

Abstract
The historic term ‘histiocytosis’ meaning ‘tissue cell’ is used as a unifying concept for diseases characterized by pathogenetic myeloid cells that share histologic features with macrophages or dendritic cells. These cells may arise from the embryonic yolk sac, fetal liver, or post-natal bone marrow. Prior classification schemes align disease designation with terminal phenotype: for example, Langerhans cell histiocytosis (LCH) shares CD207+ antigen with physiologic epidermal Langerhans cells. LCH, Erdheim-Chester disease (ECD), juvenile xanthogranoluma (JXG), and Rosai-Dorfman disease (RDD) are all characterized by pathologic ERK activation driven by activating somatic mutations in MAPK pathway genes. The title of this Primer (‘Histiocytic Disorders’) was chosen to differentiate the above diseases from Langerhans cell sarcoma and malignant histiocytosis, which are hyper-proliferative lesions typical of cancer. By comparison LCH, ECD, RDD, and JXG share some features of malignant cells including activating MAPK pathway mutations, but are not hyper-proliferative. ‘Inflammatory myelo-proliferative neoplasm’ may be a more precise nomenclature. By contrast, hemohagocytic lymphohistiocytosis (HLH) is associated macrophage activation and extreme inflammation, representing a syndrome if immune dysregulation. These diseases affect children and adults in varying proportions depending on which of the entities is involved.
Introduction
Histiocytic disorders were originally classified by histological features of pathological cells compared with presumed physiological counterparts.1, 2 The term ‘histiocyte’ (Greek for ‘tissue cell’) was applied by Aschoff and Kiyono in 19133 to designate mononuclear phagocytes, cells of myeloid origin that now include blood and tissue dendritic cells (DC) and macrophages.1, 4, 5 An updated contemporary classification proposes to classify histiocytoses not only by terminal phenotype, but also taking into account molecular lesions and clinical features.2 (Box 1)
Box 1. Classification of histiocytoses.
| C Group |
|---|
| • Cutaneous non-LCH |
| - Non-XG family: includes cutaneous RDD |
| - XG family: includes JXG |
| • Cutaneous non-LCH with major systemic component |
| H Group |
| • Primary HLH (monogenic inherited conditions; FIG. 1) |
| • Secondary HLH (non-Mendelian HLH) |
| • HLH of uncertain origin |
| L Group |
| • LCH |
| • ICH |
| • ECD |
| • Mixed LCH/ECD |
| M Groupa |
| • Primary malignant histiocytoses |
| • Secondary malignant histiocytosis (following or in association with another haematological malignancy) |
| R Group |
| • Sporadic RDD |
| - Classic RDD |
| - Extranodal RDD |
| - RDD with neoplasia or immune disease |
| - Unclassified |
| • Familial RDD |
Somatic and germlme pathogenic variants associated with Histiocytic disorders are outlined in FIG. 1. ECD, Erdheim-Chester disease; HLH, haemophagocytic lymphohistiocytosis; ICH, indeterminate cell histiocytosis; JXG, juvenile xanthogranuloma; LCH, Langerhanscell histiocytosis; RDD, Rosai-Dorfman disease; XG, xanthogranuloma.
Malignant histiocytoses are not discussed in this review, but represent hyperproliferative, dysplastic malignancies with some histological features shared with histiocytic disorders.
This Primer discusses histiocytic diseases including Langerhans cell histiocytosis (LCH), Erdheim Chester Disease (ECD), juvenile xanthogranuloma (JXG), Rosai-Dorfman disease (RDD, sometimes known as Rosai–Dorfman–Destombes disease), and hemophagocytic lymphohistiocytosis (HLH). The first four diseases have features of transformed cells, though implications of oncogenic drivers on classification as ‘cancer’ remains an area of debate. ‘Hallmark’ cancer features include oncogenic driver mutations, apoptosis resistance, self-sufficiency in growth signals and tissue invasion and metastasis.6 However, these histiocytic cells have very low proliferative index and LCH has recently been characterize with senescence-associated secretory phenotype.7 The histiocytosis discussed here rarely transform to lymphoma-like malignant diseases such as histiocytic sarcoma or malignant histiocytosis. LCH, ECD, JXG and RDD could reasonably be described as inflammatory myeloid neoplastic disorders.8, 9 By contrast, HLH is clearly a syndrome of immune dysregulation.10
LCH primarily affects children of <10 years of age, with potential to develop lesions in virtually any organ system, most frequently bone and skin. Adults are also affected, typically in the third to fifth decades of life. Clinical presentations in children and adults are highly variable, with symptoms based on sites of disease. Lesions in spleen, liver and bone marrow are considered ‘high risk’ due to the association of increased potential for death11 compared with lesions in other ‘low risk’ sites. Extent of disease is typically established by the number of lesions and organ systems involved (for example, single lesion, single system; multifocal lesions, single system; or multifocal lesions, multisystem). Patients with pituitary lesions may present with diabetes insipidus and other endocrinopathies. LCH-associated neurodegeneration (LCH-ND) is a severe complication that can arise acutely with systemic symptoms or years after the initial LCH diagnosis, with clinical signs of cerebellar dysfunction and associated abnormal MRI findings in the in cerebellum, pons and brainstem.12 Clinical responses in LCH-ND have been reported with LCH-directed chemotherapy as well as MAPK inhibitors.9, 13
ECD affects primarily adults with clinical signs and symptoms similar to LCH. Xanthelasma (yellowish deposit of cholesterol under the skin) below the eyes is a unique clinical finding. Initial evaluations and staging are similar to LCH, with attention paid to characteristic potential distribution of lesions including heart, major blood vessels, kidneys and peritoneum. ECD had a more dire prognosis than LCH before the use of IFNα and, more recently, MAPK pathway inhibitors.14, 15 Despite substantial advances in life expectancy among patients with ECD, few are cured with current therapies.
JXG primarily occurs in children of <4 years of age, with single or multiple orange-red papules on the skin (xanthogranulomas, characterized by the presence of foamy macrophages (xanthoma cells)) that usually resolve spontaneously. Systemic disease can arise in the liver, spleen, kidney, brain, bone or other sites, is typically progressive without therapy, and is potentially fatal.
The classic presentation of RDD is in children of <10 years of age, with painless, massive enlargement of cervical lymph nodes that may spontaneously resolve. Rarely, RDD may cause bone lesions, subcutaneous nodules, proptosis (protruding eyeballs), or intra-cerebral masses. RDD probably represents a variety of conditions that share a common histological phenotype, accounting for greater variability in presentation and outcomes than other histiocytoses. Patients with progressive disease or with lesions in critical locations may require systemic chemotherapy.
HLH is a syndrome of extreme immune activation. Historically, HLH has been divided into primary (or genetic or familial) and secondary forms. Primary HLH pertains to patients with predisposing inherited defects caused by pathogenetic variants in genes that compromise cellular cytotoxicity, inflammasome function, or other cellular functions. ‘Secondary’ HLH refers to patients who develop a syndrome of HLH without an underlying defect in cytotoxicity, and often occurs in association with a strong immunologic stimulus such as infection, or malignancy or autoimmune/autoinflammatory condition. Patients, typically young children but occasionally adults, present with clinical and laboratory findings reflecting pathological inflammation and are at very high risk of death without prompt initiation of immune suppression. For patients with fixed immune defects associated with HLH, allogeneic hematopoietic stem cell transplant is required for long-term survival.
This Primer discusses current practice and evolving understanding of the epidemiology, mechanisms/pathophysiology, diagnosis, screening, prevention, management, quality of life and future outlook for patients with histiocytic disorders.
Epidemiology
The Histiocytic disorders discussed in this section are rare and inconsistently documented in disease registries. High-risk LCH was tracked in the Surveillance, Epidemiology, and End Results (SEER) database, but reporting was probably incomplete. Additional resources include the International Rare Histiocytic Disorders Registry (IRHDR) that opened in 2014, the Belgian Langerhans Cell Histiocytosis Registry, and the UK Histiocytosis Registry. These registries are important efforts to capture disease experience but have variable participation, and no data are yet publicly available.
Langerhans cell histiocytosis (LCH)
The current version of the International Classification of Diseases for Oncology (ICD‐O‐3) only includes high-risk LCH. Low-risk LCH (which includes LCH not otherwise specified (NOS), LCH unifocal and LCH multifocal) is not included.16 In the USA and Canada, cancer registries collect information about all forms of LCH. Most LCH cases occur during childhood, with age-adjusted incidence rates (in children of 0–14 years of age) varying between 2.6 and 8.9 new cases per million individuals per year, globally.17–19 The French National Registry of Childhood Hematopoietic Malignancies reported a standardized incidence rate (in children of 0–14 years of age) of 5 cases per million individuals between 2000–2004, with a slight predominance among male children.17 In Sweden, a population-based study described an annual incidence rate of 8.9 per million children of 0–14 years of age.19 Review of SEER programme databases in the USA found that African-American children had a lower relative risk compared to risk of white children developing LCH (0.41), whereas Hispanic children had a 1.63-fold higher risk than non-Hispanic children.16 A WHO publication on histiocytosis stated the incidence may be increased in northern European regions compared with Asian and African regions.20 Household crowding and reduced educational level (according to area-based measures) were also associated with increased risk of LCH among children and adolescents in the USA.16 This finding may be linked to an increased risk of infections that can trigger immune dysregulation as a theoretical, but unproven, cause of LCH. A large case-control study conducted in the USA and Canada identified that neonatal infections were associated with an increased risk (OR=3.8) of multisystem LCH (but no specific pathogen was identified), whereas childhood vaccinations were associated with decreased risk of localized and systemic LCH. (OR=0.4).21 Another case-control study of children living in Los Angeles, USA area reported an association with infections in the first year of life (OR=2.8); additionally, LCH was associated with a family history of cancer (OR=2.5) and parental exposures to heavy metals granites or wood dust in the work place (OR=2.5).22 A study from the Texas Cancer Registry matched individuals in the registry with healthy individuals born on the same year and found that children born of Hispanic mothers were 51% more likely to develop LCH than children with white non-Hispanic mothers; the risk was even more pronounced when both parents were Hispanic (adjusted OR=1.80).23 A GWAS study of patients with LCH and their parents found that a variant of SMAD6 that is present with increased frequency in Hispanic populations was associated with increased risk of LCH.24 A functional link between SMAD6 and LCH is unclear, although it is intriguing that the gene is in very close proximity to MAP2K1 (encoding ERK activator kinase 1 (MEK1)), a central component of the MAPK signaling pathway. Smoking is clearly linked to pulmonary LCH in adults, but no other co-morbidities have been identified.25 Interrogation of the SEER database from 2000–2009 identified 59 adult patients with disseminated LCH, of which 51 were white, 3 were black and 4 were of other ethnicities.26 It is likely that since SEER data were limited to adult disseminated LCH, the overall incidence for adult LCH is higher than the 0.07 per million per year estimated from this study. It seems probable that genetic and/or ethnic background has a role in susceptibility to LCH through mechanisms that are currently unknown.
Erdheim Chester disease (ECD)
The global incidence or prevalence of ECD is not known; however, the number of ECD cases world-wide since 1930 is estimated to be ~1,500, by taking into account the published case reports and series, the various cohorts of patients followed both in Europe and the USA, and the numbers from the ECD Global Alliance patient’s association (assuming unique cases among registries). ECD primarily affects adults (mean age at diagnosis of 55 years), with a male preponderance (3:1)) Childhood ECD is rare (<20 reported cases world-wide)27. Pediatric ECD could be considered disseminated JXG in most cases.28 ECD, like JXG, RDD and HLH, is not reportable by cancer registries. The Histiocyte Society Rare Disease Registry is attempting to characterize submitted cases.
Juvenile Xanthogranuloma (JXG)
Data from the Kiel Tumor Registry suggest that JXG occurs at a frequency of ~1 per million children.29 The male:female ratio is 1.4:1, with 35% of patients having lesions at birth and 71% developing lesions within the first year of life 29. There is a 3:1 predominance of females among patients with JXG with disseminated lesions.30 There are several case reports of children with neurofibromatosis type 1 and JXG, consistent with the known role of MAPK pathway activation in pathogenesis of both diseases.31
Rosai–Dorfman Disease (RDD)
Rosai-Dorfman Destombes Disease (RDD) was first described in 1965 by Destombes,32 who reported four cases of children or young adults with massive lymphadenopathy. Four years later, Rosai and Dorfman analyzed 34 cases of the same entity, which they named ‘sinus histiocytosis with massive lymphadenopathy’.33 These patients had massive cervical lymphadenopathy, with a benign, but prolonged clinical course.
Since the original publication of the RDD registry, 34 which included 423 cases in 1990, there have been >1,200 additional cases reported.34 Although RDD is most frequent in children and young adults, it can occur at any age (mean age at diagnosis 20.6 years). There are no studies to define incidence of RDD in children compared with adults.33 RDD is more common in boys, and the classic presentation of chronic benign cervical lymphadenopathy was initially described in sub-Saharan African boys and may be more common in African-Americans than in other ethnic groups in the USA.35 However, the cutaneous form is more frequent in females.36
Hemophagocytic Lymphohistiocytosis (HLH)
Data on the epidemiology of HLH is scarce, which makes it difficult to summarize differences in the incidence or prevalence in different ethnic groups. The first report from Sweden estimated the primary form of HLH to occur in 1 in 50,000 live born children37 and a more recent Swedish study reported an annual incidence of 0.12 per 100,000 children <15 years of age.38 A single center study in the USA estimated a frequency of 1 case of HLH for every 3,000 general hospital admissions to a large pediatric referral hospital and a cross-sectional prevalence of 1.07 cases per 100,000 persons less than 18 years old across Texas.39, 40 A nationwide survey in Japan found an annual incidence of 1 per 800,000 individuals of all ages, with a male/female ratio of 0.94 and <5% being familial cases.41 More than 56% of the patients were under 15 years of age, and a majority of all patients had Epstein–Barr virus (EBV) or other infections. For unknown reasons, EBV-associated HLH is more common in Asia than in US and Europe.42, 43 Additionally, EBV-associated HLH in people of Asian descent and from Central or South America frequently includes atypical T cell infection that is unusual in other populations.44 Inherited defects in genes regulating effector immune function are more common in infants than older children.45 The incidence of HLH in adults is not known, but is increasingly recognized over the past decade, with cancer-associated HLH being more common in adults than children, most frequently lymphomas or hematologic malignancies.46,47
Mechanisms/pathophysiology
LCH, ECD, JXG, RDD and HLH are characterized by the accumulation of cells with macrophage and/or dendritic cell (DC) phenotype in various tissues. All of these histiocytic disorders also present with an intense inflammatory infiltrate that has a key role in generating the organ-specific lesions and clinical symptoms. However, the pathological cell may have a different origin and/or phenotype in each condition. Whereas LCH, ECD, JXG and RDD have overlapping pathophysiology with accumulation of clonal mononuclear phagocytes, the origin of HLH disease is very different, as it primarily involves activated CD8+ T cells that in turn drive macrophage activation and extreme systemic inflammation.
Langerhans cell histiocytosis
LCH lesions contain varying proportions of the clonal pathological CD1a+, CD207+ dendritic cells (Langerhans cells) within an intense inflammatory infiltrate composed of macrophages, lymphocytes, eosinophils, multinucleated giant cells and, less commonly, neutrophils and plasma cells. Given the intense inflammatory infiltrate, the nature of LCH as a reactive inflammatory disorder versus neoplastic disorder was debated.48
In 1994, two different groups identified clonality of LCH cells using X-linked polymorphic DNA probes.49, 50 However, for many years, a pathogenetic genetic lesion remained elusive.51 In 2010, a recurrent V600E mutation in the serine/threonine-protein kinase B-raf (encoded by BRAF; noted throughout this Primer as BRAFV600E) was identified in 57% of LCH lesions52. BRAF is a central kinase of the RAS-RAF-MEK-ERK mitogen-activated protein kinase (MAPK) signal-transduction pathway involved in numerous cell functions. The BRAFV600E mutation renders the MAPK pathway constitutively active. This pathway is an evolutionarily conserved signaling cascade that transmits signals from cell surface receptors that normally execute programs related to cell cycle progression, differentiation, protein translation and resistance to cell death.53 Additional, mutually exclusive, clonal somatic activating mutations in genes of the MAPK pathway, including MAP2K1, ARAF, NRAS and KRAS, as well as alternative BRAF mutations have been subsequently described in LCH lesions.54–57 (Figure 1A) In patients with multiple lesions, the same somatic mutations are identified in every lesion, and in patients who relapse, the original mutation is consistently re-identified, supporting clonal nature of LCH. MAPK pathway activation is described as a universal feature of LCH, as confirmed by consistent ERK phosphorylation, even in cases without identified MAPK activating mutations.52 58
Figure 1. Mutations found in histiocytic disorders.

Bar graphs showing the genes and mutations that have been associated with LCH, ECD, JXG, and RDD (panel A) and HLH (panel B) and the percentage of patients carrying these variants. DIAP: Dysregulated Immune Activation and Proliferation-Associated Genes , PIDD: primary immune deficiency disease; WT: wild type (no mutations found) Data from Durham 81 and Chinn 45
Earlier models considered LCH cells to originate from cutaneous Langerhans cells (LCs), mostly owing to phenotypic similarities and the fact that the intracytoplasmic Birbeck granules (which can be found in LCH cells) were originally thought to be exclusive to epidermal LC.59 Ultrastructurally, the Birbeck granule is an intra-cytoplasmic rod-shaped structure of variable length with a central, periodically striated lamella.60 In the 2000s, the Birbeck granules were associated with expression of CD207, which encodes C-type lectin domain family 4 member K, an endocytic receptor (also known as CD207 antigen or Langerin) and was also expressed in both LC and LCH cells. Birbeck granules are now thought to be subdomains of the endosomal recycling compartment that forms where Langerin accumulates, and thus provides access to the non-classical antigen-processing pathway. Where physiological Langerin expression was initially thought to be restricted to epidermal LC, subsequent studies identified multiple lineages with potential to express Langerin, including dermal dendritic cells.61, 62 (Figure 2A) Additionally, transcriptional profiling of CD207+ cells from LCH lesions identified a signature more consistent with immature myeloid precursors than with differentiated epidermal LCs.8 Together, multiple origins of CD207+ cells and the fact that the gene expression signature of LCH cells is similar to that of relatively immature myeloid cells suggested that LCH may have potential to arise from multiple hematopoietic precursors other than differentiated epidermal LC.
Figure 2. Physiopathology of Langerhans cell histiocytosis.

(A): The BRAFV600E mutation is detected in about 50% of LCH lesions. Interestingly, this mutation is also detectable in pluripotent CD34+ hematopoietic progenitor cells (HPC) in the bone marrow and most particularly in CMP and GMP that are the bone marrow precursors of dendritic cells and monocytes. In the blood from LCH patients, the mutation can be mostly detectable in monocytes and dendritic cells but not in lymphocytes and neutrophils. This myeloid skewing has been further explored using mouse models. The BRAFV600E mutation was recently expressed in murine haematopoietic stem cells. Strikingly, the mice developed a LCH disease, suggesting that the expression of the BRAFV600E mutation in multipotent HPC is sufficient to drive to LCH lesion formation. Moreover, the mutated HPC acquired a senescence programme, so called senescence associated secretory phenotype (SASP),that is characterized by cell cycle arrest and secretion of cytokines, in particular IL-1 and IL-6. These cytokines partially participated in an autocrine and paracrine fashion in the myeloid skewing of the HPC observed in LCH. Thus, the myeloid skewing observed in LCH is driven by extrinsic and intrinsic cues. (B): The senescence programme that is started in the bone marrow is conserved in tissues. Mutated mononuclear phagocytes do also secrete a lot of cytokines as part of the SASP. For example, IL-6, IL-8 and IL-1 participate in recruiting inflammatory cells to the tissue, leading to granuloma formation. Matrix metalloproteinases have a role in the tissue destruction observed in LCH lesions. Moreover, mutated mononuclear phagocytes are trapped in the tissue because of CCR7 down-regulation that impairs their migration and because of BCL-2 and BCL-xL up-regulation that make them resistant to apoptosis. The MAPK pathway is an evolutionarily conserved signaling cascade that transmits signals from cell surface receptors that normally execute programs related to cell cycle progression, differentiation, protein translation and resistance to cell death. Mutations such as the BRAFV600E lead to constitutive activation of the pathway and uncontrolled cell growth as well as other effect mentioned above. HSC: hematopoietic stem cell; MPP, multipotent progenitors; Mep, megakaryocyte erythroid progenitors; Cmp, common myeloid progenitors; Gmp, granulocyte macrophage progenitors; Clp, common lymphoid progenitors.References:7, 61, 73
Role of the cell of origin and mutations.
The discovery of the BRAFV600E mutation provided the opportunity to track LCH cells in the hematopoietic system. BRAFV600E mutation was identified not only at the tissue site but also localized to CD34+ hematopoietic progenitor cells and myeloid precursors in some patients with LCH. Ability to detect BRAFV600E in bone marrow and peripheral blood (in patients with BRAFV600E+ lesions) was associated with disseminated high-risk LCH.63, 64 The percentage of haematopoietic progenitor cells or peripheral blood mononuclear cells harboring the mutation was extremely low, <0.1% in most cases.63, 65, 66
Based on these observations, we proposed a hypothesis that clinical presentation of LCH is defined at least in part by the cell of origin and specific mutations.9 Hence, if the BRAFV600E mutation affects a very immature hematopoietic progenitor, the disease will be disseminated, whereas if the BRAFV600E mutations occurs in a mature myeloid cell, the disease may have more restricted tissue distribution. This hypothesis was validated by generating a mouse model in which the BRAFV600E mutation was constitutively expressed in CD11c+ cells (CD11c is a marker of myeloid DC progenitors and monocyte). The mice developed an aggressive LCH-like phenotype.63 Strikingly, the phenotype of the disease was less severe when the BRAFV600E mutation was expressed in CD207+ cells (CD207 is expressed on mature epidermal Langerhans cells and some dermal DC). Notably, single cell study of LCH lesions identified transcription patterns consistent with heterogeneous differentiation within LCH lesions, likely reflecting impact of local factors on pathogenic cells.67
Further, the specific mutation probably has a role in clinical presentation and outcomes. For example, two retrospective series noted increased risk of treatment failure or relapse in patients with BRAFV600E.68, 69 Similarly, BRAFV600E was highly associated with potential to develop LCH-ND.12, 64 The overall mutation burden in LCH is quite low (median of 1 mutation in an exon in a single gene per patient).58 However, adults tend to have more somatic mutations in additional genes compared to children, and in some cases LCH can arise as part of myelodysplasia resulting from clonal hematopoiesis.70 Rarely, LCH can also arise as a clonal phenotype along with other hematopoietic malignancies.71, 72
Altered signaling.
Some mechanisms downstream of MAPK activation that drive LCH pathogenesis have been described. Interestingly, BRAFV600E is not associated with increased proliferation; rather, pathogenetic DCs accumulate and persist within LCH lesions.73 BRAFV600E expression in myeloid DCs inhibits expression of C-C chemokine receptor type 7 (CCR7), which is required for antigen-presenting cells to migrate to draining lymph nodes. Further, DC in LCH lesions upregulated the expression of Bcl-2-like protein 1 (also known as BCL-xL), conferring resistance to cell death.7, 73 (Figure 2B) Additionally, by mechanisms that have not yet been defined, DCs in LCH lesions recruit activated T cells that become exhausted, contributing to pathological local and systemic inflammation.74, 75
BRAFV600E oncogene-induced senescence has been proposed as key mechanism for LCH pathophysiology.7 Indeed, in a mouse model in which the BRAFV600E mutation is expressed under the Scl promotor (Scl is expressed on short-term and long-term hematopoietic stem cells), the mutated hematopoietic stem cells have a proliferative disadvantage compared to non-mutated hematopoietic stem cells, explaining the low fraction of mutant allele observed in patients with LCH. Moreover, the mutated hematopoietic stem cells predominately differentiate toward the mononuclear phagocyte compartment in a cell intrinsic and extrinsic manner. These mutated hematopoietic progenitors present all the canonical features of senescence (low proliferative capacity, high Cyclin-dependent kinase inhibitor 2A (CDKN2A) expression, high activity of senescence-associated β-galactosidase, and secretion of multiples pro-inflammatory cytokines and matrix metallo-proteinases). This senescence program is conserved in tissues and may be responsible for key features of LCH, such as the accumulation of poorly proliferative mononuclear phagocytes in tissues and the large infiltration of immune cells and subsequent fibrotic injuries.
LCH-associated neurodegenerative disease.
A progressive LCH-associated neurodegenerative disease (LCH-ND) arises in up to 10% of patients, sometimes years after initial disease is presumed to be cured.76 Aetiology of this phenomenon has not been well understood. Earlier studies described brain biopsies with enriched CD8+ T cells at sites of neurodegeneration, leading to speculation that LCH-ND could be an autoimmune or paraneoplastic phenomenon.77 More recently, BRAFV600E+ peripheral blood mononuclear cells have been identified in patients with LCH-ND without other systemic lesions.12
Further, BRAFV600E+ microglia-like cells are identified near blood vessels in areas of neurodegeneration (in patients with history of BRAF-V600E+ lesions).12 Moreover, these cells express markers of senescence.7 Notably, two studies found BRAFV600E mutation to be highly correlated with risk of LCH-ND12, 69. Further, in patients without active systemic LCH, persistent BRAFV600E+ peripheral blood mononuclear cells were highly specific for patients with LCH-ND.12,7 A mouse model in which BRAF-V600E is expressed on yolk-sac cells that seed embryonic brain with microglia resulted in progressive neurodegeneration.78 However, LCH-ND cells arise only after (or concurrent with) systemic lesions, and the microglia-like cells do not express some features of tissue-resident microglia (for example, P2Y purinoceptor 12 (P2RY12)). Thus, we hypothesize that LCH-ND arises from hematopoietic cells, clonal to systemic LCH lesions that have potential to migrate to the brain.
Erdheim Chester Disease
ECD is characterized by macrophage-like CD68+, CD14+, CD163+, and CD1a-, CD207- cells. This immunohistochemical pattern in combination with specific clinical findings (for example, “hairy” kidney, sheath around the aorta, and tibial hyperintensity on FDG-PET, see Diagnosis, screening and prevention, below) supports the diagnosis of ECD.79 Like LCH, clonality has also been confirmed in ECD. Some patients have mixed clinical and histological features of LCH and ECD, supporting a potential shared cell of origin.79 (Figure 2A) ECD histiocytes harbor BRAFV600E or alternative mutations activating the MAPK pathway.80 Moreover, BRAFV600E mutation has also been identified in CD34+ hematopoietic progenitors from patients with ECD65, 66. (Figure 1A) Hence, LCH and ECD share many pathological features, such as infiltration of myeloid cells that carry a mutation in genes involved in the MAPK pathway, and presence of the same mutation at the hematopoietic progenitor level, but are very distinct epidemiologically and clinically. In addition to BRAFV600E in approximately 50% of ECD, recurrent MAPK gene mutations have been reported in MAP2K1, ARAF, NRAS, and KRAS, and translocations of BRAF, ALK, and NTRK1.81 (Figure 1) Interestingly, PI3K-AKT-activating mutations in PI3KCA (not mutually exclusive with MAPK pathway mutations) have also been reported in ECD.82 Expression of the BRAFV600E mutation in mouse hematopoietic stem cells and human umbilical cord stem cells transplanted into immune deficient mice gives rise to a LCH-like phenotype but not an ECD-phenotype.7, 83 Either the cell of origin of ECD is different from that of LCH or there is an additional event required to cause LCH rather than ECD. Mechanisms underlying varying histological manifestations and clinical presentations of LCH and ECD for individual patients are still not known. The CCR7 down-regulation found in dendritic cells from LCH lesions has not been studied in ECD, as CCR7 is not a key chemokine for macrophages.73 The overexpression of anti-apoptotic proteins observed in LCH as part of the senescence programme, has not been studied in ECD lesions.7, 73 The number of ECD cells within histological lesions is classically limited, but in some cases the lesions are more densely infiltrated with pathologic histiocytes. As opposed to the pediatric population with LCH (in which individuals with BRAFV600E mutation have a worse outcome than individuals with a different mutation) the prognosis of patients with ECD carrying a BRAF mutation, to date, is the same as for patients with alternative mutations.64, 84 However, BRAF-V600E is associated with increased risk of cardiovascular involvement, including aortic, cardiac and pericardial infiltration in ECD.28, 84
Juvenile Xanthogranuloma
Very little is known regarding pathogenetic mechanisms of JXG. The origin of JXG cells is unknown, but JXG cells share some common features with dermal macrophages including expression of fascin and coagulation factor XIII. There are cases of mixed LCH and JXG phenotype, supporting potential common cell of origin. Whole exome or targeted DNA and/or RNA sequencing of pediatric patients with JXG found mutations activating MAPK pathway, including recurrent BRAFV600E, NTRK1 fusions, and mutations in MAP2K1 and CSF1R.58, 81, 85 (Figure 1A). Additionally, JXG can arise in the setting of neurofibromatosis with germline NF1 mutations,31 with some case reports of JXG and juvenile myelomonocytic leukemia (JMML) in patients with NF1.86, 87 A unique group of infants with an aggressive form of JXG with spleen, liver, and bone marrow showed infiltration with histiocytes with activating ALK fusions.88
Rosai Dorfman Disease
The causes of RDD are not known. Among the histiocytoses, RDDD has relatively indistinct diagnostic criteria, based on absence of ECD, JXG or LCH-defining cell markers and presence of emperipolesis, a relatively non-specific phenomenon of trafficking by viable lymphocytes through histiocytes, a characteristic but non-specific finding. RDD probably represents heterogeneous conditions with shared terminal phenotype. However, at least some cases are associated with MAPK pathway activating mutations, including in ARAF, NRAS, KRAS, MAP2K1, and CSF1R, though overall mutation frequency is much lower than in LCH, ECD and JXG.80, 89, 90 81 (Figure 1A) Hereditary syndromes such as H-syndrome and Faisalabad histiocytosis have been reported to exhibit pathological findings that are compatible with RDD and are associated with germline mutations in SLC29A3.91–93. In addition to characteristic histiocytes, there is an abundance of plasma cells in RDD lesions, and RDD can arise along with enriched IgG4+ cells in some cases.94 It is not known if IgG4+ RDD represents a distinct sub-class of RDD or if the presence of IgG4+ cells reflects non-specific immune activation.
Hemophagocytic Lymphohistiocytosis
HLH differs substantially from the rest of the histiocytoses reviewed in this Primer as a syndrome of immune dysregulation that is classically driven by pathologic activation of cytotoxic T cells that in turn drives extreme systemic immune activation. ‘Hemophagocytic’ refers to the characteristic histologic feature of bone marrow and tissue macrophages that engulf red blood cells. HLH manifests with fevers, cytopenias and splenomegaly, along elevations of characteristic inflammatory biomarkers such as ferritin and soluble IL-2 receptor alpha (sIL-2Rα). HLH can occur for a variety of reasons. ‘Primary’ HLH refers to patients with predisposing inherited inborn errors of immunity that intrinsically cause a predisposition to the development of HLH. Some patients will develop severe HLH at a very young age, such as infants with pathogenetic mutations in PRF1 or UNC13D that lead to complete absence of perforin or Munc13–4 function, whereas other patients may develop milder HLH or develop HLH at older ages, such as patients with pathogenetic mutations in STX11 or XIAP. (Figure 1 B) Age of presentation and clinical severity of HLH has been associated with functional severity of cytotoxic gene defects. 95, 96 However, there is tremendous variability in the phenotype associated with each genetic disorder, even within families with the same specific genetic variants. ‘Secondary’ HLH is driven by persistent immune activation, such as with malignancy, infection, or autoimmune disease, rather than by an intrinsic defect in immune function.
Several inherited inborn errors of immunity associated with HLH are characterized by defective T cell and NK cell granule-mediated cytotoxicity and are often classified as Familial HLH disorders. (Box 2). Autosomal recessive mutations in PRF1 result in defective perforin, a protein carried by cytotoxic granules of NK cells and T cells that is required for forming pores in the membrane of target cells.97 Autosomal recessive mutations in UNC13D (encoding Protein unc-13 homolog D, also known as Munc13–4), STX11 (encoding Syntaxin 11), and STXBP2 (encoding Syntaxin-binding protein 2), impair the complex process whereby cytotoxic granules traffic to and fuse with the plasma membrane and extrude their contents into the extracellular space at the immunological synapse.98–100 Some conditions associated with lysosomal trafficking defects and hypopigmentation (for example, Griscelli Syndrome and Chediak-Higashi Syndrome) also affect cytotoxic lymphocyte degranulation and, patients may be at risk for developing HLH (Box 2).101, 102 An infection or other immunological trigger may initiate the syndrome of HLH in these disorders by the abnormal persistence of antigen presenting cells and expansion of hyperactive polyclonal CD8+ T cells that secrete IFN-γ, TNFα, and other cytokines, with resultant activation of macrophages leading to a vicious cycle of lymphohistiocytic proliferation and hypercytokinemia.103–105 The magnitude of pathological inflammation distinguishes HLH from other hyperinflammatory conditions.106
Cytotoxic function-deficient mice with knockout mutations in genes that are that are required for cytotoxic lymphocyte function (e.g. Prf1) develop an HLH-like disease when infected with lymphocytic choriomeningitis virus.99, 107–109 In these HLH mouse models, if CD8+ T cells were depleted or when IFN-γ was neutralized, the HLH phenotype was abrogated, whereas phenotype was not rescued by depletion of NK cells were depleted or other pro-inflammatory cytokines were neutralized, suggesting that CD8+ T cells and IFN-s are essential in HLH pathophysiology. The HLH-like phenotype is thought to result from the failure of the cytotoxic T cells to eliminate activated antigen presenting cells, leading to continued stimulation of the cytotoxic T lymphocytes. Although defects of NK cell cytotoxic activity alone are not sufficient to induce HLH in mouse models99, NK cells probably have a role in HLH pathophysiology. When NK cell cytotoxicity is conserved, the HLH phenotype is less severe, through reduction of the cytotoxic CD8+ T proliferation.99, 110 The importance of IFN-γ as master-regulator of inflammation is further supported by clinical responses to antibody therapy directed against IFN-γ in patients with HLH.111
There are additional inherited disorders that cause HLH by more complex or alternative mechanisms that can also be considered primary HLH disorders (Box 2). These disorders include X-linked lymphoproliferative disease type 1 (due to mutations in SH2D1A),112–114 X-linked lymphoproliferative disease type 2 (due to mutations in XIAP (also known as BIRC4)),115 other diseases that are often classified as EBV-susceptibility diseases that reflect T cell dysfunction, including ITK116 CD27,117 and MAGT1118), and diseases caused by mutations in CDC42119 or NLRC4120, 121. Persistent and/or extreme EBV viremia in turn can induce HLH. The full discussion of the pathophysiology of these diseases is outside the scope of this Primer, but general mechanisms are listed in Box 2. Notably, patients with XIAP deficiency and activating variants in NLRC4 have dysregulated inflammasome function, implying over-exuberant inflammasome-derived cytokine production can also drive HLH. The spectrum of genetic disorders associated with HLH is expanding, with increased recognition that a wide spectrum of inborn errors of immunity can be associated with HLH, usually in the setting of infectious immune challenges.45, 122 123 Inborn errors of metabolism can also occasionally be complicated by HLH .124 This occurrence may be due to abnormal macrophage activation due to the accumulation of non-degraded substrates. Rheumatological disorders (autoimmune and/or auto-inflammatory) with exuberant hyper-cytokinemias can also be complicated by HLH, which may be called macrophage activation syndrome (MAS) in this setting.125
Diagnosis, screening and prevention
Clinical Presentations
LCH.
Clinical presentations for LCH are widely variable depending on sites of disease and the effect on specific organ systems.9, 10, 126–128 (Figure 3 , Supplementary Figure 1) LCH most frequently manifests in the skin, where lesions may present as pruritic or ulcerative skin rashes that are easily mistaken for more common rashes, especially in infants (Figure 4A), in whom LCH can arise as skin-limited disease, or the skin can be one of many organs involved in more severe multisystem disease.129 Chronic inner ear and/or mastoid lesions may present with chronic drainage that could be confused with otitis externa. In women, LCH of the vulva can mimic other genitourinary conditions. LCH can also involve the gastrointestinal track anywhere from mouth to anus, with weight loss and chronic diarrhoea as possible signs of intestinal involvement. In the mouth, LCH can involve the gingiva, causing early eruption of deciduous teeth or loss of mature teeth. LCH frequently arises in bone, where it can cause local pain and/or mechanical damage. (Figure 4B) Back pain with vertebra plana (complete collapse of a vertebra) is a classic presentation. Adults with smoking history may present with acute pneumothorax or may develop symptoms of obstructive pulmonary disease if lung LCH and associated parenchymal damage progresses.25 (Figure 4D) Disseminated LCH can involve liver, spleen and/or bone marrow. (Figure 4 C) Initially liver disease may present with hepatomegaly and signs of acute hepatitis. Long-standing disease can progress to sclerosing cholangitis (diffuse inflammation of the bile ducts) with jaundice and other signs of liver failure. (The spleen can be diffusely infiltrated or have focal lesions, clinically manifesting as splenomegaly. Patients with bone marrow infiltration may present with clinical signs of cytopenias (bruising, anemia, and infections) and in some cases may develop hyperinflammatory symptoms of secondary HLH.130 Sudden onset of diabetes insipidus is a classic presentation of LCH involving pituitary gland. Mass central nervous system (CNS) lesions can cause focal neurological deficits and/or headache, whereas LCH-ND typically presents with signs of cerebellar dysfunction. 76, 131 (Figure 4E)
Figure 3. Clinical presentations of LCH, ECD, JXG and RDD.
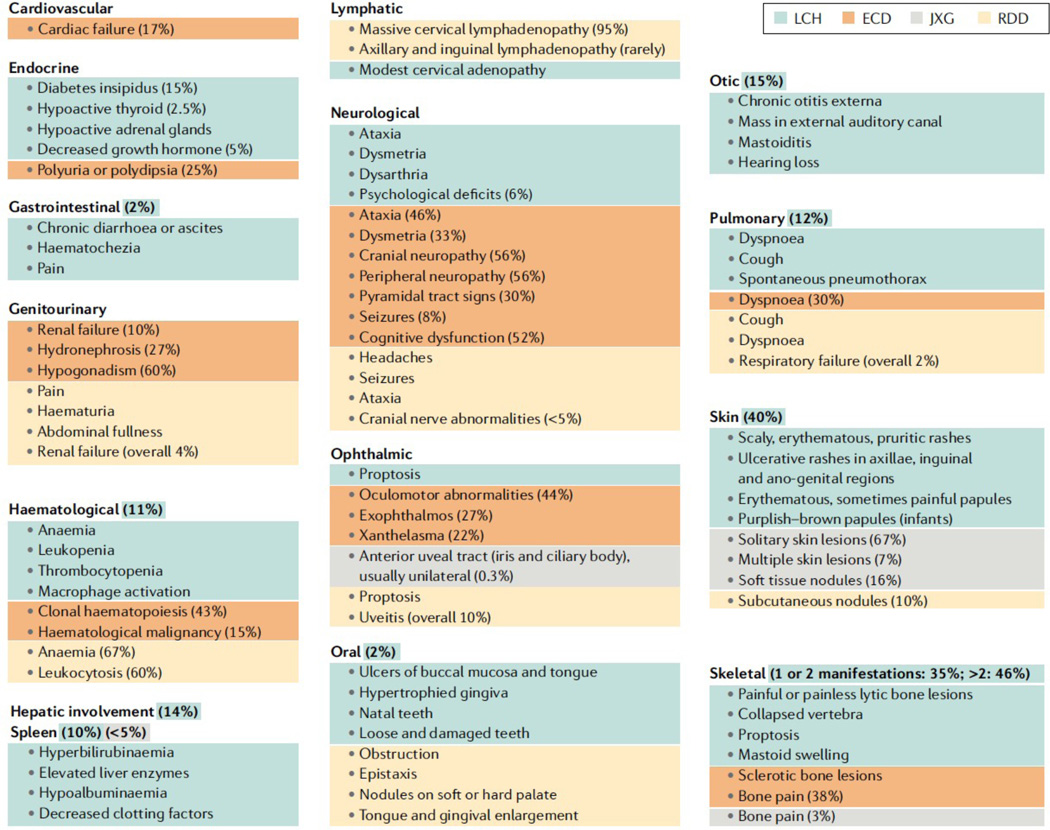
The organ systems known to be involved in histiocytic disorders. Percentages of patients reporting a specific manifestation are derived from large series studies for LCH:17; ECD: 28, 84, 151; JXG: 29, 30; RDD 33, 34
Figure 4. Manifestations of LCH.

A. Erythematous, scaly LCH rash; B. Multiple lytic skull lesions (arrows) in a patient with LCH; C. PET scan showing involvement of skull base, occiput, left fifth rib, left iliac bone, and left hemisacrum. Although the intense uptake in long bones is physiological, the scan shows hypermetabolic splenomegaly and bilateral cervical/upper abdominal lymphadenopathy.); D Pulmonary cysts and nodules (arrows) from LCH; E. Brain MRI with intense T2 signal in the dentate nuclei characteristic of neurodegenerative LCH (arrow).
Erdheim Chester Disease.
ECD can affect nearly every organ system and is frequently a multi-system disease, leading to a vast array of symptoms and signs in adults. (Figure 3) The natural history of ECD is difficult to evaluate, owing to the wide spectrum of presentations and treatments. Untreated ECD can be severe and fatal, particularly in those with multi-system disease. The rare occurrences of spontaneous regression and self-healing that can occur in LCH are not observed in patients with ECD. The mean time from symptom onset to diagnosis in a recent series of 261 patients was 2.7 years.28 ECD may be heralded decades before the diagnosis by diabetes insipidus supposedly of unknown origin.84 Iconic radiographical and clinical signs of ECD include the ‘hairy kidney’, sheath of fibrotic tissue with histiocytes around the aorta, sclerosis of long bones, right atrial pseudotumors, and xanthelesma on the eyelids (Figure 5). Like LCH, neurological involvement in ECD may have a tumoural or neurodegenerative presentation. A retrospective analysis of all CNS manifestations (both clinical and only detected by imaging) in 253 patients with ECD showed CNS involvement in 97 individuals (38%).132 A review of 74 brain MRIs identified three non-exclusive patterns: tumoural (66%), degenerative (50%) and vascular (18%).132
Figure 5. ECD clinical presentations.

A.The classical ECD finding of “hairy kidney“ (a ring of ECD tissue around the kidneys seen on CT scan of the abdomen with contrast; arrow). B. Typical sheathing of the aorta (arrow) illustrated on this contrast-enhanced CT scan of the chest.
JXG.
JXG most often presents in children with cutaneous flesh colored, yellow-orange, brown or purple macules or papules, which may occur anywhere on the body.133 (Figure 6) Some patients have isolated or very few cutaneous lesions, whereas others have hundreds, with potential to become quite large in some cases. JXG is present in one or two sites in 75% of patients.133 Extra-cutaneous sites include the liver (22%), lung (16%), soft tissue (16%), spleen (11%), eye (9%), oral cavity (7%), lymph nodes (7%), brain (7%), adrenal glands (7%), gastrointestinal tract (7%), bone marrow (7%) and heart (4%).30, 134, 135 Disseminated JXG can be aggressive and potentially fatal.
Figure 6. JXG Rash.

The rashes of JXG patients are typically yellowish, orange, or purplish papules, which can appear anywhere on the body. Most patients have fewer than a dozen scattered lesions, but some can have many more, like this patient. JXG involves the visceral organs (lung, liver, kidney, spleen, brain, pancreas, adrenal glands, or intestines) in <5% of patients and causes bone lesions in <3%.
RDD.
RDD may occur as an isolated disorder or in association with autoimmune, hereditary, and malignant diseases.136 A review of 47 patients with RDD found that most had chronic disease activity.137 Extranodal RDD has been documented in 43% of cases, with the most frequent sites being skin, soft tissue, the upper respiratory tract, multifocal bone, kidney, eye and retro-orbital tissue usually with lymphadenopathy.34 Head and neck involvement has been reported in 22% of patients, most commonly affecting the orbit, nasal cavity or the parotid gland.138 Unlike neurological LCH and ECD, neurodegenerative findings have not been reported in patients with RDD. CNS RDD are mass lesions that can be asymptomatic and, therefore, discovered incidentally, or they may arise with clinical symptoms of central endocrinophathies, seizures, or headaches.139 137
HLH.
It is important to distinguish between the syndrome of HLH and an underlying genetic disease which predisposes to HLH .140 The syndrome of HLH refers to a constellation of clinical and laboratory manifestations. Diagnostic criteria for the syndrome of HLH were established by the Histiocyte Society and include fever, splenomegaly, cytopenias, hypertriglyceridemia and/or hypofibrinogenemia, observation of hemophagocytosis, decreased NK-cell function, elevated ferritin, and elevated sIL-2Rα levels (Box 3).141 There are several genetic inborn errors of immunity which predispose patients to develop the syndrome of HLH. Such diseases can be referred to as primary or genetic HLH disorders. Other common manifestations include CNS involvement in 30–73% of patients142 (sometimes as an isolated manifestation143), and hepatitis or acute liver failure, which occurs in 90% of patients 144. Patients who develop HLH in the absence of a primary/genetic HLH disorder are often referred to as having secondary HLH, though distinctions between presumed ‘primary’ and ‘secondary’ can be blurred with infectious triggers in many ‘primary’ cases, and complex or undiscovered genetic predisposition in presumed ‘secondary’.45
Diagnostic Tests and Imaging
LCH.
Initial evaluations typically include studies to identify potential organ damage and systemic inflammation including complete blood count, liver panel, complete metabolic panel, ferritin, and prothrombin time (PT) or partial thromboplastin time (PTT).10 For screening, skeletal x-ray of the whole body with 4 views of skull, chest x-ray, and/or abdominal ultrasonography may identify lesions. We favor 18F-fluorodeoxyglucose (FDG)-PET–CT scans to define the extent of disease because of sensitivity relative to other imaging modalities and ability to define metabolic responses to therapy.145, 146 Chest CT is appropriate for pulmonary lesions, owing to need for high resolution imaging. MRI is preferred for spine and brain imaging, though maxillo-facial bone sites are best imaged by CT.131 Bone marrow evaluations are appropriate for young children or for patients presenting with cytopenias. For diagnostic procedures, excisional biopsy is preferred to fine needle aspiration or core biposies, owing to complex architecture of lesions. More limited biopsies may give an inaccurate representation of the histology, as pathological cells may not be evenly distributed in the sample. While excisional biopsies are appropriate, aggressive excisions including wide margins are contraindicated for LCH, as these can impair tissue remodeling. Unlike other malignancies or neoplastic disorders, bone has potential to repair with successful LCH therapy.127 Targeted sequencing of MAPK pathway genes may identify pathogenetic mutations that support LCH diagnosis and inform disease risk and potential therapeutic options. It is possible to evaluate for BRAFV600E+ peripheral blood mononuclear cells or bone marrow cells with high sensitivity techniques.147 Next generation sequencing approaches should take into account potential for low variant allele frequency (median 8% pathogenic cells in LCH lesions).63
There are several differential diagnoses for the LCH presentations that may require tissue biopsy to differentiate. LCH skin rashes mimic a wide variety of more common cutaneous diseases such as psoriasis, seborrhea (excessive discharge from the sebaceous glands), viral exanthema (widespread rash), and the ‘blueberry muffin’ presentation of neuroblastoma and myeloid leukemias. Orbital bone involvement causing proptosis (extreme bulging out of the eyes), or other osteolytic bone lesions may mimic sarcomas, neuroblastoma, and leukemias. Benign lesions similar to LCH include disorders of bone and connective tissue including dermal bone cysts, myofibromatosis148, fibrous dysplasia149, and cranial fasciitis.150 Hepatomegaly and elevated liver enzymes could be caused by a vast number of primary hepatic diseases. Diabetes insipidus can be idiopathic or also associated with germ cell tumors, sarcoidosis, lymphocytic hypophysitis (infiltration of the pituitary with lymphocytes), or tuberculosis. The MRI images of the LCH neurodegenerative syndrome are similar to those of patients with encephalitis, sequelae of acute disseminated encephalomyelitis (ADEM), and drug overdoses. Pneumothorax caused by a number of primary pulmonary conditions with cysts and interstitial lung abnormalities can be confused with LCH and vice versa. LCH rarely presents along with other hematologic malignancies (leukemia or lymphoma), frequently sharing pathogenic somatic mutations beyond the typical MAPK repertoire. Biologically, LCH in context of bi-phenotypic leukemia or lymphoma is likely distinct from more typical presentations.72
ECD.
Evaluations for cytopenias, renal and hepatic insufficiency, endocrinopathies, immunological assessment (T cell and B cell subsets and immunoglobulins), and inflammatory markers (sedimentation rate and C-reactive protein) should be performed.28, 151 The most frequent radiographical sign of ECD is long-bone osteosclerosis (abnormal bone density), seen in 80–95% of patients but symptomatic in only 38%.28 It was historically evaluated by bone scan, but PET–CT may be more effective and also allow extra-osseous evaluations. 152, 153 Abdominal CT scans reveal the iconic fibrotic infiltration around the kidneys (‘hairy kidneys’) in 63% of patients.28 (Figure 5A) CT scans may also reveal sheathing of the aorta and pericardial disease sometimes leading to tamponade. (Figure 5B) Right atrium pseudotumor, another classical feature of ECD, can be observed by cardiac MRI in 36% of patients.154Biopsies should be evaluated for mutations of genes in the MAPK and PI3K-AKT pathways (Figure 1).
Several diseases have features similar to ECD. Takayasu arteritis has some features comparable to aortic abnormalities in patients with ECD. However, radiological findings differ between Takayasu arteritis and ECD: the entire wall is affected in Takayasu arteritis, whereas the adventitial and periadventitial periaortic spaces, but not the wall itself, are affected in patients with ECD.155 Radiological characteristics can also help distinguishing between ECD and mediastinal and retroperitoneal fibrosis. Idiopathic retroperitoneal fibrosis is classically not circumferential, infiltrating the anterior and lateral sides of the aorta, but sparing the posterior space. IgG4-related disease, a multi-system condition that may be confused with ECD, is characterized histologically by dense lymphoplasmacytic infiltrates rich in IgG4+ plasma cells, abundant fibrosis with a storiform (cartwheel) pattern, lymphoid follicles, tissue eosinophilia and obliterative phlebitis (vascular inflammation resulting in obstruction). Serum IgG4 levels may be elevated as they are in approximately half the patients with IgG4-related disease.156 Other histiocytoses (such as RDD) should also be considered in the differential diagnosis.
JXG.
For infants with only a few cutaneous lesions (Figure 6) and no systemic symptoms or signs, evaluations should include a complete blood count, liver enzyme panel, basic metabolic panel, and ophthalmological examination. Patients with a large number of lesions and/or systemic symptoms or abnormalities of the aforementioned laboratory tests should have MRIs of the brain and abdomen to evaluate for possible systemic lesions. When cytopenias are present, bone marrow aspirates and biopsies are indicated. Targeted or whole exome sequencing may be informative. (Figure 1)
JXG lesions may be mistaken for atheromas or lipomas, or LCH. Urticaria pigmentosa, neurofibromatosis type I (NF1), and myelogenous leukemia have also been associated with JXG and can all be differentiated by biopsies.157 Patients with NF1 and JXG may have highly increased risk of developing juvenile myelomoncytic leukemia.87
RDD.
Laboratory analyses should include a complete blood count, liver function tests, basic metabolic panel, serum immunoglobulins and erythrocyte sedimentation rate.136 Staging evaluation includes a skeletal survey for osteolytic lesions and a PET scan to locate affected lymph nodes, periorbital or sinus masses, pulmonary, spleen, liver, and bone lesions. (Figure 7) Biopsy is required for diagnosis, and excisional biopsy is preferred owing to complex and heterogeneous histology. Targeted sequencing or whole exome sequencing may be informative in some cases. (Figure 1)
Figure 7. RDD clinical Presentation.

Massive cervical adenopathy
Several differential diagnoses for RDD exist. Sinus involvement or proptosis from retro-orbital RDD presents much like neuroblastoma, sarcomas, or LCH. The lytic osseous lesions of RDD are often not as symmetrical as those observed in LCH and can occur in any bone. CNS disease can mimic a meningioma. Histopathological characteristics resembling RDD have been described in 41% of patients with autoimmune lymphoproliferative syndrome type Ia, with TNFRSF6 heterozygous germline mutations affecting the FAS gene, and in patients with autoimmune hemolytic anemia, systemic lupus erythematous, and juvenile idiopathic arthritis.158, 159 RDD may arise in patients with H-syndrome and Faisalabad histiocytosis, associated with germline mutations in SLC29A3.91, 92 Some patients with the IgG4 related disease have features of RDD.160 RDD probably represents a common histological endpoint from many conditions.
HLH.
A sample diagnostic algorithm is given in Figure 8. Hemophagocytosis alone has limited diagnostic sensitivity and specificity, as it can be observed in patients without HLH and can be absent in patients with HLH.161–163 Ferritin and sIL-2Rα may be elevated in many settings, but highly elevated inflammatory markers should prompt more in depth evaluations for HLH.39, 164, 165 Inherited cytotoxic defects are associated with decreased NK cell function; however, these findings can be non-specific in very young children or temporarily depressed in acutely ill patients. Tests that evaluate perforin expression or lymphocyte CD107 (also known as Lysosome-associated membrane glycoprotein 1 upregulation) can be used to more reliably screen patients for perforin deficiency or pathogenetic variants in in the genes that are crucial for degranulation. 166, 167 Newer measurements, such as C-X-C motif chemokine 9 (CXCL9), a marker of IFNγ activity, and IL-18, which reflects inflammasome activation, may become helpful in identifying patients with pathological inflammation that can be used to reflect magnitude of pathological activation, which may distinguish HLH from other conditions.168, 169
Figure 8. Diagnostic algorithm for HLH.

Flow diagram of clinical decision making when confronted with a patient who has signs and symptoms of HLH. aSome patients with genetic predisposition to HLH may develop atypical disease presentations; bIf safe to obtain; cIf not previously performed; dFor example, consider HSV, VZV, HHV6, HIV, Parvovirus, leishmaniasis, tick-borne illnesses, mosquito-borne illnesses, histoplasmosis and TB, amongst others.
All patients with HLH should be evaluated for malignancy and infection, which are especially common in adults.170, 171 HLH can arise either at diagnosis or during periods of immune dysregulation resulting from chemotherapy or HCT.172 Leukemias and lymphomas are predominantly associated with HLH, though solid tumors have been reported.170, 173 HLH may occur in up to 1% of hematological malignancies.172 PET scan or CT–MRIs of the entire body can be helpful to rule out associated malignancies. MRI of brain and (where possible) cerebrospinal fluid evaluations can support identification of CNS pathology in patients with HLH. (Figure 9) HLH can be associated with infections varying from common triggering viruses such as EBV to virtually any viral, bacterial, fungal, mycobacterial, or parasitic infection. Rheumatological or metabolic disorders should be considered.124, 125 Patients should be evaluated for underlying genetic HLH disorders and inborn errors of immunity.45 Rapid screening tests can help define some primary HLH disorders. Flow cytometric assays are available to screen for perforin, SAP (signaling lymphocytic activation molecule (SLAM)-associated protein, also known as SH2 domain-containing protein 1A), and XIAP (X-linked inhibitor of apoptosis)deficiencies.167, 174–176 Degranulation (CD107a) assays screen for genetic diseases that compromise granule-mediated cytotoxicity.177 IL-18 levels can be useful when considering XIAP deficiency and pathological variants in NLRC4, or MAS in the setting of rheumatological disorders.168, 178, 179 Genetic HLH panels are frequently used, but broad primary immune deficiency gene panels or whole exome sequencing offer the benefit of capturing additional inborn errors of immunity or metabolic diseases.45, 180
Figure 9. HLH clinical presentations.

The organ systems known to be involved in HLH, with percentages of patients reporting a specific manifestation. The image shows brain MRI of an HLH patient with CNS involvement, with T2 signal hyperintensity and micro-cystic encephalomalacia of the cerebral white matter and cerebral cortex.
A diagnosis of the syndrome of HLH does not exclude important underlying disorders or mimicking disorders. For HLH, differential diagnoses may also include concurrent diagnoses. Patients with hyperinflammatory conditions such as those with sepsis, the systemic inflammatory response syndrome (SIRS), multi-organ failure from many causes, metabolic storage diseases, malignancies, and immune deficiencies may present with many of the signs and symptoms of HLH. The term ‘macrophage activation syndrome’ (MAS) is the subject of ongoing nomenclature debate, but typically describes pathological inflammation in the setting of autoimmune or autoinflammatory disease or cancer. MAS is diagnosed using criteria established by a collaborative effort that examined discriminating features for MAS in patients with systemic juvenile idiopathic arthritis181.
Pathology
LCH.
The diagnosis of LCH is relatively straightforward, with biopsy demonstrating pathological round CD1a+–CD207+ histiocytes having reniform nuclei with infiltrating immune cells, predominantly T cells. (Figure 10).62, 182 CD207+ cells may be abundant or scarce (median of 8% lesional cells).63 The cells should be distinguished from the physiological normal inflammatory CD207+ dendritic cells, which have a branching morphology owing to their antigen-presenting role. Relative expression of CD1a and CD207 can vary; notably, it can be dim or absent in brain, liver and bone marrow lesions.183 The presence of a macrophage phenotype with an MAPK activating mutation can be consistent with LCH despite lack of classic antigens. In one study, BRAFV600E+ cells were identified in > 50% of bone marrow aspirates clinically reported as normal.63
Figure 10. Histology characteristics of LCH, JXG and RDD.
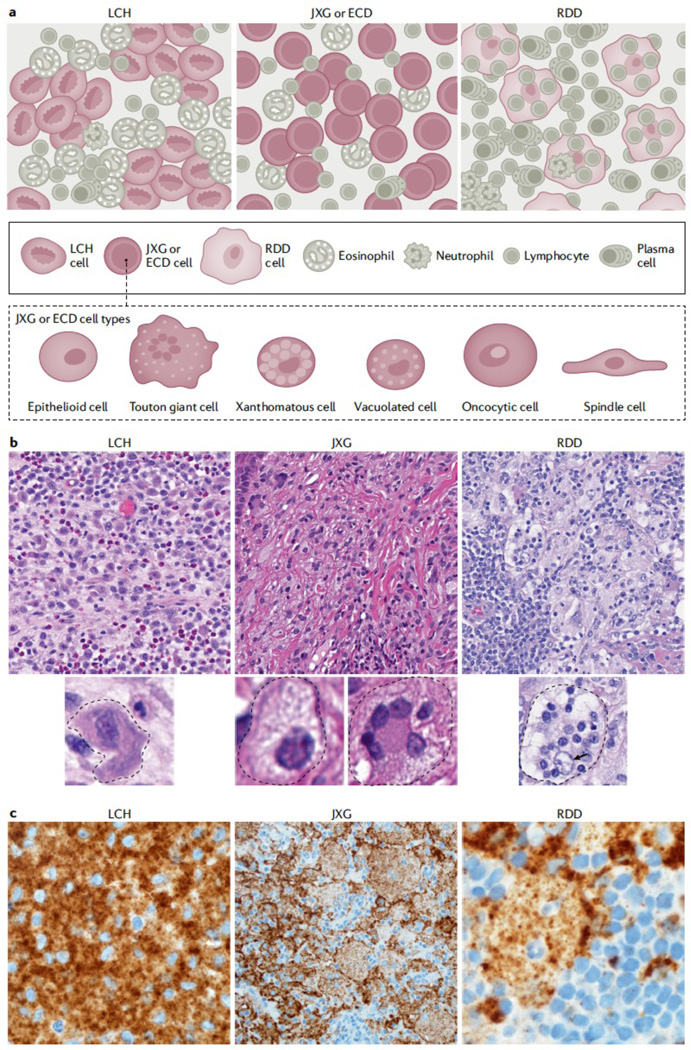
A. Although Langerhans cell histiocytosis (LCH), histiocytoses of the xanthogranuloma family (including Erdheim Chester Disease (ECD) and juvenile xanthogranuloma (JXG),), and Rosai Dorfman Disease (RDD) have certain classic morphologies and defining phenotypes, our understanding of histiocytic lesions continues to evolve and may be best conceptualized along a spectrum, sometimes with morphology and phenotypes overlapping in certain instances and often with similar mutation profiles. In fact, ECD and JXG share similar morphological cell types under the microscope, including xanthomatous cells with variable admixture of Touton giant cells, vacuolated cells, spindled cells, oncocytic cells, and epithelioid cells. B. H&E images for LCH, xanthogranuloma family, and RDD (original magnification at 400x). C Immunostaining specific for BRAF-V600E (known as BRAF-VE1, brown) Although the mutant specific immunohistochemistry for BRAF-VE1 is more commonly expressed in LCH and ECD, with strong granular cytoplasmic staining of the histiocytes, rare examples in pediatric JXG family (limited to the central nervous system of children) and isolated case reports of RDD have been identified to also harbour the mutation and express the mutant protein, which reacts to the immunostaining. Original magnification at 1000x for LCH, 400x for CNS-JXG, and 1000x for RDD. Photos made with assistance of Mr Chris Woods Cincinnati Children’s Department of Pathology.
ECD.
ECD lesions show infiltration of foamy histiocytes surrounded by fibrosis or xanthogranulomatosis. (Figure 10) Touton giant cells are frequently, but not always, observed.151 Microscopically, ECD is indistinguishable from the fibrohistiocytic JXG subtype 184, 185 and depends on distinct clinical-radiographical features for an integrated diagnosis. Immunophenotype of ECD and JXG includes expression of CD163, CD68, CD14, Factor XIIIa (less consistently in xanthomatous cells) and often fascin.85
JXG.
The classic JXG cells are large with a vacuolated cytoplasm and small round or indented nuclei. (Figure 10) Like ECD cells, these cells stain with CD163, CD14, CD68, fascin, Factor XIIIa, CD4, but not CD1a.184 A subset of JXG-like lesions may harbor ALK gene rearrangements most commonly KIF5B-ALK with TPM3-ALK also described. 88
RDD.
The diagnostic feature of RDD is a large histiocytic cell with ample pale foamy cytoplasm, often described as ‘watery-clear’, and a large hypochromatic nucleus with prominent nucleolus. (Figure 10) Histologically RDD is characterized by large histiocytes with ample pale cytoplasm, often with emperipolesis (that is, viable lymphocytes and plasma cells trafficking through cytoplasm of RDD histiocytes). The biological or clinical significance of emperipolesis is not well understood but may be a mechanism of immune evasion. The engulfed cells are found in a membrane-bound vesicle in the macrophage cytoplasm and are not phagocytosed. Emperipolesis in characteristic cells distinguishes RDD from other causes of massive lymphadenopathy. The RDD cells do not present any cytologic atypia, have very few mitoses figures, selectively stain for S100, fascin and macrophage markers (CD14, CD69, or CD163) and lack DC markers (CD1a and CD207)186 In affected lymph nodes, histiocytes are mainly located within the sinuses.184 Lesions can contain numerous plasma cells, and systematic evaluation for associated IgG4-related disease is recommended if there is an elevated plasma IgG/IgG4 levels ratio is >40%.156 The cytomorphology and immunophenotype should considered together with the pattern of tissue involvement, as small focal areas of RDD-like cells do not equate to disease.
HLH.
Bone marrow studies are important not only to evaluate for hemophagocytosis (Figure 11) but also to characterize possible bone marrow failure, malignancy, metabolic storage disease and/or infection. Hemophagocytosis is a diagnostic criterion, but has limited sensitivity and specificity. Activated macrophages, some with hemophagocytosis, may also occur in a number of diseases affecting lymph nodes, spleen, CNS, liver, bone marrow, and thymus, together with increased CD8+ T-cells.187 Staining with anti-CD68 PGM-1 clone or anti-CD163 antibodies can highlight activated macrophages in bone marrow or other tissues.
Figure 11. Histology of HLH.

Bone marrow aspirate shows a large activated histiocyte with cytoplasmic phagocytosis. (Wright-Giemsa stain, original magnification 1000x).
Management
Treatment goals and strategies for histiocytic disorders vary depending on the disease, and optimal therapeutic strategies remain uncertain for most patients with histiocytic disorders. Cytotoxic therapies may be used in attempts to cure patients with systemic LCH, ECD, JXG and RDD with therapeutic goal to clear the pathogenic clone. MAPK pathway inhibitors are being evaluated for MAPK-driven histiocytoses. Early trials with LCH and ECD demonstrate very high response rates, but disease may recur with cessation of MAPK inhibition.68, 69, 188, 189 Immune modulation may be used to control pathological inflammation and symptom management. Prompt initiation of immune suppression is central to treatment of HLH, and HCT is required to cure patients with intractable immune dysregulation.
LCH
Patients with isolated bone lesions or limited skin rash may be treated with local approaches, whereas patients with multifocal and/or CNS disease typically require systemic therapy.
Single bone lesions.
Patients with isolated bone lesions may be treated with curettage (operative removal of diseased tissue) alone, and/or with steroid injection. 190 Lesions in the facial bones or skull base are considered ‘CNS-Risk’, owing to association with developing pituitary gland disease and LCH-ND, and are, therefore, treated with systemic chemotherapy with same strategies as multifocal disease.131 For bone lesions, wide excision with ‘clean margins’ may impair bone remodeling and is not indicated. Radiation therapy is rarely used for bone lesions in children due to potential impact on growth and efficacy of alternatives, but may be applied more broadly for adults with single bone lesions that are inaccessible to surgery or interventional radiology.
Lung.
Smoking cessation, without pharmacological therapy, may be effective for some adult patients with isolated lung disease.191 However, others with extensive cysts and nodules are preferably treated with systemic chemotherapy. Additionally, such patients should be evaluated thoroughly for extra-pulmonary lesions, and patients with multi-system disease typically require chemotherapy.192
Skin.
Isolated cutaneous LCH may resolve with no treatment in some infants.129 Progressive disease may respond to oral hydroxyurea, methotrexate with or without 6-mercaptopurine (6-MP), or thalidomide.193–195 Radiation therapy and surgical excision are rarely used owing to potential toxicity and limited utility for wide-spread disease.
LCH of pituitary, parenchymal brain lesions and LCH-ND.
In patients who present with isolated diabetes insipidus, polydipsia (excessive thirst) and polyuria (excessive urination) without other signs of LCH, it may be challenging to differentiate LCH of the pituitary gland or brain from germinoma or pituitary hypophysitis (inflammation of the pituitary gland). Suspected or proven LCH lesions that progress on MRI may be treated with systemic chemotherapy.196–198 Parenchymal or neurodegenerative LCH lesions may respond to chemotherapy or MAPK inhibitors.12, 68, 199
Multifocal disease.
Treatment regimens evolved from studies by the German, Austrian, and Dutch Pediatric Leukemia Study Group and the Histiocyte Society over the past four decades. The current standard of care for multifocal site, low risk or high risk LCH is 12 months of vinblastine and prednisone, with 6-mercaptopurine (6-MP) added for high-risk patients.11 While overall survival has greatly improved over the past decades, one year progression-free survival for low-risk patients is 50% and for high-risk patients 33%. The Japanese LCH Study Group have used a multi-drug protocol including vincristine, cytarabine, prednisolone, methotrexate, and 6MP.200 The one year progression free-survival for this study was 76% for high-risk patients and 94% for low-risk patients. By 5 years the progression-free survival was 46% for high-risk and 70% for low risk. Other drugs used to treat LCH, including cytarabine, clofarabine, and cladribine, have reported efficacy for multi-site disease in case series and single-arm trials.197, 198, 201 On the basis of the current understanding of LCH deriving from a myeloid precursor, therapies including nucleoside analogs such as cytarabine, cladribine, and clofarabine, may be effective, owing to the sensitivity of myeloid malignancies to these drugs. For a similar reason, oral hydroxyurea, historically effective in myeloid neoplastic disorders, was evaluated and found to elicit responses in some patients with skin and bone LCH.193 The Histiocyte Society LCH-IV Trial is currently testing the effect of 24 versus 12 months of therapy for both multi-focal low risk and high risk LCH (NCT02205762). A multi-institutional randomized phase 3 trial is testing frontline cytarabine versus standard vinblastine–prednisone for pediatric LCH (NCT02670707).
Adult patients have been treated with similar regimens. Cladribine was one of the first reported regimens and has gained popularity in patients with pulmonary LCH.202, 203 Some groups have found vinblastine/prednisone is safe and effective in adults, though neuropathy and steroid toxicity seem more significant in adults than children.204, 205 Cytarabine monotherapy has been reported as well-tolerated and effective in case series of adults with bone disease.204 Other published approaches include MACOP-B chemotherapy and Zoledronic acid.206, 207
Recurrent or progressive LCH.
Data to guide salvage therapy strategies for LCH are primarily limited to case series and early phase trials. (Table 1) Nucleoside inhibitors alone or in combination have efficacy and toxicity correlated to dose. 197, 198, 201, 208, 209 As discussed above, mutually exclusive activating MAPK pathway mutations have been identified in almost all patients with LCH. Case series and early phase trials have reported near universal responses to MAPK pathway inhibition.9, 14, 68, 69, 80, 188, 189, 210 Many patients, even those with high risk site involvement, achieve a complete metabolic response by PET scan. Patients with the early onset of neurodegenerative changes on MRI and clinical deficits may have marked radiological improvement and variable degrees of clinical improvement; by contrast, patients with long-standing neuropathology may have limited potential for clinical improvement. Patients frequently fail to achieve complete responses with MAPK inhibition, and early reports suggest that those who do achieve clinical complete responses have a high risk of relapse with cessation of therapy. 68, 69
Table 1 |.
Treatments for progressive paediatric Langerhans cell histiocytosis
| Regimen | Number of patients | Risk group | Outcomes |
|---|---|---|---|
| Vbl/Pred/Etop/Mtx/6-MP266 | 14 | High risk | 85% PFS |
| 121 | Low risk | 76% PFS | |
| Cladribine198 | 46 | High risk | 48% OS |
| 37 | Low risk | 97% OS | |
| Vcr/A RA-C/Pred267 | 10 | Low risk | 50% PFS |
| Clofarabine229 | 4 | High risk | 33% PFS |
| 2 | Low risk | 50% PFS | |
| Clofarabine201 | 3 | High risk | 33% PFS |
| 8 | Low risk | 100% PFS | |
| Cladribine/cytarabine209 | 27 | High risk | 78% PFS |
| Thalidomide195 | 6 | High risk | 0% PFS |
| 6 | Low risk | 50% PFS | |
| Vemurafenib/dabrafenib68 | 13 | Neurodegenerative disease | 33% PFS |
| 7 | High risk | ||
| 1 | Low risk | ||
| Vemurafenib69 | 44 | High risk | 5% PFSa |
| 10 | Low risk | 60% PFS |
6-MP, 6-mercaptopurine; ARA-C, cytarabine; CR, complete response; Etop, etoposide; Mtx, methotrexate; OS, overall survival; PFS, progression-free survival; Pred, prednisone; Vbl, vinblastine; Ver, vincristine.
PFS is after stopping vemurafenib. 86% complete response after 6 weeks of treatment.
The BRAF-V600E and MEK inhibitors are generally tolerated but can cause serious adverse reactions to the skin, tendons, joints, eyes, and heart, which require dose reduction or change in therapy. Second malignancies have been described in some adult patients, but so far none in children.211 The effect of long-term MAPK inhibition in children is not known. Optimal timing and duration of MAPK inhibition versus chemotherapy is also uncertain and merits further evaluation with prospective studies.
Erdheim Chester Disease
ECD was historically considered a fatal diagnosis, with nearly 60% of patients dying within 3 years of diagnosis.154 Interferon α (IFNα)-based regimens have improved survival,212 and BRAF and MEK inhibitors have further improved outcomes.188, 213 A review of the Pitié-Salpêtrière hospital cohort reported a 5-year survival rate of 79%.84 PEGylated IFN-α is better tolerated than the non-PEGylated form. The therapeutic response of IFN based regimens (measured by PET–CT and conventional imaging) is usually much slower and less complete than that observed with targeted therapies.28, 84 Assessments should be made every 6 months. IFN-α treatment is associated with numerous adverse effects, such as fatigue and depression, affecting half of the patients.15 In cases of mild or non-severe ECD (for example, without heart and/or CNS involvement) and contraindications to or adverse effects to IFN-α, the alternative regimens include the IL-1 receptor antagonist anakinra,214 the anti-TNF-α monoclonal chimeric antibody infliximab,215 and the mammalian target of rapamycin (mTOR) inhibitor sirolimus with steroids.216
In 2012, the first patients with multi-system refractory ECD or combined ECD–LCH with the BRAFV600E mutation were successfully treated with the BRAF-V600E inhibitor vemurafenib.213 Subsequently, over 80 such patients were treated with BRAF-V600E or MEK inhibitors with excellent outcomes, as measured with the PET Response Criteria in Solid Tumors (PERCIST).14, 189, 210 These results led to FDA approval of vemurafenib for BRAF-mutated ECD. When therapy was stopped, relapses occurred after a median of six months in 75% of the patients, though re-treatment with the same MAPK inhibitors following relapse was generally effective.189 The most frequent side effects in ECD case series are similar to those noted above for LCH, including photosensitivity and keratosis pilaris, spinocellular carcinoma, melanoma, sarcoidosis-like disease, drug reaction with eosinophilia and systemic symptoms (DRESS), arthralgias, pancreatitis, and QT-segment prolongation. Myelodysplastic syndrome and pancreatic adenocarcinoma have also been reported.70, 217 Vemurafenib toxicity is usually dose limited and is the main reason for using lower doses in ECD patients than in the melanoma trials. Dabrafenib, another BRAF-V600E inhibitor, has also been reported to be effective with similar range of toxicities.218
The robust efficacy of the MEK inhibitor cobimetinib in three patients with multi-system and refractory ECD was reported in 2018.219 In 2019, cobimetinib was shown to be effective in 12 patients with ECD (overall response rate 89%).188 Treatment efficacy was not affected by genotype, and responses were observed in patients with a range of activating MAPK pathway gene mutations (ARAF, BRAF,, KRAS, RAF1, MAP2K1 and MAP2K2). The most frequent adverse effects of cobimetinib are nausea, acneiform rash, and rhabdomyolysis.189
Juvenile xanthogranuloma
Most skin-limited JXG lesions involute spontaneously, but excision of a single lesion or a limited number of lesions may be indicated in some cases, if the lesion is cosmetically unacceptable or perhaps endangers vision or impairs swallowing. Our preference for treatment of patients with systemic JXG is clofarabine. We reported three such patients were successfully treated with moderate doses of clofarabine that can typically be administered in outpatient settings.201 A range of therapies including cladribine and combined cytarabine with cladribine have also been reported with responses.220, 221 Patients with ALK mutations can be cured by treatment with crizotinib, an inhibitor of the ALK tyrosine kinase receptor, although one patient had a spontaneous recovery.88 The many therapies given to patients with JXG reflect the current lack of data to guide therapeutic strategies. Patients with JXG are eligible to participate in Cobimetinib trials discussed previously.
Rosai-Dorfman disease
Patients with the classic manifestations of RDD present with limited lymphadenopathy or subcutaneous nodules with slow progression or remissions and relapses over many years, but they typically do not require treatment. Such limited involvement is rarely life-threatening, but some patients have a fatal outcome from involvement of the brain, liver, kidney, or spleen or massive lymphadenopathy causing respiratory or vascular compromise.136, 222 Individuals with autoimmune diseases such as systemic lupus erythematosus, autoimmune hemolytic anemia, or autoimmune hepatitis are more likely to have extensive disease and a higher fatality rate than patients with classic RDD. Patients with extra-nodal disease have a protracted clinical course and require systemic chemotherapy.136, 137 No standard of care has been established, and the therapeutic approach is usually determined on a case-by-case basis.
Surgery is generally limited to biopsy, whereas debulking might be required in patients with upper airway obstruction or intracranial lesions.136 Radiotherapy has little efficacy in most cases, although it can be beneficial in refractory orbital RDD with visual disturbances.136 Systemic corticosteroids are usually helpful in decreasing nodal size and improving symptoms, but they are immunosuppressive, and most patients relapse after cessation of therapy.136
A review of 40 RDD cases from the 1970’s until the mid-1990’s found variable response with a variety of chemotherapeutic agents.222 In one report, only two of ten patients given chemotherapy reached complete remission, both with a combination of low-dose methotrexate and 6-mercaptopurine.223 There are case reports of successful treatment with a combination of prednisone, oral methotrexate, thioguanine, and leucovorin224, IFNα,225 acyclovir,226 thalidomide227 , and the tyrosine kinase inhibitor imatinib.228 Cladribine or clofarabine have been found to be effective in some cases of recurrent, refractory, or severe RDD.201, 229 The efficacy of the anti-CD20- specific monoclonal antibody rituximab has been described and seems particularly interesting in cases of associated autoimmune hemolytic anemia or systemic lupus erythematosus.230 Radiotherapy and temozolomide have been used in neurological RDD.139 A recent case series reports that MEK inhibitor cobimetinib was effective in two patients;231 MEK inhibitor could be considered for severe, refractory RDD and/or RDD with severe life threatening presentation (such as CNS RDD).
Hemophagocytic lymphohistiocytosis
The initial management of HLH hinges on suppressing the hyperinflammatory process and treating the underlying trigger. Subsequently, prompt preparation for allogeneic hematopoietic stem cell transplantation is important for patients with many of the genetic HLH disorders, or those who have refractory or relapsing disease that suspected to be a genetic disorder. HLH is typically fatal without intervention. Treatment should begin quickly, with the exception that corticosteroids or etoposide should be withheld until evaluations for malignancy are completed (if feasible), as they could interfere with the ability to diagnose hematological malignancies. Exceptions to the requirement for HLH-specific treatment include patients who have secondary HLH that is expected to improve with treatment of the underlying trigger. For instance, individuals with visceral leishmaniasis may respond to antimicrobial therapy alone, and HLH that develops with malignancy may improve with treatment of the malignancy. In some cases, these interventions will result in improvement of the secondary hyperinflammatory manifestations.
The most commonly used HLH treatments are dexamethasone and etoposide according to the HLH-94 protocol, which was studied for the treatment of HLH by the Histiocyte Society in a multi-center trial.232 Etoposide has specific activity against activated T cells, which may account for evolution of this treatment approach.233 Cyclosporine is not recommended for early treatment of HLH owing to the risk of toxicity and the observation that early initiation of cyclosporine did not improve outcomes in the HLH-2004.141 Cyclosporine may be useful to prevent disease reactivation in patients who have achieved remission.232 Intrathecal treatment is recommended for central nervous system HLH that does not completely respond to systemic therapy. Not all patients with HLH require etoposide; corticosteroids alone may suffice in patients with mild disease. An alternative approach includes corticosteroids and anti-thymocyte globulin (ATG).234 ATG is a polyclonal antibody preparation made by immunizing horses or rabbits with human thymocytes. When administered to patients, ATG depletes human T cells. Treatment with etoposide and ATG resulted in complete responses in ~50–75% of patients.141, 234, 235 Rituximab or other B-cell depleting therapy should be considered for EBV-associated HLH.236 In contrast to patients with HLH, patients with MAS associated with autoimmune or auto-inflammatory disease are typically treated with IL-1 inhibitors, corticosteroids, and cyclosporine and are not treated with etoposide unless inflammation control is insufficient with conventional therapies.237
Newer treatments for HLH include biologic agents. Emapalumab is a monoclonal antibody directed against interferon gamma that was approved by the Food and Drug Administration for refractory, recurrent, or progressive HLH based on a clinical trial that demonstrated responses in 63% of previously treated patients.111, 238 Alternative approaches to immune suppression targeting specific immune cells, immune activation pathways and/or cytokines are also under investigation.132, 238 Alemtuzumab, a monoclonal antibody against CD52, has been used as salvage therapy.239, 240 Ruxolitinib, a Janus kinase (JAK) inhibitor that blocks multiple cytokine signaling pathways, was effective in a number of patients with HLH and murine studies.241–245 Anakinra (an IL-1R antagonist) was effective in secondary HLH.246 Tadekinig alfa is a recombinant human IL-18 binding protein247 that may be effective for some forms of MAS.
Allogeneic haematopoietic stem cell transplant is generally reserved for patients with severe genetic HLH disorders, such as those caused by mutations in PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, and SH2D1A, and can also be considered for patients with refractory or relapsing HLH that is thought to be a genetic disorder of the immune system. Reduced intensity conditioning regimens were often used in the past decade owing to the high mortality associated with fully myeloablative regimens in HLH. However, reduced intensity conditioning can frequently be associated with mixed chimerism and graft failure. A national trial with reduced intensity condition resulted in relatively high overall survival, but also high rates of secondary graft failure.248 Patients are at risk of HLH recurrence if donor chimerism declines to <5–30% of whole blood or T cell populations.248, 249 New developments in reduced toxicity myeloablative approaches may improve sustained donor chimerism while still minimizing toxicity.250–252 Propective studies are required to optimize survival, engraftment, and morbidity in HCT in this high risk population.252
Global variations
Diagnosing histiocytosis in developing settings may be extremely difficult because of the limited numbers of physicians who are aware of the diagnoses and the scarce resources for radiographical and other diagnostic and molecular tests, especially regarding histochemical stains to differentiate these diseases from more common benign and malignant entities. Available treatments may be extremely limited, based on need to deliver systemic chemotherapy along with required supportive care. Oral therapy with MAPK inhibitors could be effective, but currently challenging to deliver owing to limited drug access and high cost. For HLH, the need for haematopoietic stem cell transplantation for definitive cure for many patients is also a challenge.
Quality of life
LCH
Long-term quality of life studies report long-term sequleae in the majority of LCH survivors, with risks of sequelae associated with severity and duration of disease.253, 254 Long-term morbidity may have been increased by historic uncertainty of LCH as an inflammatory disorder, leading to tolerance of ‘smoldering’ disease and practice of treating relapses symptomatically. Specific long-term challenges including behaviour problems and learning in children, especially those with brain lesions and LCH-ND. Approximately 5% of all patients with LCH develop LCH-associated changes in the cerebellum, pons or basal ganglia (detectable with MRI), and some of them develop a neurodegenerative syndrome with ataxia, dysmetria (poor handwriting), dysarthria (unclear speech), and behavioural, or learning disabilities131, 255 Even adults with isolated pulmonary disease have been reported to struggle with long-term anxiety.256 The majority of patients with diabetes insipidus will need life-long supplemental vasopressin, and half of these individuals will also need supplemental thyroid, adrenal, growth, or gonadal hormones.196 Some patients with bone lesions can lead to permanent dysfunction, especially at vertebral sites where vertebra plana can cause instability of the spine.257 Pulmonary LCH can lead to destruction of sufficient pulmonary tissue that 50–90% of patients may experience dyspnea on exertion, need supplemental oxygen, and rarely lung transplant.258–260 If sclerosing cholangitis occurs with fibrosis of large parts of the liver, liver transplant may be required.261 A caregiver for a girl with LCH shares their story (Box 4).
Erdheim Chester disease
Prior to modern therapies, life expectancy for patients with ECD was >60% 3 years after diagnosis.154 Outcomes have improved with IFN-a and MAPK inhibitor therapeutic strategies, though patients typically require ongoing therapy. Long-term morbidities arise from both chronic disease and toxicities of long-term therapy.153 A patient-reported outcome tool was developed for ECD, the Erdheim-Chester Diseases Symptom Scale (ECD-SS). Sixty-two of 63 symptoms in the inventory were endorsed by at least 1 of 50 ECD participants, and neurologic or psychologic symptoms were reported by 92%. This study highlights the unmet need of management of neuro-psychiatric symptoms in this population.262
Juvenile Xanthogranuloma
Most patients with a few JXG lesions will have spontaneous resolution, and the disease never recurs. Others with more extensive disease can have long term problems from structural damage by brain lesions or possibly hepatic failure from large tumors263. Our experience is that long-term consequences of JXG are comparable to LCH.
Rosai-Dorfman Destombes disease
Most RDD patients have no long term problems from isolated adenopathy that resolves spontaneously or with modest treatment. The chronic nature of RDD can lead to discomfort or dysfunction based on sites of disease. In general, our experience is that long-term consequences of RDD are comparable to LCH. In the largest series, 17/238 (17%) patients died as the result of complications of their disease.34
Hemophagocytic lymphohistiocytosis
HLH can substantially affect a patient’s quality of life, as it is a life-threatening condition. Treatment usually requires many weeks to months of therapy. Most patients also require treatment of an underlying trigger, and some patients develop adverse effects of HLH such as bleeding complications, organ failure, and infections, which are inadequately quantified in the literature. Most patients require hospitalization, though data regarding general length of stay is limited. Intensive care needs are common, and even with treatment, mortality remains high. Similar to other rare and serious diseases that require care at specialized centers, patients and families may have to travel to tertiary care medical centers, a requirement that places additional financial and social stressors on all those involved. For patients with an underlying genetic HLH disorder, allogeneic HCT unfortunately leads to additional hospitalization time away from home and confers significant additional mortality risk, approximately 30%.264 Late effects including organ damage, decreased motor skills, learning disabilities, and other long-term issues may be substantial. However, with successful treatment and transplantation, many patients can live normal lives.
Outlook
Inherited and acquired genetics
In all histiocytic disorders, the future will be shaped by questions and opportunities arising from molecular genetics. The discovery of somatic BRAFV600E and other mutations affecting the MAPK pathway in LCH have changed our understanding of the neoplastic histiocytic disorders LCH, ECD, JXG and RDD. For LCH, there is an association of increased risk of mortality and neurological consequences for those patients with the BRAFV600E mutation. Although most patients with LCH, ECD, JXG and RDD have only one driver mutation, others may have up to over a dozen, and each additional mutation may have a role in pathogenesis.81 In adults, these histiocytic disorders may arise as a result of the accumulation of novel mutations through clonal hematopoiesis.53 However, many questions remain concerning the origin of mutations, the range of biological effects and the clinical utility of genotyping and mutation detection as biomarkers.
HLH includes a spectrum of disorders of constitutive genetic defects, in which clinical exome sequencing has already widened the number of genotypes known to be at risk, far beyond those classical defects associated with impaired cytotoxicity. This number will continue to expand through primary immunodeficiency screening programmes, together with the increasing application of sequencing to study older children and adults with late-onset or secondary HLH.
Cell of origin question
The ‘cell of origin’ question is familiar yet problematic to the field of oncogenesis. Although it is highly attractive to pinpoint a mutational hit to a cellular compartment, by definition, neoplastic transformation completely skews developmental architecture so that it may be difficult to map a malignant clone to any healthy counterpart. In the case of histiocytosis, somatic activation of the MAPK pathway seems to confer little proliferative advantage to stem or progenitor cells, and children with multi-system7 LCH have a stable, low burden of mutated clones.58, 81 This finding is in stark contrast to the progressive clonal dominance of events such as BCR-ABL1 translocation that are ultimately leukemogenic. In the periphery, the accumulation of lesional histiocytes is clearly driven by ERK activation. Mouse models suggest that retention in the tissues and inhibition of apoptosis correlate with accumulation of histiocytes, in keeping with the typically low proliferation rate observed in clinical lesions.7, 73 Multisystem LCH and ECD are associated with the presence of somatic mutation in monocytes, myeloid dendritic cells and their precursors. Systemic involvement and the propensity to recur suggest that lesional histiocytes arise from self-renewing progenitor or stem cells in the bone marrow in these patients. By contrast, the mutation is not usually detected in the peripheral blood or bone marrow of patients with single system disease or isolated lesions.63 Spontaneous healing and complete responses to minimally cytoreductive therapy in these individuals, suggest the the mutation must arise in a non-self-renewing cell, such as a committed progenitor that seeds the periphery, producing multiple lesions or a large rapidly growing solitary mass, but the disease never becomes systemic or recurring. The potential clinical utility of peripheral blood mutation detection as an indicator of disease risk has been highlighted in several retrospective surveys.81 Integration with clinical risk factors in prospective studies is in progress.
Response to inhibitors
Much remains unknown about the consequences of ERK activation in haematopoietic progenitors. The lack of any clonal advantage is corroborated by the failure of RAF and MEK inhibitors to eradicate the neoplastic clone, despite inducing remarkable clinical improvement in almost all patients.68, 69, 189 Although inhibitor therapy can be dialled down to a low level and has been life-saving for an increasing number of infants and adults with refractory disease, the rapid relapse that occurs with treatment cessation is currently a major clinical issue. It is uncertain how long children or younger adults will tolerate inhibitor therapy, and strategies to eliminate the ‘histiocytic stem cell’ are urgently needed. Suggestions range from full intensity allograft to maintenance style cytotoxic therapy with methotrexate and 6-mercaptopurine or hydroxyurea.
The argument for a pre-natal origin
During normal development, histiocytes arise early in pre-natal life, and many cell populations, notably microglia of the brain, are capable of self-renewal and do not require continual replenishment by hematopoiesis. It is conceivable that somatic MAPK activation could arise in a mosaic fashion during development, leading to variable clinical syndromes, depending on the site and degree of mosaicism. This mechanism was proposed for self-healing congenital histiocytosis involving neonatal Langerhans cells (Hashimoto-Pritzker disease ).9, 129 A more sinister connection with neurodegenerative disease has recently been suggested by mouse models showing that microglia harbouring activating BRAF mutation cause profound neurological deficits.78 The hypothesis of congenital mosaicism has a number of attractive features, such as the ability to explain localised cephalic disease that crosses tissue boundaries (for example, the skin–bone–pituitary gland lesions characteristic of the LCH variant Hand-Schuller-Christian disease) and the well-documented association between cranio-facial lesions, posterior pituitary gland failure and neurodegeneration. However, such a mechanism does not easily account for the typically late presentation of LCH-ND, which is associated with circulating BRAF-V600E+ monocytes and a perivascular distribution of similar cells in areas of early, active injury of the brain, with scant evidence of microglial activation.12 Although a second trigger could be invoked to explain the protracted kinetics of LCH-ND following systemic symptoms, a post-mortem study argued that persistence of mutant cells in the peripheral blood combined with pathological evidence of an infiltrative process strongly favors a hematogenous mechanism of neuro-degenerative LCH.12 It should be noted that neither involvement of the haematopoietic stem cell per se nor clinical responses of neurodegeneration to inhibitor therapy exclude the possibility that congenital mosaicism is involved in pathogenesis in some way. The concurrence of BRAF mutation in histiocytosis and a second malignancy may provide a potential test case if both lineages can be traced to a common ancestral fetal cell.
Germline genetics and second hits
In both neoplastic histiocytoses and HLH, there is a complex interplay between constitutional germline genetics, somatic mutations and environmental second hits that will be fertile ground for future exploration and clinical translation. LCH and possibly ECD have ethnic proclivities that may be linked to loci that modify myeloid cell development or the inflammatory response. The discovery of somatic mutations in these disorders does not exclude a role for environmental triggers such as trauma, infection and other inflammatory insults. Smoking and alpha-1 anti-trypsin deficiency both predispose lungs to develop LCH that appears genetically indistinct from other types of LCH. Cutaneous LCH also frequently involves areas subject to trauma such as skin folds. As an example hypothesis, modulation of the threshold of TLR signalling by ERK activation may be a key mechanism linking sensing of the environment to pathogenesis,265 but the molecular details of this interaction and potential to intervene remain untested.
In HLH, the meaning of primary and secondary are being eroded and replaced by the concept that germline predisposition, immunosuppression, inflammatory triggers and somatic mutations may all lower the threshold of hyperinflammation to a critical level. The feed-forward signals that drive this process are increasingly well understood and are likely to be targetable. A humanised antibody against the archetypal macrophage activator INFγ recently became the first licensed drug for this indication. Anti-IL-1, anti-IL-6 and anti-IL-18 strategies including small molecule JAK inhibitors have also been empirically tested with the promise of widening the range of exit strategies for refractory disease, beyond the traditional route of allogeneic haematopoietic stem cell transplantation.
Classification
The histiocytoses reviewed here represent a collection of disorders that share a terminal phenotype with features shared with cells of the mononuclear phagocyte system. Advances in genomics and ontogeny over the past decade frame a model of aberrant differentiation of myeloid precursors for LCH, ECD, JXG and some cases of RDD. Debate continues if these disorders are cancer. Clearly they represent clonal disorders associated with oncogene drivers, but designation may depend on one’s definition of cancer. We believe inflammatory myeloid neoplastic disorder appropriately characterizes this group. HLH itself represents a spectrum of disorders that share common endpoint of pathological inflammation that results from some combination of inherited disorders of immune function along with inflammatory trigger(s). The current front-line standard of care for high-risk LCH still cures <50% of patients.9 Outcomes for children and adults with histiocytic disorders may be improved by treating these conditions like cancer through cooperative trials with cancer consortia networks and resources. Organized prospective trials may further inform development of precision approaches that target specific mutations and specific cells in patients with particular genetic backgrounds
Supplementary Material
Box 2 |. Inherited disorders associated with HLH268.
Familial HLH
Mechanisms of disease: defective granule-mediated cytotoxicity
- Mutated genes
- PRF1 (FHL2)
- UNC13D (FHL3)
- STX11 (FHL4)
- STXBP2 (FHL5)
Pigmentary disorders associated with HLH
Mechanisms of disease: defective granule-mediated cytotoxicity
- Mutated genes
- RA827A (Griscelli syndrome type 2)
- LYST(Chediak-Higashi syndrome)
- AP3B1 (Hermansky-Pudlak syndrome 2)
X-linked lymphoproliferative diseases
- Mutated genes; mechanisms of disease
- SH2D1A (XLPl); defective 2B4-mediated cytotoxicity, defectiveTcell restimulation-induced cell death and absent iNKT cells
- XIAP (XLP2); dysregulated NLRP3 inflammasome and increased effector cell susceptibility to cell death
Other diseases
- Mutated genes; mechanisms of disease
- NLRC4; constitutively active NLRC4 inflammasome
- CDC42; defective formation of actin-based structures; defective proliferation, migration and cytotoxicity; and increased IL-ljS and 11–18 production
EBV susceptibility disorders
- Mutated genes; mechanisms of disease
- MAGTl; defective Mg2* transporter, low NKG2D, and defective cytotoxicity
- ITK; defective tyrosine kinase function, defective cytotoxic T cell expansion and cytolytic capacity, and decreased iNKT cells
- CD27; CD27 is expressed on T cells, participates in co-stimulatory signalling, interacts with CD70 and is required for normal T cell proliferation and triggering of cytotoxicity against EBV-infected B cells; decreased iNKT cells
- CD70; CD70 is expressed by EBV-infected B cells, interacts with CD27 on T cells and is required for normal expansion and cytotoxicity of the T cells; decreased NKG2D and 2B4; decreased iNKT cells
- CTPSl; CTP synthase 1 is involved in de novo synthesis of the CTP nucleotide (critical precursor of nucleic acid metabolism) and its deficiency leads to impaired proliferation; decreased iNKT cells
- RASGRPl; RASguanyl-releasing protein 1 activates RAS, which leads to MAP kinase pathway activation; defects in T cell activation, proliferation and migration; decreased cytotoxicity; decreased iNKT cells
CTP,cytidine triphosphate; EBV, Epstein-Barrvirus; FHL, familial haemophagocytic lymphohisti-ocytosis; HLH, haemophagocytic lymphohistiocytosis; iNKT cells, invariant natural killer T cells.
Box 3 |. Clinical criteria to diagnose HLH.
The Histiocyte Society HLH-2004 treatment trial established clinical criteria for haemophagocytic lymphohistiocytosis (HLH) for the purposes of the trial. A genetic HLH disorder can be diagnosed based on the finding of a pathogenic variant (or variants) in a known causative gene. A clinical syndrome of HLH can be diagnosed if at least five out of eight clinical and laboratory criteria are present. These criteria are as follows.
Fever
Splenomegaly
Cytopenia in at least two of the following cell lineages: haemoglobin <9 g/dl (<10 g/dl in neonates), platelets (count <100 × lOVml) and neutrophils (count <1 × 107ml)
Hypertriglyceridaemia (>265 mg/dl) or hypofibrinogenaemia (<150 mg/dl)
Hyperferritinaemia (>500ng/ml)
Soluble CD25 >2,400 U/ml (or elevated compared with laboratory-defined normal ranges)
Haemophagocytosis in bone marrow, spleen, lymph nodes or liver, as observed in histological sections
Low or absent natural killer cell cytotoxicity, as measured by a chromium release assay
Of note, a genetic defect alone does not imply that a patient has an active HLH process, only that the patient has an underlying genetic predisposition to the development of the syndrome of HLH. Diagnosis of active HLH in patients with a genetic predisposition to HLH should still be made based on the presence of signs and symptoms of hyperinflammation.
Box 4 |. A patient’s story.
Our daughter was diagnosed in October 2013. The journey for her and our family has taken many turns. Before diagnosis, we had chased Langerhans cell histiocytosis symptoms that mimic other common childhood diseases for over a year. The official diagnosis was in some way a relief and terrifying at the same time. We finally had a better understandi ng of how to help her get better. Unfortunately, the lack of treatment for such a long time meant that her condition had progressed to include bone lesions throughout her body, diabetes insipidus, and involvement in her spleen. We agreed to participate in a clinical trial for her treatment. The IV chemotherapy treatment and adverse effects were difficult. It was also a struggle to handle caring fora small toddler who was frequently hospitalized, another young child who was rarely permitted into the hospital, and normal work and life responsibilities. The treatment worked and her LCH was inactive until February of 2015. She was started on a stronger IV chemotherapy for 6 months. She handled this treatment better overall, and rang the ‘cured’ bell in September of 2015. We were overjoyed! Unfortunately, she relapsed four more times, with disease ranging from skin lesions to bone lesions, to CNS involvement. Each time we worked with her doctors to find the best treatment with the least amount of adverse effects. Her treatments have included IV chemotherapy, oral chemotherapy, BRAF inhibitors, and combinations of these different treatments. Emotionally and physically it has been draining for her and the entire family. She’s now ten and has a better understanding of the implications of another relapse. She’s more willing to share if she has any unexplained pain. The entire family becomes anxious before scans and follow-up appointments. But we trust her team and are excited at all the breakthroughs in the research and treatments that happened in the past eight years.
Acknowledgements
TXCH Histiocytosis Program is supported by a research grant from the HistioCure Foundation (C.E.A. and K.L.M). The North American Consortium for Histiocytosis is supported by a Consortium Grant from St. Baldrick’s Foundation (C.E.A. and K.L.M). Additional support was received from National Institutes of Health (NIH) grants: CA154947 (M.M and C.E.A) and NCI SPORE in Lymphoma P50CA126752 (C.E.A); Cancer Research UK (CRUK) grant C30484/A21025 (to M.C.); St. Baldrick’s Foundation Innovation Grant (C.E.A); and Leukemia and Lymphoma Society TRP (C.E.A.)
Footnotes
Competing interests
K.L.M. and C.E.A. have served on advisory committees for SOBI Corporation. All other authors have no conflicts of interest relevant to this publication.
Publication notes
This version of the article has been accepted for publication, after peer review but is not the Version of Record and does not reflect post-acceptance improvements, or any corrections. The Version of Record is available online at: http://dx.doi.org/10.1038/s41572-021-00307-9. Use of this Accepted Version is subject to the publisher’s Accepted Manuscript terms of use https://www.springernature.com/gp/open-research/policies/accepted-manuscript-terms’.
Reference List
- 1.Favara BE et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med. Pediatr. Oncol 29, 157–166 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Emile JF et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 127, 2672–2681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aschoff L. & Kiyono K. Frage der grossen Mononulearn. Folia Haematol 15, 383–390 (1913). [Google Scholar]
- 4.Steinman RM & Cohn ZA Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp. Med 137, 1142–1162 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinman RM & Cohn ZA Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp. Med 139, 380–397 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D. & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Bigenwald C, Chakraborty R, & Chen ST BRAFV600E-induced senescence in hematopoietic progenitors drives Langerhans cell histiocytosis pathophysiology. Nature Medicine 27, 851–861 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen CE et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J. Immunol 184, 4557–4567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen CE, Merad M, & McClain KL Langerhans-Cell Histiocytosis. N. Engl. J. Med 379, 856–868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan MB, Allen CE, Weitzman S, Filipovich AH, & McClain KL How I treat hemophagocytic lymphohistiocytosis. Blood 118, 4041–4052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gadner H. et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood 111, 2556–2562 (2008). [DOI] [PubMed] [Google Scholar]
- 12.McClain KL et al. CNS Langerhans cell histiocytosis: Common hematopoietic origin for LCH-associated neurodegeneration and mass lesions. Cancer 124, 2607–2620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh EA, Leary S, & Longoni G. Clinical and Radiographic Improvement of Neurodegenerative Langerhans Cell Histiocytosis (ND-LCH) Following Dabrafenib in: 32nd Annual Meeting of the Histiocyte Society Dublin, Ireland October 17–19, 2016. Pediatr. Blood Cancer 63 Suppl 2, S10–S65 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Haroche J. et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol 33, 411–418 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Hervier B. et al. Treatment of Erdheim-Chester disease with long-term high-dose interferon-alpha. Semin. Arthritis Rheum 41, 907–913 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro KB, Degar B, Antoneli CB, Rollins B, & Rodriguez-Galindo C. Ethnicity, race, and socioeconomic status influence incidence of Langerhans cell histiocytosis. Pediatr. Blood Cancer 62, 982–987 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Guyot-Goubin A. et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer 51, 71–75 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Alston RD et al. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr. Blood Cancer 48, 555–560 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Stalemark H. et al. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer 51, 76–81 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Swerdlow SH, Camp E, & Harris N. WHO classification of tumours of haematopoietic and lymphoid tissues. in WHO classification of Tumours of Haematopoietic and Lymphoid Tissues. (eds. Weiss L. & Facchetti F) 470–472 (IARC, Lyon, France, 2017). [Google Scholar]
- 21.Bhatia S. et al. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr 130, 774–784 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Venkatramani R, Rosenberg S, Indramohan G, Jeng M, & Jubran R. An exploratory epidemiological study of Langerhans cell histiocytosis. Pediatr Blood Cancer 59, 1324–1326 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Peckham-Gregory EC, McClain KL, Allen CE, Scheurer ME, & Lupo PJ The role of parental and perinatal characteristics on Langerhans cell histiocytosis: characterizing increased risk among Hispanics. Ann. Epidemiol 28, 521–528 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peckham-Gregory EC et al. A genome-wide association study of LCH identifies a variant in SMAD6 associated with susceptibility. Blood 130, 2229–2232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vassallo R, Ryu JH, Colby TV, Hartman T, & Limper AH Pulmonary Langerhans’-cell histiocytosis. N. Engl. J Med 342, 1969–1978 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Goyal G. et al. Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long-term outcomes. Br. J Haematol 182, 579–581 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Tran TA et al. Erdheim-Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol. Oncol 31, 782–786 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Haroche J, Cohen-Aubart F, & Amoura Z. Erdheim-Chester disease. Blood 135, 1311–1318 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Janssen D. & Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am. J Surg. Pathol 29, 21–28 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Isaacs H Jr. Fetal and neonatal histiocytoses. Pediatr Blood Cancer 47, 123–129 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Cambiaghi S, Restano L, & Caputo R. Juvenile xanthogranuloma associated with neurofibromatosis 1: 14 patients without evidence of hematologic malignancies. Pediatr Dermatol. 21, 97–101 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Destombes P. [Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases)]. Bull. Soc. Pathol. Exot. Filiales 58, 1169–1175 (1965). [PubMed] [Google Scholar]
- 33.Rosai J. & Dorfman RF Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch. Pathol 87, 63–70 (1969). [PubMed] [Google Scholar]
- 34.Foucar E, Rosai J, & Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin. Diagn. Pathol 7, 19–73 (1990). [PubMed] [Google Scholar]
- 35.Elshikh M. et al. Disease Characteristics, Radiologic Patterns, Comorbid Diseases, and Ethnic Differences in 32 Patients With Rosai-Dorfman Disease. J Comput. Assist. Tomogr 44, 450–461 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Wang KH et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br. J Dermatol 154, 277–286 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Henter JI, Elinder G, Soder O, & Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr. Scand 80, 428–435 (1991). [DOI] [PubMed] [Google Scholar]
- 38.Meeths M, Horne A, Sabel M, Bryceson YT, & Henter JI Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer 62, 346–352 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Allen CE, Yu X, Kozinetz CA, & McClain KL Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 50, 1227–1235 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Niece JA, Rogers ZR, Ahmad N, Langevin AM, & McClain KL Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr. Blood Cancer 54, 424–428 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Ishii E. et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int. J Hematol 86, 58–65 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Marsh RA Epstein-Barr Virus and Hemophagocytic Lymphohistiocytosis. Front Immunol. 8, 1902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu XJ et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: A retrospective multicenter study. Pediatr Blood Cancer 64, (2017). [DOI] [PubMed] [Google Scholar]
- 44.Shamriz O. et al. T Cell-Epstein-Barr Virus-Associated Hemophagocytic Lymphohistiocytosis (HLH) Occurs in Non-Asians and Is Associated with a T Cell Activation State that Is Comparable to Primary HLH. J Clin Immunol.( 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chinn IK et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood 132, 89–100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schram AM & Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 125, 2908–2914 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, & Bosch X. Adult haemophagocytic syndrome. Lancet 383, 1503–1516 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Arceci RJ, Brenner MK, & Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol. Oncol Clin. North Am 12, 339–357 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Willman CL et al. Langerhans’-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N. Engl. J Med 331, 154–160 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Yu RC, Chu C, Buluwela L, & Chu AC Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 343, 767–768 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Da Costa CE et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes. Cancer 48, 239–249 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Badalian-Very G. et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 116, 1919–1923 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samatar AA & Poulikakos PI Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov 13, 928–942 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Brown NA et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E-negative Langerhans cell histiocytosis. Blood 124, 1655–1658 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Mourah S. et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur. Respir. J 47, 1785–1796 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Nelson DS et al. MAP2K1 and MAP3K1 mutations in Langerhans cell histiocytosis. Genes Chromosomes. Cancer 54, 361–368 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Chakraborty R. et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 128, 2533–2537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chakraborty R. et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 124, 3007–3015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nezelof C, Basset F, & Rousseau MF Histiocytosis X histogenetic arguments for a Langerhans cell origin. Biomedicine. 18, 365–371 (1973). [PubMed] [Google Scholar]
- 60.Birbeck MS, Breathnach AS, & Everall JD An electron microscope study of basal melanocytes and high-level clear cells (Langerhans cells) in vitiligo. Journal of Investigative Dermatology 37, 51–64 (1961). [Google Scholar]
- 61.Merad M, Ginhoux F, & Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol 8, 935–947 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Valladeau J. et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 12, 71–81 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Berres ML et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J. Exp. Med 211, 669–683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heritier S, Emile JF, & Barkaoui M. BRAF mutation correlates with high-risk langerhans cell histiocytosis and increased resistance to first line therapy. J Clin Oncol 34, 3023–3030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Durham BH et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 130, 176–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milne P. et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood 130, 167–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Halbritter F. et al. Epigenomics and Single-Cell Sequencing Define a Developmental Hierarchy in Langerhans Cell Histiocytosis. Cancer Discov. 9, 1406–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eckstein OS, Visser J, Rodriguez-Galindo C, & Allen CE Clinical responses and persistent BRAF V600E(+) blood cells in children with LCH treated with MAPK pathway inhibition. Blood 133, 1691–1694 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donadieu J. et al. Vemurafenib for Refractory Multisystem Langerhans Cell Histiocytosis in Children: An International Observational Study. J. Clin. Oncol 37, 2857–2865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen AF et al. High frequency of clonal hematopoiesis in Erdheim-Chester disease. Blood 137, 485–492 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yokokawa Y. et al. Unique clonal relationship between T-cell acute lymphoblastic leukemia and subsequent Langerhans cell histiocytosis with TCR rearrangement and NOTCH1 mutation. Genes Chromosomes. Cancer 54, 409–417 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Rodig SJ et al. Aggressive Langerhans cell histiocytosis following T-ALL: clonally related neoplasms with persistent expression of constitutively active NOTCH1. Am. J. Hematol 83, 116–121 (2008). [DOI] [PubMed] [Google Scholar]
- 73.Hogstad B. et al. RAF/MEK/extracellular signal-related kinase pathway suppresses dendritic cell migration and traps dendritic cells in Langerhans cell histiocytosis lesions. J. Exp. Med 215, 319–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sengal A. et al. Overcoming T-cell exhaustion in LCH: PD-1 blockade and targeted MAPK inhibition are synergistic in a mouse model of LCH. Blood 137, 1777–1791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senechal B. et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS. Med 4, e253 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grois N. et al. Central nervous system disease in Langerhans cell histiocytosis. J. Pediatr 156, 873–881 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Grois N, Prayer D, Prosch H, & Lassmann H. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 128, 829–838 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Mass E. et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549, 389–393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haroche J. et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol 18, e113–e125 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Diamond EL et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov. 6, 154–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Durham BH et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat. Med 25, 1839–1842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emile JF et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood 124, 3016–3019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rafiei A. et al. BRAFV 600E or mutant MAP2K1 human CD34+ cells establish Langerhans cell-like histiocytosis in immune-deficient mice. Blood Adv. 4, 4912–4917 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cohen-Aubart F. et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am. J Hematol 93, E114–E117 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Picarsic J. et al. BRAF V600E mutation in Juvenile Xanthogranuloma family neoplasms of the central nervous system (CNS-JXG): a revised diagnostic algorithm to include pediatric Erdheim-Chester disease. Acta Neuropathol. Commun 7, 168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Burgdorf WH & Zelger B. JXG, NF1, and JMML: alphabet soup or a clinical issue? Pediatr Dermatol. 21, 174–176 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Zvulunov A, Barak Y, & Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol. 131, 904–908 (1995). [PubMed] [Google Scholar]
- 88.Chang KTE et al. ALK-positive histiocytosis: an expanded clinicopathologic spectrum and frequent presence of KIF5B-ALK fusion. Mod. Pathol 32, 598–608 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Garces S. et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai-Dorfman disease. Mod. Pathol 30, 1367–1377 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee LH et al. Real-time genomic profiling of histiocytoses identifies early-kinase domain BRAF alterations while improving treatment outcomes. JCI. Insight 2, e89473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Avitan-Hersh E. et al. A case of H syndrome showing immunophenotye similarities to Rosai-Dorfman disease. Am. J Dermatopathol 33, 47–51 (2011). [DOI] [PubMed] [Google Scholar]
- 92.Rossbach HC, Dalence C, Wynn T, & Tebbi C. Faisalabad histiocytosis mimics Rosai-Dorfman disease: brothers with lymphadenopathy, intrauterine fractures, short stature, and sensorineural deafness. Pediatr Blood Cancer 47, 629–632 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Morgan NV et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS. Genet 6, e1000833 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Picarsic J. & Jaffe R. Nosology and Pathology of Langerhans Cell Histiocytosis. Hematol. Oncol Clin North Am. 29, 799–823 (2015). [DOI] [PubMed] [Google Scholar]
- 95.Ammann S. et al. Effective Immunological Guidance of Genetic Analyses Including Exome Sequencing in Patients Evaluated for Hemophagocytic Lymphohistiocytosis. J Clin Immunol. 37, 770–780 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Meeths M. et al. Pathophysiology and spectrum of diseases caused by defects in lymphocyte cytotoxicity. Exp. Cell Res 325, 10–17 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Stepp SE et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 286, 1957–1959 (1999). [DOI] [PubMed] [Google Scholar]
- 98.Feldmann J. et al. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell 115, 461–473 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Zur SU et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet 14, 827–834 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Cote M. et al. Munc18–2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest 119, 3765–3773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Menasche G. et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet 25, 173–176 (2000). [DOI] [PubMed] [Google Scholar]
- 102.Nagle DL et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat. Genet 14, 307–311 (1996). [DOI] [PubMed] [Google Scholar]
- 103.de Saint BG & Fischer A. The role of cytotoxicity in lymphocyte homeostasis. Curr. Opin. Immunol 13, 549–554 (2001). [DOI] [PubMed] [Google Scholar]
- 104.Henter JI et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood 78, 2918–2922 (1991). [PubMed] [Google Scholar]
- 105.Osugi Y. et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood 89, 4100–4103 (1997). [PubMed] [Google Scholar]
- 106.Henderson LA et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 72, 1059–1063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jordan MB, Hildeman D, Kappler J, & Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 104, 735–743 (2004). [DOI] [PubMed] [Google Scholar]
- 108.Pachlopnik SJ et al. A Griscelli syndrome type 2 murine model of hemophagocytic lymphohistiocytosis (HLH). Eur. J Immunol 38, 3219–3225 (2008). [DOI] [PubMed] [Google Scholar]
- 109.Crozat K. et al. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J Exp. Med 204, 853–863 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sepulveda FE et al. Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood 121, 595–603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Locatelli F. et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J Med 382, 1811–1822 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Sayos J. et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 395, 462–469 (1998). [DOI] [PubMed] [Google Scholar]
- 113.Coffey AJ et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet 20, 129–135 (1998). [DOI] [PubMed] [Google Scholar]
- 114.Nichols KE et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc. Natl. Acad. Sci. U. S. A 95, 13765–13770 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rigaud S. et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 444, 110–114 (2006). [DOI] [PubMed] [Google Scholar]
- 116.Huck K. et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest 119, 1350–1358 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Montfrans JM et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol. 129, 787–793 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li FY et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lam MT et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J Exp. Med 216, 2778–2799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Canna SW et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet 46, 1140–1146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Romberg N. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet 46, 1135–1139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bode SF et al. The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica 100, 978–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spinner MA et al. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 137, 638–640 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Althonaian N, Alsultan A, Morava E, & Alfadhel M. Secondary Hemophagocytic Syndrome Associated with COG6 Gene Defect: Report and Review. JIMD. Rep 42, 105–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schulert GS & Grom AA Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu. Rev. Med 66, 145–159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch. Dis. Child 75, 17–24 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Allen CE, Ladisch S, & McClain KL How I treat Langerhans cell histiocytosis. Blood 126, 26–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodriguez-Galindo C. & Allen CE Langerhans cell histiocytosis. Blood 135, 1319–1331 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Simko SJ et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J. Pediatr 165, 990–996 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chellapandian D. et al. A multicenter study of patients with multisystem Langerhans cell histiocytosis who develop secondary hemophagocytic lymphohistiocytosis. Cancer 125, 963–971 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yeh EA et al. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatr. Blood Cancer 65, (2018). [DOI] [PubMed] [Google Scholar]
- 132.Cohen-Aubart F. Central nervous system involvement in Erdheim-Chester disease: an observational cohort study. Neurology In Press, (2020). [DOI] [PubMed] [Google Scholar]
- 133.Freyer DR, Kennedy R, Bostrom BC, Kohut G, & Dehner LP Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr 129, 227–237 (1996). [DOI] [PubMed] [Google Scholar]
- 134.Collum LM, Power WJ, Mullaney J, & Farrell M. Limbal xanthogranuloma. J Pediatr Ophthalmol. Strabismus 28, 157–159 (1991). [DOI] [PubMed] [Google Scholar]
- 135.Flaitz C, Allen C, Neville B, & Hicks J. Juvenile xanthogranuloma of the oral cavity in children: a clinicopathologic study. Oral Surg. Oral Med Oral Pathol Oral Radiol. Endod 94, 345–352 (2002). [DOI] [PubMed] [Google Scholar]
- 136.Abla O. et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 131, 2877–2890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cohen-Aubart F. & et al. La maladie de Rosai-Dorfman-Destombes est une histiocytose inflammatoire polymporphe: etude phenotypique multicentrique de 47 patients. Medecine Interne 36, A40–A41 (2015). [Google Scholar]
- 138.Juskevicius R. & Finley JL Rosai-Dorfman disease of the parotid gland: cytologic and histopathologic findings with immunohistochemical correlation. Arch Pathol Lab Med 125, 1348–1350 (2001). [DOI] [PubMed] [Google Scholar]
- 139.Sandoval-Sus JD et al. Rosai-Dorfman disease of the central nervous system: report of 6 cases and review of the literature. Medicine (Baltimore) 93, 165–175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jordan MB et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: Recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr. Blood Cancer 66, e27929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bergsten E. et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 130, 2728–2738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Horne A. et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr. Treat. Options. Neurol 19, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Benson LA et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol. Neuroimmunol. Neuroinflamm 6, e560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Palazzi DL, McClain KL, & Kaplan SL Hemophagocytic syndrome in children: an important diagnostic consideration in fever of unknown origin. Clin. Infect. Dis 36, 306–312 (2003). [DOI] [PubMed] [Google Scholar]
- 145.Phillips M, Allen C, Gerson P, & McClain K. Comparison of FDG-PET scans to conventional radiography and bone scans in management of Langerhans cell histiocytosis. Pediatr. Blood Cancer 52, 97–101 (2009). [DOI] [PubMed] [Google Scholar]
- 146.Ferrell J. et al. Discrepancies between F-18-FDG PET/CT findings and conventional imaging in Langerhans cell histiocytosis. Pediatr Blood Cancer 68, e28891 (2021). [DOI] [PubMed] [Google Scholar]
- 147.Ballester LY et al. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol. Oncol 36, 307–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsuji M. et al. Solitary myofibromatosis of the skull: a case report and review of literature. Childs Nerv. Syst 20, 366–369 (2004). [DOI] [PubMed] [Google Scholar]
- 149.DiCaprio MR & Enneking WF Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg. Am. 87, 1848–1864 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Adler R. & Wong CA Cranial fasciitis simulating histiocytosis. J Pediatr 109, 85–88 (1986). [DOI] [PubMed] [Google Scholar]
- 151.Goyal G. et al. Erdheim-Chester disease: Consensus recommendations for the evaluation, diagnosis, and treatment in the molecular era. Blood 135, 1929–1945 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Arnaud L. et al. 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum. 60, 3128–3138 (2009). [DOI] [PubMed] [Google Scholar]
- 153.Diamond EL et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 124, 483–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Haroche J. et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim-Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation 119, e597–e598 (2009). [DOI] [PubMed] [Google Scholar]
- 155.Haroche J. et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore) 83, 371–392 (2004). [DOI] [PubMed] [Google Scholar]
- 156.Kamisawa T, Zen Y, Pillai S, & Stone JH IgG4-related disease. Lancet 385, 1460–1471 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Morier P, Merot Y, Paccaud D, & et al. Juvenile Xanthogranuloma and urticaria pigmentosa. Arch Dermatol. 111, 365–366 (1975). [DOI] [PubMed] [Google Scholar]
- 158.Maric I. et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am. J Surg. Pathol 29, 903–911 (2005). [DOI] [PubMed] [Google Scholar]
- 159.Vaiselbuh SR, Bryceson YT, Allen CE, Whitlock JA, & Abla O. Updates on histiocytic disorders. Pediatr Blood Cancer 61, 1329–1335 (2014). [DOI] [PubMed] [Google Scholar]
- 160.Zhang X, Hyjek E, & Vardiman J. A subset of Rosai-Dorfman disease exhibits features of IgG4-related disease. Am. J Clin Pathol 139, 622–632 (2013). [DOI] [PubMed] [Google Scholar]
- 161.Gupta A. et al. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 51, 402–404 (2008). [DOI] [PubMed] [Google Scholar]
- 162.Ho C. et al. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am. J Clin Pathol 141, 62–71 (2014). [DOI] [PubMed] [Google Scholar]
- 163.Goel S, Polski JM, & Imran H. Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann. Clin Lab Sci 42, 21–25 (2012). [PubMed] [Google Scholar]
- 164.Lehmberg K, McClain KL, Janka GE, & Allen CE Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer 61, 2101–2103 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Hayden A. et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 1, 2529–2534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bryceson YT, March ME, Ljunggren HG, & Long EO Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rubin TS et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood 129, 2993–2999 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Weiss ES et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 131, 1442–1455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lin H. et al. IFN-gamma Signature in the Plasma Proteome Distinguishes Pediatric Hemophagocytic Lymphohistiocytosis from Sepsis and SIRS. Blood Adv.( 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Riviere S. et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am. J Med 127, 1118–1125 (2014). [DOI] [PubMed] [Google Scholar]
- 171.Parikh SA, Kapoor P, Letendre L, Kumar S, & Wolanskyj AP Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin. Proc 89, 484–492 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Daver N. et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer 123, 3229–3240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lehmberg K. et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br. J. Haematol 170, 539–549 (2015). [DOI] [PubMed] [Google Scholar]
- 174.Abdalgani M. et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood 126, 1858–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gifford CE et al. Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin Cytom. 86, 263–271 (2014). [DOI] [PubMed] [Google Scholar]
- 176.Ammann S. et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp. Immunol 176, 394–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bryceson YT et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood 119, 2754–2763 (2012). [DOI] [PubMed] [Google Scholar]
- 178.Romberg N, Vogel TP, & Canna SW NLRC4 inflammasomopathies. Curr. Opin. Allergy Clin Immunol 17, 398–404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wada T. et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine 65, 74–78 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Gadoury-Levesque V. et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. 4, 2578–2594 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ravelli A. et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann. Rheum. Dis 75, 481–489 (2016). [DOI] [PubMed] [Google Scholar]
- 182.Chikwava K. & Jaffe R. Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev. Pathol 7, 607–614 (2004). [DOI] [PubMed] [Google Scholar]
- 183.Picarsic J, Egeler RM, Chikwava K, Patterson K, & Jaffe R. Histologic patterns of thymic involvement in Langerhans cell proliferations: a clinicopathologic study and review of the literature. Pediatr. Dev. Pathol 18, 127–138 (2015). [DOI] [PubMed] [Google Scholar]
- 184.Picarsic J. & Jaffe R. Pathology of Histiocytic Disorders and Neoplasms and Related Disorders. in Histiocytic Disorders (eds. Janka G. & Abla G) 3–50 (Springer, 2018). [Google Scholar]
- 185.Zelger BW, Sidoroff A, Orchard G, & Cerio R. Non-Langerhans cell histiocytoses. A new unifying concept. Am. J Dermatopathol 18, 490–504 (1996). [DOI] [PubMed] [Google Scholar]
- 186.Paulli M. et al. Immunophenotypic characterization of the cell infiltrate in five cases of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Hum. Pathol 23, 647–654 (1992). [DOI] [PubMed] [Google Scholar]
- 187.Ost A, Nilsson-Ardnor S, & Henter JI Autopsy findings in 27 children with haemophagocytic lymphohistiocytosis. Histopathology 32, 310–316 (1998). [DOI] [PubMed] [Google Scholar]
- 188.Diamond EL et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 567, 521–524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cohen AF et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood 130, 1377–1380 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Egeler RM, Thompson RC Jr., Voute PA, & Nesbit ME Jr. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr. Orthop 12, 811–814 (1992). [DOI] [PubMed] [Google Scholar]
- 191.Tazi A. et al. Relapsing nodular lesions in the course of adult pulmonary Langerhans cell histiocytosis. Am. J Respir. Crit Care Med 157, 2007–2010 (1998). [DOI] [PubMed] [Google Scholar]
- 192.Eckstein OS et al. Management of severe pulmonary Langerhans cell histiocytosis in children. Pediatr Pulmonol.(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zinn DJ et al. Hydroxyurea: a new old therapy for Langerhans cell histiocytosis. Blood 128, 2462–2465 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Minkov M. et al. Treatment of multisystem Langerhans cell histiocytosis. Results of the DAL-HX 83 and DAL-HX 90 studies. DAL-HX Study Group. Klin. Padiatr 212, 139–144 (2000). [DOI] [PubMed] [Google Scholar]
- 195.McClain KL & Kozinetz CA A phase II trial using thalidomide for Langerhans cell histiocytosis. Pediatr. Blood Cancer 48, 44–49 (2007). [DOI] [PubMed] [Google Scholar]
- 196.Donadieu J. et al. Endocrine involvement in pediatric-onset Langerhans’ cell histiocytosis: a population-based study. J Pediatr 144, 344–350 (2004). [DOI] [PubMed] [Google Scholar]
- 197.Simko SJ, McClain KL, & Allen CE Up-front therapy for LCH: is it time to test an alternative to vinblastine/prednisone? Br. J. Haematol 169, 299–301 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Weitzman S. et al. 2’-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr. Blood Cancer 53, 1271–1276 (2009). [DOI] [PubMed] [Google Scholar]
- 199.Allen CE et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr. Blood Cancer 54, 416–423 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Morimoto A. et al. Intensified and prolonged therapy comprising cytarabine, vincristine and prednisolone improves outcome in patients with multisystem Langerhans cell histiocytosis: results of the Japan Langerhans Cell Histiocytosis Study Group-02 Protocol Study. Int. J Hematol 104, 99–109 (2016). [DOI] [PubMed] [Google Scholar]
- 201.Simko SJ et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr. Blood Cancer 61, 479–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Goyal G. et al. Single-agent cladribine as an effective front-line therapy for adults with Langerhans cell histiocytosis. Am. J Hematol 96, E146–E150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lorillon G, Meignin V, & Tazi A. [Adult pulmonary Langerhans cell histiocytosis]. Presse Med 46, 70–78 (2017). [DOI] [PubMed] [Google Scholar]
- 204.Cantu MA et al. Optimal therapy for adults with Langerhans cell histiocytosis bone lesions. PLoS. One 7, e43257 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tazi A. et al. Vinblastine chemotherapy in adult patients with langerhans cell histiocytosis: a multicenter retrospective study. Orphanet. J. Rare. Dis 12, 95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Derenzini E. et al. High efficacy of the MACOP-B regimen in the treatment of adult Langerhans cell histiocytosis, a 20 year experience. BMC. Cancer 15, 879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sivendran S, Harvey H, Lipton A, & Drabick J. Treatment of Langerhans cell histiocytosis bone lesions with zoledronic acid: a case series. Int. J. Hematol 93, 782–786 (2011). [DOI] [PubMed] [Google Scholar]
- 208.Barkaoui MA et al. Long-term follow-up of children with risk organ-negative Langerhans cell histiocytosis after 2-chlorodeoxyadenosine treatment. Br. J Haematol 191, 825–834 (2020). [DOI] [PubMed] [Google Scholar]
- 209.Donadieu J. et al. Cladribine and cytarabine in refractory multisystem Langerhans cell histiocytosis: results of an international phase 2 study. Blood 126, 1415–1423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Diamond EL et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol 4, 384–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hyman DM et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J Med 373, 726–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Arnaud L. et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood 117, 2778–2782 (2011). [DOI] [PubMed] [Google Scholar]
- 213.Haroche J. et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 121, 1495–1500 (2013). [DOI] [PubMed] [Google Scholar]
- 214.Cohen-Aubart F. et al. Variability in the efficacy of the IL1 receptor antagonist anakinra for treating Erdheim-Chester disease. Blood 127, 1509–1512 (2016). [DOI] [PubMed] [Google Scholar]
- 215.Cohen-Aubart F. et al. Efficacy of infliximab in the treatment of Erdheim-Chester disease. Ann. Rheum. Dis 77, 1387–1390 (2018). [DOI] [PubMed] [Google Scholar]
- 216.Pegoraro F. et al. Long-term follow-up of mTOR inhibition for Erdheim-Chester disease. Blood 135, 1994–1997 (2020). [DOI] [PubMed] [Google Scholar]
- 217.Ruan GJ et al. Acute Pancreatitis From Treatment With BRAF Inhibitors in Erdheim-Chester Disease: A Report From 2 Tertiary Referral Centers. Pancreas 50, e6–e8 (2021). [DOI] [PubMed] [Google Scholar]
- 218.Estrada-Veras JI et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 1, 357–366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cohen AF et al. Efficacy of the MEK inhibitor cobimetinib for wild-type BRAF Erdheim-Chester disease. Br. J Haematol 180, 150–153 (2018). [DOI] [PubMed] [Google Scholar]
- 220.Stover DG, Alapati S, Regueira O, Turner C, & Whitlock JA Treatment of juvenile xanthogranuloma. Pediatr Blood Cancer 51, 130–133 (2008). [DOI] [PubMed] [Google Scholar]
- 221.Blouin P. et al. Juvenile xanthogranuloma with hematological dysfunction treated with 2CDA-AraC. Pediatr Blood Cancer 55, 757–760 (2010). [DOI] [PubMed] [Google Scholar]
- 222.Pulsoni A. et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am. J Hematol 69, 67–71 (2002). [DOI] [PubMed] [Google Scholar]
- 223.Horneff G, Jurgens H, Hort W, Karitzky D, & Gobel U. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): response to methotrexate and mercaptopurine. Med. Pediatr Oncol 27, 187–192 (1996). [DOI] [PubMed] [Google Scholar]
- 224.Scheel MM, Rady PL, Tyring SK, & Pandya AG Sinus histiocytosis with massive lymphadenopathy: presentation as giant granuloma annulare and detection of human herpesvirus 6. J Am. Acad. Dermatol 37, 643–646 (1997). [DOI] [PubMed] [Google Scholar]
- 225.Palomera L, Domingo JM, Olave T, Romero S, & Gutierrez M. Sinus histiocytosis with massive lymphadenopathy: complete response to low-dose interferon-alpha. J Clin Oncol 15, 2176 (1997). [DOI] [PubMed] [Google Scholar]
- 226.Baildam EM, Ewing CI, D’Souza SW, & Stevens RF Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): response to acyclovir. J R. Soc. Med 85, 179–180 (1992). [PMC free article] [PubMed] [Google Scholar]
- 227.Chen E, Pavlidakey P, & Sami N. Rosai-Dorfman disease successfully treated with thalidomide. JAAD. Case. Rep 2, 369–372 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Utikal J. et al. Imatinib as a treatment option for systemic non-Langerhans cell histiocytoses. Arch Dermatol. 143, 736–740 (2007). [DOI] [PubMed] [Google Scholar]
- 229.Rodriguez-Galindo C. et al. Extranodal Rosai-Dorfman disease in children. J Pediatr Hematol Oncol 26, 19–24 (2004). [DOI] [PubMed] [Google Scholar]
- 230.Namoglu EC et al. Management and outcomes of sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman Disease). Leuk. Lymphoma 61, 905–911 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Moyon Q. et al. Lung Involvement in Destombes-Rosai-Dorfman Disease: Clinical and Radiological Features and Response to the MEK Inhibitor Cobimetinib. Chest 157, 323–333 (2020). [DOI] [PubMed] [Google Scholar]
- 232.Ehl S. et al. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol. Pract. 6, 1508–1517 (2018). [DOI] [PubMed] [Google Scholar]
- 233.Johnson TS et al. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. 192, 84–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Mahlaoui N. et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics 120, e622–e628 (2007). [DOI] [PubMed] [Google Scholar]
- 235.Henter JI et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood 100, 2367–2373 (2002). [DOI] [PubMed] [Google Scholar]
- 236.Chellapandian D. et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br. J Haematol 162, 376–382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ravelli A, Grom AA, Behrens EM, & Cron RQ Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 13, 289–298 (2012). [DOI] [PubMed] [Google Scholar]
- 238.Ehl S. et al. Is neutralization of IFN-gamma sufficient to control inflammation in HLH? Pediatr Blood Cancer 68, e28886 (2021). [DOI] [PubMed] [Google Scholar]
- 239.Strout MP, Seropian S, & Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat. Rev. Clin. Oncol 7, 415–420 (2010). [DOI] [PubMed] [Google Scholar]
- 240.Marsh RA et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr. Blood Cancer 60, 101–109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Das R. et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood 127, 1666–1675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, & de Saint BG Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood 128, 60–71 (2016). [DOI] [PubMed] [Google Scholar]
- 243.Sin JH & Zangardi ML Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: First case report. Hematol. Oncol Stem Cell Ther 12, 166–170 (2019). [DOI] [PubMed] [Google Scholar]
- 244.Broglie L. et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. 1, 1533–1536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ahmed A. et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. 6, e630–e637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Eloseily EM et al. Benefit of Anakinra in Treating Pediatric Secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 72, 326–334 (2020). [DOI] [PubMed] [Google Scholar]
- 247.Gabay C. et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis 77, 840–847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Allen CE et al. Reduced-intensity conditioning for hematopoietic cell transplant for HLH and primary immune deficiencies. Blood 132, 1438–1451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hartz B. et al. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood 127, 3281–3290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Felber M. et al. Targeted busulfan-based reduced-intensity conditioning and HLA-matched HSCT cure hemophagocytic lymphohistiocytosis. Blood Adv. 4, 1998–2010 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Naik SH et al. Incorporation of thiotepa in a reduced intensity conditioning regimen may improve engraftment after transplant for HLH. Br. J Haematol 188, e84–e87 (2020). [DOI] [PubMed] [Google Scholar]
- 252.Marsh RA et al. A Comparison of Hematopoietic Cell Transplant Conditioning Regimens for Hemophagocytic Lymphohistiocytosis Disorders. J Allergy Clin Immunol.( 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Haupt R. et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer 42, 438–444 (2004). [DOI] [PubMed] [Google Scholar]
- 254.Nanduri VR, Pritchard J, Levitt G, & Glaser AW Long term morbidity and health related quality of life after multi-system Langerhans cell histiocytosis. Eur. J. Cancer 42, 2563–2569 (2006). [DOI] [PubMed] [Google Scholar]
- 255.Vrijmoet-Wiersma CM et al. Health-related quality of life, cognitive functioning and behaviour problems in children with Langerhans cell histiocytosis. Pediatr. Blood Cancer 52, 116–122 (2009). [DOI] [PubMed] [Google Scholar]
- 256.Bugnet E. et al. Psychological features of adult patients with langerhans cell histiocytosis. PLoS. One 16, e0246604 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Mammano S, Candiotto S, & Balsano M. Cast and brace treatment of eosinophilic granuloma of the spine: long-term follow-up. J Pediatr Orthop. 17, 821–827 (1997). [PubMed] [Google Scholar]
- 258.Ronceray L, Potschger U, Janka G, Gadner H, & Minkov M. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J. Pediatr 161, 129–133 (2012). [DOI] [PubMed] [Google Scholar]
- 259.Delobbe A, Durieu J, Duhamel A, & Wallaert B. Determinants of survival in pulmonary Langerhans’ cell granulomatosis (histiocytosis X). Groupe d’Etude en Pathologie Interstitielle de la Societe de Pathologie Thoracique du Nord. Eur. Respir. J 9, 2002–2006 (1996). [DOI] [PubMed] [Google Scholar]
- 260.McClain KL, Gonzalez JM, Jonkers R, De JE, & Egeler M. Need for a cooperative study: Pulmonary Langerhans cell histiocytosis and its management in adults. Med Pediatr Oncol 39, 35–39 (2002). [DOI] [PubMed] [Google Scholar]
- 261.Braier J. et al. Langerhans cell histiocytosis: retrospective evaluation of 123 patients at a single institution. Pediatr. Hematol. Oncol 16, 377–385 (1999). [DOI] [PubMed] [Google Scholar]
- 262.Diamond EL et al. A scale for patient-reported symptom assessment for patients with Erdheim-Chester disease. Blood Adv. 3, 934–938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Maeda M. et al. Long-term outcomes of children with extracutaneous juvenile xanthogranulomas in Japan. Pediatr Blood Cancer 67, e28381 (2020). [DOI] [PubMed] [Google Scholar]
- 264.Marsh RA et al. Practice pattern changes and improvements in hematopoietic cell transplantation for primary immunodeficiencies. J Allergy Clin Immunol. 142, 2004–2007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Peyret V. et al. Functional Toll-like Receptor 4 Overexpression in Papillary Thyroid Cancer by MAPK/ERK-Induced ETS1 Transcriptional Activity. Mol. Cancer Res 16, 833–845 (2018). [DOI] [PubMed] [Google Scholar]
- 266.Minkov M. et al. Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J. Pediatr 153, 700–705 (2008). [DOI] [PubMed] [Google Scholar]
- 267.Egeler RM, de KJ, & Voute PA Cytosine-arabinoside, vincristine, and prednisolone in the treatment of children with disseminated Langerhans cell histiocytosis with organ dysfunction: experience at a single institution. Med Pediatr Oncol 21, 265–270 (1993). [DOI] [PubMed] [Google Scholar]
- 268.Canna SW & Marsh RA Pediatric hemophagocytic lymphohistiocytosis. Blood 135, 1332–1343 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.