

SUMMARY
Binding of arrestin to phosphorylated G protein-coupled receptors (GPCRs) is crucial for modulating signaling. Once internalized, some GPCRs remain complexed with β-arrestins, while others interact only transiently; this difference affects GPCR signaling and recycling. Cell-based and in vitro biophysical assays reveal the role of membrane phosphoinositides (PIPs) in β-arrestin recruitment and GPCR-β-arrestin complex dynamics. We find that GPCRs broadly stratify into two groups, one that requires PIP binding for β-arrestin recruitment and one that does not. Plasma membrane PIPs potentiate an active conformation of β-arrestin and stabilize GPCR-β-arrestin complexes by promoting a fully engaged state of the complex. As allosteric modulators of GPCR-β-arrestin complex dynamics, membrane PIPs allow for additional conformational diversity beyond that imposed by GPCR phosphorylation alone. For GPCRs that require membrane PIP binding for β-arrestin recruitment, this provides a mechanism for β-arrestin release upon translocation of the GPCR to endosomes, allowing for its rapid recycling.
Graphical Abstract
In brief
By acting as allosteric regulators of arrestin conformation, membrane phosphoinositides potentiate an active conformation of arrestin and stabilize GPCR-arrestin complexes. Membrane PIPs can therefore drive GPCR conformational diversity beyond what is seen from GPCR phosphorylation alone and provide spatial control for GPCR-arrestin complex formation.
INTRODUCTION
G protein-coupled receptor (GPCR) activation and deactivation are tightly regulated, allowing them to achieve robust signaling. GPCR deactivation is a multi-step process divided into an acute and a prolonged phase (Rajagopal and Shenoy, 2018). In addition to promoting G protein engagement, agonist stimulation leads to the recruitment of GPCR kinases (GRKs), which phosphorylate the receptor and trigger recruitment of two β-arrestin proteins (βarr1 and βarr2; Komolov and Benovic, 2018). βarrs block further G protein engagement, termed acute desensitization, and can also promote GPCR internalization through clathrin-mediated endocytosis. Once internalized, GPCRs have markedly different trafficking itineraries, with some rapidly recycled to the plasma membrane (PM), while others are retained in intracellular compartments or degraded at lysosomes (Hanyaloglu and von Zastrow, 2008). The recent discovery that GPCRs can signal from intracellular compartments has led to a re-framing of GPCR signaling to include not only temporal regulation but also differences that result from spatially distinct GPCR populations (Irannejad et al., 2013, 2015; Lobingier and von Zastrow, 2019).
Although most GPCRs recruit βarrs, the nature and duration of this interaction can differ substantially, leading to the categorization of GPCRs as either “class A” (transient) or “class B” (stable) βarr binders (Oakley et al., 2000, 2001; Zhang et al., 1999). These different stabilities are also correlated with distinct rates of recycling and resensitization, with class A GPCRs generally doing both more rapidly than class B GPCRs (Oakley et al., 1999). While it is clear that a cluster of phosphorylation sites found in class B GPCRs (Oakley et al., 2001) facilitates the persistent βarr association, it remains unclear what precipitates the release of βarrs from class A GPCRs, believed to be a prerequisite for receptor dephosphorylation and recycling.
An integral part of GPCR/βarr complex formation is βarr activation. It requires recruitment to an activated and phosphorylated GPCR followed by displacement of βarr’s auto-inhibitory C terminus by the GPCR’s phosphorylated cytoplasmic tail (C-tail) or, in some instances, by its third intracellular loop (ICL3) (Sente et al., 2018; Asher et al., 2022). Once the βarr C terminus is displaced, structural rearrangements occur that allow βarr to fully engage the GPCR (Sente et al., 2018). Recent studies have shown that the resulting complex can adopt multiple distinct poses, for some of which βarr binds only the phosphorylated C-tail (tail-engaged) or for others where both the C-tail and the transmembrane (TM) core of the GPCR are bound (fully engaged) (Shukla et al., 2014; Latorraca et al., 2018; Staus et al., 2018). While full engagement is necessary for GPCR desensitization (Kumari et al., 2017), it is not required for internalization (Cahill et al., 2017). In addition to the tail and fully engaged poses, βarr itself has been shown to adopt multiple active conformations (Nuber et al., 2016; Lee et al., 2016), thought to arise from different inputs (i.e., GPCR phosphorylation patterns), which may explain how βarrs can differentially engage signaling partners to produce distinct downstream responses (Latorraca et al., 2020; Reiter et al., 2012; Song et al., 2009).
Recent observations have also expanded our understanding of βarr activation from relying on a strict 1:1 GPCR:βarr interaction to incorporate the ability of some class A GPCRs to recruit super-stoichiometric quantities of βarr in clathrin-coated structures (CCSs). Observations of “catalytically activated” βarrs led to the proposal that an interaction with membrane phosphatidylinositol 4,5-bisphosphate (PIP2) may stabilize βarrs at the PM without requiring a bound GPCR (Eichel et al., 2018). Components of the endocytic machinery, such as the clathrin adapter two complex (AP2) (Kadlecova et al., 2017), and βarrs (Gaidarov et al., 1999) have been shown to bind PIPs. These lipids help define the identity of subcellular compartments, act as coincidence detectors for protein-protein recognition, and ensure that trafficking occurs in the appropriate subcellular context (Di Paolo and De Camilli, 2006; De Matteis and Godi, 2004). Recent structural work that showed PIP2 bound at the interface between the neurotensin type 1 receptor (NTSR1) and βarr1 (Huang et al., 2020), together with these observations of “catalytic activation,” inspired us to ask the following: what role do membrane PIPs serve in mediating GPCR-βarr complex assembly?
Here, we show that some GPCRs require PIP binding for βarr recruitment. We further show that the requirement of PIPs depends on specific GPCR phosphorylation sites and broadly adheres to the previously described class A/B typology. Using in vitro biochemical and biophysical assays, we demonstrate that PIP binding contributes to the stability of GPCR-βarr complexes by stabilizing the fully engaged state of the complex. We also find that PIPs alone promote activation of βarrs, providing a biophysical understanding for catalytic activation of βarrs. Overall, our results describe how GPCRs use membrane identity, together with GPCR phosphorylation, to modulate their engagement with βarrs. This integration allows for GPCRs to regulate βarr dissociation (class A) or maintain association (class B) that allows sustained G protein signaling from intracellular compartments.
RESULTS AND DISCUSSION
A PIP-binding-deficient mutant of βarr2 (K233Q/R237Q/K251Q, henceforth 3Q; homologous mutations in βarr1 are K232Q/R236Q/K250Q) was previously shown to be impaired for internalization of the β2 adrenoceptor (β2AR) (Gaidarov et al., 1999) and failed to traffic to CCSs, despite being recruited to PM upon GPCR stimulation (Eichel et al., 2018). Based on a recent structure of the related β1 adrenoceptor (β1AR) (Lee et al., 2020), we can exclude the possibility that mutation of these residues affects binding by disrupting direct contacts to the GPCR (Figure S1A). Further, as the behavior of 3Q βarrs paralleled the effects seen when membrane PIP2 levels were pharmacologically or chemogenetically reduced (Eichel et al., 2018; Fillion et al., 2019; Tóth et al., 2012), we reasoned these mutants would be useful to test the role of membrane PIPs in βarr recruitment to GPCRs, without the pleotropic effects and challenges associated with directly altering them.
βarr PIP binding is important for desensitization of endogenous β2AR
Given the phenotype of βarr2 (3Q), we wondered whether it could desensitize β2AR (Figure 1A). We monitored real-time cAMP production elicited by endogenous β2AR (Tewson et al., 2016) in HEK293 cells lacking both βarrs (O’Hayre et al., 2017). In the absence of exogenously expressed βarr2, isoproterenol (iso) stimulation led to a sustained cAMP response. Expression of wild-type (WT) βarr2, but not βarr2 (3Q) (Figures 1B, 1C, S1B, and S1C), enhanced desensitization, suggesting that the PIP-binding function of βarrs plays an important role in GPCR desensitization and does so at endogenous levels of GPCR expression.
Figure 1. βarr PIP binding is important for recruitment to and desensitization of GPCRs.
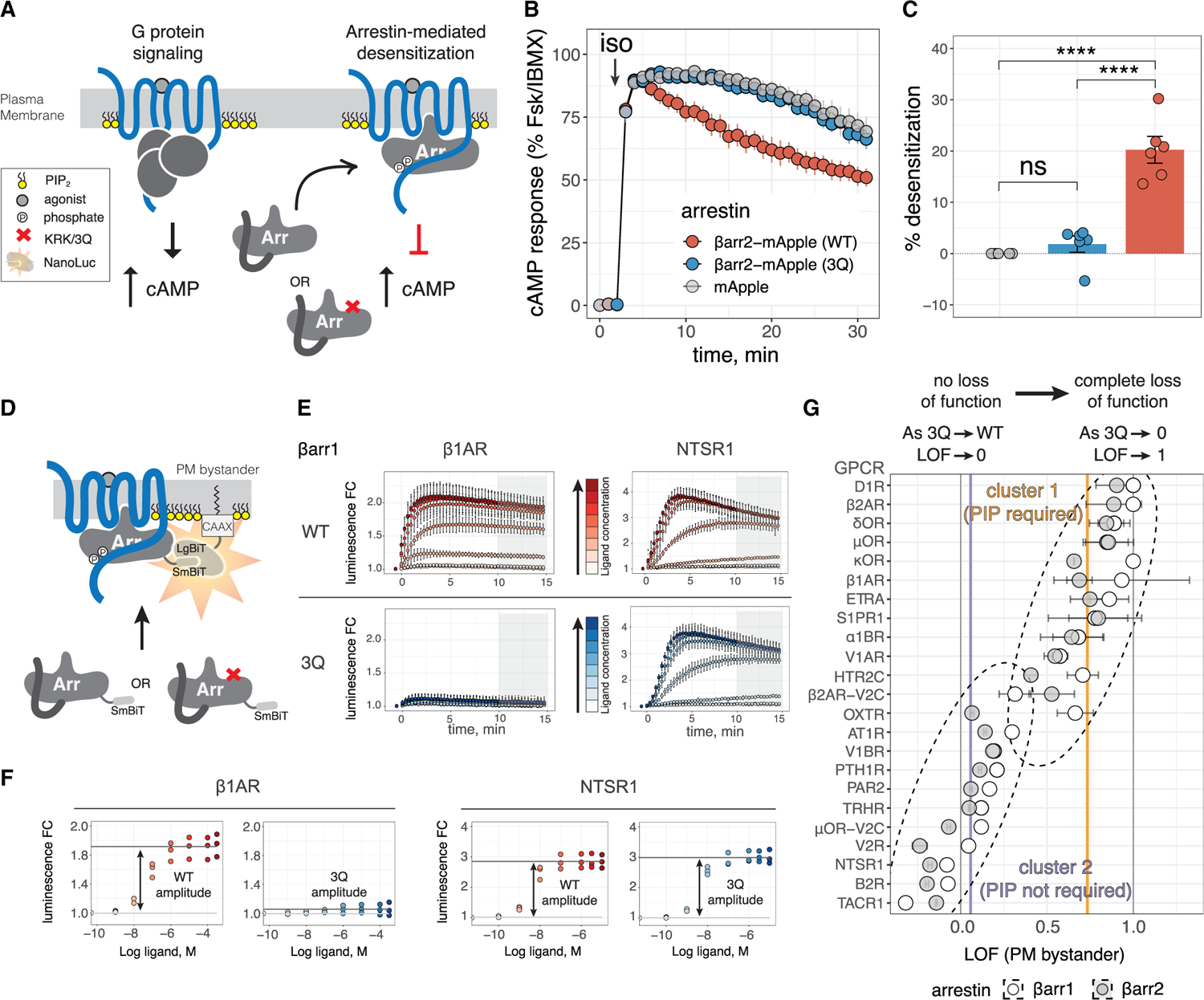
(A) Cartoon depicting GPCR signaling and desensitization. In key, “phosphate” denotes phosphorylated Ser/Thr residues.
(B) cAMP response in Δβarr1/2 HEK293 cells upon stimulation of endogenous β2AR with isoproterenol (iso). Data are normalized to response with Forskolin (Fsk) plus 3-isobutyl-1-methylxanthine (IBMX) and shown as mean ± SEM (n = 6; hereafter n denotes numbers of independent experiments).
(C) Desensitization (percentage reduction relative to mApple) measured as area under curve shown in (B). Conditions were compared by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. ****p < 0.0001; ns, p > 0.05.
(D) NanoBiT assay measuring βarr translocation to PM upon stimulation with GPCR agonist. Complementation of SmBiT and LgBiT forms a functional luciferase (see key in A).
(E) Two representative GPCRs illustrate data obtained for βarr recruitment in the NanoBiT assay. Luminescence was measured over time after agonist addition (at t = 0 min), and values are shown as fold change (FC) over vehicle treatment ± SD (n = 3–4 performed in technical duplicate). Colors denote concentration of agonist (iso for β1AR and neurotensin for NTSR1) used for stimulation. Gray boxes show the integration range for endpoint values used for CRCs.
(F) CRCs obtained from data shown in (E).
(G) LOF values obtained for 23 GPCRs. Points are LOF and error bars are standard error in LOF. Dashed ellipses denote clusters obtained from k means clustering of data. Vertical gray lines denote LOF = 0 and LOF = 1; vertical purple and orange lines are the centers of the respective clusters and correspond to LOF = 0.06 and LOF = 0.73, respectively.
GPCRs stratify into two groups based on their reliance on PIP binding for βarr recruitment
A previous study showed impaired recruitment of 3Q βarr2 to β2AR, but seemingly not to chimeric GPCR β2AR-V2C, which bears the C-tail of the vasopressin V2 receptor (V2R) (Eichel et al., 2018), suggesting that GPCRs differ in their dependence on βarr PIP-binding for recruitment. To investigate this for a wide range of GPCRs, we used a bystander NanoBiT assay (Dixon et al., 2016; Xu et al., 2022), where N-terminally SmBiT-fused βarr complements with a LgBiT fused to a PM-localizing motif (CAAX). PM recruitment of βarrs is measurable in real time in cells as a luminescence fold change (FC) (Figure 1D). We selected a set of 23 GPCRs (Table S1) and compared agonist-induced recruitment of WT βarrs with the corresponding 3Q mutants (Figure 1E). We measured endpoint luminescence FC (avg. 10–15 min post-stimulation) and generated sigmoidal concentration response curves (CRCs). From these CRCs we obtained a recruitment amplitude for each GPCR-βarr pair (Figure 1F; Data S1) and calculated a metric that reflected the relative sensitivity of βarr recruitment to the 3Q mutation, which we termed the loss-of-function index (LOF) (see STAR Methods). GPCRs with a low LOF value recruit WT and 3Q βarrs to the PM similarly and thus do not require PIP binding; GPCRs with a high LOF value show greatly diminished recruitment of 3Q βarr and require PIP binding (Figure 1G). Both WT and 3Q forms of βarr1 and βarr2 express similarly (Figure S1D; Data S1).
Although GPCRs spanned a continuum of LOF values, they broadly clustered into two groups. A k means clustering (STAR Methods) of the 57 pairs demonstrated a weak inverse correlation between the amplitude of WT βarr recruitment and LOF (Pearson correlation = −0.53; −0.43 excluding the tachykinin receptor 1 (TACR1) and B2 bradykinin receptor (B2R)) (Figure S1E), suggesting that differences in LOF were not due to lower levels of WT recruitment. Cluster 1 (high LOF, center = 0.73) and cluster 2 (low LOF, center = 0.06) included those previously classified as class A and class B GPCRs (Table S1), respectively (Oakley et al., 2000). Two chimeric GPCRs, β2AR-V2C and an analogous chimera of the mu-type opioid receptor (μOR-V2C), both showed a reduced dependence on βarr PIP-binding, compared with the original GPCRs. The vasopressin V1A receptor (V1AR), previously shown to undergo rapid recycling (Innamorati et al., 1998a, 1998b), clusters with class A GPCRs in cluster 1, whereas the vasopressin V1B receptor (V1BR) (Perkovska et al., 2018), whose proximal C-tail more closely resembles that of V2R, clusters with class B GPCRs in cluster 2. In addition, β1AR, the sphingosine 1-phosphate receptor 1 (S1PR1), delta-type opioid receptor (δOR), and kappa-type opioid receptor (kOR), all of which have been shown to either recycle rapidly or interact transiently with βarrs (Nakagawa and Asahi, 2013; Martínez-Morales et al., 2018; Trapaidze et al., 2000; Li et al., 2002), were in cluster 1. Similarly, GPCRs known to co-localize with βarrs at endosomes, including the proteinase-activated receptor 2 (PAR2) (Déry et al., 1999; DeFea et al., 2000), B2R (Khoury et al., 2014), and the parathyroid hormone/parathyroid hormone-related peptide receptor (PTH1R) (Feinstein et al., 2011), were in cluster 2. Two GPCRs, the oxytocin receptor (OXTR) and the 5-hydroxytryptamine receptor 2C (HTR2C), displayed unexpected behavior where βarr1 recruitment was more sensitive to loss of PIP binding than βarr2, with the OXTR-βarr pairs being divided between the two clusters. OXTR was previously classified as a class B GPCR (Oakley et al., 2001); however, these studies only examined βarr2. Using a direct NanoBiT assay where SmBiT and LgBiT were fused to the GPCR C terminus and βarr N terminus, respectively, we found that it paralleled recruitment measured with the PM bystander (Figure S1F; Data S1), and LOF measured in the PM bystander assay was strongly correlated to LOF measured in the direct assay (Pearson correlation = 0.88). These results show that a bystander NanoBiT assay effectively reports on the nature of βarr complexes, precluding the need to directly modify a GPCR of interest.
While our LOF term captures differences in signal efficacy, we wondered whether there was a difference in potency between WT and 3Q βarrs. We compared the EC50 values of the dose response curves generated for both PM bystander and direct recruitment and found no significant differences between WT and 3Q (Figures S1G and S1H).
Given that class B GPCRs co-localize with βarrs at endosomes, we wondered whether PIP binding affected this process. Since βarr1 and βarr2 displayed similar behavior for most GPCRs, we focused on βarr1 for these experiments (Data S1) and tested βarr2 only for selected cases (Data S1). We used an endosomal bystander LgBiT, utilizing the FYVE domain of endofin (Namkung et al., 2016), in combination with the SmBiT-βarrs. This assay robustly detected endosomal translocation; all GPCRs known to co-localize with βarrs at endosomes did so (Figure S1I). OXTR showed stronger endosomal recruitment of βarr2, compared with βarr1 (Data S1), consistent with their PM recruitment pattern (Figure 1G). By contrast, HTR2C showed a preference for βarr1 (Data S1). As expected, cluster 1 GPCRs whose ability to co-localize with βarrs at endosomes had not been described showed little signal for endosomal translocation for WT and 3Q βarrs, whereas cluster 2 GPCRs showed robust signal for recruitment of both WT and 3Q βarrs to endosomes.
Even though endpoint assessment of βarr1 recruitment to NTSR1 and other cluster 2 GPCRs was largely unaffected by the loss of the PIP-binding site, prior NTSR1 experiments found that a loss of PIP binding slowed the kinetics of βarr1 recruitment (Huang et al., 2020), suggesting PIP2 may play a role in the complexes formed with cluster 2 GPCRs. We evaluated the rate of βarr translocation to the PM in response to stimulation of cluster 2 GPCRs, using the PM bystander assay. As seen for NTSR1 (Figure 2A), other cluster 2 GPCRs showed a consistently slower association speed for 3Q than WT (Figures 2B and S2A). There also appeared to be a trend where βarr2 showed, on average, a greater reduction in rate of translocation to cluster 2 GPCRs than βarr1 (Figure S2B). These results show that even cluster 2 GPCRs are impacted by the loss of PIP binding in βarrs.
Figure 2. Loss of βarr PIP binding slows βarr recruitment to strongly coupled GPCRs.
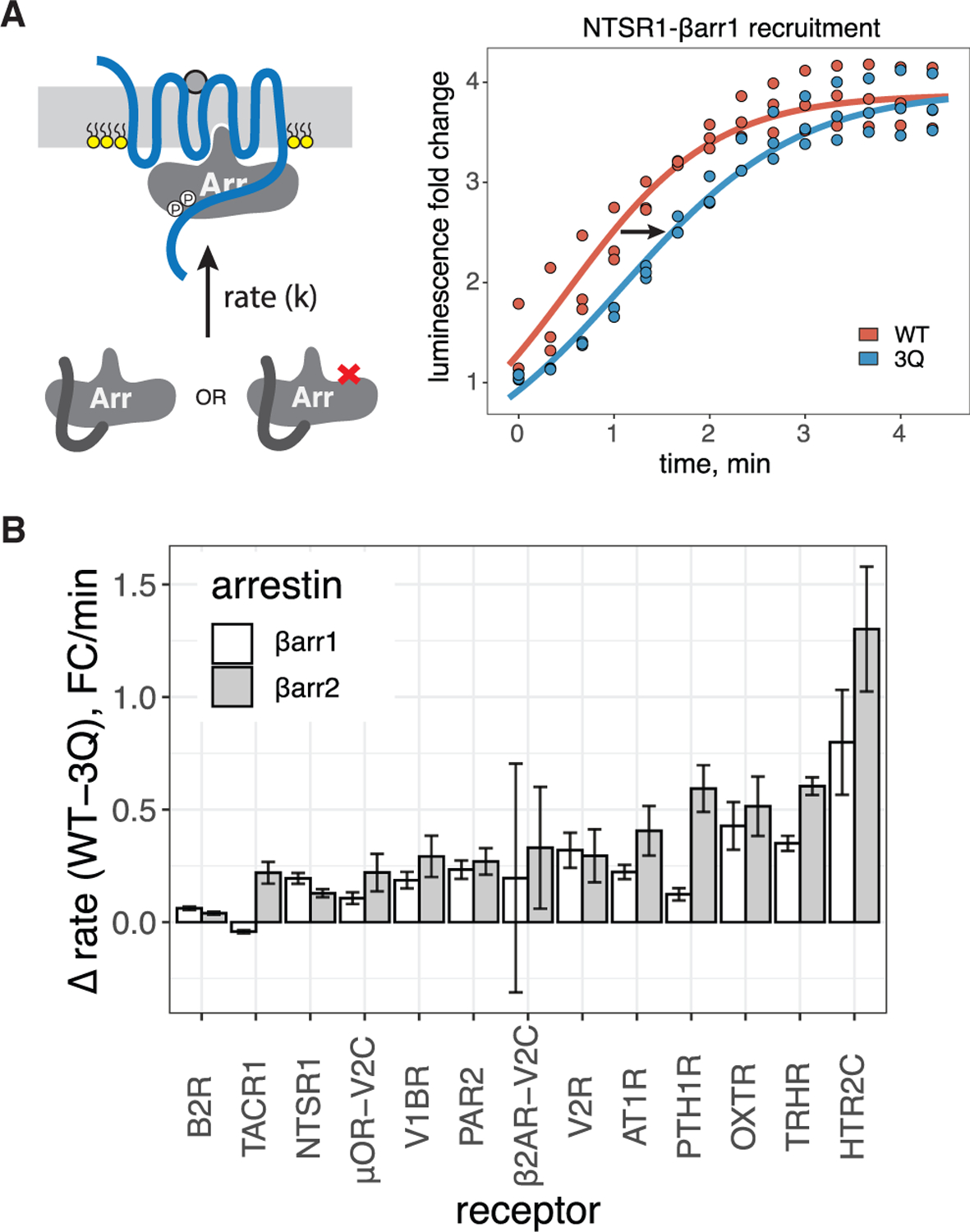
(A) Left, cartoon depicting βarr translocation kinetics. Right, representative fits of initial rates for translocation of βarr1 (WT or 3Q) to PM upon stimulation of NTSR1.
(B) Difference in initial rate between WT and 3Q βarr1/2. Data are mean ± SEM (n = 3).
These results provide several major findings. While the tested GPCRs can be divided into two groups, the reliance on PIP binding for βarr recruitment exists on a continuum. Generally, GPCRs that co-localize with βarrs at endosomes do not require the PIP-binding ability of βarrs for PM recruitment. Although these GPCRs do not require βarr PIP-binding for recruitment, the kinetics of recruitment is negatively impacted by the loss of PIP binding, suggesting that PIP-mediated interactions may stabilize GPCR-βarr complexes for all GPCRs. Finally, although βarr1 and βarr2 behave similarly for most GPCRs, there are exceptions; in much the same way that a continuum of LOF values is observed, this suggests that GPCR-βarr complexes are incredibly diverse in their sensitivity to allosteric inputs and possibly their conformational landscape.
GPCR phosphorylation patterns affect the dependence on PIP binding by βarrs
The distinction between class A and class B GPCRs was previously attributed to the presence of suitably positioned phosphosite clusters in the GPCR C-tail (Oakley et al., 2001), leading us to hypothesize that some particular GPCR phosphorylation could overcome the dependence on βarr-PIP binding for recruitment to class A GPCRs. We chose the NTSR1 as a model GPCR since WT NTSR1 strongly associated with βarrs, and the major phosphorylation cluster responsible for this phenomenon was identified in the rat ortholog (Oakley et al., 2001). We examined mutants of human NTSR1 where putative phosphorylated sites in both the C-tail (two clusters) and ICL3 were mutated to alanine (Figure 3A). All NTSR1 mutants showed similar cell-surface expression (Figure S3A), except for ICL3–4A, which was slightly reduced; however, at the level of expression used in our assays, these differences in cell-surface expression would be unlikely to impact NanoBiT responses (Figures S3B and S3C). Although WT NTSR1 is a cluster 2 GPCR, NTSR1 phosphorylation mutants were split across clusters 1 and 2 (Figure 3A; Data S1), suggesting that the loss of specific phosphorylation sites rendered βarr recruitment to NTSR1 PIP dependent.
Figure 3. NTSR1 phosphorylation patterns govern PIP dependence for βarr recruitment.
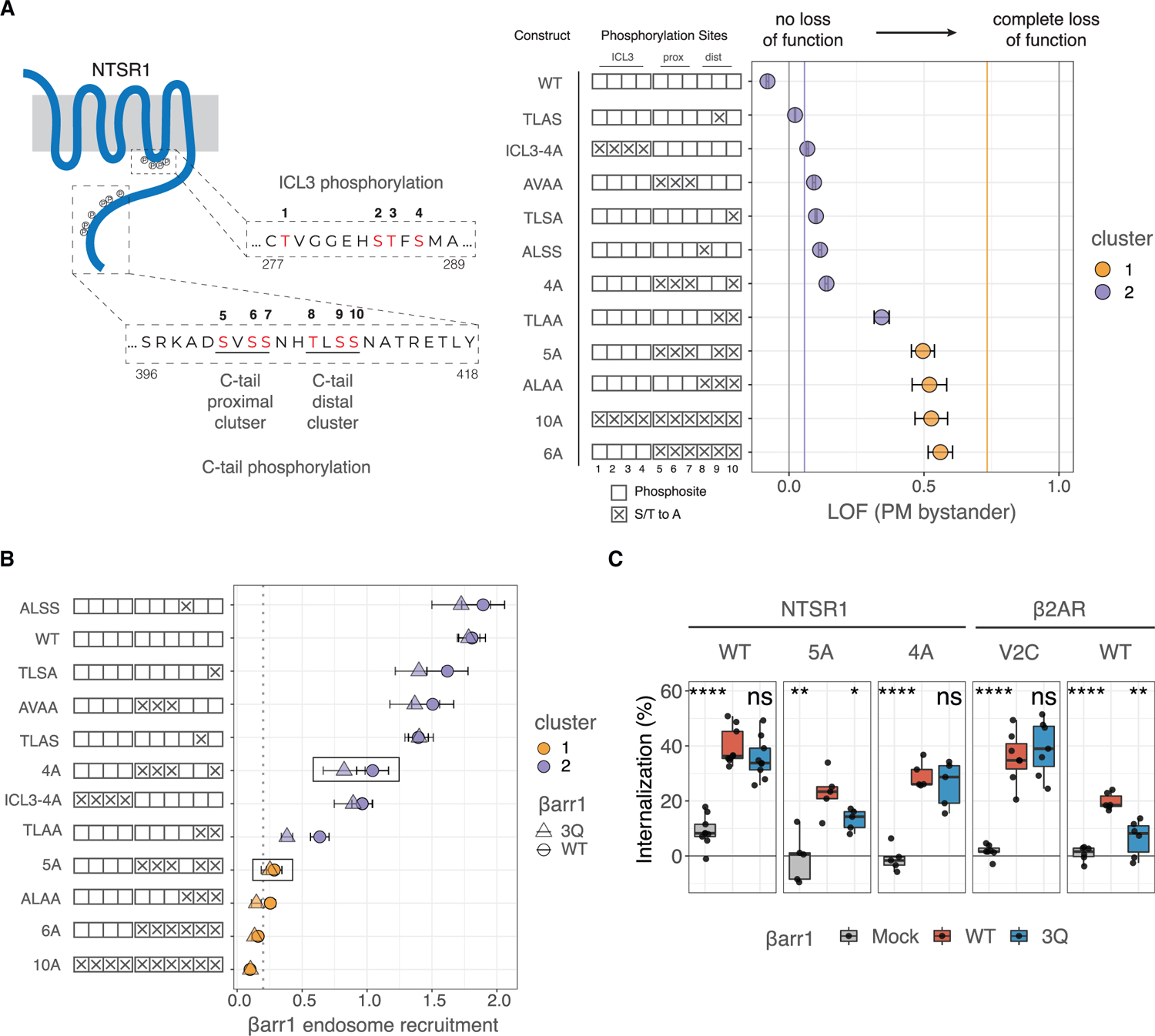
(A) Left, cartoon of human NTSR1 showing motifs in ICL3 and C terminus that are subject to phosphorylation. Phosphorylation sites examined in this study are in red (numbered 1–10). Residue numbers corresponding to the region of human NTSR1 are listed at the start and end of sequences. Construct key shows possible phosphosites as empty boxes, which when mutated to alanine are denoted by “X.” Right, LOF for recruitment of βarr1 to different NTSR1 constructs, measuring by PM bystander NanoBiT assay. Data are mean ± SEM (n = 3). Vertical gray lines denote LOF = 0 and LOF = 1; vertical purple and orange lines are the centers of clusters 1 and 2 (LOF = 0.06 and LOF = 0.73, respectively) as shown in Figure 1G.
(B) Translocation of βarr1 to endosomes upon agonist stimulation as measured by the endosomal bystander NanoBiT assay (Figure S1I). Data indicate recruitment (fold change over basal upon stimulation) for WT and 3Q βarr1, shown as circles and triangles, respectively. Shapes and error bars are mean and SEM, respectively, from n = 3. Points are colored by cluster designation obtained from k means clustering of all GPCR-βarr recruitment data.
(C) Flow-cytometry-based GPCR internalization assay. Δβarr1/2 cells expressing N-terminally FLAG-tagged NTSR1 or β2AR constructs along with βarr constructs indicated were stimulated with agonist (neurotensin or iso, respectively). Data show loss of cell-surface receptors (n = 5–10). Internalization by 3Q βarr1 and mock were each compared with WT using a two-tailed paired t test. ns, p > 0.05; *p % 0.05, **p % 0.01, ***p % 0.001, and ****p % 0.0001.
Removal of the two C-tail phosphorylation site clusters (NTSR1–6A and −10A) resulted in a dramatic reduction in βarr recruitment (Data S1), with remaining βarr recruitment being PIP dependent (Figure 3A). Despite contacts between ICL3 phosphorylation sites and βarr1 in a recent structure (Huang et al., 2020), removal of ICL3 phosphorylation sites did not affect PIP dependence (NTSR1-ICL3–4A), and similar results were obtained upon mutating the proximal phosphosite cluster (NTSR1-AVAA) as well as point mutations in the distal phosphosite cluster (NTSR1-TLSA, -ALSS, and -TLAS). However, triple mutation of the distal phosphorylation cluster (NTSR1-ALAA) led to a dramatic reduction in recruitment and an increase in PIP dependence. NTSR1–5A, bearing a single C-terminal phosphorylation site in the distal phosphosite cluster, showed PIP sensitivity comparable to NTSR-ALAA, whereas NTSR1–4A with two phosphorylatable residues in the distal phosphosite cluster showed much less PIP dependence, suggesting that two phosphorylation sites are sufficient to overcome PIP dependence. Similarly, NTSR1-TLAA, which differs from NTSR1–5A only in the addition of the proximal cluster of phosphosites, exhibits intermediate sensitivity between NTSR1–5A and −4A constructs, suggesting that a phosphorylation site from the proximal cluster may serve as a partial rescue for the absence of one in the distal cluster. Comparing the rate of βarr1 translocation to the PM for each NTSR1 construct, we found that those in cluster 2 (Figure 3A) tended to recruit βarr1 more rapidly than those in cluster 1 (Figure S3D), but all variants showed slower recruitment of 3Q compared with WT βarr1 (Figure S3E).
Using the endosomal bystander assay, we found that cluster 2 NTSR1 mutants promoted translocation of βarr1 to endosomes (Figure 3B; Data S1), whereas cluster 1 NTSR1 mutants did not. To validate the importance of the two phosphosites in the distal phosphosite cluster, we measured internalization of the NTSR1 mutants using βarr1/2-deficient HEK293 cells where either WT or 3Q βarr1 was reintroduced. WT NTSR1 was robustly internalized by both WT and 3Q βarr1. By contrast, NTSR1–5A showed a significant difference in internalization between WT and 3Q βarr1, whereas NTSR1–4A showed no difference in internalization between the two. The trend between NTSR1–5A and NTSR1–4A parallels that seen for β2AR and β2AR-V2C (Figure 3C). Together, these data show that two suitably positioned phosphorylation sites are sufficient to render βarr1 PIP binding dispensable for PM recruitment, allow robust βarr-dependent internalization, and support βarr translocation to endosomes. Given that GPCRs, such as the μOR, can have different phosphorylation patterns depending on the stimulating agonist (Just et al., 2013), we speculate that the resulting βarr complexes may also have different behaviors in cells.
PIP2 binding affects complex stability and dynamics in vitro
We next examined how PIP binding affects GPCR-βarr complex formation in vitro. Using NTSR1 as a model, we compared the ability of GRK5-phosphorylated NTSR1 (GRK5p) to form a complex with βarr1 (WT or 3Q mutant) in the presence of a soluble PIP2 derivative, diC8-PI(4,5)P2 (henceforth PIP2), by size-exclusion chromatography (SEC) (Figures 4A and 4B). Although NTSR1 is a strong coupler, complexing with full-length WT βarr1 resulted in only 25% complex following SEC, and 3Q βarr1 resulted in an even smaller amount (<5%) of complex (Figure 4C). Using a C-terminally truncated, pre-activated βarr1 (residues 1–382; ΔCT hereafter) enhanced complex formation by more than 2-fold, and this was only slightly reduced with the corresponding 3Q βarr. The LOF metric analysis quantitatively showed a greater LOF in the full-length βarr1 than in βarr1 ΔCT, suggesting that removal of the βarr1 C terminus partially overcomes the impairment in complex formation of the 3Q mutation (Figure S4A). PIP2 affinity in vitro was reduced 20× for 3Q βarr1 compared with WT (Figure S4B). Since βarr activation proceeds via release of its C terminus, we tested whether the lack of 3Q βarr1 complex formation was caused by impaired C-terminal release. We designed a Förster resonance energy transfer (FRET)-based sensor to report on βarr1 C-terminal release (Figure S4C). We introduced two cysteine residues, A12C and V387C, into a cysteine-free βarr1 background and stochastically labeled these positions with AlexaFluor 488 and ATTO647N. NTSR1 (GRK5p) robustly displaced the C terminus of both WT and 3Q βarr1 (12–387) (Figure S4D), and the displacement was comparable to that seen for a saturating concentration of a phosphorylated C-terminal peptide of the V2R (V2Rpp), which is known to completely displace the βarr1 C terminus (Shukla et al., 2013). Notably, NTSR1 (GRK5p) fully displaced the βarr1 C terminus with 10× greater potency than V2Rpp. These data show that not only does NTSR1 (GRK5p) fully displace the βarr1 C terminus, but it does so with higher efficacy than an equimolar concentration of phosphopeptide, and this is independent of the PIP binding ability of βarr1.
Figure 4. PIP binding stabilizes fully engaged GPCR-βarr complexes.
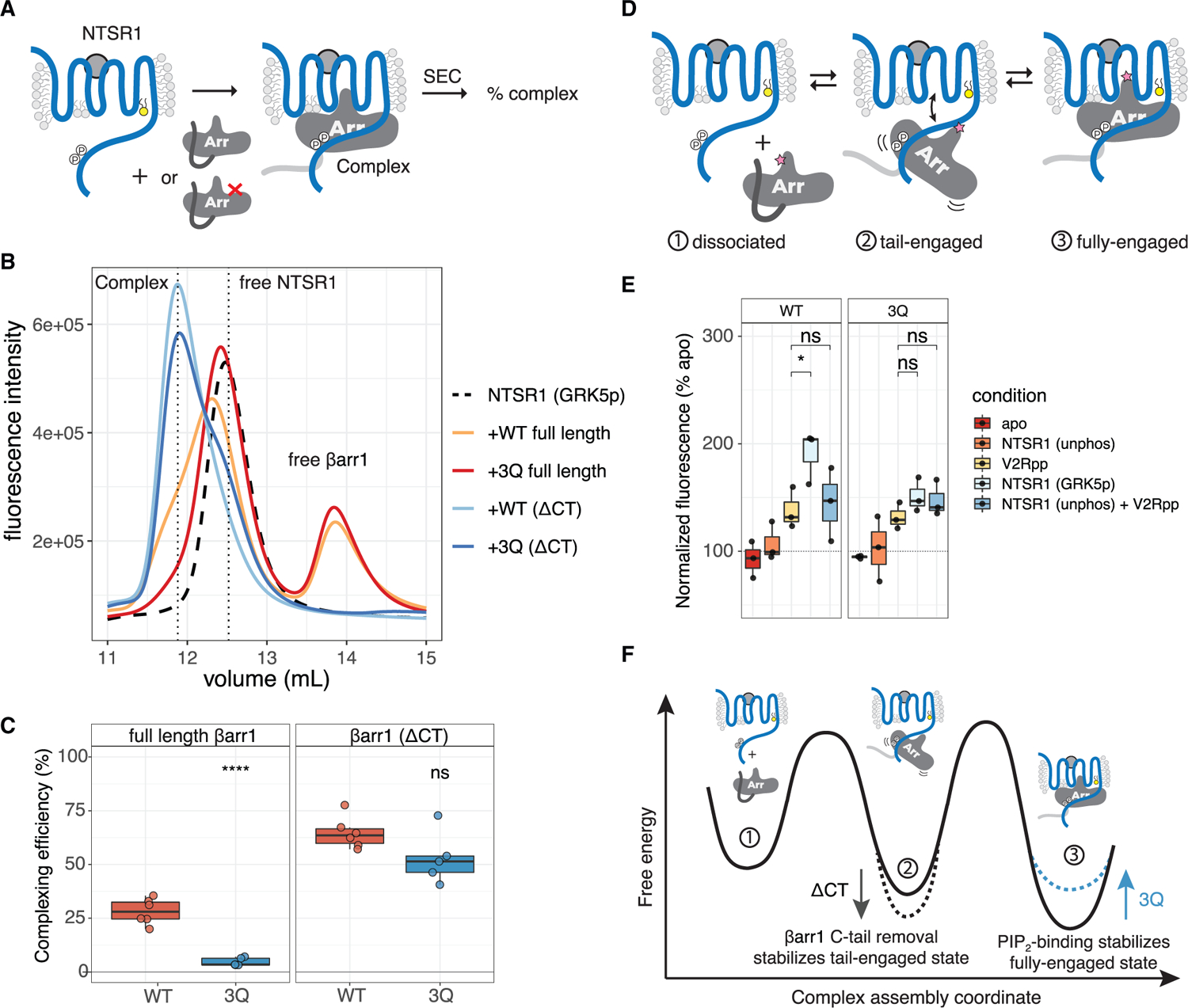
(A) Cartoon of complexing efficiency assay. SEC resolves complex from components.
(B) Representative experiment showing SEC chromatograms with vertical dashed lines indicating free NTSR1, complex, and free βarr1.
(C) Complexing efficiency for NTSR1 with the indicated βarr constructs. Individual points are shown (n = 6). Two-tailed unpaired t test used to compare conditions. ns, p > 0.05; ****p % 0.0001.
(D) Cartoon showing equilibrium of NTSR1-βarr1complex. Pink star denotes bimane probe used for experiment shown in (E).
(E) Spectra of L68bim-labeled βarr1 in complex with NTSR1. Individual points are shown (n = 3). V2Rpp-NTSR1 (GRK5p) and V2Rpp-NTSR1 (unphos) + V2Rpp were each compared by two-tailed unpaired t test. ns, p > 0.05; *p % 0.05. “Apo” indicates free βarr1; “unphos” and “GRK5p” indicate unphosphorylated and GRK5-phosphorylated NTSR1, respectively. Spectra are normalized to apo (100%) for each experiment, and the fluorescence intensity at λmax was compared.
(F) Free energy diagram illustrating how PIP binding, by stabilizing the fully engaged state of the NTSR1-βarr1 complex, slows βarr1 dissociation.
One explanation for the reduced stability of the NTSR1-βarr1 (3Q) complex may be due to a difference in the proportion of fully engaged complex being formed (Figure 4D). To test this hypothesis, similarly to the FRET sensor, we site-specifically incorporated an environmentally sensitive bimane fluorophore at position L68C on the βarr finger loop (L68bim), a region that can become buried within the GPCR TM core. A similar approach was used to report on full engagement for rhodopsin/arrestin-1 (Sommer et al., 2005, 2006) where it underwent a blue-shift and an increase in fluorescence emission as the bimane moved to the lower polarity environment of the GPCR TM core. Addition of V2Rpp alone to βarr1 (L68bim) leads to an ~50% increase in bimane fluorescence; this is thought to result from a change in environment of the bimane probe, which is from being buried in the polar βarr1 N-lobe to being solvent exposed (Latorraca et al., 2020). We speculated that the addition of intact GPCR may further increase this signal. We compared the fluorescence changes of βarr1 L68bim (WT or 3Q) upon addition of NTSR1 that was either unphosphorylated or GRK5-phosphorylated (Figure 4E). In the absence of NTSR1 phosphorylation, there was no increase in fluorescence; however, NTSR1 (GRK5p) led to a ~2-fold enhancement in fluorescence intensity for WT but a smaller 1.5-fold enhancement for 3Q. The addition of V2Rpp at a saturating concentration to the unphosphorylated NTSR1 did not result in a significant increase over phosphopeptide alone. Importantly, for βarr1 (3Q), NTSR1 (GRK5p) did not elicit a response different from that of V2Rpp alone.
Together, these data support a model of complex assembly shown in Figure 4F. Displacement of the βarr1 C terminus by the phosphorylated GPCR C-tail is rapid and reversible. The resulting tail-bound state is in equilibrium with a fully engaged state, where βarr1-PIP binding serves to stabilize this state and to thereby slow dissociation. In the context of full-length βarr1, destabilization of the fully engaged state in the 3Q mutant leads to a reduction in overall complex stability, presumably due to increased dissociation from the tail-bound state. Consistent with these findings, removal of the βarr1 C terminus, which increases the stability of the tail-bound state, resulted in a reduced impact of the 3Q mutant (Figures 4C and S4A).
PIP2, in the absence of a GPCR, triggers conformational changes in βarr1
The finding that βarrs in CCSs, even in the absence of an associated GPCR, can promote MAPK signaling (Eichel et al., 2016) suggested that βarrs can adopt an active-like conformation without remaining bound to a GPCR. While PIP2 was speculated to maintain the membrane association of βarrs (Eichel et al., 2018), the impact of PIPs on the conformational landscape of βarrs was unknown. Since PIP binding affects the dynamics of NTSR1-βarr1 complexes in vitro, we examined the effect of PIPs on βarr1 in the absence of an associated GPCR. We compared the effect of PIP2 with that of V2Rpp for promoting conformational changes in βarr1, using FRET and fluorescence reporters on the finger and gate loops (Figure 5A; Kim et al., 2013; Latorraca et al., 2020). We found that both the finger loop (Figures 5B, S5A, and S5B) and the gate loop (Figures 5C, S5C, and S5D) showed saturable conformational changes upon addition of PIP2 with the changes being smaller than those seen for V2Rpp. Importantly, the 3Q mutant responded to V2Rpp similarly as to WT βarr1 but lacked these PIP2-dependent changes. These data suggest that binding of PIP2 to the βarr1 C-lobe allosterically promotes conformational changes in key regions involved in GPCR recognition and βarr1 activation. As activation and release of the βarr1 C terminus are coupled, we wondered whether the PIP2-induced conformational changes were due to C-terminal release. Using our βarr1 C-terminal FRET sensor (Figure S4C), we found that PIP2 promoted a small movement of the C terminus (Figure 5D) but only at concentrations higher than those that saturated the finger and gate loop sensors (Figures 4B and 4C). As was the case for the other sensors, this FRET change in response to PIP2 is absent in the corresponding 3Q mutant (Figures S5E and S5F). This finding is consistent with a recent study that found little or no C-terminal displacement for βarr1 with IP6 (Chen et al., 2021). These data provide direct evidence that PIP2 affects the conformational landscape of βarr1 in the absence of a GPCR and explain how βarrs may continue to engage downstream effects after GPCR dissociation.
Figure 5. PIP2 alone promotes conformational changes in βarr1.
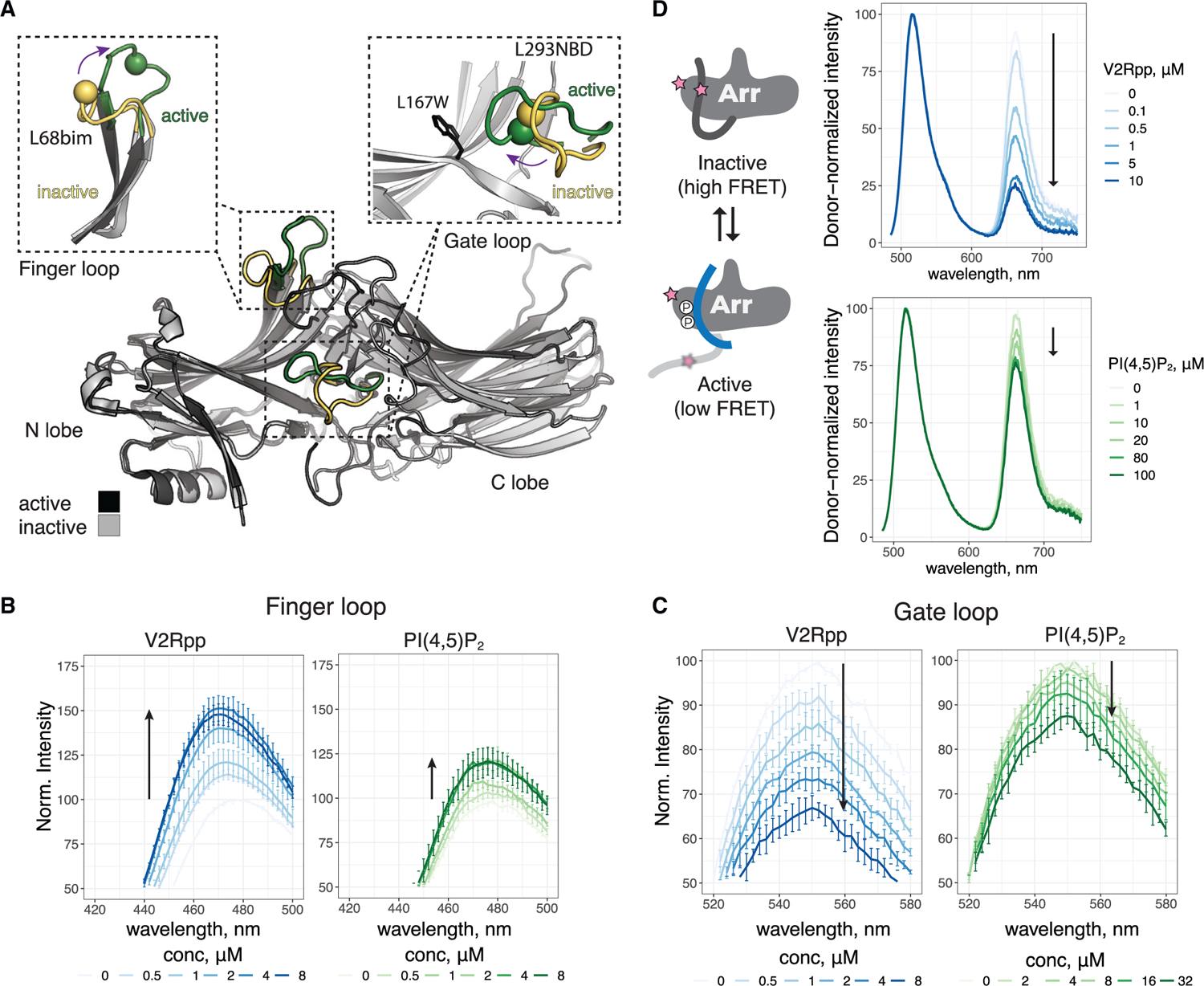
(A) Overlay of inactive (PDB: 1G4M) and active (PDB: 4JQI) βarr1. The N and C lobes of βarr1 are indicated. Activation leads to reorganization of several loops; the gate and finger loops are highlighted. Re-orientation of these loops from inactive (yellow) to active (green) is monitored by site-specific fluorescence spectroscopy. In finger loop inset, the sphere denotes Cα L68C, which is labeled with bim. In gate loop inset, the sphere denotes Cα L293C, which is labeled with NBD. The installed W residue replacing L167 that quenches 293NBD is shown.
(B) Spectra of L68bim-labeled βarr1 in response to V2Rpp and PIP2.
(C) Spectra of L167W-L293NBD-labeled βarr1 in response to V2Rpp and PIP2.
(D) Left, cartoon showing how FRET change is linked to C-terminal release. Right, spectra of AF488/AT647N-labeled βarr1 in response to V2Rpp and PIP2. In (B)–(D), arrows indicate direction of change with increasing concentration. In (B) and (C), values are mean ± SEM (n = 3), and spectra were normalized to apo for each experiment. In (D), spectra are normalized by donor intensity within a given experiment, and data are shown for a representative experiment (n = 2–4).
Intrigued by the PIP2 effect on βarr1 conformation, we wondered if other PIPs (Figure S6A), known as markers of subcellular membrane identity, could also promote conformational changes in the βarr1 finger loop. Like PI(4,5)P2, PI(3,4)P2, PI(3,5)P2, and PI(3,4,5)P3 all showed similar changes in the finger loop (Figures S6B–S6F). PI(4)P showed a weaker response, and PI(3)P and phosphatidyl glycerol (PG) did not have any effect on the βarr1 finger loop (Figures S6G–S6I). Interestingly, these results show that PM-resident PIPs (Di Paolo and De Camilli, 2006)—PI(4,5)P2, PI(3,4)P2, PI(3,4,5)P3, and to a lesser extent PI(4)P—promote conformational changes in βarr1, but that the early endosome marker PI(3)P is unable to do so (Figure S6J). PI(3,5)P2 showed a similar effect to other PIP2s but is understood to be less abundant (Hasegawa et al., 2017).
Based on contacts observed in the NTSR1-βarr1 structure (Huang et al., 2020), we speculate that PIPs bearing at least two phosphates on the inositol ring may be necessary for chelation of K232 and either R236 or K250 (Figure S6K), possibly stabilizing the active conformation of the C-lobe β strands. A single phosphate at the 4-position may be sufficient to coordinate K250 and R236, explaining the small effect seen for PI(4)P. These data show that different PIPs can promote conformational changes in βarr1 and raise the possibility of compartment-specific differences in the behavior of GPCR-βarr complexes based on their resident PIPs.
PIP2 increases the population of active βarr1
While our fluorescence experiments support PIP2-promoted conformational changes consistent with βarr activation, the lack of C-terminal release raised questions of whether these conformational changes truly reflect an increase in the population of active βarr1, as would be recognized by an intracellular binding partner. We used an engineered Fab, Fab30, which has a high affinity for the active (V2Rpp-bound) state of βarr1 (Shukla et al., 2013), as a probe for the active state of βarr1. Using surface plasmon resonance (SPR), we measured binding of Fab30 to immobilized βarr1 (Figure 5A). While Fab30 showed robust and stable binding to βarr1 in the presence of V2Rpp, it interacted only transiently in the absence of V2Rpp (Figures 6B and S7; Tables S2 and S3). Interestingly, binding was enhanced when Fab30 was co-injected with PIP2 (Figure 5B). This suggests that PIP2 increases the proportion of βarr1 in a Fab30-recognizable active-like state. We compared the effect of different additives on Fab30 binding with WT, 3Q, and ΔCT βarr1. At 1 μM, Fab30 showed 10% ± 1% (of maximum) binding to WT βarr1, compared with 57% ± 2% binding for ΔCT βarr1 (Figure 6C). This shows that Fab30 binding is favored by a conformation accessible to WT βarr1, which is enhanced by removal of the βarr1 C terminus. When Fab30 is co-injected with a saturating concentration of PIP2 (40 μM), binding to WT βarr1 increased more than 3-fold, to 34% ± 2%, compared with Fab30 alone. PIP2 had a smaller effect on βarr1 (ΔCT) but still showed increased binding from 57% to 66% ± 1%. While all three βarr1 constructs showed an increase in Fab30 binding in the presence of PIP2 (Figures S7G–S7L), the degree of enhancement was most strongly potentiated for WT βarr1 above the Kd for Fab30 (Data S1). Since removal of the βarr1 C terminus diminished the PIP2 enhancement of Fab30 binding, we reasoned that PIP2 likely acts in cis with C-terminal displacement, consistent with our FRET experiments. To test if this effect was specific to PIP2, versus other anionic lipids, we compared the ability of PG, PI(3)P, and PIP2 to enhance Fab30 binding. While PG and PI(3)P showed a small enhancement in Fab30 binding to WT, it was significantly less than that of PIP2 (Figure 6C). The difference in enhancement between PI(3)P or PG and PIP2 was absent in 3Q βarr1 and diminished for βarr1 ΔCT. Based on these and our fluorescence data, we propose that spontaneous activation of βarr1 to an active-like state capable of binding Fab30 is possible but rare (Figure 6D). V2Rpp dramatically shifts the equilibrium toward the active state by displacing the βarr1 C terminus; removal of the βarr1 C terminus is sufficient to greatly enhance the active population, even in the absence of V2Rpp or PIP2. By contrast, PIP2 does not displace the βarr1 C terminus directly but promotes its movement through an allosteric connection to the C-lobe, where PIP2 binds. While PIP2 may stabilize the same active state of βarr1 achieved with V2Rpp, albeit to a lesser extent, it may also act to stabilize an active-like state of βarr1 that is on-pathway toward activation and capable of binding Fab30, although to a lesser extent than V2Rpp-bound βarr1. Further studies will be necessary to distinguish these possibilities.
Figure 6. PIP2 enhances Fab30 binding to βarr1.
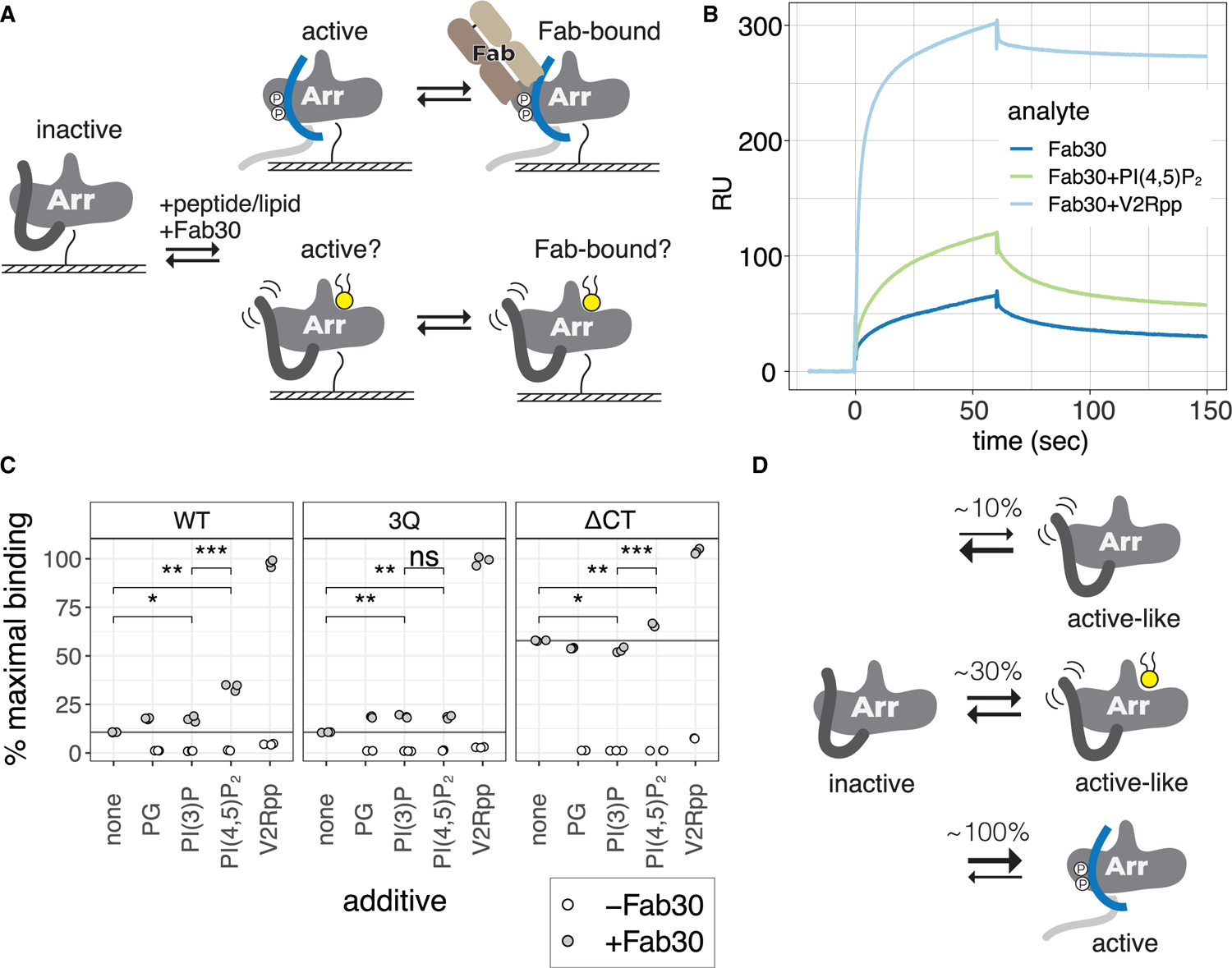
(A) Cartoon of SPR experiments, where βarr1 is immobilized via N-terminal biotinylation, and Fab30 is injected in the presence or absence of PIP2 or V2Rpp.
(B) Representative sensograms for SPR experiment with WT βarr1 immobilized.
(C) Binding of Fab30 (1 μM) to immobilized βarr1 constructs in the presence of different additives. Percentage of maximal binding is based on expected maximum response for 1:1 interaction with Fab30. Additives were each injected at 40 μM together with Fab30. Points are independent measurements (n = 3); open points show binding for additive alone. Means were compared by two-tailed unpaired t test. ns, p > 0.05; *p % 0.05, **p % 0.01, and ***p % 0.001.
(D) The proportion of active-like βarr1 increases in the presence of PIP2.
Conclusions
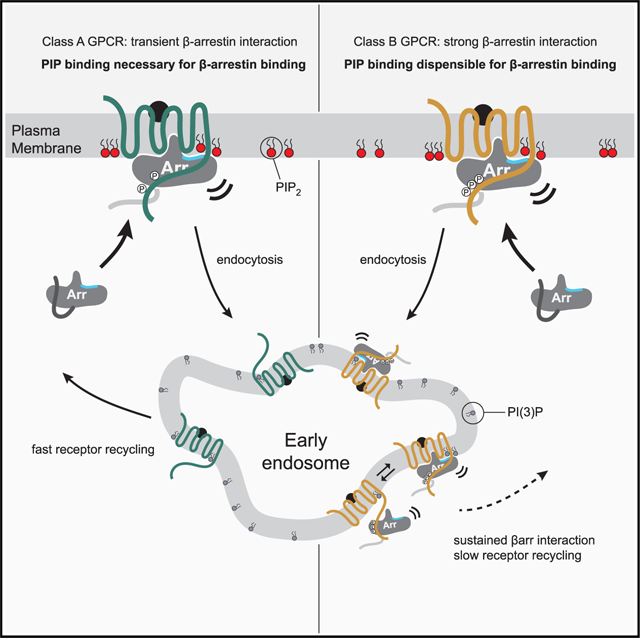
Our findings offer a molecular basis for understanding the phenotypic classification of GPCRs into class A or class B for βarr recruitment. In our model, class A GPCRs (Figure 7, left panel) require the coincident detection of membrane PIPs for recruitment to an activated and phosphorylated GPCR. Our data show that this is due to an insufficiency in phosphorylation of these GPCRs, requiring the simultaneous action of both phosphate-mediated contacts and PIP-mediated contacts to form a complex that is desensitized and internalized. A further trait of class A GPCRs is that they exhibit, to a varying degree, a catalytic activation phenotype wherein a βarr, after recruitment to an active GPCR, loses association with the GPCR but remains concentrated in CCSs. Since there is an increasing concentration gradient of PIP2 leading into CCSs (Sun et al., 2007), our data showing that PIP2 promotes conformational changes in βarr in the absence of a GPCR provide a mechanistic explanation for this phenomenon. Once a GPCR enters a CCS, endocytosis proceeds and PIP2 levels drop. We suggest that this may serve as the timing component for βarr dissociation from these PIP-dependent GPCRs (Zhang et al., 1999), making the GPCR susceptible to dephosphorylation and complete dissociation of βarr, followed by rapid recycling to the PM (Krueger et al., 1997). By contrast, class B GPCRs (Figure 7, right panel) possess phosphorylation sites that can promote association with βarrs without the need for concurrent membrane PIP binding. This allows for βarrs to co-localize with these GPCRs at endosomes. βarrs also possess loops at the edge of the C-lobe capable of interacting with the membrane in a βarr-activation-dependent manner (Lally et al., 2017). This contact likely contributes to the overall membrane-binding interface but may lack the tunability that the PIP-dependence offers class A GPCRs.
Figure 7. Model for PIP regulation of GPCR-βarr complex assembly and disassembly.
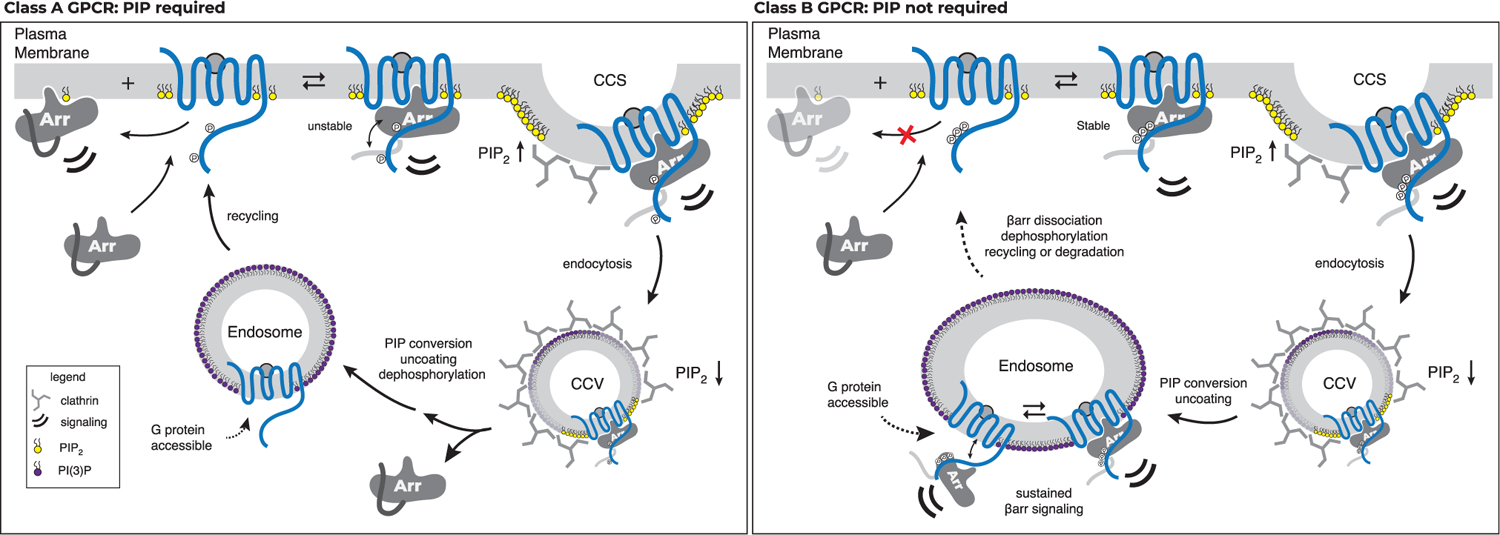
GPCRs stratify into two groups with respect to the strength of their interaction with βarrs: one group requires an interaction between βarrs and PIPs at the PM for recruitment (PIP required, left), whereas the other does not (PIP not required, right).
One question this model raises is the following: if PIP2 promotes activation of βarrs, why are βarrs not basally associated with the PM? Consistent with our SPR experiments, and the fact that a GPCR core—but not a phosphorylated C terminus—is required for βarr accumulation at CCSs (Eichel et al., 2018), we speculate that PIP binding may occur when βarrs transition to an active-like conformation at a GPCR and may stabilize this state. This is consistent with findings that GPCRs may concentrate lipids such as PIP2 in their local membrane environment (Yen et al., 2018; Song et al., 2019). Our data suggest that while PIP-mediated contacts are not necessary to maintain association, they likely affect the equilibrium between tail-engaged and fully engaged states of the complex. Tail engagement has been shown to be sufficient for MAPK signaling downstream of βarrs. We speculate that this shift in equilibrium, particularly for endosomes defined by PI(3)P, may explain how class B GPCRs, such as V2R and PTH1R, are able to remain associated with βarr while also promoting G protein signaling from endosomes (Nguyen et al., 2019; Thomsen et al., 2016).
Overall, our data offer a simple explanation for several phenotypes observed for GPCR-βarr complexes and provide a biophysical framework for understanding the interplay between phosphorylation-mediated and PIP-mediated contacts in complex assembly. A reliance on PIPs for βarr recruitment offers a robust solution for recruitment of βarrs to GPCRs, with spatial control and temporal precision. Given the interplay between PIP-dependent recruitment and phosphorylation, we believe that distinct signaling outcomes may not only be due to differences in phosphorylation alone (Latorraca et al., 2020) and can be further fine-tuned by membrane PIPs that are present in distinct subcellular locations.
Limitations of the study
These studies are limited to in-cell and in vitro experiments, each with its own caveats. In vitro experiments use GPCRs solubilized in detergent, and soluble derivatives of membrane PIPs were used (C8 versus C18 alkyl chains). In-cell experiments generally employ overexpression and were performed in HEK293 cells. To interrogate the role of membrane PIPs, we focused on a βarr mutant (3Q) rather than direct perturbation of membrane PIP levels. While indirect, this approach avoids confounding effects produced by changes to the tightly regulated pool of membrane PIPs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Brian Kobilka (kobilka@stanford.edu).
Materials availability
Plasmids generated in this study will be distributed upon request.
Data and code availability
Experimental datasets can be found in Mendeley Data: https://dx.doi.org/10.17632/ynrr4j7t4s.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
E. coli BL21(DE3) Rosetta cells (NEB) were used to express Fab30 and NiCo21(DE3) competent E. coli (NEB) were used to express all β-arrestin 1 constructs for in vitro studies.
Cell lines
β-arrestin-1/2 double knockout HEK293A cell lines (not authenticated; O’Hayre et al., 2017) were cultured in complete growth Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (UCSF Cell Culture Facility). Cultures were maintained at 37 °C in a humidity and CO2 controlled incubator. Cell line cultures were free of mycoplasma contamination.
Spodoptera frugiperda Sf9 cells (Expression systems) were used for baculovirus generation, and expression of GRK5 and NTSR1.
Trichuplusia ni Hi5 cells (Expression systems) were used for expression of Fab30.
METHOD DETAILS
Plasmid construction
For cell-based assays, we used human, full-length GPCR plasmids cloned into the pCAGGS vector or the pcDNA3.1 vectors. A list of constructs used in NanoBiT assays is provided in Table S4, including those previously described (Xu et al., 2022; Hisano et al., 2019; Huang et al., 2020; Shihoya et al., 2018; Baidya et al., 2020; Inoue et al., 2012; Kato et al., 2019; Kawakami et al., 2022). When a comparison of cell surface expression levels was necessary GPCR constructs were N-terminally tagged with a FLAG epitope. Specifically, NTSR1 was tagged with an N-terminal FLAG epitope through a linker (MDYKDDDDKGTELGS; the FLAG epitope is underlined) and inserted into the pcDNA3.1 vector. β2AR and μOR were N-terminally modified with an HA signal sequence followed by a flag epitope then a flexible linker (MKTIIALSYIFCLVFADYKDDDDKGGSGGGGSGGSSSGGG; the FLAG epitope is underlined) and inserted into the pCAGGS vector. When unspecified GPCRs are untagged. For the bystander NanoBiT-based βarr recruitment assays, human full-length βarr (βarr1 or 2; WT or 3Q) was N-terminally SmBiT-fused with the flexible linker (MVTGYRLFEEILGG SGGGGSGGSSSGG; the SmBiT is underlined) and inserted into the pCAGGS vector (SmBiT-βarr) (Baidya et al., 2020). For the PM-localizing tag, LgBiT was C-terminally fused to the CAAX motif derived from human KRAS (SSSGGGKKKKKKSKTKCVIM) through the same flexible linker (LgBiT-CAAX). For the endosome-localizing tag, LgBiT was N-terminally fused with the human Endofin FYVE domain (amino-acid regions Gln739-Lys806) again through the same flexible linker (Endo-LgBiT). For the direct NanoBiT-based βarr recruitment assay, human full-length βarr was N-terminally LgBiT-fused with the same flexible linker and inserted into the pCAGGS vector (LgBiT-βarr). GPCRs were C-terminally SmBiT-fused with the flexible linker (GGSGGGGSGGSSSGGVTGYRLFEEIL; the SmBiT is underlined) and inserted into the pCAGGS vector (GPCR-SmBiT).
mApple-N1 and βarr2 mApple were previously described (Shaner et al., 2008; Eichel et al., 2016). The βarr2 (3Q) mutant was previously described (Gaidarov et al., 1999) and generated by inserting the βarr2 (3Q) coding region from βarr2 (3Q)-EGFP (Eichel et al., 2018) into mApple-N1 using HindIII and ApaI restriction sites.
NanoBiT-β-arrestin recruitment assays
βarr recruitment to the PM was measured using a bystander NanoBiT approach with SmBiT-βarr and the LgBiT-CAAX constructs. HEK293A cells (Thermo Fisher Scientific) were seeded in a 6-cm culture dish (Greiner Bio-One) at a concentration of 2 × 105 cells per ml (4 ml per dish hereafter) in DMEM (Nissui Pharmaceutical) supplemented with 10% FBS (Gibco), glutamine, penicillin, and streptomycin, one day before transfection. The transfection solution was prepared by combining 5 μl of polyethylenimine solution (1 mg/ml) and a plasmid mixture consisting of 100 ng SmBiT-βarr, 500 ng LgBiT-CAAX and 200 ng of a target GPCR construct in 200 μl of Opti-MEM (Thermo Fisher Scientific). For the NTSR1 titration experiment, diluted volume of the FLAG-NTSR1 plasmid (13 ng to 200 ng) was transfected with 100 ng SmBiT-β-arrestin and 500 ng LgBiT-CAAX with a balanced volume of the pcDNA3.1 vector (total plasmid volume of 800 ng). After an incubation for one day, the transfected cells were harvested with 0.5 mM EDTA-containing Dulbecco’s PBS, centrifuged, and suspended in 2 ml of Hank’s balanced saline solution (HBSS) containing 0.01% bovine serum albumin (BSA fatty acid–free grade, SERVA) and 5 mM HEPES (pH 7.4) (assay buffer). The cell suspension was dispensed in a white 96-well plate (Greiner Bio-One) at a volume of 80 μl per well and loaded with 20 μl of 50 μM coelenterazine (Carbosynth), diluted in the assay buffer. After 2 h incubation at room temperature, the plate was measured for its baseline luminescence (SpectraMax L, 2PMT model, Molecular Devices). Thereafter, 20 μl of 6x ligand serially diluted in the assay buffer were manually added. The ligand used was dependent on the GPCR expressed, as described in Table S1. The plate was immediately read for the second measurement as a kinetics mode and luminescence counts recorded for 15 min with an accumulation time of 0.18 sec per read and an interval of 20 sec per round. βarr endosomal translocation was measured by following the same procedure as described above except using the SmBiT-βarr and Endo-LgBiT constructs. Similarly, direct recruitment was measured by the same protocol as described above but using LgBiT-βarr (100 ng) and C-terminally fused-SmBiT GPCR (500 ng) constructs. For every well, the recorded kinetics data were first normalized to the baseline luminescence counts.
Analysis of cell-based recruitment data
NanoBiT data were analyzed by converting kinetic data into concentration-response data by determining an average fold-change (relative to signal pre-stimulation) from 10–15 minutes post-agonist addition. At least three independent experiments were performed for each GPCR-sensor combination. Concentration-dependent data from two technical replicates for each independent experiment were collectively fit to a four-parameter log logistic function (LL2.4) provided in the drc package (v 3.0–1) (Ritz et al., 2015) of the statistical environment R (R Core Team, 2020). This equation, of the form provides pre- and post-transition values, c and d, respectively, that define the amplitude response for that assay. The related function LL.4 was used for fitting EC50 values. Cutoffs for bystander NanoBiT experiments were determined based on a limit of detection defined as triple the standard error (3s) of the response in mock-transfected cells. Amplitude values were defined as amplitude = top – bottom of fit, and amplitude error was calculated as . Converting amplitude to LOF for each assay was based on the formula: LOF = 1 – amplitude(3Q)/amplitude(WT). Errors for LOF were calculated as: . In cases where a fit failed to converge due to weak recruitment, these amplitudes and errors were set to zero. In particular, recruitment of βarr1 (3Q) to DRD1 in both PM bystander (CAAX) and direct recruitment was set to zero. The error amplitude for βarr1 (3Q) endosomal translocation assay with DRD1 was also set to zero. The error amplitude for βarr1 (3Q) endosomal translocation assay with S1PR1 was set to zero, and the “top” value of the fit was set to 1.2 based on manual inspection. Recruitment of βarr1 (3Q) to kOR was set to zero for both CAAX and endosomal translocation, as was WT for endosomal translocation and the corresponding errors. K means clustering was performed using pre-built functions in the tidyverse package (v 1.3.1) of R. The number of clusters was varied from 1 to 10 and an elbow plot of within cluster sum of squares vs k suggested the data fit well to two clusters.
For recruitment kinetics, luminescence fold-change was plotted against time, and the values from zero to five minutes (initial rate) were fit to a logistic function of the form: , where L is the curve’s maximum value, x0 is the value of the sigmoid midpoint and k is the logistic growth rate. Fitting was done using the self-starting SSlogis four parameter nls function in the tidyverse package (v 1.3.1) of R.
GPCR internalization assays
GPCR internalization assays was performed as described previously with minor modifications (Grundmann et al., 2018). βarr1/2 double knockout cells, previously described (O’Hayre et al., 2017), were seeded in 6-cm dishes at concentration of 2 × 105 cells/ml (4 mL per dish) and cultured for 1 day before transfection. The cells were transfected with 1 μg of the N-terminally FLAG-tagged NTSR1 or the β2AR construct, along with 200 ng of the WT or 3Q βarr1 or empty plasmid, using PEI transfection reagent as described above. After 1-day culture, the transfected cells were harvested by EDTA-PBS and HEPES-HBSS and, following centrifugation, the cells were suspended in 500 μL of 0.01% BSA-containing HEPES-HBSS. The cell suspension was dispensed in a 96-well V-bottom plate (100 μL per well) and mixed with 100 μL of 2 × GPCR solution ligand (2 μM neurotensin for FLAG-NTSR1 or 20 μM iso for FLAG-β2AR). After 30-min incubation in a CO2 incubator, the plate was centrifuged at 1,500 g for 5 min and the cells were washed twice with D-PBS. The cell pellets were suspended in 2% goat serum and 2 mM EDTA-containing D-PBS (blocking buffer; 100 μL per well) and incubated for 30 min on ice. After centrifugation, the cells were stained with anti-FLAG-epitope tag monoclonal antibody (Clone 1 × 106, FujiFilm Wako Pure Chemicals; 10 μg mL−1 in the blocking buffer; 25 μL per well) for 30 min on ice. After washing with D-PBS, the cells were labeled with a goat anti-mouse IgG secondary antibody conjugated with Alexa Fluor 647 (Thermo Fisher Scientific; 10 μg/mL dilution in blocking buffer; 25 μL per well) for 15 min on ice. The cells were washed once with D-PBS, resuspended in 100 μL of 2 mM EDTA-containing-D-PBS and filtered through a 40 μm filter. The fluorescently labeled cells (approximately 20,000 cells per sample) were analyzed by the EC800 flow cytometer (Sony). Fluorescent signal derived from Alexa Fluor 647 was recorded in the FL3 channel. Mean fluorescence intensity (MFI) from all recorded events was analyzed using FlowJo software (FlowJo).
Cell-surface expression analysis by flow cytometry
HEK293A cells were seeded in a 6-well culture plate at a concentration of 2 × 105 cells/ml (2 mL per dish) and cultured for 1 day before transfection. The cells were transfected with 1 μg of N-terminally FLAG-tagged GPCR construct using PEI transfection reagent as described above and cultured for 1 day. The cells were collected by adding 200 μl of 0.53 mM EDTA-containing Dulbecco’s PBS (D-PBS), followed by 200 μl of 5 mM HEPES (pH 7.4)-containing Hank’s Balanced Salt Solution (HBSS). The cell suspension was transferred to a 96-well V-bottom plate in duplicate and fluorescently labeled with the anti-FLAG epitope tag antibody and a goat anti-mouse IgG secondary antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, 10 μg per ml diluted in the blocking buffer) as described above. Live cells were gated with a forward scatter (FS-Peak-Lin) cutoff at the 390 setting, with a gain value of 1.7 and fluorescent signal derived from Alexa Fluor 488 was recorded in the FL1 channel. For each experiment, the MFI value of mutants was normalized to that of WT performed in parallel.
cAMP desensitization
βarr1/2 double knockout cells were plated in 6-cm dishes and transfected 24-hours later with the indicated DNA using Lipofectamine 2000 according to the manufacturer’s protocol. The following day, cells were transduced with baculovirus containing CMV cADDis Green Upward cAMP biosensor (Montana Molecular #U0200G) according to manufacturer’s protocol but without addition of sodium butyrate and then seeded in triplicate in a black 96-well plate (Corning cat# 3340). The following day, the cells were washed once with 37°C assay buffer [135 mM NaCl, 5 mM KCl, 0.4 mM MgCl2, 1.8 mM CaCl2, 5 mM glucose, 20 mM HEPES pH 7.4], loaded into the pre-warmed 37°C plate reader (Biotek Synergy H4), and equilibrated for five minutes. Prior to beginning the kinetic assay, mApple was read using monochromators set to Ex:568/9.0 and Em:592/13.5. Then cADDis was read using monochromators set to Ex:500/9.0 and Em:530/20.0. These fluorescent reads were used to verify similar expression across conditions within each experiment for mApple-tagged constructs and the cADDis biosensor. Three cADDis timepoints were then collected to establish baseline, the plate was ejected, isoproterenol in 37°C assay buffer was added to a final concentration of 100 nM, and the plate was returned to continue collection. Thirty minutes after isoproterenol addition, IBMX and Fsk in 37°C assay buffer were added to a final concentrations of 300 μM and 10 μM respectively. Responses were averaged across technical replicates, normalized to the maximum Fsk/IBMX response, and then averaged across biological replicates which were defined as experiments that were initiated and carried out on different days. There was no randomization, blinding, or exclusion of data. For the two clones tested, one set of experiments was performed six times, whereas the other was performed three times.
Western blotting
HEK293A cells were transfected with the SmBiT-βarr and LgBiT-CAAX constructs by following the procedure described in the NanoBiT-based βarr PM recruitment assay. After 1-day culture, the transfected cells were lysed by SDS-PAGE sample buffer (62.5 mM Tris-HCl (pH 6.8), 50 mM dithiothreitol, 2% SDS, 10% glycerol and 4 M urea) containing 1 mM EDTA and 1 mM phenylmethylsulfonyl fluoride. Lysates derived from an equal number of cells were separated by 8% SDS-polyacrylamide gel electrophoresis. Subsequently, the proteins were transferred to PVDF membrane. The blotted membrane was blocked with 5% skim milk-containing blotting buffer (10 mM Tris-HCl (pH 7.4), 190 mM NaCl and 0.05% Tween 20), then incubated with primary antibody (1 μg per mL, except anti-α-tubulin used at 1:2000 dilution) followed by secondary antibodies conjugated with horseradish peroxidase (1:2000 dilution). After blotting, membrane were soaked with an ImmunoStar Zeta reagent then imaged with a chemiluminescence imager. Band intensity was quantified by densitometry using ImageJ.
Fab30 expression and purification
Fab30 (Shukla et al., 2013) was cloned into pFastBac-dual with an octa-histidine tag added to the C-terminus of the heavy-chain subunit (with an intervening AAA linker) and a GP67 secretion signal added to the N-terminus of both the heavy- and the light-chains (Maeda et al., 2018). Fab30 was expressed by Hi5 insect cells (Expression Systems) into the media using a FastBac-derived baculovirus. Cells were infected at a density of 3×106 cells/mL and harvested 72 hrs post infection. Cells were pelleted by centrifugation, and the supernatant was transferred to a large beaker with constant stirring. Tris pH 7.5 was added to a final concentration of 50 mM, followed by NiCl2 (to 1 mM) and CaCl2 (to 5 mM). A heavy precipitate will form. Add protease inhibitors and stir at room temperature for 45 minutes. Any precipitate was removed by centrifugation, and the clarified supernatant was applied to nickel sepharose 2 mL resin/L of supernatant. The resin was washed with 100 mL of 20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, then with 100 mL of 20 mM HEPES pH 7.5, 100 mM NaCl, 20 mM imidazole. Fab30 was eluted with 20 mM HEPES pH 7.5, 100 mM NaCl, 250 mM imidazole. Pooled fractions containing Fab30 were concentrated and subjected to polishing by SEC on a Superdex 200 pg 16/600 GL column (GE Healthcare) in 20 mM HEPES, pH 7.5, 100 mM NaCl. Peak fractions were pooled and concentrated to 200–300 mM, supplemented with glycerol to 15% v/v final, and aliquots were flash-frozen and stored at −80 °C until use.
NTSR1 expression and purification
Full length human NTSR1 was modified with an N-terminal Flag tag followed by an octa-histidine tag and cloned into pFastBac1 vector. NTSR1 was expressed in Sf9 insect cells (Expression Systems) using a FastBac-derived baculovirus. Cells were infected at a density of 4×106 cells/mL and harvested 60 hrs post infection. Cells were lysed in hypotonic buffer (10 mM HEPES, pH 7.4, and protease inhibitors) and solubilized at 4 °C for 2 hours in a buffer containing 1% lauryl maltose neopentyl glycol (LMNG, Anatrace), 0.1% cholesteryl hemisuccinate tris salt (CHS, Steraloids), 0.3% sodium cholate (Sigma), 20 mM HEPES 7.4, 500 mM NaCl, 25% glycerol, iodoacetamide (to cap cysteine residues) and protease inhibitors. Insoluble debris was removed by centrifugation and the supernatant was incubated with Ni-NTA (Qiagen) resin for 1 hour at 4 °C. The resin was washed in batch with buffer containing 0.01% LMNG, 0.001% CHS, 0.003% sodium cholate, 20 mM HEPES pH 7.4, 500 mM NaCl, 10 mM imidazole and eluted with the same buffer supplemented with 200 mM imidazole, 2 mM CaCl2 and 10 μM NTS8–13 (Acetate salt, Sigma). The eluate was loaded onto M1 FLAG immunoaffinity resin and washed with buffer containing 0.01% LMNG, 0.001% CHS, 0.003% sodium cholate, 20 mM HEPES pH 7.4, 500 mM NaCl, 10 mM imidazole, 0.1 μM NTS8–13 and 2 mM CaCl2. The receptor was eluted with buffer containing 100 mM NaCl, 20 mM HEPES pH 7.4, 0.005% LMNG, 0.005% CHS, 1 μM NTS8–13, 0.2 mg/mL flag peptide (DYKDDDDK) and 5 mM EDTA. Elution fractions containing receptor were pooled and subjected to polishing by SEC on a Superdex 200 Increase 10/300 GL column (GE Healthcare) in 20 mM HEPES, pH 7.4, 100 mM NaCl, 0.0025% LMNG, 0.00025% CHS, and 0.1 μM NTS8–13. Peak fractions were pooled and concentrated to 200 μM and aliquots were flash-frozen and stored at −80 °C until use.
GRK5 expression and purification
Full length human GRK5 was modified with a C-terminal hexa-histidine tag and cloned into pVL1392 vector for baculovirus production. GRK5 was expressed and purified as previously published (Beyett et al., 2019). Briefly, Sf9 insect cells (Expression Systems) were infected with a BestBac-derived baculovirus at a density of 3.5 × 106 cells/mL and harvested 48 hours post infection. Cells were resuspended, lysed by sonication and the supernatant was applied to Ni-NTA resin. The resin was washed with lysis buffer and GRK5 eluted with lysis buffer supplemented with 200 mM imidazole. The combined eluate was then subjected to cation-exchange chromatography using a MonoS 10/100 column (GE healthcare) and eluted with a linear gradient of NaCl. Fractions containing GRK5 were combined and run on a Superdex 200 10/300 GL column (GE healthcare). GRK5 was aliquoted, flash frozen, and stored at −80 °C until use.
β-arrestin expression and purification
The parent construct for β-arrestin 1 (βarr1) is the long splice variant of human, where all cysteine residues are removed by mutation (C59V, C125S, C140L, C150V, C242V, C251V, C269S). This construct is modified with an N-terminal 6x Histidine tag, followed by a 3C protease site, a GG linker, AviTag and GGSGGS linker. The sequence was codon-optimized for expression in E. coli and cloned into a pET-15b vector. Point mutations were prepared using site-directed mutagenesis. βarr1 (ΔCT) was prepared by truncating βarr1 at residue 382. All βarr1 constructs used were prepared as follows: NiCo21(DE3) competent E. coli (NEB) were transformed, and large-scale cultures were grown in TB + ampicillin at 37°C until an OD600 of 1.0. Cells were then transferred to room temperature and induced with 25 μM IPTG when the OD600 reached 2.0. Cells were harvested 20 h post induction and resuspended in lysis buffer [50 mM HEPES pH 7.4, 500 mM NaCl, 15% glycerol, 7.13 mM 2-mercaptoethanol (BME)] to a final volume of 40 mL/L of cells. Cells were lysed by sonication and the clarified lysate applied to nickel sepharose and batch incubated for 1.5 h at 4 °C. The resin was washed with 10 column volumes of wash buffer (20 mM HEPES pH 7.4, 500 mM NaCl, 10% glycerol, 7.13 mM BME) + 20 mM imidazole, followed by 10 column volumes of wash buffer + 40 mM imidazole. The protein was then eluted with 5 column volumes of wash buffer + 200mM imidazole and dialyzed overnight in 100x volume of dialysis buffer (20 mM HEPES 7.4, 200 mM NaCl, 2 mM BME, 10% glycerol) in the presence of 1:10 (w:w) of 3C protease. The digested protein was then subjected to reverse-Nickel purification and diluted with dialysis buffer containing no NaCl to bring the NaCl concentration to 75 mM. The protein was then purified by ion exchange chromatography (mono Q 10/100 GL, GE Healthcare), followed by SEC using a Superdex 200 increase 10/300 GL column (GE Healthcare) with SEC buffer (20 mM HEPES pH 7.4, 300 mM NaCl, 10% glycerol). Purified protein was concentrated to between 100–300 μM using a 30 kDa spin concentrator and aliquots were flash-frozen in liquid nitrogen and stored at −80 °C until use.
β-arrestin labeling and biotinylation
Following SEC, elution peak fractions were pooled to a concentration of 10–20 μM and labeled with different fluorophore(s), either: monobromobimane (mBBr), N,N’-Dimethyl-N-(Iodoacetyl)-N’-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)Ethylenediamine (IANBD amide), or a 1:3 mixture of Alexa Fluor 488 C5 Maleimide and ATTO647N Maleimide, respectively. Fluorophores were dissolved to in DMSO to 25–40 mM and added at 10x molar excess over protein, then allowed to react for 1 h at room temperature prior to quenching with L-Cysteine (10x molar excess over fluorophore). The labeling reaction was further incubated for 10 minutes after cysteine addition, after which samples were spin filtered and subjected to a second round of size-exclusion chromatography, as detailed above, to remove free dye. The purified, was concentrated to between 100–300 μM using a 30 kDa spin concentrator and aliquots were flash-frozen in liquid nitrogen and stored at −80 °C until use.
Arrestins (SEC-pure) were biotinylated using recombinant BirA enzyme, according to commercial protocols (Avidity), with exception that biotinylation was carried out for 12 h at 4 °C, rather than 30 °C. After biotinylation was complete, the reaction was flowed over 100 μL (packed) of nickel Sepharose, equilibrated in arrestin SEC buffer and supplemented with 10 mM imidazole, then washed with 200 μL of the equilibration buffer. The combined flow-through and wash fractions were then purified by size-exclusion as described above.
NTSR1 in vitro phosphorylation
NTSR1 (2.5 μM) was equilibrated in phosphorylation buffer (20 mM bis-tris propane (BTP) pH 7.5, 35 mM NaCl, 5 mM MgCl2, 20 μM NTS8–13, 20 μM 08:0 PI(4,5)P2, 0.05 mM TCEP, 0.002% MNG, 0.0002% CHS) at 25 °C with gentle mixing for 1 h. GRK5 was added to the reaction to a final concentration of 200 nM, and briefly incubated while the reaction was warmed from 25 °C to 30 °C. ATP was added to a final concentration of 1 mM. Upon completion, the reaction was supplemented with CaCl2 to a final concentration of 2 mM and applied to an equilibrated M1 FLAG immunoaffinity resin and washed with buffer containing 0.004% LMNG, 0.004% CHS, 20 mM HEPES pH 7.4, 100 mM NaCl, 0.2 μM NTS8–13, 2 mM CaCl2. The receptor was eluted with buffer containing 100 mM NaCl, 20 mM HEPES pH 7.4, 0.004% LMNG, 0.004% CHS, 0.2 μM NTS8–13, 0.2 mg/mL Flag peptide, 5 mM EDTA), followed by SEC using a Superdex 200 increase 10/300 GL column (GE Healthcare) with SEC buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.004% LMNG, 0.0004% CHS).
Analytical fluorescence-detection size-exclusion chromatography
In a final volume of 20 μL, NTSR1 (4.5 μM), the respective arrestin construct (9 μM), NTS8–13 peptide (50 μM) and 08:0 PI(4,5)P2 (5 μM) were incubated in buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl, 0.004% LMNG, 0.0004% CHS and 0.2 μM NTS8–13. Using a Prominence-i LC autosampler (Shimadzu), 10 μL was injected onto a ENrich size-exclusion chromatography 650 10 × 300 column (Bio-rad) pre-equilibrated in 20 mM HEPES pH 7.4 100 mM NaCl, 0.004 % LMNG, 0.004% CHS and 0.2 μM NTS8–13, and run at a flow rate of 0.8 ml/min. Tryptophan fluorescence was monitored at λ(EX) of 280 nm and λ(EM) of 340 nm. Peaks in the obtained size-exclusion chromatograms were modeled as gaussians, deconvolved and quantified (AUC) using Magic Plot 3 (Magic Plot).
Surface plasmon resonance measurements
SPR experiments were performed using a GE Biacore T100 instrument. Approximately 300–400 resonance units (RU) of FPLC-purified biotinylated arrestin in HBS-P+ Buffer (Cytiva) were captured on an SA-chip (Cytiva), including a reference channel for online background subtraction of bulk solution refractive index and for evaluation of non-specific binding of analyte to the chip surface (Biacore T100 Control Software; Cytiva). All measurements were performed with 2-fold serial dilutions using 60 s association followed by a dissociation time of more than 240 s at 25 °C with a flow rate of at 30 μl min−1. Measurement of titrations at equilibrium were used to determine Kd values using Biacore Analysis Software (v.2.0.4, Cytiva) and fits to a total binding model were performed in GraphPad Prism 9. The concentration of V2Rpp stocks were determined by reaction with Ellman’s reagent as previously described (Latorraca et al., 2020). Regeneration was performed by 2 injections of 2 M MgCl2 for 10 s at 50 μl min−1 flow rate. In all cases regeneration resulted in a complete return to baseline. Single cycle measurements were performed as described above. All single cycle measurements were performed as triplicates and quantifications calculated to the RUmax of the individual immobilized ligands (βarr1 proteins). RUmax was defined by the expected change, corrected for mass of Fab30 relative to βarr1 and the amount of βarr1 immobilized.
Fluorescence anisotropy measurements
BODIPY-TMR phosphatidylinositol 4,5-bisphosphate (BODIPY-PIP2) (Echelon Biosciences) was dissolved to a stock concentration of 1 mM in 50 mM HEPES pH 7.4 and used at a final concentration of 4 nM in the assay. For the arrestin measurements, a two-fold dilution series of was made from a stock of βarr1 (ΔCT), yielding fourteen samples with final concentrations ranging from 150 μM to 0.02 μM. A control sample containing buffer only was included to measure the free anisotropy of BODIPY-PIP2. After mixing the BODIPY-PIP2 with arrestin or buffer, samples were incubated for 1h at room temperature prior to measurements. Samples were measured in five 20 μL replicates in a black 384-well plate on a Tecan Infinite M1000 (Tecan Life Sciences), using an excitation wavelength of 530 nm, an emission wavelength of 573 nm and bandwidths of 5 nm. The obtained data was fit using to a one-site total binding model Y = Bottom + ðtop bottomÞ=1 + 10HS logðEC50 XÞ where HS denotes the Hill-slope.
Bulk fluorescence measurements
Bulk fluorescence measurements were performed on either a Fluorolog instrument (Horiba) using FluorEssence v3.8 software and operating in photon-counting mode, or a Tecan Infinite M1000 PRO multimodal microplate reader (Tecan). Fluorolog measurements of bimane-labeled βarr1 constructs (NTSR1 experiments) were performed at final concentration of 0.4 μM βarr1 in buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl and 0.004% LMNG (w/v)/0.0004% CHS (w/v) supplemented with 4 μM NTS(8–13). The concentration of V2Rpp stocks were determined by reaction with Ellman’s reagent as previously described (Latorraca et al., 2020). For NTSR1 experiments the following concentrations were used: 4 μM NTSR1, 4.1 μM 08:0 PI(4,5)P2, 50 μM V2Rpp (depending on condition). Samples were incubated for 1 h in the dark before measurement. Fluorescence data were collected in a quartz cuvette with 135 mL of sample. Bimane fluorescence was measured by excitation at 370 nm with excitation and emission bandwidth passes of 3 nm, and emission spectra were recorded from 400 to 550 nm in 2 nm increments with 0.1 s integration time. Care was taken to extensively rinse and dry the cuvette between individual measurements. To remove background fluorescence, buffer spectra were collected using the same settings, and subtracted from each sample spectrum.
FRET measurements of AF488-AT647N-labeled βarr1 constructs were performed as described for bimane measurements, with the following differences: samples were excited at 476 nm with 3 nm excitation and 4 nm emission slit widths. Spectra were collected from 485 nm to 750 nm in 1 nm increments with 0.1 s integration time. FRET measurements in the absence of NTSR1 were performed in buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl and 0.004% LMNG (w/v)/0.0004% CHS (no NTS). FRET measurements with NTSR1 were done with 0.5 μM NTSR1 and 0.5 μM 08:0 PI(4,5)P2.
NBD spectra measured on the Tecan Infinite M1000 PRO were collected using 384-well black plates with 50 μL of sample and at a final concentration of 0.5 μM βarr1 in buffer containing 20 mM HEPES pH 7.4, 100 mM NaCl and 0.004% LMNG (w/v)/0.0004% CHS. For NBD the following instrument settings were used: excitation: 490 nm, emission 510–580 nm (1 nm steps) with 20 s read time and 400 Hz flash mode. Gain and z-position were optimized prior to reading. Bimane spectra were collected in 96-well ½ area white plates with 100 μL of sample using the following instrument settings: excitation: 370 nm, emission 420–500 nm (1 nm steps) with 20 s read time and 400 Hz flash mode.
Efret values for FRET experiments were calculated as and normalized to donor intensity within a given experiment. Scaled FRET values (apo = 100, min(FRET) = 0) were fit to a single exponential decay function using the nls function in R for EC50 values (obtained as t1/2 for decay). NS denotes concentration-dependent non-specific signal. L167W-293NBD was fit using the same function. L68bim data was fit to a total binding model , where background is a constant value. Fitting was independently performed in R and with GraphPad Prism 9 for corroboration, values reported are from Prism 9.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical tools used for each set of experiments in this study are outlined in the relevant method sub-section. In all cases, boxplots are defined as: center line, median; box range, 25–75th percentiles; whiskers denote minimum–maximum values. The t tests described above assume data are normally distributed. While individual datapoints appear consistent with this assumption but given the small sample size we did not perform explicit tests of this assumption.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-DYKDDDDK tag antibody (mouse monoclonal, clone 1E6) | FujiFilm Wako Pure Chemicals | Cat# 012–22384; RRID: AB_10659717 |
| Goat anti-mouse IgG secondary antibody, conjugated with Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A32723; RRID: AB_2633275 |
| Goat anti-mouse IgG secondary antibody, conjugated with Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A21235; RRID: AB_2535804 |
| Anti-β-Arrestin1 antibody (rabbit monoclonal, clone D8O3J) | Cell Signaling Technology | Cat# 12697; RRID: AB_2797996 |
| Anti-β-Arrestin2 antibody (rabbit monoclonal, clone C16D9) | Cell Signaling Technology | Cat# 3857; RRID: AB_2258681 |
| anti-α-tubulin antibody (mouse monoclonal, clone DM1A) | Santa Cruz Biotechnologies | Cat# sc-32293; RRID: AB_628412 |
| Anti-Rabbit IgG, conjugated with horseradish peroxidase | GE Healthcare | Cat# NA9340; RRID: AB_772191 |
| Anti-Mouse IgG, conjugated with horseradish peroxidase | GE Healthcare | Cat# NA9310; RRID: AB_772193 |
| Bacterial and virus strains | ||
| SCS1 Supercompetent Cells | Agilent Technologies | Cat# 200231 |
| E. coli NiCo21(DE3) | NEB | Cat# C2529H |
| E. coli Rosetta (DE3) | Millipore Sigma | Cat# 70954–3 |
| Biological samples | ||
| Biological samples | ||
| Fetal bovine serum | Thermo Fisher Scientific | Cat# 10270–106 |
| Goat serum | Nippon Bio-test Laboratories | Cat# 0208–01 |
| Chemicals, peptides, and recombinant proteins | ||
| Polyethylenimine (PEI) Max (MW 40,000) | Polysciences | Cat# 24765–1 |
| Opti-MEM | Thermo Fisher Scientific | Cat# 31985–070 |
| 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) | Sigma-Aldrich | Cat# H3375 |
| Ethylenediaminetetraacetic acid (EDTA) solution | Thermo Fisher Scientific | Cat# 15575–020 |
| Trypsin | Thermo Fisher Scientific | Cat# 27250–018 |
| Bovine serum albumin | SERVA Electrophoresis | Cat# 11945 |
| Coelenterazine | Carbosynth | Cat# EC14031 |
| Angiotensin II | Peptide Institute | Cat# 4001-v |
| Bradykinin | Peptide Institute | Cat# 4002-v |
| (D-Ala2,N-Me-Phe4,glycinol5)-Enkephalin (DAMGO) | Bachem AG | Cat# 4007829 |
| Dopamine | FUJIFILM Wako Pure Chemical | Cat# 040–15433 |
| Dynorphin A | Peptide Institute | Cat# 4080-v |
| Endothelin-1 | Peptide Institute | Cat# 4198-s |
| Isoproterenol | Tokyo Chemical Industry | Cat# I0260 |
| Isoproterenol | Sigma-Aldrich | Cat# I6504 |
| Methionine-Enkephalin | Peptide Institute | Cat# 4042-v |
| Neurotensin | Peptide Institute | Cat# 4029 |
| Norepinephrine | Tokyo Chemical Industry | Cat# A0906 |
| Oxytocin | Peptide Institute | Cat# 4084-v |
| PAR2 peptide | Peptide Institute | Cat# PAR-3664-PI |
| Parathyroid Hormone (PTH) | Peptide Institute | Cat# 4068-s |
| Serotonin | FUJIFILM Wako Pure Chemical | Cat# 321–42341 |
| Sphingosine 1-phosphate | Cayman Chemical | Cat# 62570 |
| Substance P | Peptide Institute | Cat# 4014-v |
| Thyrotropin-releasing hormone (TRH) | Peptide Institute | Cat# 4011 |
| [Arg8]-vasopressin (AVP) | Peptide Institute | Cat# 4085-v |
| DMEM | Nissui | Cat# 05919 |
| Penicillin G | Sigma-Aldrich | Cat# P3032 |
| Streptomycin | Thermo Fisher Scientific | Cat# 11860–038 |
| ImmunoStar Zeta | FUJIFILM Wako Pure Chemical | Cat# 291–72401 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma-Aldrich | Cat# I5879 |
| Forskolin (Fsk) | Sigma-Aldrich | Cat# F6886 |
| V2Rpp (ARGRpTPPpSLGPQDEpSCpTpTApSpSpSLAKDTSS) | Tufts University Core Facility | N/A |
| Monobromobimane (mBBr) | Thermo Fisher Scientific | Cat# M1378 |
| IANBD Amide (N,N’-Dimethyl-N-(Iodoacetyl)-N’-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)Ethylenediamine) | Thermo Fisher Scientific | Cat# D2004 |
| L-Cysteine | Sigma-Aldrich | Cat# 30089 |
| 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) | Thermo Fisher Scientific | Cat# 22582 |
| BODIPY-TMR PI(4,5)P2 | Echelon Biosciences | Cat # C-45M6 |
| FLAG peptide (DYKDDDDK) | Stanford Pan Facility | N/A |
| AlexaFluor 488 C5 maleimide | Thermo Fisher Scientific | Cat# A10254 |
| ATTO647N C2 maleimide | ATTO TEC | Cat# AD647N-41 |
| Lauryl maltose neopentyl glycol (LMNG) | Anatrace | Cat# NG310 |
| Cholesteryl Hemisuccinate (CHS) | Steraloids | Cat# C6823 |
| Benzamidine hydrochloride hydrate | Sigma-Aldrich | Cat# B6506 |
| Leupeptin | Sigma-Aldrich | Cat# 50-568-49 |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) solution | Sigma-Aldrich | Cat# 646547 |
| ESF921 culture medium | Expression Systems | Cat# 96–001 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | Cat# E5134 |
| 2-Mercaptoethanol (BME) | Sigma-Aldrich | Cat# M6250 |
| HBS-P+ Buffer 10x | Cytiva | Cat# BR100671 |
| 08:0 PI(4,5)P2 | Avanti Polar Lipids | Cat# 850185 |
| 08:0 PI(3,4)P2 | Avanti Polar Lipids | Cat# 850183 |
| 08:0 PI(3,5)P2 | Avanti Polar Lipids | Cat# 850184 |
| 08:0 PI(3,4,5)P3 | Avanti Polar Lipids | Cat# 850186 |
| 08:0 PI(3)P | Avanti Polar Lipids | Cat# 850187 |
| 08:0 PI(4)P | Avanti Polar Lipids | Cat# 850182 |
| 08:0 PG | Avanti Polar Lipids | Cat# 840433 |
| Adenosine triphosphate (ATP) | Sigma-Aldrich | Cat# A7699 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma-Aldrich | Cat# I6758 |
| Neurotensin fragment 8–13 acetate salt | Sigma-Aldrich | Cat# N5266 |
| Series S Sensor Chip SA | Cytiva | Cat# BR100531 |
| Imidazole | Sigma-Aldrich | Cat# I2399 |
| Ampicilin | Sigma-Aldrich | Cat# A9393 |
| Sodium cholate | Sigma-Aldrich | Cat# C6445 |
| HRV3C protease | EMD Millipore | Cat# 71493 |
| Critical commercial assays | ||
| PrimeSTAR HS DNA Polymerase | Takara Bio | Cat# R010B |
| NEBuilder HiFi DNA Assembly Master Mix | New England BioLabs | Cat# E2621X |
| NucleoBond Xtra Midi Plus | Macherey-Nagel | Cat# 740412.50 |
| PureYield Plasmid Miniprep System | Promega | Cat# A1222 |
| FastGene Plasmid Mini Kit | Nippon Genetics | Cat# FG-90502 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat# LT07–118 |
| Green Up cADDis cAMP Assay Kit | Montana Molecular | Cat# U0200G |
| BirA500 biotin-protein ligase standard reaction kit | Avidity | Cat #1 |
| Deposited data | ||
| All experimental data | This paper; Mendeley Data | https://dx.doi.org/10.17632/ynrr4j7t4s.1 |
| Active-state crystal structure of β-arrestin 1 bound to the phosphorylated V2R C-tail and Fab30 | Shukla et al., 2013 | PDB: 4JQI |
| Inactive-state crystal structure of β-arrestin 1 | Han et al., 2001 | PDB: 1G4M |
| neurotensin receptor and arrestin2 complex | Huang et al., 2020 | PDB: 6UP7 |
| Experimental models: Cell lines | ||
| Human: HEK293A cells | Thermo Fisher Scientific | Cat# R70507 |
| Human: ΔβArr1/2 DKO HEK293 cells | O’Hayre et al., 2017 | Clones 1 and 2 |
| Insect: Spodoptera frugiperda Sf9 cells | Expression Systems | Cat# 94–001 |
| Insect: Trichuplusia ni Hi5 cells | Expression Systems | Cat# 94–002 |
| Recombinant DNA | ||
| pFastBac-NTSR1 | Huang et al., 2020 | N/A |
| pVL-GRK5 | Huang et al., 2020 | N/A |
| pFB Dual Fab30 | This study | N/A |
| pET15b-β-arrestin-1 | Huang et al., 2020 | N/A |
| pET15b-β-arrestin-1 (ΔCT) | Huang et al., 2020 | N/A |
| pET15b-β-arrestin-1 (3Q) | This study | N/A |
| pET15b-β-arrestin-1 (3Q) (ΔCT) | This study | N/A |
| pET15b-β-arrestin-1 A12C V387C | This study | N/A |
| pET15b-β-arrestin-1 A12C V387C (3Q) | This study | N/A |
| pET15b-β-arrestin-1 L68C | Latorraca et al., 2020 | N/A |
| pET15b-β-arrestin-1 L68C (3Q) | This study | N/A |
| pET15b-β-arrestin-1 L167W L293C | Latorraca et al., 2020 | N/A |
| pET15b-β-arrestin-1 L167W L293C (3Q) | This study | N/A |
| Plasmids for NanoBiT experiments | See Table S4 (Xu et al., 2022; Hisano et al., 2019; Huang et al., 2020; Shihoya et al., 2018; Baidya et al., 2020; Inoue et al., 2012; Kato et al., 2019; Kawakami et al., 2022) | N/A |
| β-arrestin-2-mApple | Eichel et al., 2016 | N/A |
| β-arrestin-2(3Q)-mApple | This study | N/A |
| mApple-N1 | Shaner et al., 2008 | Addgene #54567 |
| Software and algorithms | ||
| GraphPad Prism, Version 8.0, 9.0 | GraphPad, Inc. | https://www.graphpad.com/ |
| Soft Max Pro, Version 7 | Molecular Devices | https://www.moleculardevices.com/products/microplatereaders#softmaxproLink |
| FlowJo software, Version 10 | FlowJo | https://www.flowjo.com/solutions/flowjo |
| PyMOL | Schrö dinger, Inc. | https://pymol.org/2/ |
| FluorEssence Version 3.8 | Horiba, Inc. | https://www.horiba.com |
| Fiji (ImageJ2) Version 2.3.0 | N/A | https://imagej.net/software/fiji/ |
| R studio Version 1.3.1093 | N/A | https://www.rstudio.com/products/rstudio/ |
| R Version 4.0.3 | The R Foundation for Statistical Computing | |
| drc package (in R) Version 3.0–1 | Ritz et al., 2015 | N/A |
| Biacore Control and Analysis Software Version 2.0.4 | Cytiva | N/A |
| MagicPlot Version 3.0 | MagicPlot Systems LLC | https://magicplot.com/ |
| Other | ||
| 96 well white plates | Greiner Bio-one | Cat# 655083 |
| 384 well black | Corning | Cat# 3575 |
| 96 well ½ area white | PerkinElmer | Cat# 6005560 |
| 96-well V-bottom plate | Greiner Bio-one | Cat# 651201 |
| 96-well plate, black | Corning | Cat# 3340 |
| Luminescence microplate reader | Molecular Devices | SpectraMax L, 2PMT |
| Flow cytometer | Sony | EC800 |
| Chemiluminescent imager | Cytiva | Amersham Imager 680 |
| BioTek Multi plate reader | BioTek | BioTek Synergy H4 |
| Tecan multimodal microplate reader | Tecan | Infinite M1000 PRO |
| SPR | Cytiva | Biacore T100 |
Highlights.
β-Arrestins coincidentally detect membrane PIPs for recruitment to GPCRs
GPCR phosphorylation affects dependence on membrane PIPs for β-arrestin recruitment
Membrane PIPs are allosteric modulators of GPCR-β-arrestin complexes
Plasma membrane PIPs alone promote β-arrestin activation
ACKNOWLEDGMENTS
We thank B. White for assistance with arrestin expression and purification; W. Huang for assistance with NTSR1 expression and purification; D. Hilger for providing BirA enzyme; Y. Seok Kim and E. Byrne for FSEC assistance; K. Sato, Y. Sugamura, S. Nakano, and A. Inoue for plasmid construction and cell-based GPCR assays; and R.S. Prosser for critical reading of the manuscript. This work was supported in part by National Institutes of Health grants K99GM147609 (to J.J.), R01NS028471 (to B.K.K.), R01AI125320 (to K.C.G.), and R01DA010711 and R01DA012864 (to M.v.Z.). Additional support to both K.C.G. and B.K.K. is provided by the Mathers Foundation. B.K.K. received additional support from the Chan-Zuckerberg Biohub. K.C.G. is supported by the Howard Hughes Medical Institute. J.J. was supported by a fellowship from the Damon Runyon Cancer Research Foundation (DRG-2318-18). B.B.-R. is a recipient of an American Heart Association Predoctoral Fellowship (19PRE34380570). F.M.H. is a recipient of a Marie Sk1odowska-Curie Individual Fellowship from the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 844622) and an American Heart Association postdoctoral fellowship (19POST34380839). M.M. was supported by an American Heart Association postdoctoral fellowship (17POST33410958). A.I. was funded by the PRIME (19gm5910013), the LEAP (20gm0010004), and the BINDS (JP20am0101095) from the Japan Agency for Medical Research and Development (AMED); KAKENHI (17K08264, 21H04791, 21H05113, JPJSBP120213501, and JPJSBP120218801) from the Japan Society for the Promotion of Science (JSPS); FOREST Program (JPMJFR215T) and JST Moonshot Research and Development Program (JPMJMS2023) from the Japan Science and Technology Agency (JST); Daiichi Sankyo Foundation of Life Science; Takeda Science Foundation; Ono Medical Research Foundation; and The Uehara Memorial Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.10.018.
DECLARATION OF INTERESTS
B.K.K. is a cofounder and consultant for ConfometRx, Inc.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- Asher WB, Terry DS, Gregorio GGA, Kahsai AW, Borgia A, Xie B, Modak A, Zhu Y, Jang W, Govindaraju A, et al. (2022). GPCR-mediated beta-arrestin activation deconvoluted with single-molecule precision. Cell 185, 1661–1675.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baidya M, Kumari P, Dwivedi-Agnihotri H, Pandey S, Chaturvedi M, Stepniewski TM, Kawakami K, Cao Y, Laporte SA, Selent J, et al. (2020). Key phosphorylation sites in GPCRs orchestrate the contribution of beta-arrestin 1 in ERK1/2 activation. EMBO Rep. 21, e49886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyett TS, Fraley AE, Labudde E, Patra D, Coleman RC, Eguchi A, Glukhova A, Chen Q, Williams RM, Koch WJ, et al. (2019). Perturbation of the interactions of calmodulin with GRK5 using a natural product chemical probe. Proc. Natl. Acad. Sci. USA 116, 15895–15900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, et al. (2017). Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 114, 2562–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhuo Y, Sharma P, Perez I, Francis DJ, Chakravarthy S, Vishnivetskiy SA, Berndt S, Hanson SM, Zhan X, et al. (2021). An eight amino acid Segment Controls Oligomerization and Preferred Conformation of the two Non-visual arrestins. J. Mol. Biol 433, 166790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis MA, and Godi A (2004). PI-loting membrane traffic. Nat. Cell Biol 6, 487–492. [DOI] [PubMed] [Google Scholar]
- Defea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, and Bunnett NW (2000). beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol 148, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déry O, Thoma MS, Wong H, Grady EF, and Bunnett NW (1999). Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. beta-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem 274, 18524–18535. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, and De Camilli P (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, et al. (2016). Nano-Luc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol 11, 400–408. [DOI] [PubMed] [Google Scholar]
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, and Von Zastrow M (2018). Catalytic activation of beta-arrestin by GPCRs. Nature 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, and Von Zastrow M (2016). Beta-arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol 18, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, and Vilardaga JP (2011). Retromer terminates the generation of cAMP by internalized PTH receptors. Nat. Chem. Biol 7, 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillion D, Devost D, and Hébert TE (2019). Measuring recruitment of beta-arrestin to G protein-coupled heterodimers using bioluminescence resonance energy transfer. Methods Mol. Biol 1957, 83–91. [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Krupnick JG, Falck JR, Benovic JL, and Keen JH (1999). Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 18, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, Rüttiger N, Ziegler N, Benkel T, Schmitt NK, et al. (2018). Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, and Schubert C (2001). Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane Translocation. Structure 9, 869–880. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, and Von Zastrow M (2008). Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol 48, 537–568. [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Strunk BS, and Weisman LS (2017). PI5P and PI(3,5)P2: minor, but essential phosphoinositides. Cell Struct. Funct 42, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y, Kono M, Cartier A, Engelbrecht E, Kano K, Kawakami K, Xiong Y, Piao W, Galvani S, Yanagida K, et al. (2019). Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J. Exp. Med 216, 1582–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, Robertson MJ, Nguyen KC, Glenn JS, Skiniotis G, et al. (2020). Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorati G, Sadeghi H, and Birnbaumer M (1998a). Transient phosphorylation of the V1a vasopressin receptor. J. Biol. Chem 273, 7155–7161. [DOI] [PubMed] [Google Scholar]
- Innamorati G, Sadeghi HM, Tran NT, and Birnbaumer M (1998b). A serine cluster prevents recycling of the V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA 95, 2222–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Ishiguro J, Kitamura H, Arima N, Okutani M, Shuto A, Higashiyama S, Ohwada T, Arai H, Makide K, et al. (2012). TGFalpha shedding assay: an accurate and versatile method for detecting GPCR activation. Nat. Methods 9, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, et al. (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Tsvetanova NG, Lobingier BT, and Von Zastrow M (2015). Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol 35, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just S, Illing S, Trester-Zedlitz M, Lau EK, Kotowski SJ, Miess E, Mann A, Doll C, Trinidad JC, Burlingame AL, et al. (2013). Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol. Pharmacol 83, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlecova Z, Spielman SJ, Loerke D, Mohanakrishnan A, Reed DK, and Schmid SL (2017). Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J. Cell Biol 216, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE, Zhang Y, Hu H, Suomivuori CM, Kadji FMN, Aoki J, Krishna Kumar K, Fonseca R, Hilger D, Huang W, et al. (2019). Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 572, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Yanagawa M, Hiratsuka S, Yoshida M, Ono Y, Hiroshima M, Ueda M, Aoki J, Sako Y, and Inoue A (2022). Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying beta-arrestin transducer bias. Nat. Commun 13, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury E, Nikolajev L, Simaan M, Namkung Y, and Laporte SA (2014). Differential regulation of endosomal GPCR/beta-arrestin complexes and trafficking by MAPK. J. Biol. Chem 289, 23302–23317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, and Sommer ME (2013). Crystal structure of pre-activated arrestin p44. Nature 497, 142–146. [DOI] [PubMed] [Google Scholar]
- Komolov KE, and Benovic JL (2018). G protein-coupled receptor kinases: past, present and future. Cell. Signal 41, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KM, Daaka Y, Pitcher JA, and Lefkowitz RJ (1997). The role of sequestration in G protein-coupled receptor resensitization. Regulation of beta2-adrenergic receptor dephosphorylation by vesicular acidification. J. Biol. Chem 272, 5–8. [DOI] [PubMed] [Google Scholar]
- Kumari P, Srivastava A, Ghosh E, Ranjan R, Dogra S, Yadav PN, and Shukla AK (2017). Core engagement with beta-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally CC, Bauer B, Selent J, and Sommer ME (2017). C-edge loops of arrestin function as a membrane anchor. Nat. Commun 8, 14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Masureel M, Hollingsworth SA, Heydenreich FM, Suomivuori CM, Brinton C, Townshend RJL, Bouvier M, Kobilka BK, and Dror RO (2020). How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell 183, 1813–1825.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, and Dror RO (2018). Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, and Luttrell LM (2016). The conformational signature of beta-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Warne T, Nehmé R, Pandey S, Dwivedi-Agnihotri H, Chaturvedi M, Edwards PC, García-Nafría J, Leslie AGW, Shukla AK, et al. (2020). Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 583, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JG, Chen C, and Liu-Chen LY (2002). Ezrin-radixin-moesin-binding phosphoprotein-50/Na+/H+ exchanger regulatory factor (EBP50/NHERF) blocks U50,488H-induced down-regulation of the human kappa Opioid receptor by enhancing its recycling rate. J. Biol. Chem 277, 27545–27552. [DOI] [PubMed] [Google Scholar]
- Lobingier BT, and Von Zastrow M (2019). When trafficking and signaling mix: how subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 20, 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Koehl A, Matile H, Hu H, Hilger D, Schertler GFX, Manglik A, Skiniotis G, Dawson RJP, and Kobilka BK (2018). Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun 9, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Morales JC, Romero-Ávila MT, Reyes-Cruz G, and García-Sáinz JA (2018). S1P1 receptor phosphorylation, internalization, and interaction with Rab proteins: effects of sphingosine 1-phosphate, FTY720-P, phorbol esters, and paroxetine. Biosci. Rep 38. BSR20181612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, and Asahi M (2013). beta1-adrenergic receptor recycles via a membranous organelle, recycling endosome, by binding with sorting nexin27. J. Membr. Biol 246, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, and Laporte SA (2016). Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun 7, 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, et al. (2019). Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex. Nat. Struct. Mol. Biol 26, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, and Hoffmann C (2016). Beta-arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, and Caron MG (1999). Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J. Biol. Chem 274, 32248–32257. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, and Caron MG (2001). Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J. Biol. Chem 276, 19452–19460. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, and Barak LS (2000). Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem 275, 17201–17210. [DOI] [PubMed] [Google Scholar]
- O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, Kawakami K, Aoki J, Messer K, Sunahara R, et al. (2017). Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci. Signal 10, eaal3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkovska S, Méjean C, Ayoub MA, Li J, Hemery F, Corbani M, Laguette N, Ventura MA, Orcel H, Durroux T, et al. (2018). V1b vasopressin receptor trafficking and signaling: role of arrestins, G proteins and Src kinase. Traffic 19, 58–82. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, and Shenoy SK (2018). GPCR desensitization: acute and prolonged phases. Cell. Signal 41, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, and Lefkowitz RJ (2012). Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol 52, 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D (2015). Dose-response analysis using R. PLoS One 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sente A, Peer R, Srivastava A, Baidya M, Lesk AM, Balaji S, Shukla AK, Babu MM, and Flock T (2018). Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat. Struct. Mol. Biol 25, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, Mckeown MR, Steinbach PA, Hazelwood KL, Davidson MW, and Tsien RY (2008). Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 5, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihoya W, Izume T, Inoue A, Yamashita K, Kadji FMN, Hirata K, Aoki J, Nishizawa T, and Nureki O (2018). Crystal structures of human ETB receptor provide mechanistic insight into receptor activation and partial activation. Nat. Commun 9, 4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, Staus DP, Hilger D, Uysal S, Huang LY, et al. (2013). Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. (2014). Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer ME, Smith WC, and Farrens DL (2005). Dynamics of arrestin-rhodopsin interactions: arrestin and retinal release are directly linked events. J. Biol. Chem 280, 6861–6871. [DOI] [PubMed] [Google Scholar]
- Sommer ME, Smith WC, and Farrens DL (2006). Dynamics of arrestin-rhodopsin interactions: acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. J. Biol. Chem 281, 9407–9417. [DOI] [PubMed] [Google Scholar]
- Song W, Yen HY, Robinson CV, and Sansom MSP (2019). State-dependent lipid interactions with the A2a receptor revealed by MD simulations using in vivo-mimetic membranes. Structure 27, 392–403.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Coffa S, Fu H, and Gurevich VV (2009). How does arrestin assemble MAPKs into a signaling complex? J. Biol. Chem 284, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staus DP, Wingler LM, Choi M, Pani B, Manglik A, Kruse AC, and Lefkowitz RJ (2018). Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in beta-arrestin coupling. Proc. Natl. Acad. Sci. USA 115, 3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Carroll S, Kaksonen M, Toshima JY, and Drubin DG (2007). PtdIns(4,5)P2 turnover is required for multiple stages during clathrin- and actin-dependent endocytic internalization. J. Cell Biol 177, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2020). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Tewson PH, Martinka S, Shaner NC, Hughes TE, and Quinn AM (2016). New DAG and cAMP sensors optimized for live-cell assays in automated laboratories. J. Biomol. Screen 21, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, et al. (2016). GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth DJ, Tóth JT, Gulyás G, Balla A, Balla T, Hunyady L, and Várnai P (2012). Acute depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate impairs specific steps in endocytosis of the G-protein-coupled receptor. J. Cell Sci 125, 2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapaidze N, Gomes I, Bansinath M, and Devi LA (2000). Recycling and resensitization of delta opioid receptors. DNA Cell Biol. 19, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Ikuta T, Kawakami K, Kise R, Qian Y, Xia R, Sun MX, Zhang A, Guo C, Cai XH, et al. (2022). Structural basis of sphingosine-1-phosphate receptor 1 activation and biased agonism. Nat. Chem. Biol 18, 281–288. [DOI] [PubMed] [Google Scholar]
- Yen HY, Hoi KK, Liko I, Hedger G, Horrell MR, Song W, Wu D, Heine P, Warne T, Lee Y, et al. (2018). PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Barak LS, Anborgh PH, Laporte SA, Caron MG, and Ferguson SS (1999). Cellular trafficking of G protein-coupled receptor/beta-arrestin endocytic complexes. J. Biol. Chem 274, 10999–11006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental datasets can be found in Mendeley Data: https://dx.doi.org/10.17632/ynrr4j7t4s.1