Grant GD, and Perou CM (2006). Common markers of proliferation. Nat. Rev. Cancer 6, 99–106. [DOI] [PubMed] [Google Scholar]
- 65.Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. [DOI] [PubMed] [Google Scholar]
- 67.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, and Craig DW (2016). Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat. Rev. Genet. 17, 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.PCAWG Transcriptome Core Group, Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, Lehmann KV, Liu F, Shiraishi Y, et al. (2020). Genomic basis for RNA alterations in cancer. Nature 578, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2022). Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. Preprint at bioRxiv. 10.1101/720243. [DOI] [Google Scholar]
- 71.Pacini C, Dempster JM, Boyle I, Gonçalves E, Najgebauer H, Karakoc E, van der Meer D, Barthorpe A, Lightfoot H, Jaaks P, et al. (2021). Integrated cross-study datasets of genetic dependencies in cancer. Nat. Commun. 12, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- 74.Srivas R, Shen JP, Yang CC, Sun SM, Li J, Gross AM, Jensen J, Licon K, Bojorquez-Gomez A, Klepper K, et al. (2016). A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol. Cell 63, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabach Y, Golan T, Hernández-Hernández A, Messer AR, Fukuda T, Kouznetsova A, Liu J-G, Lilienthal I, Levy C, and Ruvkun G (2013). Human disease locus discovery and mapping to molecular pathways through phylogenetic profiling. Mol. Syst. Biol. 9, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dolled-Filhart M, Roach C, Toland G, Stanforth D, Jansson M, Lubiniecki GM, Ponto G, and Emancipator K (2016). Development of a companion diagnostic for Pembrolizumab in non-small cell lung cancer using immunohistochemistry for programmed death ligand-1. Arch. Pathol. Lab Med. 140, 1243–1249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All transcriptomic and clinical information including treatment and outcome information of the datasets used in the paper are publicly available as individually described in Table S1. For ease of usage, all datasets analyzed in this manuscript can be found in https://github.com/PangeaResearch/enlight-data. See Key Resources Table for full details.
Any additional data required to reanalyze the data reported in this paper is available from the lead contact upon request.
A web service that generates EMS for the drugs described in this study as well as for any new data supplied by the user is available at https://ems.pangeabiomed.com/.