


Abstract
Rph1, a Cys2-His2 zinc finger protein, binds to an upstream repressing sequence of the photolyase gene PHR1, and represses its transcription in response to DNA damage in Saccharomyces cerevisiae. In this report, we have demonstrated that the phosphorylation of Rph1 protein was increased in response to DNA damage. The DNA damage-induced phosphorylation of Rph1 was missing in most damage checkpoint mutants including rad9, rad17, mec1 and rad53. These results indicate that Rph1 phosphorylation is under the control of the Mec1-Rad53 damage checkpoint pathway. Rph1 phosphorylation required the kinase activity of Rad53 since it was significantly decreased in rad53 checkpoint mutant. Furthermore, loss of other kinases including Dun1, Tel1 and Chk1, which function downstream of Mec1, did not affect the Rph1 phosphorylation. This contrasts with the derepression of Crt1-regulated genes, which requires both Rad53 and Dun1 protein kinases. These results imply that post-translational modification of Rph1 repressor is regulated by a potentially novel damage checkpoint pathway that is distinct from the RAD53-DUN1-CRT1 cascade implicated in the DNA damage-dependent transcription of ribonucleotide reductase genes.
INTRODUCTION
The DNA-damage checkpoint is a mechanism that detects abnormal DNA structures and generates a signal that arrests the cell cycle for subsequent repair of DNA damage. However, recent evidence suggests that the checkpoint pathway has more roles (1). It participates in the control of activation of DNA repair pathways (2–4), activation of transcriptional programs (5) and maintenance of genome instability (6), in addition to the control of cell cycle arrest.
Disruption of the checkpoint pathways, which results in increased mutagenesis and genomic instability, is considered to be important at the early stages of carcinogenesis (7). The best evidence for the existence of a link between the checkpoints and cancer comes from studies of ATM, the gene mutated in the cancer susceptibility syndrome Ataxia Telangiectasia (8), and of the Ataxia Telangiectasia and rad-related (ATR) genes. These genes encode homologs of yeast MEC1 and TEL1 checkpoint proteins (9).
In the last several decades, numerous studies in yeast have led to the understanding of the molecular mechanisms of the DNA damage checkpoint pathways. Many of the key players have been identified in the budding yeast Saccharomyces cerevisiae. In addition, their structural and functional counterparts in the fission yeast Schizosaccharomyces pombe and human cells have been identified, indicating that the checkpoint controls seem to have been highly conserved during evolution (10–17).
In S.cerevisiae, DNA-damage signal modifiers or sensors consist of RAD9 and RAD24 epistasis group of genes, including RAD17, RAD24, MEC3 and DDC1 (10,13,18). MEC1 and RAD53 are essential genes that can act as transducers of the checkpoint signal. MEC1 is a member of the phosphatidylinositol 3-kinase family that includes TEL1, fission yeast RAD3, mammalian ATM, ATR and DNA-dependent protein kinase (DNA-PK) (5,17). Rad53 is the homolog of fission yeast Cds1 and mammalian Chk2. Rad53 is a serine/threonine protein kinase that is phosphorylated and activated in response to DNA damage, and is required for prevention of replication after DNA damage and inhibition of mitotic entry before completion of DNA replication (19).
A set of DNA repair-related genes has been known to be transcriptionally induced in response to DNA-damaging agents (20–22). These genes can be divided into two classes. One includes RAD2, RAD51, RAD54, PHR1 and MAG1, which function directly in the repair of damaged DNA. The other includes POL1, RNRs and CDC8, which function primarily in nucleotide metabolism and DNA synthesis. PHR1 encodes a photoreactivating enzyme and its transcription is induced by a large number of different DNA-damaging agents including UV-irradiation, 4-nitroquinoline oxide, methyl methanesulfonate (MMS), nitrosoguanidine, bleomycin and cis-diamminedichloroplatinin (II) (23,24). Although photolyase plays a key role in the repair of pyrimidine dimers, it seems that PHR1 expression is regulated by a global damage response pathway rather than by a dimer-specific or UV-specific pathway (24).
RPH1 (repressor of PHR1) was isolated as a DNA damage-responsive repressor acting through the URSPHR1, an upstream repressing sequence of PHR1 in S.cerevisiae. It has been demonstrated that derepression of PHR1 enhances light-dependent repair of UV-induced DNA damage (25). Therefore, it is of interest to investigate the relationship between the DNA-damage checkpoint pathway and the Rph1-dependent damage inducible repair as this may serve as a model system with which to understand the regulatory mechanisms underlying the transcriptional responses. In this study, we demonstrate that Rph1 repressor is phosphorylated in response to DNA damage and that this modification is mediated by the DNA damage checkpoint pathway. Furthermore, we reveal that Rad53 protein kinase is required for Rph1 phosphorylation. Since the DNA damage-dependent phosphorylation of Rph1 was independent of DUN1 and thus is regulated in a manner distinct from the previously characterized Rad53-Dun1 cascade, this may represent a novel Rad53-dependent pathway involved in the regulation of a damage-responsive transcriptional repressor.
MATERIALS AND METHODS
Yeast and bacterial strains
Strains used in this study are shown in Table 1. The Escherichia coli strains DH5α and BL21 were grown in LB broth with ampicillin (100 µg/ml) as required. Yeast cell cultures were grown in complete YPD or minimal (0.67% yeast nitrogen base, 2% glucose) media (25). Plasmids were propagated in the bacterial strain DH5α and introduced into yeast cells by lithium acetate transformation (25).
Table 1. Plasmids and strains used in this study.
| Plasmids/strains | Description/genotype | Reference/source |
|---|---|---|
| PJH1504 | PRS316+GAL-:rad53D339A | S. E. Lee and J. Haber |
| PJH1509 | PRS316+GAL-:RAD53 | S. E. Lee and J. Haber |
| W303-1a | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; ura3 | N. F. Lowndes |
| rad9 | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; rad9::ura3 | N. F. Lowndes |
| mec3 | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; mec3::ura3 | N. F. Lowndes |
| rad24 | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; rad24::ura3 | N. F. Lowndes |
| rad17 | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; rad17::ura3 | N. F. Lowndes |
| rad9/24 | MATa; ade2-3; can1-100; his3-11; leu2-3,115; trp1-12; rad9::his3; rad24::ura3 | N. F. Lowndes |
| mec1-1 | segregant from a cross between W303-1b and RGY39(mec1-1; tel1::ura3;mcdc13 or 15 | N. F. Lowndes |
| Y301 | MATa; sad1-1, can1-100, ade2-1; his3-11,15; leu2-3, 112; trp1-1; ura3-1 | S. J. Elledge |
| GBS1734 | MATa; ade2-101;his3-Δ200;leu2-Δ1;lys2-801amber;rph1Δ::TRP1 trp1-Δ63;ura3-52 | Y. K. Jang, L. Wang and G. B. Sancar (25) |
| RDKY3731 | MATa; ura3-52, leu2Δ1, trpΔ63, his3Δ200, lys2-Bgl, hom3-10, ade2Δ1, ade8, hxt13::URA3, tel::HIS3 | K. Myung and R. Kolodner |
| RDKY3739 | MATa; ura3-52, leu2Δ1, trpΔ63, his3Δ200, lys2-Bgl, hom3-10, ade2Δ1, ade8, hxt13::URA3, dun1::HIS3 | K. Myung and R. Kolodner |
| RDKY3745 | MATa; ura3-52, leu2Δ1, trpΔ63, his3Δ200, lys2-Bgl, hom3-10, ade2Δ1, ade8, hxt13::URA3, chk1::HIS3 | K. Myung and R. Kolodner |
Preparation of GST fusion proteins and yeast cell extracts
Escherichia coli BL21 was used for expression of glutathione S-transferase (GST) fusion proteins. Cells were grown to an OD595 of 0.5, at which point isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.5 mM and the cells were grown for an additional 3 h at 27°C before harvesting. Yeast total cell extract was prepared as described below. Cells were harvested by centrifugation at an OD595 of 0.5–0.7, resuspended to a concentration of ∼5 × 108 cells/ml in lysis buffer containing 100 mM Tris–HCl (pH 8.0), 1 mM dithiothreitol (DTT), 20% glycerol and 1 mM phenylmethylsulfonyl fluoride and then disrupted with glass beads for 5 min at 4°C. The extracts were clarified by centrifugation at 12 000 g in a microfuge at 4°C for 15 min, and the concentration of total protein was determined by the Bradford reaction using the Bio-Rad reagents. Fusion proteins were incubated with glutathione–Sepharose 4B (Amersham Pharmacia Biotech) for 3 h, washed and eluted with 10 mM reduced glutathione in 50 mM Tris–HCl (pH 8.0).
Polyclonal antibodies against Rph1
Recombinant GST–Rph1 fusion protein (25) was expressed in E.coli using pGEX vector, purified on glutathione–Sepharose beads and used to immunize rabbits. Antisera were tested for reactivity against the purified protein, wild-type S.cerevisiae cell extracts and rph1 null mutant cell extracts. When compared with pre-immune serum, the antibody clearly recognized the Rph1-specific band, which could also be detected in the wild-type cells. For purification of the antibody, we removed antibacterial antibodies from the antisera and then utilized GST-affinity and antigen-affinity purification steps (26). The purified antibody was finally concentrated with Centricon 30 (Amicon). Immunoblot analysis with the purified anti-Rph1 antibody showed that the Rph1-specific band disappeared in rph1 null mutant cells compared to wild-type.
Immunoblot analysis
Eighty micrograms of total proteins were boiled in sodium dodecyl sulfate (SDS) sample buffer and loaded onto 6% SDS–polyacrylamide gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated in Blotto (5% fat free milk powder in PBS) plus anti-Rph1 antisera (1:1000 dilution), washed in PBS-T (0.1% Tween-20 in PBS), and then reacted with a peroxidase-conjugated secondary antibody (1:8000 dilution). Immunodetection was accomplished using HP-conjugated secondary antibodies and the enhanced chemiluminescence method.
In vitro kinase assay
Kinase reactions were carried out with total lysate of each cell strain, and GST–Rph1 bound to glutathione–Sepharose beads in incomplete kinase buffer (50 mM Tris–HCl, pH 7.5 and 5 mM MgCl2) supplemented with 1 mM DTT, 50 µM ATP and 3 µCi [γ-32P]ATP (3000 Ci mmol-1; NEN). Reactions were incubated at 25°C for 5 min, stopped by mixing with SDS–PAGE loading buffer and the proteins were separated by SDS–PAGE. The radiolabeled proteins were Coomassie-stained, destained and dried onto 3MM papers and visualized by autoradiography.
In vivo labeling and phosphoamino acid analysis
pEG-KT is a GST tagging vector for use in yeast (27). EG-RPH1 was generated from the full RPH1 open reading frame inserted between the SmaI-SalI sites of pEG-KT. EG-RPH1 expressing cells and wild-type cells were grown in a low phosphate medium to an OD595 of 0.5–0.7, resuspended in fresh low phosphate medium containing 1 mCi of 32P-orthophosphate (NEX053H, NEN) and incubated at 30°C for 1 h. After preparation of total cell lysates, immunoprecipitation was performed using either anti-GST antibody (Amersham Pharmacia) or anti-Rph1 antibody. The GST–Rph1 fusion protein and endogeneous Rph1 protein were separated by SDS–PAGE and transferred onto PVDF membranes.
Two-dimensional phosphoamino acid analysis was conducted by excising the appropriate 32P-labeled band from the PVDF membrane and subjecting each one to hydrolysis in 5.7 M HCl for 2 h at 110°C. Following lyophilization, the samples were resuspended in 0.8 µl of pH 1.9 buffer [2.5% (v/v) formic acid and 7.8% (v/v) acetic acid] containing phosphotyrosine, phosphoserine and phosphothreonine standards at a final concentration of 5 mg/ml. Each sample (1500 c.p.m.) was loaded onto cellulose thin-layer plates (Merck, Whitehouse Station, NJ). The first dimensional electrophoresis was performed at 500 V for 55 min in pH 1.9 running buffer. The second dimension was performed at 530 V for 28 min in pH 3.5 running buffer. The plates were then dried, sprayed with 0.25% (w/v) ninhydrin in acetone and baked at 65°C for 20 min. The 32P-labeled threonine, serine and tyrosine were detected by autoradiography and compared with localization of unlabeled standards.
RESULTS
Rph1 protein is phosphorylated in response to DNA damage
In this study, we aimed to understand how RPH1 could regulate transcriptional induction of PHR1 in response to DNA damage. First, northern blot analysis was performed to determine whether or not the level of RPH1 transcripts responded to DNA damage. The results showed that the RPH1 transcripts were slightly induced in response to DNA damaging agents, and that the transcriptional level of RPH1 in checkpoint mutants were not significantly different to those in wild-type cells (data not shown). Next, we investigated whether Rph1 protein could be modified upon DNA damage and whether such modification was dependent on damage checkpoints. To examine changes in the mobility shift of Rph1 protein upon DNA damage, immunoblot analysis was performed. Rph1 protein was detected as several bands, representing patterns of phosphoprotein (Figs 1 and 2). Upon treatment with the DNA damaging agent the pattern of phosphorylation was changed with an increase in the slower migrating forms (Figs 1A and 3A). To demonstrate that the mobility shift of Rph1 resulted from phosphorylation, calf intestine alkaline phosphatase was added to wild-type cell extracts from the mock- and damage-treated cells. As shown in Figure 1B, Rph1 migrated as a single band rather than multiple bands after the alkaline phosphatase treatment. These results demonstrated that phosphorylation of Rph1 protein was induced by DNA damage.
Figure 1.

Rph1 is phosphorylated in response to DNA damage and its phosphorylation occurs on both Ser and Thr residues. (A) Time course analysis of Rph1 phosphorylation after UV-irradiation. Wild-type (WT) cells (W303) were treated with UV (100 J/m2) and total cell extracts were prepared at intervals for 120 min. Eighty micrograms of total cell lysates were applied for immunoblotting. (B) Abolishment of Rph1 phosphorylation by treatment with alkaline phosphatase. The electrophoresis mobility shift of Rph1 protein disappeared with treatment with 60 U of calf intestine alkaline phosphatase (Takara, Japan). –, mock treatment; +, alkaline phosphatase treated. (C) Expression of GST–Rph1 was induced in a selective medium containing 2% galactose. GST–Rph1 overexpressing cells were resuspended in radiolabeled orthophosphate-containing media followed by 1 h incubation. The radiolabeled GST–Rph1 fusion proteins were eluted by immunoprecipitation with α-GST antibody and analyzed by phosphoamino acid assay. (D) Endogeneous Rph1 was radiolabeled with orthophosphate for 1 h as described above. The labeled Rph1 protein was eluted by immunoprecipitation with an anti-Rph1 antibody and analyzed by phosphoamino acid assay. S, Ser; T, Thr; arrow, loading origin.
Figure 2.
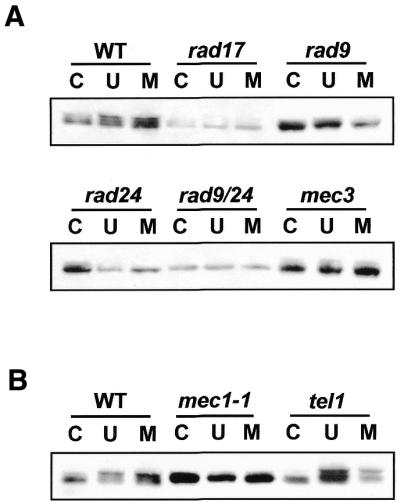
Loss of checkpoint genes affects phosphorylation of Rph1. Individual cultures from WT (W303), rad17, rad9, rad24, rad9/24, mec3 (A) and mec1, tel1 (B) damage checkpoint mutants cells were grown to mid-log phase, irradiated with UV (100 J/m2) or treated with MMS (0.1%) at 30°C for 1 h and the cell lysates were extracted for immunoblotting. Rph1 phosphorylation appeared to be different patterns in several checkpoint mutants. C, no damage; U, UV-irradiated (100 J/m2); M, MMS-treated (0.1%).
Figure 3.

Rph1 phosphorylation is dependent on Rad53 protein kinase. (A) Damage-dependent phosphorylation of Rph1 was checked in the rad53 defective mutant. Wild-type cell and rad53 mutant cell lysates were used for immunoblotting. (B) In vitro kinase assay was performed with GST–Rph1 fusion protein expressed in E.coli. GST–Rph1 proteins bound to glutathione–Sepharose were incubated with total cell lysates treated with UV (100 J/m2) or MMS (0.1%) at 30°C for 1 h and then the degree of phosphorylation was examined by using in vitro kinase assay. (C) Immunoblot analysis showed that Rph1 phosphorylation was diminished in rad53-D339A (a kinase dead mutant) but recovered in Rad53-op (Rad53-overexpressing). C, no damage; U, UV-irradiated (100 J/m2); M, MMS-treated (0.1%).
Rph1 protein is phosphorylated on Ser/Thr residues
To identify the phosphorylated residues on Rph1, GST–Rph1 expressing plasmids were introduced into rph1 null mutant cells and were analyzed after metabolic labeling of the cells with inorganic 32P. GST–Rph1 proteins were immunoprecipitated with anti-GST antibody and then subjected to two-dimensional phosphoamino acid analysis. The results showed that the GST–Rph1 fusion protein was phosphorylated on serine (Ser) and threonine (Thr) residues (Fig. 1C). To exclude a possibility that the phosphorylation occurred on GST, wild-type yeast cells were similarly labeled and the endogenous Rph1 protein was immunoprecipitated with an anti-Rph1 polyclonal antibody. The endogenous Rph1 was also phosphorylated on Ser and Thr residues (Fig. 1D). Taken together, we concluded that Ser and Thr residues of Rph1 were phosphorylated in vivo.
The damage checkpoint pathway directs phosphorylation of Rph1 protein
As previously mentioned, the phosphorylation of Rph1 in response to DNA damage raised the possibility that Rph1 phosphorylation might be mediated by the DNA damage checkpoint pathways. To test this hypothesis, immunoblot analysis was carried out with total cell extracts derived from several damage checkpoint mutants. As shown in Figure 2A, the Rph1 phosphorylation was diminished in extracts of cells mutated in a range of checkpoint genes including rad9, rad17, rad24 and mec3. Interestingly, the degree of Rph1 phosphorylation was different in tel1 mutant extracts when compared to wild-type and was absent in mec1 (Fig. 2B). TEL1 genetically interacts with MEC3, MEC1, RAD53 and many genes involved in the S phase checkpoint and downstream signal transduction pathways (6,15,16). Unlike Mec1, the loss of Tel1 kinase does not abolish the phosphorylation of Rph1 in response to DNA damage. These observations indicated that DNA damage induced the phosphorylation of Rph1 through the damage checkpoint pathway.
Rph1 is phosphorylated by Rad53-dependent checkpoint pathway
Since Rad53, Chk1 and Dun1 are protein kinases that function downstream of Mec1, we next examined whether they might be required for Rph1 phosphorylation. Although Rph1 had a basal level of phosphorylation in the absence of damage, its damage-induced phosphorylation was clearly diminished in rad53 mutants but not in chk1 or dun1 mutants (Fig. 3A). Furthermore, reduction of Rph1 phosphorylation was observed when rad53 mutant extracts were used in an in vitro kinase assay performed on GST–Rph1 fusion protein(s) bound to glutathione–Sepharose. As seen in Figure 3B, GST–Rph1 was phosphorylated in the untreated cell extracts and its phosphorylation was increased in extracts derived from UV-irradiation or MMS-treated cells. However, the damage-induced phosphorylation of Rph1 was not detected in rad53 mutant cells. In order to examine whether the Rad53 kinase activity was required for Rph1 phosphorylation, we compared the degree of phosphorylation in cells overexpressing Rad53 or in Rad53-D339A cells, a kinase dead mutant. Rad53-op, a Rad53 overexpressing cell, served as a positive control and rad53-D339A, the kinase-defective mutant, served as a negative control for the Rad53 kinase activity. Rph1 phosphorylation was significantly reduced in rad53-D339A, but restored in Rad53-op (Fig. 3C). These results strongly indicated that Rad53 kinase was required for phosphorylation of Rph1 upon DNA damages and implied a direct involvement of the damage checkpoints in the phosphorylation.
DISCUSSION
In the present work, we report that Rph1, a damage-responsive repressor of PHR1, is phosphorylated in response to DNA damage and that the DNA-damage checkpoint pathway regulates its phosphorylation. Ser and Thr residues on Rph1 protein were identified as phosphorylated in vivo. Interestingly, Rad53 protein kinase, one of the key central signal transducers, was required for the Rph1 phosphorylation.
Several recent reports have suggested that the DNA damage checkpoint is linked directly or indirectly to the DNA damage-dependent transcriptional response and to the delay of cell cycle progression (20,21,25,28,29). Particularly, several studies demonstrated that most of the damage-checkpoint genes, including RAD9, MEC1 and RAD53 of S.cerevisiae, are involved in the induction of a large regulon of >15 genes whose roles are in DNA repair and metabolism. This indicates that the DNA damage regulon (DDR) might be reminiscent of the SOS response in bacteria (20,21). In addition, it has been demonstrated that the damage-responsive transcription of PHR1 is dependent on Rad53 but not on Dun1 kinase or Tup1 corepressor (24,25). Hence, it would be very interesting to understand the regulation mechanisms of the DDR in yeast. A recent study has revealed that Dun1 Ser/Thr kinase is involved in the transcriptional activation of RNR2 and has delineated a pathway by which the damage signal is transmitted through the checkpoint and transcriptional response apparatus (30). The repressor protein Crt1 was found to bind to X boxes on the promoters of RNR2 and RNR3 genes and to cooperate with the Tup1-Ssn6 corepressor. Hyperphosphorylation of Crt1 by MEC1-RAD53-DUN1 cascade forced the protein to dissociate from the X boxes, leading to derepression of RNR2 transcription (28). However, the RAD53-DUN1-CRT1 cascade appeared to control only a small set of genes, including RNR2 and RNR3 but not UBI4 or PHR1, the well-known damage-inducible genes. Thus, it has been suggested that not only are there multiple DNA damage-responsive regulators, but also that the signal transduction cascade involved in the regulation of DDR differs depending on the promoter context of the target.
Recently, Rph1 and Gis1 have been identified as the transcriptional regulators that bind to PHR1’s upstream repressing sequence (URS) as mentioned previously (25). To understand in detail the mechanism of Rph1 action in the damage-responsive transcription of PHR1, we investigated a possible link between Rph1 protein modification and the damage checkpoint. We show that Rph1 phosphorylation is increased in response to DNA damage and is dependent on most of the checkpoint proteins including Rad9, Rad17, Rad24, Mec3, Mec1 and the Rad53 protein kinase, but not on Dun1, Tel1 or Chk1. In northern blot analysis, the damage-induced transcript level of PHR1 was decreased in a rad53 mutant in response to DNA damage (24; data not shown). These data strongly support the idea that the damage-dependent transcription of PHR1 was dependent on Rad53 but not on Dun1 (24). Therefore, the RAD53-RPH1 pathway, in addition to RAD53-DUN1-CRT1 cascade, seems to be another mechanism in the DDR regulation in yeast. In S.cerevisiae, at least four different proteins, Rph1, Gis1, Crt1 and Swi6, are known as transcriptional regulators of the damage-inducible DNA repair genes (25,28,31,32). Transcriptional regulation of PHR1 mediated by Rph1 and Gis1 differs in one important aspect from that by Crt1. PHR1 expression is Tup1 independent and requires both Rph1 and Gis1 repressors. Cooperation of Crt1 with Tup1-Ssn6 corepressor is involved in the damage-dependent expression of RNR2 gene. However, Rph1, Gis1 and Crt1 are similar in that they are all damage-responsive repressors. In addition, the derepression mediated by both Crt1 and Rph1 appears to require their Rad53-dependent hyperphosphorylation. Thus, the derepression mechanism via Rph1 hyperphosphorylation might share mechanistic similarities with that of Crt1. In response to DNA damage, cells activate a checkpoint cascade, which leads to Mec1-dependent activation of the Rad53 protein kinase. Our unpublished observation (E.M.Kim, Y.K. Jang and S.D.Park) suggests that Rad53 does not physically interact with Rph1 as judged by coimmunoprecipitation assay in our hands, implying the existence of another kinase or molecule in the RAD53-RPH1 pathway. Hyperphosphorylation of Rph1 possibly leads to dissociation of Rph1 from PHR1’s URS as in the derepression by Crt1. Although the biological role of the damage-dependent phosphorylation of Rph1 is not clearly understood yet, our gel mobility shift assay showed that the binding ability of Rph1 to URSPHR1 was slightly decreased in response to DNA damage, implying a relation between the phosphorylation and the DNA binding capacity (E.M.Kim, Y.K.Jang and S.D.Park, unpublished results). Moreover, we have found that Rph1 is a nuclear protein and its localization is regulated in response to UV-irradiation by using a fluorescence microscope to examine the GFP–Rph1 fusion protein. Furthermore, the UV damage-dependent redistribution of Rph1 is not affected by the loss of Rad53 protein, suggesting the potential involvement of a Rad53-dependent damage checkpoint pathway in the DNA-damage-dependent redistribution of Rph1 (E.M.Kim, Y.K.Jang and S.D.Park, unpublished observation). Once the phosphorylation site is identified, the mechanism of Rph1 action can be investigated by making appropriate mutants.
The present work defines a potentially novel pathway in which the damage-checkpoint controls the DNA-damage- dependent transcription of a DNA repair gene through Rad53-dependent hyperphosphorylation of Rph1 repressor.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs O. Hwang and K. Myung for their critical reading and comments on this manuscript and Mr Hyunsoo Kim for his excellent technical assistance. We are grateful for the hospitality of Dr Achille Pellicioli (Istituto F.I.R.C., Milano, Italy) for the RAD53-myc plasmid. We thank Drs S. J. Elledge (Baylor College of Medicine, Houston, TX), N. F. Lowndes (National University of Ireland, Ireland), S. E. Lee (University of Texas at San Antonio, Houston, TX), J. Haber (Department of Biology, Brandeis University, USA) and R. Kolodner (University of California at San Diego, USA) for S.cerevisiae strains. We also thank Dr M. J. Gait and the two anonymous reviewers, who helped improve this paper considerably. This work was supported in part by a grant for the Leading Scientists from the Korea Science and Engineering Foundation (2001) to S.D.P. E.M.K. and S.D.P. were supported by Research Fellowship BK21 from the Korean Ministry of Education. Y.K.J. is a recipient of postdoctoral fellowship (1998) of the Korea Research Foundation.
REFERENCES
- 1.Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]
- 2.Cortez D., Wang,Y., Qin,J. and Elledge,S.J. (1999) Requirement of ATM-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science, 286, 1162–1166. [DOI] [PubMed] [Google Scholar]
- 3.Gatei M., Young,D., Cerosaletti,K.M., Desai-Mehta,A., Spring,K., Kozlov,S., Lavin,M.F., Gatti,R.A., Concannon,P. and Khanna,K. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nature Genet., 25, 115–119. [DOI] [PubMed] [Google Scholar]
- 4.Zhao S., Weng,Y.C., Yuan,S.S., Lin,Y.T., Hsu,H.C., Lin,S.C., Gerbino,E., Song,M.H., Zdzienicka,M.Z., Gatti,R.A., Shay, J.W., Ziv,Y., Shiloh,Y. and Lee,E.Y. (2000) Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature, 405, 473–477. [DOI] [PubMed] [Google Scholar]
- 5.Elledge S.J. (1996) Cell cycle checkpoints: preventing an identity crisis. Science, 274, 1664–1672. [DOI] [PubMed] [Google Scholar]
- 6.Myung K., Datta,A. and Kolodner,R.D. (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell, 104, 397–408. [DOI] [PubMed] [Google Scholar]
- 7.Hartwell L.H. and Kastan,M.B. (1994) Cell cycle control and cancer. Science, 266, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 8.Morgan S.E. and Kastan,M.B. (1997) p53 and ATM: cell cycle, cell death and cancer. Adv. Cancer Res., 71, 1–25. [DOI] [PubMed] [Google Scholar]
- 9.Brown E.J. and Baltimore,D. (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev., 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- 10.Foiani M., Pellicioli,A., Lopes,M., Lucca,C., Ferrari,M., Liberi,G., Muzi Falconi,M. and Plevani,P. (2000) DNA damage checkpoints and DNA replication controls in Saccharomyces cerevisiae. Mutat. Res., 451, 187–196. [DOI] [PubMed] [Google Scholar]
- 11.Carr A.M. (1995) DNA structure checkpoints in fission yeast. Semin. Cell Biol., 6, 65–72. [DOI] [PubMed] [Google Scholar]
- 12.Kaufmann W.K. and Paules,R.S. (1996) DNA damage and cell cycle checkpoint. FASEB J., 10, 238–247. [DOI] [PubMed] [Google Scholar]
- 13.Longhese M.P., Foiani,M., Muzi-Falconi,M., Lucchini,G. and Plevani,P. (1998) DNA damage checkpoint in budding yeast. EMBO J., 17, 5525–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulovich A.G., Toczyski,D.P. and Hartwell,L.H. (1997) When checkpoints fail. Cell, 88, 315–321. [DOI] [PubMed] [Google Scholar]
- 15.Rhind N. and Russell,P. (1998) Mitotic DNA damage and replication checkpoints in yeast. Curr. Opin. Cell Biol., 10, 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart E. and Enoch,T. (1996) S-phase and DNA-damage checkpoints:a tale of two yeasts. Curr. Opin. Cell Biol., 8, 781–787. [DOI] [PubMed] [Google Scholar]
- 17.Weinert T. (1998) DNA damage and checkpoint pathways: molecular anatomy and interactions with repair. Cell, 94, 555–558. [DOI] [PubMed] [Google Scholar]
- 18.Paciotti V., Clerici,M., Lucchini,G. and Longhese,M.P. (2000) The checkpoint protein Ddc2, functionally related to S.pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev., 14, 2046–2059. [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuoka S., Huang,M. and Elledge,S.J. (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science, 282, 1893–1897. [DOI] [PubMed] [Google Scholar]
- 20.Aboussekhra A., Vialard,J.E., Morrison,D.E., de la Torre-Ruiz,M.A., Cernakova,L., Fabre,F. and Lowndes,N.F. (1996) A novel role for the budding yeast RAD9 checkpoint gene in DNA-damage dependent transcription. EMBO J., 15, 3912–3922. [PMC free article] [PubMed] [Google Scholar]
- 21.de la Torre-Ruiz M.A., Green,C.M. and Lowndes,N.F. (1998) RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J., 17, 2687–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jelinsky S.A. and Samson,L.D. (1999) Global response of Saccharomyces cerevisiae to an alkylating agent. Proc. Natl Acad. Sci. USA, 96, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebastian J., Kraus,B. and Sancar,G.B. (1990) Expression of the yeast PHR1 gene is induced by DNA-damaging agents. Mol. Cell. Biol., 10, 4630–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sancar G.B. (2000) Enzymatic photoreactivation: 50 years and counting. Mutat. Res., 451, 25–37. [DOI] [PubMed] [Google Scholar]
- 25.Jang Y.K., Wang,L. and Sancar,G.B. (1999) RPH1 and GIS1 are damage-responsive repressors of PHR1. Mol. Cell. Biol., 19, 7630–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harlow E. and Lane D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 27.Mitchell D.A., Marshall,T.K. and Deschenes,R.J. (1993) Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast, 9, 715–722. [DOI] [PubMed] [Google Scholar]
- 28.Huang M., Zhou,Z. and Elledge,S.J. (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell, 94, 595–605. [DOI] [PubMed] [Google Scholar]
- 29.Kiser G.L. and Weinert,T.A. (1996) Distinct roles of yeast MEC and RAD checkpoint genes in transcriptional induction after DNA damage and implications for function. Mol. Biol. Cell., 7, 703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Z. and Elledge,S.J. (1993) DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell, 75, 1119–1127. [DOI] [PubMed] [Google Scholar]
- 31.Ho U., Mason,S., Kobayashi,R., Hoekstra,M. and Andrews,B. (1997) Role of the casein kinase I isoform, Hrr25 and the cell cycle- regulatory transcription factor, Sbf, in the transcriptional response to DNA damage in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sidorova J.M. and Breeden,L.L. (1997) Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes Dev., 11, 3032–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]