

Abstract
TNF‐stimulated gene (TSG‐6) was reported to suppress hypertrophic scar (HS) formation in a rabbit ear model, and the overexpression of TSG‐6 in human HS fibroblasts (HSFs) was found to induce their apoptotic death. The molecular basis for these findings, however, remains to be clarified. HSFs were subjected to TSG‐6 treatment. Treatment with TSG‐6 significantly suppressed HSF proliferation and induced them to undergo apoptosis. Moreover, TSG‐6 exposure led to reductions in collagen I, collagen III, and α‐SMA mRNA and protein levels, with a corresponding drop in proliferating cell nuclear antigen (PCNA) expression indicative of impaired proliferative activity. Endoplasmic reticulum (ER) stress was also suppressed in these HSFs as demonstrated by decreases in Bip and p‐IRE1α expression, downstream inositol requiring enzyme 1 alpha (IRE1α) ‐Tumor necrosis factor receptor associated factor 2 (TRAF2) pathway signalling was inhibited and treated cells failed to induce NF‐κB, TNF‐α, IL‐1β, and IL‐6 expression. Overall, ER stress was found to trigger inflammatory activity in HSFs via the IRE1α‐TRAF2 axis, as confirmed with the specific inhibitor of IRE1α STF083010. Additionally, the effects of TSG‐6 on apoptosis, collagen I, collagen III, α‐SMA, and PCNA of HSFs were reversed by the IRE1α activator thapsigargin (TG). These data suggest that TSG‐6 administration can effectively suppress the proliferation of HSFs in part via the inhibition of IRE1α‐mediated ER stress‐induced inflammation (IRE1α/TRAF2/NF‐κB signalling).
Keywords: endoplasmic reticulum stress, hypertrophic scar fibroblasts, inflammation, IRE1α, TSG‐6
1. INTRODUCTION
Hypertrophic scar (HS) development is a common outcome in the context of wound healing and tissue repair, particularly in individuals healing from surgery, trauma, or burns. These scars can cause psychological distress for affected patients in addition to contributing to functional disorders associated with related symptoms such as pain, stiffness, and pruritus. A range of approaches to HS treatment have been designed to date, including cryotherapy, radiation therapy, laser‐based treatment, pressure therapy, intralesional steroid administration, and surgical excision. These treatments, however, are often associated with high recurrence rates and the potential for adverse outcomes, yielding unsatisfactory results. 1 , 2 There thus remains a pressing need for a robust and reliable approach to HS treatment.
Fibroblasts are the primary cell type responsible for HS formation, with HS fibroblasts (HSFs) exhibiting enhanced proliferative capabilities. When activated, fibroblasts can undergo differentiation to yield rapidly proliferating myofibroblasts that secrete a range of extracellular matrix (ECM) proteins including various collagens. When the normal homeostatic balance between the degradation and production of ECM proteins is disrupted, this can drive HS formation. In contrast, when fibroblasts remain in a relatively inactive and non‐proliferative state in which they produce only limited quantities of secreted ECM proteins, collagens in wound sites can be readily degraded and myofibroblasts ultimately undergo apoptotic death, contributing to scarless wound healing. 3 , 4 , 5 , 6 Further studies exploring the mechanisms governing HSF proliferation and excessive ECM protein secretion thus have the potential to define novel molecular targets that can be leveraged when developing effective treatments to protect against HS formation.
While the mechanisms underlying HS development are incompletely characterised, inflammation is known to play a central role in this process. Both excessive and prolonged inflammation can contribute to such scar formation, with overly robust inflammation leading to the infiltration of the wound site by inflammatory cells that produce a range of inflammatory cytokines including interleukin 1β (IL‐1β), IL‐6, and tumour necrosis factor‐α (TNF‐α). These factors, in turn, drive fibroblasts to proliferate and secrete excessive levels of collagen, thus facilitating HS formation. 4 , 7 , 8 Anti‐inflammatory therapies thus represent a promising approach to preventing or treating HS development. 9 , 10 , 11 , 12
The endoplasmic reticulum (ER) is a critical organelle that regulates a wide range of physiological and pathological processes. Under conditions of stress, misfolded protein accumulation within the ER can drive the induction of the unfolded protein response, which is coordinated by three key sensor proteins (PERK, ATF6, and IRE1α). In the absence of ER stress, these proteins are bound by the ER chaperone GRP78, also known as Bip (binding immunoglobulin protein). ER stress leads to the dissociation of Bip from these three proteins such that it may bind to misfolded proteins, thereby leading to PERK, IRE1α, and ATF6 activation. 13 , 14 , 15 Using a rat ear model of HS formation, Kim et al. previously demonstrated that treatment using the ER stress inhibitor TUDCA was sufficient to significantly reduce scar development, highlighting a role for ER stress in this pathological process. 16 Multiple studies have also revealed a link between ER stress and inflammation in the context of metabolic disorders, infection, autoimmunity, and neurodegenerative disease. 13 , 14 , 15 Of the three ER sensor proteins, IRE1α has been most closely associated with the inflammatory response. 13 , 14 , 15 For example, inhibiting IRE1α was sufficient to alleviate inflammation and associated pulmonary damage in a murine ventilator‐induced lung injury (VILI) model. 17 Prior work has shown that excisional wound healing was associated with the upregulation of IRE1 in a murine model of HS formation, with small molecule‐mediated IRE1 inhibition being sufficient to reduce scar formation. 18 IRE1α signalling may thus be linked to inflammatory activity and play a key role in the context of HS development, with therapeutic approaches that can regulate IRE1α‐associated inflammation thus representing a promising approach to suppressing HS development.
The TNF‐stimulated gene 6 (TSG‐6) glycoprotein is upregulated in many cell types following exposure to proinflammatory cytokines or other factors, whereupon it can suppress inflammation both in vitro and in vivo. 19 , 20 In a rabbit ear model system, TSG‐6 has been reported to suppress HS formation, and the overexpression of TSG‐6 in human HSFs induces their apoptotic death. 10 , 21 The mechanistic role that TSG‐6 plays in HS formation, however, remains to be fully clarified. Human adipose‐derived mesenchymal stem cell‐derived TSG‐6 has recently been shown to alleviate severe acute pancreatitis by inhibiting ER stress and associated inflammation. 22 As such, this study was developed with the hypothesis that TSG‐6 can serve as a regulator of human HSF proliferation through its ability to suppress ER stress‐associated inflammation via the IRE1α signalling axis.
Here, the ability of TSG‐6 administration to inhibit the proliferation of human HSFs while inducing their apoptotic death and suppressing their collagen deposition activity was assessed in vitro. Moreover, the ability of TSG‐6 to influence HSF functionality via modulating IRE1α signalling and inflammatory pathways was assessed.
2. MATERIALS AND METHODS
2.1. Cell culture
Human HSFs (cat no. CM‐1387) and normal human skin fibroblasts (HFFs, cat no. SCSP‐109) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and were cultured in DMEM (Thermo Fisher, IL, USA) containing 10% FBS (Thermo Fisher) and 1% (v/v) penicillin/streptomycin (Shanghai biyuntian Biotechnology Co., Ltd, China) in a humidified 37°C 5% CO2 incubator.
2.2. CCK‐8 assay
HFFs and HSFs were plated in 96‐well plates (1 × 10 5 /well) in triplicate, after which they were treated with a range of TSG‐6 concentrations (0, 10, 100, 200, 400 ng/mL). After 0, 24, 48, 72, or 96 h, 10 μL of CCK‐8 reagent (Sigma‐Aldrich, USA) was added per well, and absorbance at 450 nm was then assessed with a microplate reader (ThermoFisher, USA).
2.3. Apoptosis analyses
A flow cytometry‐based approach was used to detect HFF and HSF apoptosis with a commercial apoptosis detection kit based on provided directions. Initially, cells were harvested with trypsin (Thermo Fisher Scientific, IL, USA), after which they were centrifuged for 10 min at 1200 rpm, rinsed with PBS, and stained at room temperature with Annexin V‐FITC (BD, USA) and propidium iodide (Sigma‐Aldrich, USA). A flow cytometer (Becton Dickinson, USA) was then used to detect apoptotic cells.
2.4. Immunofluorescent staining
After appropriate treatment, cells were fixed using 4% paraformaldehyde, permeabilized for 15 min with 0.1% Triton X‐100, and blocked for 30 min with 5% goat serum. Following incubation overnight with anti‐p‐IRE1α, 1:150, Abcam, UK at 4°C, cells were probed with secondary Cy3‐ or AF488‐conjugated anti‐IgG (1:100, Shanghai biyuntian Biotechnology Co., Ltd, China) for 90 min. Cells were then washed thrice with PBS, counterstained for 5 min with DAPI (4,6‐diamidino‐2‐phenylindole dihydrochloride), and imaged with a laser fluorescence microscope (Olympus, Tokyo, Japan). All data were analysed by an investigator blinded to experimental groupings.
2.5. Western immunoblotting
RIPA buffer (Shanghai biyuntian Biotechnology Co., Ltd, China) containing 1 mM PMSF (Shanghai biyuntian Biotechnology Co., Ltd, China) was used to lyse cells on ice for 30 min, after which samples were centrifuged for 10 min at 12000 rpm at 4°C. Protein levels in the resultant supernatants were then assessed with a BCA Protein Assay Kit (Takara, Japan), and 30 ug of protein per sample was separated via 10% SDS‐PAGE and transferred onto PVDF membranes (Shanghai biyuntian Biotechnology Co., Ltd, China). Blots were blocked with 5% BSA and then incubated overnight with appropriate primary antibodies (anti‐collagen I, Affinity, AF7001, 1:500; anti‐collagen III, Affinity, 22 734‐1‐AP, 1:500; anti‐proliferating cell nuclear antigen (PCNA), Proteintech, 10 205‐2‐AP, 1:5000; anti‐α‐SMA, Affinity, AF1032, 1:500; anti‐Bip, Proteintech, 66 574‐1‐Ig; anti‐p‐IRE1α, Affinity, AF7150, 1:2000; anti‐TRAF2, Proteintech, 26 846‐1‐AP, 1:1000; anti‐NF‐κBp65, CST, 8242S, 1:1000; anti‐ GAPDH, 60004‐1‐lg, 1:100000) at 4°C. Blots were then washed thrice with TBST, incubated for 90 min with appropriate secondary antibodies at room temperature, washed three more times, and protein bands were then detected using ECL solution (Thermo, USA). ImageJ (v 1.53a, NIH, MD, USA) was then used for densitometric analyses, with GAPDH being used for normalisation.
2.6. ELISAs
Levels of interleukin 1β (IL‐1β), IL‐6, and tumour necrosis factor‐α (TNF‐α) were quantified in supernatants collected from appropriately treated HSFs using commercial ELISA kits (Shanghai enzyme Biotechnology Co., Ltd, China) based on provided directions. 9
2.7. Real‐time PCR
TRIzol (Biosharp, China) was used to isolate RNA from cultured cells, after which an ultraviolet spectrophotometer was used to measure RNA purity and concentrations. cDNA was then synthesised using the PrimeScript™ RT reagent Kit (Takara, China) with the following settings: 1 h at 37°C, 5 min at 85°C, followed by storage at −20°C. Real‐Time (RT‐PCR) was performed using the iTaq universal SYBR Green Supermix (Bio‐Rad, Hercules, CA, USA). For qPCR amplification, the following settings were used: 95°C for 300 s; 40 cycles of 95°C for 20 s, 55°C for 20 s, and 72°C for 20 s. The temperature was then lowered to 60°C and samples were heated to 95°C to denature DNA. Relative gene expression was assessed via the 2−△△Ct method, with GAPDH as a normalisation control. Utilised primers are listed in Table 1.
TABLE 1.
qRT‐PCR primer sequences
| Target gene | Primers | Sequence (5′‐3′) |
|---|---|---|
| GAPDH | F | CAACGAATTTGGCTACAGCA |
| R | AGGGGTCTACATGGCAACTG | |
| Collagen I | F | CTGCTGGACGTCCTGGTGAA |
| R | ACGCCTGTCCAGCAATACCTTGAG | |
| Collagen III | F | CCCACTATTATTTTGGCACAACAG |
| R | AACGGATCCTGAGTCACAGACA | |
| α‐SMA | F | AGGGACTAATGGTTGGAATGG |
| R | CAATCTCACGCTCGGCAGTAG | |
| PCNA | F | AACCTGCAGAGCATGGACTC |
| R | TATCCGCGTTATCTTCGGCC | |
| NF‐κB p65 | F | TGGCCCCTATGTGGAGATCA |
| R | GTATCTGTGCTCCTCTCGCC | |
| Bip | F | GACAAGAAGGAGGACGTGGG |
| R | GCATCGCCAATCAGACGTTC | |
| TRAF2 | F | GGAGGCATCCACCTACGATG |
| R | GGGAGAAGATGGCGGGTATG |
2.8. Statistical analysis
Data are means ± standard error of the mean (SEM). Data were compared via one‐way ANOVAs and t‐tests using SPSS v 22.0 (IL, USA). P < 0.05 was the threshold of significance.
3. RESULTS
3.1. The impact of TSG‐6 on HSF proliferation
Initially, the impact of TSG‐6 on HFF and HSF proliferation was examined via CCK‐8 assay following treatment with various TSG‐6 concentrations (0, 10, 100, 200, 400 ng/mL) for a range of time points (0, 24, 48, 72, and 96 h) (P > 0.05). While low doses of TSG‐6 failed to inhibit HSF proliferation at the tested time points, treatment with 100 ng/mL of TSG‐6 significantly inhibited the proliferative activity of these cells (P < 0.05), and such inhibition was enhanced with higher doses and over longer treatment periods (P < 0.05). As such, TSG‐6 can disrupt the ability of HSFs to proliferate in a dose‐ and time‐dependent fashion, with peak inhibition being evident following treatment with 200 ng/mL of TSG‐6 for 48 h (Figure 1A). These treatment conditions were thus used in all subsequent experiments. In contrast, TSG‐6 failed to suppress HFF proliferation (P > 0.05) (Figure 1B).
FIGURE 1.
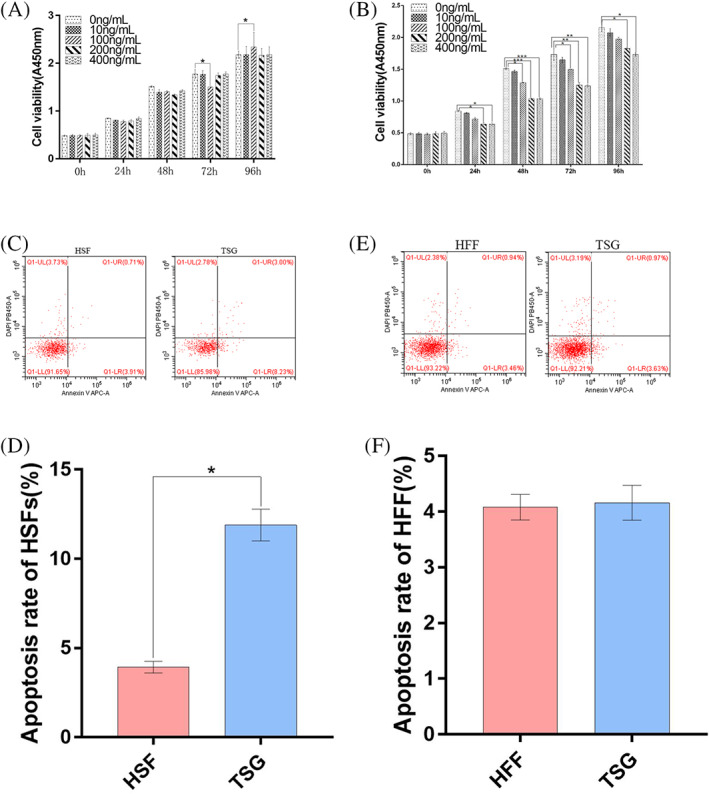
The impact of TSG‐6 on the proliferation and apoptotic death of HFFs and HSFs. (A), (B) The impact of TSG‐6 on the proliferation of HFFs (A) and HSFs (B) was assessed via CCK‐8 assay following treatment at a range of concentrations (0, 10, 100, 200, 400 ng/mL) for 0, 24, 48, 72 and 96 h. (C) Representative HSF apoptosis rates following treatment for 48 h with 200 ng/mL of TSG‐6. (D) Quantification of HSF apoptosis rates. (E) Representative HFF apoptosis rates following treatment for 48 h with 200 ng/mL of TSG‐6. (F) Quantification of HFF apoptosis rates. Data are means ± SD (n = 3/group). *P < 0.05, **P < 0.01, ***P < 0.01 vs the 0 ng/mL group
3.2. TSG‐6 promotes the apoptotic death of HSFs
A flow cytometry approach was next used to examine the ability of TSG‐6 to induce apoptotic HSF death (Figure 1C‐F). Rates of apoptotic death were significantly elevated in HSFs treated with TSG‐6 relative to untreated HSFs (Figure 1C,D) (P < 0.05), whereas TSG‐6 failed to induce HFF apoptosis (Figure 1E,F) (P > 0.05). As such, TSG‐6 can promote the apoptotic death of HSFs without any corresponding effect on HFFs.
3.3. TSG‐6 inhibits the expression of proliferation‐ and fibrosis‐associated proteins in HSFs
Next, Western blotting and RT‐PCR approaches were used to examine the expression of the fibrosis‐related markers collagen I, collagen III, and α‐SMA. Cells were assigned to the HSF and HSF + TSG‐6 (TSG) groups. TSG‐6 treatment led to significant reductions in both mRNA and protein levels of these three fibrosis‐associated targets in HSFs (Figure 2A‐D, G‐I) (P < 0.05). As HSF proliferation is closely tied to HS formation, the inhibition of such proliferative activity is of potential therapeutic benefit. As such, RT‐PCR and Western blotting approaches were next used to assess the expression of the proliferation marker PCNA Relative to untreated HSFs, those that had been treated with TSG‐6 exhibited lower PCNA mRNA and protein levels (Figure 2E,F,J) (P < 0.05), suggesting that TSG‐6 can markedly disrupt HSF proliferation.
FIGURE 2.
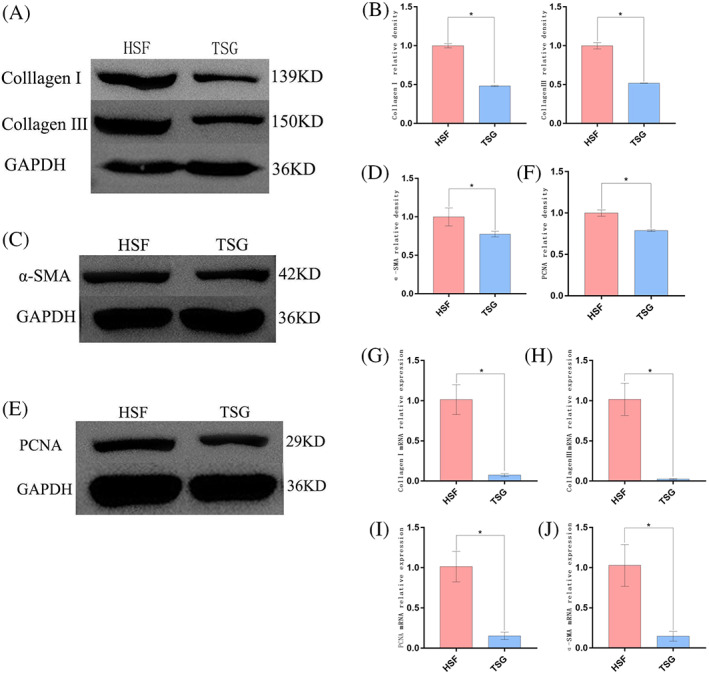
The impact of TSG‐6 on fibroblast‐associated protein expression and HSF proliferation. (A), (B) Western blotting analysis of Collagen I and Collagen III levels. (C), (D) Western blotting analysis of α‐SMA levels. (E), (F) Western blotting analysis of PCNA levels. (G) to (J) Collagen I, Collagen III, PCNA, and a‐SMA expression were analysed via RT‐PCR after TSG‐6 treatment. Data are means ± SD (n = 3/group). *P < 0.05 vs the HSF group
3.4. TSG‐6 treatment of HSFs suppresses ER stress, IRE1α, and inflammatory signalling
To establish the relationship between ER stress and HSF characteristics, Western blotting were used to analyse the ER stress‐related marker proteins Bip and p‐IRE1α in HSFs and HFFs, RT‐PCR for Bip, revealing significantly higher levels in both of these markers in HSFs relative to HFFs (Figure 3A‐C) (P < 0.05), thus suggesting that HSFs exhibit the basal induction of ER stress. When HSFs were treated with TSG‐6, this led to a dramatic reduction in Bip mRNA levels as well as a drop in Bip and p‐IRE1α protein levels relative to those observed in untreated HSFs (P < 0.05) (Figure 4A‐C), consistent with the ability of TSG‐6 to alleviate ER stress. These results were also supported by immunofluorescent staining data demonstrating that treatment with TSG‐6 led to significant reductions in p‐IRE1α fluorescence intensity relative to untreated HSFs (P < 0.05) (Figure 4D‐E).
FIGURE 3.
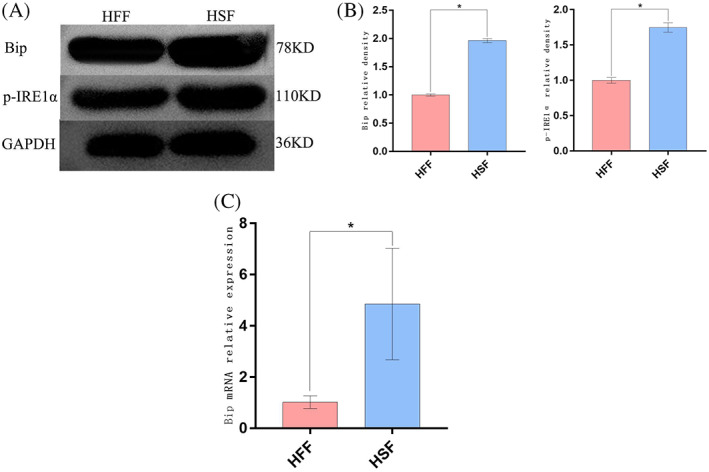
Analysis of ER stress‐related protein expression. (A), (B) Bip and p‐IRE1α levels were detected via Western blotting. (C) Bip mRNA levels were detected via RT‐PCR. Data are means ± SD (n = 3/group). *P < 0.05 vs the HFF group
FIGURE 4.
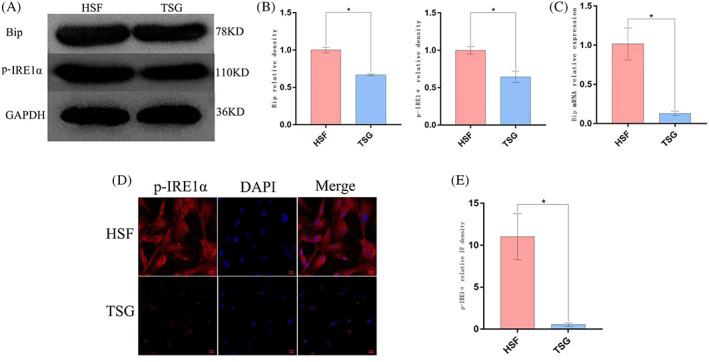
The impact of TSG‐6 on ER stress in HSFs. (A), (B) Bip and p‐IRE1α levels were detected via Western blotting. (C) Bip mRNA levels were detected via RT‐PCR. (D) p‐IRE1α levels were detected via immunofluorescent staining. (E) Quantification of the immunofluorescence intensity p‐IRE1α. Data are means ± SD (n = 3/group). *P < 0.05 vs the HSF group. (Magnification ×400). Scale bar: 20 μm
The effects of TSG‐6 administration on signalling activity downstream of IRE1α was next examined in these HSFs. Cells were next assigned to the HFF, HSF, and HSF + TSG‐6 (TSG) groups. Relative to HFFs, HSFs exhibited significant increases in TRAF2 and NF‐κB p65 mRNA and protein levels (P < 0.05) (Figure 5A‐B), consistent with the activation of IRE1α signalling activity in these cells. These changes were reversed, however, when HSFs were treated with TSG‐6, supporting its ability to suppress IRE1α signalling within HSFs (P < 0.05) (Figure 5C). To expand on these results and to gauge the anti‐inflammatory efficacy of TSG‐6 treatment, proinflammatory IL‐1β, IL‐6, and TNF‐α levels were detected via ELISA. Relative to HFFs, HSFs produced significantly higher levels of all three of these cytokines (P < 0.05) (Figure 5D), while TNF‐α, IL‐1β, and IL‐6 secretion were significantly inhibited following TSG‐6 treatment (P < 0.05).
FIGURE 5.
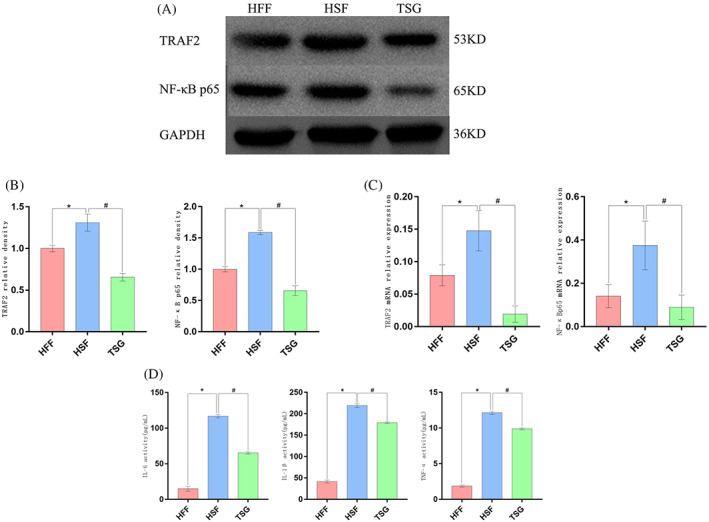
The impact of TSG‐6 on signalling downstream of IRE1α and proinflammatory cytokine induction in HSFs. (A‐B) TRAF2 and NF‐κBp65 levels were detected via Western blotting. (C) TRAF2 and NF‐κBp65 mRNA levels were assessed via RT‐PCR. (D) IL‐1β, IL‐6, and TNF‐α levels were measured via ELISA. *P < 0.05 vs the HFF group, # P < 0.05 vs the HSF group.
3.5. TSG‐6 inhibits IRE1α pathway signalling to disrupt ER stress‐mediated inflammatory and proliferative activity in HSFs
To more fully characterise the association between IRE1α pathway activity and inflammation in HSFs, these cells were treated using STF083010 (25 μM, Hefei zhenwo Biomedical Technology Co., Ltd, China) to specifically inhibit IRE1α. Cells were assigned to the HSF and HSF + STF (STF) groups. Relative to untreated HSFs, those that had been treated with STF083010 exhibited significant suppression of TRAF2 mRNA expression with concomitant suppression of p‐IRE1α and TRAF2 protein expression and corresponding reductions in the levels of downstream NF‐κB p65, TNF‐α, IL‐1β, and IL‐6 (P < 0.05 vs HSF groups) (Figure 6A‐D). These data thus confirmed that inhibiting IRE1α activation in HSFs was sufficient to ablate inflammatory signalling mediated by the IRE1α‐TRAF2 axis.
FIGURE 6.
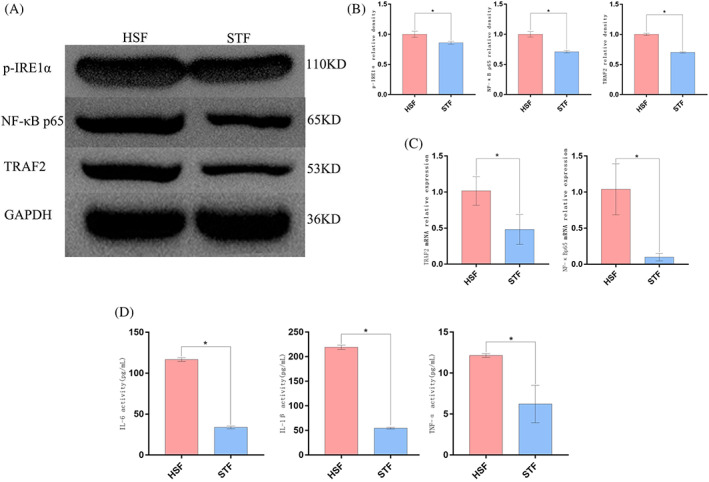
ER stress induces inflammatory activity via the IRE1α signalling pathway in HSFs. The specific inhibitor STF083010 was used to inhibit IRE1α pathway activity. (A), (B) p‐IRE1α, TRAF2, and NF‐κBp65 levels were detected via Western blotting. (C) TRAF2, and NF‐κBp65 mRNA levels were detected via RT‐PCR (D) IL‐1β, IL‐6, and TNF‐α levels were measured via ELISA. Data are means ± SD (n = 3/group). *P < 0.05 vs the HSF group
To additionally characterise the mechanism of action underlying the antiproliferative and anti‐inflammatory effects of TSG‐6 treatment, thapsigargin (TG) (1 μM, Shanghai siding Biotechnology Co., Ltd, China) was used to treat these cells as a means of specifically activating IRE1α signalling. Cells were next assigned to the HSF, HSF + TSG‐6 (TSG), and HSF + TSG‐6 + TG (TG) groups. Apoptosis rates for HSFs treated in the TG group were significantly reduced relative to those for cells treated in the TSG group (P < 0.05), with corresponding increases in collagen I, collagen III, α‐SMA, and PCNA mRNA and protein levels (P < 0.05). These data thus suggested that the activation of the ER stress pathway can markedly ablate the effects of TSG‐6 on HSF proliferation, apoptosis, and fibrosis‐related molecules supporting a model wherein these therapeutic effects are mediated by the IRE1α‐TRAF2‐ NF‐κBp65 signalling axis. As such, inhibiting the IRE1α pathway through treatment with TSG‐6 may represent an effective means of suppressing HSF proliferation, potentially thereby protecting against HS development (Figure 7).
FIGURE 7.
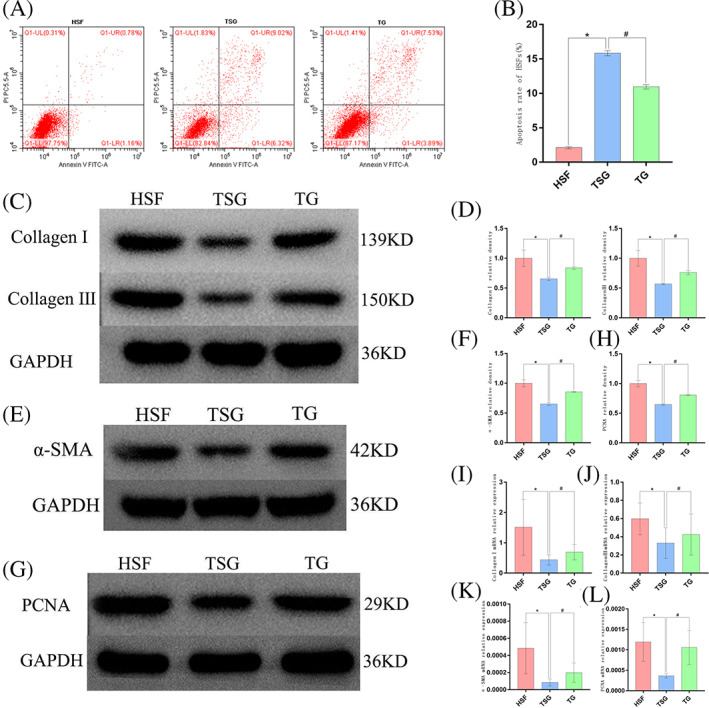
The impact of TSG‐6 on HSF proliferation and apoptosis was suppressed following TG treatment‐mediated ER stress induction. (A) Representative flow cytometry plots from analyses of HSF apoptosis rates. (B) Quantification of HSF apoptosis rates. (C), (D) Western blotting analysis of Collagen I and Collagen III levels. (E), (F) Western blotting analysis of α‐SMA levels. (G‐H) Western blotting analysis of PCNA levels. (I‐L) Collagen I, Collagen III, PCNA, and a‐SMA expression were analysed via RT‐PCR. Data are means ± SD (n = 3/group). *P < 0.05 vs HSF group, # P < 0.05 vs TSG group
4. DISCUSSION
TSG‐6 is a well‐known secretory factor responsible for immunomodulation, which is expressed in many cell types in response to stimulation by several proinflammatory mediators. Furthermore, TSG‐6 has been shown to play important roles in reducing inflammatory responses in the context of lung injury, corneal injury, skin wounding, peritonitis, and pancreatitis. 19 , 20 In a prior analysis of a rabbit ear model system, TSG‐6 was reported to suppress HS formation, 10 and the overexpression of TSG‐6 in human HSFs was found to induce their apoptotic death. 21 The molecular basis for these findings, however, remains to be clarified.
A large body of evidence supports a link between the proliferation and ECM‐secreting activities of fibroblasts and HS formation, with this loss of normal homeostatic balance between the proliferation and apoptosis of these activated fibroblasts ultimately contributing to the formation of a pronounced scar during the wound healing and tissue remodelling process. 3 , 4 , 5 , 6 Fibroblasts have thus emerged as an important target for anti‐cicatricial therapy. 23 , 24 , 25 Here, human HSFs were used as a model to study the therapeutic benefits associated with TSG‐6 treatment, revealing that such treatment was associated with significant increases in the rate of HSF apoptotic death, in line with prior evidence. 21 Similarly, a CCK‐8 assay revealed that treatment with TSG‐6 could significantly suppress HSF proliferation. Moreover, Western blotting and RT‐PCR revealed that TSG‐6 treatment was associated with reduced HSF expression of PCNA, which is a DNA polymerase delta auxiliary protein that is expressed at high levels in proliferating cells wherein it plays a key role in cell cycle progression and replication. 26 , 27 This result was consistent with the observed TSG‐6‐mediated induction of apoptotic HSF death and CCK‐8 assay findings. Following cutaneous injury, fibroblasts surrounding the wound site can secrete collagens and other protein components of the ECM, thereby facilitating tissue repair and healing processes. Both type I and type III collagen are important ECM components that are associated with HS development, 4 , 28 , 29 while the expression of α‐SMA is associated with the differentiation of fibroblasts into myofibroblasts, making it a valuable marker for myofibroblasts that is correlated with scar formation and ECM protein deposition. 3 , 4 , 29 As such, collagen I, collagen III, and α‐SMA are important fibrosis‐associated proteins that play a pathogenic role in the context of HS development. 23 , 30 Here, TSG‐6 treatment was found to readily inhibit the expression of all three of these markers at the mRNA and protein levels, consistent with its ability to inhibit excessive ECM deposition mediated by HSFs, thus supporting the anti‐cicatricial effects of such treatment. Similarly, our previous experiments indicated that TSG‐6 could reduce the deposition of collagen I and collagen III in a rabbit ear model, in line with the present study. 10
Inflammatory activity is thought to play an integral role in HS formation, with high levels of inflammatory cells and mediators, including IL‐1β, IL‐6, and TNF‐α, serving to alter the proliferation, differentiation, and collagen deposition activity of fibroblasts. 4 , 7 In cases of fetal or oral mucosal wound healing, which are not subject to HS formation, marked reductions in inflammatory cell infiltration and proinflammatory cytokine production have been reported. 31 , 32 The administration of anti‐inflammatory treatments to wounded tissues at an early time point has the potential to protect against scar formation while driving more rapid healing responses, with cytokines serving as central regulators of HS‐related inflammation. 7 , 9 , 10 , 11 , 12 Here, TNF‐α, IL‐1β, and IL‐6 expression levels were found to be significantly elevated in HSFs under basal conditions, while TSG‐6 treatment suppressed the expression of all three of these cytokines, in line with prior evidence from a rabbit ear HS model system in which TSG‐6 was able to prevent scar formation in part via suppressing IL‐1β, IL‐6, and TNF‐α expression. 10
In a prior study using a rabbit ear model of HS formation, Kim et al. found that treatment with the ER stress inhibitor TUDCA was sufficient to significantly reduce scar formation, suggesting that ER stress plays an important role in this process. 16 Prior evidence also suggests that in a murine model of HS formation, IRE1 is upregulated in the context of excisional wound healing, and small molecule inhibition of IRE1 is sufficient to suppress HS development. 18 Here, the relationship between ER stress and HSF characteristics was assessed by measuring Bip and p‐IRE1α levels in these cells, revealing that ER stress was induced in HSFs as evidenced by increases in the expression of both of these proteins. Treatment of these cells with TSG‐6, however, significantly alleviated such ER stress, in line with a prior report. 22
Several prior studies have documented a close relationship between ER stress and inflammation, with the interplay between these pathways contributing to the pathogenesis of a range of diseases and disorders. 13 , 14 IRE1α in particular has been found to be associated with inflammation, as TRAF2 can be recruited to the cytoplasmic domain of p‐IRE1α under conditions of ER stress, with the resultant IRE1α‐TRAF2 complex serving to promote NF‐κB pathway signalling, thus propagating an inflammatory state within the cell. 17 , 33 NF‐κB (p65) serves as a core transcriptional regulator of inflammation‐related gene expression, 34 , 35 with growing evidence suggesting that NF‐κB activation can promote pro‐inflammatory gene expression, thereby triggering inflammatory cascades that drive HS development. 9 , 11 , 12 Furthermore, NF‐κB acts as an important link between ER stress and inflammatory response. 15 , 33 While ER stress can thus induce IRE1α‐mediated inflammatory NF‐κB signalling, no prior studies have reported on the link between this signalling axis and HS formation. As these prior data suggested a potential role for this IRE1α‐TRAF2‐NF‐κB signalling pathway in HS formation, STF083010 was herein utilised as a specific IRE1α inhibitor, resulting in the marked suppression of IRE1α‐TRAF2 pathway signalling, leading to the consequent downregulation of NF‐κB, TNF‐α, IL‐1β, and IL‐6 in treated cells. Previously, it has been demonstrated that TSG‐6 can play an important role in anti‐inflammatory responses and ECM remodelling by inhibiting the activation of NF‐κB signalling. 36 Furthermore, TSG‐6 secreted by human adipose tissue‐derived mesenchymal stem cells ameliorated severe acute pancreatitis via ER stress downregulation in mice. 22 Additionally, both NF‐κB activity and inflammatory responses were suppressed by TSG‐6. Therefore, TSG‐6 may regulate inflammatory response through ER stress.
Together, the results of this analysis suggest that ER stress can induce inflammatory signalling in HSFs via the IRE1α‐TRAF2‐NF‐κB axis, with TSG‐6 serving to inhibit signalling via this pathway, thereby blunting downstream inflammation. As such, the ability of TSG‐6 to suppress HSF proliferation may be tied to ER stress‐associated inflammatory IRE1α signalling. To further confirm this link between TSG‐6 treatment and the suppression of cicatricial activity, these cells were treated with TG to specifically activate IRE1α. Consistent with these other results, the activation of IRE1α signalling reversed the beneficial impacts of TSG‐6 administration on HSF proliferation, apoptosis, and fibrosis‐related protein expression. As such, TSG‐6 prevented HSF proliferation at least in part through the inhibition of ER stress‐related inflammation through the IRE1α signalling axis.
There are some limitations to this study. For one, these results were restricted to in vitro analyses, and further in vivo validation of these results will thus be essential. Moreover, whether TSG‐6 can additionally suppress HSF proliferation through mechanisms unrelated to ER stress remains to be established.
5. CONCLUSIONS
In summary, these data suggest that treatment with TSG‐6 can suppress the proliferation and promote their apoptotic death of HSFs in vitro of HSFs in vitro. This anti‐proliferation and fibrosis‐related molecules activity were linked to the inhibition of ER stress‐induced inflammatory activity mediated via the IRE1α‐TRAF2‐NF‐κB signalling axis. As such, TSG‐6 may offer potential value for the development of novel alternative therapies for the treatment or prevention of HS formation.
AUTHOR CONTRIBUTIONS
Li Ma: Conceptualization, Methodology, Software, Writing – original draft. Lei Hua: Visualisation, Methodology, Investigation. Wenyuan Yu: Visualisation, Investigation. Li Ke: Investigation, Data curation, Visualisation. Liang‐yong Li: Conceptualization, Methodology, Writing – review & editing, Supervision.
CONFLICT OF INTEREST
The authors declare that there are no competing interests associated with the manuscript.
ACKNOWLEDGEMENTS
The research was supported by Natural Science Foundation of Anhui Province (grant number: 1908085MH250) and the Natural Science Research Foundation of Colleges and Universities in Anhui Province (grant number: KJ2017A286).
Ma L, Hua L, Yu W, Ke L, Li L. TSG‐6 inhibits hypertrophic scar fibroblast proliferation by regulating IRE1α/TRAF2/NF‐κB signalling. Int Wound J. 2023;20(4):1008‐1019. doi: 10.1111/iwj.13950
Li Ma and Lei Hua contributed equally to this work.
Funding information the Natural Science Research Foundation of Colleges and Universities in Anhui Province, Grant/Award Number: KJ2017A286; Natural Science Foundation of Anhui Province, Grant/Award Number: 1908085MH250
Contributor Information
Li Ke, Email: keli_kl@163.com.
Liang‐yong Li, Email: yffslly@163.com.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supproting the findings of this study are available in the article.
REFERENCES
- 1. Barone N, Safran T, Vorstenbosch J, Davison PG, Cugno S, Murphy AM. Current advances in hypertrophic scar and keloid management. Semin Plast Surg. 2021;35:145‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elsaie ML. Update on management of keloid and hypertrophic scars: a systemic review. J Cosmet Dermatol. 2021;20:2729‐2738. [DOI] [PubMed] [Google Scholar]
- 3. Xue M, Zhao R, March L, Jackson C. Dermal fibroblast heterogeneity and its contribution to the skin repair and regeneration. Adv Wound Care (New Rochelle). 2022;11:87‐107. [DOI] [PubMed] [Google Scholar]
- 4. Coma M, Frohlichova L, Urban L, et al. Molecular changes underlying hypertrophic scarring following burns involve specific deregulations at all wound healing stages (inflammation, proliferation and maturation). Int J Mol Sci. 2022;22:897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greaves NS, Ashcroft KJ, Baguneid M, Bayat A. Current understanding of molecular and cellular mechanisms in fibroplasia and angiogenesis during acute wound healing. J Dermatol Sci. 2013;72:206‐217. [DOI] [PubMed] [Google Scholar]
- 6. Zhu Z, Ding J, Shankowsky HA, Tredget EE. The molecular mechanism of hypertrophic scar. J Cell Commun Signal. 2013;7:239‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang ZC, Zhao WY, Cao Y, et al. The roles of inflammation in keloid and hypertrophic scars. Front Immunol. 2020;11:603187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eming SA, Martin P, Tomic‐Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6:265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma L, Li LY, Zhao TL. Anti‐inflammatory effects of ginsenoside Rg3 on the hypertrophic scar formation via the NF‐kappaB/IkappaB signaling pathway in rabbit ears. Pharmazie. 2020;75:102‐106. [DOI] [PubMed] [Google Scholar]
- 10. Wang H, Chen Z, Li XJ, Ma L, Tang YL. Anti‐inflammatory cytokine TSG‐6 inhibits hypertrophic scar formation in a rabbit ear model. Eur J Pharmacol. 2015;751:42‐49. [DOI] [PubMed] [Google Scholar]
- 11. Xie F, Teng L, Xu J, et al. Interleukin‐10 modified bone marrow mesenchymal stem cells prevent hypertrophic scar formation by inhibiting inflammation. Pharmazie. 2020;75:571‐575. [DOI] [PubMed] [Google Scholar]
- 12. Zhou Y, Hua T, Weng X, Ma D, Li X. Calcitonin gene‐related peptide alleviates hypertrophic scar formation by inhibiting the inflammation. Arch Dermatol Res. 2022;314:53‐60. [DOI] [PubMed] [Google Scholar]
- 13. Amen OM, Sarker SD, Ghildyal R, Arya A. Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: therapeutic and molecular approach. Front Pharmacol. 2019;10:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chipurupalli S, Samavedam U, Robinson N. Crosstalk between ER stress, autophagy and inflammation. Front Med (Lausanne). 2021;8:758311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shi M, Chai Y, Zhang J, Chen X. Endoplasmic reticulum stress‐associated neuronal death and innate immune response in neurological diseases. Front Immunol. 2021;12:794580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim S, Lee SE, Yi S, et al. Tauroursodeoxycholic acid decreases keloid formation by reducing endoplasmic reticulum stress as implicated in the pathogenesis of keloid. Int J Mol Sci. 2021;22:10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye L, Zeng Q, Dai H, et al. Endoplasmic reticulum stress is involved in ventilator‐induced lung injury in mice via the IRE1alpha‐TRAF2‐NF‐kappaB pathway. Int Immunopharmacol. 2020;78:106069. [DOI] [PubMed] [Google Scholar]
- 18. Boyko TV, Bam R, Jiang D, et al. Inhibition of IRE1 results in decreased scar formation. Wound Repair Regen. 2017;25:964‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Day AJ, Milner CM. TSG‐6: a multifunctional protein with anti‐inflammatory and tissue‐protective properties. Matrix Biol. 2019;78‐79:60‐83. [DOI] [PubMed] [Google Scholar]
- 20. Milner CM, Day AJ. TSG‐6: a multifunctional protein associated with inflammation. J Cell Sci. 2003;116:1863‐1873. [DOI] [PubMed] [Google Scholar]
- 21. Li XY, Li T, Li XJ, Wang JN, Chen Z. TSG‐6 induces apoptosis of human hypertrophic scar fibroblasts via activation of the Fas/FasL Signalling pathway. Folia Biol (Praha). 2018;64:173‐181. [DOI] [PubMed] [Google Scholar]
- 22. Li Q, Song WJ, Ryu MO, et al. TSG‐6 secreted by human adipose tissue‐derived mesenchymal stem cells ameliorates severe acute pancreatitis via ER stress downregulation in mice. Stem Cell Res Ther. 2018;9:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bai X, He T, Liu J, et al. Loureirin B inhibits fibroblast proliferation and extracellular matrix deposition in hypertrophic scar via TGF‐beta/Smad pathway. Exp Dermatol. 2015;24:355‐360. [DOI] [PubMed] [Google Scholar]
- 24. Gong YF, Zhang XM, Liu F, et al. Inhibitory effect of recombinant human endostatin on the proliferation of hypertrophic scar fibroblasts in a rabbit ear model. Eur J Pharmacol. 2016;791:647‐654. [DOI] [PubMed] [Google Scholar]
- 25. Guo Q, Li Y, Chen Y, et al. Beta‐Elemene induces apoptosis by activating the P53 pathway in human hypertrophic scar fibroblasts. IUBMB Life. 2022;74:508‐518. [DOI] [PubMed] [Google Scholar]
- 26. Boehm EM, Gildenberg MS, Washington MT. The many roles of PCNA in eukaryotic DNA replication. Enzyme. 2016;39:231‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Hong WL, Li ZM, Zhang QY, Zeng K. The mechanism of miR‐222 targets matrix metalloproteinase 1 in regulating fibroblast proliferation in hypertrophic scars. Aesthet Plast Surg. 2021;45:749‐757. [DOI] [PubMed] [Google Scholar]
- 28. Armour A, Scott PG, Tredget EE. Cellular and molecular pathology of HTS: basis for treatment. Wound Repair Regen. 2007;15(Suppl 1):S6‐S17. [DOI] [PubMed] [Google Scholar]
- 29. Nabai L, Pourghadiri A, Ghahary A. Hypertrophic scarring: current knowledge of predisposing factors, cellular and molecular mechanisms. J Burn Care Res. 2020;41:48‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao D, Wang Y, Du C, et al. Honokiol alleviates hypertrophic scar by targeting transforming growth factor‐beta/Smad2/3 signaling pathway. Front Pharmacol. 2017;8:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaw TJ, Kishi K, Mori R. Wound‐associated skin fibrosis: mechanisms and treatments based on modulating the inflammatory response. Endocr Metab Immune Disord Drug Targets. 2010;10:320‐330. [DOI] [PubMed] [Google Scholar]
- 32. Karppinen SM, Heljasvaara R, Gullberg D, Tasanen K, Pihlajaniemi T. Toward understanding scarless skin wound healing and pathological scarring. F1000Res. 2019;8:787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Junjappa RP, Patil P, Bhattarai KR, Kim HR, Chae HJ. IRE1alpha implications in endoplasmic reticulum stress‐mediated development and pathogenesis of autoimmune diseases. Front Immunol. 2018;9:1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell JP, Carmody RJ. NF‐kappaB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. 2018;335:41‐84. [DOI] [PubMed] [Google Scholar]
- 35. Rothschild DE, McDaniel DK, Ringel‐Scaia VM, Allen IC. Modulating inflammation through the negative regulation of NF‐kappaB signaling. J Leukoc Biol. 2018;103:1131‐1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang H, Tian W, Wang S, et al. TSG‐6 secreted by bone marrow mesenchymal stem cells attenuates intervertebral disc degeneration by inhibiting the TLR2/NF‐kappaB signaling pathway. Lab Investig. 2018;98:755‐772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supproting the findings of this study are available in the article.