

Abstract
Cancer therapies trigger diverse cellular responses, ranging from apoptotic death to the acquisition of persistent therapy-refractory states such as senescence. Tipping the balance towards apoptosis could improve treatment outcomes regardless of therapeutic agent or malignancy. We found that inhibition of mitochondrial protein BCL-xL increased the propensity of cancer cells to die following treatment with a broad array of oncology drugs, including mitotic inhibitors and chemotherapy. Functional precision oncology and OMIC analyses suggested that BCL-xL inhibition redirected the outcome of p53 transcriptional response from senescence to apoptosis which likely occurred via caspase-dependent down-modulation of p21 and downstream cytostatic proteins. Consequently, the addition of BCL-2/xL inhibitor strongly improved melanoma response to senescence-inducing drug targeting mitotic kinase AURKA in mice and patient-derived organoids. This study uncovers crosstalk between mitochondrial apoptotic pathway and cell cycle regulation that can be targeted to augment therapeutic efficacy in cancers with wild-type p53.
Keywords: Senescence, senogenic, senolytic, apoptosis, melanoma, patient-derived organoids, Aurora kinase, BCL-2, BCL-xL, alisertib, paclitaxel, navitoclax, p53, p21, BAX
INTRODUCTION
Metastatic melanomas are treated with immune checkpoint inhibitors and therapies targeting the oncogenic MAPK pathway. However, many tumors are intrinsically resistant or acquire resistance to standard-of-care therapies1–3, underscoring the need for novel treatment options. The development of new therapies for melanoma is challenged by the relative resistance of melanocytes to apoptosis, a characteristic attributed to the high expression of pro-survival mitochondrial proteins BCL-2 and BCL-xL4,5. Pro-survival BCL-2 proteins prevent mitochondrial outer membrane permeabilization (MOMP) and subsequent apoptosis6. It has been suggested that high BCL-2/xL expression protects melanocytes following damaging UV exposure to ensure uninterrupted melanin production. However, it also enables melanomas to escape cell death under various cancer treatments, including traditional chemotherapy7.
Damaged tumor cells that escape therapy-induced death are likely to become senescent. Cellular senescence is a type of stress response characterized by stable cell cycle arrest and increased secretion of immune modulators, growth factors, and other molecules, a phenotype known as the senescence-associated secretory phenotype (SASP)8. While therapy-induced senescence can limit tumor growth by inhibiting malignant cell proliferation 9 and promoting immune surveillance of the tumor10,11, it does not permanently eliminate cancer cells; this creates a significant risk of tumor relapse. Furthermore, senescent cells can negatively impact the long-term outcome of patients by promoting proliferation and invasiveness of neighboring non-senescent cells12–14, leading to chemotherapy resistance15, and giving rise to stem-like cells responsible for post-therapy tumor recurrence16. Additionally, the senescent-like stress response induced by Wnt5A in melanoma cells has been linked with melanoma invasion17. These factors provide a strong rationale to develop strategies for redirecting cancer therapy response from senescence to apoptosis.
Senescent cells can be uniquely vulnerable to certain drugs termed senolytics18–20. A dual inhibitor of BCL-2 and BCL-xL, navitoclax (ABT263), exhibits senolytic activity in several cell types21,22. It is currently in late-stage clinical development for the treatment of myelofibrosis and Phase I-II clinical trials in solid malignancies, including a study of navitoclax in combination with BRAFV600E and MEK inhibitors in melanoma (NCT01989585). Considering the implication of BCL2/xL in melanocytes’ resistance to apoptosis and senescent cell survival, we hypothesized that inhibition of BCL2/xL in melanoma cells undergoing therapy would promote apoptosis and limit senescence, resulting in improved treatment outcomes. To test this hypothesis, we examined the ability of navitoclax to enhance melanoma response to a variety of existing and emerging oncology agents, including senescence-inducing drugs.
We have previously demonstrated that a small molecule inhibitor of Aurora Kinase A (AURKA) alisertib induces senescence in melanoma cells23,24. In preclinical studies, alisertib inhibited growth of melanoma patient-derived xenografts (PDXs) and mouse tumors, but did not cause tumor regression23–26. Alisertib has been evaluated in several Phase I-III clinical trials where it demonstrated an acceptable safety profile27–31 and an evidence of anti-tumor activity, although it mainly induced disease stabilization in patients with solid tumors32,33. Here we used alisertib as a prototypical senescence-inducing drug to test whether BCL2/xL inhibition with navitoclax can convert a senescence-type response into apoptosis. Molecular mechanisms governing this fate switch were interrogated using genomic, transcriptomic, and proteomic profiling of cells and tumors treated with senescence-inducing therapy in the absence or presence of BCL-2/xL inhibition. Furthermore, we employed aggressive mouse melanomas and a diverse set of melanoma patient-derived organoids to determine whether the senescence-to-apoptosis fate switch on a cellular level can lead to an improved therapeutic outcome.
RESULTS
BCL-2/xL inhibition promoted cell death in response to select oncology agents
We hypothesized that inhibition of pro-survival BCL-2 family proteins would promote therapy-induced cell death and limit therapy-induced senescence. To test this, human melanoma cells were treated with various drugs, including chemotherapeutics and kinase inhibitors, in the absence or presence of BCL2/xL inhibitor navitoclax (hereafter referred to as BCLi). Following 24h treatment, the percentages of dead and senescent cells were quantified using propidium iodide and SA-β-galactosidase assay, respectively (Fig. 1A). BCLi alone induced modest cell death response (2.1%, Table S1, Fig. 1B, select representative data are shown in Fig. 1C). In the absence of BCLi, only 3 out of 43 tested treatments induced >4.2% cell death (double the percentage induced by BCLi alone). Notably, 15 out of 43 treatments induced >4.2% cell death in the presence of BCLi. These drugs were termed “BCLi-enhanced” (Fig. 1D). Surprisingly, the percentages of senescent cells remaining after drug treatment were minimally affected by BCLi addition (Fig. 1E). However, “BCLi-enhanced” drugs were more potent inducers of senescence compared to “unaffected” drugs (Fig. 1F). This data suggest that BCLi did not selectively kill senescent cells but rather increased the likelihood of cells dying during treatment with senescence-inducing drugs.
Figure 1. BCL-2/xL inhibition promoted cell death in response to a variety of oncology agents.
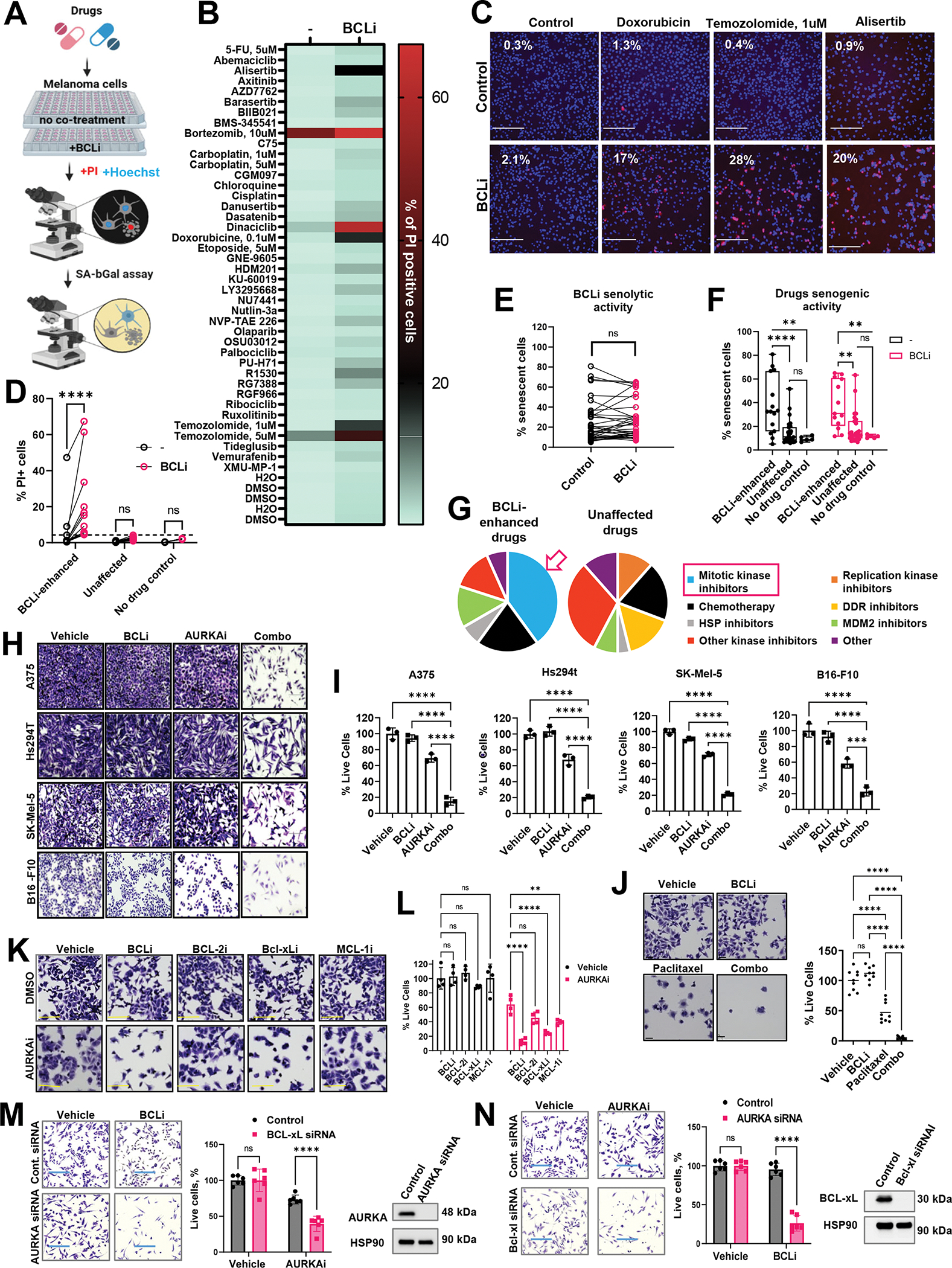
(A) Schematic of the drug screen design. (B) Percentages of dead PI-positive cells in cells treated for 24h with indicated drugs in the presence or absence of BCLi navitoclax. Median values from 6 quantified images are shown per each condition. N=6. (C) Representative microscopy images from the library screen shown in B. Numbers in the top left corners indicate the percentages of apoptotic cells. Scale bar 275μm. (D) Identification of drug screen hits. Drugs that, combined with BCLi, induced at least double the percentage of cell death induced by BCLi alone (indicated by the punctate line) were designated as “BCLi-enhanced.”, and all others as “Unaffected.” Statistics using 2-way ANOVA with Sidak’s multiple comparison test. (E) Pairwise comparison of the percentages of senescent cells with and without BCLi addition. Statistical analysis using paired t-test. (F) Comparison of the percentages of senescent cells induced by BCLi-enhanced and unaffected drugs with and without BCLi. Statistics as in D. (G) Drug categories enriched in BCLi-enhanced and unaffected drug groups. (H) Representative images of crystal violet-stained melanoma cells treated with 1μM navitoclax (BCLi), 1μM alisertib (AURKAi), or both drugs combined for 24 h. Scale bar 200 μm. (I) Cell count from the experiment in H. Data is shown as percentages of cells relative to vehicle control. Statistical comparisons using one-way ANOVA with Dunnett’s post-test. N=3. (J) Representative images of crystal violet-stained A375 cells after 24h treatment with vehicle, 1μM navitoclax (BCLi), 1μM paclitaxel, or both drugs combined for 24 h. Scale bar 50μm. Cell quantification relative to vehicle-treated cells is shown on right. Statistical comparisons using one-way ANOVA with Tukey’s test. N=9 random fields from 3 individual wells (3 images/well). (K) Representative images of crystal violet-stained A375 cells after 24h treatment with 1μM navitoclax (BCL-2/xLi), 1μM venetoclax (BCL-2i), 1μM A-1155463 (BCL-xLi), 1μM A-1210477 (MCL-1i) in the presence or absence of 1μM alisertib (AURKAi). Scale bar 150μm. (L) Relative cell numbers from the experiment in K. Data are presented in percentages relative to untreated cells (Vehicle/-) with statistical analysis using 2-way ANOVA with Sidak’s post-test. Experiment was repeated three times with consistent results. (M) Left: representative images of crystal violet-stained Hs294T cells transfected with AURKA or non-targeting siRNA and treated with 1μM navitoclax (BCL-2/xLi) for 24h. Scale bar 275μm. Experiment was performed with three biological replicates. Middle: quantification from 6 random fields per condition and one-way ANOVA statistical analysis with Sidak’s post-test. Right: efficiency of target knockdown determined by western. (N) Same as M, except BCL-xL-targeting siRNA was used and cells were treated with 1μM alisertib (AURKAi) for 24h. All panels: Ns P > 0.05, * P≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001. When applicable, p-values were adjusted for multiple comparisons. See also Fig. S1.
We noted that many “BCLi-enhanced” drugs belonged to a category of mitotic kinase inhibitors (Fig. 1G). In fact, all tested mitotic kinase inhibitors were enhanced by BCLi, suggesting that the enhancement by BCLi could be broadly applicable to this class of drugs, including agents potentially emerging in the future. Among the mitotic kinase inhibitors we tested, Aurora kinase A inhibitor alisertib (hereafter AURKAi) was the most clinically advanced (phase II-III trials). Therefore, we chose this drug for further validation and mechanistic studies reasoning that this work could provide a basis for a subsequent clinical study. Four human and mouse melanoma cell lines were treated with AURKAi in the absence or presence of BCLi. The cell numbers were moderately reduced by single-agent AURKAi. In contrast, cultures treated with AURKAi and BCLi combination displayed up to 90% cell reduction (Fig. 1H, I).
Enhancing effects of BCLi was reproduced with other drugs. Based on the enrichment of mitotic inhibitors and chemotherapy among “BCLi-enhanced” drugs, we postulated that BCLi would enhance an FDA-approved anti-mitotic chemotherapy drug paclitaxel. Indeed, nearly all cells were eliminated by combined paclitaxel and BCLi treatment, while about 50% of cells survived after paclitaxel monotherapy (Fig. 1J).
BCLi navitoclax is a broad-acting inhibitor targeting BCL-2 and BCL-xL. To identify protein that inhibited drug-induced melanoma cell death, we used specific inhibitors of BCL2 and BCL-xL. We also used an inhibitor of MCL1, a pro-survival BCL-2 family protein not targeted by BCLi. The BCL-xL inhibitor was more effective at enhancing AURKAi response than BCL-2 and MCL1 inhibitors (Fig. 1K, L). The data pattern was minimally affected when numbers of cells remaining after each tested combination was normalized to single-agent treatment with corresponding BCL-2 family inhibitor (Fig. S1). These data implicate BCL-xL as the primary inhibitor of cell death response to AURKAi.
To validate that the anti-tumor activity of AURKAi and BCLi combination was not an off-target effect of tested dugs, we used RNAi to deplete endogenous AURKA and BCL-xL in melanoma cells. Western blot showed effective knockdown (KD) for both proteins (Fig. 1M, N). In agreement with drug studies, AURKA-KD cells were more sensitive to BCL-xL inhibition, and BCL-xL-KD cells were more sensitive to AURKA inhibition, compare to corresponding non-KD cells (Fig. 1M, N).
Combined AURKAi and BCLi treatment induced apoptosis associated with mitochondrial depolarization and caspase activation
To define the mechanism whereby BCLi augmented drug response, we analysed apoptosis and proliferation in alisertib and navitoclax-treated A375 melanoma cells using PARP cleavage and BrdU incorporation assay. Single agent therapies with BCLi and AURKAi induced PARP cleavage in a small percent of cells. In contrast, combined administration of BCLi and AURKAi prominently increased percentages of cells with PARP cleavage, indicating apoptosis (Fig. 2A, top panel, Fig. 2B, left panel). Remaining populations of live cells identified by lack of PARP cleavage and intact DNA content displayed reduced percentages of BRDU+ cells which were similar to that in single-agent AURKAi-treated cells (Fig. 2A, bottom panels, Fig. 2B, right panel). These data indicate that BCLi promoted apoptosis, but not cell cycle arrest. We confirmed the induction of apoptosis by AURKAi and BCLi combination using an Annexin V binding assay (Fig. 2C, D). In accordance with cell cycle arrest and senescence induction, melanoma cells treated with AURKAi displayed decreased expression of mitosis marker phospho-histone H3 (Fig. 2E), increased expression of senescence-associated β-galactosidase (Fig. 2F), and secreted higher levels of IL6 and IL8 (Fig. 2G), which are common markers of senescence-associated secretory phenotype. Notably, a few cells remaining after treatment with combination therapy displayed similar rates of senescence to AURKAi-treated cells based on SA-β-gal staining and phospho-H3 (Fig. 2E, F). Altogether these data suggest that the majority of cells treated with single-agent AURKAi survived and underwent senescence, while the addition of BCLi to AURKAi increased the proportion of cells that died from apoptosis.
Figure 2. Treatment combining AURKAi and BCLi induced apoptosis associated with mitochondrial depolarization and caspase activation.
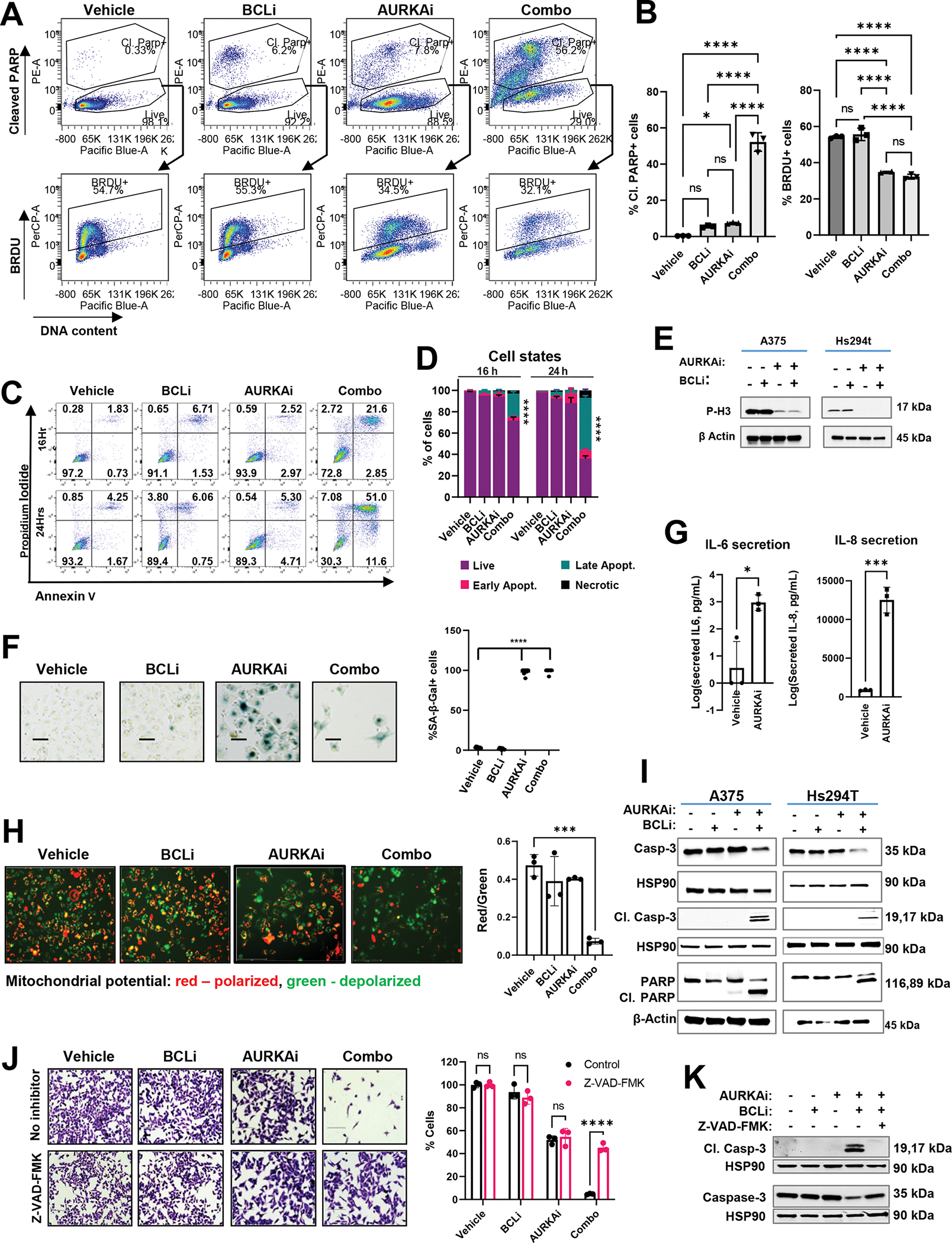
(A) Representative flow cytometry plots from PARP cleavage and BrdU incorporation assay. A375 cells were treated with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi) or both drugs combined for 24 hours and pulsed with BrdU for 5 hours. BRDU+ cells were gated on live cells (cells with cleaved PARP and with sub-G1 DNA content were gated out). (B) Statistical comparison of the percentages of BRDU+ cells and cells with cleaved PARP from A. Statistics by one-way ANOVA with Tukey’s adjustments. N=3. (C) Representative results of Annexin V/PI staining detecting cells in early (bottom right quadrants) and late (top right quadrants) apoptosis. (D) Quantification of experiment in C. Data are presented as mean ± standard deviation for three replicates, 2-way ANOVA with Dunnett’s post-test. (E) Representative results of the senescence-associated (SA) β-galactosidase activity in cells treated as in A for 48 hours. Scale bar 150μm. (F) Quantification of SA-β-Gal positive cells from E, 9 fields from 3 biological replicates. Experiment was repeated two times with consistent results. Statistical analysis using one-way ANOVA with Tukey’s post-test. (G) ELISA of secreted IL-6 and IL-8 in the conditioned media from A375 cells treated with vehicle or 1μM AURKAi alisertib for 24h. (H) Representative results of JC1 staining. JC1 aggregates (red) and monomeric JC1 (green) indicate healthy polarized mitochondria and dysfunctional depolarized mitochondria, respectively. Scale bar 150μm. Quantified data and one-way ANOVA is shown on the right. N=3. (I) Western blot of indicated apoptotic proteins in A375 and Hs294t cells treated as in A for 24 hrs. Three independent experiments were performed with consistent results. (J) Crystal violet staining of A375 cells treated as described in A for 24 hrs in the presence or absence of pan-caspase inhibitor Z-VAD-FMK (20μg/ml). Scale bar 200 μm. Cell count is shown on the right. N=3. Statistics by ANOVA with Tukey/s post-test. (K) Western blot of cleaved and total caspase 3 in A375 cells treated as in K. Two independent experiments were performed. All panels: Ns - P > 0.05, * - P≤ 0.05, ** - P ≤ 0.01, *** - P ≤ 0.001, **** - P ≤ 0.0001. P-values were adjusted for multiple comparisons. See also Fig. S2.
Transmission electron microscopy (TEM) was used to examine the morphological features of melanoma cells treated with BCLi and AURKAi. Combination treatment, but not single-agent treatments, induced several apoptosis hallmarks, including cell shrinkage, blebbing, nuclear fragmentation, chromatin condensation, and the appearance of membrane-bound apoptotic bodies (Fig. S2A). We also noted altered mitochondrial morphology in combo-treated cells, where mitochondria appeared smaller and had disrupted and poorly-defined cristae (Fig. S2B). These changes are consistent with the mitochondrial fragmentation and cristae remodeling that occur during the mitochondrial outer membrane permeabilization (MOMP), a part of the intrinsic apoptotic pathway34,35.
Mitochondrial depolarization is another critical step of intrinsic apoptosis. We used the potential-sensitive fluorescent probe JC-1 to analyze the mitochondrial membrane potential (ΔΨm) modulation after treatment. JC-1 forms aggregates in healthy polarized mitochondria that emit red fluorescence. In cells with depolarized mitochondria, JC-1 remains diffused within the cytoplasm and emits green fluorescence. Cells treated with BCLi and AURKAi combined exhibited a lower red/green fluorescence ratio than single-agent and vehicle-treated cells, indicating mitochondrial depolarization (Fig. 2H).
MOMP and depolarization trigger the release of mitochondrial contents that activates caspases, which are proteases that cleave cellular proteins during programmed cell death35. AURKAi and BCLi treatment increased cleavage of executioner caspase 3 in A375 and Hs294T melanoma cells (Fig. 2J). Combination-treated cells also exhibited cleavage of PARP, which is a substrate of caspases and a marker of apoptotic cell death (Fig. 2I). To determine whether caspase activation was required for cell death, we used caspase inhibitor Z-VAD-FMK. Inhibition of caspases rescued cells treated with AURKAi and BCLi combination (Fig. 2J). Furthermore, Z-VAD-FMK abrogated caspase 3 cleavage in combination-treated cells (Fig. 2K), underscoring the role of caspases in BCLi- and AURKAi-mediated apoptosis.
BCLi and AURKAi combination treatment killed melanoma organoids with wild type p53
To test AURKAi and BCLi combination using the functional precision oncology approach, we established 19 Patient-Derived Organoids (PDOs) using the fine-needle aspiration (FNA) technique developed by our group36–38. Tumor cells withdrawn by FNA were cultured in a 3D matrix until tumor spheroids formed (Fig. 3A). Remarkable histological similarity was observed between PDOs and matched patient tumors (Fig. 3B). We routinely test cancer-related mutations in clinical specimens we use to generate PDOs using targeted DNA sequencing 39,40. Based on this information, our PDO cohort included tumors with BRAF V600E (42%), mutations in NRAS (15%), and TP53 (31%), and contained tumors from patients relapsed on BRAF and immunotherapy (Table S2), which is consistent with melanoma genetic and treatment landscape.
Figure 3. Combined AURKAi and BCLi treatment induced cell death in melanoma PDOs with wild-type p53.
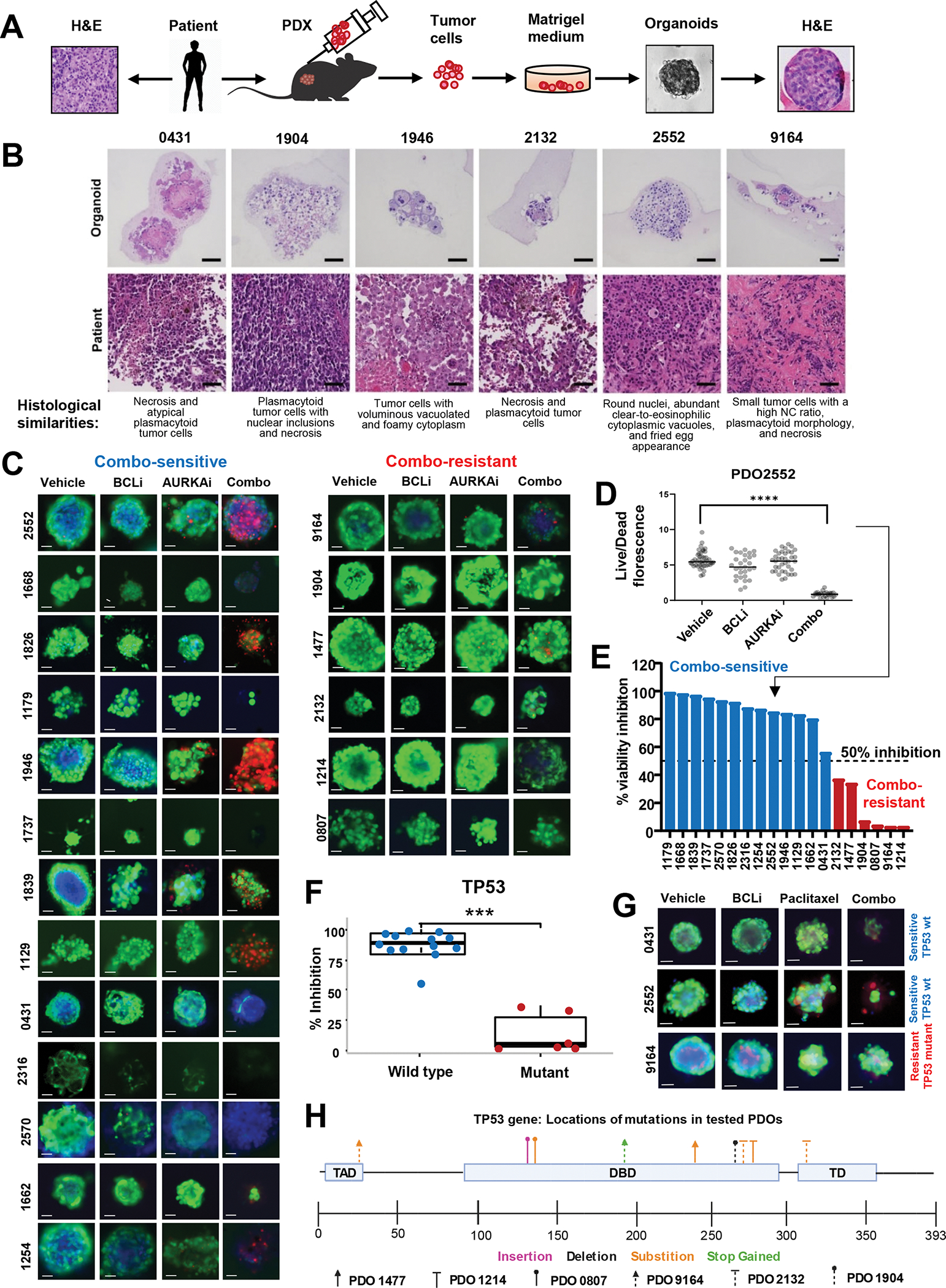
(A) Schematic of the protocol for PDO generation using the fine-needle aspiration technique. (B) H&E staining of indicated PDOs and corresponding original tumor samples. Scale bar 50μm. (C) Representative images of PDOs from 19 distinct melanoma patients treated with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi), or both drugs combined for 72 hrs and stained with calcein-AM (live cells), propidium iodide (PI, dead cells), and Hoechst 33342 (DNA dye). Scale bar 50μm. (D) Quantification of the ratios of green/red (live/dead) fluorescent signal in individual organoids for PDO2552 (top left panels in C). See Fig. S1 for data from other 18 PDO models. N=1–40. Statistics using one-way ANOVA with Tukey’s post-test. (E) Stratification of PDOs into sensitive to AURKAi and BCLi co-treatment (combo-sensitive, blue bars) and combo-resistant (red bars) based on data from C. Punctate line indicates a sensitivity cut-off defined as 50% viability inhibition after combo treatment compared to vehicle control. (F) Association of TP53 mutational status and response to alisertib/navitoclax combination based on combo-induced viability inhibition shown in E. Correlation was queried using a two-sample t-test. Blue and red colors of data points represent combo-sensitive and combo-resistant PDOs. See Fig. S2 for response correlation with other genetic markers. (G) Same as C, except PDOs were treated with 0.1μM paclitaxel instead of alisertib. Scale bar 50uM. Blue and red text indicates TP53 mutational status. (H) Schematic representation of the location and types of TP53 mutations present in 19 PDOs used in C. PDOs identified by a unique arrowhead shape while mutation type is color-coded. All panels: Ns - P > 0.05, * - P≤ 0.05, ** - P ≤ 0.01, *** - P ≤ 0.001, **** - P ≤ 0.0001. P-values were adjusted for multiple comparisons. See also Fig. S3, 4.
To measure therapeutic response in PDOs, we used two bioprobes: Calcein AM (live cells emit green fluorescence) and Propidium Iodide (dead cells emit red fluorescence). Fig. 3C shows representative images of BCLi- and AURKAi-treated organoids from 19 tested models. We quantified the live/dead ratio as the ratio of cumulative green and red fluorescent signals for each imaged organoid. Representative results for one model are shown in Fig. 3D, and complete results are in Fig. S3A. Combined alisertib and navitoclax treatment significantly reduced live/dead ratios in the majority of the tested PDOs (Fig. S3A). In order to quantitatively compare AURKAi and BCLi treatment responses across tested models, we calculated the % viability inhibition using the formula: (live/dead ratio in combo)/(live/dead ratio in vehicle)*100. A cut-off value of 50% inhibition was used to stratify PDOs into combo-sensitive (13 models) and combo-resistant (6 models) (Fig. 3E).
To uncover potential genetic biomarkers of response to AURKAi and BCLi combination therapy, we compared genetic alterations in combo-sensitive and -resistant models (Table S2, Fig. S4A). Strikingly, TP53 mutations were strong predictors of resistance (p<0.001). All combo-sensitive PDOs retained wild-type TP53, and all combo-resistant PDOs had TP53 mutations (Fig. 3F). A similar pattern was observed for paclitaxel and BCLi combination treatment, where PDOs with wild-type TP53 responded well, while PDOs with mutant TP53 were resistant (Fig. 3G, Fig. S4B). We also found that responsive PDOs had a higher prevalence of CDK4 mutations; however, their predictive power was lower compared to TP53 mutations (Fig. S4A, Table S2).
We further studied the TP53 mutations in combination-resistant PDOs. Each PDO had 1–3 “loss-of-function” type mutations, such as deletions, insertions, and gains of a premature stop codon (Fig. 3H, Table S3). Notably, all PDOs had at least one mutation within the DNA binding domain essential for p53’s role as a transcription factor. This pattern indicates that the transcriptional activity of p53 is important for cell death induced by BCLi addition.
Combination treatment with AURKAi and BCLi blocked melanoma progression in vivo without severe toxicities
We used an aggressive B16-F10 murine melanoma model to test AURKAi and BCLi therapy in vivo. While single-agent treatments had minimal effect on tumor growth, the combination of AURKAi and BCLi effectively blocked tumor progression (Fig. 4A–C). Histologically, tumors from BCLi-treated mice were similar to control tumors and contained necrotic areas consistent with an aggressive growth. Tumors from AURKAi- and combination-treated mice had a distinct phenotype characterized by high-grade pleomorphism and enlarged cell size consistent with senescence (Fig. 4D). Lamins are key structural components of the nuclear lamina, a filament meshwork within the nuclear membrane essential for maintaining nuclear shape and integrity41. Senescent cells are reported to display decrease in Lamin B1 or uneven distribution of lanims along the nuclear rim along with abnormal nuclear morphology 42–44. Further confirming senescence, we observed an increased proportion of cells with disrupted Lamin B1 nuclear outlines in AURKAi and combination-treated tumors (Fig. 4E). Notably, tumors in alisertib and combination groups displayed similar proliferation rates based on Ki67 staining (Fig. 4D), suggesting that improved efficacy of AURKAi in combination with BCLi was not due to enhanced proliferation arrest, but increased cell death. Accordingly, we observed an increase of cleaved caspase 3 staining in combination therapy group compared to single-agent treatments, although the overall percentage of apoptotic cells was relatively low (Fig. S5A). This could be due to the prompt clearance of apoptotic cells within the tumor microenvironment.
Figure 4. Combination treatment with AURKAi and BCLi blocked melanoma progression in vivo without severe toxicities.
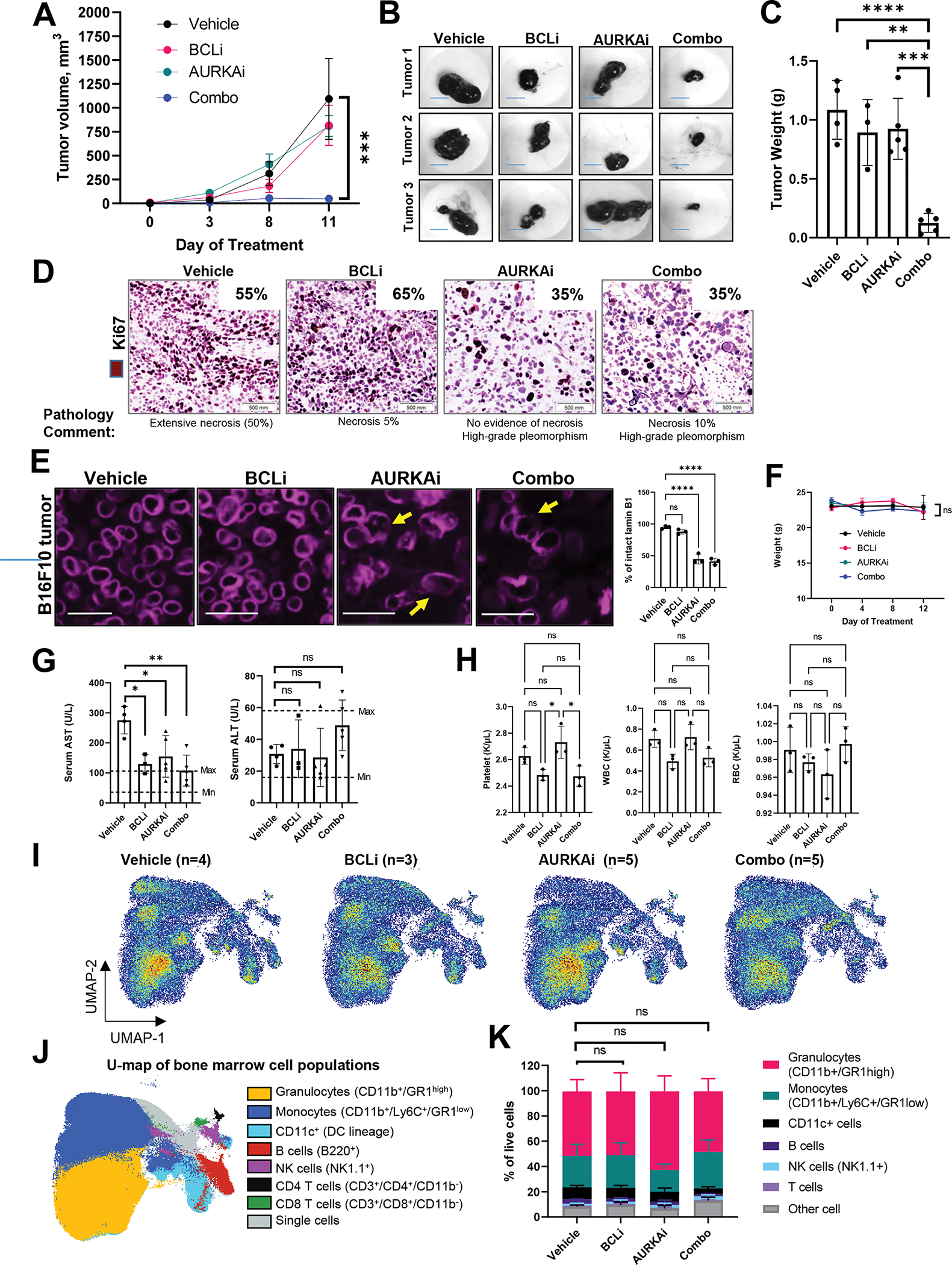
(A) The growth of B16F10 melanoma tumors. Mice were treated with 100 mg/kg navitoclax (BCLi), 25 mg/kg of alisertib (AURKAi), a combination of both drugs (same doses), or drug vehicles for 11 days by daily oral gavage. Data are presented as means ± SEM. N=5 mice per group. Statistical analysis using 2-way ANOVA. Main treatment effects compared to vehicle control were analyzed using Dunnett’s test. (B) Representative images of tumors in A. Scale bar 8mm. (C) Tumor weight from the experiment in A. Statistics using ANOVA with Dunnett’s test. N=3–5 mice. (D) Representative images of Ki67 IHC in tumors shown in A. Inserts indicate the percentages of Ki67+ cells across the slides determined by a dermatopathologist. (E) Immunofluorescence (IF) staining of Lamin B1 in B16F10 tumors shown in A. Arrows indicate representative cells with disrupted Lamin B1 nuclear border. Scale bar 25 μm. Statistical analysis using ANOVA with Dunnett’s test. (F) Body weight changes during the treatment. Statistical analysis as in A. (G) Serum levels of AST and ALT in mice shown in A. Data are presented as individual data points, means ± SEM. Statistical analysis using 1-way ANOVA with Tukey’s post-test. N=3–5 mice. (H) Blood counts of platelet, white and red blood cells (WBC and RBC). Statistical analysis was performed using ANOVA with Dunnett’s post-test. (I) Effect of indicated treatments on bone marrow cell populations in mice shown in A analyzed by spectral cytometry and UMAP dimension reduction algorithm. Samples were concatenated based on treatment. (J) Main bone marrow cell populations were identified based on marker expression indicated in parenthesis and projected on two UMAP dimensions shown by overlay plots. (K) Bone marrow composition in mice from the indicated treatment group. Statistical analysis using 2-way ANOVA. Main treatment effects compared to vehicle control were analyzed using Dunnett’s multiple comparisons test. All panels: Ns - P > 0.05, * - P≤ 0.05, ** - P ≤ 0.01, *** - P ≤ 0.001, **** - P ≤ 0.0001. P-values were adjusted for multiple comparisons. See also Fig. S5.
We also tested whether AURKAi and BCLi treatment induced toxicities in mice. Mice in all treatment displayed similar body weight that was not significantly impacted during the course of treatment indicating lack of severe gastrointestinal toxicities (Fig. 4F). In addition, serum levels of liver enzymes AST and ALT were within the normal range in mice treated with AURKAi and BCLi combination (Fig. 4G). Thrombocytopenia is a common side effect of BCLi navitoclax observed in clinic45,46. Therefore, we performed blood count in non-tumor bearing mice treated with AURKAi and BCLi single therapies and combined. There was a numerical decrease in platelets in BCLi and combination groups that was not significant compared to vehicle, but significant compared to AURKAi only group (Fig. 4H, left panel). There was no significant difference in platelets between combo and BCLi-only groups, indicating that while BCLi affects platelets, addition of AURKAi to BCLi does not further exacerbate this effect. White and red blood cell counts were not significantly affected in any of the treatment groups (Fig. 4H, middle and right panel). Finally, we did not detect significant changes in bone marrow composition (Fig. 4I, J, K, Fig. S5B), indicating the lack of severe myelotoxicities. To validate lack of toxicities in human non-malignant cells, we obtained skin fibroblasts from 3 human donors and treated them with AURKAi and BCLi. No significant impact on cell numbers was detected after 24hrs of treatment (Fig. S5C, D).
AURKAi treatment activated the p53 transcriptional program
To gain further insight into the mechanism of the anti-tumor activity of dual AURKAi and BCLi treatment, we performed RNA sequencing of tumors shown in Fig. 4A. Despite a dramatic difference in size, tumors from single-agent AURKAi and AURKAi-BCLi combination-treated mice exhibited highly similar transcriptomes as they clustered together on a heat map of differentially expressed genes (DEGs, Fig. 5A). Out of 50601 tested transcripts, there were 546 DEGs modulated by AURKAi and 677 DEGs modulated by combo. Of note, 378 DEGs overlapped between these treatments, which constituted a statistically significant overlap based on Fisher’s exact test (p<0.0001). Furthermore, principal component analysis (PCA) indicated similarity of AURKAi- and combination-treated tumors, which clustered together on a PCA plot (Fig. 5B). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed an enrichment of DEGs related to p53 pathways in combo and AURKAi treatments with a similar pattern of mRNA modulation (Fig. 5C, D). These data demonstrate similar induction of the p53 transcriptional response by AURKAi and AURKAi-BCLi combination treatments. Western blot analysis of p53 protein expression confirmed this conclusion (Fig. 5E).
Figure 5. AURKAi activated the p53 transcriptional program.
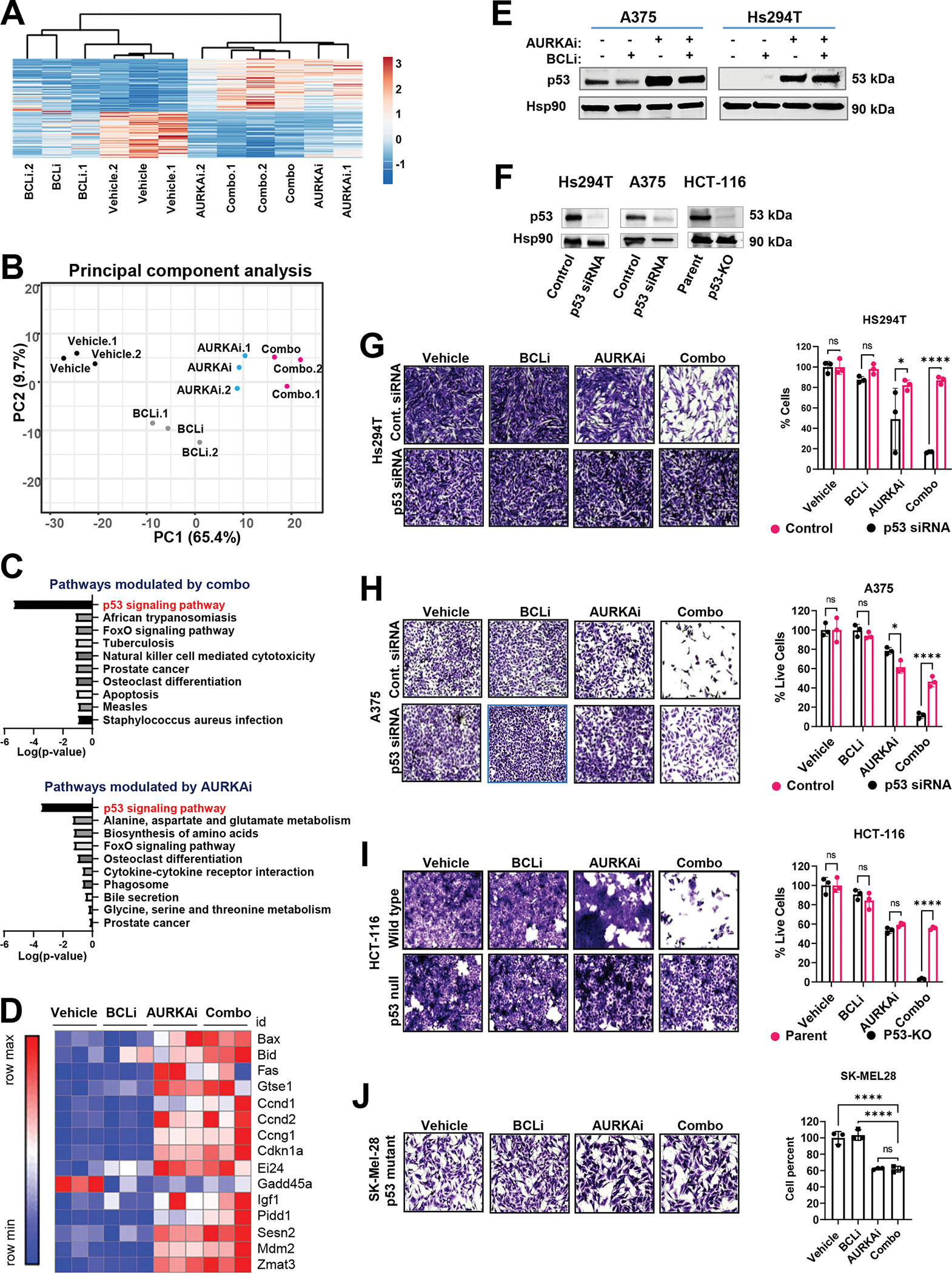
(A) RNAseq of tumors shown in Fig. 4A. The heat map of genes modulated by either combined AURKAi and BCLi treatment or corresponding individual treatments. N=3. Rows are centered; unit variance scaling is applied to rows. Both rows and columns are clustered using correlation distance and average linkage. (B) Results of the principal component analysis of gene expression data sin A. (C) Top 10 KEGG pathways enriched within the genes differentially expressed after combo (top panel) and alisertib (bottom panel) treatments. Log10-transformed Benjamini-adjusted p-values were plotted. (D) The heat map shows relative mRNA expression of genes within the KEGG p53 signaling pathway across. “Row max” corresponds to the highest expression value of a protein in this row across all tested samples. “Row min” is expression in the sample with lowest expression value. (E) Western blot analysis of p53 protein in melanoma cells treated with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi) or combination for 24 hrs. Three independent experiments were performed with consistent results. (F) Western blot shows the efficiency of p53 knockdown/knockout. (G) Representative images and quantificationof crystal violet-stained Hs294T melanoma cells transfected with p53 siRNA or non-targeting siRNA and treated as described in E. Scale bar 200μm. N=3. Statistical analysis using one-way ANOVA with Sidak’s post- test. Two independent experiments were performed with consistent results. (H) Same as G, except A375 cells were used. (I) Representative images and quantification of crystal violet-stained HCT-116 wild-type and p53-null cells treated as in E. Scale bar 200μm. Statistical analysis as in G. (J) Representative images and quantification of Sk-Mel-28 cells with mutated TP53 treated as in E. Scale bar 200μm. Statistical analysis using one-way ANOVA with Dunnett’s test. (All panels) Ns - P > 0.05, * - P≤ 0.05, ** - P ≤ 0.01, *** - P ≤ 0.001, **** - P ≤ 0.0001. P-values were adjusted for multiple comparisons.
We tested the functional significance of p53 activation using p53 knockdown and knockout (Fig. 5F). Loss of endogenous p53 rescued cells from AURKAi and BCLi-induced death (Fig. 5G, H, I). Similarly, SK-Mel28 melanoma cells expressing mutated TP53 were resistant to AURKAi and BCLi treatment (Fig. 5J). Altogether, these data demonstrate that AURKAi-mediated activation of p53 is required for BCLi-mediated apoptosis but is not sufficient to induce apoptosis on its own.
Network analysis identified p53-regulated cell cycle inhibitor p21 as an important hub connecting proteins differentially affected by AURKAi and AURKAi and BCLi combination
Our data so far demonstrated that AURKAi activated p53 transcriptional program, which led to senescence in the absence of BCLi and to apoptosis when BCLi was added. Therefore, we hypothesized that BCLi modulated p53 response on a post-transcriptional level. We employed a reverse-phase protein array (RPPA) detecting 447 proteins involved in the regulation of cell cycle, apoptosis, and other cancer-relevant processes to identify proteins significantly modulated by either AURKAi, BCLi, or combination treatments (Fig. 6A). In contrast to the transcriptomic data, cells treated with AURKAi and BCLi combination displayed distinct proteome compared to AURKAi-treated cells, as indicated by the PCA (Fig. 6B).
Figure 6. The addition of BCLi caused extensive proteome remodeling in AURKAi-treated cells.
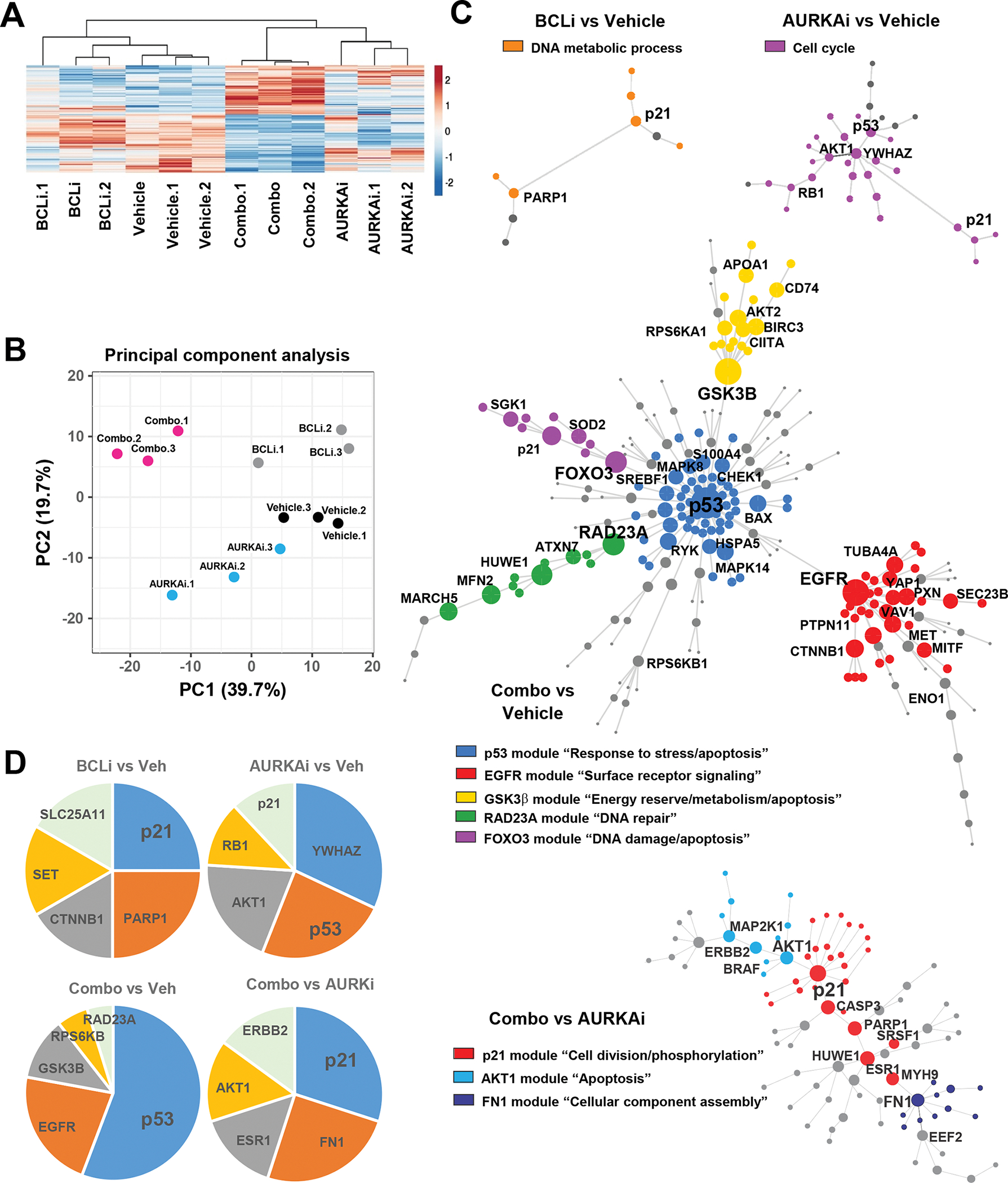
Reverse phase proteomic analysis (RPPA) of A375 melanoma cells treated with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi) or both drugs for 24 hrs. (A) Heat map shows proteins significantly modulated by AURKAi and BCLi treatment. N=3. Rows are centered; unit variance scaling is applied to rows. Rows and columns are clustered using correlation distance and average linkage. (B) Principal component analysis of protein expression data shown in A. (C) Protein networks constructed based on data in A. Circles represent individual proteins differentially expressed in pairwise-compared groups. Connecting lines represent known/validated protein-protein interactions. The size of individual circles reflects how many connections this protein has with other network proteins. Colors indicate distinct modules (groups of interconnected proteins), and legends indicate GO annotations enriched within this module. (D) Relative connectivity of top five most connected proteins within the networks shown in C.
To identify key proteome reprogramming events associated with a senescence-to-apoptosis fate switch, we constructed networks from proteins affected by BCLi, AURKAi, or combination treatment (Fig. 6C). The networks modulated by BCLi, and AURKAi single-agent treatments were relatively simple due to the limited number of proteins affected by these conditions (Fig. 6C, top panels). In contrast, proteins differentially modulated by AURKAi-BCLi dual treatment compared to vehicle treatment formed a complex network comprising five sub-networks (modules). The most integral protein in the AURKAi-BCLi-regulated network was p53, which was also the main hub of the largest central module that connected four distal modules. This module was enriched with GO annotations relevant to stress and apoptosis. Distal modules were enriched for surface receptor signaling, energy and metabolism, DNA repair, DNA damage, and apoptosis annotations (Fig. 6C, middle panel). Finally, a network of proteins differentially expressed in cells treated with AURKAi only and AURKAi+BCLi combination was enriched for GO terms related to cell division and apoptosis, reflecting the distinct cellular responses induced by these treatments. The algorithm also identified a module enriched with proteins involved in a cellular component assembly that may reflect distinct cytoskeleton changes in senescent and apoptotic cells (Fig. 6C, bottom panel). Connectivity analysis was performed to identify most influential proteins within the networks (Fig. 6D). This analysis identified several proteins, including p21, FN1, ERBB2, AKT1, and ESR1. We chose to focus on p21, a key mediator of p53-induced cell cycle arrest and senescence, which was the most connected protein in the network (Fig. 6D).
Post-transcriptional regulation of p53 target genes determines therapeutic cell fate
In silico network modeling identified p21 as an influencer of proteome differences between senescence- and apoptosis-committed cells, therefore we performed a series of functional assays to validate its role in cell fate regulation. Western blot analysis showed that p21 protein was induced by AURKAi but downregulated after the addition of BCLi (Fig. 7A). Of note, addition of BCL-xLi, but not BCL-2i or MCLi also abrogated AURKAi-mediated p21 induction, which aligned with the ability of these agents to improve AURKAi efficacy (Fig. 1K). In contrast to protein levels, p21 mRNA levels were comparable in AURKAi and combination-treated cells (Fig. 7B), indicating that p21 downregulation occurred on a post-transcriptional level. To determine whether p21 downregulation resulted from decreased protein stability, we used cycloheximide to block protein synthesis. We found that p21 degradation was accelerated by the BCLi and AURKAi and BCLi combination treatment (Fig. 7C). Of note, there were no statistically significant differences in p21 degradation in the presence of proteasome inhibitor MG132 (Fig. 7C, right lanes, Fig. S6A), suggesting that non-proteasomal mechanisms regulate p21 protein stability. It has been reported that p21 is a target for proteolytic cleavage by activated caspases 47–50. Since caspases were activated by the AURKAi and BCLi combination therapy (Fig. 2J), we tested whether caspase activity was important for BCLi-mediated downregulation of p21 protein. Co-treatment with pan-caspase inhibitor Z-VAD-FMK abrogated p21 downregulation in AURKAi and BCLi-treated cells (Fig. 7D), indicating that BCLi-mediated p21 degradation is likely to be caspase-dependent.
Figure 7. Differential modulation of p53 targets regulated cellular response to AURKAi and BCLi therapy.
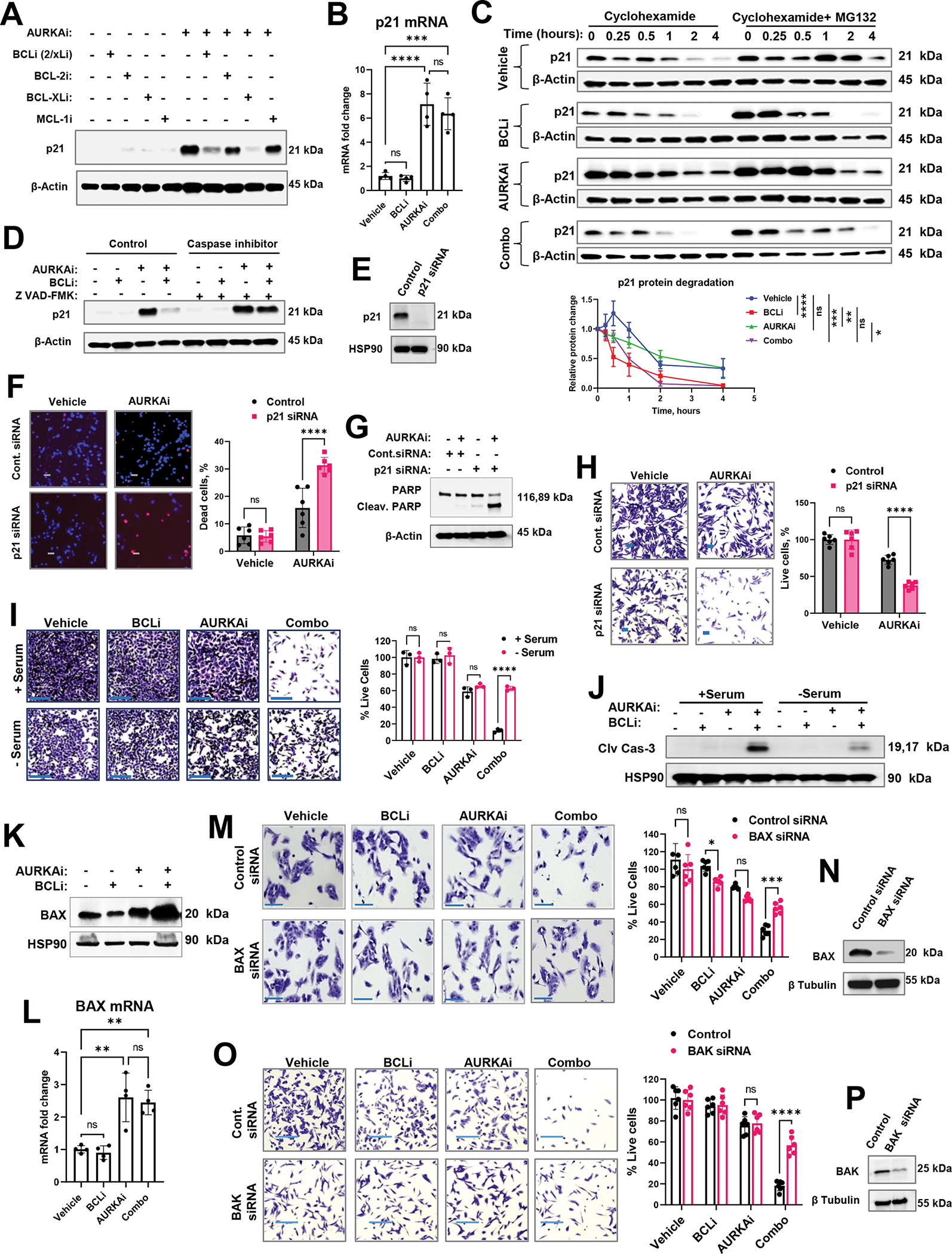
(A) Western blot of p21 in A375 melanoma cells treated with 1μM navitoclax (BCL-2/xLi), 1μM venetoclax (BCL-2i), 1μM A-1155463 (BCL-xLi), 1μM A-1210477 (MCL-1i) in the presence or absence of 1μM alisertib (AURKAi) for 24hrs. Experiment was repeated three times with consistent results. (B) Real-time PCR of relative CDKN1A (p21) mRNA expression in A375 cells after 24hrs of treatment with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi), or combination of both drugs. The experiment was repeated three times with 2 replicates each. Statistical comparison using one-way ANOVA with Tukey’s post- test. (C) Western blot analysis of p21 degradation. A375 melanoma cells were treated as in B for 16hrs followed by the addition of Cycloheximide (150μg/ml) ± MG-132 (1μM). Cell lysates were collected at indicated time points (0.25h – 4h). P21 bands from 3 independent experiments were quantified using densitometry and normalized to corresponding actin bands and plotted on the graph below. The square root transformation was used to correct for heteroscedasticity. Statistical analysis using 2-way ANOVA with Tukey’s post-test. (D) Western blot in A375 cells treated as in B for 24hrs in the absence or presence of pan-caspase inhibitor Z-VAD-FMK (20 μg/ml). Three independent experiments were performed with consistent results. (E) Western blot analysis of p21 knockdown. (F) Representative images of Hs294T cells transfected with p21-targeting or non-targeting siRNA, treated with vehicle or 1μM alisertib (AURKAi) for 24hrs, and stained with Hoechst and PI. Scale bar 50μm. Experiment was performed with 3 biological replicates. Right: data quantification from 6 random fields (n=6) and statistics by 2-way ANOVA with Sidak’s post-test. (G) Western blot analysis of cleaved PARP in Hs294T cells treated as in F. (H) Cristal violet staining in Hs294T cells treated as in F. Statistical analysis as in F. (I) Representative images and quantification of crystal violet-stained A375 cells treated as in B for 24hrs in serum-containing (+ serum, control culture condition) and serum-free (- serum, cell cycle-arresting conditions) media. Scale bar 200μm. Statistical comparisons using one-way ANOVA with Sidak’s post-test. N=3 biological replicates. (J) Western blot analysis of cleaved caspase 3 in A375 cells treated as described in I. Experiment was repeated twice with consistent results. (K) Western blot of BAX protein expression in A375 cells treated with vehicle, 1μM navitoclax (BCLi), 1μM alisertib (AURKAi), or their combination for 16hrs. Three independent experiments were performed with consistent results. (L) Real-time PCR analysis of BAX mRNA in A375 cells treated as in B for 24hrs. Experiment was repeated two times with two replicates each. Statistical analysis using one-way ANOVA with Tukey’s post-test. N=4. (M) Representative images of crystal violet-stained A375 cells transfected with non-targeting and BAX-targeting siRNA. Scale bar 150μm. Two independent experiments were performed with consistent results. Right: quantified cell numbers and statistical comparison using one-way ANOVA with Sidak’s post-test. N=6. (N) Western blot for BAX siRNA efficiency. (O-P) Same as M and N, except BAK-specific siRNA was used. All panels: Ns - P > 0.05, * - P≤ 0.05, ** - P ≤ 0.01, *** - P ≤ 0.001, **** - P ≤ 0.0001. P-values were adjusted for multiple comparisons. See also Fig. S6.
To test whether p21 downregulation enabled apoptotic response to AURKAi, we knocked down endogenous p21 in melanoma cells using siRNA (Fig. 7E). AURKAi-treated p21-knockdown (p21-KD) cells displayed an increased rate of cell death (Fig. 7F) and PARP cleavage (Fig. 7G) compared to wild-type cells. Accordingly, fewer p21-KD cells remained after treatment with AURKAi compared to control p21 wild type cells (Fig. 7H). Similarly, p21-knockout HCT116 cells cells displayed more cell death after AURKAi compared to p21 wild-type cells (Fig. S6B). Interestingly, the total numbers of cells following AURKAi treatment were comparable in p21 wild type and KO cells (Fig. S6C), which was likely due to the failure of p21-KO cells to undergo cell cycle arrest upon AURKAi treatment (Fig. S6D). Fig. S6E illustrates the loss of p21 protein in p21 KO cells.
Since cell cycle arrest is a main function of p21, we tested if the arrested state itself protected cells from therapy-induced apoptosis. The serum withdrawal approach was used to induce p21-independent cell cycle arrest. Cells cultured in serum-free conditions were resistant to cell death induced by BCLi-AURKAi co-treatment (Fig. 7I), despite the evidence of caspase activation (Fig. 7J). This data indicates that BCLi-mediated downregulation of p21 may facilitate cell death by preventing cell cycle arrest.
Finally, we investigated downstream effectors of p53-dependent apoptosis enabled by p21 degradation. p53-regulated pro-apoptotic mitochondrial protein BAX was induced on protein and mRNA levels in AURKAi and AURKAi/BCLi-treated cells (Fig. 7K, L). Down modulation of BAX using RNAi abrogated AURKAi/BCLi-induced cell death (Fig. 7M). These findings were confirmed using isogenic BAX wild-type and knockout HCT116 cells (Fig. S6F, G). BAX induces apoptosis by assembling into oligomers that delineate pores to permeabilize the mitochondrial outer membrane (MOM)51. BAK is a homolog of BAX with similar function, therefore we predicted that BAK loss will limit AURKAi and BCLi-mediated apoptosis, similar to BAX loss. Indeed, melanoma cells transfected with BAK siRNA were less sensitive to AURKAi and BCLi combination then control siRNA-expressing cells (Fig. 7O, P). We also tested the involvement of another pro-apoptotic p53 target PUMA. PUMA mRNA was induced by AURKAi treatment without notable changes to the protein expression (Fig. S6H, I). Knockdown and knockout of PUMA gene BBC3 had minimal effect of cell viability upon AURKAi and BCLi treatment (Fig. S6J–O). Based on these data, we concluded that BAX and BAK mediate p53-dependent apoptosis induced by AURKAi and BCLi treatment.
DISCUSSION
The ideal outcome of cancer therapy is a complete response where cancer cells are permanently eliminated from the body. However, cancer cells can often survive treatment by adopting cell-cycle arrest or senescence fates, which limit therapeutic benefit and worsen a patient’s long-term prognosis. Identifying key drivers of therapeutic fate choice can improve our ability to eliminate tumors and is an unmet clinical need. Our study demonstrated that inhibition of the pro-survival mitochondrial protein BCL-xL can direct cancer cells towards adopting an apoptotic cell fate upon drug treatment by modulating the output of p53 transcriptional response. These findings reveal a rational strategy to use pharmacological inhibitors of BCL-xL to promote apoptosis during cancer treatment. We have demonstrated the efficacy of this approach by combining BCLi with an emerging cancer therapeutic alisertib (AURKAi) that induces melanoma cell senescence when used as a monotherapy. The combination displayed superior anti-tumor activity in vivo without major toxicities to normal organs. We confirmed the translational potential of alisertib and navitoclax combination using a functional precision oncology approach52. To this end, a “preclinical trial” of AURKAi and BCLi combination was conducted with nineteen melanoma patient-derived organoids. These models recapitulate the biology and therapeutic response of patients’ tumors53–60.
By testing the combination of AURKAi and BCLi on a set of genetically diverse melanoma PDOs, we were able to uncover the dependence of apoptotic response on the presence of wild-type transcriptionally-competent p53. All TP53 wild-type PDOs were sensitive to AURKAi and BCLi treatment regardless of their BRAF mutational status and treatment history, while all mutated tumors were resistant, implicating p53 mutational status as a biomarker for patient selection. Notably, melanoma tumors often retain wild-type p53 (about 80%) 61, suggesting that most melanoma patients could benefit from the AURKAi and BCLi combination. We also found colon cancer cells to be sensitive to AURKAi and BCLi combination. These data suggest that therapeutically combining AURKAi and BCLi may benefit patients with various cancer types.
Our drug screening data demonstrated that BCLi can enhance activity of several other drugs, including all mitotic kinase inhibitors we tested. These findings match well with our current knowledge that prolonged mitotic inhibition activates p53 as a part of mitotic surveillance pathway regulated by USP28 and p53BP162–66. Cells arrested in mitosis have two mutually exclusive cell fates: cell death from mitotic catastrophe or mitotic slippage where cells escape from mitosis and return to interphase in a tetraploid state67,68. The fate choice is determined by the balance of pro-survival and pro-apoptotic BCL-2 family proteins69,70. Accordingly, a synergistic activity of anti-mitotic drugs and BCL-xL has been reported in various cancer cell types71,72.
P53 is a stress-induced transcription factor that transactivates genes regulating apoptosis, cell cycle and other cellular processes73. Several studies showed that activation of p53 promotes death in senescent cells25,74. Intriguingly, our RNAseq data revealed that the p53 transcriptional program was activated equally in alisertib-treated cells and cells treated with alisertib and navitoclax combined. These findings indicate that p53 activation itself is not a decisive factor of therapeutic fate. Indeed, it is well documented that activation of p53 can lead to either cell death or survival via cell cycle arrest and damage repair75. However, our knowledge of how cell fate upon p53 activation is determined remains limited. A recent study showed that mitochondrial apoptotic priming determines whether cells undergo cell cycle arrest or apoptosis upon p53 re-expression76, implicating mitochondrial polarization as a major player in the regulation of p53 response. In agreement with these findings, we show that targeting pro-survival BCL-2 family proteins, which increase mitochondrial apoptotic priming, triggers cell death in the context of AURKAi-mediated p53 activation.
Cell cycle arrest and apoptosis are mutually exclusive cellular processes. The outstanding question is, what are the mechanisms that abrogate pro-survival cell cycle arrest processes when mitochondrial outer membrane potential drops below the critical threshold? Our study implicates p21 protein stability as a critical factor. P21 is a p53-regulated inhibitor of cell cycle kinases. Our data suggest that p21 protein is rapidly degraded in AURKAi+BCLi-treated cells in a caspase-dependent manner, and this leads to apoptosis induction. Thus, p21-regulated processes may counteract apoptosis processes even after caspase activation has occurred. This intriguing idea aligns well with the view that caspase activity and caspase substrate cleavage can occur without cell death. In fact, non-apoptotic caspase activity has been implicated in regulation of many cellular processes, including development, innate immune response, tissue regeneration, cell-fate determination, stem-cell differentiation, and neural activation77–82. In contrast to p21, BAX, a pro-apoptotic target of p5383–85, is spared by BCLi treatment. BAX oligomerizes to form pores in the mitochondrial outer membrane leading to MOMP and caspase activation86. In the context of AURKAi and BCLi treatment, BAX-dependent apoptosis coincided with degradation of p21, implicating the balance of these factors in melanoma therapeutic fate regulation.
Based on experimental findings, we proposed a working model where alisertib, paclitaxel, and other cancer drugs activate p53 transcriptional programs in melanoma cells. Due to the high activity of BCL-xL, which inhibits the pro-apoptotic p53 effector BAX, p53 activation translates into a p21-mediated cytostatic response. Inhibition of BCL-xL permits BAX-mediated MOMP, resulting in caspase-mediated cleavage of p21 that restricts protective senescence-initiating processes during apoptosis. Our findings argue for using BCL-xL inhibitors to augment therapeutic responses in p53 wild-type cancers.
Limitations of the study
RPPA analysis comparing proteins in senescence- and apoptosis-committed cells is limited to a few hundred proteins. This limited our ability to assess the full extent of p21-associated network. Therefore, while the data we collected provides an insight into cell fate regulation in the context of cancer therapy, more work needs to be done to dissect the exact mechanism of how p21 activity counteracts cells death.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anna Vilgelm (anna.vilgelm@osumc.com).
Materials availability
Patient-derived organoids generated in this study will be shared by a lead contact upon request pending an execution of an MTA.
Data and code availability
Access to the original data. RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is GSE212127. RPPA proteomics data have been deposited to BioStudies. Accession number is S-BSST919.
Access to the original code. This paper does not report original code.
Access to any additional information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human melanoma cells A375, Hs294T, Sk-Mel5, B16F10, Sk-Mel28 were purchased from the American Type Culture Collection. Colorectal HCT116 cells with p53 KO were provided by Alexander Zaika (University of Miami). HCT116 cell lines with wild type or knockout of BAX and PUMA were provided by Bert Vogelstein (John Hopkins University). The HCT116 wild type and p21 knockout (p21−/−) cell lines were purchased from Horizon Discovery (HD R02–035). Cells were cultured in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco) at 37°C. Cell lines have not been authenticated.
Patient-derived organoids and fibroblasts
Patient-derived organoids were generated using fine –needle aspiration (FNA) from patient-derived xenografts generated previously25,39,40. Briefly, to establish PDX tumors in mice, frozen fragments of tumor tissue cryopreserved in liquid nitrogen with 10% DMSO were thawed at room temperature and washed ones with sterile PBS. Small incision was made in the betadine-disinfected skin on the back of a nude mouse and a piece of tumor tissue of 1–2 mm in diameter was placed subcutaneously into the incision. Wounds were closed using surgical clips. Mice were monitored until the tumors reached approximately 10 mm in diameter. FNAs were performed on PDX tumors using a 25-gauge needle as described previously [36]. Detailed protocol for PDO generation, growth, passaging, and cryopreservation can be found here [37, 38]. Organoids were cultured in complete media containing DMEM-F12 (Gibco), 15% FBS (Gibco), 1X B27-supplement (Gibco), and 5% Matrigel (Corning) in ultra-low attachment plates at 37°C. Growth of organoids was typically observed after 1–2 weeks in culture. Sex of PDOs is indicated in the Table S2. Human skin biopsies were obtained from CTCL patients under IRB #2020C0056. Sex, age, treatment, or any other patient and disease related information was not collected. To obtain human fibroblasts, skin biopsies were enzymatically digested with liberase TL (Roche Cat#05401020001) for 90 minutes and dissociated cells were filtered through a 70 μM cell strainer. The resulting cells were plated in tissue culture-treated 6-well plates. After about one week in culture, media was removed and attached fibroblasts were collected by trypsinization. Fibroblasts were cultured in standard cell culture conditions in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco).
Mice
All animal procedures were approved by the IACUC at Vanderbilt and OSU. Patient-derived xenografts (PDX) had been generated previously39 [31, 65]. Tumors were propagated by serial passaging in 6–10 week old female NSG mice (NOD-SCID IL2Rgammanull) and 6–10 week old female nude mice (Foxn1nu) and used to generate PDOs. To assess AURKAi and BCLi treatment efficacy in vivo, 6–10 week old female C57Bl/6 mice were injected subcutaneously with 50,000 B16F10 cells. Treatment was initiated when tumors were palpable. Mice were euthanized if tumors exceeded 15 mm in diameter or became perforated.
METHOD DETAILS
Cell viability and senescence-associated (SA) β-galactosidase assays
Cells were seeded at 0.2×106 - 0.3×10 cells per well in flat-bottom 6-well plates and allowed to attach overnight prior to treatment ad treated as described in figure legends. For crystal violet staining, cells were fixed with 100% methanol (Sigma) for 10 minutes and stained with 1% (w/v) crystal violet (Sigma) for 15 minutes, followed by at least three washes with water. The crystal violet stain was visualized using phase-contrast microscopy. Cells were counted with ImageJ software. For graphical presentation, cell counts from each experiment were shown as percentages relevant to a control sample (no drug, control siRNA, wild type, etc.). Histochemical staining of cells for Senescence-associated (SA) β-galactosidase activity was performed using a kit (BD) according to the manufacture’s protocol.
Flow Cytometry
Apoptosis, DNA Damage, and Cell Proliferation Kit (BD) measured BrdU incorporation and PARP cleavage. Cells were pulsed with 50 mM of BrdU during the final 5 hours of culture and processed according to the manufacture’s protocol. Annexin V staining was performed using the FITC Annexin V Apoptosis Detection Kit (BD). A375 cells were plated at 0.2×106 - 0.3×10 cells/well in 6-well flat-bottom plates cells and treated with drugs the following day. Experiments were performed in triplicate. Results were analyzed using FlowJo software. For bone marrow analysis, femurs were flushed with PBS, and cell suspensions were stained with fluorescent antibodies. Data were acquired using Cytek Aurora spectral flow cytometer and analyzed in OMIQ.
Transfection
Transfections were performed in A375 and Hs294T cells using Lipofectamine RNAi Max (Thermofisher) according to the manufacturer’s protocol. We used siRNA targeting p53 (Santa Cruz Biotechnology), BAX (Cell Signaling), PUMA (Cell Signaling), AURKA (Cell Signaling), p21 (Cell Signaling), Bak (Cell Signaling), BCL-xL (Thermo Fisher), and control non-targeting siRNA (Cell Signaling).
Western blot
Cell lysates were prepared by washing cells once with cold PBS followed by lysis with RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitors (Sigma-Aldrich). Protein concentrations in lysates were determined using Bradford Protein Assay reagent (Bio-Rad) according to the manufacturer’s protocol. All primary antibodies (caspase 3, cleaved-caspase3, cleaved-PARP, p21, BAX, Bak, BCLxl, HSP90, and β-actin) were purchased from Cell Signaling except for p53, which was from Sana Cruz Biotechnology. All primary antibodies were hybridized overnight at 4°C at 1:500 dilutions except for housekeeping proteins β-actin, β-tubulin, and HSP90, which were used at 1:2000 dilution. Secondary antibodies (Cell Signaling) were used at a dilution of 1:10,000 for 2 hours at room temperature. Densitometry analysis was performed using ImageJ.
Immunohistochemcal and immunofluorescent tissue staining and electron Microscopy
Organoids and patient tumors were submitted to Translational Pathology Shared Resource (Vanderbilt) and Comparative Pathology and Digital Imaging Shared Resource (OSU) for H&E and Ki67 staining. Slides were evaluated in a blind manner by a dermatopathologist, who provided pathology comments and estimation of necrosis area and KI67 positivity. For IF staining, FFPE tissues were sectioned at 5μm thickness, deparaffinized in xylenes, rehydrated, and antigen retrieval was performed using either 1xTarget Retrieval pH 6.0 (Dako) or 1x EDTA Decloaker pH 8.4 (Biocare Medical) in a pressure cooker for 2 minutes at 110°C. Sections were then permeabilized for 20 minutes in 1X Tris-Buffered Saline+0.01%Tween-20 (TBST). Blocking solution (1xTBS+0.3%TritonX100+10%Donkey Serum+5%BSA) was applied followed by endogenous peroxidase quenching with 3% H2O2 before the addition of primary Cleaved Caspase-3 antibodies (1:400, Cell Signaling 9661S). Slides receiving anti-mouse antibodies were also blocked with ReadyProbes Mouse on Mouse IgG Blocking Solution prior to addition of Lamin B1 (1:1000, Cell Signaling), After washing in TBST, AlexaFluor-conjugated secondary antibodies were added (anti-rabbit 594 and anti-rat 594, Invitrogen). Nuclei were counterstained with Hoescht (1:15000, Invitrogen 33342) and slides mounted with SlowFade Gold antifade mountant (Invitrogen). For electron microscopy, A375 cells were treated with experimental drugs for 16 hours. Fixation, dehydration, embedding, sectioning, and staining were done by the cell imaging shared resource at Vanderbilt using a standardized protocol. Imaging was performed on the Philips/FEI T-12 transmission electron microscope.
Fluorescent viability assay in PDOs
An equal number of PDOs were seeded in an ultra-low attachment 96 well plate. Drugs or drug vehicles (DMSO) were added to culture wells in triplicate, and PDOs were incubated for 72 hours. After 48 hours of drug dosing, propidium iodide was added to the wells at a final concentration of 50 μg/ml. After 24 additional hours of culturing, Hoechst 33342 and Calcein AM (Thermo Fisher Scientific) were added at a final concentration of 10 μg/ml and 5μM, respectively. Organoids were incubated in standard cell culture conditions at 37C with 5% CO2 for an additional two hours. Images were taken on an inverted fluorescent microscope (EVOS™ M7000, Thermo Fisher). The live/dead ratios were calculated by quantification of images in Image J. RGB images were split into three channels. Live cells were evaluated in the Green Channel, and dead cells were evaluated in the Red Channel based on fluorescence intensity in the organoid region. Organoid regions were identified in a trans channel image.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA sequencing data generation and analysis
Targeted DNA sequencing was described previously39,40. RNAseq data was generated by OSUCCC GSR using ribo-depleted total transcriptome library generation approach. Input RNA quality and quantity was assessed using the Agilent 2100 Bioanalyzer RNA Nanochip (Agilent Technologies, Santa Clara, CA) and Qubit Fluorometer (Thermo Fisher), respectively. The RNA integrity number (RIN) values for all samples exceeded the minimum requirement of 3 and the RNA concentrations were equal or greater than 100ng/uL. Ribo-depleted total transcriptome libraries were generated with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB #E7760) and NEBNext rRNA Depletion Kit (NEB #E7405) with an input amount of 100 ng total RNA per sample. Library fragmentation profile and amount was assessed by the Agilent Bioanalyzer HS DNA chip and Qubit Fluorometer, respectively. Libraries were pooled and sequenced on an Illumina NovaSeq SP flowcell in paired-end 100 bp format (Illumina, San Diego, CA) to a minimum read yield ~70 million paired reads (equivalent to ~35 million clusters).
Individual FASTQ files were trimmed for adapter sequences and filtered for a minimum quality score of Q20 using AdapterRemoval v2.2.0. Preliminary alignment using HISAT2 v2.0.6 was performed to a composite reference of rRNA, mtDNA, and PhiX bacteriophage sequences obtained from NCBI RefSeq. Reads aligning to these references were excluded in downstream analyses. Primary alignment was performed against the mouse genome reference GRCm38p4 using HISAT2. Gene expression values for genes described by the GENCODE Gene Transfer Format (GTF) release M14 (mouse) were quantified using the featureCounts tool of the Subread package v1.5.1 58 in stranded mode.
RNAseq data were normalized on log2 scale. To identify genes differentially expressed between treatment groups (DEGs), the log2 fold change between the average normalized expression in compared groups were calculated for each transcript and compared using Wald test. P-values were adjusted for multiple comparison using Benjamini-Hochberg method. Gene was considered a DEG if adjusted p-value < 0.05. To construct a heatmap, normalized counts of genes differentially expressed in vehicle and combo samples were uploaded into ClustVis web portal 87. Individual genes were organized in rows, and treatment conditions in columns. Rows were centered; unit variance scaling was applied to rows. Both rows and columns were clustered using correlation distance and average linkage. For PCA, unit variance scaling was applied to rows; SVD with imputation was used to calculate principal components. X and Y axis show principal component 1 and principal component 2. Heatmap showing expression of p53 target genes heatmap was constructed using Morpheus using their standard configuration (https://software.broadinstitute.org/morpheus). No clustering was performed. A relative color scheme was chosed that uses the minimum and maximum values in each row to convert values to colors. RNAseq pathways were analyzed using DAVID online computational platform 88. List of DEGs associated with AURKAi and Combination treatments were uploaded into DAVID and processed using Functional Annotation Clustering tool and GOTERM_BP_DIRECT annotation category.
Proteomics analysis and construction of protein networks
For the analysis of cell proteome, A375 cell pellets were submitted to MD Anderson Cancer Center RPPA core for lysis and analysis of 447 proteins using reverse phase protein array (RPPA) technology 89. We analyzed three biological replicates for each treatment condition. Normalized linear (Normlinear) expression data from RPPA analysis was then used to calculate PCA, plot the heat map, and identify proteins differentially-expressed between treatment group. To construct PCA plots and heatmaps, expression data was uploaded to ClustVis web portal. Individual proteins were organized in rows, and treatment conditions in columns. For PCA, unit variance scaling was applied to rows; SVD with imputation was used to calculate principal components. X and Y axis show principal component 1 and principal component 2. For heatmap, rows were centered; unit variance scaling was applied to rows. Both rows and columns were clustered using correlation distance and average linkage.
Protein expression between treatment groups was compared in GraphPadPrism using multiple unpaired t-test with individual variances computed for each protein. Two-stage step-up method was used to identify differentially expressed proteins and the FDR threshold was set at 10%. For each comparison, a list of proteins that passed the FDR threshold was uploaded to OmicsNet and network analysis of one list of molecules was performed using the “Proteins” option. STRING database of known and predicted protein-protein interactions was selected for network creation and Streiner Forest (PCSF) network tool was applied to simplify the network by identifying subnetwork enriched with input values. The networks were visualized in 2D. WalkTrap algorithm was used to identify modules within the networks and GO:BP database was used for pathway enrichment analysis.
Statistical analysis
Continuous variables were expressed as mean with standard deviation. Statistical methods and tests were indicated in the figure legends. Welch’s t-test, one-way, two-way, or repeated-measures ANOVA were used to compare two samples and multiple samples, respectively. All tests were two-tailed, p values ≤ 0.05 were considered statistically significant, and p values were adjusted for multiple comparisons. The statistical analyses were performed using STATA, version 16 (StataCorp LLC), R 3.6.0, and GraphPad Prism.
Supplementary Material
Table S1. Drug screen results. Related to Figure 1.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Aurora A (D3E4Q) Rabbit mAb | Cell signaling | 14475 |
| Bcl-xL (54H6) Rabbit mAb | Cell signaling | 2764 |
| p-Histone H3 (S10) Rabbit | Cell signaling | 9701S |
| Caspase-3 Rabbit | Cell signaling | 9662S |
| Cleaved Caspase-3 Rabbit | Cell signaling | 9661S |
| PARP Rabbit | Cell signaling | 9542S |
| CD45 | BD Biosciences | 564279 |
| CD3 | BD Biosciences | 563024 |
| CD4 | BD Biosciences | 741050 |
| CD8 | Biolegend | 100725 |
| B220 | Biolegend | 103209 |
| NK1.1 | Biolegend | 108714 |
| FoxP3 | Biolegend | 126404 |
| CD69 | Invitrogen | 11-0691-85 |
| PD-1 | Biolegend | 135228 |
| Ki67 | Invitrogen | 58-5698-82 |
| CD11b | Biolegend | 101267 |
| CD11c | BD Biosciences | 749040 |
| GR1 | Biolegend | 108426 |
| Ly6C | Biolegend | 128036 |
| I-A/I-E | Biolegend | 107622 |
| F4/80 | Invitrogen | 17-4801-80 |
| CCR5 | Invitrogen | 46-1951-82 |
| CD103 | BD Biosciences | 564322 |
| PD-L1 | BD Biosciences | 740614 |
| Viability | Invitrogen | 50-112-9035 |
| KLRG1 | BD Biosciences | 746353 |
| CD80 | BD Biosciences | 741956 |
| Fixable Viability Dye eFluor™ 780 | Invitrogen | 50-112-9035 |
| Anti-p53 Antibody (DO-1) | Santacruz Biotechnology | SC-126 |
| Bax Rabbit Antibody | Cell signaling | 2772S |
| Bak Rabbit | Cell signaling | 3814S |
| Puma Rabbit mAb | Cell signaling | 12450S |
| p21 Waf1/Cip1 rabbit antibody | Cell signaling | 2947S |
| β-actin rabbit antibody | Cell signaling | 4970S |
| β-tubulin rabbit antibody | Cell signaling | 2146S |
| HSP90 rabbit antibody | Cell signaling | 4874S |
| Anti-Lamin B1 Antibody (B-10) | Santa Cruz Biotechnology | SC-374015 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, | Invitrogen | A-11012 |
| Bacterial and Virus Strains | ||
| NA | ||
| Biological Samples | ||
| NA | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s modified Eagle’s medium/F12 | Gibco | 11330032 |
| GlutaMAX | Gibco | 35050061 |
| Gibco™ Penicillin-Streptomycin | Gibco | 15-140-122 |
| Fetal Bovine Serum | Gibco | 16000036 |
| B-27™ Supplement (50X) | Gibco | 17504044 |
| DMEM | Gibco | 11965-092 |
| Opti-MEM | Gibco | 31985-070 |
| Crystal violet | Aqua solution, Inc | C8126 |
| Hoechst 33342 | Invitrogen | H3570 |
| Propodium Iodide | Invitrogen | P3566 |
| Calcine AM | Sigma | 564061 |
| Senescence β-Galactosidase Staining Kit | Sigma | CS0030 |
| 5-FU | MedChemexpress | HY-90006 |
| Abemaciclib | MedChemexpress | HT-16297/CS-1229 |
| Alisertib | Adooq Bioscience | A10004 |
| Axitinib | Adooq Bioscience | A10103 |
| AZD 7762 | Cayman chemical company | 11491 |
| Barasertib | Selleckchem | S1147 |
| BIIB 021 | Adooq Bioscience | 848695-25-0 |
| BMS-345541 | Selleckchem | S8044 |
| Bortezomib | Adooq Bioscience | A10160 |
| C-75 | Adooq Bioscience | A15331 |
| Carboplatin | Adooq Bioscience | A10182 |
| CGM097 | Novartis | N/A |
| Choloquinine | Adooq Bioscience | 16445 |
| Cisplatin | Adooq Bioscience | A10221 |
| Danusertib | Adooq Bioscience | A10715 |
| Dasatenib | Adooq Bioscience | BMS-354825 |
| Dinacicclib | Adooq Bioscience | SCH 727965 |
| Doxorubicin | Adooq Bioscience | A14403 |
| Etoposide | Selleckchem | S1225 |
| GNE-9605 | Adooq Bioscience | A14149 |
| HDM201 | Novartis | N/A |
| KU-60019 | Selleckchem | S1570 |
| LY3295668 | Selleckchem | S8782 |
| NU7441 | ApeXbio | A8315 |
| Nutlin-3a | Selleckchem | S8059 |
| NVP-TAE 226 | Adooq Bioscience | A11507 |
| Olaparib | Cayman chemical company | 10621 |
| OSU03012 | Adooq Bioscience | 742112-33-0 |
| Palbociclib | LC Laboratories | P-7766 |
| PU-H71 | Selleckchem | S8039 |
| R1530 | Adooq Bioscience | A13111 |
| RG7388 | Roche | N/A |
| RGF966 | Adooq Bioscience | A13218-10Mm-D |
| Ribociclib | Adooq Bioscience | A22303 |
| Ruxolitinib | Adooq Bioscience | A11041-10Mm-D |
| Temozolomide, 1uM | Santa Cruz Biotechnology | SC-203292 |
| Tideglusib | Adooq Bioscience | A11592 |
| Vemurafenib | Adooq Bioscience | A10739 |
| XMU-MP-1 | Adooq Bioscience | A 16228 |
| DMSO | Sigma | D8418 |
| Paclitaxel | Adooq Bioscience | A10689 |
| Navitoclax | MedChemexpress | HY-10087 |
| Venetoclax | LC Laboratories | V-3579 |
| A1155463 | Adooq Bioscience | A16112 |
| A-1210477 | Adooq Bioscience | A15545 |
| Apoptosis, DNA damage and cell proliferation kit | BD Biosciences | 51-9007685AK |
| FITC Annexin V Apoptosis Detection Kit with PI | BD Biosciences | 556547 |
| JC-1 Mitochondrial Membrane Potential Dye | Thermo Fisher | 3168 |
| Human IL-8 (CXCL8) Mini TMB ELISA Development Kit | Peprotech | 900-TM18 |
| Human IL-6 Mini TMB ELISA Development Kit | Peprotech | 900-TM16 |
| Z-VAD-FMK | Abcam | ab120487 |
| Matrigel, Corning™ Matrigel™ Membrane Matrix | Corning | 354234 |
| Calcein, AM, cell-permeant dye | Thermo fisher Scientific | C1430 |
| Tumor tissue dissociation kit | Miltenyi Biotech | 130-096-730 |
| FOXP3/ Transcription Factor Staining Buffer Set | Invitrogen | 00-5523-00 |
| SYBR-green master mix | BioRad | 1725270 |
| RNA-easy mini kit | Qiagen | 74104 |
| iScript cDNA synthesis kit | BioRad | 1708890 |
| Lipofectamine™ RNAiMAX Transfection Reagent | Invitrogen | 13778-075 |
| Dako TRS, Citrate pH 6 | Agilent Technologies | S236984-2 |
| Triton™ X-100 | Sigma | X100 |
| NEBNext Ultra II Directional RNA Library Prep Kit | Illumina | NEB #E7760 |
| NEBNext rRNA Depletion Kit | Illumina | NEB #E7405 |
| Ethylenediaminetetraacetic acid (EDTA) | Thermo scientific | J15694-AP |
| ACK lysing buffer | KD medical | RGF-3015 |
| Phosphate Buffer saline | corning | 46-013-CM |
| Super Signal West Pico PLUS Chemiluminescent Substrate | Thermo scientific | 34580 |
| Cycloheximide | Sigma | 01810 |
| MG-132 | Adooq | A11043 |
| Laemmli SDS sample buffer | Alfa Aesar | J60015 |
| RIPA buffer | Sigma | R0278 |
| Halt protease and phosphatase single use inhibitor cocktail | Thermo scientific | 78442 |
| 10X Tris/Glycine/SDS Buffer | Bio-Rad | 1610772 |
| Tris Buffer Saline | Fisher Bioreagents | BP2471-1 |
| Tween 20 | Fisher Bioreagents | BP 337-500 |
| Trans Blot Turbo RTA transfer kit | Biorad | 170-4270 |
| Bovine serum Albumin | Fisher Bioreagents | BP9706-100 |
| Protein assay dye reagent concentrate | Bio-Rad | 5000006 |
| Tween 80 | Sigma | P1754 |
| Polyethylene glycol400 | Alfa Aesar | B21992 |
| Saline solution | Growcells | MSDW-1000 |
| Reverse Phase Protein Array (RPPA) | Core Facility at MD Anderson Cancer Center (Houston, TX) | NA |
| Deposited Data | ||
| RNA-seq data | GEO | GSE212127 |
| RPPA data | BioStudies | S-BSST919 |
| Experimental Models: Cell Lines | ||
| A375 | American Type Culture Collection | CRL-1619 |
| Hs294T | American Type Culture Collection | HTB-140 |
| Sk-Mel5 | American Type Culture Collection | HTB-70 |
| B16F10 | American Type Culture Collection | CRL-6475 |
| Sk-MeL28 | American Type Culture Collection | HTB-72 |
| HCT-116 Control | Alexander Zaika (University of Miami) | N/A |
| HCT-116 P53 KO | Alexander Zaika (University of Miami) | N/A |
| HCT-116 P21 KO | Horizon Discovery | HDR02-035 |
| HCT-116 Control | Bert Vogelstein (John Hopkins University) | N/A |
| HCT-116 BAX-KO | Bert Vogelstein (John Hopkins University) | N/A |
| HCT-116 PUMA-KO | Bert Vogelstein (John Hopkins University) | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/Sz | The Jackson Laboratory | 005557 |
| Nude Mice (NU/J) | The Jackson Laboratory | 002019 |
| C57BL/6 mice | Charles river | 556 |
| Oligonucleotides | ||
| Silencer AURKA siRNA | Cell signaling | 8883S |
| Silencer select siRNA BCL2L1 | Invitrogen | 4427038, ID-S196 |
| Signal Silence® p21 Waf1/Cip1 siRNA I | Cell signaling | 6456S |
| Signal Silence® p21 Waf1/Cip1 siRNA II | Cell signaling | 6558S |
| P53 siRNA | Santa Cruz Biotechnology | Sc-29435 |
| Single Silencer Bax si RNA | Cell signaling | 6321S |
| Single Silencer Bak si RNA | Cell signaling | 6486S |
| Single Silencer Control si RNA | Cell signaling | 6568S |
| Single Silencer Puma si RNA | Cell signaling | 12845S |
| Primer M Bax F 5’TGAAGACAGGGGCCTTTTTG | Sigma | N/A |
| Primer M Bax R 5’AATTCGCCGGAGACACTCG | Sigma | N/A |
| Primer CDKN1A F 5’GTCTTGTACCCTTGTGCC | Sigma | N/A |
| Primer CDKN1A R 5’GGTAGAAATCTGTCATGCTGG | Sigma | N/A |
| Software and Algorithms | ||
| ImageJ | NIH Image | https://imagej.nih.gov/ij/index.html |
| Graphpad’s Prism 7.03 software | GraphPad | https://www.graphpad.com/ |
| The Database for Annotation, Visualization, and Integrated Discovery (DAVID) software | Laboratory of Human Retrovirology and Immunoinformatics (LHRI) | https://david.ncifcrf.gov |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus |
| cBioPortal for Cancer Genomics version 3.6.3 | Memorial Sloan Kettering Cancer Center | https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf |
| STATA,version 16 | stataCorp LLC | N/A |
| R (version3.3.0.) | Bioconductor | https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf |
| OMIQ | Dotmatics | https://www.omiq.ai |
| Network creation and Streiner forest (PCSF) network tool | OmicsNet | https://www.omicsnet.ca/ |
| Heat map and principal component analysis | ClustVis | https://biit.cs.ut.ee/clustvis/ |
| Other | ||
| N/A | ||
Acknowledgments
The authors would like to thank Kerry Vazquez for administrative support, Kevin Weller at the OSU IMDP for training in spectral cytometry, and Pearlly Yan and Kyle Shin for processing RNAseq data. This work was supported by the grants from NIH (R37 CA233770–01, to AEV; CA116021and CA116021-S1 to AR; NCI K12CA090625 to VLW; NCI R01CA272875 to VLW; and 1K08 CA240901–01A1 to VLW), BCRF (IIDRP-16–001 to AEV), DOD (W81XWH2210019 to AEV) the Department of Veterans Affairs (5101BX000196–04 to AR; and Senior Research Career Scientist Award, to AR), and the ACS (133934-CSDG-19–216-01-TBG and RSG-22–084-01-MM to VLW). Support for VICC and OSU core facilities used in this study was provided by P30 CA68485 and P30CA016058, respectively. Library preparation and sequencing performed by OSUCCC Genomics Shared Resource (Supported by P30CA016058).
Footnotes
Conflict of Interest
The authors declare no competing interests.
References
- 1.Patel H, Yacoub N, Mishra R, White A, Long Y, Alanazi S, and Garrett JT (2020). Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers (Basel) 12. 10.3390/cancers12020482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. (2017). Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390, 1853–1862. 10.1016/S0140-6736(17)31601-X. [DOI] [PubMed] [Google Scholar]
- 3.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369, 122–133. 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jhappan C, Noonan FP, and Merlino G (2003). Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 22, 3099–3112. 10.1038/sj.onc.1206450. [DOI] [PubMed] [Google Scholar]
- 5.Anvekar RA, Asciolla JJ, Missert DJ, and Chipuk JE (2011). Born to be alive: a role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front Oncol 1. 10.3389/fonc.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kale J, Osterlund EJ, and Andrews DW (2018). BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ 25, 65–80. 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soengas MS, and Lowe SW (2003). Apoptosis and melanoma chemoresistance. Oncogene 22, 3138–3151. 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Kohli J, and Demaria M (2020). Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. 10.1016/j.trecan.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Ewald JA, Desotelle JA, Wilding G, and Jarrard DF (2010). Therapy-induced senescence in cancer. J Natl Cancer Inst 102, 1536–1546. 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velarde MC, Demaria M, and Campisi J (2013). Senescent cells and their secretory phenotype as targets for cancer therapy. Interdisciplinary topics in gerontology 38, 17–27. 10.1159/000343572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagiv A, and Krizhanovsky V (2013). Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 14, 617–628. 10.1007/s10522-013-9473-0. [DOI] [PubMed] [Google Scholar]
- 12.Perez-Mancera PA, Young AR, and Narita M (2014). Inside and out: the activities of senescence in cancer. Nat Rev Cancer 14, 547–558. 10.1038/nrc3773. [DOI] [PubMed] [Google Scholar]
- 13.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 7, 165–176. 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun X, Shi B, Zheng H, Min L, Yang J, Li X, Liao X, Huang W, Zhang M, Xu S, et al. (2018). Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis 9, 260. 10.1038/s41419-018-0303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dean JL, McClendon AK, and Knudsen ES (2012). Modification of the DNA damage response by therapeutic CDK4/6 inhibition. The Journal of biological chemistry 287, 29075–29087. 10.1074/jbc.M112.365494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, and Pillai MR (2011). Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. The Journal of biological chemistry 286, 37813–37829. 10.1074/jbc.M110.200675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, et al. (2015). Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res 28, 184–195. 10.1111/pcmr.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, George J, Danson S, and Kirkland JL (2020). Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers (Basel) 12. 10.3390/cancers12082134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niedernhofer LJ, and Robbins PD (2018). Senotherapeutics for healthy ageing. Nat Rev Drug Discov 17, 377. 10.1038/nrd.2018.44. [DOI] [PubMed] [Google Scholar]
- 20.Kirkland JL, and Tchkonia T (2017). Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28. 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435. 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Deursen JM (2019). Senolytic therapies for healthy longevity. Science 364, 636–637. 10.1126/science.aaw1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Hawkins OE, Su Y, Vilgelm AE, Sobolik T, Thu YM, Kantrow S, Splittgerber RC, Short S, Amiri KI, et al. (2013). Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence. EMBO Mol Med 5, 149–166. 10.1002/emmm.201201378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilgelm AE, Johnson CA, Prasad N, Yang J, Chen SC, Ayers GD, Pawlikowski JS, Raman D, Sosman JA, Kelley M, et al. (2016). Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. J Natl Cancer Inst 108, djv406. 10.1093/jnci/djv406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, et al. (2015). Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 75, 181–193. 10.1158/0008-5472.CAN-14-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilgelm AE, Cobb P, Malikayil K, Flaherty D, Andrew Johnson C, Raman D, Saleh N, Higgins B, Vara BA, Johnston JN, et al. (2017). MDM2 Antagonists Counteract Drug-Induced DNA Damage. EBioMedicine 24, 43–55. 10.1016/j.ebiom.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson MA, Mahoney MR, Tap WD, D’Angelo SP, Keohan ML, Van Tine BA, Agulnik M, Horvath LE, Nair JS, and Schwartz GK (2016). Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann Oncol 27, 1855–1860. 10.1093/annonc/mdw281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, Jung J, Burack R, Zhou X, Leonard EJ, et al. (2014). Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol 32, 44–50. 10.1200/JCO.2012.46.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X, et al. (2012). Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol 127, 63–69. 10.1016/j.ygyno.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 30.Barr PM, Li H, Spier C, Mahadevan D, LeBlanc M, Ul Haq M, Huber BD, Flowers CR, Wagner-Johnston ND, Horwitz SM, et al. (2015). Phase II Intergroup Trial of Alisertib in Relapsed and Refractory Peripheral T-Cell Lymphoma and Transformed Mycosis Fungoides: SWOG 1108. J Clin Oncol 33, 2399–2404. 10.1200/JCO.2014.60.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haddad TC, D’Assoro A, Suman V, Opyrchal M, Peethambaram P, Liu MC, Goetz MP, and Ingle JN (2018). Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine-resistant, ER+ metastatic breast cancer. Breast Cancer Res Treat 168, 639–647. 10.1007/s10549-017-4616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tayyar Y, Jubair L, Fallaha S, and McMillan NAJ (2017). Critical risk-benefit assessment of the novel anti-cancer aurora a kinase inhibitor alisertib (MLN8237): A comprehensive review of the clinical data. Crit Rev Oncol Hematol 119, 59–65. 10.1016/j.critrevonc.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Borisa AC, and Bhatt HG (2017). A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur J Med Chem 140, 1–19. 10.1016/j.ejmech.2017.08.045. [DOI] [PubMed] [Google Scholar]
- 34.Sun MG, Williams J, Munoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, and Frey TG (2007). Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol 9, 1057–1065. 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- 35.Tait SW, and Green DR (2013). Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol 5. 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilgelm AE, Bergdorf K, Wolf M, Bharti V, Shattuck-Brandt R, Blevins A, Jones C, Phifer C, Lee M, Lowe C, et al. (2020). Fine-Needle Aspiration-Based Patient-Derived Cancer Organoids. iScience 23. 10.1016/j.isci.2020.101408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phifer CJ, Bergdorf KN, Bechard ME, Vilgelm A, Baregamian N, McDonald OG, Lee E, and Weiss VL (2021). Obtaining patient-derived cancer organoid cultures via fine-needle aspiration. STAR Protoc 2, 100220. 10.1016/j.xpro.2020.100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergdorf K, Phifer C, Bharti V, Westover D, Bauer J, Vilgelm A, Lee E, and Weiss V (2020). High-throughput drug screening of fine-needle aspiration-derived cancer organoids. STAR Protoc 1, 100212. 10.1016/j.xpro.2020.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shattuck-Brandt RL, Chen SC, Murray E, Johnson CA, Crandall H, O’Neal JF, Al-Rohil RN, Nebhan CA, Bharti V, Dahlman KB, et al. (2020). Metastatic Melanoma Patient-Derived Xenografts Respond to MDM2 Inhibition as a Single Agent or in Combination with BRAF/MEK Inhibition. Clin Cancer Res. 10.1158/1078-0432.CCR-19-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vilgelm AE, Saleh N, Shattuck-Brandt R, Riemenschneider K, Slesur L, Chen SC, Johnson CA, Yang J, Blevins A, Yan C, et al. (2019). MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci Transl Med 11. 10.1126/scitranslmed.aav7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vergnes L, Peterfy M, Bergo MO, Young SG, and Reue K (2004). Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 101, 10428–10433. 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Righolt CH, Zatreanu DA, and Raz V (2013). Quantification of the Spatial Organization of the Nuclear Lamina as a Tool for Cell Classification. ISRN Mol Biol 2013, 374385. 10.1155/2013/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matias I, Diniz LP, Damico IV, Araujo APB, Neves LDS, Vargas G, Leite REP, Suemoto CK, Nitrini R, Jacob-Filho W, et al. (2022). Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 21, e13521. 10.1111/acel.13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freund A, Laberge RM, Demaria M, and Campisi J (2012). Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23, 2066–2075. 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Vos S, Leonard JP, Friedberg JW, Zain J, Dunleavy K, Humerickhouse R, Hayslip J, Pesko J, and Wilson WH (2021). Safety and efficacy of navitoclax, a BCL-2 and BCL-XL inhibitor, in patients with relapsed or refractory lymphoid malignancies: results from a phase 2a study. Leuk Lymphoma 62, 810–818. 10.1080/10428194.2020.1845332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, et al. (2010). Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol 11, 1149–1159. 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gervais JL, Seth P, and Zhang H (1998). Cleavage of CDK inhibitor p21(Cip1/Waf1) by caspases is an early event during DNA damage-induced apoptosis. J Biol Chem 273, 19207–19212. 10.1074/jbc.273.30.19207. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Fujita N, and Tsuruo T (1999). Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 18, 1131–1138. 10.1038/sj.onc.1202426. [DOI] [PubMed] [Google Scholar]
- 49.Tyagi A, Singh RP, Agarwal C, and Agarwal R (2006). Silibinin activates p53-caspase 2 pathway and causes caspase-mediated cleavage of Cip1/p21 in apoptosis induction in bladder transitional-cell papilloma RT4 cells: evidence for a regulatory loop between p53 and caspase 2. Carcinogenesis 27, 2269–2280. 10.1093/carcin/bgl098. [DOI] [PubMed] [Google Scholar]
- 50.Moosavi MA, and Yazdanparast R (2008). Distinct MAPK signaling pathways, p21 up-regulation and caspase-mediated p21 cleavage establishes the fate of U937 cells exposed to 3-hydrogenkwadaphnin: differentiation versus apoptosis. Toxicol Appl Pharmacol 230, 86–96. 10.1016/j.taap.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Cosentino K, and Garcia-Saez AJ (2017). Bax and Bak Pores: Are We Closing the Circle? Trends Cell Biol 27, 266–275. 10.1016/j.tcb.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Letai A, Bhola P, and Welm AL (2022). Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 40, 26–35. 10.1016/j.ccell.2021.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao H, Korn JM, Ferretti S, Monahan JE, Wang YZ, Singh M, Zhang C, Schnell C, Yang GZ, Zhang Y, et al. (2015). High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 21, 1318–1325. 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 54.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nature medicine 17, 1514–1520. nm.2454 [pii] 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao X, Liu Z, Yu L, Zhang Y, Baxter P, Voicu H, Gurusiddappa S, Luan J, Su JM, Leung HC, and Li XN Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro-Oncology 14, 574–583. nos061 [pii] 10.1093/neuonc/nos061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krepler C, Xiao M, Spoesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, et al. (2015). Personalized pre-clinical trials in BRAF inhibitor resistant patient derived xenograft models identify second line combination therapies. Clinical Cancer Research. 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerbel RS (2003). Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: better than commonly perceived-but they can be improved. Cancer biology & therapy 2, S134–139. [PubMed] [Google Scholar]
- 58.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, Kalyandrug S, Christian M, Arbuck S, Hollingshead M, and Sausville EA (2001). Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. British journal of cancer 84, 1424–1431. 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scholz CC, Berger DP, Winterhalter BR, Henss H, and Fiebig HH (1990). Correlation of drug response in patients and in the clonogenic assay with solid human tumour xenografts. Eur J Cancer 26, 901–905. [DOI] [PubMed] [Google Scholar]
- 60.Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, Schipper L, Hoes L, Vis DJ, van de Haar J, et al. (2019). Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med 11. ARTN eaay2574 10.1126/scitranslmed.aay2574. [DOI] [PubMed] [Google Scholar]
- 61.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lambrus BG, and Holland AJ (2017). A New Mode of Mitotic Surveillance. Trends Cell Biol 27, 314–321. 10.1016/j.tcb.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haschka M, Karbon G, Fava LL, and Villunger A (2018). Perturbing mitosis for anti-cancer therapy: is cell death the only answer? EMBO Rep 19. 10.15252/embr.201745440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, and Oegema K (2016). 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214, 155–166. 10.1083/jcb.201604081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, and Holland AJ (2016). A USP28–53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214, 143–153. 10.1083/jcb.201604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fong CS, Mazo G, Das T, Goodman J, Kim M, O’Rourke BP, Izquierdo D, and Tsou MF (2016). 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5. 10.7554/eLife.16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gascoigne KE, and Taylor SS (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122. 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 68.Huang HC, Shi J, Orth JD, and Mitchison TJ (2009). Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 16, 347–358. 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sinha D, Duijf PHG, and Khanna KK (2019). Mitotic slippage: an old tale with a new twist. Cell Cycle 18, 7–15. 10.1080/15384101.2018.1559557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A, and Fava LL (2015). The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun 6, 6891. 10.1038/ncomms7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bennett A, Sloss O, Topham C, Nelson L, Tighe A, and Taylor SS (2016). Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol 6. 10.1098/rsob.160134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi J, Zhou Y, Huang HC, and Mitchison TJ (2011). Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res 71, 4518–4526. 10.1158/0008-5472.CAN-10-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vousden KH, and Lane DP (2007). p53 in health and disease. Nat Rev Mol Cell Biol 8, 275–283. 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 74.Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, Desprez PY, Benz C, Davalos AR, and Campisi J (2018). Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep 8, 2410. 10.1038/s41598-018-20000-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hafner A, Bulyk ML, Jambhekar A, and Lahav G (2019). The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol 20, 199–210. 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- 76.Sanchez-Rivera FJ, Ryan J, Soto-Feliciano YM, Clare Beytagh M, Xuan L, Feldser DM, Hemann MT, Zamudio J, Dimitrova N, Letai A, and Jacks T (2021). Mitochondrial apoptotic priming is a key determinant of cell fate upon p53 restoration. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2019740118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakajima YI, and Kuranaga E (2017). Caspase-dependent non-apoptotic processes in development. Cell Death Differ 24, 1422–1430. 10.1038/cdd.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feinstein-Rotkopf Y, and Arama E (2009). Can’t live without them, can live with them: roles of caspases during vital cellular processes. Apoptosis 14, 980–995. 10.1007/s10495-009-0346-6. [DOI] [PubMed] [Google Scholar]
- 79.Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM, Tavernarakis N, Penninger J, Madeo F, and Kroemer G (2008). No death without life: vital functions of apoptotic effectors. Cell Death Differ 15, 1113–1123. 10.1038/cdd.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galluzzi L, Kepp O, Trojel-Hansen C, and Kroemer G (2012). Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep 13, 322–330. 10.1038/embor.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuranaga E (2012). Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells 17, 83–97. 10.1111/j.1365-2443.2011.01579.x. [DOI] [PubMed] [Google Scholar]
- 82.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, and Vandenabeele P (2007). Caspases in cell survival, proliferation and differentiation. Cell Death Differ 14, 44–55. 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 83.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, and Moll UM (2012). p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548. 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, and Green DR (2005). PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309, 1732–1735. 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 85.Green DR, and Kroemer G (2009). Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130. 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kalkavan H, and Green DR (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25, 46–55. 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 43, W566–570. 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 89.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, and Kornblau SM (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5, 2512–2521. 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Drug screen results. Related to Figure 1.
Data Availability Statement
Access to the original data. RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession number is GSE212127. RPPA proteomics data have been deposited to BioStudies. Accession number is S-BSST919.
Access to the original code. This paper does not report original code.
Access to any additional information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.