

Abstract
Tebentafusp is a first-in-class immunotherapy agent that comprises an engineered T-cell receptor targeting a gp100 epitope presented by human leukocyte antigen-A*02:01 cells, fused to an anti-CD3 single-chain variable fragment. Tebentafusp is both the first bispecific T-cell engager to show efficacy in the treatment of advanced solid cancer and the first anti-cancer treatment to demonstrate an overall survival benefit in patients with uveal melanoma (UM). This review article will focus on the clinical development of tebentafusp, the mechanism of action and resultant evolution of the management of advanced UM.
Keywords: gp100, ImmTAC, metastatic, tebentafusp, uveal melanoma
Introduction
Uveal melanoma (UM) is a rare cancer, with an approximate worldwide incidence of around 6500 cases per year, mainly affecting Caucasian patients.1,2 Treatment options for primary lesions include radiation (brachytherapy or proton beam therapy) or surgical enucleation. Approximately half of the patients will develop metastatic disease,3 and patients remain at lifelong risk of relapse. Most commonly, relapse occurs in the liver (~90%) with less common sites for metastatic spread including the lung, bone and skin.4 Risk of relapse is greatest at 1–5 years from initial diagnosis and is higher for patients over 50 years of age.5 Risk factors for relapse include larger size of primary tumour, higher mitotic count and the presence of two genetic alterations: monosomy 3 and 8q amplification.6,7 Genomic analysis can assist in risk stratification for relapse, aiding in surveillance decisions.
Despite significant improvements in the management of metastatic cutaneous melanoma (CM) over the last decade, similar improvements for patients with UM have been less forthcoming, and until recently the median survival for metastatic UM remained less than 1 year.8 No clinical trials of systemic agents have demonstrated significant survival advantages for patients to date (Table 1). Based on these trials, standard-of-care (SOC) treatment options have remained limited to the use of immune checkpoint inhibitors (ICPIs) which demonstrate only a very modest improvement in survival for most patients treated. When treatment responses do occur, they are often short-lived. Possible reasons for this include the low tumour mutational burden observed in UM,9 low programmed death-ligand 1 (PD-L1) expression10 and the immunosuppressive tumour microenvironment,11 as summarised in Figure 1.
Table 1.
Summary of past clinical trials in UM.
| Reference | Phase | Year | No. UM patients per arm | Location | Therapeutic agent(s) | mPFS (m) (95% CI) | mOS (m) (95% CI) |
|---|---|---|---|---|---|---|---|
| Cytotoxic chemotherapy | |||||||
| Schmittel et al.12 | II | 2005 | 19 | Germany | Gemcitabine + treosulfan + cisplatin | 3.0 (1.8–3.1) | 7.7 (1.9–13.8) |
| Schmittel et al.13 | II | 2006 | 24 versus 24 | Germany | Gemcitabine plus treosulfan (GeT) versus Treosulfan (T) | GeT: 3 (1.1–4.9) versus T: 2 (1.7–2.3) | |
| Homsi et al.14 | II | 2010 | 22 | United States | Docosahexaenoic acid–paclitaxel | 9.8 | |
| Leyvraz et al.15 | III | 2014 | 86 versus 85 | Multiple | Fotemustine (systemic (IV) versus HIA) | IV: 3.5 (2.0–4.1) versus HIA: 4.5 (4.1–6.0) | IV: 13.8 (10.2–17.2) versus HIA: 14.6 (10.2–15.4) |
| Piperno-Nuemnann et al.16 | II | 2016 | 35 | France | Temozolomide + bevacizumab | 3 (2.8–6) | 10 (8–15) |
| Small molecule inhibitors | |||||||
| Penel et al.17 | II | 2008 | 13 | France | Imatinib | 10.8 | |
| Mahipal et al.18 | Pilot | 2012 | 20 | United States | Sunitinib | 4.2 | 8.2 |
| Falchook et al.19 | I | 2012 | 16 | United States | Trametinib | 1.8 (1.8–3.7) | |
| Sacco et al.20 | II | 2013 | 38 versus 36 | International | Sunitinib (S) versus dacarbazine (D) | S: 2.76 (2.63–4.67) versus D: 3.88 (2.7–8.06) | S: 6.35 (3.29–8.42) versus D: 8.65 (6.25–13.55) |
| Carvajal et al.21 | II | 2014 | 50 versus 51 | International | Selumetinib versus Investigator choice chemotherapy | 3.9 (2.1–5.3) versus 1.8 (1.0–2.1) | 11.8 (9.8–15.7) versus 9.1 (6.1–11.1) |
| Mouriaux et al.22 | II | 2016 | 32 | International | Sorafenib | 7.8 | |
| Piperno-Neumann et al.23 | I | 2020 | 153 | International | Protein kinase C inhibitor AEB071 | 3.5 (2.5–3.6) | |
| Shoushstari et al.24 | I | 2021 | 24 | United States | Sotrastaurin (PKC inhibitor) and alpelisib (PI3Kα inhibitor) | 2 | 6 |
| ICPIs | |||||||
| Maio et al.25 | II | 2013 | 82 | Italy | Ipilimumab | 3.6 (2.8–4.4) | 6.0 (4.3–7.7) |
| Zimmer et al.26 | II | 2015 | 53 | Germany | Ipilimumab | 2.8 (2.5–2.9) | 6.8 (3.7–8.1) |
| Joshua et al.27 | II | 2015 | 11 | Canada | Tremelimumab | 2.9 (2.8–3) | 12.8 (3.8–19.7) |
| Pelster et al.28 | II | 2020 | 35 | United States | Ipilimumab/nivolumab | 5.5 (3.4–9.5) | 19.1 (9.6–NR) |
| Piulats Rodriguez et al.29 | II | 2021 | 52 | Spain | Ipilimumab/nivolumab (GEM-1402) | 3.0 (2.0–4.1) | 12.7 (7.1–18.3) |
Included studies were prospective in design and included a minimum of 10 patients with UM.
CI, confidence interval; HIA, hepatic intra-arterial; ICPIs, immune checkpoint inhibitors; m, months; mOS, median overall survival; mPFS, median progression-free survival; NR, not reached; UM, uveal melanoma.
Figure 1.

Theories behind poor ICPI response in UM.9–11,30
ICPI, immune checkpoint inhibitor; UM, uveal melanoma.
Due to the generally poor outcomes for UM, the recommendation for patients with metastatic UM is to be offered clinical trials wherever possible.31 International guidance also recommends consideration of locoregional management of liver disease including resection, ablation or stereotactic management in those patients for whom this is clinically and technically feasible.31,32 However, with the recent approval of tebentafusp, the treatment landscape of metastatic UM is finally set to change.
ImmTACs
Immune mobilising monoclonal T-cell receptors against cancer (ImmTACs) are a class of bispecific T-cell engagers. Tebentafusp is the first ImmTAC shown to demonstrate a survival benefit in any solid tumour.33 ImmTACs combine a soluble T-cell receptor (TCR) domain fused to an effector activating domain. In the case of tebentafusp, this is an anti-CD3 single-chain variable fragment (scFv). The anti-CD3 scFv domain recruits and activates CD3+ T cells, while the TCR domain recognises peptides of interest presented by human leukocyte antigen (HLA) complexes. The two domains are linked by a disulphide bond (Figure 2(a)). As is the case with all T-cell therapeutic platforms, chosen epitopes should be expressed at high level by the tumour but minimally expressed elsewhere to minimise on-target, off-tumour toxicity. Furthermore, in contrast to antibody-based therapies, ImmTACs have the advantage of being able to target intracellular epitopes, where 90% of the neo-antigen pool are thought to be derived. While natural TCRs tend to possess low antigen affinity, ImmTAC TCRs are affinity enhanced to increase TCR binding to target. The T cells activated in the tumour microenvironment are not necessarily tumour specific but are directed to the tumour cell target by the ImmTAC. ImmTACs can therefore direct tumour-specific toxicity of non-tumour-specific T cells. T-cell activation induces the release of pro-inflammatory cytokines and cytolytic agents which ultimately leads to cell lysis. In turn, cell lysis may encourage ‘epitope spreading’ which can further stimulate T-cell activation,34 as well as PD-L1 upregulation within the tumour microenvironment.35
Figure 2.
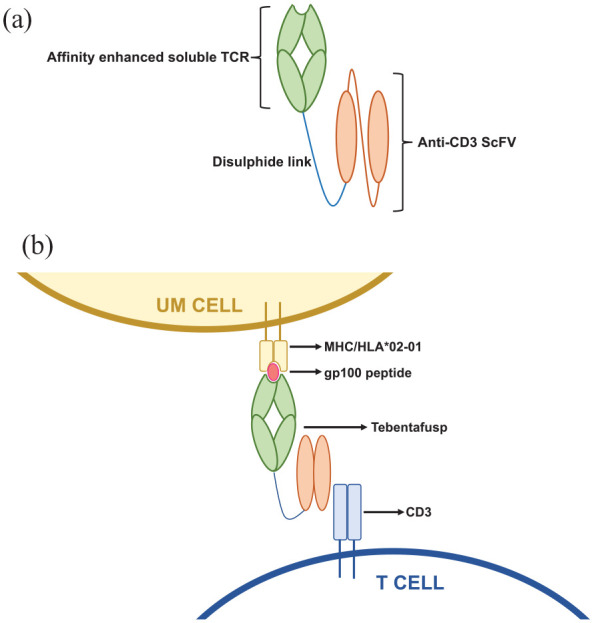
(a) Structure of ImmTAC – a soluble TCR domain and an anti-CD3 scFv domain joined by a disulphide linker. (b) Schematic demonstrating the mechanism of action of tebentafusp: The TCR domain binds HLA-A*02:01-positive melanoma cells presenting a melanoma-associated antigen gp100-derived peptide, and the anti-CD3 scFv recruits T cells.
HLA, human leukocyte antigen; ImmTAC, immune mobilising monoclonal T-cell receptors against cancer; scFv, single-chain variable fragment; TCR, T-cell receptor.
The main limitation of ImmTACs is the HLA restricted antigen presentation of the TCR target peptide. Given that differing HLA haplotypes will bind and present different peptides to TCRs, HLA restricted ImmTAcs will only be effective in a population expressing the relevant HLA haplotype. However, it has been argued that this is not unlike the restricted use of drugs targeting specific genetic alterations.36 In the case of tebentafusp, patients must have the HLA-A*02:01 allele. There is substantial heterogeneity in the representation of all HLA-A*02 subtypes worldwide, with prevalence higher in European and North American Caucasian populations (~30%), and lower in Black and South Asian populations (~20–25%). HLA-A*02:01 is the most common HLA-A*02 variant seen in Caucasian patients (the population most frequently affected by UM) but is significantly less prevalent in some ethnic groups including South Asians.37,38 Despite this, HLA restriction leaves a significant proportion of patients ineligible for treatment. A second possible drawback of these treatments is potential variability in target epitope expression. Identification of tumour-associated peptide antigen targets which are highly expressed on the tumour population is therefore desirable. The main advantages and limitations of ImmTACs are summarised in Table 2.
Table 2.
Summary of the advantages and disadvantages of ImmTACs over other immunotherapy treatment modalities.
| Advantages | Disadvantages |
|---|---|
| Ability to target peptides derived from intracellular targets | HLA restriction excludes patients |
| Ability to recruit non-tumour-specific T cells | On-target, off-tumour side effects with potential for significant toxicity |
| Can be produced on relatively large scale | Limited number of tumour-specific antigen targets so far |
| No pre-conditioning/prior immunosuppression required | Limited scope for preclinical testing in animal models |
| Heterogeneity of target expression |
HLA, human leukocyte antigen; ImmTAC, immune mobilising monoclonal T-cell receptors against cancer.
Tebentafusp
Tebentafusp redirects T cells towards HLA-A*02:01-positive UM cells presenting a melanoma-associated antigen glycoprotein 100 (gp100)-derived peptide (antigen gp100280–288)39 (Figure 2(b)). Gp-100 is a melanocyte-specific molecule involved in the maturation of melanosomes; organelles responsible for transporting melanin. Gp100 has been investigated in early phase trials as a therapeutic target for the treatment of both cutaneous and UM using approaches including peptide vaccines and adoptive cell transfer. Although results from these trials are generally encouraging, tebentafusp is the first gp100-targeted treatment to gain regulatory approval,40 following the publication of positive phase I–III trial data (Table 3).
Table 3.
Summary of trials utilising tebentafusp.
| Trial | Phase | Year | Number of patients (UM/CM) | Therapeutic agent(s) | mPFS (m) (95% CI) | mOS (m) (95% CI) |
|---|---|---|---|---|---|---|
| Tebentafusp (ImmTAC) | ||||||
| IMCgp100-01 (Middleton et al.41) | I | 2016 | 61 CM, 19 UM | IMCgp100 (tebentafusp) | NA | 33.4 (13.9–47.2) |
| IMCgp100-102 (Carvajal et al.42 ) | I/II | 2020 | 127 UM | IMCgp100 (tebentafusp) | 2.8 (2–3.7) | 16.8 (12.9–21.3) |
| IMCgp100-202 (Nathan et al.33) | III | 2021 | 252 UM versus 126 UM | Tebentafusp (T) versus Investigator choice SOC (dacarbazine, ipilimumab or pembrolizumab) | T: 3.3 (3.0–5.0) versus SOC: 2.9 (2.8–3.0) | T: 21.7 (18.6-28.6) versus. SOC: 16 (9.7–18.4) |
CI, confidence interval; CM, cutaneous melanoma; ImmTAC, immune mobilising monoclonal T-cell receptors against cancer; m, months; mOS, median overall survival; mPFS, median progression-free survival; NA, not available; SOC, standard of care; UM, uveal melanoma.
IMCgp-100-01 was the first-in-human, phase I trial of tebentafusp, involving 84 patients with CM and 18 UM patients.41 Of these 18 patients, 3 patients (16.7%) had a partial response to treatment, with a further 8 patients (44.4%) demonstrating stable disease. These results demonstrated promising clinical activity of tebentafusp, and the study also established a recommended phase II dose. The toxicity profile of tebentafusp was in line with the postulated mechanism of action.
Following this, the IMCgp100-102 single-arm phase I–II trials recruited 127 patients who had previously undergone treatment with at least one prior line of therapy for metastatic UM.43 For the whole cohort, while median progression-free survival (PFS) was just 2.8 months, median overall survival (OS) was 16.8 months. The overall response rate (ORR) by RECIST 1.144 was 5%, but 44% of those patients with measurable disease (n = 116) had some degree of reduction in the size of target lesions, and the 12-month OS was 86% for this subgroup of patients. This compares to previously demonstrated OS of 37% at 12 months seen in a systematic review of second-line UM treatments.45 In line with the phase I trial data, toxicity was predictable, manageable and decreased in severity and frequency over the first few doses. In this trial, development of rash appeared to be a positive predictor of treatment response,46 but the association was subsequently found not to be significant in a multi-variant analysis following publication of phase III trial data.33
A larger phase III study, IMCgp100-20233 was then conducted with a primary endpoint of OS rather than ORR. A further co-primary endpoint was overall survival in patients who developed a rash. In this randomised controlled trial, treatment-naïve UM patients were randomised 2:1 to receive tebentafusp as a first-line therapy (n = 252) versus clinician choice of SOC treatment (n = 126); pembrolizumab (n = 103), ipilimumab (n = 16) or dacarbazine (n = 7).
Eligible patients required at least one measurable lesion and a performance status of 0 or 1. Treatment with tebentafusp could continue beyond initial radiological disease progression in the absence of clinical progression, clinical deterioration and dose-limiting toxicity. However, patients were not allowed to continue treatment beyond the second response scan if this also demonstrated continued progressive disease (PD). The rationale for this was based on established evidence that patients may derive benefit from immunotherapy agents even after initial radiological appearances of PD (pseudoprogression). Similarly, patients in the trial receiving pembrolizumab or ipilimumab as SOC were allowed to continue beyond initial progression if they met the aforementioned criteria.
At initial interim analysis after a median of 14.1 months of follow-up, a significant improvement in OS was observed for patients receiving tebentafusp; 21.7 months [confidence interval (CI): 18.6–28.6] compared to 16.0 months (CI: 9.7–18.4) for patients receiving SOC [hazard ratio (HR): 0.51, p < 0.001]. Interestingly, the improvement in PFS was much less pronounced for patients treated with tebentafusp compared with patients treated with SOC (3.3 months versus 2.9 months, HR: 0.73, 95% CI: 0.58–0.94, p = 0.01). An even greater overall survival benefit with tebentafusp was observed in patients for whom the best response was PD (OS of 15.3 months versus 6.5 months, HR for death 0.43).33 These observations imply tebentafusp induces a change in the tumour micro-environment which is profound enough to reduce the rate of tumour progression to deliver a significant survival benefit but in a way that is not sensitively measurable by standard radiological techniques.
Although radiological responses did not correlate well with survival benefit, emerging data on reduction in ctDNA in patients on the 102 expanded phase II study showed linear correlation between the degree of ctDNA reduction and improvement of overall survival.43 More than 90% of patients being treated in the second-line and third-line settings had detectable ctDNA which correlated with tumour burden. The extent of reduction in ctDNA levels in patients treated with tebentafusp was closely correlated with OS benefit. This observation has led to the suggestion that serial measurement of ctDNA may represent a more clinically accurate marker of response than scan assessment in patients receiving ImmTACs,47 especially in the first few months of treatment.
One criticism of the IMCgp100-202 study design has been that patients in the SOC arm were not permitted treatment with doublet immunotherapy (ipilimumab/nivolumab). Two single-arm phase II studies have reported outcomes in UM patients treated with ipilimumab/nivolumab.28,48 One-year landmark OS in these studies was not however superior to that seen in the SOC arm of IMCgp100-202 (52% and 56% versus 58.5%). A cross-trial data comparison was performed between GEM-1402 and IMCgp100-202,48 using statistical methods to adjust for imbalances in the patient variables between the two trials. Although no significant difference was observed between pembrolizumab and ipilimumab/nivolumab (HR: 0.74, 95% CI: 0.45–1.21), tebentafusp demonstrated superior OS over ipilimumab/nivolumab (HR: 0.51, 95% CI: 0.32–0.79).29 Interestingly, an analysis of outcomes for patients on the two ipilimumab/nivolumab phase II studies compared to matched historical controls treated implied that the only patients who have benefit from this combination are the rare group of UM patients who have disease limited to extra-hepatic sites.49
Pharmacokinetics and pharmacodynamics
Pharmacodynamic studies of tebentafusp have demonstrated increased levels of pro-inflammatory cytokines including CXCL10, CXCL11, interleukin (IL)-6, interferon gamma (IFN-γ), tumour necrosis factor alpha (TNFα) after treatment. Serum levels of IFN-γ, TNFα, IL-2 and Il-10 are seen to increase to up to 10-fold that of pre-treatment levels in the 8–24 h after drug administration.35 Levels then fall to almost pre-treatment levels prior to the next dose administration. The amplitude of serum cytokine elevation is also observed to be smaller after three doses have been administered.35
Tumour tissue studies show increases in CD3+, CD4+ and CD8+ lymphocyte infiltration and increased expression of the cell death marker cleaved caspase 3,35 suggesting clear evidence of an increase in tumor-infiltrating lymphocytes (TILs) following treatment, with associated tumour cell death. In a sub-study within the phase I clinical trial, pre- and post-treatment tumour biopsies from 11 patients (8 CM, 1 UM, 1 acral melanoma and 1 lentiginous melanoma) were compared. An at least twofold increase in the number of intratumoral T cells following treatment was observed in 8 out of the 11 patients (73%). In the single UM patient included in the sub-study, a low level of intratumoural T cells was observed pre-treatment, which increased noticeably following treatment with tebentafusp.35
Pharmacokinetic studies demonstrate that tebentafusp has a half-life of around 7.5 h (FDA, 2022). Studies suggest that body weight, age, gender, renal or liver dysfunction do not significantly impact this. To date however, patients with severe renal function (creatinine clearance < 30 ml/min) or moderate to severe liver dysfunction (total bilirubin > 1.5 × ULN) have been excluded from studies. Roughly, one-third of patients in the reported clinical trials are noted to develop anti-tebentafusp antibodies.33 To date, there is no evidence that outcomes are worse for patients who developed antibodies. The use of tebentafusp has not been found to cause reduced tumoral gp100 expression.35
Dosing considerations
In the phase I trial, there were two treatment arms. In arm 1, patients received weekly treatment with a dose escalation from 20 µg (cycle 1 day 1) to 30 µg (cycle 1 day 8) to 68 µg (cycle 1 day 15). This approach was adopted to reduce the frequency of higher-grade cytokine release syndrome (CRS) during therapy induction. In arm 2, patients received daily treatment for 4 days every 3 weeks with doses ranging from 10 to 50 µg between participants. The recommended phase II dose was 68 µg.
Acute toxicity tends to peak around 4–6 h after drug administration and for this reason, treatment delivery should ideally be early in the day, in a clinician-supported treatment delivery facility, with an extended monitoring period available for patients receiving the first few doses of treatment.
Adverse events and toxicity management
Adverse events (AEs) can be grouped into two main categories (Table 4): cytokine-mediated toxicity and skin toxicity. Cytokine-release-mediated AEs include pyrexia, chills and hypotension, usually occurring some hours after drug administration and are generally short-lived. In contrast to this, skin toxicities relate to the on-target off-tumour effect of gp100 expression in normal melanocytes and take somewhat longer to resolve – usually days. Most toxicities occur during the first cycle of treatment. In the phase III trial, Grade 3 and 4 AEs were seen in 44% of patients in the tebentafusp arm versus 17% in the control arm. There were no treatment-related deaths and only 2% of patients discontinued treatment with tebentafusp due to treatment-related AEs. This compared to a discontinuation rate of 5% in the control group.
Table 4.
AEs observed following tebentafusp administration.33
| Toxicity | All grade (%) | G1–2 (%) | G3–4 (%) |
|---|---|---|---|
| Cytokine mediated | |||
| CRS | 89 | 88 | 1 |
| Pyrexia | 76 | 72 | 4 |
| Chills | 47 | 46 | <1 |
| Hypotension | 38 | 35 | 3 |
| Skin toxicity | |||
| Rash | 83 | 65 | 18 |
| Pruritus | 69 | 65 | 4 |
| Erythema | 23 | 23 | 0 |
| Other toxicity | |||
| Nausea | 43 | 42 | 1 |
| Fatigue | 41 | 38 | 3 |
| AST elevation | 19 | 15 | 4 |
AEs, adverse events; AST, aspartate aminotransferase; CRS, cytokine release syndrome.
Cytokine-mediated AEs
CRS is defined by the presence of hypotension, hypoxia and pyrexia. Almost 90% of patients treated with tebentafusp experience CRS of any grade, usually within a few hours of drug administration. 99% of CRS observed was Grade 1 or 2 (G1-2).
Patients with G1-2 CRS may develop fever, hypotension, tachycardia and rigours. These are seen mostly in the first 2–12 h following administration, peaking 4–6 h post-dose. Patients on antihypertensive agents should omit these for 48 h prior to, and 24 h after, their first few of tebentafusp, with the timing of restarting antihypertensives dependent on their clinical condition. Early detection of hypotension is vital; a baseline blood pressure (BP) measurement should be taken prior to commencement of therapy based on at least three separate measurements taken over a period of at least several minutes. BP should then be checked at least every 2 h after treatment. Patients may be managed with delivery of antipyretic agents and intravenous (IV) fluids. In cases of hypotension refractory to IV fluid administration, IV glucocorticoid administration may be required, under the guidance of a senior clinician. A suggested management algorithm for cytokine-mediated hypotension is outlined in Figure 3(a).
Figure 3.

(a) Management algorithm for cytokine-mediated hypotension following tebentafusp administration. Patients on antihypertensives should omit these for 48 h before, and 24 h after treatment with tebentafusp for the first few doses. (b) Stepwise management of pyrexia.
SBP = systolic blood pressure, IV = intravenous, PO = oral, TDS = three times daily, QDS = four times daily, CI = contraindication.
Due to the potential for life-threatening CRS to occur, it is important that centres ensure adequate support and training for the clinical team. Establishing a clear protocol of management for the described immune toxicity is vital. In centres with critical care links, it is important that these teams are educated on CRS management. Careful patient selection is an important consideration, with consideration paid to the renal and cardiac function of patients eligible for tebentafusp who may require aggressive fluid resuscitation. Given the role for corticosteroids in the management of fluid refractory CRS, patients with inadequate adrenal function, or on long-term corticosteroids should have their baseline steroid dose doubled prior to and immediately following drug administration. A proposed management algorithm for severe or fluid refractory CRS is outlined in Figure 3(b).
Skin toxicity
The most common skin toxicities include rash, pruritus and erythema, and most often occur within the first few days of drug administration.35 The majority are low grade (65%) but higher grade or refractory cases have been seen (18%). In general, skin toxicity is easily managed with supportive measures. Non-pharmacological interventions to manage mild (G1) skin toxicity include fans and cold showers, while for persistent or higher-grade (>G2) toxicity, pharmacological interventions including antihistamines, emollients and topical steroids may be of use. In management of pruritus, calamine containing topical therapies, emollients and antihistamines may have utility (Figure 4).
Figure 4.

Management of skin toxicity (pruritus). IV = intravenous, PO = oral, QDS = four times daily, PRN = as needed.
Other toxicities
Fluctuations in liver function tests may be noted either due to inflammation related to drug action or clinical disease progression. In cases of Grade 3 liver dysfunction, tebentafusp should be held until results return to Grade 1 or normal ranges. Management of liver dysfunction should be in line with local management guidelines for drug-induced liver injury.
Conclusions and future directions
There is scope to build upon the initial success of tebentafusp with combination studies underway, investigating tebentafusp together with ICPIs. Current studies include tebentafusp alone versus tebentafusp with durvalumab ± tremelimumab (NCT02535078). These studies may uncover ways to increase ICPI activity in other tumours which have been up until now considered immunologically ‘cold’. Of note, in the phase I dose escalation study, among patients who received ICPIs following tebentafusp, there were a group who had a superior than expected response to ICPIs.42 This may be due to upregulation of PD-1 and PD-L1; epitope spreading by tebentafusp and potentially an increased presence of TILs. ICPIs in UM may therefore possess greater efficacy if given following tebentafusp delivery. To investigate this, a study is proposed which will evaluate the benefit from pembrolizumab and lenvantinib when given prior to versus following tebentafusp (NCT05282901). The final results of UM trials investigating other agents, including nivolumab with relatlimab (a LAG3-directed ICPI) (NCT04552223), and darovasertib, a PKC inhibitor, either alone or in combination with crizotinib (NCT03947385), are awaited, and may guide future combination study designs.
Future trials could also evaluate the role of tebentafusp in the adjuvant treatment of patients diagnosed with high-risk UM.50 These studies, if successful, could dramatically change the treatment landscape for patients with UM. Despite this, just as is the case with adjuvant ICPI treatment, attention must be paid to the risks and benefits of such treatments given the potential quality of life impact on patients; not only because of toxicity risk, but also because of the relatively intense drug delivery schedule.
Liquid biopsies, including ctDNA, are finding an ever-expanding role in management of malignant disease.51 The tebentafusp clinical trials published to date suggest that monitoring of ctDNA levels may yet become a more accurate method of response assessment that conventional imaging, especially in the months immediately following treatment initiation.47 In a wider clinical context, ctDNA monitoring has the potential to become a commonly used tool to guide clinical decision-making in all melanoma patients, and future trials should include translational sub-studies to investigate the utility of this.52 TebeMRD (NCT05315258) aims to establish whether treating uveal and CM patients who develop molecular relapsed disease after definitive treatment (assessed via serial measurements of ctDNA), can prevent or delay the appearance of macroscopic disease.
A limitation to the use of tebentafusp is the requirement for high-intensity monitoring, This, coupled with the weekly dosing schedule may be difficult to replicate in healthcare settings where oncology services may be far from the population they serve or in poor-income countries. An area of further research/consideration is to assess whether CRS effects are predictable or preventable without loss of efficacy. This may reduce the need for initial admissions.
As well as a potentially expanded role for tebentafusp, other bispecific immunotherapy agents are now being developed for use in other solid tumours. An immTAC targeting a MAGE-4 peptide (IMC-C103C) is being investigated for use in malignancies such as upper gastrointestinal, and head and neck cancers,53 and early phase studies are underway investigating IMC-F106C, an immTAC targeting PRAME, a tumour antigen overexpressed in tumours including squamous cell carcinoma, small-cell lung cancer, endometrial carcinoma as well as melanoma.54 IMC-F106C is being studied both as a single agent and in combination with both ICPIs and tebentafusp (NCT04262466). Limitations of both these agents, like tebentafusp, is their restriction to HLA*0201-positive individuals. Strategies to overcome this limitation and account for population HLA diversity, such as employing non-polymorphic HLA molecules, may circumvent this problem in the future.39 Outside of use in malignancies, other bispecifics are in development which may be utilised in the management of HIV or Hepatitis B.55,56 Finally, in addition to targeting gp100 in UM, ongoing research should look to identify other druggable targets in this rare disease. Emerging evidence suggests an evolving role for human endogenous retroviruses (HERV) in the molecular pathogenesis of UM. Specific HERV loci are identified in primary UM and may represent potential neoantigenic targets for future immunotherapeutics.57
The recent approval of tebentafusp is an exciting step forward not just in the field of cancer immunotherapy but also in the treatment of UM; a rare, aggressive and notoriously treatment-resistant malignancy. The clinical applications for tebentafusp are likely to expand as combination and adjuvant studies are conducted and reported on. In the future, wider clinical applications for ImmTAC agents and other bispecific antibody platforms may further change the already varied treatment landscape for a wide range of solid tumour malignancies.
Acknowledgments
None.
Footnotes
ORCID iD: Paul D. Nathan  https://orcid.org/0000-0002-2327-3250
https://orcid.org/0000-0002-2327-3250
Contributor Information
Sarah Howlett, Mount Vernon Cancer Centre, Northwood, Middlesex, UK.
Thomas J. Carter, Mount Vernon Cancer Centre, Northwood, Middlesex, UK
Heather M. Shaw, Mount Vernon Cancer Centre, Northwood, Middlesex, UK University College London Hospital, London, UK.
Paul D. Nathan, Mount Vernon Cancer Centre, Rickmansworth Road, Northwood, Middlesex HA6 2RN, UK.
Declarations
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Author contribution(s): Sarah Howlett: Writing – original draft; Writing – review & editing.
Thomas J. Carter: Writing – original draft; Writing – review & editing.
Heather M. Shaw: Conceptualisation; Supervision; Writing – review & editing.
Paul D. Nathan: Conceptualisation; Supervision; Writing – review & editing.
Funding: The authors confirmed that no financial support was received for the research, authorship, and/or publication of this article
The authors declare that there is no conflict of interest.
Availability of data and material: Not applicable.
References
- 1. Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 2009; 93: 1129–1131. [DOI] [PubMed] [Google Scholar]
- 2. Mahendraraj K, Lau CS, Lee I, et al. Trends in incidence, survival, and management of uveal melanoma: a population-based study of 7,516 patients from the Surveillance, Epidemiology, and End Results database (1973–2012). Clin Ophthalmol 2016; 10: 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Damato BE, Dukes J, Goodall H, et al. Tebentafusp: T cell redirection for the treatment of metastatic uveal melanoma. Cancers 2019; 11: 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lorigan J, Wallace S, Mavligit G. The prevalence and location of metastases from ocular melanoma: imaging study in 110 patients. AJR Am J Roentgenol 1991; 157: 1279–1281. [DOI] [PubMed] [Google Scholar]
- 5. Kath R, Hayungs J, Bornfeld N, et al. Prognosis and treatment of disseminated uveal melanoma. Cancer 1993; 72: 2219–2223. [DOI] [PubMed] [Google Scholar]
- 6. Robertson AG, Shih J, Yau C, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017; 32: 204–220.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eleuteri A, Taktak AF, Coupland SE, et al. Prognostication of metastatic death in uveal melanoma patients: a Markov multi-state model. Comput Biol Med 2018; 102: 151–156. [DOI] [PubMed] [Google Scholar]
- 8. Eskelin S, Pyrhönen S, Hahka-Kemppinen M, et al. A prognostic model and staging for metastatic uveal melanoma. Cancer 2003; 97: 465–475. [DOI] [PubMed] [Google Scholar]
- 9. Helgadottir H, Höiom V. The genetics of uveal melanoma: current insights. Appl Clin Genet 2016; 9: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Javed A, Arguello D, Johnston C, et al. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 2017; 9: 1323–1330. [DOI] [PubMed] [Google Scholar]
- 11. Krishna Y, McCarthy C, Kalirai H, et al. Inflammatory cell infiltrates in advanced metastatic uveal melanoma. Hum Pathol 2017; 66: 159–166. [DOI] [PubMed] [Google Scholar]
- 12. Schmittel A, Scheulen ME, Bechrakis NE, et al. Phase II trial of cisplatin, gemcitabine and treosulfan in patients with metastatic uveal melanoma. Melanoma Res 2005; 15: 205–207. [DOI] [PubMed] [Google Scholar]
- 13. Schmittel A, Schmidt-Hieber M, Martus P, et al. A randomized phase II trial of gemcitabine plus treosulfan versus treosulfan alone in patients with metastatic uveal melanoma. Ann Oncol 2006; 17: 1826–1829. [DOI] [PubMed] [Google Scholar]
- 14. Homsi J, Bedikian AY, Papadopoulos NE, et al. Phase 2 open-label study of weekly docosahexaenoic acid–paclitaxel in patients with metastatic uveal melanoma. Melanoma Res 2010; 20: 507–510. [DOI] [PubMed] [Google Scholar]
- 15. Leyvraz S, Piperno-Neumann S, Suciu S, et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): a multicentric randomized trial. Ann Oncol 2014; 25: 742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piperno-Neumann S, Diallo A, Etienne-Grimaldi M-C, et al. Phase II trial of bevacizumab in combination with temozolomide as first-line treatment in patients with metastatic uveal melanoma. Oncologist 2016; 21: 281–282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Penel N, Delcambre C, Durando X, et al. O-Mel-Inib: a Cancero-pole Nord-Ouest multicenter phase II trial of high-dose imatinib mesylate in metastatic uveal melanoma. Invest New Drugs 2008; 26: 561–565. [DOI] [PubMed] [Google Scholar]
- 18. Mahipal A, Tijani L, Chan K, et al. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res 2012; 22: 440–446. [DOI] [PubMed] [Google Scholar]
- 19. Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol 2012; 13: 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sacco JJ, Nathan PD, Danson S, et al. Sunitinib versus dacarbazine as first-line treatment in patients with metastatic uveal melanoma. J Clin Oncol, 2013; 31(15) suppl: 9031–9031. [Google Scholar]
- 21. Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 2014; 311: 2397–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mouriaux F, Servois V, Parienti J-J, et al. Sorafenib in metastatic uveal melanoma: efficacy, toxicity and health-related quality of life in a multicentre phase II study. Br J Cancer 2016; 115: 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piperno-Neumann S, Larkin J, Carvajal RD, et al. Genomic profiling of metastatic uveal melanoma and clinical results of a phase I study of the protein kinase C inhibitor AEB071genomic profiling of metastatic UM in a phase I trial of AEB071. Mol Cancer Ther 2020; 19: 1031–1039. [DOI] [PubMed] [Google Scholar]
- 24. Shoushtari AN, Khan S, Komatsubara K, et al. A phase Ib study of sotrastaurin, a PKC inhibitor, and alpelisib, a PI3Kα inhibitor, in patients with metastatic uveal melanoma. Cancers 2021; 13: 5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maio M, Danielli R, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann Oncol 2013; 24: 2911–2915. [DOI] [PubMed] [Google Scholar]
- 26. Zimmer L, Vaubel J, Mohr P, et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS One 2015; 10: e0118564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joshua AM, Monzon JG, Mihalcioiu C, et al. A phase 2 study of tremelimumab in patients with advanced uveal melanoma. Melanoma Res 2015; 25: 342–347. [DOI] [PubMed] [Google Scholar]
- 28. Pelster MS, Gruschkus SK, Bassett R, et al. Nivolumab and ipilimumab in metastatic uveal melanoma: results from a single-arm phase II study. J Clin Oncol 2021; 39: 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez JP, Piperno-Neumann S, Rutkowski P, et al. 823P a propensity score weighted comparison of tebentafusp or pembrolizumab versus combination ipilimumab and nivolumab in untreated metastatic uveal melanoma. Ann Oncol 2022; 33: S924. [Google Scholar]
- 30. Kaunitz GJ, Cottrell TR, Lilo M, et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab Invest 2017; 97: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Network NCC. NCCN clinical practice guidelines in oncology: uveal melanoma. Version 1.2019. National Comprehensive Cancer Network, 2019. [DOI] [PubMed] [Google Scholar]
- 32. Nathan P, Cohen V, Coupland S, et al. Uveal melanoma UK national guidelines. Eur J Cancer 2015; 51: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 33. Nathan P, Hassel JC, Rutkowski P, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med 2021; 385: 1196–1206. [DOI] [PubMed] [Google Scholar]
- 34. Bossi G, Buisson S, Oates J, et al. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol Immunother 2014; 63: 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Middleton MR, McAlpine C, Woodcock VK, et al. Tebentafusp, a TCR/anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma antitumor responses mediated by tebentafusp in melanoma. Clin Cancer Res 2020; 26: 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oates J, Hassan NJ, Jakobsen BK. ImmTACs for targeted cancer therapy: why, what, how, and which. Mol Immunol 2015; 67: 67–74. [DOI] [PubMed] [Google Scholar]
- 37. Hammond MG, du Toit ED, Sanchez-Maza A. Genetic diversity of HLA: functional and Medical implications. Anthropology report for sub-Saharan Africa (EDK Press) 1997; 345–352. [Google Scholar]
- 38. Mehra N, Jaini R, Rajalingam R, et al. Molecular diversity of HLA-A* 02 in Asian Indians: predominance of A* 0211. Tissue Antigens 2001; 57: 502–507. [DOI] [PubMed] [Google Scholar]
- 39. Lowe KL, Cole D, Kenefeck R, et al. Novel TCR-based biologics: mobilising T cells to warm ‘cold’tumours. Cancer Treat Rev 2019; 77: 35–43. [DOI] [PubMed] [Google Scholar]
- 40. Martinez-Perez D, Viñal D, Solares I, et al. Gp-100 as a novel therapeutic target in uveal melanoma. Cancers 2021; 13: 5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Middleton MR, Steven NM, Evans TJ, et al. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific t cell redirector with solid tumour activity: results from the FIH study in melanoma. J Clin Oncol, 2016; 34(15) suppl: 3016–3016. [Google Scholar]
- 42. Carvajal RD, Butler MO, Shoushtari AN, et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: a phase 2 trial. Nat Med 2022; 28: 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carvajal RD, Nathan P, Sacco JJ, et al. Phase I study of safety, tolerability, and efficacy of tebentafusp using a step-up dosing regimen and expansion in patients with metastatic uveal melanoma. J Clin Oncol 2022; 40: 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–247. [DOI] [PubMed] [Google Scholar]
- 45. Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res 2019; 29: 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sacco J, Carvajal R, Butler M, et al. 64MO a phase (ph) II, multi-center study of the safety and efficacy of tebentafusp (tebe) (IMCgp100) in patients (pts) with metastatic uveal melanoma (mUM). Ann Oncol 2020; 31: S1442–S1443. [Google Scholar]
- 47. Shoushtari A, Collins L, Espinosa E, et al. 1757O early reduction in ctDNA, regardless of best RECIST response, is associated with overall survival (OS) on tebentafusp in previously treated metastatic uveal melanoma (mUM) patients. Ann Oncol 2021; 32: S1210. [Google Scholar]
- 48. Piulats JM, Espinosa E, de la Cruz Merino L, et al. Nivolumab plus ipilimumab for treatment-naive metastatic uveal melanoma: an open-label, multicenter, phase II trial by the Spanish multidisciplinary melanoma group (GEM-1402). J Clin Oncol 2021; 39: 586–598. [DOI] [PubMed] [Google Scholar]
- 49. Piulats Rodriguez J, Patel S, Espinosa E, et al. Nivolumab and ipilimumab (N plus I) is active in patients (pts) with metastatic uveal melanoma (mUM) with extra-hepatic only involvement: pooled analysis from 2 phase II trials. Ann Oncol 2020; 31: S764. [Google Scholar]
- 50. Liu AW, Wei AZ, Maniar AB, et al. Tebentafusp in advanced uveal melanoma: proof of principle for the efficacy of T-cell receptor therapeutics and bispecifics in solid tumors. Expert Opin Biol Ther 2022; 22: 997–1004. [DOI] [PubMed] [Google Scholar]
- 51. Wan JC, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017; 17: 223–238. [DOI] [PubMed] [Google Scholar]
- 52. Tivey A, Britton F, Scott J-A, et al. Circulating tumour DNA in melanoma—clinic ready? Curr Oncol Rep 2022; 24: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishihara M, Kageyama S, Miyahara Y, et al. MAGE-A4, NY-ESO-1 and SAGE mRNA expression rates and co-expression relationships in solid tumours. BMC Cancer 2020; 20: 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moureau S, Vantellini A, Schlosser F, et al. IMC-F106C, a novel and potent immunotherapy approach to treat PRAME expressing solid and hematologic tumors. Cancer Res 2020; 80: 5572. [Google Scholar]
- 55. Fergusson JR, Wallace Z, Connolly MM, et al. Immune-mobilizing monoclonal T cell receptors mediate specific and rapid elimination of hepatitis B–infected cells. Hepatology 2020; 72: 1528–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang H, Buisson S, Bossi G, et al. Elimination of latently HIV-infected cells from antiretroviral therapy-suppressed subjects by engineered immune-mobilizing T-cell receptors. Mol Ther 2016; 24: 1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bendall ML, Francis JH, Shoushtari AN, et al. Specific human endogenous retroviruses predict metastatic potential in uveal melanoma. JCI Insight 2022; 7: e147172. [DOI] [PMC free article] [PubMed] [Google Scholar]