

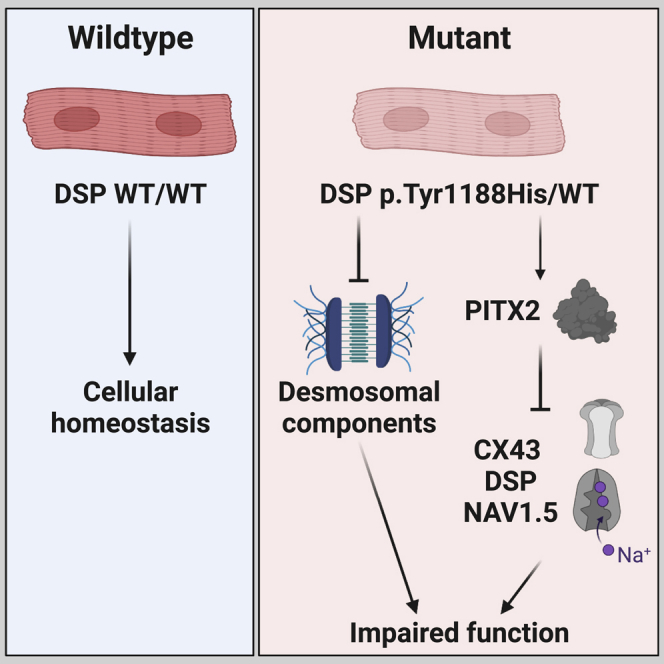
Summary
Arrhythmogenic cardiomyopathy (ACM) is an inherited progressive disease characterized by electrophysiological and structural remodeling of the ventricles. However, the disease-causing molecular pathways, as a consequence of desmosomal mutations, are poorly understood. Here, we identified a novel missense mutation within desmoplakin in a patient clinically diagnosed with ACM. Using CRISPR-Cas9, we corrected this mutation in patient-derived human induced pluripotent stem cells (hiPSCs) and generated an independent knockin hiPSC line carrying the same mutation. Mutant cardiomyocytes displayed a decline in connexin 43, NaV1.5, and desmosomal proteins, which was accompanied by a prolonged action potential duration. Interestingly, paired-like homeodomain 2 (PITX2), a transcription factor that acts a repressor of connexin 43, NaV1.5, and desmoplakin, was induced in mutant cardiomyocytes. We validated these results in control cardiomyocytes in which PITX2 was either depleted or overexpressed. Importantly, knockdown of PITX2 in patient-derived cardiomyocytes is sufficient to restore the levels of desmoplakin, connexin 43, and NaV1.5.
Keywords: arrhythmogenic cardiomyopathy, induced pluripotent stem cells, PITX2, desmoplakin, desmosome, cardiomyocyte, function
Graphical abstract
Highlights
-
•
The p.Tyr1188His mutation in desmoplakin drives arrhythmogenic cardiomyopathy
-
•
Mutant cardiomyocytes show a global decline in intercalated disc proteins
-
•
Mutant cardiomyocytes show induced levels of the transcriptional repressor PITX2
-
•
Suppression of PITX2 restores expression of desmoplakin, connexin 43, and NaV1.5
Here, van Kampen and colleagues investigated the consequences of a novel missense mutation in desmoplakin, identified in a patient diagnosed with arrhythmogenic cardiomyopathy. The authors observed reduced levels of intercalated disc proteins and a prolongation of the action potential duration in mutant cardiomyocytes. Furthermore, they identified aberrant PITX2 signaling as potential disease characteristic, as knockdown could restore the molecular phenotype.
Introduction
Desmosomes are essential multiprotein complexes localized at the intercalated disc, physically connecting the intermediate filament (IF) networks of neighboring cardiomyocytes (Vermij et al., 2017). Besides providing resilience against mechanical forces during cardiac contraction, the desmosomes also play a crucial role in signal transduction (Chen et al., 2014; Garcia-Gras et al., 2006). The desmosomes encompass five essential components, the cadherins desmoglein (DSG) and desmocollin (DSC), the armadillo proteins plakoglobin (JUP) and plakophillin-2 (PKP2), and desmoplakin (DSP). The classical view of desmosomes is that they operate as a single functioning structure. However, recent studies demonstrated a close connection between desmosomes, adherens junctions, gap junctions, and ion channels, together forming the area composita (Jansen et al., 2012; Veeraraghavan and Gourdie, 2016; Vermij et al., 2017).
Mutations in desmosomal genes are frequently identified as an underlying cause of cardiovascular disease, notably in patients diagnosed with arrhythmogenic cardiomyopathy (Basso et al., 2018; Marcus et al., 1982). This progressive condition is characterized by loss of cardiomyocytes, gradual replacement of the myocardium by fibro-fatty deposits, and life-threatening ventricular arrhythmias (Hoorntje et al., 2017). Patients bearing mutations in DSP frequently present a complex clinical phenotype characterized by a wide range of cardiac abnormalities (Gao et al., 2020; Wang et al., 2022). To date, a number of pivotal studies focused on the role of DSP in arrhythmogenic cardiomyopathy (ACM) pathogenesis. In one study, cardiomyocyte-specific overexpression of the DSP p.Arg2834His mutation resulted in increased levels of apoptotic cardiomyocytes, lipid accumulation, cardiac fibrosis, and dysfunction (Yang et al., 2006). Interestingly, and in concordance with the clinical phenotype, these mice displayed accelerated ACM pathogenesis when exposed to endurance exercise, which could be linked to aberrant Wnt/β-catenin signaling (Martherus et al., 2016). Similarly, cardiomyocyte-restricted deletion of one Dsp allele in mice led to accumulation of JUP in the nucleus, where it competes with β-catenin, subsequently affecting Wnt-signaling pathways (Garcia-Gras et al., 2006). In addition to an impaired Wnt/β-catenin signaling axis, diminished DSP levels lead to cardiac conductance abnormalities due to mislocalization and reduced levels of connexin 43 (CX43) and sodium voltage-gated channel, alpha subunit 5 (NaV1.5) at the intercalated disc, indicating that DSP plays a role in maintaining CX43 and NaV1.5 stability in cardiomyocytes (Gomes et al., 2012; Gusev et al., 2020; Lyon et al., 2014; Zhang et al., 2013). Even though these studies significantly increased our understanding of DSP-driven ACM, current treatments focus on relieving the symptoms rather than curing the disease due to a lack of therapeutic targets.
To contribute to a better understanding of the pathomolecular mechanisms at play, we used human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes to extensively study a novel heterozygous missense mutation in DSP (gene: c.3562T>C; protein: p.Tyr1188His), identified in a patient with the clinical diagnosis of ACM. Compared with wild-type cardiomyocytes, we observed a prolonged action potential duration (APD) and reduced expression of desmosomal components, which was paralleled by abnormal levels of CX43 and NAV1.5. Strikingly, the transcription factor paired-like homeodomain 2 (PITX2), which acts as a repressor of structural and ion channel-related genes, was induced in these cells. Knockdown of PITX2 in mutant cardiomyocytes led to restoration of DSP, CX43, and NaV1.5. Together, we identified DSP c.3562T>C as a novel pathogenic mutation and demonstrated that aberrant levels of PITX2 in response to mutant DSP might contribute to the electrophysiological abnormalities often seen in patients with ACM.
Results
A novel genetic missense mutation in DSP alters its homodimerization properties
In a patient diagnosed with ACM and experiencing monomorphic ventricular tachycardia, abnormal repolarization, and akinesia of the right ventricular apex (Table S1), we identified a novel missense mutation within the DSP gene (c.3562T>C) that translates to an amino acid substitution of a conserved tyrosine to a histidine (p.Tyr1188His; Figures 1A and 1B). No other mutations in cardiac genes were detected. As this mutation resides within the homodimerization domain (ROD domain) of DSP, we hypothesized that this would affect its homodimerization properties (Green et al., 1990; O’Keefe et al., 1989). To study the consequences of this DSP p.Tyr1188His mutation on homodimerization, we generated three constructs; two in which wild-type DSP is fused to either FLAG (DSP-WT:FLAG) or GFP (DSP-WT::GFP) and one encoding mutant DSP fused to GFP (DSP p.Tyr1188His::GFP; Figure 1C). Next, we transfected these constructs into HEK293 cells and performed FLAG pull-down assays after 48 h. Immunoblotting against FLAG and DSP revealed that mutant DSP protein can still interact with WT DSP, albeit with a lower binding affinity when compared with DSP-WT::FLAG co-transfected with DSP-WT::GFP (Figure 1D). These data indicate that the novel missense mutation in DSP affects the homodimerization properties of DSP molecules.
Figure 1.

The novel missense mutation p.Tyr1188His in desmoplakin affects its homodimerization properties
(A) Schematic of DSP displaying the functional domains.
(B) Alignment of the protein sequence-of-interest for the indicated species. The height of the orange and red bars above each amino acid indicates the degree of conservation.
(C) Overview of the constructs used to assess the binding properties between wild-type (WT) and mutant DSP molecules.
(D) Representative immunoblots for FLAG, GFP, and vinculin (VIN) after co-immunoprecipitation of DSP-WT::FLAG in HEK293 cells transfected with the indicated conditions.
(A–C) The novel mutation is either indicated in red or with an arrow. IF, intermediate filament; IP, immunoprecipitation.
Heterozygous DSP p.Tyr1188His hiPSC-derived cardiomyocytes display reduced desmosomal protein levels and impaired function
To further understand the molecular consequences of this novel mutation, we reprogrammed patient skin fibroblasts to obtain hiPSCs bearing the heterozygous DSP c.3562T>C mutation (Table S2). Next, we corrected the mutant allele utilizing CRISPR-Cas9 in combination with a single-stranded DNA template, yielding an isogenic control line (Figures 2A and S1A). Hereinafter, we refer to these lines as Pa. DSPWT/WT and Pa. DSPp.Tyr1188His/WT. An additional synonymous mutation (blocking mutation) was included in the template to prevent recutting by Cas9 (Figure S1B). No genomic changes were observed for the top three predicted off-targets; ferredoxin 2 (FDX1L), integrator complex subunit 8 (INTS8), and zinc finger FYVE-type containing 27 (ZFYVE27; Figure S1C). The pluripotency markers Nanog homeobox (NANOG), POU class 5 homeobox 1 (OCT3/4), and SRY-box transcription factor 2 (SOX2) were expressed in Pa. DSPWT/WT and Pa. DSPp.Tyr1188His/WT hiPSCs (Figure S1D), and no changes in the karyotype were identified (Figures S1E and S1F). Directed differentiation of hiPSCs to cardiomyocytes yielded comparable percentages (90%–98%) of cardiac troponin T positive cells for both lines (Figure S2A). DSP, JUP, and PKP2 correctly localized to the cell periphery in 1-month-old mutant cardiomyocytes (Figures 2B, S2B, and S2C). However, molecular analyses revealed a significant reduction in DSP protein levels in mutant cells, whereas the mRNA levels were unaffected (Figures 2C–2E and S2D). Immunoblot analysis for desmosomal components DSC, DSG, JUP, and PKP2 showed a significant decline for all proteins in Pa. DSPp.Tyr1188His/WT hiPSC-derived cardiomyocytes compared with the isogenic control (Figures 2F and 2G). Since the patient displayed abnormalities in the cardiac conduction system including repolarization irregularities and arrhythmias, we performed electrophysiology assays on mutant hiPSC-derived cardiomyocytes. We observed a prolonged APD at 50% and 90% of repolarization in mutant cardiomyocytes compared with control (Figure 2H). Together, cardiomyocytes bearing the novel DSPp.Tyr1188His mutation show reduced desmosomal protein levels and a prolonged APD.
Figure 2.

Heterozygous Pa. DSPp.Tyr1188His/WT hiPSC-derived cardiomyocytes display reduced desmosomal protein levels and a prolonged action potential duration
(A) Sanger sequencing traces of the patient-derived and corrected isogenic control hiPSC lines. The DNA template used for CRISPR-Cas9-targeting, correcting the DSPp.Tyr1188His mutation (C > T), is shown. Intended mutation is indicated in green.
(B–H) Molecular and functional analyses on 1-month-old hiPSC-derived cardiomyocytes obtained from three independent experiments.
(B) Representative immunostainings for DSP. DSP in red; cardiac troponin T (TNNT2) in green; DAPI in blue. Scale bar: 10 μm.
(C) Gene expression levels of DSP normalized to GUS.
(D) Representative immunoblots for DSP.
(E) Quantification of DSP protein levels normalized to VIN.
(F) Representative immunoblots for DSC, DSG, JUP, and PKP2.
(G) Quantification of the desmosomal protein levels. Values normalized to VIN.
(H) Action potential duration (APD) measured at 50% and 90% of cardiomyocyte repolarization (Pa. DSPWT/WT, n = 50 cell clusters; Pa. DSPp.Tyr1188His/WT, n = 58 cell clusters).
Data are plotted as mean. The dots in (C), (E), (G), and (H) represent technical replicates, whereas the color of each dot indicates the experimental origin (3 independent experiments; 4–34 technical replicates). Significance has been assessed by a two-tailed unpaired Student’s t test or two-tailed Mann-Whitney test when data were not normally distributed (∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns, not significant).
Wnt-signaling- and ion-handling-related processes are affected in Pa. DSPp.Tyr1188His/WT cardiomyocytes
Next, we aimed to identify affected signaling cascades that could contribute to the observed phenotype. To this end, we performed mRNA sequencing on 1-month-old Pa. DSPWT/WT and Pa. DSPp.Tyr1188His/WT cardiomyocytes. Mutant cardiomyocytes displayed a different expression profile compared with the isogenic control (Figures 3A and 3B), with 662 up- and 843 downregulated genes (log2 fold change >1 and <−1; adjusted p value [p-adj] < 0.05; Figure 3B). Gene Ontology (GO) analysis of upregulated genes identified non-canonical Wnt signaling (GO: 0035567) as enriched term (Figure 3C). Interestingly, Wnt signaling has previously been linked to ACM pathogenesis (Garcia-Gras et al., 2006). Among the gene hits for this term frizzled-2 (FZD2) and secreted frizzled-related protein 4 (SFRP4) were identified, for which the induced expression levels were validated in three independent differentiations (Figure 3D). Enriched GO terms for the downregulated genes included “cell adhesion” (GO: 0007155), “signal transduction” (GO: 0007165), and “regulation of ion transmembrane transport” (GO: 0034765; Figure 3E). Strikingly, gap junction alpha-1 protein (GJA1) and SCN5A, encoding for CX43 and NaV1.5, were among the significantly downregulated genes, which was validated in three additional batches of cardiomyocytes (Figure 3F). The aberrant expression of GJA1 and SCN5A suggests that the DSPp.Tyr1188His/WT mutation not only affects structural components but also cellular processes related to signal propagation and contraction.
Figure 3.

Gene networks related to Wnt signaling and ion handling are dysregulated in heterozygous Pa. DSPp.Tyr1188His/WT hiPSC-derived cardiomyocytes
(A–F) mRNA sequencing analyses on 1-month-old hiPSC-derived cardiomyocytes.
(A) Principal-component analysis (PCA) for Pa. DSPWT/WT and Pa. DSPp.Tyr1188His/WT cardiomyocytes.
(B) Volcano plot showing the up- and downregulated genes (fold change (log2) >1 and <−1; p-adj < 0.05) in Pa. DSPp.Tyr1188His/WT and control cardiomyocytes. The top five up- and downregulated genes are indicated.
(C) Gene Ontology analysis on the upregulated (fold change (log2) >1) genes.
(D) Validation of FZD2 and SFRP4, both belonging to the “non-canonical Wnt-signaling pathway” term.
(E) Gene Ontology analysis on the downregulated (fold change (log2) <−1) genes.
(F) Validation of GJA1 and SCN5A, both belonging to the “regulation of ion transmembrane transport” term. mRNA sequencing was performed on four replicates obtained from one hiPSC differentiation.
The dots in (D) and (F) represent technical replicates, whereas the color of each dot indicates the experimental origin (3 independent experiments; 4–8 technical replicates). Gene expression data are normalized to GUS and plotted as mean. Significance has been assessed by a two-tailed unpaired Student’s t test (∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). GO: 0007187, G protein-coupled receptor signaling pathway coupled to cyclic nucleotide second messenger.
Knockin hiPSC-derived DSPp.Tyr1188His/WT cardiomyocytes corroborate findings observed in patient-derived cardiomyocytes
In an effort to exclude confounding effects, such as the presence of second genomic hits, we generated an independent knockin (KI) hiPSC line bearing the DSPp.Tyr1188His/WT mutation. We used a healthy hiPSC line and followed the same targeting process as described above and added a second single-stranded DNA template containing only the blocking mutation (Figures S3A and 4A). Hereafter, we refer to these lines as KI. DSPWT/WT and KI. DSPp.Tyr1188His/WT. Amplification of potential off-target sites did not reveal any editing events (Figure S3B). Furthermore, chromosome integrity was unaffected, and the pluripotency markers NANOG, OCT3/4, and SOX2 were expressed in both lines (Figures S3C–S3E). Directed differentiation of control and KI. DSPp.Tyr1188His/WT cells yielded comparable percentages (75%–82%) of cardiac troponin T-positive cells (Figure S4A). Molecular analyses revealed normal localization of DSP, JUP, and PKP2 in 1-month-old mutant cardiomyocytes (Figures S4B–S4D). Similar to Pa. DSPp.Tyr1188His/WT, a decline in DSP protein levels was observed in the KI. DSPp.Tyr1188His/WT cardiomyocytes (Figures 4C and 4D). Interestingly, also the mRNA levels of DSP were reduced, which is in contrast to the patient-derived line (Figures 4B and S4E). Western blot analysis revealed a significant decline for DSC and JUP, whereas DSG and PKP2 were expressed at a similar level compared with the isogenic control (Figures 4E and 4F). Importantly, the APD at 50% and 90% of cardiomyocyte repolarization was also prolonged in KI. DSPp.Tyr1188His/WT cardiomyocytes (Figure 4G). mRNA sequencing on 1-month-old KI. DSPp.Tyr1188His/WT and control cardiomyocytes revealed a distinct expression profile for mutant cardiomyocytes with 1,378 and 2,499 genes up- and downregulated, respectively (log2 fold change >1 and <−1; p-adj < 0.05; Figures 5A and S5A). GO term analysis for the differentially expressed genes revealed “regulation of transcription” and “ion handling” as enriched terms (Figures S5B and S5D). Significant upregulation of the transcription factors forkhead box C2 (FOXC2) and lipopolysaccharide induced TNF factor (LITAF) was confirmed in three additional batches of differentiated cardiomyocytes (Figure S5C). In line with our observations in the patient-derived line, we noticed dysregulation of ion channels including potassium inwardly rectifying channel subfamily J member 2 (KCNJ2) and SCN5A (Figure S5E). Together, characterization of the KI. DSPp.Tyr1188His/WT line corroborated the data collected from Pa. DSPp.Tyr1188His/WT cardiomyocytes, demonstrating that this novel missense mutation in DSP is responsible for eliciting the observed molecular and functional changes.
Figure 4.

hiPSC-derived KI. DSPp.Tyr1188His/WT cardiomyocytes corroborate findings observed in patient-derived cardiomyocytes
(A) Sanger sequencing traces of the control and knockin hiPSC lines. The two different DNA templates used to introduce the intended mutation are depicted. Intended mutation is indicated in green.
(B–G) Molecular and functional analyses on 1-month-old hiPSC-derived cardiomyocytes obtained from three independent experiments.
(B) Gene expression levels of DSP normalized to GUS.
(C) Representative immunoblots for DSP.
(D) Quantification of DSP protein levels normalized to VIN.
(E) Representative immunoblots for DSC, DSG, JUP, and PKP2.
(F) Quantification of the desmosomal protein levels. Values normalized to VIN.
(G) APD measured at 50% and 90% (KI. DSPWT/WT, n = 48 cell clusters; KI. DSPp.Tyr1188His/WT, n = 81 cell clusters) of cardiomyocyte repolarization.
Data are plotted as mean. The dots in (B), (D), (F), and (G) represent technical replicates, whereas the color of each dot indicates the experimental origin (3 independent experiments; 4–60 technical replicates). Significance has been assessed by a two-tailed unpaired 2Student’s t test or two-tailed Mann-Whitney test when data were not normally distributed (∗p < 0.05, ∗∗∗∗p < 0.0001, ns, not significant).
Figure 5.

Pa. DSPp.Tyr1188His/WT and KI. DSPp.Tyr1188His/WT cardiomyocytes display impaired ion-handling
(A) PCA reveals different gene expression profiles of KI. DSPWT/WT and KI. DSPp.Tyr1188His/WT cardiomyocytes.
(B–F) Combined analyses of the Pa. DSPp.Tyr1188His/WT (patient) and KI. DSPp.Tyr1188His/WT (knockin) mRNA sequencing datasets.
(B and C) Venn diagram showing the overlap of the up- (B; fold change (log2) >1) and downregulated (C; fold change (log2) <−1) genes.
(D and E) Gene Ontology analyses on the up- (D) and downregulated (E) genes overlapping between the two datasets.
(F) STRING: functional protein association network for the gene ontology terms “regulation of ventricular cardiac muscle cell depolarization” and “regulation of ion transmembrane transport.” Clustered based on kmeans. The thickness of each line indicates the level of confidence (data supported) and the colors to which cluster each component belongs.
(G–H) Representative immunoblots for CX43 and NaV1.5 in Pa. DSPp.Tyr1188His/WT (G) and KI. DSPp.Tyr1188His/WT (H) and isogenic control cardiomyocytes.
(I) Quantification of CX43 and NaV1.5 protein levels. Values normalized to VIN. Data are plotted as mean.
The dots in (I) represent technical replicates, whereas the color of each dot indicates the experimental origin (3 independent experiments; 4–6 technical replicates). Significance has been assessed by a two-tailed unpaired Student’s t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Combinatorial mRNA sequencing analysis of Pa. and KI. DSPp.Tyr1188His/WT cardiomyocytes reveals impaired cardiac muscle cell depolarization
To identify the molecular changes specifically caused by the novel DSPp.Tyr1188His mutation and not due to the genetic background of the lines, we performed a combinatorial analysis on the differentially expressed genes obtained from the patient (Figure 3) and KI (Figure S5) datasets. In addition to these gene lists, we also included a set of differentially expressed genes obtained from an mRNA sequencing analysis in which we combined all WT and mutant samples from both datasets (“combined patient and KI”). We identified a subset of genes that were consistently differentially expressed between the patient and KI datasets (Figures 5B and 5C). Next, we performed GO analyses on the 104 up- and 275 downregulated genes shared between all comparisons (Figures 5B–5E). The significantly upregulated genes were associated with the Wnt-signaling pathway, whereas the shared downregulated genes were enriched for “regulation of ion transmembrane transport” and “regulation of ventricular cardiac muscle cell depolarization” (Figures 5D and 5E). Next, we performed a string-db analysis using the genes within the GO terms “regulation of ion transmembrane transport” and “regulation of ventricular cardiac muscle cell depolarization,” accentuating the presence of genes fundamental for cardiomyocyte function such as GJA1 and SCN5A. Reduced expression of GJA1 and SCN5A was validated in three additional differentiations of 1-month-old Pa. DSPp.Tyr1188His/WT and KI. DSPp.Tyr1188His/WT cardiomyocytes (Figures 3F, S5E, and S5F). Immunoblot analysis confirmed that CX43 and NaV1.5 protein levels were reduced in Pa. DSPp.Tyr1188His/WT and KI. DSPp.Tyr1188His/WT cardiomyocytes compared with control cells (Figures 5G–5I). Together, we robustly identified impaired expression of cardiac ion-handling-related components as a consequence of the novel DSPp.Tyr1188His mutation.
PITX2 levels are increased in DSPp.Tyr1188His/WT cardiomyocytes and repress expression of structural and ion-handling genes
The observation that many ion channels, indispensable for proper cardiomyocyte function, were dysregulated in Pa. and KI. DSPp.Tyr1188His/WT cardiomyocytes prompted us to investigate potential upstream effectors. Martin and coworkers previously reported that the transcription factor PITX2 dictates a gene network in mouse postnatal atrial cardiomyocytes encompassing channel and calcium-handling genes as well as genes involved in stabilizing cell-cell junctions (Tao et al., 2014). The authors combined chromatin immunoprecipitation sequencing and transcriptomics on conditional Pitx2 knockout mice, demonstrating that a loss of Pitx2 in cardiomyocytes results in upregulation of Dsp, Gja1, and Scn5a, indicating that PITX2 acts as a repressor for these genes. In humans, mutations in genomic loci adjacent to or within PITX2, thereby affecting its expression, predisposes the heart to atrial arrhythmias (Chinchilla et al., 2011; Gudbjartsson et al., 2007; Herraiz-Martínez et al., 2021; Kaab et al., 2008; Ouwerkerk et al., 2020). Based on these findings, we hypothesized that dysregulation of PITX2 in DSPp.Tyr1188His/WT cardiomyocytes might contribute to the observed reduction of DSP, CX43, and NaV1.5. mRNA sequencing analyses and subsequent validation experiments revealed an induction of PITX2 in Pa. and KI. DSPp.Tyr1188His/WT cardiomyocytes (Figure 6A), which was confirmed at the protein level (Figures 6B–6D). To see whether PITX2 could evoke a similar response in an independent model, we used lentivirus to overexpress PITX2 (lenti-PITX2) for 7 days in healthy hiPSC-derived control cardiomyocytes. PITX2 levels were induced approximately 20-fold compared with the baseline condition (Figures S6A and S6C). The PITX2 targets DSP, GJA1, and SCN5A were all repressed in lenti-PITX2-treated cardiomyocytes (Figures S6B and S6C). On the protein level, we could confirm the induction of PITX2 and reduced levels of DSP and CX43 (Figures 6E and 6F). Functionally, we observed a significant prolongation of the APD at 90% of cardiomyocyte repolarization (Figure 6G). On the contrary, knockdown of PITX2 gene expression in control cardiomyocytes induced the expression of DSP, GJA1, and SCN5A (Figures S6D–S6F), which was confirmed by immunoblotting for PITX2, DSP, and CX43 (Figures S6G and S6H). The APD was shortened in siPITX2-treated cardiomyocytes at 50% and 90% of repolarization compared with control (Figure S6I). These results demonstrate that PITX2 is induced in Pa. and KI. DSPp.Tyr1188His/WT cardiomyocytes and that PITX2 represses genes important for cardiomyocyte function.
Figure 6.

Paired-like homeodomain 2 levels are increased in mutant cardiomyocytes and repress expression of DSP, CX43, and NaV1.5
(A–D) Molecular analyses on 1-month-old Pa. DSPp.Tyr1188His/WT, KI. DSPp.Tyr1188His/WT, and isogenic control cardiomyocytes.
(A) Gene expression levels for PITX2 normalized to GUS.
(B and C) Representative immunoblots for PITX2 in Pa. DSPp.Tyr1188His/WT (B) and KI. DSPp.Tyr1188His/WT (C) cardiomyocytes.
(D) Quantification of PITX2 protein levels normalized to VIN.
(E–G) Molecular and functional analyses on 1-month-old control hiPSC-derived cardiomyocytes treated with either empty viral particles or particles encoding for PITX2 (lenti-PITX2).
(E) Representative immunoblots for PITX2, DSP, CX43, and NaV1.5. (F) Protein levels of PITX2, DSP, CX43, and NaV1.5 normalized to VIN.
(G) APD measured at 50% and 90% (control, n = 66 cell clusters; lenti-PITX2, n = 64 cell clusters) of cardiomyocyte repolarization.
Data are plotted as mean. The dots in (A), (D), (F), and (G) represent technical replicates, whereas the color of each dot indicates the experimental origin (3 independent experiments; 4–29 technical replicates). Significance has been assessed by a two-tailed unpaired Student’s t test or two-tailed Mann-Whitney test when data were not normally distributed (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns, not significant). KI, knockin; Pa., patient.
Suppression of PITX2 in Pa. DSPp.Tyr1188His/WT cardiomyocytes restores expression of structural and ion-handling genes
After identifying PITX2 as potential upstream effector of DSP, GJA1, and SCN5A, we speculated that knockdown of PITX2 in Pa. DSPp.Tyr1188His/WT cardiomyocytes could restore the observed phenotype. To investigate this, we treated 1-month-old mutant and isogenic control cardiomyocytes with scramble small interfering RNA (siRNA) or siRNA targeting PITX2 for 72 h. PITX2 levels were reduced by approximately 90% in both control and mutant cardiomyocytes, which led to a significant increase in GJA1 expression in Pa. DSPp.Tyr1188His/WT cardiomyocytes (Figures S7A and S7B). On the protein level, we observed restoration of DSP, CX43, and NaV1.5 upon silencing of PITX2 in mutant cardiomyocytes (Figures 7A and 7B). To explore if PITX2 induction is unique to the novel DSPp.Tyr1188His mutation or whether it is a more general phenomenon for mutations in DSP, we assessed the levels of PITX2 in hiPSC-derived cardiomyocytes bearing a known disease-causing mutation in DSP (KI. DSPp.Arg1113X/WT). Compared with control cells, mutant cardiomyocytes display reduced levels of DSP (Figures 7C and S7C). Interestingly, this reduction is accompanied by elevated levels of PITX2, which was confirmed at the protein level (Figures 7C–7E). Altogether, knockdown of the repressor PITX2 in Pa. DSPp.Tyr1188His/WT cardiomyocytes restores expression of genes important for cardiomyocyte function. Additionally, KI. DSPp.Arg1113X/WT cardiomyocytes show induced levels of PITX2, suggesting a more general mechanism.
Figure 7.

RNAi-mediated knockdown of paired-like homeodomain 2 in Pa. DSPp.Tyr1188His/WT cardiomyocytes rescues the expression of structural and ion-related units
(A and B) Molecular analyses on 1-month-old Pa. DSPWT/WT and Pa. DSPp.Tyr1188His/WT cardiomyocytes treated with scramble siRNA or siRNA targeting PITX2 for 72 h. Data are obtained from 3–4 independent experiments.
(A) Representative immunoblots for PITX2, DSP, CX43, NaV1.5, and VIN.
(B) Quantification for PITX2, DSP, CX43, and NaV1.5 normalized to VIN.
(C–E) Molecular analyses on 1-month-old KI. DSPWT/WT and KI. DSPp.Arg1113X/WT cardiomyocytes. Data are obtained from three independent experiments.
(C) Gene expression levels for DSP and PITX2 normalized to GUS.
(D) Representative immunoblots for PITX2 and VIN.
(E) Quantification of PITX2 protein levels normalized to VIN.
Data are plotted as mean. The dots in (B), (C), and (E) represent technical replicates, whereas the color of each dot indicates the experimental origin (3–4 independent experiments; 3–6 technical replicates). For (B), significance has been assessed on log-transformed data using an ordinary two-way ANOVA followed by a Tukey’s multiple comparisons test (single pooled variance; alpha = 0.05). For (C) and (E), a two-tailed unpaired Student’s t test was applied (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns, not significant).
Discussion
Through the use of matching patient-derived (Pa. DSPp.Tyr1188His/WT) and KI (KI. DSPp.Tyr1188His/WT) hiPSC-derived cardiomyocytes, we demonstrated that this novel DSP mutation evokes molecular and functional changes that can be linked to ACM pathogenesis. This implies that this mutation is causal for the observed phenotype in the patient clinically diagnosed with ACM.
Molecular analyses of DSPp.Tyr1188His/WT hiPSC-derived cardiomyocytes revealed a reduction in desmosomal proteins, an observation previously reported in ventricular tissue obtained from patients with ACM (Asimaki et al., 2009). Interestingly, this pathomolecular characteristic seems to be a hallmark of ACM, as it is not evident in other forms of cardiac disease (Asimaki et al., 2009; Bueno-Beti and Asimaki, 2021). Besides reduced protein levels, we also observed lower mRNA levels for several components of the intercalated disc. This is in contrast to a recent study from our lab in which we only observed an effect on the protein level in an in vitro and in vivo KI model of an ACM-driving PKP2 mutation, indicating that the DSPp.Tyr1188His/WT mutation evokes a different molecular response (H. Tsui, S.J.K., S.J.H., V. Meraviglia, W.B.H., S. Casini, P. van der Kraak, A.V., X. Yin, M. Mayr, A. Bossu, G. Marchal, J.M.-K., J. Eding, D. Versteeg, K. Bezstarosti, J.A.G., S. Klaasen, L. van Laake, J. Demmers, G. Kops, C. Mummery, T.A.B.V., C. Remme, M. Bellin, E.R., unpublished data). In support of this, a change in the transcriptomic landscape was demonstrated through dysregulation of genes associated with ion handling, Wnt signaling, and transcriptional activity. Of note, compared with control cardiomyocytes, KI. DSPp.Tyr1188His/WT cardiomyocytes showed reduced DSP mRNA levels, whereas Pa. DSPp.Tyr1188His/WT cardiomyocytes displayed similar expression levels. Moreover, we consistently observed higher protein levels for PITX2 in the KI line compared with the patient-derived line. Since DSP is one of the potential downstream targets of the repressor PITX2, it is conceivable that the repressive effect on DSP expression is augmented in KI. DSPp.Tyr1188His/WT cardiomyocytes compared with patient-derived cardiomyocytes (Tao et al., 2014). More experiments are required to pinpoint the exact underlying molecular mechanism.
We further showed that mutant DSPp.Tyr1188His/WT cardiomyocytes exhibit lower levels of the gap junction protein CX43 and NaV1.5, which in turn may be causal for the observed prolongation of the action potential. These observations are in line with previous studies that demonstrated diminished CX43 and NaV1.5 levels in explanted cardiac material isolated from patients diagnosed with ACM (Asimaki et al., 2009; Bueno-Beti and Asimaki, 2021; Jansen et al., 2012). In addition, studies revealed redistribution of CX43 to the long axis of cardiomyocytes upon desmosomal protein deficiency, which in turn may act as an arrhythmogenic substrate (Bueno-Beti and Asimaki, 2021; Oxford et al., 2007; Zhang et al., 2013). The observed reduction and redistribution of proteins crucial for electrical signal propagation in cardiomyocytes may be explained at two levels. Firstly, as the desmosomes are interconnected with adherens junctions, gap junctions, and ion channels, it is conceivable that desmosomal mutations do not only affect the integrity of desmosomes but also of the linked protein complexes (Vermij et al., 2017). Specifically, proper localization of CX43 and NaV1.5 to cell-cell junctions seem to be highly dependent on a functional desmosome (Cerrone et al., 2014; Gusev et al., 2020; Jansen et al., 2012; Lyon et al., 2014; Sato et al., 2009; Zhang et al., 2013). Secondly, influential transcriptional programs such as Hippo and Wnt signaling are partly regulated by proteins residing at the intercalated discs in cardiomyocytes (Chen et al., 2014; Conti et al., 2004; Garcia-Gras et al., 2006; Guo et al., 2020). Desmosomal instability may therefore affect these signaling cascades and in turn affect the expression of structural and ion-handling-related genes. For instance, CX43 (GJA1) is a target of canonical Wnt signaling (Ai et al., 2000).
Here, we uncovered that aberrant expression of PITX2 in mutant DSPp.Tyr1188His/WT cardiomyocytes contribute to the observed phenotype. Interestingly, mutations within PITX2, which physically interacts with FOXC1 and FOXC2, have previously been linked to the ocular conditions Axenfeld-Rieger syndrome and glaucoma (Acharya et al., 2011). In the heart, PITX2 is predominantly expressed in the left atria and well known for its role in atrial fibrillation; however, it has also been detected in ventricular tissue (Chinchilla et al., 2018; Furtado et al., 2011; Ouwerkerk et al., 2020; Tao et al., 2016; Torrado et al., 2014). Moreover, it has been proposed that atrial disease can be a subentity of heart failure induced by ventricular abnormalities (Coats et al., 2021). Mikhailov and coworkers demonstrated that PITX2 expression is reactivated in the ventricular failing myocardium of patients experiencing systolic heart failure (Torrado et al., 2014). Likewise, PITX2 is induced after myocardial infarction in Hippo-deficient mouse ventricles, subsequently activating expression of genes associated with the electron transport chain and reactive oxygen species scavengers (Tao et al., 2016). Through modulation of PITX2 expression levels in mutant Pa. DSPp.Tyr1188His/WT cardiomyocytes, we demonstrate reactivation of genes important for ion handling and signal propagation, including SCN5A and GJA1. Previously, PITX2 has been linked to Hippo and Wnt signaling, cascades reported to be frequently dysregulated in the setting of ACM (Basu and Roy, 2013; Briata et al., 2003; Garcia-Gras et al., 2006; Tao et al., 2016). We did notice enrichment for terms related to (non)-canonical Wnt signaling in our transcriptome datasets, which might provide a link between mutations in DSP and PITX2 induction. Together, these findings imply that PITX2 may play a role in the malfunctioning ventricular myocardium. Further research is required to unravel the exact mechanisms leading to PITX2 induction in cardiomyocytes bearing mutations in DSP.
In conclusion, our results reveal that the novel DSPp.Tyr1188His mutation evokes a pathological response in cardiomyocytes. We observed reduced desmosomal protein levels, which were accompanied by a reduction in CX43 and NaV1.5. Functionally, mutant cardiomyocytes displayed a prolonged APD that could be linked to induced levels of the repressor PITX2. Indeed, knockdown of PITX2 in patient-derived cardiomyocytes could alleviate the observed repressive effects. Even though the mutation-induced molecular changes are well defined in this study, the electrophysiological properties are not because of technical limitations inherent of the followed methodology. Follow-up studies should implement patch-clamp experiments to further facilitate our understanding of the electrical alterations caused by the mutation. Together, our data underscore the advantages of combining patient and KI hiPSC-derived cardiomyocytes, bearing mutations in the endogenous locus, to identify a potential novel therapeutic target for the treatment of ACM.
Experimental procedures
Resource availability
Corresponding author
For further information, please contact Eva van Rooij (e.vanrooij@hubrecht.eu).
Materials availability
Materials and additional details can be made available from the corresponding author upon reasonable request.
Co-immunoprecipitation
HEK293T cells grown on a 145 cm2 dish until 60%–70% confluency were transfected with the indicated plasmids using polyethylenimine. After 48 h, protein was isolated using a mild lysis buffer. Magnetic beads coated with monoclonal anti-FLAG M2 were used to pull down protein complexes containing DSP-WT:FLAG molecules. Samples were analyzed on a 7% SDS-PAGE gel using the antibodies listed in Table S4.
Cardiomyocyte cultures
The genomic integrity and pluripotency status of genetically modified hiPSCs was assessed by means of targeted sequencing, immunofluorescence, and karyo sequencing. To initiate directed differentiation toward cardiomyocytes, hiPSCs were cultured on Geltrex-coated plates in Essential 8 Medium until 80%–90% confluency, Next, medium was refreshed with RPMI-1640-Medium-GlutaMAX-Supplement-HEPES supplemented with human recombinant albumin, L-Ascorbic Acid 2-Phosphate, and CHIR99021 (cardio differentiation medium with CHIR). After 48 h, medium was refreshed with cardio differentiation medium with IWP2. After 48 and 96 h, cells were refreshed with plain cardio differentiation medium. From day 8 onward, cells were kept in RPMI-1640-Medium-GlutaMAX-Supplement-HEPES supplemented with B-27-Supplement-serum free. hiPSC-derived cardiomyocyte cultures were subsequently analyzed by fluorescence-activated cell sorting (FACS; BD Biosciences, FACSCalibur) for the percentage of cardiomyocytes (positive for cardiac Troponin T).
Molecular assays
hiPSC-derived cardiomyocytes were seeded at a density of 100,000 cells/cm2 for downstream applications. We analyzed the RNA, protein, and functional properties of 1-month-old cardiomyocytes either kept under baseline conditions, transfected with control siRNA or an siRNA targeting PITX2, or infected with control virus or lenti-PITX2. For the functional experiments, cells were grown in clusters and treated with FluoVolt and Powerload for 15 min at 37°C. During measurements, cells were immersed in a solution containing, in mM, NaCl (130), KCl (4), CaCl2 (1.8), MgCl2 (1.2), NaHCO3 (18), HEPES (10), and glucose (10) (pH 7.4). A custom-build microscope with a 10× objective was used for the recordings. APDs were corrected for the beating rate using an adjustment of the Fredericia formula: APDcorrected = APD/(∛(60/BPM)).
mRNA sequencing
mRNA-sequencing was performed on 1-month-old hiPSC-derived cardiomyocytes. RNA libraries were prepared with the TruSeq Stranded mRNA polyA kit (Illumina) according to the manufacturer’s protocol. Strand-specific single-end 75 bp reads were generated on an Illumina NextSeq 500 system. Reads were checked for their quality using FastQC and aligned against the human genome (assembly GRCh37) using STAR (STAR_2.4.2a). Differential expression was calculated using DESeq2 v.1.2 with pooled dispersion estimates. Differentially expressed genes were further analyzed for functional enrichment using the STRING v.11.5 database. No changes were made to the default settings. Homo sapiens served as background. Only the molecular function (GO-MF), biological process (GO-BP), cellular component (GO-CC), and KEGG data sources were considered for enrichment analysis.
Human material
The study fulfilled the Dutch criteria of the code of proper use of human tissue. Written informed consent was obtained for the generation and use of the patient-derived hiPSCs.
Statistical analysis
The number of samples (n) used in each experiment is indicated in the legend of each figure. Data are presented as mean. Statistical analyses were performed using PRISM (GraphPad Software v. 9). Outliers were identified using the ROUT method (Q = 5%) and were removed if present. For comparison of two groups, data were tested for normality using the Kolmogorov-Smirnov test with Dallal-Wilkinson-Lillie for p value method (alpha = 0.05). Significance has been assessed by a two-tailed unpaired Student’s t test or two-tailed Mann-Whitney test when data were not normally distributed. For comparisons of four groups with two variables (genotype and treatment), data were tested for homoscedasticity (Spearman’s rank correlation test; alpha = 0.05) and Gaussian distribution (Kolmogorov-Smirnov test; alpha = 0.05). In case these requirements were not met, a log transformation was applied. Data were then tested for significance using an ordinary two-way ANOVA, followed by a Tukey’s multiple comparisons test (single pooled variance; alpha = 0.05). Correlation and significance between proteins have been assessed by non-parametric Spearman correlation (two-tailed; alpha = 0.05). Differences were considered statistically significant when p <0.05. Asterisks indicate statistical significance (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Author contributions
S.J.v.K. designed and performed all experiments related to hiPSC targeting using CRISPR-Cas9. S.J.v.K., C.J.B., and E.v.R. designed all molecular experiments related to phenotyping of the hiPSC-derived cardiomyocytes. S.J.v.K., S.J.H., E.K., E.W.S., and J.M.-K. maintained the hiPSC lines and performed molecular experiments on hiPSC-derived cardiomyocytes. S.J.v.K. and C.J.B. performed mRNA sequencing analyses. W.B.v.H., B.G., T.P.d.B., and T.A.B.v.V. designed and performed the electrophysiology experiments. S.J.v.K., I.P., and H.d.R. designed and performed the experiments regarding co-immunoprecipitation and cloning. L.W.v.L. and J.A.G. provided the human skin biopsies and clinical data. S.J.v.K. and E.v.R. planned all experiments, performed data analyses, and wrote the manuscript.
Acknowledgments
We thank V. Sigurdsson and the department of dermatology, UMC Utrecht, for help with skin biopsies. We thank Farhad A. Moqadam and Ana Rita Leitoguinho for the design of the gRNAs. We also thank Sjoerd J. Klaasen and Geert J.P.L. Kops for their help with the karyo sequencing procedure. This work was supported by funds from the Dutch Cardiovascular Alliance with the support of the Dutch Heart Foundation, DCVA2017-18 ARENA-PRIME (E.v.R.) and CVON2018-30 PREDICT2 (W.B.v.H. and T.A.B.v.V.); the Vici grant from the Dutch Research Council (NWO), project 09150181910020 (E.v.R.); and by the Fondation Leducq Transatlantic Network of Excellence, 17CVD02 (E.v.R.).
Conflicts of interest
The authors declare no competing interests.
Published: March 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2023.01.015.
Supplemental information
Data and code availability
The mRNA sequencing datasets have been deposited with the Gene Expression Omnibus repository under accession numbers GEO: GSE208213 and GSE208212 for the patient and KI lines, respectively.
References
- Acharya M., Huang L., Fleisch V.C., Allison W.T., Walter M.A. A complex regulatory network of transcription factors critical for ocular development and disease. Hum. Mol. Genet. 2011;20:1610–1624. doi: 10.1093/hmg/ddr038. [DOI] [PubMed] [Google Scholar]
- Ai Z., Fischer A., Spray D.C., Brown A.M.C., Fishman G.I. Wnt-1 regulation of connexin43 in cardiac myocytes. J. Clin. Invest. 2000;105:161–171. doi: 10.1172/JCI7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimaki A., Tandri H., Huang H., Halushka M.K., Gautam S., Basso C., Thiene G., Tsatsopoulou A., Protonotarios N., McKenna W.J., et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- Basso C., Pilichou K., Bauce B., Corrado D., Thiene G. Diagnostic criteria, genetics, and molecular basis of arrhythmogenic cardiomyopathy. Heart Fail. Clin. 2018;14:201–213. doi: 10.1016/j.hfc.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Basu M., Roy S.S. Wnt/β-catenin pathway is regulated by PITX2 homeodomain protein and thus contributes to the proliferation of human ovarian adenocarcinoma cell, SKOV-3. J. Biol. Chem. 2013;288:4355–4367. doi: 10.1074/jbc.M112.409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briata P., Ilengo C., Corte G., Moroni C., Rosenfeld M.G., Chen C.Y., Gherzi R. The Wnt/β-catenin→Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol. Cell. 2003;12:1201–1211. doi: 10.1016/s1097-2765(03)00407-6. [DOI] [PubMed] [Google Scholar]
- Bueno-Beti C., Asimaki A. Histopathological features and protein markers of arrhythmogenic cardiomyopathy. Front. Cardiovasc. Med. 2021;8:746321. doi: 10.3389/fcvm.2021.746321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrone M., Lin X., Zhang M., Agullo-Pascual E., Pfenniger A., Chkourko Gusky H., Novelli V., Kim C., Tirasawadichai T., Judge D.P., et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.N., Gurha P., Lombardi R., Ruggiero A., Willerson J.T., Marian A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014;114:454–468. doi: 10.1161/CIRCRESAHA.114.302810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchilla A., Daimi H., Lozano-Velasco E., Dominguez J.N., Caballero R., Delpón E., Tamargo J., Cinca J., Hove-Madsen L., Aranega A.E., Franco D. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011;4:269–279. doi: 10.1161/CIRCGENETICS.110.958116. [DOI] [PubMed] [Google Scholar]
- Chinchilla A., Esteban F.J., Lozano-Velasco E., Hernandez-Torres F., Dominguez J.N., Aránega A.E., Franco D. Ventricular chamber-specific pitx2 insufficency leads to cardiac hypertrophy and arrhythmias. bioRxiv. 2018 doi: 10.1101/253062. Preprint at. [DOI] [Google Scholar]
- Coats A.J.S., Heymans S., Farmakis D., Anker S.D., Backs J., Bauersachs J., Boer R.A.D., Jelena Č., Cleland J.G.F., Dobrev D., et al. Atrial disease and heart failure : the common soil hypothesis proposed by the Heart Failure Association of the European Society of Cardiology. Eur. Heart J. 2021;43:1635. doi: 10.1093/eurheartj/ehab834. [DOI] [PubMed] [Google Scholar]
- Conti M.A., Even-Ram S., Liu C., Yamada K.M., Adelstein R.S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J. Biol. Chem. 2004;279:41263–41266. doi: 10.1074/jbc.C400352200. [DOI] [PubMed] [Google Scholar]
- Furtado M.B., Biben C., Shiratori H., Hamada H., Harvey R.P. Characterization of Pitx2c expression in the mouse heart using a reporter transgene. Dev. Dyn. 2011;240:195–203. doi: 10.1002/dvdy.22492. [DOI] [PubMed] [Google Scholar]
- Gao S., Puthenvedu D., Lombardi R., Chen S.N. Established and emerging mechanisms in the pathogenesis of arrhythmogenic cardiomyopathy: a multifaceted disease. Int. J. Mol. Sci. 2020;21:6320. doi: 10.3390/ijms21176320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gras E., Lombardi R., Giocondo M.J., Willerson J.T., Schneider M.D., Khoury D.S., Marian A.J. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes J., Finlay M., Ahmed A.K., Ciaccio E.J., Asimaki A., Saffitz J.E., Quarta G., Nobles M., Syrris P., Chaubey S., et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 2012;33:1942–1953. doi: 10.1093/eurheartj/ehr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K.J., Parry D.A., Steinert P.M., Virata M.L., Wagner R.M., Angst B.D., Nilles L.A. Structure of the human desmoplakins. Implications for function in the desmosomal plaque. J. Biol. Chem. 1990;265:11406–11407. [PubMed] [Google Scholar]
- Gudbjartsson D.F., Arnar D.O., Helgadottir A., Gretarsdottir S., Holm H., Sigurdsson A., Jonasdottir A., Baker A., Thorleifsson G., Kristjansson K., et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- Guo H., Lu Y.W., Lin Z., Huang Z.P., Liu J., Wang Y., Seok H.Y., Hu X., Ma Q., Li K., et al. Intercalated disc protein Xinβ is required for Hippo-YAP signaling in the heart. Nat. Commun. 2020;11:4666. doi: 10.1038/s41467-020-18379-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev K., Khudiakov A., Zaytseva A., Perepelina K., Makeenok S., Kaznacheyeva E., Kostareva A. Impact of the DSP-H1684R genetic variant on ion channels activity in iPSC-derived cardiomyocytes. Cell. Physiol. Biochem. 2020;54:696–706. doi: 10.33594/000000249. [DOI] [PubMed] [Google Scholar]
- Herraiz-Martínez A., Tarifa C., Lozano-Velasco E., Jiménez-Sábado V., Casabella S., Hernández-Torres F., Daimi H., Vázquez Ruiz de Castroviejo E., Delpón E., Caballero R., et al. Novel PITX2 homeodomain-contained mutations from ATRIAL fibrillation patients deteriorate calcium homeostasis. Hearts. 2021;2:251–271. [Google Scholar]
- Hoorntje E.T., Te Rijdt W.P., James C.A., Pilichou K., Basso C., Judge D.P., Bezzina C.R., Van Tintelen J.P. Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017;113:1521–1531. doi: 10.1093/cvr/cvx150. [DOI] [PubMed] [Google Scholar]
- Jansen J.A., Noorman M., Musa H., Stein M., De Jong S., Van Der Nagel R., Hund T.J., Mohler P.J., Vos M.A., Van Veen T.A., et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab S., Darbar D., van Noord C., Dupuis J., Pfeufer A., Newton-Cheh C., Schnabel R., Makino S., Sinner M.F., Kannankeril P.J., et al. Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation. Eur. Heart J. 2008;30:813–819. doi: 10.1093/eurheartj/ehn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon R.C., Mezzano V., Wright A.T., Pfeiffer E., Chuang J., Banares K., Castaneda A., Ouyang K., Cui L., Contu R., et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014;23:1134–1150. doi: 10.1093/hmg/ddt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus F.I., Fontaine G.H., Guiraudon G., Frank R., Laurenceau J.L., Malergue C., Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- Martherus R., Jain R., Takagi K., Mendsaikhan U., Turdi S., Osinska H., James J.F., Kramer K., Purevjav E., Towbin J.A. Accelerated cardiac remodeling in desmoplakin transgenic mice in response to endurance exercise is associated with perturbed wnt/β-catenin signaling. Am. J. Physiol. Heart Circ. Physiol. 2016;310:H174–H187. doi: 10.1152/ajpheart.00295.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe E.J., Erickson H.P., Bennett V. Desmoplakin I and desmoplakin II. Purification and characterization. J. Biol. Chem. 1989;264:8310–8318. [PubMed] [Google Scholar]
- Ouwerkerk A.F.V., Hall A.W., Kadow Z.A., Lazarevic S., Reyat J.S., Tucker N.R., Nadadur R.D., Bosada F.M., Bianchi V., Ellinor P.T., et al. Epigenetic and transcriptional networks underlying atrial fibrillation. Circ. Res. 2020;127:34–50. doi: 10.1161/CIRCRESAHA.120.316574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford E.M., Musa H., Maass K., Coombs W., Taffet S.M., Delmar M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007;101:703–711. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- Sato P.Y., Musa H., Coombs W., Guerrero-Serna G., Patiño G.A., Taffet S.M., Isom L.L., Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G., Kahr P.C., Morikawa Y., Zhang M., Rahmani M., Heallen T.R., Li L., Sun Z., Olson E.N., Amendt B.A., et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Zhang M., Li L., Bai Y., Zhou Y., Moon A.M., Kaminski H.J., Martin J.F. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ. Cardiovasc. Genet. 2014;7:23–32. doi: 10.1161/CIRCGENETICS.113.000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrado M., Franco D., Hernández-Torres F., Crespo-Leiro M.G., Iglesias-Gil C., Castro-Beiras A., Mikhailov A.T. Pitx2c is reactivated in the failing myocardium and stimulates Myf5 expression in cultured cardiomyocytes. PLoS One. 2014;9:e90561. doi: 10.1371/journal.pone.0090561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R., Gourdie R.G. STORM-based quantitative assessment of sodium channel localization relative to junctional proteins within the cardiac intercalated disk. Microsc. Microanal. 2016;22:1032–1033. [Google Scholar]
- Vermij S.H., Abriel H., van Veen T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017;113:259–275. doi: 10.1093/cvr/cvw259. [DOI] [PubMed] [Google Scholar]
- Wang W., Murray B., Tichnell C., Gilotra N.A., Zimmerman S.L., Gasperetti A., Scheel P., Tandri H., Calkins H., James C.A. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. Europace. 2022;24:268–277. doi: 10.1093/europace/euab183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Bowles N.E., Scherer S.E., Taylor M.D., Kearney D.L., Ge S., Nadvoretskiy V.V., DeFreitas G., Carabello B., Brandon L.I., et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Res. 2006;99:646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Deng C., Rao F., Modi R.M., Zhu J., Liu X., Mai L., Tan H., Yu X., Lin Q., et al. Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL-1 cardiomyocytes. Mol. Med. Rep. 2013;8:780–786. doi: 10.3892/mmr.2013.1594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mRNA sequencing datasets have been deposited with the Gene Expression Omnibus repository under accession numbers GEO: GSE208213 and GSE208212 for the patient and KI lines, respectively.