

Abstract
X-linked adrenoleukodystrophy (ALD) is a rare inherited neurological disorder that poses considerable challenges for clinical management throughout the lifespan. Although males are generally more severely affected than females, the time course and presentation of clinical symptoms is otherwise difficult to predict. Opportunities to improve outcomes for individuals with ALD are rapidly expanding due to the introduction of newborn screening programs for this condition and an evolving treatment landscape. The aim of this comprehensive review is to synthesize current knowledge regarding the neurocognitive and mental health effects of ALD. This review provides investigators and clinicians with context to improve case conceptualization, inform prognostic counseling, and optimize neuropsychological and mental health care for patients and their families. Results highlight key predictive factors and brain-behavior relationships associated with the diverse manifestations of ALD. The review also discusses considerations for endpoints within clinical trials and identifies gaps to address in future research.
Keywords: adrenomyeloneuropathy, adrenal insufficiency, neuropsychology, behavior, psychiatric, assessment
1. Introduction
X-linked adrenoleukodystrophy (ALD) is a rare neurologic disease with highly variable clinical presentation. Although earlier epidemiological studies estimated the incidence of ALD at 1:16,800 births,1 newborn screening (NBS) programs in several states provide a range of updated estimates from 1:3,878 to 1:51,081 births.2–6 ALD is caused by pathogenic variants in the ABCD1 gene (OMIM 300371) located on Xq28, resulting in accumulation of very-long chain fatty acids in tissues and plasma that can contribute to a diverse set of phenotypes that evolve across the lifespan.
Clinical expression of ALD is more severe in males than in females. Specifically, most males develop adrenal insufficiency requiring lifelong monitoring.7 About 35–40% of boys with ALD develop a severe childhood cerebral form characterized by rapidly progressing brain inflammation and demyelination.8 Risk for developing childhood cerebral ALD (CCALD) is highest between 3 and 12 years of age.9 CCALD can be halted by treatment with hematopoietic cell transplantation (HCT), with better survival and reduced neurologic disability if performed earlier in the disease course.10,11 While roughly two-thirds of males with ALD reach adulthood without cerebral disease, they are at risk for developing an adult form of cerebral ALD (ACALD). Additionally, nearly all males with ALD and some females are at risk for developing myeloneuropathy due to spinal cord disease later in life. This symptom presentation, often referred to as adrenomyeloneuropathy (AMN) in males, presents with slowly progressing myelopathy and peripheral neuropathy, with symptoms including sphincter dysfunction, muscle stiffness and weakness in the legs, and neuropathic pain. As myeloneuropathy progresses, it typically leads to the development of functional disabilities such as the need for assistance in walking.
Aside from sex differences in symptom presentation, it is not currently possible to predict which manifestations of ALD will develop based on genotype or other biomarkers.12–14 The same ABCD1 variant can result in remarkably divergent clinical courses. Once clinical signs of cerebral ALD emerge, the disease is often in advanced stages, with demyelinating lesions affecting multiple brain regions. There is limited treatment efficacy for patients with advanced disease since HCT does not reverse the injury to cerebral myelin and may even accelerate disease progression.10 The dramatic difference in outcomes associated with earlier treatment, along with advocacy from affected families, prompted the development of NBS programs for ALD and the addition of ALD to the federal Recommended Uniform Screening Panel in 2016.15 As of December 2022, a total of 30 U.S. states and the District of Columbia had begun screening male and female newborns for ALD; most states have been screening for less than 5 years.16
As more individuals with ALD are identified through NBS and have the opportunity to receive earlier management and treatment, consideration of the long-term neurocognitive and mental health effects of ALD grows increasingly relevant. Along with NBS, improvements in HCT as well as newer treatments (e.g., gene therapy) hold promise to continue to improve the outlook for patients with cerebral ALD.17 These major advances have enabled a shift in focus from methods to keep patients alive and prevent neurological devastation toward goals such as maximizing neurocognitive function, mental health and quality of life. This review aims to integrate the literature reporting on these important functional outcomes among ALD patients. We highlight key brain-behavior connections and discuss the role of neurocognitive measurement in predicting and evaluating treatment outcomes. This review also provides clinicians with recommendations for improving interdisciplinary patient care based on literature findings, reveals gaps in the literature, and identifies needs for further research.
Methods
2.1. Search Strategy
A medical librarian created the literature search strategy after clarifying goals and defining selection criteria with the research team. The search strategy was built in Ovid MEDLINE using Medical subject headings (MeSH) and keywords (Supplemental Table 1); no limits were applied to database searches. Hand searching with review of relevant conference proceedings and reference lists from published articles was used as a supplement. The initial search was completed in February 2021 and updated in October 2022.
2.2. Selection Criteria
Studies were included if they met the following criteria: 1) peer-reviewed; 2) empirical study; 3) contained three or more patients with a clinical diagnosis of ALD; and 4) included quantitative measures of neuropsychological function. Quantitative measures included neurocognitive testing across a wide range of domains (e.g., intellectual functioning, language, memory, attention, etc.); standardized patient, caregiver or clinician measures of emotional or psychiatric symptoms; and questionnaires assessing adaptive functioning or quality of life. Studies were excluded if they were review articles or were not available in English. Studies that focused solely on neurologic symptoms/dysfunction, neuroimaging findings, or self-report of cognitive complaints were not included.
2.3. Search Results
Duplicate references were removed and 308 abstracts were uploaded to Rayyan18 for independent screening by two researchers (EIP and EM). Studies selected for full-text review were obtained as PDFs and reviewed by the research team. Conflicts were resolved by consensus. Thirty-two group studies (>3 patients) met inclusion criteria (Table 1). Additional case reports and references were identified during the full text phase that did not meet inclusion criteria but contained pertinent information, and are integrated in this review where relevant data were unavailable within the selected studies.
Table 1.
Published cohort studies measuring neurocognition and mental health in individuals with ALD
| Publication | n | Age Range (years) | Gender | Phenotype | Therapy | Neuropsychological Domains Assessed |
|---|---|---|---|---|---|---|
| Beam et al. (2007) | 12 | 0–15 | M | CCALD | HCT | Intellectual abilities+, adaptive functioning, language, motor |
| Beckmann et al. (2018) | 16 | 8.0–20.0 | M | CCALD | HCT | Quality of life |
| Bougnères et al. (2021) | 4 | 3.5–19.5 | M | CCALD | GT | Intellectual abilities+ |
| Buermans et al. (2019) | 33 | 19–71 | M | Adult asymptomatic ALD | -- | Language, memory, visual-motor/visual construction, executive functions, psychomotor speed |
| Cox et al. (2006) | 52 | 2.1–14.6 | M | Child asymptomatic ALD | -- | Intellectual abilities+, adaptive functioning, language, academic achievement, visual/perceptual abilities, visual-motor/visual construction, memory, attention, executive functions |
| Edwin et al. (1990) | 57 | M = 38.2, SD = 12.4 | M/F | AMN, ACALD, females with ALD | -- | Intellectual abilities+, attention, executive functions, language, motor, facial recognition, visuospatial judgment, memory |
| Edwin et al. (1996) | 84 | M = 33.7, SD = 10.3 | M | AMN, ACALD | -- | Intellectual abilities+, language, motor, fine motor, facial recognition, visuospatial judgment, visual construction, learning and memory, executive functions |
| Engelen et al. (2012) | 46 | 22–76 | F | Females with ALD | -- | Quality of life, psychiatric symptoms |
| Furushima et al. (2009) | 6 | 6.3–14.8 | M | Child asymptomatic ALD | -- | Intellectual abilities+, visual/perceptual abilities, visual construction |
| Furushima et al. (2015) | 3 | 10–13 | M | CCALD | -- | Intellectual abilities+ |
| Gassas et al. (2011) | 6 | 5.3–13.3 | M | CCALD | HCT | Adaptive functioning, psychiatric symptoms |
| Gupta et al. (2021) | 14 | 5–16 | M | CCALD | HCT | Intellectual abilities+, psychiatric symptoms |
| Kaga et al. (2009) | 8 | 3.3–14.6 | M | Child asymptomatic ALD | -- | Intellectual abilities+, visual/perceptual abilities, visual construction |
| Kühl et al. (2017) | 14 | 21–48 | M | ACALD | HCT | Intellectual abilities+ |
| Kühl et al. (2018) | 36 | 4.2–15.4 | M | CCALD | HCT | Intellectual abilities+ |
| Lyon-Caen et al. (1991) | 6 | 31–59 | M/F | AMN, ACALD, females with ALD | -- | Intellectual abilities+, language, memory |
| Matsukawa et al. (2020) | 12* | 18–45 | M | Adolescent CALD, ACALD, AMN | HCT | Intellectual abilities+, activities of daily living |
| McKinney et al. (2013) | 8 | M = 7.9, SD = 1.55 | M | CCALD | HCT | Intellectual abilities+, adaptive functioning |
| McKinney et al. (2016) | 10 | 5–14 | M | CCALD | HCT | Intellectual abilities+ |
| Miller et al. (2011) | 60 | 4–23.3 | M | CCALD | HCT | Intellectual abilities+ |
| Moser et al. (1991) | 372 | < 3 to adulthood | M | All ALD phenotypes | Some HCT | Intellectual abilities+, language, visual/perceptual abilities, visual construction, visual-motor, facial recognition, visuospatial judgment, fine motor, learning and memory, attention, executive functions |
| Peters et al. (2004) | 94 | 4.9–18.6 | M | CCALD | HCT | Intellectual abilities+ |
| Pierpont et al. (2017) | 62 | 4–16 | M | CCALD | HCT | Intellectual abilities+, attention, learning and memory, fine motor, visual-motor |
| Pierpont et al. (2018) | 65 | 4–16 | M | CCALD | HCT | Intellectual abilities+, fine motor, visual-motor, adaptive functioning |
| Pierpont et al. (2020) | 36 | 4.0–16.1 | M | CCALD | HCT | Intellectual abilities+, fine motor, visual-motor, psychiatric symptoms |
| Riva et al. (2000) | 15 | 5.6–17.3 | M | CCALD, child asymptomatic ALD | -- | Intellectual abilities+, executive functions, language, visual/perceptual abilities, visual-motor, working memory |
| Salsano et al. (2014) | 12 | 24–62 | M | Adolescent CALD, ACALD, AMN, AI, and asymptomatic adults | -- | Attention, executive functions, memory, working memory, visual/perceptual abilities, visual-motor, visual construction, abstract reasoning |
| Schäfer et al. (2021) | 172 | 18–80 | M/F | AMN, ACALD, arrested ACALD | -- | Intellectual abilities+, attention, learning and memory, executive functions, processing speed |
| Shapiro et al. (1995) | 36 | 5–15 | M | CCALD | HCT | Intellectual abilities+, auditory processing, visual/perceptual abilities, language, memory, motor, attention, executive functions, academic achievement |
| Shapiro et al. (2000) | 12 | 3.6–18.2 | M | CCALD | HCT | Intellectual abilities+, auditory processing, visual/perceptual abilities, language, memory, motor, attention, processing speed, academic achievement |
| Tran et al. (2017) | 8* | 2.1–17 | M | Adrenal insufficiency, CCALD | Some HCT | Intellectual abilities+, visual-motor, psychiatric symptoms |
| Walterfang et al. (2007) | 10 | 27.3–56.3 | M/F | AMN | -- | Attention, memory, language, executive function, visual/perceptual abilities, psychiatric symptoms |
Note: AMN, adrenomyeloneuropathy; CCALD, childhood cerebral adrenoleukodystrophy; ACALD, adult cerebral adrenoleukodystrophy (may also be referred to as “cerebral AMN”); AI, adrenal insufficiency; ALD, adrenoleukodystrophy; HCT, hematopoietic cell transplantation (allogeneic); GT, gene therapy (autologous)
These numbers reflect a sub-cohort of patients within a larger study for whom neuropsychological data were reported
Intellectual abilities refers to a broad set of neurocognitive abilities conventionally measured on IQ tests, which may be further separated into more specific domains (e.g., verbal reasoning, visual reasoning, fluid reasoning, working memory, processing speed)
Results were categorized by the phenotypes that can be expressed in individuals with ALD at different life stages. Although the majority of published studies reporting on neuropsychological functioning have focused on children and adolescents, studies of adults with ALD were also included to provide lifespan developmental context. Since the experiences of males and females differ considerably, they are discussed separately. In addition to the neuropsychological burden that ALD places on patients, it is also critical to acknowledge the impact of this disease on family members. Therefore, a final section briefly reviews issues related to the impact of ALD on the broader caregiver and sibling support system.
3. Males with ALD
3.1. Neurodevelopment in early childhood
In the absence of demyelinating lesions, neurodevelopment in infancy and early childhood has been presumed to be normal in males with ALD.19,20 Overt neurological signs attributable to CCALD are rarely evident prior to 3 years of age, with 19 months reported as the earliest known age of radiological disease onset.21,22 In a few reported cases, neurodevelopmental or neuropsychiatric symptomatology (e.g., autism spectrum disorder, anxiety, inattention) were present at an early age and preceded neurological symptoms by some years.23,24 However, no studies have prospectively examined whether there is greater susceptibility to these symptoms among individuals with ALD in the absence of cerebral lesions, or if these cases simply reflect the expected rate of neurodevelopmental disorders in the general population.
Boys with ALD who have not developed white matter lesions or neurological symptoms of CCALD are sometimes described as neurologically “asymptomatic,” although this could also be considered a “pre-symptomatic” period as patients may subsequently develop any combination of adrenal insufficiency, cerebral disease and/or spinal cord pathology. Four studies have examined neurocognitive function in neurologically asymptomatic boys with ALD in early to middle childhood. In the largest of these studies, Cox and colleagues reported that a substantial majority of boys with ALD who had normal MRIs also scored within normal limits (defined as > −2SD) across IQ, academic, and adaptive measures as well as five other neurocognitive domains.25 Notably, while this study’s methodology of creating summary scores from multiple tests within each domain likely reduced spurious findings from single tests, it also may have reduced the sensitivity of the study to detect subtle deficits (e.g., scores that were mildly below average or were a reduction from previous scores) or more selective deficits (e.g., low scores within a more specific domain of function).
Among a smaller group of neurologically asymptomatic boys with ALD who had no evidence of CCALD on MRI, deficits on tasks of visual perception and reasoning were noted, as well as associated neurophysiological differences (i.e., an increased amplitude of visual evoked potentials).26,27 Riva and colleagues reported that boys with neurologically asymptomatic ALD exhibited deficits relative to normative samples on a test of word retrieval and scored low average on a test of verbal fluency.28 Notably, most boys (7 of 8) in this asymptomatic group had some white matter findings on MRI, and 6 patients later experienced progression of cerebral disease. Consequently, the observed deficits reported in this study may represent early manifestations of cerebral disease rather than pre-clinical markers.
3.2. Childhood onset cerebral adrenoleukodystrophy (CCALD)
3.2.1. Onset of cerebral disease
A recent meta-analysis reported the median age of diagnosis of CCALD as 7 years, with 90% of cases identified between ages 3 and 12 years of age.29 When reviewing neuropsychological outcomes, the variability in the location of cerebral lesions is an important consideration, as different regions govern different skill areas. Disease most often initiates in the splenium of the corpus callosum with further advancement into parietal-occipital white matter (~80% of cases); the genu of the corpus callosum with subsequent frontal white matter involvement (15–17%); or rarely, in another location such as the projection fibers, cerebellum or basal ganglia (<5%).30–32 Lesions arising in the corpus callosum can progress rapidly, particularly in younger patients, although in a small subset of patients cerebral disease spontaneously arrests after an initial period of deterioration.31,33 Severity of demyelination in CCALD has conventionally been measured using the Loes score,34 a rating system that quantifies the location and extent of cerebral lesions on magnetic resonance imaging (MRI) using a 0–34 point scale. Scores of 10 or higher have often been considered “high risk” for poorer treatment response and outcomes.10
Due to a lack of prospective longitudinal studies, existing data regarding neurocognitive changes associated with disease onset are extrapolated from retrospective observations of patients who have already developed cerebral disease. When prospective surveillance enables identification of CCALD at a very early stage (Loes score ≤ 2), neurocognitive deficits are typically absent or subtle (i.e., mildly abnormal) on standard neurocognitive testing.35 Beyond this stage, deficits can commonly be measured in neurocognitive domains that depend on efficient white matter connections or on posterior brain regions, including processing speed, sustained attention, visual perception and reasoning, fine motor speed and dexterity, and verbal fluency.26,36,37 Across a variety of neurocognitive measures, the degree of deficit is associated with the severity of the white matter abnormality on MRI (Figure 1).33 Behavioral concerns may also develop at an early stage; caregiver ratings of psychiatric symptomatology may reflect concerns regarding inattention, hyperactivity and impulsive behaviors.35 Other neurocognitive functions such as language, memory and working memory, which rely more substantially on frontal and temporal cortex as well as striatal circuits,38 may not reflect the extent of cerebral involvement at the outset of disease.37
Figure 1.
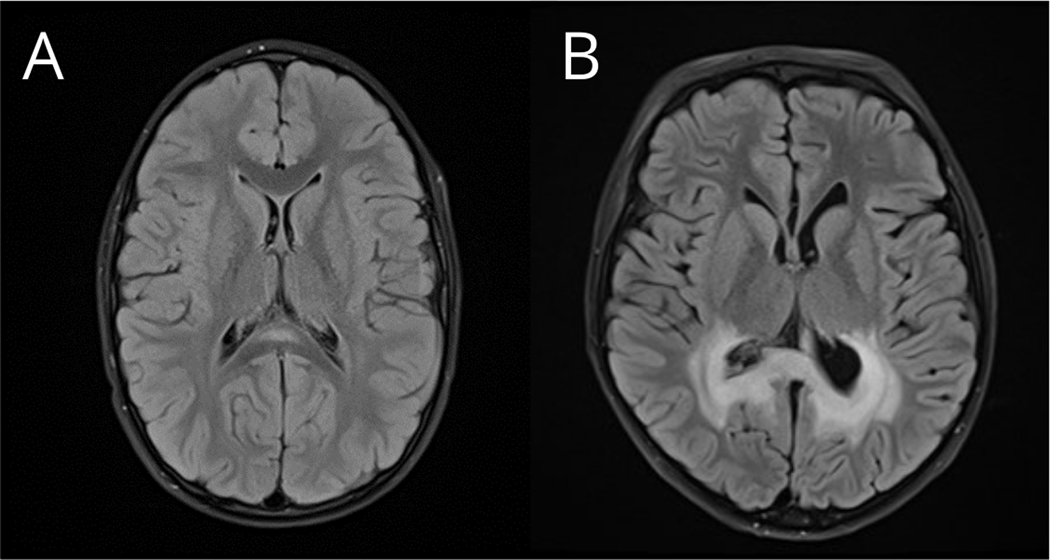
A) T2 axial FLAIR brain MRI showing a small region of demyelination in the splenium of the corpus callosum (Loes score = 1). This patient with CCALD showed no clinical deficits on neuropsychological testing when examined prior to treatment at 7.4 years of age. B) T2 axial FLAIR brain MRI from another patient with CCALD showing a larger region of demyelination involving parietal-occipital white matter (Loes score = 7). This patient showed clinical neuropsychological deficits including slowed processing speed and moderate visual-motor impairment when evaluated prior to treatment at 7.9 years of age.
In the absence of prospective surveillance, cerebral disease onset may not be detected until disease is more advanced. Case studies indicate that initial clinical symptoms are often non-specific and mimic behavioral or emotional disorders such as attention deficit hyperactivity disorder (ADHD) and anxiety.19,39 As a result, misdiagnosis is common and contributes to delays in disease identification in communities without NBS. In some instances, symptoms are first noted at school, such as problems with spatial orientation, hand-eye coordination, and difficulty learning and participating in academic work.19 Cases have been reported where auditory processing issues were the first noticeable clinical sign of CCALD.40 Boys may also present with heightened emotional lability, including irritability, agitation and oppositionality.19,41,42 Symptoms such as sleep disturbance, blunted affect, reduced spontaneous speech, and poor eye contact have also been described in case reports as preceding neurocognitive decline.43
3.2.2. Neuropsychological outcomes after allogeneic HCT for CCALD
HCT has been effective in treating CCALD since the early 1990s and is presently the standard of care.44,45 Successful treatment typically halts the long-term advancement of cerebral demyelination, although it is common to observe some degree of clinical progression in the months following HCT as disease stabilizes.11,32,46,47 In terms of neurocognitive outcomes, studies have consistently demonstrated that HCT performed early in the disease process results in better preservation of abilities. Shapiro and colleagues were the first to report long-term neurocognitive outcomes after HCT among a set of 8 boys with CCALD treated before disease was advanced.46 Followed 5 to 10 years from HCT, these patients showed generally stable scores across a variety of neuropsychological measures. A study of long-term functional status found that when patients are treated with HCT at an early stage, outcomes such as school completion, employment and independence in daily living activities can be attainable.48
Several recent studies expand upon these initial findings and illustrate a fairly narrow window of opportunity to perform HCT without risk for persistent neurocognitive impairment. Pierpont et al. analyzed long-term outcomes of 62 patients who received HCT at a relatively early stage of CCALD considered “standard risk” (Loes < 10).37 Nearly all patients survived with relatively few major neurological disabilities (e.g., spastic gait, swallowing difficulty). Nevertheless, at the most recent follow up evaluation (median of 4.2 years after HCT), 67% of patients had developed severe impairment in at least one neurocognitive domain. Higher pre-HCT Loes scores were associated with lower neurocognitive test scores at the time of transplant as well greater risk for a declining performance trajectory in the 5 years following treatment. A subsequent study focused on a subset of patients who, due to knowledge of their ALD status early in life, were treated with lower severity disease (Loes ≤ 5).35 Most boys whose cerebral disease was limited to the corpus callosum and immediately adjacent white matter (i.e., Loes ≤ 2) at the time of HCT functioned within normal limits on neurocognitive and psychiatric measures administered 2 years after HCT. Performance in this sub-group was demonstrably better on tests of processing speed and fine motor dexterity than those with a greater extent of CCALD (Loes 2.5–4.5). Those with the smallest lesions also showed fewer symptoms of inattention and hyperactivity on psychiatric rating scales, underscoring the importance of early detection of very small demyelinating lesions and rapid intervention for preserving mental health as well as cognition.
Because HCT does not repair damaged myelin, neurocognitive and psychiatric impairments acquired in the pre-HCT period do not routinely improve after treatment.37 Functions compromised by the existing white matter injury can interfere with acquisition of new knowledge or skills, resulting in worsening of standardized neurocognitive test scores over time (i.e., falling further behind healthy same-aged peers). This pattern has been frequently observed in patients when CCALD lesions extend into cerebral cortex.35,37 In the context of successful engraftment and stabilization of the lesion on MRI, these changes do not usually reflect continued deterioration in function, but rather the failure to maintain the rate of skill development that is typical.
The long-term impact of CCALD on brain function after HCT differs across neuropsychological domains. The profile of strengths and weaknesses observed in pediatric patients is closely tied to the nature and location of damage to the neural substrate. Since CCALD in most cases involves myelin loss in neurons in posterior brain regions, problems on tasks requiring visual processing are common, such as visual-spatial reasoning or visual-motor integration.37,49 Other processing difficulties such as poor sustained attention, slowed processing speed, and reduced reaction time are also frequently observed.35,37 In contrast, several studies have demonstrated that verbal intellectual functioning, language skills and memory are more likely to remain intact among individuals with CCALD following HCT.36,37,46 Although most studies have analyzed neuropsychological outcomes of HCT recipient cohorts without regard to demyelination pattern, some information is emerging regarding outcomes for patients with less common variants of CCALD.50,51 Gupta et al. compared a small group of patients with lesions exclusively in frontal structures to those with lesions of only parietal-occipital structures.51 Neurocognitive outcomes were generally similar across the groups, with a slightly increased risk for working memory impairments among the patients with frontal lesions. Reflecting the importance of the frontal lobes for emotional and behavioral regulation, higher rates of severe psychiatric impairment were observed among the patients with frontal lesions, including problems such as hyperactivity, aggression, and atypical behaviors (e.g., poor social boundaries, tangential speech, destructive behaviors). This finding aligns with a report by Kaga et al. noting considerable ADHD-like behavior among three patients with frontal lesions26 Patients with a frontal pattern were also more likely have been prescribed stimulant medication for treatment of these behaviors.51
As the use of HCT for CCALD has increased in recent decades, there has been intensified focus on finding more precise predictors of neurocognitive outcomes. A variety of MRI markers (regional cerebral blood volume, diffusion tensor imaging metrics, intensity of score for active neuroinflammation, i.e., “gadolinium enhancement score”) have been associated with neurological, neurocognitive and motor outcomes.52–55 At the functional level, early studies reported that deficits in visual reasoning at baseline assessment were a predictor of neurocognitive decline after HCT.10,56 A more recent study revealed that poor performance on pegboard tasks of fine motor speed/dexterity at the pre-HCT assessment was predictive of greater impairment in adaptive functioning at follow-up.57 Thus, in addition to MRI severity metrics, neurocognitive tests involving speed, coordination, and execution of visual-motor tasks are important prognostic indicators to estimate functional capacities and independence in daily life of patients after HCT.
Aspects of the treatment regimen have also been examined in connection with neurocognitive outcomes. Conditioning for HCT involves use of chemotherapy and/or radiation, which has known long-term effects on neurocognitive function in pediatric patients.58–60 High-dose total body irradiation as part of the preparative conditioning has been specifically shown to be a risk factor for poorer neurocognitive outcomes in CCALD patients, including reduced working memory and adaptive function.37,57 On the other hand, comparisons related to the source of stem cells (i.e., bone marrow vs. umbilical cord blood) have found no clear association between the graft source and neurocognitive outcomes.10,37
When cerebral disease has progressed to an advanced stage prior to treatment (Loes >10), severe neurocognitive and adaptive impairment is the typical outcome for HCT survivors. In long-term follow up of 23 patients who survived treatment for CCALD with advanced disease, more than half were unable to walk independently, read or write, make phone calls, or bathe without assistance at their most recent evaluation.57 In a study examining the quality of life of boys after HCT, boys treated with advanced CCALD had poorer mobility and perceived cognitive functioning than healthy boys and boys treated with early stage CCALD.61 Limitations in mobility and function were associated with increased symptoms of depression, anger and anxiety in this sample.
3.2.3. Neuropsychological outcomes after gene therapy for CCALD
Initial attempts to treat CCALD through lentiviral hematopoietic cell gene therapy have produced mixed results. An initial single-center feasibility trial with long-term follow up of 4 patients who received gene therapy reported that 3 boys (Loes scores: 2.25, 2.5, and 7) showed major neurocognitive decline (e.g., unable to produce sentences, cortical blindness) between 9 and 60 months post treatment.62 The fourth patient, who was treated at an earlier age relative to the other patients (4.4 years old) and had a relatively low Loes score at baseline (2.5) with only parietal-occipital involvement, maintained stable, intact verbal and nonverbal intellectual functioning 8.3 years after treatment. An interim analysis of a larger international phase 2/3 trial reported that 15 of 17 patients (88%) were alive and free of major functional disability (e.g., cortical blindness, seizures, incontinence, wheelchair dependence, etc.) at a median of 29.4 months after receiving gene therapy.63 Accelerated approval for this lentiviral-mediated gene therapy product was recently granted by the Food and Drug Administration based on these data and another phase 3 study. Although neurocognitive test results have yet to be published from these trials, preliminary analyses of the phase 2/3 trial data have indicated a clear pattern of more favorable and stable neurocognitive outcomes among gene therapy recipients with the lowest pre-treatment Loes scores (i.e., ≤ 2) than those with higher scores.64
3.3. Adult non-cerebral phenotype and adrenomyeloneuropathy
Adrenomyeloneuropathy (AMN) is a common phenotype affecting most males with ALD during adulthood, emerging most often in the 3rd or 4th decade of life.8 Generally, in the absence of cerebral involvement, men with ALD or AMN tend to have intact intellectual functioning.65,66 While overall IQ is typically preserved, mild or isolated impairments in specific neurocognitive domains have been noted, most frequently within the domains of executive function,66–68 visuoconstruction,67,69 verbal fluency,67,69 and memory.68–70 Psychomotor slowing has also been reported.67,68 In a sample of 33 men with ALD and no cerebral involvement, Buermans and colleagues reported that 6 patients (18%) had borderline to impaired scores in two cognitive domains and 3 patients (9%) had borderline or impaired scores in three cognitive domains.67 Neither age nor the presence of minor MRI abnormalities (i.e., vascular lesions) were associated with neurocognitive test performance. Schäfer and colleagues compared neurocognitive status in a large cohort (n=117) of men and women categorized as having either pure AMN (Loes score of 0) or AMN with corticospinal tract involvement but no cerebral contrast enhancement (Loes score < 2).65 No clear differences were found between these two groups. Taken together, these studies suggest that patients diagnosed with AMN without cerebral involvement should not be assumed to be free of cognitive challenges, even if widespread or severe cognitive impairment is not typical.
As with other ALD phenotypes, mental health is an important component of care for individuals with AMN, but has received little research attention. A study by Walterfang et al. investigated psychiatric symptoms among a small cohort of 10 AMN patients, reporting a high prevalence of mood and anxiety disorders (60%).66 Major depressive episodes were especially common, and the group as a whole had self-reported depression scores in the mild to moderate range. Broader psychiatric symptoms were minimal. The level of psychopathology was not associated with a history of hypoadrenalism or mineralcorticoid replacement, suggesting that these findings did not primarily reflect a neuroendocrine effect.
3.4. Adult-onset cerebral adrenoleukodystrophy (ACALD)
3.4.1. Onset of ACALD
Although primary cerebral demyelination with onset in adulthood is relatively rare (2–5%),8,71,72 estimates indicate that between 19 and 63% of men with AMN will develop secondary cerebral demyelinating lesions, typically within 10 years after onset of AMN symptoms.72–74 Patients are likely to show a rapid decline once patterns of cerebral demyelination are confirmed. As would be expected, men with ACALD show notable neurocognitive impairment relative to those without active cerebral involvement.65 Common areas of deficit include processing speed, visual-spatial perception and reasoning, and memory (both verbal and visual).65,69 Performance on measures of attention, verbal fluency, and verbal encoding have also measured below average in patients with ACALD compared to normative data.65 Executive functions may be profoundly impaired.69 As in the childhood form, language and verbal reasoning is generally spared in earlier stages of disease, but declines as the disease progresses to more advanced stages.69
Aligned with the findings in CCALD, the severity and location of white matter lesions are linked with neuropsychological profiles in ACALD. Specifically, Schäfer et al. reported associations between MRI severity and performance on measures dependent on processing speed.65 While processing speed deficits occurred irrespective of the pattern of localization, deficits in other cognitive domains were more closely tied to specific patterns of cerebral demyelination. Adults with lesions in parietal-occipital white matter demonstrated greater deficits in tasks involving visual construction abilities, while individuals with frontal lesions showed greater deficits in executive functions.65
Psychiatric disturbance is a prominent concern among men with ACALD, with a review of 34 case reports revealing that psychiatric symptomatology was described in 56% of patients.75 Symptoms such as emotional lability, agitation, aggression, and irritability may emerge as initial clinical signs, and are sometimes present for years before demyelinating disease is detected.76,77 These non-specific symptoms can be difficult to distinguish from primary psychiatric disorders such as bipolar disorder or schizophrenia.77,78 As the disease progresses, psychiatric symptoms increase in severity. Patients may experience depression, character/personality disturbances, mania (disinhibition, emotional lability, increased spending, hypersexuality, loudness, and perseveration), obsessive-compulsive features, and psychosis (e.g., visual hallucinations).75,79–81
3.4.2. Neuropsychological outcomes after allogeneic HCT for ACALD
Similar to the findings in children, HCT at an early stage is the only treatment with demonstrated long-term efficacy in arresting disease progression from ACALD.32 Better functional outcomes have been observed among individuals who: 1) present with less severe radiological disease;79,80 2) have no or mild evidence of cognitive deficits and/or good preservation of activities of daily living and motor function prior to HCT,32,79,80 and 3) have no or limited involvement of the internal capsule, cerebellum or thalamus.32,80 Findings have been mixed with regard to the superiority of various stem cell sources and conditioning regimens for predicting long-term functional outcomes.32,79,80
During the early post-HCT period, men with ACALD commonly experience acute cognitive effects (e.g., disorientation, attention deficit), exacerbation of motor disability (e.g., ataxia, dysarthria, lack of ambulation, bladder disturbance), and/or behavioral changes (e.g., severe depression, behavioral disinhibition, psychosis).32,80 In a study of 14 adult patients (aged 21–48 years), significant changes in intellectual functioning and/or behavior were observed in all but one patient during the months immediately following HCT.32 Although some of these effects are transient and resolve within 6 to 12 months, enduring consequences to interpersonal relationships may occur.32
After the immediate post-transplant period, ACALD patients often stabilize, and long-term outcomes remain favorable for a subset. Among patients who survived beyond the first 6 months post-HCT, Kühl and colleagues reported that 3 (27%) showed continued progressive deterioration and died from ACALD; 3 (27%) showed moderate neurocognitive decline; and 5 (45%) showed stable neurocognitive functioning during a median of 5 years of follow-up.32 For patients whose neurocognitive function stabilized, continued MRI lesion progression was uncommon but AMN symptoms persisted. Another study reported stable cognitive functions in 9/11 (82%) survivors of HCT for ACALD, but these outcomes were based on a retrospective scoring system rather than quantitative neurocognitive testing.80 Across studies, evidence of enduring psychiatric symptoms post-transplant is observed in some patients, including mood symptoms and episodes of psychosis.32,80 Quality of life can be quite varied as some patients are able to remain active, relatively independent, and employed, while others require substantial additional support in their daily life and are forced to retire or discontinue work in their professions.32,79,80
3.5. Impact of endocrine dysfunction on neuropsychological status
Approximately 80% of boys and men with ALD develop primary adrenal insufficiency (also known as Addison’s disease) within their lifetime, with the first decade of life representing a period of heightened risk.7 Adrenal insufficiency may be the first clinical manifestation of ALD, particularly in young boys under the age of 6, and the vast majority of boys with CCALD have adrenal insufficiency at the time of diagnosis.82 There is a lack controlled research in this population examining the effects of adrenal insufficiency on neuropsychological function separately from other ALD phenotypes. Therefore, to consider the potential effects of adrenal insufficiency on cognition and mental health, we briefly review studies of primary adrenal insufficiency more generally.
Studies investigating neurocognitive functioning in individuals with primary adrenal insufficiency in other (i.e., non-ALD) adult populations have shown evidence of mild neurocognitive deficits, mainly in verbal learning and memory,83,84 visual memory,84 episodic memory,85 attention,86 and some executive functions.84 Individuals with primary adrenal insufficiency generally perform similarly to matched healthy controls on measures of verbal intelligence, reasoning, working memory, autobiographical memory, processing speed, or psychomotor speed.83–86 Acute changes in mood, behavior, and mental status (e.g., delirium, psychosis) can be associated with untreated adrenal insufficiency and adrenal crisis, but these psychiatric symptoms typically resolve with hydrocortisone replacement.87 Depression, anxiety, and other mood changes are known side effects of hydrocortisone replacement;88 individuals with primary adrenal insufficiency have been shown to experience more depressive symptoms than matched healthy controls.83 Impairments in quality of life have also been documented, even with the benefit of hydrocortisone replacement.89
In extrapolating these findings to consider the potential impact of adrenal insufficiency on neuropsychological functioning in males with ALD, it seems plausible or perhaps likely that acute effects on mood and psychiatric health could be observed in ALD patients experiencing untreated adrenal insufficiency. Further, adrenal insufficiency could contribute to chronic, milder effects on neurocognitive function and mental health. This could explain, at least in part, some of the mild deficits observed in some AMN patients relative to healthy adults reported in the literature. Although these effects depart from the severe impact of cerebral demyelination (CCALD and ACALD) on neuropsychological outcomes both in quality (i.e., which domains are most affected) and severity, they further underscore the importance of lifelong adrenal monitoring among males with X-ALD.
In addition to adrenal androgen deficiencies, primary testicular dysfunction is known to be a manifestation of disease among men with ALD. However, a recent review found sparse reports of the effects of low testosterone in ALD.90 Potential contribution of low testosterone to cognition and mental health might be estimated from the broader literature on low testosterone, which can be associated with depression and lower scores on neurocognitive testing, including memory, processing speed, attention, visuospatial, and language skills.91–93 However, the weight of this contribution to the neuropsychological presentation of adults with ALD is presently unclear and worth further investigation.
4. Females with ALD
Adrenal insufficiency and cerebral involvement are extremely rare among girls and women with ALD.94 More commonly, signs and symptoms of spinal cord dysfunction develop later in life, varying in severity from mildly impaired vibration sense or overresponsive reflexes with minimal functional disability to paraparesis requiring a wheelchair.95 This myeloneuropathy emerges in an age-dependent manner.96–98 Engelen and colleagues reported that the frequency of neurological symptoms (i.e., myelopathy, peripheral neuropathy) among adult women with ALD was 67%, with a dramatic increase in frequency of symptoms with age (from 18% among women under age 40 to 88% among women over the age of 60).98 Gait disturbance, sensory symptoms, and incontinence (both urinary and bowel) were also common complaints. In terms of neurocognitive function, an early report indicated that the average full scale IQ of a cohort of 16 women with ALD was within the average range (M=103, SD=10.6),68 a finding which was recently replicated in a larger cohort of 47 women.65 Despite broadly intact intellectual functioning, a greater number of female patients with AMN than expected may experience deficits in working memory and executive function.65,68
Regarding the impact of ALD on quality of life among women, neurologically symptomatic women report reduced physical functioning and role limitations (e.g., reduced work productivity, limitations in daily activities as a result of physical health) relative to asymptomatic women.98 However, there were no measurable differences in terms of emotional or mental health status. The experience of females with ALD may be impacted considerably by their life stage at the time of diagnosis and the manner in which they learn about their status as having an X-linked condition. A recent study of females with X-linked conditions, including a high proportion of participants with ALD, found that many women (~90%) in this community felt that they had insufficient access to knowledgeable health care providers and medical information about their condition.99 A high proportion indicated the desire for more comprehensive counseling regarding their risk for symptoms as well as increased access to research studies, treatments and reproductive methods.
5. Impact of ALD on caregivers and family members
The psychological impact of diagnosis and management of ALD on parents and other caregivers can vary dramatically depending on the age of their child and the symptom presentation(s) that emerge, as well as availability of expert providers and treatment options. Among parents of children with ALD identified via NBS, considerable distress and anxiety may be experienced upon first learning of their child’s diagnosis, especially when the diagnosis is unfamiliar to the health care provider delivering the news.100 Follow-up appointments with genetic counselors and other medical professionals knowledgeable about ALD can help alleviate concerns and provide reassurance for some parents.100 A key source of stress frequently cited in qualitative interviews with parents whose child tested positive for ALD or other metabolic conditions on NBS is a lack of up-to-date, relevant, and culturally and linguistically sensitive educational materials about the child’s condition.101 Additional tools for communicating with the child, their siblings, and extended family were noted to be important. In some cases, NBS programs may also lead to the diagnosis of other individuals in the family, such as siblings or extended family members who are at risk, creating additional challenges and psychological impacts.100 Despite these notable sources of uncertainty and stress, there is also emerging evidence of potential psychological benefits from NBS. Boychuk et al. found that among caregivers of patients with ALD and other metabolic diseases, those who were alerted of their child’s diagnosis from NBS results experienced fewer symptoms of depression compared to caregivers whose child’s diagnosis was identified due to emergent clinical symptoms.102 These reassuring results suggest that the potential benefits of early identification may outweigh the risks for many families.
For families living in locations where NBS programs have yet to be adopted, parent experiences may reflect considerable concern regarding a long diagnostic journey and/or delays to diagnosis.103 Even after a diagnosis is made, lack of knowledge among health care professionals can contribute to ongoing stress among some parents. Mothers also reported significant feelings of powerlessness with regard to limited or unsatisfactory treatment options for their male children, especially those who were diagnosed at an advanced stage.103 This sentiment of frustration with lack of options has also been echoed among mothers of daughters with ALD, as the future risks and implications of this diagnosis for females can be challenging to ascertain.100
Caregiving for a child with a chronic illness can take a toll on caregivers’ mental health. Higher rates of depression, anxiety, mental and physical distress have been found among parents of patients with CCALD relative to healthy adults.104 Mothers may be particularly susceptible to depression, especially younger mothers and those whose children were more recently diagnosed. Guilt over being a genetic carrier weighs on some parents, as well as living with the uncertainty regarding disease progression.100,103–105 Romantic and spousal relationships may also be impacted in various ways. Relationship strain can be especially salient for those experiencing complicated grief from losing a family member (e.g., son, brother) to ALD.106 Parents reported using a variety of coping strategies for managing the emotional impact of ALD, including seeking external support from family members, relying on their faith or religion, seeking additional information about the disease and possible treatments, and remaining hopeful.100,103
While unaffected siblings often play key roles in families impacted by a pediatric chronic illness such as ALD, their perspectives are often overlooked in research. Parents of children with ALD have reported that their other children may also experience different parenting practices (e.g., attention, discipline practices) than their affected children.100 Research on the experiences of siblings with other rare metabolic neurological disorders suggest that they may be expected to take on additional caregiving responsibilities for their affected sibling, have less individualized time with their parents, have fewer opportunities to socialize with peers, and feel a wide range of emotions from pride to sadness and worry.107 For patients requiring HCT, the availability of a donor source from a family member may also contribute to family system stress. About 20–30% of transplants for cerebral forms of ALD have historically involved a matched family member as the donor, most often a sibling.10,32,50,80 Although the experiences of sibling donors have not been studied specifically within ALD, research in other populations has indicated that they can face stressors including a lack of clear understanding about the procedure, internal and external pressure to donate, guilt over transplant complications, and anxiety.108,109 Siblings may also endure social isolation owing to the sibling’s immunosuppression protocols. Ensuring adequate social and emotional support for siblings is critical for promoting healthy adjustment, building strong family connections, and empowering them to be strong advocates for their affected siblings.107
6. Discussion
ALD remains a complicated disease with wide-ranging presentation and severity of symptoms. The unpredictability of disease course and broad impacts on both the individual and their support system make clinical care challenging. NBS programs are an essential tool to increase early detection of disease manifestations, but adoption of NBS across the U.S. and other nations continues to progress slowly and there are many complexities to management of screen-positive cases.6,110,111 Evaluation of the success of these early detection efforts will depend on increasing the quality of measurement approaches. Historically, for patients with cerebral ALD, improving survival and freedom from major functional disability were important benchmarks to evaluate therapy relative to the natural history. However, as earlier identification of cerebral ALD becomes more widespread and novel treatment approaches are assessed relative to HCT, it will also be critical to consider the extent to which these care management approaches impact the neurocognitive and mental health burden of ALD.
Findings of this review identify specific neurocognitive domains that represent useful clinical endpoints when investigating the comparative effects of monitoring and therapeutic approaches. Specifically, for individuals with CCALD and ACALD, global metrics of neurocognition such as overall intellectual functioning (e.g., full scale IQ) may be too broad to capture localized, disease-related effects among patients with early disease; these global metrics will mask important strengths and weaknesses in each patient’s functioning. The brain-behavior relationships outlined in this review highlight that examination of specific subdomains is needed to evaluate effects of ALD on brain function. Measures of processing speed, for example, may represent a sensitive indicator of myelin integrity even in relatively early stages. Slowed processing is seen in both children and adults with cerebral disease, and in patients with a variety of demyelination patterns. Because parietal-occipital presentation is the most common demyelination pattern, especially in children, visual perceptual and visual reasoning abilities are also a sensitive indicator of cerebral disease burden for larger cohort studies, and can assist with prediction of outcomes. Changes in fine motor dexterity are also common in cerebral forms of ALD, but are also observed in men with AMN and therefore it may be challenging to distinguish phenotype-specific effects among adult patients. Attention problems, hyperactivity and executive functions may be additional important indicators of disease burden, but can be more difficult to measure with objective testing and may rely on parent or clinician-report scales. Finally, when considering outcomes among patients with more advanced disease, measures of adaptive functioning and quality of life may better capture the range of functioning as compared to direct neurocognitive testing which can be challenging for patients with profound visual, auditory or behavioral impairment to complete.
This review also underscores that across a variety of phenotypes, including AMN, psychiatric symptomatology is a prominent consequence of ALD. Despite these findings, very few controlled observational studies or clinical trials have utilized quantitative measures of psychiatric symptoms as endpoints. Among patients with cerebral forms of ALD, severe psychiatric conditions are commonly observed, and are especially apparent among individuals with a frontal pattern and those with ACALD. When present, these symptoms nearly always persist after treatment and can have a profound effect on the trajectory of patients’ lives, limiting interpersonal relationships and the ability to develop or maintain independent daily functions. Although symptoms may differ in quality and severity, consideration of the emotional burden is also important for individuals with AMN. The sparse literature that exists suggest that symptoms of anxiety and depressed mood may be seen at a higher rate than in unaffected individuals. Importantly, anxiety or mood-related symptoms in AMN patients may not be direct sequelae of a disease process, but rather may be attributable to related factors (e.g., medications, feelings surrounding changes in functional abilities or quality of life, stressful life events). Nevertheless, inclusion of mental health related metrics in observational studies and trials will enable a more comprehensive perspective of ALD.
6.1. Gaps and suggestions for future research
Decades of research have provided a foundation enabling further questions to be asked, and there remain several gaps to fill in future investigation, outlined in Table 2. Broadly, we highlight the need for longitudinal and quantitative measurement of neurocognitive, mental health, and quality of life outcomes across ALD phenotypes, harmonization of neuropsychological and neuroimaging protocols across research sites and centers to increase sample sizes and generalizability of findings, and increased attention and study of the impact of ALD on women, caregivers, and other family members.
Table 2.
Summary of needs for investigations of neurocognition and mental health in ALD
| Phenotype | Needs |
|---|---|
| Neurologically asymptomatic |
|
| Cerebral ALD (CCALD & ACALD) |
|
| Myeloneuropathy |
|
| Caregivers/family |
|
| All phenotypes |
|
6.2. Implications for neuropsychological and mental health care
As there is currently no cure for ALD that avoids potentially complex and life-impacting complications, it will be important for care teams to guide families in coping with the disease and its many effects. An ideal care model will include resources for educational and/or occupational supports, rehabilitation, mental health treatment, and management of the practical concerns that arise (e.g., navigating insurance coverage; covering financial costs; supporting the potential need to relocate for treatment). Below we provide suggestions for evaluation and care at different stages of the ALD journey.
6.2.1. Early childhood monitoring
If available, prospective neurodevelopmental screening offers opportunities to establish an assessment of a patient’s baseline functioning prior to the potential onset of cerebral disease and identify any pre-existing neurodevelopmental differences that may not be associated with ALD. These evaluations can support a comprehensive care approach, providing an opportunity to further educate parents, caregivers, and siblings about the disease and the need for ongoing surveillance. For many boys who were identified via newborn screening, neurodevelopmental screening may provide reassurance that development is “on track,” consistent with absence of lesions on MRI.
6.2.2. Pre-treatment evaluation and care
Assessment of neurocognitive function prior to initiating therapy for cerebral ALD can help to quantify disease severity, and along with MRI results, can provide important information to assess the utility of transplant and estimate likely functional outcomes after treatment.10,57,61,80 Specifically, tests that measure speed and coordination of visual-motor functions (e.g., tests of fine motor dexterity or visual perceptual skills) administered pre-treatment have been shown to be predictive of level of independence in daily living after treatment.10,57 For men with ACALD, quantifying AMN symptom burden may also aid in prediction of HCT outcomes.32 In some early-identified cases, patients may present to treatment with above average neurocognitive abilities, providing for a greater buffer against deficits associated with cerebral disease. In communicating results of these evaluations, clinicians can help set expectations for patients and families regarding areas where the patient is experiencing or may experience functional changes.
6.2.3. Post-treatment and long-term evaluation and care
After treatment for cerebral disease, evaluation by neuropsychologists and other mental health professionals facilitates targeted clinical, academic, home, occupational, and/or community-focused recommendations. Clinicians can encourage access to outpatient and/or school-based therapeutic interventions (e.g., occupational, physical, and speech/language therapy), which are critical for retention of functional skills and for targeting specific deficits. In the pediatric context, feedback from evaluations can help educate school personnel about this uncommon disease and how to help a student adapt in the academic setting. Involvement of special education professionals and interventionists can help to accommodate any changes in visual and auditory processing, communication, and mobility using adaptive devices and equipment and ensure maximal academic and community participation. Implementation of accommodations and behavioral techniques to support attention, emotional, and behavioral regulation in the classroom may also be warranted.
Mental health professionals within an interdisciplinary clinical team are in a unique position to recommend or provide treatment approaches to improve well-being. Specifically, individual or family-based psychotherapies can offer opportunities to process and learn skills for coping with ALD across the lifespan. In some cases, the affected individual and their family members may need space to process feelings of grief associated with changes in a patient’s functioning. Psychotherapy also provides parents and caregivers an opportunity to develop a diverse set of tools for managing any challenging behaviors that emerge, and to learn strategies for reducing stress. In some cases, medications are warranted to manage a patients’ symptoms, and referral to psychiatric care is needed.
Declines associated with advanced stages of cerebral disease are devastating, and will continue to be observed in males with ALD in the absence of universally implemented early detection programs and diligent follow-up. Such patients will require additional adaptations to support preservation of skills and quality of life for the longest timeframe possible. During advanced stages of disease, social workers and other clinicians can help connect parents, caregivers, and family members with governmental programs that may offer home and community-based support for families. These may include disability and waiver programs, nursing care, in-home personalized assistance, respite care, palliative care, and case management services.
6.2.4. Support for myeloneuropathy
For both men and women with myeloneuropathy, screening for mental health and psychiatric concerns, even when related to physical symptom burden and coping, may be a useful consideration in clinical practice. Although there are frustratingly few therapeutic options to treat the symptoms of spinal cord disease, medications can be used to addressed spasticity and neuropathic pain, and referrals can be made to specialists in rehabilitation (e.g., physical therapists), continence care, and pain management.45 Specific interventions such as yoga or functional electrical stimulation may also be helpful at maintaining best possible function, including improved agility, balance, and walking capabilities, which are necessary for occupational and social functioning, and quality of life.112,113
6.2.5. Support for caregivers, siblings, and other family members
Emerging literature sheds light on the substantial caregiving and family burden associated with ALD, as well as important gaps that need to be addressed—namely, the need for better education among healthcare providers regarding ALD, increased insight regarding the perspectives of girls and women with ALD, a better understanding of the impact of ALD on family members (especially fathers and siblings), adequate psychological support for all family members, and access to accurate and comprehensive information for families.
6.2.6. Addressing barriers to care
Managing a chronic illness such as ALD requires extensive investment of time and resources, as well as access to high-quality health care. Some individuals and families experience psychosocial or systemic barriers that prevent access to or utilization of health care, and there has been increased attention to these disparities in access to treatments for rare disease (https://www.rarediseasediversity.org/). Geographic location (e.g., what state or region one lives in), access to comprehensive or specialized care centers, type of medical insurance in the U.S., and availability of a matched donor are just a few factors that may affect quality of care among individuals with ALD. Accordingly, it is important for clinicians to be aware of the many ways in which health care disparities contribute to greater psychological strain for some families. Efforts to optimize access to clinical care and support resources, discussed in Table 3, are critical for enhancing quality of life among affected families.
Table 3.
Crucial points for optimizing access and clinical care for ALD
| Domain | Target | Needs |
|---|---|---|
|
| ||
| Mental health | Caregivers and family |
|
| Pediatric patients |
|
|
| Adult patients |
|
|
| Females with ALD |
|
|
|
| ||
| Supportive care | Caregivers and family |
|
|
| ||
| Access and service utilization | Families receiving newborn screening or other presymptomatic diagnosis |
|
| Families and patients undergoing treatment |
|
|
| Families and patients in advanced stages of cerebral ALD, including those stabilized with significant functional impairments |
|
|
| Families and patients in advanced stages of myeloneuropathy |
|
|
6.3. Conclusions
Considerable advances have been made in recent decades in understanding how different components of ALD diagnosis, disease course and treatment affect neurocognition and mental health. The ability of investigators and care teams to improve the everyday experience of patients and families will increase as additional progress is made toward optimizing early detection of cerebral disease and expanding treatment options, utilizing sensitive measurement tools to capture relevant outcomes, harmonizing protocols and care standards with consensus-based approaches,29,45,114 and improving understanding of predictive and modifiable factors in the disease course. Access to informed, interdisciplinary care at ALD centers of excellence115 and effective dissemination of knowledge about ALD will further impact the experience of families coping with this challenging disease.
Supplementary Material
Synopsis.
To inform future investigations, clinical trials, and multidisciplinary care practices, this comprehensive review synthesizes current knowledge regarding the neurocognitive and mental health effects of ALD across the lifespan.
Acknowledgements
The authors wish to express gratitude to the families who have participated in research studies to better understand X-ALD.
Funding:
Funding for this research was provided by ALD Alliance, the National Institutes of Health’s National Center for Advancing Translational Sciences KL2TR002492, the National Institutes of Health’s National Institute for Neurological Disorders and Stroke K23NS123258 and the University of Minnesota Department of Pediatrics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing Interest Statement:
Elizabeth Pierpont, Ashley Isaia, Erin McCoy, Sarah Jane Brown, and Ashish Gupta declare that they have no conflict of interest. Julie Eisengart served as an advisory board member for bluebird bio.
Ethics Approval: No ethics approval was required for this literature review article.
Informed Consent: Not applicable.
Patient consent: No personally identifying information is contained in this article.
Compliance with Ethical Standards
The authors declare no conflicts of interest with regard to this manuscript. No research with human participants or animals was carried out in preparation of this review paper.
Data availability statement:
Data sharing is not applicable to this article as no datasets were generated or analyzed in the study.
References
- 1.Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. [PubMed] [Google Scholar]
- 2.Wiens K, Berry SA, Choi H, et al. A report on state-wide implementation of newborn screening for X-linked Adrenoleukodystrophy. Am J Med Genet A. 2019;179(7):1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matteson J, Sciortino S, Feuchtbaum L, Bishop T, Olney RS, Tang H. Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up. Int J Neonatal Screen. 2021;7(2). [DOI] [PMC free article] [PubMed]
- 4.Eng L, Regelmann MO. Adrenoleukodystrophy in the era of newborn screening. Curr Opin Endocrinol Diabetes Obes. 2020;27(1):47–55. [DOI] [PubMed] [Google Scholar]
- 5.Hall PL, Li H, Hagar AF, Jerris SC, Wittenauer A, Wilcox W. Newborn Screening for X-Linked Adrenoleukodystrophy in Georgia: Experiences from a Pilot Study Screening of 51,081 Newborns. Int J Neonatal Screen. 2020;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S, Clinard K, Young SP, et al. Evaluation of X-Linked Adrenoleukodystrophy Newborn Screening in North Carolina. JAMA Netw Open. 2020;3(1):e1920356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huffnagel IC, Laheji FK, Aziz-Bose R, et al. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J Clin Endocrinol Metab. 2019;104(1):118–126. [DOI] [PubMed] [Google Scholar]
- 8.Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turk B, Mallack E, Adang L, et al. Consensus Guidelines: MRI Surveillance of Children with Presymptomatic Adrenoleukodystrophy (P4.6–047). Neurology. 2019;92(15 Supplement):P4.6–047. [Google Scholar]
- 10.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971–1978. [DOI] [PubMed] [Google Scholar]
- 11.Raymond GV, Aubourg P, Paker A, et al. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2019;25(3):538–548. [DOI] [PubMed] [Google Scholar]
- 12.Sutovsky S, Kolnikova M, Petrovic R, et al. Differing clinical presentations of two unrelated cases of X-linked adrenoleukodystrophy with identical mutation Y296C in the ABCD1 gene. Neuro endocrinology letters. 2014;35(5):411–416. [PubMed] [Google Scholar]
- 13.Korenke GC, Fuchs S, Krasemann E, et al. Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype. Annals of neurology. 1996;40(2):254–257. [DOI] [PubMed] [Google Scholar]
- 14.Sobue G, Ueno-Natsukari I, Okamoto H, et al. Phenotypic heterogeneity of an adult form of adrenoleukodystrophy in monozygotic twins. Annals of neurology. 1994;36(6):912–915. [DOI] [PubMed] [Google Scholar]
- 15.Kemper AR, Brosco J, Comeau AM, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. 2017;19(1):121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salzman R, Kemp S, 2022. https://adrenoleukodystrophy.info/clinical-diagnosis/ald-newborn-screening.
- 17.Gupta AO, Raymond G, Pierpont EI, et al. Treatment of cerebral adrenoleukodystrophy: allogeneic transplantation and lentiviral gene therapy. Expert Opin Biol Ther. 2022;22(9):1151–1162. [DOI] [PubMed] [Google Scholar]
- 18.Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan-a web and mobile app for systematic reviews. Syst Rev. 2016;5(1):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ievers CE, Brown RT, McCandless SE, Devine DE. Case studies: psychological test findings for two children with X-linked adrenoleukodystrophy. Journal of Developmental and Behavioral Pediatrics. 1999;20(1):31–35. [DOI] [PubMed] [Google Scholar]
- 20.Incecik F, Herguner MO, Mert G, et al. X-linked adrenoleukodystrophy in a 6-year-old boy initially presenting with psychiatric symptoms. The Turkish journal of pediatrics. 2014;56(6):651–653. [PubMed] [Google Scholar]
- 21.Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3(3):140–151. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda T, Kawahara Y, Miyauchi A, et al. Low donor chimerism may be sufficient to prevent demyelination in adrenoleukodystrophy. JIMD Rep. 2022;63(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vawter-Lee MM, Hallinan BE, Burrow TA, Spaeth CG, Arthur TM. A Novel Catastrophic Presentation of X-Linked Adrenoleukodystrophy. JIMD reports. 2015;24:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garside S, Rosebush PI, Levinson AJ, Mazurek MF. Late-onset adrenoleukodystrophy associated with long-standing psychiatric symptoms. The Journal of clinical psychiatry. 1999;60(7):460–468. [DOI] [PubMed] [Google Scholar]
- 25.Cox CS, Dubey P, Raymond GV, Mahmood A, Moser AB, Moser HW. Cognitive evaluation of neurologically asymptomatic boys with X-linked adrenoleukodystrophy. Arch Neurol. 2006;63(1):69–73. [DOI] [PubMed] [Google Scholar]
- 26.Kaga M, Furushima W, Inagaki M, Nakamura M. Early neuropsychological signs of childhood adrenoleukodystrophy (ALD). Brain & development. 2009;31(7):558–561. [DOI] [PubMed] [Google Scholar]
- 27.Furushima W, Inagaki M, Gunji A, Inoue Y, Kaga M, Mizutani S. Early signs of visual perception and evoked potentials in radiologically asymptomatic boys with X-linked adrenoleukodystrophy. J Child Neurol. 2009;24(8):927–935. [DOI] [PubMed] [Google Scholar]
- 28.Riva D, Bova SM, Bruzzone MG. Neuropsychological testing may predict early progression of asymptomatic adrenoleukodystrophy. Neurology. 2000;54(8):1651–1655. [DOI] [PubMed] [Google Scholar]
- 29.Mallack EJ, Turk BR, Yan H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J Inherit Metab Dis. 2021;44(3):728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61(3):369–374. [DOI] [PubMed] [Google Scholar]
- 31.Liberato AP, Mallack EJ, Aziz-Bose R, et al. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology. 2019;92(15):e1698–e1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kühl J-S, Suarez F, Gillett GT, et al. Long-term outcomes of allogeneic haematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Brain: A Journal of Neurology. 2017;140(4):953–966. [DOI] [PubMed] [Google Scholar]
- 33.Mallack EJ, van de Stadt S, Caruso PA, et al. Clinical and radiographic course of arrested cerebral adrenoleukodystrophy. Neurology. 2020;94(24):e2499–e2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15(9):1761–1766. [PMC free article] [PubMed] [Google Scholar]
- 35.Pierpont EI, Nascene DR, Shanley R, et al. Neurocognitive benchmarks following transplant for emerging cerebral adrenoleukodystrophy. Neurology. 2020;95(5):e591–e600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis. 1995;18(4):413–429. [DOI] [PubMed] [Google Scholar]
- 37.Pierpont EI, Eisengart JB, Shanley R, et al. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 2017;74(6):710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Esposito M, Postle BR. The cognitive neuroscience of working memory. Annu Rev Psychol. 2015;66:115–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiersma RE, Gupta AO, Lund TC, et al. Primary Adrenal Insufficiency in a Boy with Type I Diabetes: The Importance of Considering X-linked Adrenoleukodystrophy. Journal of the Endocrine Society. 2022;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furushima W, Kaga M, Nakamura M, Gunji A, Inagaki M. Auditory agnosia as a clinical symptom of childhood adrenoleukodystrophy. Brain Dev. 2015;37(7):690–697. [DOI] [PubMed] [Google Scholar]
- 41.Muranjan M, Karande S, Sankhe S, Eichler S. Childhood cerebral X-linked adrenoleukodystrophy with atypical neuroimaging abnormalities and a novel mutation. J Postgrad Med. 2018;64(1):59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salsano E, Gambini O, Giovagnoli AR, Farina L, Uziel G, Pareyson D. Effectiveness of valproate for the treatment of manic-like behavior in X-linked adrenoleukodystrophy. Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2012;33(5):1197–1199. [DOI] [PubMed] [Google Scholar]
- 43.Guler AS, Fis NP, Berkem M. X-linked adrenoleukodystrophy in a 7-year-old boy presenting with psychiatric symptoms. European Child & Adolescent Psychiatry. 2011;20(5):275–276. [DOI] [PubMed] [Google Scholar]
- 44.Aubourg P, Blanche S, Jambaque I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322(26):1860–1866. [DOI] [PubMed] [Google Scholar]
- 45.Engelen M, van Ballegoij WJC, Mallack EJ, et al. International Recommendations for the Diagnosis and Management of Patients With Adrenoleukodystrophy: A Consensus-Based Approach. Neurology. 2022. [DOI] [PMC free article] [PubMed]
- 46.Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356(9231):713–718. [DOI] [PubMed] [Google Scholar]
- 47.Yalcin K, Celen SS, Daloglu H, et al. Allogeneic hematopoietic stem cell transplantation in patients with childhood cerebral adrenoleukodystrophy: A single-center experience “Better prognosis in earlier stage”. Pediatr Transplant. 2021;25(4):e14015. [DOI] [PubMed] [Google Scholar]
- 48.Gassas A, Raiman J, White L, Schechter T, Clarke J, Doyle J. Long-term adaptive functioning outcomes of children with inherited metabolic and genetic diseases treated with hematopoietic stem cell transplantation in a single large pediatric center: parents’ perspective. J Pediatr Hematol Oncol. 2011;33(3):216–220. [DOI] [PubMed] [Google Scholar]
- 49.Tran C, Patel J, Stacy H, et al. Long-term outcome of patients with X-linked adrenoleukodystrophy: a retrospective cohort study. Eur J Paediatr Neurol. 2017;21(4):600–609. [DOI] [PubMed] [Google Scholar]
- 50.Kühl JS, Kupper J, Baque H, et al. Potential risks to stable long-term outcome of allogeneic hematopoietic stem cell transplantation for children with cerebral X-linked adrenoleukodystrophy. JAMA Netw Open. 2018;1(3):e180769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta AO, Nascene DR, Shanley R, et al. Differential outcomes for frontal versus posterior demyelination in childhood cerebral adrenoleukodystrophy. J Inherit Metab Dis. 2021. [DOI] [PMC free article] [PubMed]
- 52.McKinney AM, Nascene D, Miller WP, et al. Childhood cerebral X-linked adrenoleukodystrophy: diffusion tensor imaging measurements for prediction of clinical outcome after hematopoietic stem cell transplantation. AJNR Am J Neuroradiol. 2013;34(3):641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKinney AM, Benson J, Nascene DR, et al. Childhood Cerebral Adrenoleukodystrophy: MR Perfusion Measurements and Their Use in Predicting Clinical Outcome after Hematopoietic Stem Cell Transplantation. AJNR Am J Neuroradiol. 2016;37(9):1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beam D, Poe MD, Provenzale JM, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13(6):665–674. [DOI] [PubMed] [Google Scholar]
- 55.Miller WP, Mantovani LF, Muzic J, et al. Intensity of MRI gadolinium enhancement in cerebral adrenoleukodystrophy: a biomarker for inflammation and predictor of outcome following transplantation in higher risk patients. AJNR Am J Neuroradiol. 2016;37(2):367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104(3):881–888. [DOI] [PubMed] [Google Scholar]
- 57.Pierpont EI, McCoy E, King KE, et al. Post-transplant adaptive function in childhood cerebral adrenoleukodystrophy. Ann Clin Transl Neurol. 2018;5(3):252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah AJ, Epport K, Azen C, et al. Progressive declines in neurocognitive function among survivors of hematopoietic stem cell transplantation for pediatric hematologic malignancies. J Pediat Hematol Onc. 2008;30(6):411–418. [DOI] [PubMed] [Google Scholar]
- 59.Wu NL, Krull KR, Cushing-Haugen KL, et al. Long-term neurocognitive and quality of life outcomes in survivors of pediatric hematopoietic cell transplant. J Cancer Surviv. 2021. [DOI] [PMC free article] [PubMed]
- 60.Parris KR, Russell KM, Triplett BM, Phipps S. Neurocognitive functioning in long-term survivors of pediatric hematopoietic cell transplantation. Bone Marrow Transplant. 2021;56(4):873–882. [DOI] [PubMed] [Google Scholar]
- 61.Beckmann NB, Miller WP, Dietrich MS, Orchard PJ. Quality of life among boys with adrenoleukodystrophy following hematopoietic stem cell transplant. Child Neuropsychol. 2018;24(7):986–998. [DOI] [PubMed] [Google Scholar]
- 62.Bougneres P, Hacein-Bey-Abina S, Labik I, et al. Long-Term Follow-Up of Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. Hum Gene Ther. 2021;32(19–20):1260–1269. [DOI] [PubMed] [Google Scholar]
- 63.Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pierpont EI, King K, Duncan CN, et al. Neurocognitive outcomes after elivaldogene autotemcel (eli-cel, Lenti-D) in the ALD-102 gene therapy study. Abstract presented at Child Neurology Society. 2022; Cincinnati, OH.
- 65.Schäfer L, Roicke H, Fischer M, Suhnel A, Kohler W. Cognitive functions in adult-onset phenotypes of X-linked adrenoleukodystrophy. Ann Neurol. 2021. [DOI] [PubMed]
- 66.Walterfang MA, O’Donovan J, Fahey MC, Velakoulis D. The neuropsychiatry of adrenomyeloneuropathy. CNS spectrums. 2007;12(9):696–701. [DOI] [PubMed] [Google Scholar]
- 67.Buermans N, van den Bosch SJG, Huffnagel IC, et al. Overall intact cognitive function in male X-linked adrenoleukodystrophy adults with normal MRI. Orphanet J Rare Dis. 2019;14(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edwin D, Speedie L, Naidu S, Moser H. Cognitive impairment in adult-onset adrenoleukodystrophy. Molecular and chemical neuropathology. 1990;12(3):167–176. [DOI] [PubMed] [Google Scholar]
- 69.Edwin D, Speedie LJ, Kohler W, Naidu S, Kruse B, Moser HW. Cognitive and brain magnetic resonance imaging findings in adrenomyeloneuropathy. Annals of neurology. 1996;40(4):675–678. [DOI] [PubMed] [Google Scholar]
- 70.Lyon-Caen O, Benoit N, Carreau V, et al. Cognitive function in adult adrenoleukodystrophy: comparison with leukoaraiosis and multiple sclerosis. Developmental neuroscience. 1991;13(4):251–253. [DOI] [PubMed] [Google Scholar]
- 71.Moser HW, Loes DJ, Melhem ER, et al. X-Linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31(5):227–239. [DOI] [PubMed] [Google Scholar]
- 72.van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001;49(2):186–194. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki Y, Takemoto Y, Shimozawa N, et al. Natural history of X-linked adrenoleukodystrophy in Japan. Brain Dev. 2005;27(5):353–357. [DOI] [PubMed] [Google Scholar]
- 74.de Beer M, Engelen M, van Geel BM. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology. 2014;83(24):2227–2231. [DOI] [PubMed] [Google Scholar]
- 75.Rosebush PI, Garside S, Levinson AJ, Mazurek MF. The neuropsychiatry of adult-onset adrenoleukodystrophy. The Journal of neuropsychiatry and clinical neurosciences. 1999;11(3):315–327. [DOI] [PubMed] [Google Scholar]
- 76.Galvao ACR, Machado-Porto GCL, Porto FHG, Lucato LT, Nitrini R. Adult-onset adrenoleukodystrophy presenting as a psychiatric disorder: MRI findings. Dement Neuropsychol. 2012;6(4):290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mehta AM, Prabhu M, Krishnan G. Adult-onset adrenoleukodystrophy presenting with status epilepticus and psychosis. BMJ Case Rep. 2021;14(11). [DOI] [PMC free article] [PubMed]
- 78.Shamim D, Alleyne K. X-linked adult-onset adrenoleukodystrophy: Psychiatric and neurological manifestations. SAGE Open Med Case Rep. 2017;5:2050313X17741009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsukawa T, Yamamoto T, Honda A, et al. Clinical efficacy of haematopoietic stem cell transplantation for adult adrenoleukodystrophy. Brain Commun. 2020;2(1):fcz048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waldhüter N, Kohler W, Hemmati PG, et al. Allogeneic hematopoietic stem cell transplantation with myeloablative conditioning for adult cerebral X-linked adrenoleukodystrophy. J Inherit Metab Dis. 2019;42(2):313–324. [DOI] [PubMed] [Google Scholar]
- 81.Salsano E, Marotta G, Manfredi V, et al. Brain fluorodeoxyglucose PET in adrenoleukodystrophy. Neurology. 2014;83(11):981–989. [DOI] [PubMed] [Google Scholar]
- 82.Korenke GC, Roth C, Krasemann E, Hufner M, Hunneman DH, Hanefeld F. Variability of endocrinological dysfunction in 55 patients with X-linked adrenoleucodystrophy: clinical, laboratory and genetic findings. Eur J Endocrinol. 1997;137(1):40–47. [DOI] [PubMed] [Google Scholar]
- 83.Schultebraucks K, Wingenfeld K, Heimes J, Quinkler M, Otte C. Cognitive function in patients with primary adrenal insufficiency (Addison’s disease). Psychoneuroendocrinology. 2015;55:1–7. [DOI] [PubMed] [Google Scholar]
- 84.Tiemensma J, Andela CD, Biermasz NR, Romijn JA, Pereira AM. Mild cognitive deficits in patients with primary adrenal insufficiency. Psychoneuroendocrinology. 2016;63:170–177. [DOI] [PubMed] [Google Scholar]
- 85.Henry M, Thomas KG, Ross IL. Episodic memory impairment in Addison’s disease: results from a telephonic cognitive assessment. Metab Brain Dis. 2014;29(2):421–430. [DOI] [PubMed] [Google Scholar]
- 86.Klement J, Hubold C, Hallschmid M, et al. Effects of glucose infusion on neuroendocrine and cognitive parameters in Addison disease. Metabolism. 2009;58(12):1825–1831. [DOI] [PubMed] [Google Scholar]
- 87.Anglin RE, Rosebush PI, Mazurek MF. The neuropsychiatric profile of Addison’s disease: revisiting a forgotten phenomenon. J Neuropsychiatry Clin Neurosci. 2006;18(4):450–459. [DOI] [PubMed] [Google Scholar]
- 88.Clinic Mayo, https://www.mayoclinic.org/drugs-supplements/hydrocortisone-oral-route/side-effects/drg-20075259?p=1. Accessed 4/14/2022 2022, April 14.
- 89.Tiemensma J, Andela CD, Kaptein AA, et al. Psychological morbidity and impaired quality of life in patients with stable treatment for primary adrenal insufficiency: cross-sectional study and review of the literature. Eur J Endocrinol. 2014;171(2):171–182. [DOI] [PubMed] [Google Scholar]
- 90.Kachwala I, Regelmann MO. Monitoring for and Management of Endocrine Dysfunction in Adrenoleukodystrophy. Int J Neonatal Screen. 2022;8(1). [DOI] [PMC free article] [PubMed]
- 91.Testosterone Beauchet O. and cognitive function: current clinical evidence of a relationship. Eur J Endocrinol. 2006;155(6):773–781. [DOI] [PubMed] [Google Scholar]
- 92.Giagulli VA, Guastamacchia E, Licchelli B, Triggiani V. Serum Testosterone and Cognitive Function in Ageing Male: Updating the Evidence. Recent Pat Endocr Metab Immune Drug Discov. 2016;10(1):22–30. [DOI] [PubMed] [Google Scholar]
- 93.Afsar B. Relationship between total testosterone, cognitive function, depressive behavior, and sleep quality in chronic kidney disease patients not on dialysis. Clin Exp Nephrol. 2013;17(1):59–65. [DOI] [PubMed] [Google Scholar]
- 94.Fatemi A, Barker PB, Ulug AM, et al. MRI and proton MRSI in women heterozygous for X-linked adrenoleukodystrophy. Neurology. 2003;60(8):1301–1307. [DOI] [PubMed] [Google Scholar]
- 95.Moser HW, Moser AB, Naidu S, Bergin A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci. 1991;13(4–5):254–261. [DOI] [PubMed] [Google Scholar]
- 96.Horn MA, Retterstol L, Abdelnoor M, Skjeldal OH, Tallaksen CM. Adrenoleukodystrophy in Norway: high rate of de novo mutations and age-dependent penetrance. Pediatr Neurol. 2013;48(3):212–219. [DOI] [PubMed] [Google Scholar]
- 97.Habekost CT, Schestatsky P, Torres VF, et al. Neurological impairment among heterozygote women for X-linked Adrenoleukodystrophy: a case control study on a clinical, neurophysiological and biochemical characteristics. Orphanet J Rare Dis. 2014;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Engelen M, Barbier M, Dijkstra IM, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137(Pt 3):693–706. [DOI] [PubMed] [Google Scholar]
- 99.Choi J, Kane T, Propst L, Spencer S, Kostialik J, Arjunan A. Not just carriers: experiences of X-linked female heterozygotes. J Assist Reprod Genet. 2021. [DOI] [PMC free article] [PubMed]
- 100.Schwan K, Youngblom J, Weisiger K, Kianmahd J, Waggoner R, Fanos J. Family Perspectives on Newborn Screening for X-Linked Adrenoleukodystrophy in California. Int J Neonatal Screen. 2019;5(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Minnesota Department of Health Newborn Screening and Children & Youth with Special Health Needs Protocol Evaluation Workgroup. Considerations for Public Health Newborn Screening Follow-up for X-ALD, MPS I, and Pompe Disease: Recommendations Based on Stakeholder Collaboration. In: Health MDo, ed2018.
- 102.Boychuk NA, Mulrooney NS, Kelly NR, Goldenberg AJ, Silver EJ, Wasserstein MP. Parental Depression and Anxiety Associated with Newborn Bloodspot Screening for Rare and Variable-Onset Disorders. Int J Neonatal Screen. 2022;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee TY, Li CC, Liaw JJ. The lived experience of Taiwanese mothers of a child diagnosed with adrenoleukodystrophy. J Health Psychol. 2014;19(2):195–206. [DOI] [PubMed] [Google Scholar]
- 104.Kuratsubo I, Suzuki Y, Shimozawa N, Kondo N. Parents of childhood X-linked adrenoleukodystrophy: high risk for depression and neurosis. Brain Dev. 2008;30(7):477–482. [DOI] [PubMed] [Google Scholar]
- 105.McGowan R. Beyond the disorder: one parent’s reflection on genetic counselling. J Med Ethics. 1999;25(2):195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lipton SK. Marital interaction and marital consensus as they are associated with grief intensity in parents who have experienced and/or anticipate the death of a child with adrenoleukodystrophy. Ph.D., Ann Arbor, University of Maryland, Baltimore; 1990.
- 107.Grant N, Von Handorf R, Karaa A, Skotko BG. The experiences and support needs of siblings of people with mucopolysaccharidosis. Am J Med Genet A. 2021;185(11):3418–3426. [DOI] [PubMed] [Google Scholar]
- 108.Bauk K, D’Auria JP, Andrews A, Presler CM. The pediatric sibling donor experience in hematopoietic stem cell transplant: an integrative review of the literature. J Pediatr Nurs. 2013;28(3):235–242. [DOI] [PubMed] [Google Scholar]
- 109.Wiener LS, Steffen-Smith E, Fry T, Wayne AS. Hematopoietic stem cell donation in children: a review of the sibling donor experience. J Psychosoc Oncol. 2007;25(1):45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moser AB, Seeger E, Raymond GV. Newborn Screening for X-Linked Adrenoleukodystrophy: Past, Present, and Future. Int J Neonatal Screen. 2022;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bailey DB. Early Intervention and Newborn Screening Parallel Roads or Divergent Highways? Infant Young Child. 2021;34(1):3–16. [Google Scholar]
- 112.Muhammad CM, Moonaz SH. Yoga as Therapy for Neurodegenerative Disorders: A Case Report of Therapeutic Yoga for Adrenomyeloneuropathy. Integr Med (Encinitas). 2014;13(3):33–39. [PMC free article] [PubMed] [Google Scholar]
- 113.Goodison W, Baron F, Seary C, Murphy E, Lachmann R, Stevenson VL. Functional electrical stimulation to aid walking in patients with adrenomyeloneuropathy: A case study and observational series. JIMD Rep. 2022;63(1):11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Regelmann MO, Kamboj MK, Miller BS, et al. Adrenoleukodystrophy: Guidance for Adrenal Surveillance in Males Identified by Newborn Screen. J Clin Endocrinol Metab. 2018;103(11):4324–4331. [DOI] [PubMed] [Google Scholar]
- 115.Orchard PJ. Cellular Therapy in Rare Childhood Neurologic Disease: Lessons, Outcomes, and Access. J Child Neurol. 2018;33(14):877–881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed in the study.