


Abstract
The ability of cell-free extracts to correct DNA mismatches has been demonstrated in both prokaryotes and eukaryotes. Such an assay requires a template containing both a mismatch and a strand discrimination signal, and the multi-step construction process can be technically difficult. We have developed a three-step procedure for preparing DNA heteroduplexes containing a site-specific nick. The mismatch composition, sequence context, distance to the strand signal, and the means for assessing repair in each strand are adjustable features built into a synthetic oligonucleotide. Controlled ligation events involving three of the four DNA strands incorporate the oligonucleotide into a circular template and generate the repair-directing nick. Mismatch correction in either strand of a prototype G·T mismatch was achieved by placing a nick 10–40 bp away from the targeted base. This proximity of nick and mismatch represents a setting where repair has not been well characterized, but the presence of a nick was shown to be essential, as was the MSH2/MSH6 heterodimer, although low levels of repair occurred in extract defective in each protein. All repair events were inhibited by a peptide that interacts with proliferating cell nuclear antigen and inhibits both mismatch repair and long-patch replication.
INTRODUCTION
The human DNA mismatch repair (MR) pathway contributes several important functions to the maintenance of genomic stability (reviewed in 1,2). One such role is the correction of base–base mismatches and insertion/deletion errors that arise during DNA replication. Although the overall mechanism remains unresolved, a heterodimer of MutS homologs is required for mismatch binding, and it is presumed that a mechanism exists to target mismatch correction to the newly synthesized strand, assuming that mismatches represent biosynthetic errors. The demonstration that MutS homologs interact with proliferating cell nuclear antigen (PCNA), and that PCNA participates in a step prior to strand excision in mismatch correction (3,4), places mismatch recognition activities at the site of replication, as well as offering a plausible mechanism for strand discrimination (5).
The ability to assess mismatch correction in vitro, using extracts from human cell lines and appropriate heteroduplex templates (6,7), represents an important step toward defining the participants in MR and reconstituting the pathway. A competent substrate must contain both a DNA mismatch and a device that allows strand discrimination; when assaying human cell extracts, this has invariably been a single-strand nick. The choice of a nick derives from the fact that mismatch correction in Escherichia coli demonstrably passes through an intermediate where one strand is nicked at a hemimethylated GATC site (8), and such a nick relieves the requirement for the MutH protein that introduces the nick. A single-strand break, present in a variety of sequence contexts and distances from the mismatch, is capable of strongly biasing mismatch excision from the discontinuous strand.
Two primary approaches for assessing MR in vitro have been developed. In one, a DNA mismatch is embedded within two overlapping restriction sites, and a site-specific nick is positioned within a range of roughly 100–1000 bp away in either strand (7,9). When such a substrate is exposed to HeLa nuclear extract and recovered, the extent of restriction site sensitivity is used as a diagnostic tool for MR. Typically, a strand bias of ∼10:1 is found, where the discontinuous (nicked) strand is 10-fold more likely to be excised and resynthesized. In the second approach (6), mismatch excision and strand resynthesis results in a protein product that reconstitutes β-galactosidase activity, expressed from an M13 phage template. The heteroduplex DNA is first treated with a cellular extract prepared from human cells, then recovered and transformed into a MR-defective strain of E.coli. A careful analysis of the relative number of blue and white phage colonies can be used to represent the frequency of MR events and the strand bias.
Preparation of mismatch-containing DNA molecules with a site-specific nick is usually performed using a pair of closely related phage molecules (7,10). When the double-stranded (replicative) form is linearized and denatured in the presence of a single-stranded partner that differs only at the mismatch site, the annealed product contains both a mismatch and a single-strand nick at the digestion site. In this approach, the DNA molecules are normally derived from bacteriophage molecules such as M13 or f1 phage. This facilitates isolation of both the single- and double-stranded partner, although the site of heterology must be constructed. Alternatively, a commercially available enzyme that nicks supercoiled plasmid DNA in a site- and strand-specific manner has been used to generate single-stranded linear molecules useful in heteroduplex formation (11).
Finally, it should be noted that the human mismatch recognition proteins are competent to recognize specific types of DNA lesions, such as methylated bases (12), cisplatin adducts (12–14), and other lesions (15,16). Processing of these lesions by the MR pathway has been implicated in the cytotoxic response to certain anticancer drugs (17). Although the mechanism is often cited as a futile processing cycle (18,19), limited mechanistic information about processing in vitro has been reported (20,21). In short, a strategy that allows for construction of a substrate containing a site-specific lesion and a unique, strand-specific nick, where each required site may be positioned in either strand, is not available.
We have developed a ligation-based protocol appropriate for construction of DNA heteroduplexes that can be completed in <2 days using commercially available reagents. The product molecules contain a site-specific single-strand nick capable of directing the repair of a nearby mismatch using the continuous strand as a template. While the approach is best characterized and perhaps best suited for small-scale preparations (≤5 µg product), it should allow for the rapid construction of a diverse set of heterologies. In addition to mismatch construction, we suggest that such a strategy is appropriate for the construction of specific DNA lesions, in either strand, placed in a context that may be scored for processing by the MR pathway. Such a capacity is not inherent to more traditional methodologies (6,7,11). Finally, in comparison with commonly used protocols for heteroduplex substrate preparation (6,7), our method is relatively expedient.
MATERIALS AND METHODS
Plasmids and reagents
A derivative of plasmid pET11a (Stratagene) was the primary template used to construct the heteroduplexes described in this paper, although pBR322, pBluescript, M13mp18 and unmodified pET11a have also been used successfully (data not shown). All restriction enzymes, T4 ligase and the Klenow fragment of DNA polymerase I were purchased from New England Biolabs. Exonuclease V was purchased from Amersham Pharmacia and Bsp106I was obtained from Stratagene. Chemical and biochemical reagents were purchased from Sigma Biochemicals unless otherwise specified. A derivative of f1MR1 (7) where the repair-directing nick was placed 141 bp away from the G·T mismatch was included as a control to demonstrate the specificity and efficiency of MR under published conditions (7). The High Pure PCR Product Purification kit (Boehringer), which binds large DNA fragments at the expense of small DNA molecules and proteins, was used where specified to isolate DNA intermediates.
A peptide fragment derived from p21 (22) that inhibits PCNA function was purchased from the American Peptide Company. The supplier estimated its purity to be ≥70%, and its concentration in aqueous solution was determined using a calculated extinction coefficient (23) of 1280 M–1 cm–1 at 280 nm, based on the single contributing amino acid present (tyrosine). Treatment of the peptide (1 mM) with a stock solution of trypsin (150 µg/ml final concentration) was used to degrade the peptide when a control solution lacking the activity was required. The added trypsin was inactivated by a stoichiometric treatment with soybean trypsin inhibitor or by heat inactivation (80°C for 20 min). These treatments alone did not affect MR activity.
Construction of pET11aΔH
Plasmid pET11a was modified prior to use in this protocol for two functional reasons. First, because the final constructs frequently incorporate a mispair within a HindIII site to allow an analysis of mismatch correction, this unique plasmid site was excised in order to remove any ambiguity about the origin of HindIII sensitivity. Secondly, the distance separating the BamHI and EcoRI sites was reduced to 28 bp, facilitating removal of the small intervening piece as described below. In order to achieve these modifications, pET11a was digested with Bsp106I and BamHI and the resulting overhangs filled in using the Klenow fragment of DNA polymerase I. The molecule was recircularized using T4 ligase to yield the product pET11aΔH, which retains its BamHI site and is now 5384 bp in size. The plasmid typically was amplified during growth in Luria broth by chloramphenicol treatment (24) and isolated using the appropriate scale Qiagen purification kit. The supercoiled form was further purified by ultracentrifugation in a cesium chloride density gradient containing ethidium bromide (24).
The oligonucleotides (Operon Technologies Inc.) used in this work were resuspended in 10 mM Tris–HCl at pH 8 containing 1 mM EDTA (TE). The DNA concentration was determined by measuring the absorbance at 260 nm using a Cary 50 spectrophotometer, assuming that 1 OD unit corresponds to 33 µg/ml. The oligonucleotide sequences used to construct mismatches are as follows: oligo (1), 5′-GCCGGAGATCGAAGCTTTCGAGC; oligo (2), 5′-AATTGCTCGAGAGCTTCGATCTCCGGC; oligo (3), 5′-CCGGCAGATCGCTCGAGAGCTTC; oligo (4), 5′-AATTGAAGCTTTCGAGCGATCTGCCGG; oligo (5), 5′-GCCGGAGATCTAAGCTTTCGAGGCAATGCCGCCTCATTGCAGCGACGTAAGCC; oligo (6), 5′-AATTGGCTTACGTCGCTGCAATGAGGCGGCATTGCCTCGAGAGCTTAGATCTCCGGC; oligo (7), 5′-GCCGGAGATCGAAGCTTTCGAGGCAATGCCGCC; oligo (8), 5′-AATTGGCGGCATTGCCTCGAGAGCTTCGATCTCCGGC; oligo (9), 5′-GCCGGAGATCTAAGCTTCATGGTACAGCAGCTAGCCCAGTAGGATGAAGTCGACC; oligo (10), 5′-AATTGGTCGACTTCATCCTACTGGGCTAGCTGCTGTACCATGGAGCTTAGATCTCCGGC.
A descriptive nomenclature was chosen for the plasmid products containing each of the heteroduplexes. For example, substrate made with oligo pair 1/2 is referred to as T·G-10H, where T·G indicates the mismatch composition. A nick is formed 10 bp away in the strand containing the G, and H represents the HindIII site that is diagnostic for nick-directed MR. Similarly, the 3/4 pair yields a G·T-10X (or G·T-10A), indicating that the nick now resides 10 bp away in the strand containing the T, and XhoI (or AvaI) can be used to score mismatch correction. The 5/6 pair is therefore T·G-40H and 7/8 is T·G-20H. In all cases, the strand polarity reading from nick to mismatch is 5′→3′.
The heteroduplex oligonucleotides were prepared as follows for a representative small-scale synthesis (15 µg plasmid). One of the oligo pairs described above was annealed using 1 nmol of each partner in 50 µl of TE supplemented with sodium chloride at a final concentration of 50 mM. The mixture was incubated in a beaker of water at 80°C and allowed to come to room temperature over ∼30 min. Each heteroduplex has a single GATC site located 6 bp from the blunt end, which was digested with DpnII (50 U/nmol oligo in a 200 µl volume for 2 h at 37°C). This yields a 5′-GATC extension complementary to those generated by BamHI digestion (see below), but intermolecular ligation will not reconstitute a BamHI site. The reaction was heated for 15 min at 65°C to quench DpnII activity before the unwanted fragments of 6 bp were removed by ultrafiltration. For the shorter oligo pairs (1/2, 3/4 and 7/8), the samples were retained using a Microcon 10 membrane (Amicon), whereas the longer oligo pairs (5/6 and 9/10) were retained with a Microcon 30 membrane. Following the initial concentration, the products were diluted to 500 µl with TE, concentrated to a volume of <50 µl and the process repeated twice. Measuring the absorbance of the filtrate at 260 nm, which becomes undetectable as the small fragment is removed, was used to monitor the separation.
Substrate construction
Plasmid pET11aΔH, typically 15 µg (4.2 pmol) at a concentration of 200 ng/µl, was linearized with BamHI (2 U/µg DNA). The choice of 15 µg was dictated by the amount of plasmid DNA that could be retained reliably by one High Pure DNA binding membrane, which retains plasmid DNA but poorly binds fragments <100 bp in length. The reaction was incubated for 10 min at 37°C in T4 ligase buffer (New England Biolabs) containing 100 µg/ml bovine serum albumin, 75 mM KCl and the heteroduplex oligo recovered after DpnII digestion (estimated to be a ∼100-fold molar excess over the plasmid ends). The reaction was then cooled on ice, whereupon 100 cohesive end ligation units of T4 ligase per microgram of plasmid (1500 U in this example) were added and the reaction incubated at 16°C overnight. The proteins and excess duplex oligos were removed from the reaction using the High Pure kit following manufacturer instructions. In the examples where the oligonucleotide pairs are long (pair 5/6) the dimeric oligo duplexes approach 100 bp and are less efficiently removed.
Following the initial ligation of a heteroduplex DNA onto each end of pET11aΔH, the purified product was digested with EcoRI (2 U/µg) for 1 h at 37°C. Again, the High Pure kit was used to remove the protein components and the short DNA fragment generated in the reaction (28 bp plus the length of the duplex oligo after DpnII digestion). In the case of substrates made with oligo pair 5/6 or 9/10, the digestion product was further purified using size exclusion chromatography by passage over Sephacryl S500 resin (Amersham Phamacia) in order to reliably remove DNA fragments up to ∼400 bp. The sample was loaded in a minimal volume (≤100 µl) and applied to a 1.1 ml column prepared in 1 ml graduated, disposable plastic pipette (Falcon). The resin was equilibrated and the DNA eluted in TE buffer containing 100 mM NaCl. The column bed was ∼20 cm, supported by a small plug of silanized glass wool (Supelco). When the distance separating the two key restriction sites was up to 400 bp, such as pBR322 or unmodified pET11a, the S500 chromatography was essential for removing the intervening DNA.
A final ligation, which creates the nicked circular substrate, was carried out at a DNA concentration of 10 ng/µl in T4 ligase buffer supplemented with 50 mM NaCl, EcoRI (2 U/µg plasmid), T4 ligase (100 U/µg plasmid) and 100 µg/ml bovine serum albumin. Ligations were incubated at 20°C for 3 h, since no further ligation was apparent during longer incubations (data not shown). The substrate was recovered by ethanol precipitation in the presence of 300 mM sodium acetate using a Beckman ultracentrifuge and a SW41 swinging bucket rotor at 20 000 r.p.m. (68 500 g) for 30 min. The pellets were washed with 80% ethanol and resuspended in 100 µl of TE. Linear molecules, including unreacted monomeric DNA, linear dimers and multimers, were removed in a 1 h digestion at 37°C using 0.25 U/µg DNA of Exonuclease V from Micrococcus leuteus as described by the manufacturer, except that 10 mM MgCl2 was used and β-mercaptoethanol was omitted from the buffer. The enzyme was inactivated using a 10 min incubation at 65°C, and the buffer, nucleotide monophosphates and small DNA fragments were removed by centrifugation through a Microcon YM-100 spin column (Amicon) followed by two 500 µl TE washes.
The purity and concentration of the substrates were established by running small samples on a 1% agarose gel, staining with ethidium bromide and comparing the product band with known amounts of pET11aΔH. The final yield ranged from 10 to 20% (1.5–3 µg nicked circular heteroduplex DNA), where the shorter heteroduplex oligos gave better yields. On a larger scale, where 60 µg of plasmid DNA was carried through the above protocol by dividing each High Pure purification step into four aliquots, the final yield for the T·G-10H substrate was 7% (4.2 µg product DNA).
Sequencing reactions to establish the identity of plasmid pET11aΔH and the product heteroduplexes were performed using an ABI (Applied Biosystems) sequencing kit. Products of the sequencing reactions were purified using a centrifugation column containing G-50 resin (Edge Biosystems) prior to sequence analysis. The oligonucleotide 5′-CGAGATCTCGATCCCGCG was chosen to anneal to the strand that ultimately harbors the nick in the inserted oligo heteroduplex. This allows for 134 bp of the plasmid sequence to be read prior to encountering the BamHI site. In the case of the pET11aΔH construct, sequencing verified the blunt end ligation described above. When nicked heteroduplexes were tested, the sequence revealed inclusion of the single duplex product that ended abruptly at the nick site, plus an unpaired adenine base added by the sequencing polymerase.
MR assays
MR assays were performed as described by Holmes et al. (7), with the following modifications. Mispaired substrate (80 ng, or 22.5 fmol) was incubated with 45 µg of HeLa nuclear extract at 37°C for 20 min, where the final concentration of KCl was adjusted to 110 mM. For repair assays using nuclear extracts derived from LoVo cells, 50 µg of protein were used, in the presence or absence of 100 ng of purified MutSα (25).
In order to assess site-specific mismatch correction using restriction enzymes, two enzymes are normally used (10,26). One enzyme digests at the mismatch site only after mismatch correction, but all molecules are linearized so that mismatch correction results in two characteristic DNA fragments. StyI represents a unique site in pET11aΔH that is useful as the linearizing enzyme, and 2 U per assay (in Buffer 2 from New England Biolabs) was sufficient to cut all of the isolated plasmid. In the same digestion reaction, sensitivity at the mismatch site to either HindIII or XhoI (1 U per assay), depending on the mismatch, yielded two fragments that were used to determine the extent of MR. Alternatively, because AvaI digests both at a unique site remote from the mismatch and its recognition sequence overlaps with XhoI, it can be used as the sole enzyme to assess mismatch correction when excision and resynthesis regenerates an XhoI site.
The DNA digestion products used to assess MR were separated on a 1% agarose gel (15 cm) in 1× TAE for 210 V h, stained with ethidium bromide and the image captured and inverted using a ChemiImager 4000 cooled CCD camera (Alpha Innotech). The NIH Image software, version 1.62, was used to determine the relative percentage of DNA fragments in a given lane. These values were used to calculate the extent of MR for each experiment in femtomoles, assuming that the recovered DNA molecules are representative of the population entering the extract.
Cell lines and nuclear extract preparation
HeLa and LoVo cells were obtained from the National Cell Culture Center (Minneapolis, MN), and nuclear extracts were prepared as previously described by Holmes et al. (7). Protein concentrations were determined using a Bradford assay (Bio-Rad) using BSA as the standard. Human MutSα was purified from HeLa cells as described by Drummond et al. (25).
RESULTS
Strategy for controlling nick and mismatch placement
The ability to assay mismatch correction in vitro depends on the presence of both a site-specific mismatch and a strand-specific nick. Therefore, a versatile strategy for substrate construction depends upon the ability to control these parameters. In our approach, a carefully designed oligonucleotide duplex determines each targeted feature (Fig. 1). The mismatch site is set by sequence choice, and is normally arranged within overlapping restriction sites that allow for scoring of base correction in either strand (7,26). The number of base pairs intervening between the mismatch and the hydroxylated 5′-extension sets the separation between the mismatch and the repair-directing nick. From a practical standpoint, this distance is limited by the inability to faithfully synthesize longer oligonucleotides. The nick site itself is formed because the 5′-OH moiety fails to participate in the final ligation step of the protocol, as described below.
Figure 1.

Essential features in oligonucleotide design. Oligonucleotide pairs were chosen such that three key features are generated upon annealing. First, a site-specific mismatch is embedded within overlapping restriction site sequences that allows for assessment of strand-specific mismatch correction (10). Secondly, a restriction site (DpnII in this example) is embedded close to one end of the duplex so that digestion leaves a 5′-phosphorylated overhang compatible with the template plasmid. Thirdly, the opposite oligo duplex end presents a 5′-OH that sets the distance (n base pairs) between the mismatch and the strand-specific nick that directs mismatch correction. The digested molecules (lane 2) efficiently dimerize upon ligase treatment (lane 4), but do not form higher-order multimers.
The next essential design element requires generation of a phosphorylated DNA extension with high fidelity. This is critical because the ultimate success of this strategy requires three efficient strand joining reactions. A 5′-GATC overhang is used in the prototype strategy presented here, which will be used to attach the oligo to a complementary overhang on the targeted plasmid by ligation in both DNA strands. Enzymatic digestion of oligonucleotides using DpnII (Fig. 1) proved to be more reliable than attempting to quantitatively phosphorylate a comparable synthetic oligonucleotide (data not shown). Only correctly annealed sequences are susceptible to digestion, and the purified fragments participate reliably in dimerization reactions without forming multimers.
Our overall strategy for constructing circular heteroduplex molecules is given in Figure 2. It depends upon the ability to directionally introduce a designed heteroduplex oligonucleotide, described above, into a target plasmid molecule. Any plasmid that affords two reliable, unique restriction sites within ∼400 bp may be used, but the approach can be simplified if the two sites are within ∼50 bp. Therefore, most plasmids with multiple cloning sites are reasonable choices. However, because the assay for MR used here depends on ethidium bromide staining of the template and digestion products after recovery from human cell nuclear extracts, a larger DNA molecule (>5 kb) facilitates staining and quantifying bands. Less DNA is required, and the fragments diagnostic for repair are not obscured by RNA carried through from the extract that escapes RNase treatment. For this work, we constructed a derivative of pET11a by removing a small, non-essential portion of the plasmid to yield a 5.4 kb plasmid we called pET11aΔH (Materials and Methods). This design brings the two primary digestion sites (BamHI and EcoRI) within 28 bp to facilitate substrate preparation.
Figure 2.

Strategy for constructing nicked heteroduplexes. A mismatch-containing oligonucleotide duplex (Fig. 1) is ligated into a template plasmid molecule (1). Linearization of the plasmid (2) in the presence of the heteroduplex oligo, T4 ligase and restriction enzyme (BamHI) allows ligation of the small fragments onto each DNA end as a dead-end complex (3), because the BamHI site is eliminated. Re-ligation of BamHI-generated plasmid ends yields a molecule competent for a second digestion, returning them to the substrate pool. In the next step, digestion with EcoRI removes one ligation product and generates a ligation-competent DNA end (4). After removal of the smaller fragment, an intramolecular ligation reaction generates the nicked circular product (5). Unwanted linear molecules are removed by digestion with Exonuclease V (Materials and Methods).
Scheme for the construction of mismatch- or lesion-containing substrates
The sequence of events required for heteroduplex construction is given in Figure 2, and the efficiency of these steps is reflected in Figure 3. The acceptor plasmid (pBR322 in this example) is first cut with BamHI in the presence of a 100-fold molar excess of the heteroduplex oligonucleotide ends described above, T4 ligase and ATP at 16–20°C. Because these oligos contain a phosphorylated 5′-GATC overhang complementary to the BamHI-generated ends, several ligation outcomes are possible. The desired and ultimate outcome for most plasmid molecules is the ligation of an oligo duplex onto each free end (Fig. 2, structure 3 and Fig. 3, lane 3). This is a dead-end reaction because ligation eliminates the BamHI site. When two plasmid ends ligate into linear multimers or circular products, they regenerate the BamHI site. Such products can be returned for another attempt at ligation with an oligonucleotide, because the BamHI retains partial activity under the conditions of the ligation. In the absence of oligonucleotides, the primary product is a linear, monomeric plasmid (data not shown). When the heteroduplex oligos are ligated together, they also form a dead-end dimeric product (Fig. 1).
Figure 3.
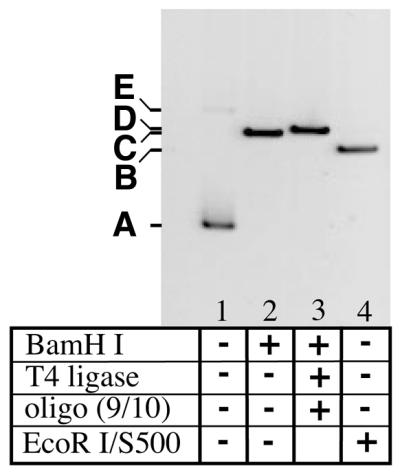
Intermediates in the ligation-based protocol. The plasmid pBR322 was used to illustrate the individual steps in the protocol for two reasons. Its smaller size (4361 bp) makes it easier to see small changes in molecular weight, and the large distance separating EcoRI and BamHI (377 bp) is useful to illustrate the excision of such a fragment. Lane 1, 100 ng pBR322, which is primarily supercoiled (band A) after CsCl banding but contains a small amount of nicked circle (band E). Lane 2, treatment with 2 U BamHI/µg DNA yields a linear plasmid (band B). Lane 3, addition of a 40-fold excess of heteroduplex oligo ends and T4 DNA ligase (100 U/µg plasmid DNA) yields band D. Lane 4, digestion with EcoRI, followed by size-exclusion chromatography on Sephacryl S500 resin (band C). An equivalent fraction of the total volume from each step was loaded, so that the relative staining intensities reflect relative yield.
The second step requires removal of one plasmid end bearing a heteroduplex oligonucleotide and concomitant generation of a ligation-competent DNA end appropriate for cyclization. In the constructs reported here, the heteroduplex oligo was designed to have a 5′-(HO)AATT overhang complementary to that generated by digestion at the unique EcoRI site. The smaller DNA fragment containing the second and unwanted heteroduplex site was removed using the High Pure resin binding step in cases where the excised fragment was small (e.g. pET11aΔH). In cases where the heteroduplex oligo incorporated was large (≥40 bp), a size exclusion chromatography step (Sephacryl S500) was required to facilitate the removal of larger DNA fragments (Fig. 3). These include both digestion products and persistent dimerized duplexes from the original ligation. In cases where the separation between the two restriction sites was larger, such as in pBR322 (375 bp), the size exclusion chromatography step was essential (Fig. 3).
A final ligation reaction allows the modified plasmid, with complementary EcoRI overhangs, to circularize and create a unique nick site (Figs 2 and 4). The nick is generated because the 5′-AATT overhang originally present in the oligonucleotide heteroduplex lacks a 5′ phosphate and cannot be sealed by ligation. The complementary, intramolecular EcoRI end generated by plasmid digestion contains a 5′-phosphate and allows the molecules to circularize. By performing the ligation at low DNA concentrations in the presence of EcoRI, the formation of the nicked circular species by an intramolecular ligation is favored (Fig. 4). Multimeric species that reconstitute EcoRI sites can be re-digested, whereas nicked circles or linear multimers involving the 5′-AATT overhang contributed by the heteroduplex oligo represent dead-end products because the EcoRI site is destroyed. It should be re-emphasized that any surviving oligonucleotide-based DNA fragments, such as dimers formed in the primary ligation, present 5′-(HO)AATT overhangs and can fatally inhibit the ring closure step.
Figure 4.

Optimization of the ring closure ligation. A modified pET11aΔH intermediate (Fig. 1, structure 4) was incubated with T4 DNA ligase using DNA concentrations decreasing from 100 to 10 ng/µl (see Materials and Methods for conditions). This LS has one DNA end generated by EcoRI digestion and one contributed by the T·G-10H oligonucleotide heteroduplex. The supercoiled (SC) pET11aΔH plasmid loaded in the first and last lanes contains a small amount of NC molecules that serves as a close marker for the mobility of the desired product. At 100 ng/µl DNA, the predominant products are multimers that migrate more slowly. At 10 ng/µl template, the desired NC heteroduplex represents up to 30% of the products, and the formation of linear multimers is reduced. For each lane representing a sample of a ligation reaction, an equivalent sample was treated with Exonuclease V, which degrades linear molecules and reveals the circular products. Note that a trace amount of CD is formed in this experiment.
Figure 4 examines the dependence of the final ligation on the DNA concentration. At 100 µg/ml, most of the linear substrate (LS) is taken up in linear multimeric species. Intramolecular reactions that give a nicked circular product (NC) are favored at the most dilute concentration shown in Figure 4 (10 µg/ml), but a larger fraction of the starting material persists. More dilute conditions did not substantially improve the product yield, and DNA recovery was rendered more difficult. The ligation reactions appear complete after 3 h at 20°C, and further addition of ligase does not significantly affect the outcome (data not shown). This suggests that the surviving DNA ends are unreactive, either due to the loss of the 5′-phosphate or because an undesired heteroduplex oligo monomer or dimer survived to the final step and ligated to the plasmid end. It should also be noted that the product mixture often contains a detectable amount of a circular dimeric product (CD in Fig. 4), in yields up to ∼3% of the exonuclease-resistant products. These molecules effectively represent a template with two sites that can be scored for MR activity. We were unable to resolve whether this small population of molecules contributed to the overall extent of mismatch correction.
In order to isolate the desired product, the dilute ligation reaction was concentrated by ethanol precipitation and the unwanted linear molecules were selectively digested by treatment with Exonuclease V (Fig. 4). This reaction distinguishes linear products, which can be digested, from the resistant circular products. The dNMPs, small oligonucleotide products and buffer were then removed by repeated ultrafiltration and buffer exchange. This yields a heteroduplex with a site- and strand-specific nick without any steps that require gel purification. We have characterized this approach for small-scale synthesis (15–60 µg starting material have been tested) in overall yields of 7–20%. However, there is no inherent reason prohibiting larger scale syntheses.
Mismatch correction is nick directed
In order to demonstrate that cell free extracts correct the mispaired substrates, we assayed them using nuclear extracts prepared from HeLa cells (7). Such extracts have been shown to repair approximately half of the circular substrates containing a single-strand nick 125 bp removed from a G·T mismatch when 24 fmol (100 ng) was assayed (7). As in our substrates, the polarity of the DNA strand reading from nick to mismatch was 5′→3′. The unrepaired molecules apparently succumb to a competing ligation reaction that seals the nick and precludes further repair (9; K.Iams and J.T.Drummond, unpublished data). As previously described by Holmes et al. (7), substrates were introduced into HeLa nuclear extracts, recovered by phenol extraction and precipitation, and the extent of repair directed toward either strand quantitated by restriction analysis.
Figure 5 shows the results of a MR assay on T·G-10H (a G·T mismatch with a nick placed 10 bp 5′ from the mismatch in the G-containing strand) and G·T-10X (a G·T mismatch with a similar nick in the T-containing strand). When T·G-10H is the substrate (lanes 1–4), nick-directed repair is predicted to remove the bases surrounding the G, and strand resynthesis will restore a TA base pair and HindIII sensitivity. When G·T-10X is the substrate (lanes 5–8), nick-directed repair and resynthesis will generate a GC base pair and XhoI sensitivity. For each template analyzed, the substrates were linearized, and the two smaller fragments (3032 and 2398 bp) represent the products of site-specific mismatch correction. Neither scoring enzyme (HindIII or XhoI) appreciably digested at the mismatch site without extract treatment (see the odd numbered lanes). Following HeLa exposure, the base present in the nicked strand was selectively removed in up to 50% of the substrate molecules (lanes 2 and 6). Mismatch removal and resynthesis in the strands without the nick (lanes 4 and 8) occurred in ∼4% (1 fmol) of the substrate molecules shown here, but was not always detectable.
Figure 5.

Strand-specific processing of G·T mismatches. Heteroduplexes (80 ng, 22.5 fmol) containing a G·T mismatch and a 5′-nick placed 10 bp away in either strand were assayed for mismatch correction in each strand. All molecules were linearized with StyI or AvaI, which cuts at a site remote from the mismatch (not shown). In the absence of HeLa treatment (odd numbered lanes) neither scoring enzyme (HindIII or XhoI; refer to the sequence information above each gel) substantially digested at the mismatch site; linear molecules are primarily observed. When the substrates were incubated with HeLa nuclear extract for 20 min under assay conditions (Materials and Methods), the base located in the nicked strand (indicated by double lines in the sequence above the gels) was partially excised and replaced. Resynthesis through this site restores HindIII or XhoI sensitivity, depending on the substrate (lanes 2 and 6) that was used to quantitate strand-specific MR (7,10). For the T·G-10H substrate (left), 35% (7.7 fmol) of the DNA is repaired, whereas for the G·T-10X, 50% of the molecules (11.3 fmol) are repaired. The putatively continuous strand remains largely insensitive to such treatment (lanes 4 and 8).
The mismatch correction described here has other hallmarks of previously characterized MR using HeLa nuclear extracts. Most importantly, the ratio of base removal in the nicked strand compared with the putatively continuous strand ranged from 7.7:1 to 10:1. This strand specificity is consistent with previously reported ratios (7) and may underestimate the true strand bias, because no correction was made for the small degree of digestion in the absence of HeLa treatment. The observed strand bias also supports one premise of the construction strategy, i.e. two intermolecular ligations must occur in the primary joining reaction between plasmid and oligo to yield a continuous junction.
In addition to the strand bias, all of the mismatch processing occurs within the 20 min time frame of the assay. No further mismatch correction in either strand is detectable over several hours. This shows that the observed DNA excision and resynthesis events depend upon the presence of the nick, which can be sealed by ligase activity present in the nuclear extract and terminate MR (9). The mixture of repaired and unrepaired substrates recovered from HeLa extract treatments in this work were found to be predominantly covalently closed, full-length molecules, as judged by denaturing gel electrophoresis (K.Iams and J.T.Drummond, unpublished observation).
Dependence of mismatch processing on MutSα (MSH2/MSH6)
One new characteristic of the substrates constructed by this ligation-based approach is that the separation between nick and mismatch is reduced, and is now 10–40 bp. Traditionally, MR activity is assayed on substrates where the separation between nick and mismatch is ≥125 bp. The ‘long patch’ capability is a hallmark of the pathway, where base–base mismatch correction events depend upon the presence of a functional MSH2/MSH6 heterodimer (MutSα). To determine whether the observed repair is MutSα-dependent, we repeated the assays using nuclear extracts derived from LoVo cells. LoVo is defective in repair of base–base mismatches (among others) due to a partial deletion of the MSH2 gene (27). No MSH2 protein is observed in LoVo extracts and the MSH6 partner is apparently proteolyzed, but MR activity can be restored to LoVo by complementation with MutSα in vitro (25). The dependence of mismatch correction on added MutSα was tested for G·T heteroduplexes where the mismatch and repair-directing strand break were positioned 10, 40 or 141 bp apart.
As shown in Figure 6, we observed that a fraction of the repair events were independent of MutSα, and that increasing the distance separating mismatch and nick increased the requirement for MutSα. When the separation was 141 bp, in a substrate prepared using traditional methodology (7), LoVo extracts failed to correct the mismatch without added MutSα (Fig. 6). When the separation was 40 bp (T·G-40H), >80% of the repair events depended upon added MutSα. In these assays, MutSα-independent repair becomes difficult to detect, and we use and recommend this distance for a standard assay. When the distance was reduced to 10 bp, a substantial fraction of mismatch removal and resynthesis events (35%) were MutSα independent. However, in all cases the repair events are dependent upon the presence of the single-strand nick and conclude within the 20 min assay time course.
Figure 6.

Dependence of repair events on the distance separating nick and mismatch. Each lane represents a repair assay performed with 50 µg of nuclear extract from the LoVo cell line, defective in hMSH2 and MR activity (25). Where indicated, 300 ng of purified MutSα (MSH2/MSH6) was added (even numbered lanes). The three substrates used were: T·G-10H, T·G-40H and a derivative of f1MR1 containing a T·G mismatch (7) with the same nick orientation (5′) so that HindIII could be used to assess mismatch correction in each case.
It should be noted that the relative assignment of MutSα dependence as a function of separation between nick and mismatch presumes that the two classes of repair events occur independently. It is plausible that, when MutSα is added to LoVo extracts, all the mismatch correction events now proceed via the MutSα-dependent pathway. We were unable to distinguish the two classes of repair when MutSα is present, such as in MR-proficient extracts derived from HeLa. We suggest that heteroduplex substrates prepared using this approach, where the nick and mismatch are 40 bp apart, are appropriate for assays to characterize MR activity, so long as the caveat is maintained that a small fraction of the mismatch correction events might represent a competing nick-directed process.
Disrupting interactions with PCNA can inhibit proximal nick-directed repair events
The observation that MutSα-independent mismatch processing increases as the repair-directing nick is moved toward the mismatch suggests the possibility that a mismatch-independent and nick-directed excision simply removes one base partner of mismatch. It is also possible that the MutSα-independent mismatch correction falls outside the traditional ‘long patch’ pathway and represents excision and resynthesis of a smaller tract size. In principle, such a ‘short patch’ repair event could be independent of PCNA activity. Therefore, we asked whether the MutSα-independent repair events required functional PCNA, which is essential for mismatch correction at an unspecified step prior to tract excision as well as for long-patch resynthesis (3,4). The apparent requirement for DNA polymerase δ (28) in the large tract resynthesis during MR is consistent with a role for PCNA at this step.
In order to characterize the contribution of PCNA to the repair events we monitored, we performed assays in the presence and absence of a peptide derived from p21 called PCNA-binding protein (PBP) (3,22). Both p21 protein and PBP have been shown to interfere with PCNA function. As previously reported by Umar et al. (27), we found that 90 µM PBP was sufficient to inhibit all detectable MR activity present in HeLa nuclear extracts when the repair-directing nick was 328 bp away from the mismatch (data not shown). This inhibition was abolished when the peptide, which is rich in cationic amino acids, was treated with trypsin and the protease activity quenched with either stoichiometric soybean trypsin inhibitor or by heat inactivation.
When the MutSα-independent repair activity found in LoVo was challenged with 90 µM PBP, mismatch correction was blocked (Fig. 7). Similarly, all MutSα-dependent repair events were inhibited by this treatment (compare lanes 4 and 5). A parallel result was obtained when the salt concentration of the assay was raised to 225 mM, which is sufficient to inhibit the ‘long patch’ MR events and human polymerase δ under defined conditions (28), but is less likely to inhibit the gap filling activity of polymerase β (29). In summary, all the mismatch excision events we observe appear to be PCNA dependent, consistent with mismatch correction when the nick and mismatch are widely separated. We are unable to distinguish the specific site of inhibition with these tools, since PCNA has been implicated at more than one stage in mismatch correction. Specifically, the MutSα-dependent repair can be inhibited at a step prior to tract excision in the presence of PBP (27), so inhibition of MR does not necessarily reflect disruption of PCNA’s contribution to long-patch replication.
Figure 7.

Nick-directed repair events are inhibited by PBP. Each lane represents an assay using LoVo nuclear extracts. In the absence of added MutSα, a low level of mismatch correction is observed (lane 1). This processing was inhibited by 225 mM KCl (lane 2) and by 90 µM PBP derived from p21 (PBP, lane 3). PBP also inhibited the MutSα-dependent repair events that occurred under standard assay conditions where the extract was complemented with MutSα (lane 4). For comparison, lane 5 shows the amount of repair (44%, or 9.9 fmol) that occurs in the absence of added PBP.
DISCUSSION
In this paper we present a straightforward protocol for constructing DNA heteroduplexes that are competent to serve as substrates in an assay for human MR. Such substrates are most commonly prepared by denaturing linear double-stranded phage DNA and allowing them to anneal to an excess of a circular single-stranded molecule that differ in sequence only within the bases comprising the mismatch. The annealing process introduces a nick at the site where the duplex was linearized. In our approach, we introduce a short heteroduplex DNA into a targeted plasmid by sequential, directional ligation steps. The reactions are carried out in an overall yield of 7–20%, and the procedure may be completed within 2 days using commercially available reagents. In our hands, the yield is comparable to that obtained using the phage-based methodology described above (10).
In addition to the ease of preparation, the possibilities for sequence content of mismatch-containing templates are diverse. Because the mismatches are constructed by annealing two oligonucleotides, any base–base mismatch is immediately accessible, as are base insertions. We have applied this strategy successfully to G·T, G·G, G·A and A·C heteroduplexes (data not shown), although construction of other mismatches or base insertions has not been attempted. Similarly, the distance separating the mismatch and nick is also readily controlled by oligonucleotide design. Greater distances appear to suppress the MutSα-independent processing of mismatches, although in our experience the efficiency of the final ring closure reaction diminishes. Finally, the sequence content around each critical site is readily controlled, which represents a variable in eukaryotic MR that remains unexplored in a systematic manner.
Although only substrates containing DNA mismatches composed of the four standard bases (G, A, T and C) are described here, the real value of this approach may lie in incorporating site-specific DNA lesions or base analogs. For example, processing of DNA lesions by the MR pathway has been implicated for specific heterologies (17,30) that resemble mismatches in an undefined manner. Lesions such as O6-methylguanine and cisplatin covalently attached to adjacent guanines are recognized by human MutSα, and excision of O6-methylguanine has been demonstrated in one case (20). Lesion- and MR-dependent synthesis has also been shown following plasmid treatment with O6-methylguanine (31). We have successfully prepared heteroduplexes containing 8-oxoguanine in each strand and tested these lesions as templates for the human MR pathway (E.D.Larson and J.T.Drummond, unpublished observation). We suggest that any DNA lesion that can be incorporated into an oligonucleotide sequence during its synthesis, or added specifically to an oligonucleotide in a chemical reaction, can be incorporated into a template to determine whether it can be processed by the human MR pathway. The only requirements are that the lesion must remain indifferent to nearby enzymatic digestion or ligation reactions, and that a method exists to assess mismatch processing.
Although we report here a protocol that is reliable in our hands, there are many strategic variations for substrate construction that remain untried. For example, we relied on 5′-GATC (BamHI) and 5′-AATT (EcoRI) extensions for ligation events, since they may be generated reliably by enzymes that do not possess substantial phosphatase or exonuclease activity. We did not test a broad range of restriction enzymes, although alternatives such as the 3′-CATG overhang generated by KpnI digestion was equally effective in the final ligation step and substrate construction (data not shown). Additionally, all of the substrates presented in this work contained a nick where the strand polarity from nick to mismatch reads 5′→3′. It has been shown that human DNA MR has a bidirectional capability (9), and it is equally capable of accessing DNA nicks with a 3′→5′ polarity with respect to the mismatch. Construction of such heterologies with this approach requires that, in the final circularization reaction, the strand bearing the 5′-OH must be contributed by the plasmid instead of the oligonucleotide. We have not pursued this extension because we have found no detectable differences in the efficiency of repair of 5′- versus 3′-nicked substrates prepared by traditional methodologies in a similar sequence context (data not shown).
Using heteroduplex templates prepared using the methodology described here, we found that mismatch correction was dependent upon the presence of the nick and displayed other characteristics of the ‘long patch’ MR pathway. However, in the absence of mismatch recognition activity (in the LoVo nuclear extracts), a fraction of the molecules (∼6%) were apparently ‘repaired’ in a nick-directed fashion. It is reasonable but not safe to assume that when the entire MR machinery is present, it competes with and may suppress this processing. When interpreting the extent of nick-directed mismatch correction using restriction analysis, it is important to recognize that a small fraction of the ‘repair’ events might not represent processing by the MR pathway. This potential for MutSα-independent events diminishes as the distance separating the nick and mismatch increases, and maximizing this distance is recommended.
This work illustrates two capabilities of the human MR pathway that, to the best of our knowledge, have not been reported. First, nicked substrates prepared by the traditional denaturation/reannealing methodology can be covalently closed by ligation, and therefore possess a 5′-phosphate. By definition, our substrates possess a 5′-OH at the nick site, which is processed by nuclear extracts to a comparable extent. This suggests that when DNA nicks are present as a strand signal, the phosphorylation state does not contribute substantially to the efficiency of repair. Secondly, mismatch correction occurs primarily by a MutSα- and PCNA-dependent pathway, even when the mismatch and repair-directing nick are only 10 bp apart. We have no proof that this represents precisely the same protein machinery as when the separation is much larger, but no distinguishing characteristics have yet been found. As previously noted (32), the proximity of nick and mismatch represents a physical situation where the mismatch binding heterodimer alone effectively spans both sites, based on a DNase I footprint analysis of mismatch binding by MSH2/MSH6 (33). This represents a setting where it is less clear how mismatch recognition might be coupled to downstream repair events that depend on accessing the nick as a strand discrimination signal.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to recognize the excellent cell culture service provided by the National Cell Culture Center (Minneapolis, MN). In addition, we would like to thank the NIH Genetics, Cellular and Molecular Sciences Training Grant GM 07757-21 for supporting E.D.L., the American Cancer Society (84-002-16-IRG) and the National Institutes of Health (RO1-CA79906) for supporting this research.
REFERENCES
- 1.Buermeyer A.B., Deschenes,S.M., Baker,S.M. and Liskay,R.M. (1999) Mammalian DNA mismatch repair. Annu. Rev. Genet., 33, 533–564. [DOI] [PubMed] [Google Scholar]
- 2.Kolodner R.D. and Marsischky,G.T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 3.Umar A., Buermeyer,A.B., Simon,J.A., Thomas,D.C., Clark,A.B., Liskay,R.M. and Kunkel,T.A. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell, 87, 65–73. [DOI] [PubMed] [Google Scholar]
- 4.Gu L., Hong,Y., McCulloch,S., Watanabe,H. and Li,G.M. (1998) ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res., 26, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleczkowska H.E., Marra,G., Lettieri,T. and Jiricny,J. (2001) hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev., 15, 724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas D.C., Roberts,J.D. and Kunkel,T.A. (1991) Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem., 266, 3744–3751. [PubMed] [Google Scholar]
- 7.Holmes J. Jr, Clark,S. and Modrich,P. (1990) Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl Acad. Sci. USA, 87, 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lahue R.S. and Modrich,P. (1988) Methyl-directed DNA mismatch repair in Escherichia coli. Mutat. Res., 198, 37–43. [DOI] [PubMed] [Google Scholar]
- 9.Fang W.H. and Modrich,P. (1993) Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem., 268, 11838–11844. [PubMed] [Google Scholar]
- 10.Su S.S., Lahue,R.S., Au,K.G. and Modrich,P. (1988) Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem., 263, 6829–6835. [PubMed] [Google Scholar]
- 11.Wang H. and Hays,J.B. (2000) Preparation of DNA substrates for in vitro mismatch repair. Mol. Biotechnol., 15, 97–104. [DOI] [PubMed] [Google Scholar]
- 12.Duckett D.R., Drummond,J.T., Murchie,A.I., Reardon,J.T., Sancar,A., Lilley,D.M. and Modrich,P. (1996) Human MutSα recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl Acad. Sci. USA, 93, 6443–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mello J.A., Acharya,S., Fishel,R. and Essigmann,J.M. (1996) The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem. Biol., 3, 579–589. [DOI] [PubMed] [Google Scholar]
- 14.Yamada M., O’Regan,E., Brown,R. and Karran,P. (1997) Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res., 25, 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li G.M., Wang,H. and Romano,L.J. (1996) Human MutSα specifically binds to DNA containing aminofluorene and acetylaminofluorene adducts. J. Biol. Chem., 271, 24084–24088. [PubMed] [Google Scholar]
- 16.Wang H., Lawrence,C.W., Li,G.M. and Hays,J.B. (1999) Specific binding of human MSH2.MSH6 mismatch-repair protein heterodimers to DNA incorporating thymine- or uracil-containing UV light photoproducts opposite mismatched bases. J. Biol. Chem., 274, 16894–16900. [DOI] [PubMed] [Google Scholar]
- 17.Fink D., Aebi,S. and Howell,S.B. (1998) The role of DNA mismatch repair in drug resistance. Clin. Cancer Res., 4, 1–6. [PubMed] [Google Scholar]
- 18.Bignami M., O’Driscoll,M., Aquilina,G. and Karran,P. (2000) Unmasking a killer: DNA O(6)-methylguanine and the cytotoxicity of methylating agents. Mutat. Res., 462, 71–82. [DOI] [PubMed] [Google Scholar]
- 19.Li G.M. (1999) The role of mismatch repair in DNA damage-induced apoptosis. Oncol. Res., 11, 393–400. [PubMed] [Google Scholar]
- 20.Duckett D.R., Bronstein,S.M., Taya,Y. and Modrich,P. (1999) hMutSα- and hMutα-dependent phosphorylation of p53 in response to DNA methylator damage. Proc. Natl Acad. Sci. USA, 96, 12384–12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J., Gu,L., Wang,H., Geacintov,N.E. and Li,G.M. (1999) Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis. Mol. Cell. Biol., 19, 8292–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warbrick E., Lane,D.P., Glover,D.M. and Cox,L.S. (1995) A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr. Biol., 5, 275–282. [DOI] [PubMed] [Google Scholar]
- 23.Gill S.C. and von Hippel,P.H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem., 182, 319–326. [DOI] [PubMed] [Google Scholar]
- 24.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982)Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Drummond J.T., Li,G.M., Longley,M.J. and Modrich,P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 26.Parsons R., Li,G.M., Longley,M.J., Fang,W.H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 27.Umar A., Boyer,J.C., Thomas,D.C., Nguyen,D.C., Risinger,J.I., Boyd,J., Ionov,Y., Perucho,M. and Kunkel,T.A. (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem., 269, 14367–14370. [PubMed] [Google Scholar]
- 28.Longley M.J., Pierce,A.J. and Modrich,P. (1997) DNA polymerase δ is required for human mismatch repair in vitro. J. Biol. Chem., 272, 10917–10921. [DOI] [PubMed] [Google Scholar]
- 29.Wang T.S., Sedwick,W.D. and Korn,D. (1975) Nuclear deoxyribonucleic acid polymerase. Further observations on the structure and properties of the enzyme from human KB cells. J. Biol. Chem., 250, 7040–7044. [PubMed] [Google Scholar]
- 30.Drummond J.T., Anthoney,A., Brown,R. and Modrich,P. (1996) Cisplatin and adriamycin resistance are associated with MutLα and mismatch repair deficiency in an ovarian tumor cell line. J. Biol. Chem., 271, 19645–19648. [DOI] [PubMed] [Google Scholar]
- 31.Ceccotti S., Aquilina,G., Macpherson,P., Yamada,M., Karran,P. and Bignami,M. (1996) Processing of O6-methylguanine by mismatch correction in human cell extracts. Curr. Biol., 6, 1528–1531. [DOI] [PubMed] [Google Scholar]
- 32.Larson E.D. and Drummond,J.T. (2001) Human mismatch repair and G/T mismatch binding by hMutSαin vitro is inhibited by adriamycin, actinomycin D and nogalamycin. J. Biol. Chem., 276, 9775–9783. [DOI] [PubMed] [Google Scholar]
- 33.Gradia S., Acharya,S. and Fishel,R. (1997) The human mismatch recognition complex hMSH2–hMSH6 functions as a novel molecular switch. Cell, 91, 995–1005. [DOI] [PubMed] [Google Scholar]