


Abstract
Rous sarcoma virus pre-mRNA contains an element known as the negative regulator of splicing (NRS) that acts to inhibit viral RNA splicing. The NRS binds serine/arginine-rich (SR) proteins, hnRNP H and the U1/U11 snRNPs, and appears to inhibit splicing by acting as a decoy 5′ splice site. Deletions within the gag gene that encompass the NRS also lead to increased read-through past the viral polyadenylation site, suggesting a role for the NRS in promoting polyadenylation. Using NRS-specific deletions and mutations, we show here that a polyadenylation stimulatory activity maps directly to the NRS and is most likely dependent upon SR proteins and U1 and/or U11 snRNP. hnRNP H does not appear to mediate splicing control or stimulate RSV polyadenylation, since viral RNAs containing hnRNP H-specific mutations were spliced and polyadenylated normally. However, the ability of hnRNP H mutations to suppress the read-through caused by an SR protein mutation suggests the potential for hnRNP H to antagonize polyadenylation. Interestingly, disruption of splicing control closely correlated with increased read-through, indicating that a functional NRS is necessary for efficient RSV polyadenylation rather than binding of an individual factor. We propose a model in which the NRS serves to enhance polyadenylation of RSV unspliced RNA in a process analogous to the stimulation of cellular pre-mRNA polyadenylation by splicing complexes.
INTRODUCTION
A fundamental step in the maturation of eukaryotic RNA involves the removal of introns from primary transcripts through the process of pre-mRNA splicing (1). Eukaryotic viruses often exploit this process as a means to increase their coding potential and to regulate gene expression. In the case of a simple retrovirus, Rous sarcoma virus (RSV), four genes are expressed through regulated splicing (2) (Fig. 1). Two genes, env and src, are the products of alternative splicing whereby a single 5′ splice site is utilized with two distinct 3′ splice sites. Two additional genes, gag and pol, are encoded by the unspliced RNAs which escape splicing and are transported to the cytoplasm. This property is in contrast to the processing of the majority of host pre-mRNAs, which are generally spliced to completion. A substantial pool of unspliced RNA is required as progeny genome and mRNA and the balance is weighed heavily against splicing, with nearly 80% of the RSV RNA remaining unspliced. To account for this, RSV has evolved a number of independent mechanisms to ensure a large pool of unspliced RNA. Among these is a unique RNA element that acts to inhibit splicing to both the env and src 3′ splice sites (3,4) (Fig. 1) and is referred to as the negative regulator of splicing (NRS). NRS deletions or mutations cause increased splicing of viral RNAs and impaired viral replication (3–5).
Figure 1.

RSV and the NRS. The RSV genome is shown with the relative positions of the long terminal repeats (LTRs) and the gag, pol, env and src genes shown (not to scale). Alternative splicing is indicated by lines between the 5′ and 3′ splice sites (ss). The location of the NRS is shown within the gag gene and is expanded below with the positions of key sequence features noted. The overlapping binding site for the U1 and U11 snRNPs is located at nt 913–926 and is denoted by a black box. The 5′ portion of the NRS (nt 701–797) is lightly shaded and the region where hnRNP H binds (nt 740–770) is darkly shaded. The relative positions of SR protein and hnRNP H binding are indicated.
The NRS can also inhibit the splicing of heterologous introns (3,6,7), which has facilitated the analysis of its component elements. It is divided into two functionally distinct essential regions (6), termed NRS5′ and NRS3′. Despite their role in splicing inhibition, both regions interact with splicing factors (7,8). NRS3′ contains overlapping binding sites for the U1 and U11 small nuclear ribonucleoproteins (snRNPs) (5,9,10), factors which recognize 5′ splice sites of major (U2-dependent) and minor (U12-dependent) introns (1,11,12). NRS5′ is purine-rich and binds members of the serine/arginine-rich (SR) family of splicing factors (8), which play important roles in all steps of the splicing process, including the recruitment of snRNPs to splice sites (13–18). The current model of NRS function proposes that SR proteins bound to NRS5′ recruit U1 snRNP to NRS3′. Through U1, the NRS can then interact with a downstream 3′ splice site, leading to formation of a non-functional spliceosome-like complex which sequesters the splice site and prevents interaction with the authentic 5′ splice site (9,10).
In addition to its role in splicing inhibition, some evidence suggests that the NRS may stimulate RSV polyadenylation (19). Deletion of a 606 nt region in the gag gene that contains the NRS results in an increase in viral read-through RNA (19). Upstream polyadenylation enhancer elements are common in viral RNAs and are generally thought to either recruit regulatory factors or alter RNA secondary structure (20). However, the relevance of the NRS to RSV polyadenylation has remained undefined.
Recently, we showed that hnRNP H also binds to the NRS (21). Heterogeneous nuclear ribonucleoproteins (hnRNPs) are an abundant family of nuclear factors that bind nascent RNA transcripts and aid in their maturation to mRNAs (22). hnRNP H has been implicated in several diverse aspects of pre-mRNA processing, including splicing stimulation (23,24), splicing repression (25) and polyadenylation stimulation (26,27). Based on this diversity of function, it was of interest to determine the role of hnRNP H in NRS splicing or polyadenylation control.
We show here that a polyadenylation stimulatory activity maps to the NRS and does not require hnRNP H but is dependent upon SR protein and snRNP-binding sites. Additional mutational analysis suggested that hnRNP H binding may antagonize this activity and impair RSV polyadenylation. We suggest a model in which the spliceosome-like inhibitory complex that forms on the NRS serves to stimulate polyadenylation of unspliced viral RNA similarly to how spliceosomal complexes influence polyadenylation of spliced cellular RNAs.
MATERIALS AND METHODS
Plasmid constructs
RSV DNA segments were from the Prague C strain (28) using the sequence coordinates of Schwartz et al. (29). To generate mutant proviruses, NRS deletion and point mutations were generated either by PCR or site-directed mutagenesis (Morph kit; 5 Prime-3 Prime) in the Prague C SacII fragment [nt 543–1806, in pBluescriptII KS(+); Stratagene], either directly or in a MroI–SphI fragment (nt 703–1011) which was then shuttled into the SacII fragment, and transferred to a plasmid containing a proviral clone of the Prague A strain (pJTM14; kindly provided by C. M. Stoltzfus, University of Iowa, IA). The pJTM14 plasmid has been described previously (19) and consists of the Prague A strain provirus cloned upstream of the chloramphenicol acetyltransferase (cat) gene followed by the SV40 early polyadenylation signal. The ΔSacII mutant was generated by digesting pJTM14 with SacII, gel purification and re-ligation. The ΔNRS construct was generated by digesting the Prague C SacII fragment with MroI and SphI, blunting with T4 DNA polymerase, re-ligation and shuttling into pJTM14. The RG11 mutant was made by shuttling the MS version of this mutation (9) into pJTM14 as described above. To generate plasmids for yeast three-hybrid analysis, NRS 740–797×2 was excised from p3Z(740–797)×2 (21) with KpnI and HincII, blunted with T4 DNA polymerase and subcloned into the SmaI site of pIIIA/MS2-2 (provided by M. Wickens, University of Wisconsin, Madison, WI) (30,31). To express Gal4–hnRNP H fusion proteins, a NcoI–BamHI fragment containing the hnRNP H cDNA from pET15b-H (a gift from D. Black, University of California, Los Angeles, CA) was shuttled into the same sites of pACT2 (Clontech) to generate pACT2-hnRNP H. For constructs used in splicing enhancer assays, pDsx-NRS5′ has been reported (32) and additional constructs were made by cloning NRS 740–770 PCR fragments containing KpnI and XbaI sites into the pDsx plasmid (a gift from R. Reed, Harvard Medical School, Cambridge, MA) (33) using a shuttling procedure described previously (32). These PCR fragments were also cloned into pGEM-3Z (Promega) for electrophoretic mobility shift assays. Plasmids used for U1 selection experiments were generated by cloning NRS 740–1011 PCR fragments into pGEM-3Z at the KpnI and XbaI sites. Full-length NRS RNA (nt 701–1011) was produced from plasmid p3ZKXMS, which was described previously (9). All constructs were confirmed by DNA sequencing and all primer sequences are available upon request.
Yeast three-hybrid analysis
The yeast three-hybrid system was used as described (30,31). Briefly, a plasmid expressing a hybrid RNA consisting of the MS2 coat protein-binding site fused to NRS 740–797 arranged in tandem (pIIIA/MS2-2/NRS740–797×2) was transformed into the yeast strain YBZ-1 (reagents generously provided by M. Wickens), which expresses a LexA–MS2 fusion protein and a HIS3 reporter under the control of a promoter containing the LexA operator. The yeast were next transformed with library plasmids containing HeLa cDNA sequences fused to the Gal4 activation domain in pACT2 (Clontech) and plated on standard yeast medium (34) containing 0.3× adenine and 0.5 mM 3-aminotiazole (3-AT) but lacking leucine and histidine. The plates were incubated at 30°C for 1–2 weeks. Positive clones were identified by growth and library plasmids were isolated and sequenced.
Electrophoretic mobility shift assays
pGEM-3Z plasmids containing NRS 740–770 fragments were linearized with XbaI, transcribed in vitro with T7 RNA polymerase and [32P]UTP and gel purified before use. An aliquot of 2.5 fmol labeled RNA was incubated at 30°C for 30 min in 25 µl of 13 mM HEPES pH 7.9, 70 mM KCl, 20 mM creatine phosphate, 0.4 mM ATP, 3 mM MgCl2, 0.16 mM DTT, 2% glycerol, 0.08 mM EDTA and 1 µg tRNA in the presence or absence of 5 µg HeLa nuclear extract. Heparin (Sigma) was then added to a concentration of 5 mg/ml and the reaction was incubated at 30°C for an additional 10 min. The completed reaction was separated on a 4% polyacrylamide (29:1) non-denaturing gel and, following electrophoresis, the gel was dried and visualized using a phosphorimager (Molecular Dynamics Storm 860). Supershifts were performed as above except after 15 min of the initial incubation, 1 µl of hnRNP H antiserum (a gift from D. Black) was added to the reactions.
In vitro splicing assays
All pDsx-NRS plasmids were linearized with MluI, transcribed in vitro with T7 RNA polymerase and [32P]UTP in a capping reaction and gel purified before use. Splicing reactions were performed using HeLa nuclear extract under standard conditions (32). The reactions were then phenol extracted, ethanol precipitated and separated on an 8 M urea–8% polyacrylamide (19:1) gel and visualized with a phosphorimager. Bands were quantitated, normalized for uridine content, and the percentage of spliced RNA was determined from the total of unspliced plus spliced RNA.
Affinity selections
pGEM-3Z plasmids containing NRS 701–1011 or 740–1011 fragments were linearized with XbaI and transcribed in vitro in the presence of biotin-11-UTP (20% of total UTP). RNAs were incubated under splicing conditions (35) with ATP in HeLa nuclear extract for 30 min at 30°C. Streptavidin–agarose beads were added and mixed at 300 mM KCl for 1 h at 4°C and then washed extensively at 300 mM KCl. Samples were treated with proteinase K to release bound nucleic acids which were then phenol/chloroform extracted, ethanol precipitated and separated on an 8 M urea–8% polyacrylamide (19:1) gel. RNA was then electroblotted to a ZetaProbe membrane (Bio-Rad), hybridized with a riboprobe corresponding to the U1 snRNA and visualized and quantitated using a phosphorimager.
Transfection of CEFs and RNA analysis
Secondary chicken embryonic fibroblasts (CEFs) were grown in medium 199 (M199; Gibco BRL) containing 2% tryptose-phosphate broth (Gibco BRL), 1% fetal calf serum (HyClone), 1% heat-inactivated chicken serum (Gibco BRL) and 1% penicillin–streptomycin (Gibco BRL). Transfections were performed using 2 µg/ml plasmid DNA in M199 containing 200 µg/ml DEAE–dextran and after 4 h the cells were subjected to a 5–6 min 10% DMSO shock. After 40–48 h, cells were harvested and total RNA was prepared using the RNeasy Mini kit (Qiagen). RNase protection assays were performed by hybridizing 2.5 µg RNA with 106 c.p.m. of a riboprobe derived from RSV nt –218 to 630 that spans the viral LTR and 5′ portion of the gag gene [transcribed from p5′XH1 (4), kindly provided by C. M. Stoltzfus]. Following an overnight incubation at 57°C, the samples were digested at 30°C for 1 h with 25 µg/ml RNase A (Boehringer Mannheim) and 50 U/ml RNase T1 (Calbiochem), digested with proteinase K, phenol extracted, ethanol precipitated, separated on an 8 M urea–4% polyacrylamide (19:1) gel and visualized with a phosphorimager. Bands were quantitated, normalized for uridine content and the percentages of unspliced and read-through RNA were calculated based on the unspliced plus spliced RNA and read-through plus processed RNA totals, respectively. Statistical analysis was performed on multiple samples using Student’s t-test (see figure legends).
RESULTS
A functional NRS is required for stimulation of RSV polyadenylation
In addition to splicing regulation, previous reports that deletions encompassing the NRS lead to poly(A) read-through suggested that the NRS may be involved in stimulating RSV polyadenylation (19). To address a possible role for the NRS in polyadenylation, proviral clones with various NRS deletions were constructed in the Prague A strain of RSV and transfected into CEFs. RNA was collected and first analyzed for splicing by RNase protection assays using a probe that spans the viral LTR and 5′ splice site (Fig. 2A). Wild-type RSV produced 56% unspliced RNA (Fig. 2B, lane 3) and, as reported previously, this percentage was modestly yet significantly (P < 0.002) reduced by deletion of an ∼1.3 kb SacII fragment that encompasses the NRS (3) (Fig. 2B, lane 9). A more drastic reduction was seen with a specific NRS deletion (Fig. 2B, lane 8) and by deletion of NRS nt 712–798, in whole or part (Fig. 2B, lanes 4–6). Splicing control was also impaired by a mutation (RG11) that eliminates both U1 and U11 binding and abolishes NRS function in a heterologous intron (7) (Fig. 2B, lane 7). Thus, all of the mutations affected NRS activity, as expected. Taking advantage of the fact that the probe spans the viral LTR and therefore can also be used to examine RNA read-through (Fig. 2A), the NRS mutations were further evaluated for effects on RSV polyadenylation (Fig. 2B). Normal read-through past the viral polyadenylation site was observed with the wild-type virus (16%) and, consistent with previous reports (19), this percentage was increased to 38% when the SacII fragment was deleted (Fig. 2B, compare lanes 3 and 9). A poly(A) stimulatory activity maps directly to the NRS, since all specific NRS deletions increased read-through as did the NRS-specific RG11 mutation (Fig. 2B, lanes 4–8). All increases were statistically significant (P < 0.02) when compared to wild-type levels. Based on these observations, we conclude that a functional NRS is required for efficient RSV polyadenylation. To assess which NRS-binding factors might play roles in splicing control, polyadenylation stimulation or both, mutations designed to differentially affect SR protein and hnRNP H binding were constructed and tested (below).
Figure 2.

A functional NRS is required for efficient RSV polyadenylation. (A) Schematic diagram of the constructs used in splicing and polyadenylation control assays. These constructs contain the RSV Prague A strain genome (not drawn to scale) fused to the cat gene which is followed by the SV40 early polyadenylation signal. The relative positions of the four viral genes are shown and the NRS is indicated by a black box. Viral transcription begins in the viral 5′ LTR (dark line) and continues to the polyadenylation site within the 3′ LTR, where it can either undergo cleavage or read-through (dashed line). The region encompassed by the riboprobe used in these studies is shown below the diagram and the sizes of the probe and the protected fragments are indicated. This probe can be utilized for analyzing both splicing and polyadenylation, since it spans both the 5′ splice site and the 5′ LTR (which is duplicated at the 3′ end). (B) RNase protection assay using RNA from transfected CEFs. Total RNA from CEFs transfected with the indicated proviral DNA was hybridized to the riboprobe described in (A). Protected fragments were separated on a denaturing gel and a phosphorimager was used for visualization and quantitation. Probe, a sample of unprocessed probe; Mock, assay performed using RNA from mock-transfected CEFs. The positions of unspliced (US), spliced (S), read-through (RT) and processed (Pro) RNAs are shown. Because the ΔSacII construct lacks nt 543–630, the unspliced protected band is slightly smaller and is indicated (USΔSacII). Bands were quantitated and normalized for uridine content. The percentages of unspliced and read-through RNA were calculated from the total of spliced plus unspliced RNA and read-through plus processed RNA, respectively. The average percentages of unspliced and read-through RNA from at least three separate transfections and protection assays are shown below the lanes with corresponding standard deviations. Significant deviations (P < 0.02) from wild-type (WT) values are indicated in bold.
SR proteins and hnRNP H bind the NRS in vivo
Previously we showed that hnRNP H interacts with the NRS between nt 740 and 770 in vitro (21) (Fig. 1). This same region has previously been identified as a potent splicing enhancer (32) and recent in vitro studies indicated that SR proteins also bind there (L.M.McNally and M.T.McNally, unpublished observations). Because these studies were done in vitro and deletion of these putative binding sites led to read-through transcripts, we sought to confirm that SR proteins and hnRNP H could also bind this region in vivo. A yeast three-hybrid analysis (30,31) was performed using a subfragment of NRS5′ (nt 740–797) that contains the overlapping hnRNP H- and SR protein-binding sites. To increase the likelihood of detecting weak interactions, the sequence was arranged in tandem. Over one million transformants were screened and 84 positive clones were isolated (Fig. 3A). The majority of these clones encoded four proteins, one of which was nucleolin, which had previously been identified biochemically as non-specifically interacting with this region in HeLa cell extracts (21). Another isolate coded for purine-rich element-binding protein α (Purα), which is involved in cellular transcription, translation and cell growth and binds purine-rich nucleic acids (36). Due to the purine-rich nature of NRS5′ and the fact that Purα is not implicated in RNA processing (36), the binding of this protein is likely also non-specific. The remaining clones encoded the SR proteins SC35, 9G8 and SRp40. SRp40 has previously been shown to specifically bind the NRS (8; L.M.McNally and M.T.McNally, unpublished observations) whereas SC35 does not bind specifically in vitro (L.M.McNally and M.T.McNally, unpublished observations). 9G8 binding has not yet been tested. While it is unclear whether these proteins can mediate NRS activity, the results confirm that SR proteins can interact with this region in vivo in yeast.
Figure 3.

SR proteins and hnRNP H interact with the NRS in vivo in a yeast three-hybrid assay. (A) Summary of results from a three-hybrid screen using NRS 740–797 RNA in tandem (740–797×2) as bait for proteins expressed from a HeLa cDNA library. Positive clones are ordered relative to the number of times they were isolated. SR proteins are indicated in bold. (B) Three-hybrid results using the Gal4 activation domain (Gal4AD) alone or fused to hnRNP H (hnRNP H) and expressed with NRS 740–797×2 in either the sense (+) or antisense (–) orientation. Individual colonies were streaked on medium lacking leucine and uracil to confirm viability of the transformants or on medium lacking histidine to assess RNA-mediated activation of the HIS3 reporter gene. Results shown are representative of four independent colonies selected from each transformation.
Although SR proteins were identified, hnRNP H was not detected in this selection. To address this, hnRNP H was tested directly and found to specifically interact with NRS5′ (Fig. 3B). Yeast strains expressing hnRNP H have a significantly increased doubling time (B.L.Fogel and M.T.McNally, unpublished observations) such that hnRNP H clones were most likely not identified because the colonies did not achieve sufficient size to be detected in the time frame of the selection. Taken together, these data indicate that both hnRNP H and three different SR proteins interact with this portion of NRS5′ in vivo under the conditions of the yeast three-hybrid assay. Either class of protein could conceivably contribute to the influence of the NRS on RSV polyadenylation. Interestingly, proteins directly involved in polyadenylation were not identified in the screen.
hnRNP H binding maps to G tracts within NRS 740–770
Since hnRNP H and SR proteins bind within the same region in vitro and in vivo, mutations that would differentially disrupt the binding of each were sought to dissect which factor might mediate the polyadenylation effect of the NRS. Electrophoretic gel shift analysis was used to map recombinant hnRNP H binding within NRS nt 740–770. Consistent with its identification as a poly(rG)-binding protein (37,38), the results indicated that hnRNP H binding requires two separate short G tracts within NRS 740–770 (Fig. 4A and data not shown). Mutation of both of these G tracts (mut-H) was sufficient to completely eliminate formation of hnRNP H-containing complexes in HeLa nuclear extract, as evidenced by supershift analysis (Fig. 4B, compare lanes 1–3 with 10–12). In addition, since this region contains three sequence repeats that are similar to SR protein consensus binding sites (18) (Fig. 4A), two mutants were engineered that were predicted to interfere with SR binding while leaving hnRNP H binding intact. One of these mutants had no effect on hnRNP H binding (mut-AT) whereas the other, mut-SR, bound only weakly (Fig. 4B, lanes 4–9), thus indicating that at least one nucleotide outside the G tracts contributes to hnRNP H binding (Fig. 4A). Identical results were obtained with all mutants in vitro using recombinant protein and in vivo using the yeast three-hybrid assay (data not shown). With the hnRNP H-binding properties of these mutants determined, they were next tested for their ability to bind SR proteins in two distinct functional assays.
Figure 4.

Mapping of the hnRNP H binding site and representative hnRNP H mutants. (A) Summary of hnRNP H binding studies using NRS nt 740–770. Residues that are essential for hnRNP H binding are indicated in bold, non-essential residues are shown in lower case and unconfirmed residues are in italic. Bases that were not tested are in normal type. The positions of three repeated sequence elements that resemble SR consensus binding sites are underlined. The sequences of the four 740–770 mutants described in this study are shown with the corresponding mutations indicated. (B) Electrophoretic mobility shift assays were performed using 2.5 fmol individual NRS 740–770 RNAs and 5 µg HeLa nuclear extract (NE) in the presence or absence of hnRNP H antiserum (Ab). The positions of free, shifted and supershifted RNAs are indicated.
Mutations designed to affect SR protein binding decrease splicing enhancer activity of and U1 binding to NRS 740–770
Since NRS5′ harbors splicing enhancer activity, splicing enhancer assays were performed to determine the effect of the above mutations on SR protein function (Fig. 5). An SR protein-binding mutant would be expected to reduce enhancer activity. NRS sequences were fused to a weak splicing substrate derived from the Drosophila doublesex gene (33,39) and increases in spliced RNA were evaluated using in vitro splicing assays. Since hnRNP H has been previously reported to be a functional component of a splicing enhancer complex (23,24), this assay would also determine whether hnRNP H might collaborate with SR proteins in NRS enhancer function. NRS 740–770 proved to be an effective splicing enhancer, resulting in 33% spliced RNA, although it was not as potent as the entire 5′-region, which yielded 40% spliced product (Fig. 5, compare lanes 4–6 with 1–3). The mut-H mutant also showed enhancer activity that was comparable to the non-mutant 740–770 (30% versus 33% spliced product) (Fig. 5, compare lanes 7–9 with 4–6). In contrast, decreased activity was observed with both mut-AT and mut-SR, as expected (11% and 5% spliced product, respectively), which were predicted to reduce SR protein binding (Fig. 5, lanes 10–15). The results with mut-H in particular indicate that hnRNP H is not essential for in vitro splicing enhancer function, although a minor role cannot be excluded, since mut-SR impacts on hnRNP H binding and shows reduced splicing relative to mut-AT.
Figure 5.

Splicing enhancer activity of NRS5′ mutants. Splicing enhancer assays were performed using the indicated in vitro transcribed doublesex–NRS RNAs incubated in HeLa nuclear extract under splicing conditions for either 1 or 2 h. The positions of the precursor, spliced and intermediate RNA forms are depicted, with the larger NRS5′ forms on the left and the smaller NRS 740–770 forms on the right. NRS sequences are indicated by a black box. Numbers beneath each set of lanes correspond to the percentage of spliced RNA (calculated from the total of unspliced plus spliced RNA) after 2 h. Results are the average of two independent experiments and the corresponding standard deviations are indicated in parentheses.
Because the above assay works through the recruitment of factors to a suboptimal 3′ splice site, we also tested the effect of the mutations on the recruitment of U1 snRNP to the NRS. It has been shown that U1 snRNP is essential for NRS-mediated splicing inhibition (5,9,10) and that NRS5′ and SR proteins are required for U1 recruitment (9). To restrict effects to mutations in 740–770, the upstream SR protein-binding site (nt 701–739) was removed and the hnRNP H- and SR protein-specific mutants were tested in U1 recruitment assays (Fig. 6). Biotinylated NRS RNAs were incubated in nuclear extract, isolated by affinity selection with streptavidin and U1 snRNP binding was assessed by northern blotting. When nt 701–739 were removed from NRS5′, U1 recruitment was only decreased by ∼40% (Fig. 6, compare lanes 3 and 7), indicating that nt 740–797 do function in this assay. Recruitment was further decreased with both mut-AT and mut-SR, but was unaffected by the mut-H mutation (Fig. 6, compare lanes 4–6 with 3). We conclude that hnRNP H does not play a role in this aspect of NRS activity and that SR proteins are most likely responsible for U1 snRNP recruitment in vitro. Collectively, the results also confirm that mut-SR and mut-AT are functional SR protein-binding mutants of varying severity.
Figure 6.
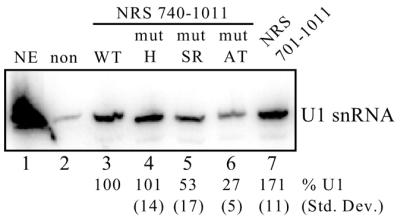
U1 snRNP recruitment to NRS mutants. Equal moles of the indicated biotinylated RNAs were incubated in HeLa nuclear extract and affinity selected with streptavidin–agarose beads. Bound snRNAs were then extracted with phenol/chloroform, separated on a denaturing polyacrylamide gel, electroblotted to a nylon membrane and hybridized to a U1 riboprobe. Non-biotinylated NRS RNA (non) was used to determine background binding to the beads whereas the full-length NRS (nt 701–1011) was used to determine optimal U1 binding (lane 7). To avoid any contributions from the upstream SR protein-binding site, the experimental RNAs lacked the primary SR protein-binding site (nt 740–1011, lanes 3–6). NE, U1 snRNA marker extracted from 3 µl of nuclear extract. Bands were quantitated using a phosphorimager and the average percentage of U1 snRNA selected from three separate experiments is shown below each lane with the corresponding standard deviation. The position of the U1 snRNA is shown on the right. Binding to the wild-type 740–1011 RNA was set at 100% (lane 3).
Effect of point mutations on splicing inhibition and polyadenylation
To extend the above results and determine whether the hnRNP H- and SR protein-specific mutations affected splicing inhibition and/or polyadenylation, proviral clones harboring the NRS 740–770 mutations were tested by RNase protection as described above. Neither the mut-H nor mut-AT mutations, singly or in combination, exhibited any significant effect on splicing control (Fig. 7, lanes 7–9). The presence of the upstream SR protein-binding region (nt 701–739) likely accounts for the lack of effect of the mut-AT mutation, while the lack of effect with the mut-H mutation indicates that hnRNP H is not essential for splicing inhibition. Likewise, polyadenylation was unaffected by the hnRNP H-specific mutation (Fig. 7, lane 7), suggesting that hnRNP H is not responsible for the stimulatory activity mapped to the NRS above. However, a modest yet significant (P < 0.002) increase in read-through was observed with the SR protein-binding mutant mut-AT (Fig. 7, lane 8). Interestingly, this effect could be suppressed in combination with the hnRNP H-specific mutation (mut-ATH; Fig. 7, lane 9). While many explanations are possible, one interpretation is that the increased read-through seen with the mut-AT mutation is not caused by decreased SR protein binding but rather by increased hnRNP H binding. Because normal polyadenylation levels were restored when mut-AT was combined with the hnRNP H-specific mutation, it is possible that binding of hnRNP H to the NRS in the context of the mut-AT mutant impairs RSV polyadenylation.
Figure 7.
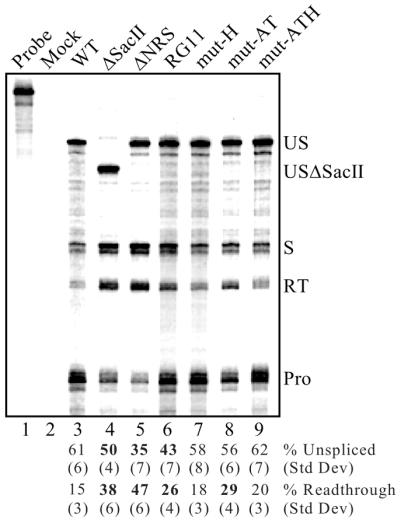
Effect of SR protein- and hnRNP H-specific mutations on splicing and read-through. RNA from CEFs transfected with the corresponding RSV mutants was used for RNase protection assays as described in Figure 6. Protected fragments were separated on a denaturing gel and a phosphorimager was used for visualization and quantitation. Probe, a sample of unprocessed probe; Mock, assay performed using RNA from mock-transfected CEFs. The positions of unspliced (US and USΔSacII), spliced (S), read-through (RT) and processed (Pro) RNAs are shown. Bands were quantitated, normalized for uridine content and the average percentages of unspliced and read-through RNA from six to eight separate transfections and protection assays are shown below the lanes with corresponding standard deviations. Significant deviations (P < 0.002) from wild-type (WT) values are indicated in bold.
DISCUSSION
Inefficient polyadenylation is a hallmark of retroviral replication and in part accounts for their ability to transduce cellular genes. Chimeric RNAs composed of retroviral genomic sequences fused to downstream cellular sequences are produced by poly(A) site read-through and can be packaged into virus particles. Reverse transcription in a subsequent infection can incorporate the cellular RNA sequences into proviral DNA, where they become a permanent constituent of the viral genetic material. The cis elements and trans-acting factors that govern RSV polyadenylation usage have not been rigorously defined, but an element in gag has previously been implicated in promoting polyadenylation (19). We show here that the stimulatory activity maps to the NRS and that it is intimately linked to NRS-mediated splicing inhibition.
Our finding that specific deletions and mutations within the NRS decrease polyadenylation is consistent with studies of a recombinant avian leukosis virus, EU8, which causes rapid onset B cell lymphomas in chickens (40). The determinant for this effect mapped to a 42 nt deletion (nt 735–776) within the NRS (40) that removes the region to which SR proteins and hnRNP H bind (21; L.M.McNally and M.T. McNally, unpublished observations). The deletion results in impaired splicing control and leads to aberrant splicing into read-through RNAs and disease from the inappropriate production of a truncated form of c-myb protein (40,41). As read-through transcripts are involved in this process, the deletion likely causes a decrease in polyadenylation efficiency and suggests a connection between NRS-mediated splicing control and viral polyadenylation in this system.
Increased read-through due to the RG11 mutation shows that snRNP binding is important for optimal polyadenylation. However, other factors are likely involved, since deletions in the upstream purine-rich region also resulted in read-through. SR proteins are likely candidates, since deletion of their binding region, NRS5′, was as effective as a complete NRS deletion and removal of subdomains within NRS5′ had intermediate effects (Fig. 2). Additionally, hnRNP H was an attractive candidate for polyadenylation control, since it was previously shown to interact within nt 740–770 of NRS5′ (21) and a highly related protein, hnRNP H′, has been shown to stimulate SV40 3′-end processing (26). To distinguish between the potential contributions of SR proteins and hnRNP H, mutations designed to differentially affect the binding of each were generated and three distinct assays indicated that mutations mut-H and mut-AT were specific for hnRNP H binding and SR protein function, respectively. These results also suggest that hnRNP H does not play a significant role in the splicing enhancer or snRNP recruitment activity that is mediated by NRS5′. Analysis of RNA from cells transfected with proviral clones harboring these mutations showed no effect on splicing inhibition. Thus, consistent with the in vitro assays, hnRNP H does not appear to play a major role in NRS-mediated splicing inhibition and, in addition, examination of read-through RNA indicated that it is not responsible for the improved polyadenylation efficiency imparted by the NRS. However, SR protein binding to this region may influence polyadenylation, since increased read-through was observed with mut-AT.
Interestingly, analysis of the mut-ATH double mutant suggests that hnRNP H may negatively influence NRS-stimulated polyadenylation, since the hnRNP H mutation suppressed read-through caused by the SR protein-binding mutation. One interpretation is that loss of SR protein binding per se is not responsible for increased read-through but rather serves to increase hnRNP H binding. Perhaps, under normal conditions, hnRNP H binding may be limited by competition with SR proteins. When the SR protein site is mutated or in cells expressing varying level of these factors, hnRNP H binding may be improved, causing inefficient polyadenylation. It is unclear what the biological relevance of such an effect could be, but it is interesting to note that the hnRNP H-binding site is conserved in all known NRS-containing avian viruses (data not shown), which may reflect an important role in the viral life cycle.
While a polyadenylation stimulatory activity mapped to the NRS, the data indicate that no discrete element within the NRS or trans-acting factor is uniquely responsible for optimal polyadenylation, but rather NRS splicing inhibition is required. Support for this stems from the observation that all mutations that decreased splicing inhibition also caused increased read-through. We therefore propose a model based on the evidence that the NRS functions similarly to a 5′ splice site (10) (Fig. 8). During the normal processing of cellular pre-mRNAs, polyadenylation and splicing are linked (20). In the exon definition model for pre-mRNA splicing (42,43) components of the splicing and polyadenylation machineries are thought to functionally bridge, and thus define, the terminal exon. Considerable evidence indicates that such interactions enhance polyadenylation, presumably through the interaction of specific splicing factors with components of the polyadenylation machinery (20,44–48). In RSV, the spliceosome-like inhibitory complex that forms on the NRS (10,35,49) may act similarly to stimulate the polyadenylation of viral RNA that does not undergo splicing. Because the viral 3′ splice sites are generally weak (50–52), formation of the non-productive NRS complex could stabilize the binding of splicing factors and thereby promote polyadenylation by improving exon definition interactions. Future studies will evaluate this model and address the mechanism by which hnRNP H may interfere with the association between the splicing and polyadenylation machineries.
Figure 8.

Model for NRS-mediated stimulation of RSV polyadenylation. (A) Spliceosome contribution to polyadenylation. A generic cellular pre-mRNA is shown with the 5′ and 3′ splice sites of a terminal intron indicated. In cellular RNA processing the terminal 3′ splice site and the polyadenylation site define the last exon and factors associated with the spliceosome act to stimulate polyadenylation (large arrow; see Discussion). (B) NRS contribution to RSV polyadenylation. The NRS is shown between authentic viral splice sites. Depicted are SR proteins that bind to nt 701–770 and initiate NRS complex formation. The spliceosome-like inhibitory complex (NRS complex) that forms between the NRS and a viral 3′ splice site is indicated. The NRS may serve to stabilize the binding of splicing factors to the weak viral 3′ splice site, which can then either recruit or stabilize the polyadenylation complex (large arrow) and thereby enhance polyadenylation of viral unspliced RNA. A potential competition between SR proteins and hnRNP H for binding to this region is reflected by an arrow. hnRNP H binding may be restricted when SR proteins are present. In contrast, if SR proteins are absent or limiting, hnRNP H binding may increase and in some way disrupt the ability of the NRS to act efficiently on the polyadenylation complex (thin line). In both cases the NRS complex is competent for splicing inhibition.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank C. Martin Stoltzfus for providing the pJTM14 and p5′XH1 plasmids and for helpful discussions. We thank Marvin Wickens for providing the yeast three-hybrid reagents, Dara Frank for contributing the HeLa cDNA library, and Roy Long for reagents, yeast protocols and many helpful discussions. We thank Doug Black for the pET15b-H plasmid and for hnRNP H antiserum, and Steve Munroe for comments on the manuscript. This research was supported by Public Health Service Grant R01 CA78709 from the National Cancer Institute.
REFERENCES
- 1.Moore M.J., Query,C.C. and Sharp,P.A. (1993) In Gesteland,R. and Atkins,J. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 303–357.
- 2.Coffin J.M. (1996) In Fields,B.N., Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 3rd Edn. Raven Press, New York, NY, pp. 1767–1847.
- 3.Arrigo S. and Beemon,K. (1988) Regulation of Rous sarcoma virus RNA splicing and stability. Mol. Cell. Biol., 8, 4858–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoltzfus C.M. and Fogarty,S.J. (1989) Multiple regions in the Rous sarcoma virus src gene intron act in cis to affect the accumulation of unspliced RNA. J. Virol., 63, 1669–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hibbert C.S., Gontarek,R.R. and Beemon,K.L. (1999) The role of overlapping U1 and U11 5′ splice site sequences in a negative regulator of splicing. RNA, 5, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McNally M.T., Gontarek,R.R. and Beemon,K. (1991) Characterization of Rous sarcoma virus intronic sequences that negatively regulate splicing. Virology, 185, 99–108. [DOI] [PubMed] [Google Scholar]
- 7.Gontarek R.R., McNally,M.T. and Beemon,K. (1993) Mutation of an RSV intronic element abolishes both U11/U12 snRNP binding and negative regulation of splicing. Genes Dev., 7, 1926–1936. [DOI] [PubMed] [Google Scholar]
- 8.McNally L.M. and McNally,M.T. (1996) SR protein splicing factors interact with the Rous sarcoma virus negative regulator of splicing element. J. Virol., 70, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNally L.M. and McNally,M.T. (1999) U1 small nuclear ribonucleoprotein and splicing inhibition by the Rous sarcoma virus negative regulator of splicing element. J. Virol., 73, 2385–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook C.R. and McNally,M.T. (1999) Interaction between the negative regulator of splicing element and a 3′ splice site: requirement for U1 small nuclear ribonucleoprotein and the 3′ splice site branch point/pyrimidine tract. J. Virol., 73, 2394–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarn W.Y. and Steitz,J.A. (1996) A novel spliceosome containing U11, U12 and U5 snRNPs excises a minor class (AT-AC) intron in vitro. Cell, 84, 801–811. [DOI] [PubMed] [Google Scholar]
- 12.Kolossova I. and Padgett,R.A. (1997) U11 snRNA interacts in vivo with the 5′ splice site of U12-dependent (AU-AC) pre-mRNA introns. RNA, 3, 227–233. [PMC free article] [PubMed] [Google Scholar]
- 13.Kohtz J.D., Jamison,S.F., Will,C.L., Zuo,P., Luhrmann,R., Garcia-Blanco,M.A. and Manley,J.L. (1994) Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature, 368, 119–124. [DOI] [PubMed] [Google Scholar]
- 14.Fu X.D. (1995) The superfamily of arginine/serine-rich splicing factors. RNA, 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- 15.Manley J.L. and Tacke,R. (1996) SR proteins and splicing control. Genes Dev., 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- 16.Reed R. (1996) Initial splice-site recognition and pairing during pre-mRNA splicing. Curr. Opin. Genet. Dev., 6, 215–220. [DOI] [PubMed] [Google Scholar]
- 17.Wang J., Takagaki,Y. and Manley,J.L. (1996) Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes Dev., 10, 2588–2599. [DOI] [PubMed] [Google Scholar]
- 18.Graveley B.R. (2000) Sorting out the complexity of SR protein functions. RNA, 6, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller J.T. and Stoltzfus,C.M. (1992) Two distant upstream regions containing cis-acting signals regulating splicing facilitate 3′-end processing of avian sarcoma virus RNA. J. Virol., 66, 4242–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J., Hyman,L. and Moore,C. (1999) Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation and interrelationships with other steps in mRNA synthesis. Microbiol. Mol. Biol. Rev., 63, 405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fogel B.L. and McNally,M.T. (2000) A cellular protein, hnRNP H, binds to the negative regulator of splicing element from Rous sarcoma virus. J. Biol. Chem., 275, 32371–32378. [DOI] [PubMed] [Google Scholar]
- 22.Dreyfuss G., Matunis,M.J., Pinol-Roma,S. and Burd,C.G. (1993) hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 23.Chou M., Rooke,N., Turck,C.W. and Black,D.L. (1999) hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol. Cell. Biol., 19, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markovtsov V., Nikolic,J.M., Goldman,J.A., Turck,C.W., Chou,M. and Black,D.L. (2000) Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol. Cell. Biol., 20, 7463–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C.D., Kobayashi,R. and Helfman,D.M. (1999) Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat β-tropomyosin gene. Genes Dev., 13, 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bagga P.S., Arhin,G.K. and Wilusz,J. (1998) DSEF-1 is a member of the hnRNP H family of RNA-binding proteins and stimulates pre-mRNA cleavage and polyadenylation in vitro. Nucleic Acids Res., 26, 5343–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veraldi K.L., Arhin,G.K., Martincic,K., Chung-Ganster,L., Wilusz,J. and Milcarek,C. (2001) hnRNP F influences binding of a 64-kilodalton subunit of cleavage stimulation factor to mRNA precursors in mouse B cells. Mol. Cell. Biol., 21, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meric C. and Spahr,P.F. (1986) Rous sarcoma virus nucleic acid-binding protein p12 is necessary for viral 70S RNA dimer formation and packaging. J. Virol., 60, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz D.E., Tizard,R. and Gilbert,W. (1983) Nucleotide sequence of Rous sarcoma virus. Cell, 32, 853–869. [DOI] [PubMed] [Google Scholar]
- 30.SenGupta D.J., Zhang,B., Kraemer,B., Pochart,P., Fields,S. and Wickens,M. (1996) A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl Acad. Sci. USA, 93, 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B., Kraemer,B., SenGupta,D., Fields,S. and Wickens,M. (1998) In Smith,C.W.J. (ed.), RNA:Protein Interactions: A Practical Approach. Oxford University Press, New York, NY, pp. 195–216.
- 32.McNally L.M. and McNally,M.T. (1998) An RNA splicing enhancer-like sequence is a component of a splicing inhibitor element from Rous sarcoma virus. Mol. Cell. Biol., 18, 3103–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Staknis D. and Reed,R. (1994) SR proteins promote the first specific recognition of pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol. Cell. Biol., 14, 7670–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Cook C.R. and McNally,M.T. (1998) SR protein and snRNP requirements for assembly of the Rous sarcoma virus negative regulator of splicing complex in vitro. Virology, 242, 211–220. [DOI] [PubMed] [Google Scholar]
- 36.Gallia G.L., Johnson,E.M. and Khalili,K. (2000) Pura: a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res., 28, 3197–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matunis M.J., Xing,J. and Dreyfuss,G. (1994) The hnRNP F protein: unique primary structure, nucleic acid-binding properties and subcellular localization. Nucleic Acids Res., 22, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honore B., Rasmussen,H.H., Vorum,H., Dejgaard,K., Liu X., Gromov,P., Madsen,P., Gesser,B., Tommerup,N. and Celis,J.E. (1995) Heterogeneous nuclear ribonucleoproteins H, H′ and F are members of a ubiquitously expressed subfamily of related but distinct proteins encoded by genes mapping to different chromosomes. J. Biol. Chem., 270, 28780–28789. [DOI] [PubMed] [Google Scholar]
- 39.Tian M. and Maniatis,T. (1993) A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell, 74, 105–114. [DOI] [PubMed] [Google Scholar]
- 40.Smith M.R., Smith,R.E., Dunkel,I., Hou,V., Beemon,K.L. and Hayward,W.S. (1997) Genetic determinant of rapid-onset B-cell lymphoma by avian leukosis virus. J. Virol., 71, 6534–6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang W., Kanter,M.R., Dunkel,I., Ramsay,R.G., Beemon,K.L. and Hayward,W.S. (1997) Minimal truncation of the c-myb gene product in rapid-onset B-cell lymphoma. J. Virol., 71, 6526–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berget S.M. (1995) Exon recognition in vertebrate splicing. J. Biol. Chem., 270, 2411–2414. [DOI] [PubMed] [Google Scholar]
- 43.Black D.L. (1995) Finding splice sites within a wilderness of RNA. RNA, 1, 763–771. [PMC free article] [PubMed] [Google Scholar]
- 44.Niwa M., Rose,S.D. and Berget,S.M. (1990) In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev., 4, 1552–1559. [DOI] [PubMed] [Google Scholar]
- 45.Nesic D., Cheng,J. and Maquat,L.E. (1993) Sequences within the last intron function in RNA 3′-end formation in cultured cells. Mol. Cell. Biol., 13, 3359–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wassarman K.M. and Steitz,J.A. (1993) Association with terminal exons in pre-mRNAs: a new role for the U1 snRNP? Genes Dev., 7, 647–659. [DOI] [PubMed] [Google Scholar]
- 47.Lutz C.S. and Alwine,J.C. (1994) Direct interaction of the U1 snRNP-A protein with the upstream efficiency element of the SV40 late polyadenylation signal. Genes Dev., 8, 576–586. [DOI] [PubMed] [Google Scholar]
- 48.Cooke C., Hans,H. and Alwine,J.C. (1999) Utilization of splicing elements and polyadenylation signal elements in the coupling of polyadenylation and last-intron removal. Mol. Cell. Biol., 19, 4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cook C.R. and McNally,M.T. (1996) Characterization of an RNP complex that assembles on the Rous sarcoma virus negative regulator of splicing element. Nucleic Acids Res., 24, 4962–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu X.D., Katz,R.A., Skalka,A.M. and Maniatis,T. (1991) The role of branchpoint and 3′-exon sequences in the control of balanced splicing of avian retrovirus RNA. Genes Dev., 5, 211–220. [DOI] [PubMed] [Google Scholar]
- 51.McNally M.T. and Beemon,K. (1992) Intronic sequences and 3′ splice sites control Rous sarcoma virus RNA splicing. J. Virol., 66, 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L. and Stoltzfus,C.M. (1995) A suboptimal src 3′ splice site is necessary for efficient replication of Rous sarcoma virus. Virology, 206, 1099–1107. [DOI] [PubMed] [Google Scholar]