

Abstract
MEK is a canonical effector of mutant KRAS; however, MEK inhibitors fail to yield satisfactory clinical outcomes in KRAS-mutant cancers. Here, we identified mitochondrial oxidative phosphorylation (OXPHOS) induction as a profound metabolic alteration to confer KRAS-mutant non-small cell lung cancer (NSCLC) resistance to the clinical MEK inhibitor trametinib. Metabolic flux analysis demonstrated that pyruvate metabolism and fatty acid oxidation were markedly enhanced and coordinately powered the OXPHOS system in resistant cells after trametinib treatment, satisfying their energy demand and protecting them from apoptosis. As molecular events in this process, the pyruvate dehydrogenase complex (PDHc) and carnitine palmitoyl transferase IA (CPTIA), two rate-limiting enzymes that control the metabolic flux of pyruvate and palmitic acid to mitochondrial respiration were activated through phosphorylation and transcriptional regulation. Importantly, the co-administration of trametinib and IACS-010759, a clinical mitochondrial complex I inhibitor that blocks OXPHOS, significantly impeded tumor growth and prolonged mouse survival. Overall, our findings reveal that MEK inhibitor therapy creates a metabolic vulnerability in the mitochondria and further develop an effective combinatorial strategy to circumvent MEK inhibitors resistance in KRAS-driven NSCLC.
Key words: KRAS-mutant lung cancer, MEK inhibitors, Drug resistance, Metabolic rewiring, Mitochondrial oxidative phosphorylation, Pyruvate dehydrogenase complex, Carnitine palmitoyl transferase IA
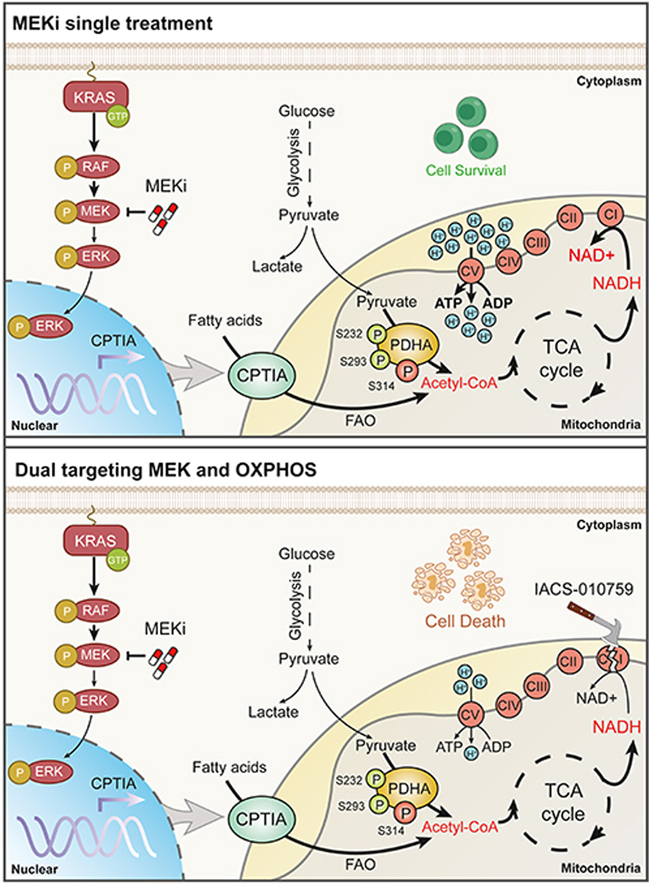
Graphical abstract
Mitochondrial oxidative phosphorylation induction was a major metabolic rewiring event to confer KRAS-mutant NSCLC resistance to MEK inhibitors.
1. Introduction
Lung cancer is the leading cause of global cancer-related deaths1, of which non-small cell lung cancer (NSCLC) is the predominant type, accounting for approximately 80%–85% of all lung cancers2. Oncogenic KRAS, one of the most commonly mutated oncogenes in NSCLC (∼30%), remains refractory to targeted therapies to date2,3. Recently, the discovery of a new pocket under the effector-binding switch II region of KRAS(G12C) has enabled the successful direct targeting of KRAS(G12C) variants4. However, the acquired resistance of KRAS-addicted tumor cells to new drugs limits their efficacy5.
Despite efforts to directly target mutant KRAS, most therapeutic approaches have focused on RAS downstream pathways as a more tractable alternative6,7. Multiple lines of evidence have highlighted that the RAF–MEK–ERK kinase cascade (MAPK) is a critical effector pathway underlying mutated RAS. Small inhibitors targeting this pathway have been successfully developed. Although some are US Food and Drug Administration (FDA)-approved, most therapeutic approaches have limited efficacy and are poorly tolerated at doses needed to sufficiently extinguish RAS signaling in tumors8. MEK inhibitors (MEKi) are currently used for BRAF-mutant melanoma and neurofibromatosis; however, clinical trials for KRAS-mutant NSCLC are less encouraging9,10. The failure of MEKi in NSCLC is due to multiple mechanisms, including secondary MEK mutations11, immune escape12, reactivation of the MAPK pathway by RAF dimerization13, or compensatory induction of RAS-related pathways14, 15, 16, which inevitably leads to the development of therapy resistance and disease recurrence. These molecular discoveries have successfully promoted the clinical application of MEKi in combination with other targeted interventions17,18. However, it is difficult to restore the sensitivity of KRAS-mutant NSCLC cells to MEKi. Further studies regarding the resistance mechanisms of MEKi and exploration of rational treatment strategies are required to augment the response to MEKi-based therapy.
Metabolic plasticity is a key feature of cancer cells, which orchestrates their metabolism to meet the high need for energy and building blocks during growth or stress adaptation19,20. EGFR inhibitors have been reported to enhance cystine uptake and glutathione de novo synthesis, protecting cells from killing21. Recent studies have indicated that mitochondrial function contributes to the intrinsic and acquired resistance of certain types of cancer to targeted therapies. BRAF inhibitors reduce tumor glycolysis while inducing mitochondrial oxidative phosphorylation (OXPHOS) by triggering the expression of a mitochondrial biogenesis gene signature, thereby conferring resistance of BRAFV600E-mutant melanoma to BRAF inhibitors22,23. MET inhibitors enhance mitochondrial OXPHOS and fatty acid oxidation (FAO) in glioblastoma, resulting in acylcarnitine accumulation and reduced therapeutic efficacy24. This evidence that drug-resistant tumor cells rely more on mitochondrial OXPHOS and less on glycolysis challenges the contention that tumor cells are usually characterized by the Warburg effect, that is, the production of ATP mostly from glycolysis and not oxidative phosphorylation, even under conditions of high oxygen availability. However, it remains unclear whether mitochondrial function and metabolic flexibility contribute to MEKi resistance in KRAS-mutant NSCLC cells.
In this study, we report a pharmacologically actionable vulnerability in which increased OXPHOS is required for KRAS-mutant NSCLC cells to evade MEKi therapy. Activated OXPHOS in the mitochondria is dependent on the activation of pyruvate dehydrogenase complex (PDHc) and carnitine palmitoyl transferase IA (CPTIA), two rate-limiting enzymes that govern the metabolic flux of pyruvate and palmitic acid to power the tricarboxylic acid (TCA) cycle. Our findings further exploited an effective combinatorial strategy to sensitize KRAS-mutant NSCLC cells to MEKi by targeting OXPHOS using IACS-010759, a novel and potent mitochondrial complex I inhibitor that is currently in clinical testing.
2. Materials and methods
2.1. Animal experiments
C57BL/6 and BALB/cA nude mice were purchased from National Rodent Laboratory Animal Resources (Shanghai, China). LSL-KrasG12D/+; Trp 53fl/fl (KP) mice on a C57BL/6J background were purchased from the Jackson Laboratory. Animals were housed in pathogen-free microisolator under a 12 h light/dark cycle and provided with mouse chow and sterile water. All animal experiments were approved by East China Normal University (Shanghai, China) and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee.
For the H460, Calu-1, A549, and A549/TR xenograft mouse models, 3 × 106 cells were suspended in 100 μL of phosphate-buffered saline (PBS) and injected subcutaneously into the flanks of BALB/c nude mice. When the tumor volume reached approximately 100 mm3, the mice were randomized and treated with vehicle (0.5% sodium carboxymethylcellulose), trametinib (1 mg/kg, orally, dissolved in 0.5% sodium carboxymethylcellulose), or IACS-010759 (5 mg/kg, orally, dissolved in 0.5% sodium carboxymethylcellulose). The tumor volume was measured every other day in a blinded manner using calipers and evaluated according to Eq. (1):
| Tumor volume (mm3) = (Length × Width2) × 0.52 | (1) |
Mice were euthanized when the animal experiments reached the end or when the tumor volume exceeded approximately 1500 mm3. The tumors were removed, weighed, and snap-frozen in liquid nitrogen for further analysis.
For the patient-derived xenograft (PDX) mouse model, LAC001 and LAC003 tumor tissues were derived from two patients with poorly differentiated lung adenocarcinoma harboring KRASG12V and KRASG12C mutations, respectively. Tumor tissues were implanted into mice to establish the PDXs as described previously25. After tissue inoculation, the mice were monitored until the tumor volume reached approximately 100 mm3. Mice were treated with 0.5 mg/kg trametinib once daily by orally administration. Tumors were monitored for trametinib resistance, which was defined as marked tumor growth in the presence of continued trametinib therapy. Approximately eight weeks after oral administration, the tumors were aseptically resected from the trametinib-resistant group and minced into small pieces (3 mm in diameter). Then, a piece of tumor was implanted into the flanks of BALB/c nude mice. When the tumor volume reached approximately 100 mm3, the mice were randomly divided into two groups to receive 1 mg/kg trametinib or 1 mg/kg trametinib plus 5 mg/kg IACS-010759 for 21 days. Tumors were measured every other day using electronic calipers. On Day 21, the mice were sacrificed, and the tumor tissues were excised and weighed after the final dose.
For the KP mouse model, 8-week-old KP mice were anesthetized with isoflurane in a gas chamber. Adeno-Cre (HanBio, Shanghai, China) at a dose of 2.5 × 107 PFU in a total volume of 125 μL was introduced into the mice. Twelve weeks after virus inhalation, the lungs were imaged using a Quantum GX micro-CT imaging system (PerkinElmer, Waltham, MA, USA) to confirm tumor formation. Subsequently, mice were randomized into two groups, one of which was treated with trametinib at a dosage of 0.5 mg/kg/day. Multifocal adenocarcinomas in mice were assessed during the treatment. By Week 6, the tumors in the trametinib treatment arm showed significant growth, indicating the emergence of drug resistance. Subsequently, the mice in the vehicle and trametinib groups were further divided into two groups, followed by treatment with vehicle or IACS-010759. On Day 21, the tumors were measured using a Quantum GX micro-CT imaging system after the final dose. Mice were supported under basic animal care, with no further drug intervention until death. Survival rates were calculated using the Kaplan–Meier method. Statistical significance was assessed using the log-rank tests.
2.2. Cell lines and culture
The cell lines of H460, Calu-1, H441, H292, A549, H23, H1944, and H358 were obtained from the Chinese Academy of Sciences (Shanghai, China). Trametinib resistant-A549 and H23 cells (A549/TR and H23/TR) were established, maintained, and authenticated. The cell lines were maintained in the appropriate culture medium supplemented with 10% fetal bovine serum (FBS, ThermoFisher Scientific, Waltham, USA) and 100 U/mL penicillin/streptomycin. The culture medium and KRAS mutation status for each cell line are listed in Supporting Information Table S1. For 13C labeled tracing and metabolomic profiling assays, 10% dialyzed FBS (S12850; Atlanta Biologicals Inc. Flowery Branch, USA) was used. All cell lines were authenticated by short tandem repeat analysis and routinely tested for mycoplasma contamination using a Myco-LumiTM Luminescent Mycoplasma Detection Kit (C0297, Beyotime, Shanghai, China) at least semi-monthly in culture.
2.3. RNA-seq analysis
Total RNA was extracted using RNA TRIzol Reagent (R4801, Magen, Shanghai, China) according to the manufacturer's instructions. RNA sequencing was performed by Novogene Company Limited (Beijing, China) using IIIumina Hiseq 3000 platform (pair end 150 bp analysis). Differential expression was analyzed using DESeq2. Pathway analysis was performed using the KEGG pathway database.
2.4. Metabolomic profiling
Cells (3 × 106) were collected and resuspended in 800 μL ice-cold 80% methanol and 20% ddH2O. The samples were vigorously vortexed, placed in liquid N2 for 15 min to freeze, and then thawed on ice for 15 min. The freeze-thaw cycle was repeated twice. Samples were subsequently centrifuged at 13,500×g for 20 min to pellet cell debris, lipids, and proteins. The supernatant was evaporated, and the resulting metabolites were resuspended. The samples were analyzed by Novogene Company Limited.
2.5. Oxygen consumption rate (OCR)
OCR was determined using an XF-96 extracellular flux analyzer (Agilent, Santa Clara, USA) as previously described26. Lung tumor cells were seeded in 96-well plates using Cell-Tak (Corning, New York, NY, USA) at a density of 3 × 104 or 1.5 × 104 cells/well, respectively. The cells were incubated in Seahorse XF RPMI medium buffer (1 mmol/L sodium pyruvate, 10 mmol/L glucose and 2 mmol/L glutamine). The Cell Mito Stress Test Kit (103015–100, Agilent) was used to measure cellular mitochondrial function, and 180 μL of Seahorse medium plus 20 μL each of 1 μmol/L oligomycin (Oligo), 1 μmol/L trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP), and 0.5 μmol/L rotenone (Rot) were automatically injected to determine the OCR, according to the manufacturer's instructions. OCR values were used to compute basal respiration and maximal respiration using Wave 2.4 and Excel Macro Report Generator 3.0.3 provided by Seahorse Biosciences. The formulas used were as follows, Eqs. (2), (3):
| Basal respiration = (Last rate before the first injection) – (Non-mitochondrial respiration rate) | (2) |
| Maximal respiration = (Maximum rate after FCCP injection) – (Non-mitochondrial respiration rate) | (3) |
2.6. Measurement of redox stress
ROS production was measured using a ROS assay kit (S0033, Beyotime). Cells were incubated with 10 mmol/L 2,7-dichlorodihydrofluorescein diacetate DCFH-DA at 37 °C for 20 min. Cells were then washed thrice in PBS and subjected to LSR Fortessa (BD Biosciences, Franklin Lakes, NJ, USA).
The cellular levels of glutathione (GSH) and oxidized glutathione (GSSG) were evaluated using a GSH and GSSG Assay Kit (S0053, Beyotime) according to the manufacturer's protocol. The results of the GSH/GSSG ratio are shown as the mean ± standard error of mean (SEM) of biological triplicates.
2.7. 13C-labeled glucose/palmitate/glutamine tracing
Methanol extraction was used to prepare metabolomic samples. Briefly, 1 × 106 cells were seeded in 6 cm dishes and refreshed with complete medium for 24 h. After quickly aspirating the cell culture medium, cells were gently rinsed with PBS. Next, 500 μL of lysis buffer (methanol:acetonitrile:H2O, 4:4:2) was added into the dishes placed on dry ice and incubated for 30 min at −75 °C to quench metabolism and perform extraction. All cells were scraped from dishes at −75 °C and transferred into tubes. Another 500 μL of lysis buffer was added to the dishes for the second extraction step. All cell contents were scraped and transferred to tubes. The mixture was then centrifuged at 13,500 × g for 15 min at 4 °C. Subsequently, the soluble extract was completely dried using a vacuum centrifuge. For 13C labeled glucose/palmitate/glutamine tracing assay, cells were treated with 13C6 labeled glucose or 13C16 labeled palmitate for 12 h, or 13C5 labeled glutamine for 1 h before extraction. The metabolite intensities were analyzed using GC–MS.
2.8. Transmission electron microscopy
Cells cultured in 6-well plates were fixed with a solution containing 2.5% glutaraldehyde in 0.1 mol/L Sorenson's buffer (0.1 mol/L H2PO4, 0.1 mol/L HPO4 (pH 7.2)) for at least 1 h. Cells were then treated with 1% OsO4 in 0.1 mol/L Sorenson's buffer for 1 h and stained using 1% tannic acid. After dehydration using an ethanol series, the cells were embedded in a mixture of Lx-112 (Ladd Research Industries, Williston, USA) and Embed-812 (EMS, Fortwashington, USA). All sections were observed under a HT7700 electron microscope (Hitachi, Chiyoda, Japan).
2.9. mtDNA quantification
Total DNA was isolated from the cells using a TIANamp Genomic DNA kit (DP304, Tiangen Biotech, Beijing, China). The samples were adjusted to a final concentration of 1 ng/μL. Nuclear and mitochondrial DNA contents were analyzed by reverse transcription-quantitative PCR analysis (RT-qPCR), as described previously27. The mtDNA content was determined by normalizing the mitochondrial DNA abundance [tRNA-Leu (UUR)] to nuclear DNA (β2M) abundance. The primers used for RT-qPCR assays are listed in Supporting Information Table S2.
2.10. PDHc activity
PDHc activity was measured using a pyruvate dehydrogenase enzyme activity microplate assay kit (ab109902; Abcam). Briefly, samples were lysed in a detergent solution provided by the manufacturer. The protein concentration in the supernatant was measured using a bicinchoninic acid protein assay kit (BCA, ThermoFisher Scientific). Protein (3 mg) was loaded into 96 well plate, and PDHc activity was measured according to the manufacturer's protocol. The PDHc activity was calculated based on the change in absorption (ΔOD) over a specific time frame (ΔT).
2.11. Acetyl-CoA analysis
Acetyl-CoA was quantified using an acetyl-CoA assay kit (MAK039, Sigma), according to the manufacturer's instructions. Briefly, 2 × 106 cells were harvested from each well. The samples were deproteinized by PCA precipitation. The samples were centrifuged at 13,500 × g for 10 min to remove the insoluble material. The supernatant was neutralized to pH 6–8 with a 3 mol/L potassium bicarbonate solution, vortexed and cooled on ice for 5 min. The potassium bicarbonate solution was centrifuged, and the supernatants were collected. The end-point assay was performed, and the fluorescence intensity (λExcitation, 535 nm; λEmission, 587 nm) was measured using a plate reader (Molecular Devices).
2.12. Cytosol/mitochondria fractionation
Cytosolic and mitochondrial fractions were prepared from cells using the Cell Mitochondria Isolation Kit (C3601, Beyotime), according to the manufacturer's protocol. Briefly, the cells were collected, homogenized with a glass homogenizer in mitochondrial isolation buffer on ice, and pelleted by centrifugation. The remaining supernatant was used as the cytosolic fraction. Mitochondrial pellets were lysed with lysis buffer, and the extracted mitochondrial proteins were used to detect PDHc activity.
2.13. Mapping of PDHA modification sites
Mass spectrometry (MS) was performed to identify PDHA modification sites, as previously described28. Briefly, PDHA was immunoprecipitated from trametinib-treated or untreated H460 cells. The samples were digested with trypsin (Promega, Madison, USA) overnight at 37 °C and desalted using a reversed-phase C18 Sep-Pak cartridge (Millipore, Billerica, USA). The digest was analyzed by LC–MS/MS on a Q-Exactive mass spectrometer (ThermoFisher Scientific). Proteins were identified by searching the fragment spectra against the UniProt human database using MaxQuant software (version 1.4.1.2) and variable modifications of serine, threonine, and tyrosine phosphorylation.
2.14. RNA isolation and reverse transcription-quantitative PCR analysis
Total RNA was extracted using RNA TRIzol Reagent (R4801, Magen) and subjected to reverse transcription using Hifair II 1st Strand cDNA Synthesis SuperMix (11120ES60, Yeasen, Shanghai, China). RT-qPCR analysis was performed according to the Hieff qPCR SYBR Green Master Mix (11201ES03, Yeasen) manufacturer's instructions, using a 96-well Thermal iCycler (Bio-Rad, Hercules, USA). RT-qPCR assays were performed in triplicate and the relative amount of cDNA was calculated using the comparative CT method with β-actin as the control. The primers are listed in Supporting Information Table S2.
2.15. Plasmids and transfection
cDNAs encoding full-length human PDHA were cloned into pCDH vectors (RRID: Addgene_72266). Point mutations in PDHA were generated by site-directed mutagenesis using a GeneArt Site-Directed Mutagenesis System Kit (A13282, Invitrogen, Waltham, USA). All the expression constructs were verified by DNA sequencing. Plasmid transfection was performed using Lipo 8000™ reagent (C0533, Beyotime).
2.16. RNA interference
CPTIA knockdown was performed using small interfering RNAs. The siRNA sequences used are listed in Table S2. Briefly, cells were transfected with 200 pmol siRNA for 48 h. The cells were either treated with the appropriate agents for the cell proliferation assay or lysed for immunoblot analysis to determine the knockdown efficiency.
2.17. CRISPR editing
PDHA was silenced in H460 and H441 cells by CRISPR editing using the lentiCRISPR v2 with sgRNA sequences targeting PDHA. Oligos were phosphorylated, annealed, and ligated into the lentiCRISPR v2 backbone digested with Esp3I (ER0451, ThermoFisher Scientific), which was then transformed into NcmDH5α competent cells (MD101-2, New Cell & Molecular Biotech, Suzhou, China), isolated, and verified by sequencing. H460 and H441 cells were transduced and selected using 1.5 μg/mL puromycin. The depletion effect was verified using immunoblotting analysis. The sgRNAs targeting sequences are shown in Table S2.
2.18. Immunoblotting analysis
Immunoblotting assays were performed using standard methods. Briefly, cell lysates or tumor tissue extracts were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with a protease and phosphatase inhibitor cocktail (MedChemExpress, Monmouth Junction, USA). The protein concentration in the supernatant was determined using a BCA kit. Equal amounts (∼50 μg) of proteins were separated on 8%–15% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Millipore). The membranes were blocked with 5% bovine serum albumin for 1 h at room temperature and immunoblotted with primary antibodies at 4 °C overnight. The membranes were washed with Tris-buffered saline containing Tween 20 and incubated with secondary antibodies for 1 h at room temperature. Visual signals were detected using an LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, USA). The antibodies used are listed in Supporting Information Table S3.
2.19. Immunoprecipitation
Cells were lysed in ice-cold immunoprecipitation buffer (150 mmol/L NaCl, 65 mmol/L Tris-HCl, 5 mmol/L EDTA, and 0.1% Triton X-100, pH7.5) containing a protease and phosphatase inhibitor cocktail. Cell lysates (∼1 mg) were incubated overnight with magnetic beads pre-incubated with the indicated antibodies. The beads were washed thrice in immunoprecipitation buffer and prepared for immunoblot analysis.
2.20. Cell viability assay
Cells were seeded onto 96-well plates at a density of 3000–5000 cells per well and allowed to adhere overnight. Cells were treated with various concentrations of the indicated drugs for 72 h. Cell viability was determined using a CellTiter 96 cell proliferation assay kit (G3580, Promega) according to the manufacturer's instructions. For drug synergy analysis, cells were treated with single agents or their fixed-ratio combination for 72 h. CI values were calculated using the CalcuSyn software, version 2 (Biosoft). Combinations with a CI value of less than 1 were considered synergistic, and a CI value of less than 0.7 indicated a strong synergy.
2.21. Clonogenic assay
Related cells were plated onto 12-well plates (2000–3000 cells per well) and allowed to adhere overnight. The cells were then treated with the indicated drugs for an additional 7–10 days. The remaining cells were fixed with cold 4% paraformaldehyde, stained with 0.2% crystal violet, and then photographed. For the analysis of cell viability of the cultured colonies, the stain was dissolved in 10% acetic acid and transferred to 96-well plates. Absorbance was measured at a wavelength of 595 nm. The relative cell viability was calculated by setting the untreated group as 100%.
2.22. Apoptosis assay
Cells were seeded in 6 cm dishes and incubated with the indicated drugs for 72 h. The cells were stained with propidium iodide and annexin V using an apoptosis detection kit (556547; BD Biosciences, Franklin Lakes, NJ, USA). Flow cytometric analysis was performed using LSR Fortessa software.
2.23. Statistics
Data are expressed as the mean ± SEM. One-way ANOVA or two-tailed Student's t-test was used to compare the means among groups. The log-rank test was used to compare the Kaplan–Meier survival curves. Statistical tests were performed using Microsoft Excel or GraphPad Prism software version 8.0. Statistical significance was set at P < 0.05.
2.24. Data availability
The RNA-seq and metabolomics data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GEO: GSE197555 and the Mendeley Data repository (https://data.mendeley.com/datasets/54xr9bsxd2/1), respectively.
3. Results
3.1. MEK inhibition activates mitochondrial oxidative metabolism
Several inhibitors targeting the MAPK pathway are FDA-approved (e.g., BRAF and MEK inhibitors) but poorly extinguish RAS signaling in tumors, partly due to drug toxicity and pervasive drug resistance29,30. This study aimed to understand how KRAS-driven NSCLC cells evade MEKi therapy and escape cell death. First, we tested the in vitro efficacy of two clinical MEKi drugs, trametinib and selumetinib, using a panel of KRAS-mutant NSCLC cell lines. Some cell lines displayed primary resistance to MEK inhibitors (MEKi), while others were more susceptible (Fig. 1A). Concentrations of 200 nmol/L and 30 μmol/L were selected to define the resistance thresholds for trametinib and selumetinib, because they are likely to be clinically achievable and relevant31.
Figure 1.

MEK inhibition activates mitochondrial oxidative metabolism. (A) Cell sensitivity to the MEK inhibitor trametinib and selumetinib. KRAS-mutant NSCLC cells were treated with trametinib (Tram) or selumetinib (Selu) at gradient concentrations for 72 h, and cell viability was measured. IC50 values are represented as mean ± SEM of three independent experiments with three technical replicates each. Resistant cell lines are marked in green, and sensitive cell lines are marked in brown. (B) Metabolite set enrichment analysis. H460 and A549 cells were treated with trametinib at their respective 1/2 IC50 values for 24 h and then harvested for metabolomic profiling determination. Metabolite set enrichment analysis was performed to identify dysregulated metabolic pathways. The corresponding enrichment pathways (n = 6) are shown. (C) Abundance of major metabolites in the glycolysis and tricarboxylic acid (TCA)-oxidative phosphorylation (OXPHOS) pathways. H460 and A549 cells were exposed to trametinib at their respective 1/2 IC50 values for 24 h. Cell lysates were subjected to LC/MS analysis. Heatmap showed change of metabolites in the glycolysis and the TCA-OXPHOS pathway in trametinib-treated cells versus untreated cells. Metabolite abundance was normalized by cell number. NADH, nicotinamide adenine dinucleotide; NAD+, oxidized nicotinamide adenine dinucleotide. Values are scaled as indicated (2 to −2; n = 6).
KRAS plays a critical role in rewiring metabolism by increasing anabolic needs and stress adaptation25. Therefore, this study assessed whether the difference in the sensitivity of KRAS-mutant NSCLC cells to MEKi was due to altered metabolism. We performed metabolomic analysis (polar and non-polar) to compare the metabolomic profiles between resistant H460 cells and sensitive A549 cells upon exposure to trametinib or vehicle. Enrichment pathway analysis of metabolites revealed that oxidative metabolism pathways, such as OXPHOS and the TCA cycle, were significantly enriched in trametinib-treated H460 cells, whereas carbohydrate metabolism pathways, including glycolysis and gluconeogenesis, were preferentially enriched in trametinib-treated A549 cells (Fig. 1B). Similar enrichment of the TCA cycle pathway was also observed in another resistant cell line H441 as compared with that in another sensitive cell line H1944 (Supporting Information Fig. S1A). We further analyzed metabolite abundance and observed an accumulation of TCA cycle- and OXPHOS-related metabolites and depletion of glycolysis-related metabolites in H460 and H441 cells upon treatment; however, such metabolic rewiring events did not occur in treated A549 and H1944 cells that are sensitive to MEK inhibition (Fig. 1C and Fig. S1B), suggesting that trametinib might preferentially trigger a metabolic shift from glycolysis to mitochondrial OXPHOS in primary resistant cells. In contrast to the metabolite accumulation of mitochondrial OXPHOS in treated H460 cells, this pathway-associated genes were slightly altered according to the transcriptomic profiling (Fig. S1C and Supporting Information Table S4), indicating that trametinib-mediated oxidative metabolism rewiring relied mainly on metabolite control rather than on transcriptional regulation in the mitochondria. Based on these results, we speculated that mitochondrial OXPHOS induction might be crucial for the growth of KRAS-mutant NSCLC cells in the context of MEK inhibition.
3.2. Increased mitochondrial oxidative metabolism is associated with MEKi resistance
To determine whether MEKi increases mitochondrial oxidative metabolism, we examined the oxygen consumption rate (OCR), an indicator of mitochondrial respiration, in trametinib-treated KRAS-mutant NSCLC cell lines. Our results showed that trametinib and selumetinib treatment resulted in a much higher cellular OCR with increased basal and maximal respiratory capacity in MEKi-resistant cell lines (H460, Calu-1, and H441), but not in MEKi-sensitive cell lines (A549, H23, and H1944) (Fig. 2A and B, and Supporting Information Fig. S2A–S2F). Moreover, we established isogenic lineages with acquired resistance to trametinib using the sensitive A549 and H23 cell lines (Fig. 2C). After long-term treatment exposure, the A549 and H23 lineage derivatives A549/TR and H23/TR showed little, if any, sensitivity to trametinib (Fig. S2G), indicating an acquired-resistance phenotype. Compared with A549 and H23 parental cells (A549/P and H23/P), A549/TR and H23/TR cells exhibited a significant increase in OCR (Fig. 2D, Fig. S2H and S2I). In agreement with these in vitro observations, short-term administration of trametinib significantly elevated the OCR levels in H460 and Calu-1 xenograft tumors in vivo (Fig. 2E and F, Fig. S2J and S2K), but not in A549 xenograft tumors (Fig. 2G and Fig. S2L). A significant increase in OCR levels was also observed in patient-derived xenograft (PDX) tumors after prolonged exposure to trametinib (Fig. 2H and Fig. S2M). These results suggest that MEK inhibition enhances OXPHOS in both primary and acquired resistant cells in vitro and in vivo.
Figure 2.

Increased mitochondrial oxidative metabolism is associated with MEKi resistance. (A, B) The levels of mitochondrial oxygen consumption rate (OCR) in MEKi-treated NSCLC cells. MEKi-resistant (H460, Calu-1, and H441) (A) and MEKi-sensitive cells (A549, H23, and H1944) (B) were treated with trametinib or selumetinib at their respective 1/2 IC50 values for 6 h. Oligo, FCCP, and Rot were injected at the indicated time points. OCR values were normalized by cell number. The representative trace of OCR values from a mitochondrial stress test is shown. Values are expressed as the mean ± SEM of three technical replicates, representative of three independent experiments with similar results. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001; ns, not significant, by one-way ANOVA with Tukey's multiple-comparisons test. (C) Schematic of resistant cell generation. (D) OCR analysis in A549/TR and parental cells. A representative trace of OCR values from a mitochondrial stress test is shown. Data represent the mean ± SEM of three technical replicates, representative of three independent experiments with similar results. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (E–G) OCR analysis in trametinib-treated tumors. During 1-week treatment with trametinib, tumors were isolated, followed by OCR analysis. Values are expressed as the mean ± SEM of three independent biologically samples. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (H) OCR analysis in PDX/TR and parental xenograft tumors. Data represent the mean ± SEM of three independent biologically samples. ∗P < 0.05, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test.
Furthermore, we found that the OXPHOS induced by trametinib was accompanied by a simultaneous increase in redox stress, as indicated by a decreased reduced/oxidized glutathione (GSH/GSSG) ratio, increased reactive oxygen species (ROS) production, and enhanced ROS-responsive gene expression (NQO1, GCLC, SOD1, GSTA4, GSTM4, SOD2, and HMOX1) in resistant cells after MEKi treatment (Supporting Information Fig. S3A–S3C). Similar results were obtained for the acquired resistant A549/TR cells compared with the parental cells (Fig. S3D–S3F). In summary, these data indicate that mitochondrial OXPHOS levels and redox stress are profoundly augmented by trametinib treatment, which is positively linked to a poor response to trametinib in KRAS-mutant NSCLC.
3.3. MEK inhibition favors carbon incorporation in the TCA cycle
To determine how metabolic reprogramming is orchestrated in resistant cells and how the enrichment of the TCA cycle metabolites is mediated, we performed 13C-labeled glucose-, palmitic acid-, and glutamine-tracing experiments to gain insights into their metabolic fate in A549/P and A549/TR cells. In the mitochondria, acetyl coenzyme A (acetyl-CoA) from glycolysis and FAO coordinately power the TCA cycle, thereby enhancing the OXPHOS system. During glycolysis, glucose carbons (U-13C-glucose) are metabolized to pyruvate, which can subsequently be converted to acetyl-CoA by PDHc, or to oxaloacetate by pyruvate carboxylase (PC), resulting in either m+2 citrate (PDHc reaction) or m+3 citrate (PC reaction). Compared to parental cells, we noted a substantial increase in carbon labeling of TCA cycle intermediates in acquired resistant A549 cells, including citrate, succinate, fumarate, malate, α-ketoglutarate, and isocitrate (Fig. 3A–D, Supporting Information Fig. S4A and S4B). The labeling of aspartate from glucose carbons was also increased (Fig. S4C). In contrast to TCA cycle intermediates, glucose labeling of glycolytic intermediates, such as pyruvate and lactate, was comparable between A549/P and A549/TR cells (Fig. S4D and S4E). In addition to glucose, palmitic acid carbon (U-13C-palmitic acid) can also be converted to acetyl-CoA (m+2) by FAO. We found that palmitic acid oxidation increased with enhanced labeling of citrate, α-ketoglutarate, succinate fumarate, and malate in A549/TR cells compared to that in A549/P cells (Fig. 3E–G, Fig. S4F and S4G). Glutamine carbon (U-13C-glutamine) is another energy source for the TCA cycle, which can be converted to α-ketoglutarate (m+5) through glutaminase and glutamate dehydrogenase. In contrast to the increased metabolic flux of glucose and palmitic acid into mitochondria for oxidative metabolism, we noted an overall decrease in carbon labeling of TCA cycle metabolites and aspartate originating from glutamine (Fig. 3H–J and Fig. S4H–S4L). These findings suggest a dependence of the TCA cycle on carbons derived from glucose and palmitic acid rather than glutamine in the context of chronic MEK inhibition. A summary of the metabolic pathways illustrating the carbon flow from glucose, glutamine and palmitic acid to the TCA cycle is shown (Fig. 3K).
Figure 3.

MEK inhibition favors carbon incorporation in the TCA cycle. (A–D) The enrichment of isotope derived from 13C6-labeled glucose from the TCA cycle in A549/P and A549/TR cells. Cells were cultured in the presence of 10 mmol/L 13C6-labeled glucose and 2 mmol/L glutamine supplemented with 10% dialyzed FBS for 12 h in PRIM-1640 media devoid of glucose, glutamine, and pyruvic acid. The relative percentages of the isotopologues for each metabolite are shown. (E–G) The enrichment of isotope derived from 13C16-labeled palmitic acid from the TCA cycle in A549/P and A549/TR cells. Cells were cultured in the presence of 10 mmol/L glucose, 2 mmol/L glutamine, and 200 μmol/L 13C16-labeled palmitic acid supplemented with dialyzed 10% FBS for 12 h in PRIM-1640 media devoid of glucose, glutamine, and pyruvic acid. The relative percentages of the isotopologues for each metabolite are shown. (H–J) The enrichment of isotope derived from 13C5-labeled glutamine from the TCA cycle in A549/P and A549/TR cells. Cells were cultured in the presence of 10 mmol/L glucose and 2 mmol/L 13C5-labeled glutamine supplemented with 10% dialyzed FBS for 1 h in PRIM-1640 media devoid of glucose, glutamine, and pyruvic acid. The relative percentages of the isotopologues for each metabolite are shown. (K) Summary of reactions related to tracing experiments. Green circles indicate 13C carbons from glucose, purple circles indicate 13C carbons from palmitic acid, whereas brown circles represent 13C carbons from glutamine. Black circles are used to display 12C carbons. All data represent the mean ± SEM of biological triplicates. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001; ns, not significant, by unpaired, two-sided Student's t test.
3.4. PDHc activation empowers trametinib-resistant cells to survive
We tested whether the increase in mitochondrial oxidative metabolism after MEK inhibition is attributed to altered mitochondrial function. We found that the number of mitochondria (Supporting Information Fig. S5A and S5B) and DNA content (Fig. S5C) were only slightly affected in primary and acquired resistant cells after trametinib exposure. Moreover, trametinib treatment minimally affected the expression of mitochondrial electron transport chain (ETC) complex components (Fig. S5D). These data suggest that the trametinib-mediated increase in mitochondrial OXPHOS is independent of mitochondrial mass.
Since the carbon flux of glucose and palmitic acid to the TCA cycle was notably increased, while the transcription of OXPHOS-related genes was slightly affected in treated resistant cells, we hypothesized that the increased mitochondrial oxidative metabolism induced by trametinib might be dependent on the rate-limiting enzymes involved. Pyruvate is the primary link between glycolysis and the TCA cycle, and its metabolic fate is mainly controlled by PC and PDHc32,33. PC is a biotin-dependent enzyme that catalyzes the carboxylation of pyruvate into oxaloacetate, an essential metabolite in the TCA cycle. First, we tested PC abundance in trametinib-treated cells and observed little difference across the cell lines (Supporting Information Fig. S6A), thereby excluding the involvement of PC in MEKi-mediated mitochondrial flexibility. Notably, the activity of PDHc, the rate-limiting enzyme complex that catalyzes pyruvate to acetyl-coenzyme A, was significantly increased by trametinib in the whole-cell lysates of resistant cells (H460, Calu-1, and H441), whereas its activity was unchanged or even decreased in treated-sensitive cells (A549, H23, and H1944; Fig. 4A). As PDHc mainly functions in the mitochondria, we subsequently extracted mitochondrial lysates and found that trametinib treatment led to a specific increase in PDHc activity in the mitochondria of H460 cells, but not A549 cells (Fig. 4B and Fig. S6B). Acquired resistant cells also displayed a higher PDHc activity than their parental counterparts (Fig. 4C and Fig. S6C). Increased PDHc activity was consistently observed in trametinib-treated Calu-1 xenograft tumors compared to that in vehicle-treated xenograft tumors (Fig. 4D). As a result of PDHc activation, the acetyl-CoA content was significantly increased in both whole-cell and mitochondrial lysates of H460 cells, but not in A549 cells exposed to trametinib (Fig. 4E). Acetyl-CoA induction was further confirmed in acquired resistant cells in vitro (Fig. 4F) and in Calu-1 xenograft tumors in vivo (Fig. 4G).
Figure 4.

PDHc activation empowers MEKi-resistant cells to survive. (A, B) Relative PDHc activity in whole-cell and mitochondrial lysates of KRAS-mutant NSCLC cells. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. Whole-cell (A) and mitochondria (B) lysates were subjected to PDHc activity measurement using an enzyme activity kit. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (C) PDHc activity in A549/TR cells and parental cells. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (D) PDHc activity in Calu-1 xenograft tumors. Tumors were isolated after 1-week administration of trametinib. PDHc activity was further examined. Values are expressed as the mean ± SEM of three independent biologically samples. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (E) Relative acetyl-CoA levels in trametinib-treated H460 and A549 cells. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. Lysates of cell and isolated mitochondria were subjected to acetyl-CoA analysis using a commercial kit according to the manufacturer's instructions. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (F) Relative acetyl-CoA level in A549/TR cells and parental cells. Data represent the mean ± SEM of biological triplicates. ∗∗P < 0.01, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (G) Acetyl-CoA level in trametinib-treated and vehicle-treated Calu-1 xenograft tumors. After 1-week trametinib treatment, tumor tissues were isolated, followed by acetyl-CoA testing. Values are expressed as the mean ± SEM of three independent biologically samples. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (H) PDHA knockout potentiated trametinib efficacy in H460 cells. Left panel, relative viability of the culture colonies; right panel, PDHA knockout efficiency in H460 cells by immunoblot analysis. Data are shown as the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test. (I, J) PDHc inhibition by devimistat re-sensitized the inhibitory effects of trametinib in H460 cells, as indicated by the growth curves (I) and relative viability of cultured colonies (J). Data are shown as the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test.
PDHc is composed of PDH (E1), dihydrolipoamide transacetylase (E2) and dihydrolipoamide dehydrogenase (E3), of which E1 subunit α (PDHE1α, PDHA) is the critical rate-limiting catalytic subunit34. To delineate the association between PDHc activity and MEKi resistance, we genetically knocked out PDHA and found that PDHA loss robustly inhibited resistant cell growth in the context of trametinib (Fig. 4H and Fig. S6D). We further applied devimistat, a small-molecule PDHc inhibitor, along with various concentrations of trametinib, and examined the antigrowth effects of drug combinations in resistant cells. As expected, PDHc blockade effectively potentiated the cytotoxic effects of trametinib (Fig. 4I and Fig. S6E). Furthermore, devimistat-sensitized trametinib suppressed the clonogenic growth of resistant H460 and H441 cells (Fig. 4J and Fig. S6F). In summary, these results indicate that PDHc plays a substantial role in conferring MEKi resistance in KRAS-mutant NSCLC.
3.5. MEK inhibition alters PDHA phosphorylation status
Although PDHc is crucial for pyruvate metabolism and TCA cycle maintenance, its activation and regulation in a specific tumor context remain elusive. It has been reported that PDHA phosphorylation at S293 and S232 serves as a negative signal for PDHc activation35. A recent study has reported that PDHA phosphorylation at S295 and S314 is essential for its activity36. To gain insight into the mechanism of PDHc activation in conferring MEKi resistance, we performed immunoprecipitation coupled with tandem mass spectrometry to investigate the post-translational modifications of PDHA. Our results demonstrated that the residues S314, S293, and S232 in PDHA were phosphorylated in trametinib-treated H460 cells compared to the untreated control (Fig. 5A). To verify this finding, we developed phosphorylated PDHA antibodies that target S232 and S314 (Supporting Information Fig. S7A). Immunoblot analysis showed that PDHA phosphorylation at S314 was increased, while phosphorylation at S293 and S232 was reciprocally inhibited in primary resistant cells (H460, Calu-1, and H441) upon trametinib treatment (Fig. 5B). Intriguingly, sensitive cells (A549, H23, and H1944) exhibited an inverse PDHA phosphorylation pattern. Elevated phosphorylation at S314 and coincidently decreased phosphorylation at S293 and S232 were consistently observed in A549/TR and H23/TR cells compared with their parental lineages (Fig. 5B), implying a MEKi sensitivity-specific effect. Moreover, trametinib treatment led to a marked increase in S314 phosphorylation and a simultaneous decrease in S293 and S232 phosphorylation in H460 and Calu-1 xenograft tumors in vivo (Fig. 5C).
Figure 5.

MEK inhibition alters PDHA phosphorylation status. (A) Phosphorylated peptides in PDHA were identified by MS analysis. Raw spectra and phospho-peptide are shown. (B) Effects of trametinib on the phosphorylation of PDHA in KRAS-mutant NSCLC cells. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. Treated cells were collected and probed with antibodies against phosphorylated PDHA (S314, S293, and S232) total PDHA, and β-actin, respectively. Trametinib-resistant cell lines are marked in green and trametinib-sensitive cell lines are marked in brown. Images are representative of three independent experiments with similar results. (C) S314, S293, and S232 phosphorylation levels of PDHA in H460 and Calu-1 xenograft tumors. Tumors were isolated on Day 7 after trametinib treatment. Immunoblot analysis for phosphorylated PDHA (S314, S293, and S232) and β-actin expression were conducted. Three biologically independent samples per group are shown. (D) Effects of trametinib on the PDHK1–PDP2 axis in KRAS-mutant NSCLC cells. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. Treated cells were collected and probed with antibodies against PDHK1, PDP2, and β-actin, respectively. (E) Colony formation assay. H460 cells with or without PDHA sgRNA and with or without reconstituted expression of PDHA WT, PDHA S314A, PDHA S293D, or PDHA S232D were treated with trametinib for 7–10 days. Clonogenic assays was conducted, where culture medium was replaced every other day. Representative clonogenic images are shown. Data are presented as the mean of three independent experiments. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test.
Mitochondrial PDHA is phosphorylated and inactivated by PDH kinase 1 (PDHK1) and reversely regulated by pyruvate dehydrogenase phosphatase 2 (PDP2)33,35. To further delineate how PDHA phosphorylation is modulated, we examined the effects of MEKi exposure on PDHK1 and PDP2 expression. We observed downregulated PDHK1 expression and upregulated PDP2 expression in trametinib-treated primary resistant cells, whereas PDHK1 and PDP2 expression was almost inversely regulated in trametinib-treated sensitive cells (Fig. 5D). Significant differences were also observed in the acquired resistant cell pairs. Notably, PDHA impairment by the overexpression of constitutively inactive PDHA S314A, S293D, and S232D mutants significantly enhanced the killing effects of trametinib in resistant H460 and H441 cells (Fig. 5E, Fig. S7B and S7C), reinforcing the functional role of PDHA in rendering KRAS-mutant NSCLC cells resistant to MEK inhibition. Taken together, these data indicate that resistant cells evade trametinib killing by augmenting PDHc activity through the PDHK1/PDP2 axis.
3.6. CPTIA transactivation confers trametinib resistance
In addition to pyruvate metabolism, the crucial role of FAO in facilitating cancer survival and metabolic adaptation has been increasingly recognized22. Given that palmitic acid fueled the TCA cycle and enhanced mitochondrial oxidative phosphorylation (Fig. 3), we next characterized the role of FAO in trametinib resistance. To this end, we performed RNA sequencing analysis in KRAS-mutated NSCLC cells exposed to trametinib and further investigated the differentially expressed genes across the treated cell lines. Our results showed a remarkable upregulation of FAO-associated genes in treated resistant H460 and H441 cells, but not in treated sensitive A549 cells (Fig. 6A), suggesting a link of FAO and trametinib sensitivity. As the rate-controlling step of FAO, carnitine palmitoyltransferase I (CPTI) is the only enzyme family that catalyzes long-chain fatty acyl-CoAs to acylcarnitines for translocation across the mitochondrial membrane. The CPTI family of proteins comprises three subtypes: CPTIA, CPTIB, and CPTIC, among which CPTIA is the most prevalent member that accomplishes mitochondrial β-oxidation and is associated with tumor development and drug resistance37,38. Surprisingly, we noticed an increased in CPTIA mRNA expression in treated resistant H460 and H441 cells, but an inverse phenomenon in treated sensitive A549 cells (Fig. 6A). We confirmed these results using immunoblot and RT-qPCR analysis and found that trametinib treatment substantially promoted CPTIA protein and mRNA expression selectively in primary and acquired resistant cells (Fig. 6B and C). Moreover, striking differences in CPTIA expression were also observed in resistant xenograft tumors from trametinib-treated mice compared to those from vehicle-treated mice (Fig. 6D), indicating that aberrant CPTIA expression was associated to a poor response to trametinib. Next, we elucidated the underlying mechanism by which CPTIA expression increased. Accumulating evidence has demonstrated that several transcription factors, including PPARs (PPARα, PPARδ, and PPARγ), PGC-1α, and CEBPB, can modulate CPTIA transcription by direct binding to its promoter22,39. Following to this direction, we noticed differential expression of transcription factors between resistant and sensitive cells according to our RNA-sequencing data (Fig. 6A) and further confirmed the results using RT-qPCR analysis. We found that the expression of PPARα, PPARδ, PPARγ, PGC-1α, and CEBPB were upregulated by trametinib. Compared with the untreated control, the fold increase in the expression of PPARα, PPARδ, PPARγ, PGC-1α, and CEBPB in trametinib-treated H460 cells was much higher than that in trametinib-treated A549 cells (Fig. 6E). A549/TR cells consistently showed a significantly higher expression of these transcription factors compared with A549/P cells (Supporting Information Fig. S8A). To further dissect the mechanism of CPTIA transactivation, we performed a focused mini-screen using siRNAs targeting these reported transcription factors, and our results showed that PPARs, PGC-1α, or CEBPB individual silencing significantly reduced CPTIA mRNA expression in trametinib-treated H460 cells and A549/TR cells (Fig. 6F and Fig. S8B). Collectively, these data suggest a stimulating effect of multiple transcription factors on CPTIA expression in trametinib-resistant cells.
Figure 6.

CPTIA transactivation confers MEKi resistance. (A) Heat map illustrating expression of fatty acid oxidation (FAO)-associated gene signature in H460, H441, and A549. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. Total mRNA was isolated from treated cells and sequenced. Z-scores were calculated based on counts of exon model per million mapped reads. FAO-related genes were identified by a cutoff of P < 0.05, n = 3. (B) Effects of trametinib on CPTIA mRNA (up) and protein (down) expression levels in KRAS-mutant NSCLC cells. Cells were treated with trametinib at their respective 1/2 IC50 values for 24 h. CPTIA expression was detected by RT-qPCR and immunoblot assays. MEKi-resistant cell lines are marked in green, and MEKi-sensitive cell lines are marked in brown. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (C) CPTIA mRNA (up) and protein (down) expression levels in acquired resistant cell pairs (A549/P and A549/TR; H23/P and H23/TR). Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (D) Trametinib upregulated CPTIA expression in H460 and Calu-1 xenograft tumors. Tumors were isolated after 1-week trametinib treatment. RT-qPCR and immunoblot analysis assays were detected. Values are expressed as the mean ± SEM of three independent biologically samples. ∗∗P < 0.01, by unpaired, two-sided Student's t test. (E) Log2 ratio values of fold change for PPARα, PPARδ, PPARγ, PGC-1α, and CEBPB transcripts in A549 and H460 cells before and after trametinib treatment. Cells were exposed to trametinib at their respective 1/2 IC50 values for 24 h. RT-qPCR analysis was further carried out. (F) CPTIA mRNA expression levels. H460 cells were treated with trametinib for 24 h after PPARs, PGC-1α, or CEBPB genetical silencing. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test. (G) CPTIA silencing enhanced the killing effects of trametinib in H460 cells. Left panel, relative viability of the culture colonies; right panel, CPTIA knockdown efficiency in H460 cells by immunoblot analysis. Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test. (H, I) CPTIA inhibition by etomoxir increased the sensitivity of H460 cells to trametinib, as shown by the growth curves (H) and relative clonogenic viability (I). Data represent the mean ± SEM of biological triplicates. ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test.
To delineate the association between CPTIA and MEKi resistance, we investigated whether direct CPTIA silencing sensitizes cancer cells to MEKi treatment. We either genetically interfered with CPTIA expression or pharmacologically inhibited CPTIA activity in resistant cells and evaluated their effects on trametinib sensitivity. Our results showed that CPTIA deficiency augmented the therapeutic efficacy of trametinib in H460 and H441 cells (Fig. 6G and Fig. S8C). The addition of etomoxir, an irreversible inhibitor of CPTIA, effectively suppressed the viability and clonogenic growth of these resistant cells (Fig. 6H and I, Fig. S8D and S8E). Taken together, our data highlight an essential role of CPTIA-dependent FAO contributing to trametinib resistance.
3.7. Targeted inhibition of OXPHOS synergizes with trametinib in vitro
Since trametinib promoted PDHc- and CPTIA-mediated mitochondrial oxidative metabolism, satisfying the energy demand in resistant cells and enabling them to survive, we investigated whether blocking mitochondrial OXPHOS could augment the killing effects of trametinib. Given the well-known safety profile and clinical benefits of IACS-010759, a potent OXPHOS inhibitor that targets complex I of the mitochondrial electron transport chain, we explored the sensitizing effects of IACS-010759 on trametinib in vitro. Our results showed that concurrent inhibition of OXPHOS and trametinib produced a strong synergy (CI < 0.7) in H460, Calu-1, and H441 cells (Fig. 7A). Moreover, pretreatment with different concentrations of IACS-010759 significantly augmented the cytotoxic effects of trametinib (Fig. 7B). As expected, co-treatment with IACS-010759 and trametinib notably inhibited the colony-formation ability of resistant cells by triggering apoptosis (Fig. 7C and D). These data strengthen the importance of mitochondrial OXPHOS in promoting the resistance of KRAS-mutant NSCLC cells to trametinib, creating pharmacologically actionable vulnerability in vitro.
Figure 7.

Targeted inhibition of OXPHOS synergizes with trametinib in vitro. (A) Synergistic interaction between trametinib and IACS-010759 in MEKi-resistant cells. H460, Calu-1, and H441 were treated with various concentrations of the indicated inhibitors for 72 h. The concentrations of trametinib or IACS-010759 were used in a 2-fold dilution series (1.56, 3.13, 6.25, 12.5, 25, and 50 μmol/L for trametinib; 0.63, 1.25, 2.5, 5.0, 10, and 20 nmol/L for IACS-010759). Relative cell viability was measured. Data represent the mean ± SEM of biological triplicates. CI values at each concentration were calculated using CalcuSyn software. (B) IACS-010759 enhanced the efficacy of trametinib in MEKi-resistant cells. H460, Calu-1, and H441 cells were treated with concentration gradients of trametinib with or without IACS-010759 for 72 h. Cell viability curves are shown. Data represent the mean ± SEM of biological triplicates. (C) Inhibition of clonogenic viability by the combined regimen. H460, Calu-1, and H441 cells were treated with trametinib, IACS-010759, or their combination as indicated. Representative long-term clonogenic images (up) and quantified clonogenic growth inhibition results (down) are shown. (D) Quantification of apoptotic cells in H460 and H441 cells analyzed by flow cytometry. Cells were treated with 500 nmol/L trametinib, 20 nmol/L IACS-010759 and their combinations for 72 h. Percentage of positive cells was analyzed. Values are expressed as the mean ± SEM of three replicates. ∗∗P < 0.01, and ∗∗∗P < 0.001, one-way ANOVA with Tukey's multiple-comparisons test.
3.8. Concurrent inhibition of MEK and OXPHOS impede resistant tumor growth in vivo
Next, we explored the therapeutic efficacy of IACS-010759 and trametinib cotreatment in mice harboring resistant tumors. Following the schedule described in the trametinib clinical trial, we selected a 3-week schedule regimen and found that IACS-010759 significantly enhanced the antitumor effects of trametinib on the growth of H460 xenograft tumors (Fig. 8A), without causing significant loss of body weight in mice (Supporting Information Fig. S9A; n = 6 per group). In addition, the combination therapy did not result in severe systemic toxicity, as the serum levels of alanine amino transferase, aspartate amino transferase, blood urea nitrogen, creatinine, and other biochemical factors, were marginally affected in the treated mice at the end of therapy (Supporting Information Table S5). Furthermore, trametinib activated OXPHOS, as evidenced by increased OCR levels, whereas the addition of IACS-010759 suppressed OXPHOS induction and led to apparent tumor regression (Fig. 8B). Immunoblot analysis of tumor lysates further revealed that PDHA activation (S314 phosphorylation) and CPTIA expression were significantly increased by trametinib treatment, whereas IACS-010759 addition significantly blocked these molecular events (Fig. 8C).
Figure 8.

Concurrent inhibition of MEK and OXPHOS impede resistant tumor growth in vivo. (A) Tumor volume and tumor weight of H460 xenografts (n = 6 per group). Mice bearing H460 xenograft tumors were treated with vehicle, trametinib (1 mg/kg body weight), and trametinib plus IACS-010759 (5 mg/kg body weight) for indicated times. Left panel, tumor growth curves; the mean AUC of tumor volume (AUCTV) on Day 21 is shown in the inset; right panel, tumor weight at the end of therapy, and each dot represents a tumor from individual mouse. Data represent the mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test. (B) OCR levels in vehicle-, trametinib- and combo-treated H460 xenograft tumors (n = 6 per group). Values are expressed as the mean ± SEM of three independent biologically samples. ∗∗P < 0.01, and ∗∗∗P < 0.001, by one-way ANOVA with Tukey's multiple-comparisons test. (C) Immunoblot analysis of p-PDHA (S314) and CPTIA in H460 xenograft tumors. At the end of the treatment, xenograft tumors were isolated from mice and tumor lysates were immunoblotted. Four biologically independent samples per group are shown. (D) Tumor volume (left panel) and tumor weight (right panel) of A549/TR xenograft tumors (n = 6 per group). Mice bearing A549/TR xenografts were treated with trametinib (1 mg/kg body weight) or trametinib plus IACS-010759 (5 mg/kg body weight) for 21 days. Data represent the mean ± SEM. ∗∗P < 0.01, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (E) Tumor volume (left panel) and tumor weight (right panel) of LAC001 PDX/TR xenografts (n = 8 per group). Data represent the mean ± SEM. ∗∗P < 0.01, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (F) Tumor volume (left panel) and weight (right panel) of LAC003 PDX/TR xenografts (n = 8 per group). Data represent the mean ± SEM. ∗∗P < 0.01, and ∗∗∗P < 0.001, by unpaired, two-sided Student's t test. (G) Representative tumor image and tumor volume of trametinib-resistant KP tumors. KP mice bearing established resistant tumors were treated with vehicle, trametinib (1 mg/kg body weight), IACS-010759 (5 mg/kg body weight) or trametinib plus IACS-010759. Mice were scanned by micro-CT after treatment (left panel). Tumor volume at the endpoint of the indicated treatments is shown (right panel). Areas marked by red dotted lines indicate lung tumors. H, heart. ∗P < 0.05, and ∗∗P < 0.01, by one-way ANOVA with Tukey's multiple-comparisons test. (H) Kaplan–Meier survival analysis of KP mice (n = 6 per group). Med., median. ∗P < 0.05, and ∗∗P < 0.01, by log-rank tests.
Additionally, we established a xenograft mouse model using resistant A549/TR cells. Tumor growth and burden were significantly reduced in the combination group compared with those in the trametinib-treated mice during the dosing period (Fig. 8D), along with a much lower cellular OCR level with decreased basal respiration and maximal respiratory capacity (Fig. S9B; n = 6 per group). The co-administration of trametinib and IACS-010759 was well tolerated, as weight loss and systematic toxicity were within acceptable limits (Fig. S9C and Table S5).
LAC001 and LAC003 PDX-based models of KRAS-mutant lung cancer, more clinically relevant mouse models, were developed to simulate the development of resistance and evaluate the antitumor activity of the combined treatment. Approximately 100 mm3 of growing PDX tumors was exposed to trametinib until tumor expansion, producing trametinib-resistant PDX (PDX/TR) tumors. Mice bearing PDX/TR tumors were then administered trametinib or the combination regimen. Our results showed that LAC001 (n = 8 per group) and LAC003 (n = 7 per group) PDX/TR xenograft tumors grew robustly in the presence of trametinib, whereas IACS-010759 addition significantly constrained tumor growth (Fig. 8E and F) and OCR levels (Fig. S9D and S9E). Notably, the combination of trametinib and IACS-010759 was tolerated in mice (Fig. S9F and S9G and Table S5).
To explore the benefits of trametinib plus IACS-010759 on mouse survival, we simulated the development of trametinib resistance in LSL-KrasG12D/+; Trp 53fl/fl mice. Once acquired resistance occurred in the trametinib-stressed tumors, the mice were regrouped randomly and treated according to schedule (Fig. S9H and S9I; n = 6 per group). Administration of the drug pair impeded tumor growth to a significantly greater extent than either treatment alone, as shown by representative micro-CT images and tumor volume (Fig. 8G). Compared with the vehicle, trametinib monotherapy had a limited effect on mouse survival, whereas the addition of IACS-010759 significantly prolonged survival, with an added median survival benefit of 84 days (Fig. 8H). These findings support the notion that the trametinib plus IACS-010759 combination regimen is effective against trametinib-resistant lung tumors.
4. Discussion
Several MAPK pathway inhibitors are FDA-approved, including BRAF and MEK kinase inhibitors, which are poorly tolerated and less effective in the treatment of KRAS-mutant tumors8. This emphasizes the need to explore combined regimens to potentiate MAPKi. Understanding metabolic alterations associated with drug resistance may accelerate the development of novel treatment strategies. This study identified a non-genetic mechanism through which KRAS-driven NSCLC cells evade trametinib therapy. Trametinib-resistant cells rewired a metabolic switch by enhancing PDHc- and CPTIA-mediated metabolic flux of pyruvate and palmitic acid to the TCA cycle, enhancing mitochondrial OXPHOS and thereby escaping apoptosis in the context of MEK inhibition. We further revealed the phosphorylation modification of PDHA and the translational regulation of CPTIA in the control of therapy-induced metabolic plasticity. Finally, we proposed a mechanism-based combination therapy involving the concurrent inhibition of MEK and OXPHOS, which led to potent regression of KRAS-mutant NSCLC in several preclinical tumor mouse models.
Metabolic reprogramming is a hallmark of cancer that has been extensively studied over the last decade; however, how therapy reprograms metabolism in drug-resistant or relapsed tumors has drawn less attention. Cancer cells primarily utilize glycolysis and bypass the TCA cycle to boost the supply of metabolic intermediates used in anabolic processes, which is known as the Warburg effect. However, accumulating evidence indicate that tumor cells display adaptive mitochondrial reprogramming to compensate for decreased glycolysis when evading drug treatment under certain circumstances. BRAF inhibitors have been reported to render BRAF-mutated melanomas addicted to OXPHOS40,41. Increased FAO is also required for BRAF-mutant melanoma to survive MAPKi-induced metabolic stress22. To the best of our knowledge, the role of mitochondrial biogenetic reprogramming in the adaptation of KRAS-mutant lung cancer cells to targeted therapy has not yet been described. In this study, we report a non-mutational mechanism underlying MEKi resistance in KRAS-mutant NSCLC, characterized by increased mitochondrial oxidative metabolism. Similar to BRAF-mutated melanoma, KRAS-mutated NSCLC responded to trametinib by regulating mitochondrial flexibility to satisfy energy demands while adapting to the treatment. During this adaptation, acetyl-CoA derived from pyruvate metabolism and FAO powered the TCA cycle and further enhanced the OXPHOS system. Notably, OXPHOS activation was a determinant of KRAS-mutant NSCLC resistance to MEK inhibition, as targeted inhibition of OXPHOS significantly augmented trametinib sensitivity in both primary and acquired resistance tumor models in vitro and in vivo. Our findings and those of recently published studies21,24, support the notion that the mechanisms underlying metabolic plasticity may represent a more attractive therapeutic target for circumventing drug resistance.
This study revealed that PDHc-dependent glycolysis cooperated with CPTIA-dependent FAO to activate the OXPHOS system in resistant cells, which determined their sensitivity to MEK pharmacological inhibition. PDHc is a mitochondrial gatekeeper enzyme that connects glycolysis and the tricarboxylic acid cycle by converting pyruvate to acetyl-CoA in the mitochondria. Although dysregulation of PDHc leads to multiple metabolic disorders and neurodegeneration42, its function and regulation in different cellular contexts and stages of cancer development remain unclear. Several studies have demonstrated that PDHc acts as an oncogenic factor. Phosphorylation of PDHA, a subunit of PDHc, by AMP-activated protein kinase (AMPK) facilitates tumor metastasis36. Genetic and pharmacological inactivation of PDHA inhibits prostate tumor development43. This study demonstrated, for the first time, that PDHc enabled KRAS-driven NSCLC resistance to MEK inhibition, revealing a newly discovered role of PDHc in tumor cell adaptation to treatment, and that targeting PDHc may potentially tackle drug resistance. A more recent study reported that cytosolic PDHA can be phosphorylated at S327 by ERK2 and translocated into the mitochondria, thereby promoting resistance to immunotherapy and lung cancer growth44. However, the effect of phosphorylation at S327 on the PDHc activity remains unclear. It is generally accepted that S293 and S232 phosphorylation of PDHA serves as a negative signal for PDHc activation. Accordingly, the removal of phosphorylated serine residues increases PDHc activity35. Moreover, phosphorylation of PDHA on two residues, S295 and S314, is essential for PDHc activation and TCA cycle maintenance in cancer metastasis, where S295 and S314 phosphorylation has been discovered in an in vitro kinase assay36. In this study, we characterized the phosphorylation events that mediate PDHc function in trametinib-treated resistant cells and highlighted their cellular roles in inducing metabolic shifts. Notably, we discovered a coordinated model of PDHc activation in which PDHA S314 phosphorylation levels were increased, and PDHA S293 and S232 phosphorylation levels were simultaneously decreased. According to a published studies, phosphorylation of PDHA S293 and S232 is typically regulated by the PDHK/PDP axis35, and PDHA S314 phosphorylation is mediated by AMPK36. We speculate that aberrant phosphorylation patterns of PDHA in specific biological conditions may dependent on altered kinase activity of its upstream regulators. However, more biochemical and biological studies are needed to further understand the mode of PDHA phosphorylation site interactions and their respective upstream regulators during therapy-driven metabolic rewiring.
As a rate-limiting mitochondrial enzyme of FAO that directly controls the transport of fatty acids into the mitochondria, CPTIA is overexpressed in cancer cells and contributes to tumor development37. A recent study has revealed that BRAF-mutated melanoma displays increased levels of PPARα- and CPTIA-dependent FAO to evade the killing effects of BRAF inhibitors22. In our study, we found that increased FAO was required for KRAS-mutant NSCLC cells to survive MKEi in a CPTIA-dependent manner. Our findings, together with those of others22, suggest that the dependency on FAO might be a common event and represents a general metabolic vulnerability in MAPKi resistant cells. Previous studies have shown that PPARs (transcriptional coactivators) form complexes with β-catenin to induce CPTIA expression22. PGC-1α binds to CEBPB, a member of the transcription factor family of CEBP, to transactivate CPTIA, resulting in sustained FAO activation39. In our study, we found that the expression of several transcription factors (PPARs, PGC-1α, and CEBPB) was simultaneously increased in treated resistant cells, implying that they may coordinately upregulate CPTIA expression through transcriptional reprogramming.
In addition to our findings, recent evidence also emphasizes that many tumor cell subpopulations rely heavily on OXPHOS for bioenergetics40 and biosynthesis45,46, indicating that OXPHOS inhibitors are potential oncological therapeutics. Biguanides, such as metformin, have been evaluated for the treatment of diabetes and metabolic disorders, suggesting that targeting OXPHOS for clinical benefits will be safe47. However, metformin and other drugs targeting oxidative metabolism have pharmacological limitations. Inadequate efficacy and unaffordable toxicity (i.e., biguanides, atovaquone and arsenic trioxide)48, ‘off-target’ effects (i.e., rotenone)49, and poor pharmacokinetic profiles (i.e., oligomycin)50 hamper the application of these early mitochondrial inhibitors in cancer treatment. Accordingly, the results of these clinical trials have also been disappointing51. In our study, we proposed a regimen of the combined use of trametinib and IACS-010759, a new clinal-grade, highly potent small-molecule inhibitor of complex I of the mitochondrial electron transport chain52, 53, 54, to improve the efficacy of MEKi in the treatment of KRAS-mutant NSCLC. IACS-010759 has been reported to efficiently reduce high-risk neuroblastoma55, SWI/SNF mutant lung tumors56, triple-negative breast cancer57, and cancer stem cells58,59, and potentiate targeted therapies in BRAF-mutant melanoma41. Our results showed that low-dose combination therapy with trametinib and IACS-010759 was quite effective and well tolerated in different KRAS-mutant NSCLC mouse models in vivo, conferring a remarkable survival benefit without affecting physiological indices. Given that targeting OXPHOS complexes is an emerging and attractive approach to disrupt altered metabolism and overcome drug resistance in tumor-specific contexts, a series of novel OXPHOS inhibitors is currently being developed60,61.
5. Conclusions
We found that clinical MEK inhibitors profoundly increase mitochondrial OXPHOS and metabolic flexibility selectively in MEKi-resistant KRAS-mutant NSCLC as compared with MEKi-sensitive counterparts. Differential activity of PDHc and CPTIA, two rate-limiting enzymes that control the metabolic flux of pyruvate and palmitic acid to mitochondrial respiration, was observed between MEKi-resistant and -sensitive cancer cells. Based on these findings, we proposed a model in which tumor cells take advantage of metabolic vulnerability in the mitochondria to adapt to MEK inhibition stress and facilitate their survival. Exploiting this vulnerability using rational combinational strategies by co-targeting mitochondrial oxidative phosphorylation renders KRAS-mutant NSCLC cells more responsive to MEKi therapy.
Acknowledgments
We thank Dr. Min Huang (Shanghai Institute of Materia Medical, Chinese Academy of Sciences, Shanghai, China) for technical supports of glucose/palmitate/glutamine tracing experiments. This work was sponsored by the National Natural Science Foundation of China (82122045, 82073073, 81874207, and 81872418), Innovative Research Team of High-level Local Universities in Shanghai (SHSMU-ZDCX20210802, China), MOE Key Laboratory of Biosystems Homeostasis & Protection (Zhejiang University, China), Science and Technology Commission of Shanghai Municipality (21S11902000, China), Jointed PI Program from Shanghai Changning Maternity and Infant Health Hospital (11300-412311-20033, China), ECNU Construction Fund of Innovation and Entrepreneurship Laboratory (44400-20201-532300/021, China), and the ECNU multifunctional platform for innovation (011 and 004, China).
Author contributions
Juanjuan Feng and Zhengke Lian designed and performed the experiments, analyzed the data and wrote the manuscript. Xinting Xia, Yue Lu, Kewen Hu, Yunpeng Zhang, Yanan Liu, Longmiao Hu, and Kun Yuan performed experiments and revised the manuscript. Kewen Hu provided technical support and revised the manuscript. Xiufeng Pang and Zhenliang Sun led the project, analyzed the data, and wrote the manuscript.
Conflicts of interest
The authors have declared that no conflict of interest exists.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.10.023.
Contributor Information
Zhenliang Sun, Email: zhenliang@smu.edu.cn.
Xiufeng Pang, Email: xfpang@bio.ecnu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Moore A.R., Rosenberg S.C., McCormick F., Malek S. RAS-targeted therapies: is the undruggable drugged?. Nat Rev Drug Discov. 2020;19:533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu P., Wang Y., Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B. 2019;9:871–879. doi: 10.1016/j.apsb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.FDA approves first kras inhibitor: sotorasib. Cancer Discov. 2021;11:OF4. doi: 10.1158/2159-8290.CD-NB2021-0362. [DOI] [PubMed] [Google Scholar]
- 5.Awad M.M., Liu S., Rybkin, Arbour K.C., Dilly J., Zhu V.W., et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan M.B., Corcoran R.B. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol. 2018;15:709–720. doi: 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 7.Liu F., Yang X., Geng M., Huang M. Targeting ERK, an Achilles' heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B. 2018;8:552–562. doi: 10.1016/j.apsb.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drosten M., Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. 2020;37:543–550. doi: 10.1016/j.ccell.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Gandara D.R., Leighl N., Delord J.P., Barlesi F., Bennouna J., Zalcman G., et al. A phase 1/1b study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12:556–566. doi: 10.1016/j.jtho.2016.11.2218. [DOI] [PubMed] [Google Scholar]
- 10.Janne P.A., Mann H., Ghiorghiu D. Study design and rationale for a randomized, placebo-controlled, double-blind study to assess the efficacy and safety of selumetinib in combination with docetaxel as second-line treatment in patients with kras-mutant advanced non-small cell lung cancer (SELECT-1) Clin Lung Cancer. 2016;17:e1–e4. doi: 10.1016/j.cllc.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Caunt C.J., Sale M.J., Smith P.D., Cook S.J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 12.Hong A., Piva M., Liu S., Hugo W., Lomeli S.H., Zoete V., et al. Durable suppression of acquired MEK inhibitor resistance in cancer by sequestering MEK from ERK and promoting antitumor T-cell Immunity. Cancer Discov. 2021;11:714–735. doi: 10.1158/2159-8290.CD-20-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones G.G., Del Rio I.B., Sari S., Sekerim A., Young L.C., Hartig N., et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat Commun. 2019;10:2532. doi: 10.1038/s41467-019-10367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manchado E., Weissmueller S., Morris JPt, Chen C.C., Wullenkord R., Lujambio A., et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–651. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wee S., Jagani Z., Xiang K.X., Loo A., Dorsch M., Yao Y.M., et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 16.Kitajima S., Asahina H., Chen T., Guo S., Quiceno L.G., Cavanaugh J.D., et al. Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS. Cancer Cell. 2018;34:439–452.e6. doi: 10.1016/j.ccell.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudreau P.O., Lee J.J., Heymach J.V., Gibbons D.L. Phase I/II trial of immunotherapy with durvalumab and tremelimumab with continuous or intermittent MEK inhibitor selumetinib in NSCLC: early trial report. Clin Lung Cancer. 2020;21:384–388. doi: 10.1016/j.cllc.2020.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arbour K.C., Manchado E., Bott M.J., Ahn L., Tobi Y., Ni A.A., et al. Phase 1 clinical trial of trametinib and ponatinib in patients with NSCLC harboring KRAS mutations. JTO Clin Res Rep. 2022;3 doi: 10.1016/j.jtocrr.2021.100256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf D.A. Is reliance on mitochondrial respiration a “chink in the armor” of therapy-resistant cancer?. Cancer Cell. 2014;26:788–795. doi: 10.1016/j.ccell.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosc C., Selak M.A., Sarry J.E. Resistance is futile: targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab. 2017;26:705–707. doi: 10.1016/j.cmet.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Zhang K.R., Zhang Y.F., Lei H.M., Tang Y.B., Ma C.S., Lv Q.M., et al. Targeting AKR1B1 inhibits glutathione de novo synthesis to overcome acquired resistance to EGFR-targeted therapy in lung cancer. Sci Transl Med. 2021;13 doi: 10.1126/scitranslmed.abg6428. [DOI] [PubMed] [Google Scholar]
- 22.Aloia A., Mullhaupt D., Chabbert C.D., Eberhart T., Fluckiger-Mangual S., Vukolic A., et al. A fatty acid oxidation-dependent metabolic shift regulates the adaptation of BRAF-mutated melanoma to MAPK inhibitors. Clin Cancer Res. 2019;25:6852–6867. doi: 10.1158/1078-0432.CCR-19-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang G., Frederick D.T., Wu L., Wei Z., Krepler C., Srinivasan S., et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016;126:1834–1856. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Nguyen T.T.T., Shang E., Mela A., Humala N., Mahajan A., et al. MET inhibition elicits PGC1alpha-dependent metabolic reprogramming in glioblastoma. Cancer Res. 2020;80:30–43. doi: 10.1158/0008-5472.CAN-19-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu K., Li K., Lv J., Feng J., Chen J., Wu H., et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest. 2020;130:1752–1766. doi: 10.1172/JCI124049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro P., Bueno M.J., Zagorac I., Mondejar T., Sanchez J., Mouron S., et al. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Rep. 2016;15:2705–2718. doi: 10.1016/j.celrep.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 27.Wang K.Z., Zhu J., Dagda R.K., Uechi G., Cherra S.J., 3rd, Gusdon A.M., et al. ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson's disease. Mitochondrion. 2014;17:132–140. doi: 10.1016/j.mito.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X., Jiang Y., Meisenhelder J., Yang W., Hawke D.H., Zheng Y., et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA Cycle in tumorigenesis. Mol Cell. 2016;61:705–719. doi: 10.1016/j.molcel.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatzivassiliou G., Haling J.R., Chen H., Song K., Price S., Heald R., et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 30.Lito P., Saborowski A., Yue J., Solomon M., Joseph E., Gadal S., et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann M.H., Gmachl M., Ramharter J., Savarese F., Gerlach D., Marszalek J.R., et al. BI-3406, a potent and selective SOS1–KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 2021;11:142–157. doi: 10.1158/2159-8290.CD-20-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sellers K., Fox M.P., Bousamra M., 2nd, Slone S.P., Higashi R.M., Miller D.M., et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–698. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplon J., Zheng L., Meissl K., Chaneton B., Selivanov V.A., Mackay G., et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109–112. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 34.Patel M.S., Nemeria N.S., Furey W., Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615–16623. doi: 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woolbright B.L., Rajendran G., Harris R.A., Taylor J.A., 3rd Metabolic flexibility in cancer: targeting the pyruvate dehydrogenase kinase:pyruvate dehydrogenase axis. Mol Cancer Ther. 2019;18:1673–1681. doi: 10.1158/1535-7163.MCT-19-0079. [DOI] [PubMed] [Google Scholar]
- 36.Cai Z., Li C.F., Han F., Liu C., Zhang A., Hsu C.C., et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol Cell. 2020;80:263–278.e7. doi: 10.1016/j.molcel.2020.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu Q., Zeng F., Liu X., Wang Q.J., Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226. doi: 10.1038/cddis.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ricciardi M.R., Mirabilii S., Allegretti M., Licchetta R., Calarco A., Torrisi M.R., et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–1929. doi: 10.1182/blood-2014-12-617498. [DOI] [PubMed] [Google Scholar]
- 39.Wu H., Liu B., Chen Z., Li G., Zhang Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1alpha/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 2020;11:233. doi: 10.1038/s41419-020-2426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haq R., Shoag J., Andreu-Perez P., Yokoyama S., Edelman H., Rowe G.C., et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vashisht Gopal Y.N., Gammon S., Prasad R., Knighton B., Pisaneschi F., Roszik J., et al. A novel mitochondrial inhibitor blocks MAPK pathway and overcomes MAPK inhibitor resistance in melanoma. Clin Cancer Res. 2019;25:6429–6442. doi: 10.1158/1078-0432.CCR-19-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown G. Pyruvate dehydrogenase deficiency and the brain. Dev Med Child Neurol. 2012;54:395–396. doi: 10.1111/j.1469-8749.2012.04266.x. [DOI] [PubMed] [Google Scholar]
- 43.Chen J., Guccini I., Di Mitri D., Brina D., Revandkar A., Sarti M., et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet. 2018;50:219–228. doi: 10.1038/s41588-017-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y., Zhao M., Gao H., Yu G., Zhao Y., Yao F., et al. MAPK signalling-induced phosphorylation and subcellular translocation of PDHE1alpha promotes tumour immune evasion. Nat Metab. 2022;4:374–388. doi: 10.1038/s42255-022-00543-7. [DOI] [PubMed] [Google Scholar]
- 45.Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sanchez N., Marchesini M., et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elgendy M., Ciro M., Hosseini A., Weiszmann J., Mazzarella L., Ferrari E., et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A–GSK3beta–MCL-1 Axis. Cancer Cell. 2019;35:798–815.e5. doi: 10.1016/j.ccell.2019.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Pollak M. Targeting oxidative phosphorylation: why, when, and how. Cancer Cell. 2013;23:263–264. doi: 10.1016/j.ccr.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 49.Yarmohammadi F., Wallace Hayes A., Najafi N., Karimi G. The protective effect of natural compounds against rotenone-induced neurotoxicity. J Biochem Mol Toxicol. 2020;34 doi: 10.1002/jbt.22605. [DOI] [PubMed] [Google Scholar]
- 50.Yang H., van der Stel W., Lee R., Bauch C., Bevan S., Walker P., et al. Dynamic modeling of mitochondrial membrane potential upon exposure to mitochondrial inhibitors. Front Pharmacol. 2021;12 doi: 10.3389/fphar.2021.679407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kordes S., Pollak M.N., Zwinderman A.H., Mathot R.A., Weterman M.J., Beeker A., et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 52.Molina J.R., Sun Y., Protopopova M., Gera S., Bandi M., Bristow C., et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036–1046. doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- 53.Chung I., Serreli R., Cross J.B., Di Francesco M.E., Marszalek J.R., Hirst J. Cork-in-bottle mechanism of inhibitor binding to mammalian complex I. Sci Adv. 2021;7 doi: 10.1126/sciadv.abg4000. :eabg4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsuji A., Akao T., Masuya T., Murai M., Miyoshi H. IACS-010759, a potent inhibitor of glycolysis-deficient hypoxic tumor cells, inhibits mitochondrial respiratory complex I through a unique mechanism. J Biol Chem. 2020;295:7481–7491. doi: 10.1074/jbc.RA120.013366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson N.M., Qin X., Finan J.M., Lam A., Athoe J., Missiaen R., et al. Metabolic enzyme DLST promotes tumor aggression and reveals a vulnerability to OXPHOS inhibition in high-risk neuroblastoma. Cancer Res. 2021;81:4417–4430. doi: 10.1158/0008-5472.CAN-20-2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lissanu Deribe Y., Sun Y., Terranova C., Khan F., Martinez-Ledesma J., Gay J., et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat Med. 2018;24:1047–1057. doi: 10.1038/s41591-018-0019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans K.W., Yuca E., Scott S.S., Zhao M., Paez Arango N., Cruz Pico C.X., et al. Oxidative phosphorylation is a metabolic vulnerability in chemotherapy-resistant triple-negative breast cancer. Cancer Res. 2021;81:5572–5581. doi: 10.1158/0008-5472.CAN-20-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Janiszewska M., Suva M.L., Riggi N., Houtkooper R.H., Auwerx J., Clement-Schatlo V., et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sancho P., Burgos-Ramos E., Tavera A., Bou Kheir T., Jagust P., Schoenhals M., et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 60.Xue D., Xu Y., Kyani A., Roy J., Dai L., Sun D., et al. Discovery and lead optimization of benzene-1,4-disulfonamides as oxidative phosphorylation inhibitors. J Med Chem. 2022;65:343–368. doi: 10.1021/acs.jmedchem.1c01509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue D., Xu Y., Kyani A., Roy J., Dai L., Sun D., et al. Multiparameter optimization of oxidative phosphorylation inhibitors for the treatment of pancreatic cancer. J Med Chem. 2022;65:3404–3419. doi: 10.1021/acs.jmedchem.1c01934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and metabolomics data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GEO: GSE197555 and the Mendeley Data repository (https://data.mendeley.com/datasets/54xr9bsxd2/1), respectively.