

Abstract
The efficiency of drug biosynthesis depends on different transcriptional regulatory pathways in Streptomyces, and the protein degradation system adds another layer of complexity to the regulatory processes. AtrA, a transcriptional regulator in the A‐factor regulatory cascade, stimulates the production of daptomycin by binding to the dptE promoter in Streptomyces roseosporus. Using pull‐down assays, bacterial two‐hybrid system and knockout verification, we demonstrated that AtrA is a substrate for ClpP protease. Furthermore, we showed that ClpX is necessary for AtrA recognition and subsequent degradation. Bioinformatics analysis, truncating mutation, and overexpression proved that the AAA motifs of AtrA were essential for initial recognition in the degradation process. Finally, overexpression of mutated atrA (AAA‐QQQ) in S. roseosporus increased the yield of daptomycin by 225% in shake flask and by 164% in the 15 L bioreactor. Thus, improving the stability of key regulators is an effective method to promote the ability of antibiotic synthesis.
Keywords: AtrA, ClpP, ClpX, daptomycin, degradation, S. Roseosporus
1. INTRODUCTION
Daptomycin, a novel cyclic lipopeptide antibiotic, is synthesized from its biosynthetic gene cluster‐dptEFABCD (dpt) in Streptomyces roseosporus (Miao et al., 2005). AtrA, downstream of the A‐factor cascade, links regulatory pathways to the dpt expression by binding to dptEp (Mao et al., 2015; Zheng et al., 2019). AtrA is highly conserved in Streptomyces and homologs regulate synthesis of antibiotics in other species. For example, in Streptomyces griseus, AtrA binds the upstream of strR at positions −117 to −142 relative to its transcriptional start point and positively regulates streptomycin production (Hirano et al., 2008). Moreover, AtrA stimulates the production of pristinamycin by binding spbR/papR5 in Streptomyces pristinaespiralis (Wang et al., 2015). However, AtrA‐based regulation is complex. In Streptomyces globisporus, AtrA binds to sgcR1/sgcR2 to promote the production of heptaene, afterwards heptaene inhibits the binding of AtrA to the promoters (Li et al., 2015). Such negative feedback is also known in Streptomyces coelicolor. Actinorhodin is not only a product positively regulated by AtrA binding to actII‐ORF4 but also acts as an inhibitor (Uguru et al., 2005). Additionally, Avel, an AtrA homolog in Streptomyces avermitilis, is involved in primary metabolism such as substrate transport and amino acid metabolism (Liu et al., 2019). Based on previous studies, AtrA is an important part of a complex regulatory network and its function is controlled by a feedback system.
The degradation of proteins in Streptomyces is mainly carried out by AAA+ proteases like ClpP, Lon, and FtsH (Neuwald et al., 1999), which is similar to most bacteria. In addition, Streptomyces has a unique proteasome and the proteasome specifically degrades proteins with the premise that Pup is covalently attached to the lysine of substrates (Boubakri et al., 2015; Nagy et al., 1998; Pearce et al., 2008), These degradation systems together form a rigorous regulatory network that facilitates proteome changes during metabolic shifts. ClpP is a serine protease that it is often encoded by multiple alleles in actinomycetes (De Crécy‐Lagard et al., 1999; Kahne & Darwin, 2021). To degrade substrates, ClpP works together with proteins from a class of ATPase, such as ClpA, ClpC, or ClpX, which can consume ATP to facilitate activation (Gominet et al., 2011). Among them, the ClpXP complex is relatively widely studied because of its conserved structure and diversity of substrate recognition. According to the alignment of the recognition sequences, it is known that ClpXP protease prefers alanine, arginine and lysine residues (Fei et al., 2020; Flynn et al., 2003; Levchenko et al., 2000). ClpP is indispensable for regulation in Streptomyces. For example, in Streptomyces lividans, deletion of clpP produced a bald phenotype (De Crécy‐Lagard et al., 1999). Moreover, AdpA as a central regulator is also able to bind to the promoter of clpP1 and clpP2, and lack of active ClpP peptidase leads to a decrease in the amount of AdpA (Guyet et al., 2013; Guyet et al., 2014). Additionally, SigT, which is involved in morphological differentiation in Streptomyces coelicolor, can be degraded by ClpXP after addition of a SsrA label (−AAXXXXXALAA) (Mao et al., 2013). Overexpression of ClpX can activate the production of actinorhodin in S. lividans and enhance the yield of actinorhodin in S. coelicolor (De Crécy‐Lagard et al., 1999; Mao et al., 2013). Although the above‐mentioned studies on ClpXP are valuable, systematic and in‐depth research on the degradation mechanism of ClpXP in Streptomyces has not been reported, including the diversity of its substrates and the specificity of its recognition sites. Importantly, the potential of changes to regulator degradation and the subsequent influence on secondary metabolite production has not been shown.
Here, we identified AtrA as a new substrate of ClpXP in S. roseosporus and revealed its multiple AAA motifs as essential recognition sites. Overexpression of mutated atrA successfully enhanced the yield of daptomycin. Our findings provide novel insights into regulatory pathway robustness. Furthermore, we used protein degradation as a tool to influence the protein level of important regulators in Streptomyces, which could be applied for increased production of antibiotics.
2. RESULTS
2.1. AtrA is tightly regulated in vivo
Based on a previous study, AtrA is a key regulator in S. roseosporus, which can bind to the co‐transcriptional start site of the daptomycin BGC, and positively regulate the production of daptomycin (Mao et al., 2015). However, subsequent overexpression of AtrA in the WT (AtrAoe) showed no increase in daptomycin production in shake flasks (Figure 1a), which was inconsistent with our hypothesis. Hence, we needed to verify the previous experimental results. After we complemented ΔatrA with atrA‐egfp with endogenous promoter (AtrA‐eGFP), we were able to observe green fluorescent mycelia under the microscope (Figure 1b). This suggested that the endogenous promotor is sufficient for gene expression and detectable AtrA‐eGFP protein levels. Through quantitative real‐time PCR (qRT‐PCR), we found that the transcript level of atrA was indeed increased in AtrAoe in comparison to WT (Figure 1c). Meanwhile, Western blot showed that atrA could not be successfully overexpressed in WT, only a faint signal was detected in a 24 h sample (Figure 2f). This indicated that AtrA may be post translationally regulated in vivo through degradation. In order to verify whether AtrA protein level was regulated by proteases, we purified AtrA from Escherichia coli harboring pET‐32a‐atrA and incubated it with cell lysates, which were collected from the fermentation broth of WT at 24 h. Most of AtrA underwent degradation within the first 5 min of incubation (Figure 1d).
FIGURE 1.
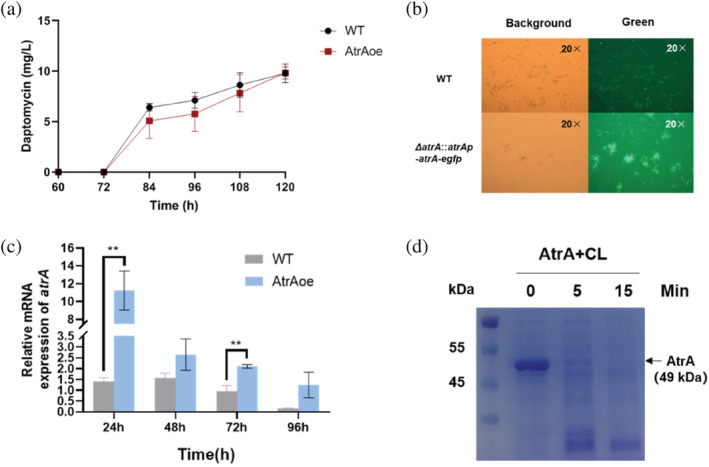
AtrA is tightly regulated in S. roseosporus. (a) HPLC analysis of daptomycin fermentation levels in AtrAoe strain and WT. (b) pSET‐152 (atrAp‐atrA‐egfp) was used to complement ΔatrA strain. One microliter of YEME fermentation broth was taken at 24 h, diluted 10 times and observed under OLYMPUS BX51 microscope. Objective magnification is 20×. (c) qRT‐PCR was used to detect the changes of atrA transcription levels in WT and AtrAoe strain. The strains were fermented in YEME medium for 24, 48, 72, and 96 h to extract RNA, respectively. (d) AtrA (pET‐32a) was incubated with cell lysate of WT, which was fermented for 24 h, at 30°C in vitro. Samples after different incubation times were analyzed on a 10% SDS‐PAGE. Forty‐nine kilodaltons represented the molecular weight of AtrA. CL represented cell lysate.
FIGURE 2.
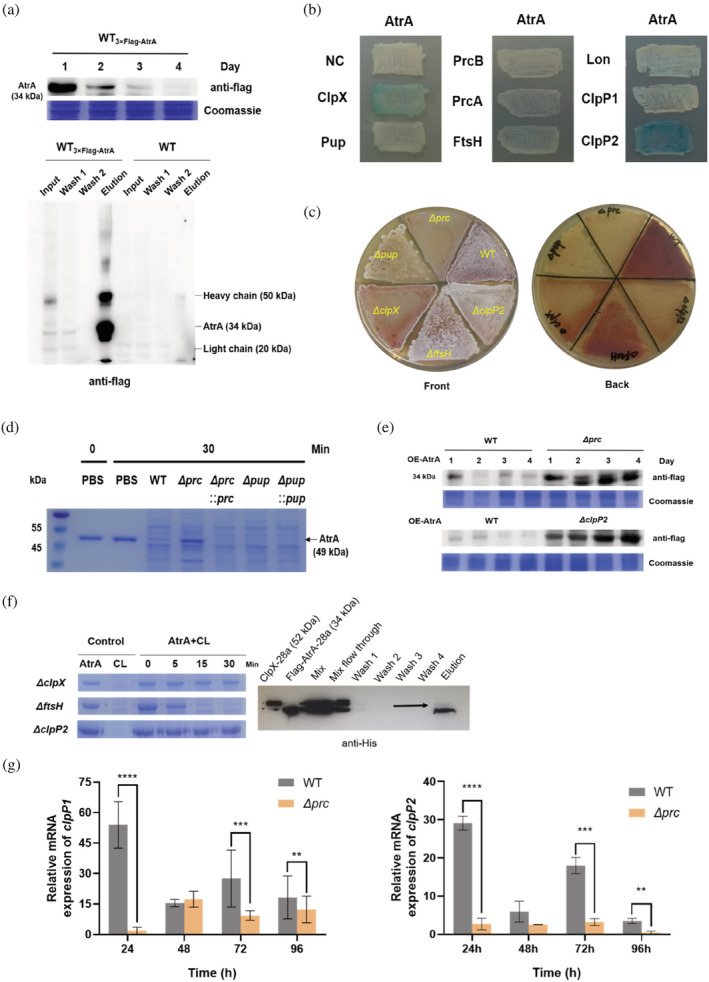
Proteases screening and validation in vivo and in vitro. (a) 3×Flag‐linker‐AtrA replaced AtrA in situ after homologous recombination in WT (WT3×Flag‐AtrA), and the endogenous protein level of AtrA was detected by Western blot. In vivo pull‐down assays were performed at 24 h. WT was used as a control. After affinity purification the samples were analyzed by Western blot, and the interacting proteins were identified by LC–MS/MS. Thirty‐four kilodaltons represented the molecular weight of AtrA. (b) Bacterial two‐hybrid system was used to screen interacting proteins. The atrA gene was cloned into pUT‐18, and clpP1, clpP2, lon, ftsH, clpX, prcA, and prcB were cloned into pKT‐25. The empty plasmids were used as a negative control. (c) Phenotypes of the WT, Δprc, ΔclpP2, ΔftsH, Δpup, and ΔclpX strains grown on R5 medium at 30°C. Images were taken at 10 days. (d) AtrA (pET‐32a) was incubated with cell lysates of WT, Δprc, Δpup, and complemented strains (Δprc::prc, Δpup::pup) at 30°C, respectively, and samples were analyzed on a 10% SDS‐PAGE. Forty‐nine kilodaltons represented the molecular weight of AtrA. (e) Western blot analysis of the overexpression of atrA in WT, Δprc and ΔclpP2. Thirty‐four kilodaltons represented the molecular weight of AtrA. (f) AtrA (pET‐32a) was incubated with cell lysates of WT, ΔftsH, ΔclpX and ΔclpP2 at 30°C, respectively, samples were analyzed on a 10% SDS‐PAGE. pET‐28a was used to produce AtrA with 3×Flag at the N‐terminus and ClpX. Four micrograms AtrA, 4 μg ClpX and 3 mM ATP were mixed and incubated at 30°C for 4 h in vitro. AtrA was adsorbed by Flag affinity gel, and proteins were detected with anti‐His antibody after elution. Thirty‐four kilodaltons represented the molecular weight of AtrA and 52 kDa represented the molecular weight of ClpX. (g) qRT‐PCR was used to detect the changes of clpP1 and clpP2 transcription levels in WT and Δprc. The strains were fermented in YEME medium for 24, 48, 72, and 96 h to extract RNA. The asterisk (**) indicated the degree of significance.
2.2. Degradation of AtrA is directly regulated by ClpXP and indirectly by the proteasome
In order to explore which protease is responsible for the degradation of AtrA, we used a strain with 3×Flag‐atrA in situ replacement (WT3×Flag‐AtrA) for pull‐down assay in vivo, and collected mycelia at 24 h which showed the highest protein level (Figure 2a). The WT served as a control. After affinity purification, the eluted proteins were digested and the peptides in the mixture were identified by LC–MS/MS. Notably, the proteasome (Prc) consists of PrcB and PrcA in S. roseosporus. Comparing the result of LC–MS/MS with proteases and their signal molecules contained in Streptomyces, prcB, prcA, ftsH, clpP1, and clpP2 were identified in the elution fraction from WT3×Flag‐AtrA (Table S1). Among them, clpP2 also appeared in the pull‐down result of WT. Since the results of pull‐down assays in vivo often show false positives, we used bacterial two‐hybrid system (BACTH) as complementary experiments in vitro. Finally, clpX and clpP2 were identified as potential interaction partners through the blue color of the colonies (Figure 2b). We verified all the above results by gene deletions, including pup, the important signal molecule of the proteasome. Phenotypes of the Δprc and the Δpup became bald and lost the ability to produce red pigment. The metabolism of red pigment of the ΔclpP2 and the ΔclpX were also affected. Whereas the white aerial hyphae of ΔclpX grew poorly. Among them, the knockout of ftsH had minimal effect on the phenotype (Figure 2c). Incubation assays showed that the deletion of the proteasome could prevent the degradation of AtrA in vitro, but the knockout of pup did not (Figure 2d). Moreover, overexpression of atrA in the Δprc (Δprc‐atrAoe) strain was indeed successful (Figure 2e). According to the previous studies, the premise of the specific degradation of the proteasome is that Pup must be covalently bound to the lysine of the substrate (Striebel et al., 2009). To further verify whether AtrA is degraded by the proteasome, we used LC–MS/MS detection of AtrA peptides after incubation with the cell lysate of Δprc and the result showed no molecular weight migration at the lysine sites (Table S2). In addition, mutating two lysine sites K104 and K132 (K‐Q/K‐R/K‐A) of AtrA was not sufficient to prevent degradation in vitro (Figure S1). On the other hand, incubation of AtrA with cell lysates of ΔclpP2, ΔclpX, and ΔftsH strains in vitro (Figure 2f), showed that ClpXP was the key protease for degradation of AtrA. Furthermore, atrA can be successfully overexpressed in ΔclpP2 strain (ΔclpP2‐atrAoe) (Figure 2e). Next, the pull‐down assay of Flag‐tagged AtrA with ClpX showed their natural binding in vitro. The incubation of AtrA with ΔclpP1 indicated that ΔclpP1 strain gave a similar effect to ΔclpP2, which reinforced the suggestion that ClpXP was involved, since it is assumed to function in the cell as the ClpXP1P2 complex, so that either clpP1 or clpP2 deletion was expected to impair its activity (Figures 2f and S2). Thus, we speculated that the proteasome might regulate the ClpP protease and therefore both knockout strains can achieve the overexpression of AtrA. The growth phenotype of the Δprc was much more defective than the ΔclpP2 growth phenotype (Figure 2c), and our previous transcriptomic and proteomic studies on the Δprc also indicate that the proteasome plays a central regulatory role in S. roseosporus (Xu et al., 2022). qRT‐PCR result verified the above speculation. The deletion of the proteasome led to a significant decrease in the transcription level of clpP1 and clpP2, therefore, Δprc could also block the degradation of AtrA through reduced levels of ClpP (Figure 2g).
2.3. Multiple AAA motifs are essential for the degradation of AtrA
Although the deletion of clpP2, clpX, and prc prevented the degradation of AtrA, HPLC results showed no production of daptomycin at 120 h in the fermentation broth of ΔclpP2‐atrAoe, ΔclpX‐atrAoe, and Δprc‐atrAoe (Figure S3). This suggested that the proteasome and proteases are indispensable for the secondary metabolism of S. roseosporus, and knocking out those genes was not feasible to promote antibiotic production. We wondered if we could prevent AtrA degradation by mutating its cleavage sites. After raising the concentration of AtrA in the degradation reaction in vitro, the cleavage bands were cut out for MS identification (Figure 3a). According to the results, we inferred that the degradation band‐3 contained two similar size peptides (Figure S4). Due to the limitations of trypsin digestion, we could only identify one site for preferential cleavage–the peptide bond of the asparagine (N34) and glycine (G35) (Figure 3e). After mutating asparagine (N) to glutamine (Q), we incubated AtrA (N‐34Q) with the cell lysate of WT again in vitro and found a new cleavage band (Figure S5). It implied that the cleavage sites were interchangeable. Meanwhile, we tested the degradation direction by incubating AtrA with (+TGA) and without a stop codon (−TGA) in the cell lysate of WT. The AtrA with the stop codon only had a His tag at the N‐terminus, while the AtrA without the stop codon had His tags at both the N‐terminus and the C‐terminus. Based on the difference in the number of His tags, different degradation directions would lead to differences in the display of degradation bands. Western blot detected AtrA (−TGA) containing two major degradation bands (Deg 1 + Deg 2) after incubating with WT cell lysate, while another degradation band (Deg 3) was detected in both reactions AtrA (+TGA) and AtrA (−TGA) for 10 min (Figure 3b), which indicated that AtrA was mainly degraded from the N‐terminus to the C‐terminus. And we speculated that a small amount of AtrA might be degraded from the C‐terminus. However, two degradation bands could be observed at 0 min, and we assumed that the reaction rate was very fast, resulting in immediate degradation after mixing. Hence, we incubated AtrA without cell lysate and confirmed the absence of cleavage products (Figure S6). Since the previous strategies did not work, we decided to block the degradation of AtrA by mutating its recognition motif. Truncation mutations of AtrA showed that the degradation of AtrA was greatly hindered when the N‐terminal 59 amino acid were deleted (Figure 3c). All the results from truncating experiments showed that AtrA would be degraded eventually. In order to improve AtrA stability, our analysis of the amino acid sequence of AtrA found that the first 30 amino acids of the N‐terminus contained three repetitive SAAA motifs, which were likely to be an important part of recognition sites for ClpX (Figure 3e). Therefore, three SAAA motifs of AtrA were mutated to AQQQ (AtrA(SAAAm)) (Figure 3d). Its half‐life was increased by about 10 min compared to AtrA, but AtrA (SAAAm) was still degraded in the end, which was consistent with the previous truncation mutation results. This observation let us to the conclusion that the N‐terminus of AtrA has the main degradation recognition sites, and the C‐terminus may also have a recognition site. Notably, ClpX can recognize sequences at the N‐terminus or C‐terminus. Surprisingly, in the last 20 amino acids an AAAS motif was found, and we speculated that multiple AAA might constitute an essential recognition motif. However, after mutating AAA to QQQ (AtrA (AAAm)), AtrA turned out to be still degraded in the cell lysate of WT (Figure 3d). Thus, it forced us to reexamine the entire plasmid sequence, and considered that the S‐tag (MKETAAAKFERQHMPS‐) of pET‐32a not only contained a AAA motif, but also the first five amino acids were very similar to the N‐terminus of OmpA (MKKTA‐), a ClpX substrate (Flynn et al., 2003). To reduce interference from these factors, atrA was transferred into pET‐28a. Finally, it could be shown that the mutation of AAA to QQQ successfully prevented the degradation of AtrA (Figure 3d). Nevertheless, AtrA (SAAAm) expressed from pET‐28a degraded faster than AtrA (SAAAm) expressed from pET‐32a. We surmised that it might be caused by some long tags encoded in pET‐32a, such as the TrxA‐tag, which increased the molecular weight of the fusion protein and might affect the rate of degradation. In addition, through bioinformatics analysis, we found many proteins in the proteome of S. roseosporus containing the AAA motif, but only AtrA contained three consecutive sequences (Figure S7 and Table S3). We screened for proteins that have a AAA motif in the first 20 amino acids at the N‐terminus and last 20 amino acids of the C‐terminus, and identified 31 proteins. Among them, six proteins were regulators, including ArpA, the regulator of the A‐factor regulatory cascade, and 25 were proteins of unknown function, suggesting that such motifs might be universally associated with protein homeostasis in the proteome of S. roseosporus.
FIGURE 3.
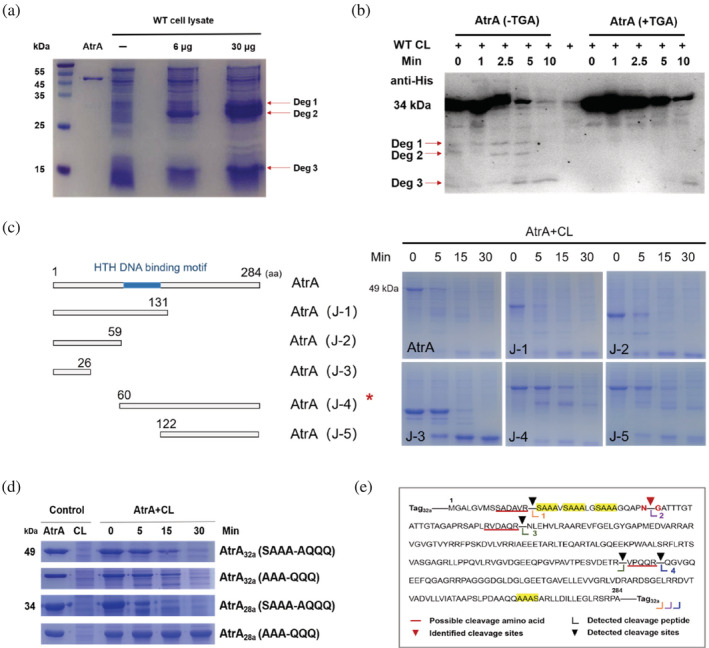
Identification of degradation bands and analysis of recognition regions of AtrA. (a) Different amounts of AtrA (pET‐32a) were incubated with cell lysate of WT at 30°C for 10 min, and samples were analyzed on a 15% SDS‐PAGE. Forty‐nine kilodaltons represented the molecular weight of AtrA. (b) AtrA with stop codon (AtrA+TGA) and without stop codon (AtrA‐TGA) were incubated with cell lysate of WT at 30°C for 0, 1, 2.5, 5, and 10 min, respectively. The samples were analyzed by Western blot. Thirty‐four kilodaltons represented the molecular weight of AtrA. (c) Schematic diagram of AtrA truncation mutants. AtrA truncation mutants were incubated with cell lysate of WT at 30°C, and samples were analyzed on a 15% SDS‐PAGE. Amino acid is abbreviated as aa. (d) Different AtrA mutants (SAAA motifs were mutated to AQQQ and AAA motifs were mutated to QQQ) purified from pET‐28a and pET‐32a were incubated with cell lysate of WT and reactions were carried out at 30°C. Thirty‐four kilodaltons and 49 kDa represented the molecular weight of AtrA purified from different vectors. (e) Schematic representation of AtrA degradation patterns.
2.4. Overexpression of AtrA (AAAm) enhances the yield of daptomycin
After mutating two kinds of motifs, Western blot results showed that AtrA (AAAm) and AtrA (SAAAm) could work for the overexpression of AtrA. According to the gray scale, the expression level of AtrA (AAAm) was slightly higher than that of AtrA (SAAAm) at 72 and 96 h (Figure 4a). However, even blocking the degradation pathway did not result in high AtrA protein levels as expected. We inferred two possibilities. On one hand, ermEp* as a strong promoter may be nonspecifically bound by some proteins in S. roseosporus, resulting in the inability to maintain a high activity through binding of AtrA. On the other hand, different types and concentration of proteins might be increased in the later growth stage, and the concentration of AtrA would be relatively reduced. In terms of the phenotype of fermentation medium, the AtrA (AAAm) strain produced more red pigment than AtrA (SAAAm) at 72 h. It indicated that the metabolism of the strain was more vigorous, and the corresponding synthetic antibiotics might be more (Figure 4b). HPLC results showed that the production of daptomycin of the AtrA (AAAm) strain was increased by about 225% compared with WT, which was slightly higher than the yield of the AtrA (SAAAm) strain (Figure 4b). Based on the property that AtrA can bind to the promoter of the BGC, it was found that AtrA (AAAm) was more effective than AtrA (SAAAm) in increasing the transcription level of dptEp by qRT‐PCR (Figure 4c). Electrophoretic mobility shift assay (EMSA) experiments showed that AtrA (AAAm) and AtrA (SAAAm) could still bind to atrAp and dptEp (Figure 4d). Therefore, the AtrA (AAAm) overexpression strain would be used for follow‐up research. Although the influence of AtrA on the primary metabolism of S. roseosporus is not clear, the dry weight of AtrA (AAAm) strain mycelia also changed (Figures S8 and S9). Thus, the concentration of daptomycin based on the dry weight of mycelia was used to precisely describe the yield (Figure 4e). Finally, the AtrA (AAAm) strain was cultured in a 15 L bioreactor and was fed decanoic acid at 72 h. The yield of daptomycin increased from 84 mg/L to 233 mg/L compared with WT at 264 h (Figure 4f). And due to the promotion of transcription of the BGC, the AtrA (AAAm) strain started to produce daptomycin as early as 72 h, whether in the shake flask or the bioreactor fermentation, which correspondingly prolonged the fermentation cycle. According to our previous work (Lyu et al., 2022), enhancing the transcription of dptE had little effect on the start point of daptomycin production. Thus, AtrA may also influence production of daptomycin precursors in S. roseosporus and lead to an earlier start of daptomycin production.
FIGURE 4.
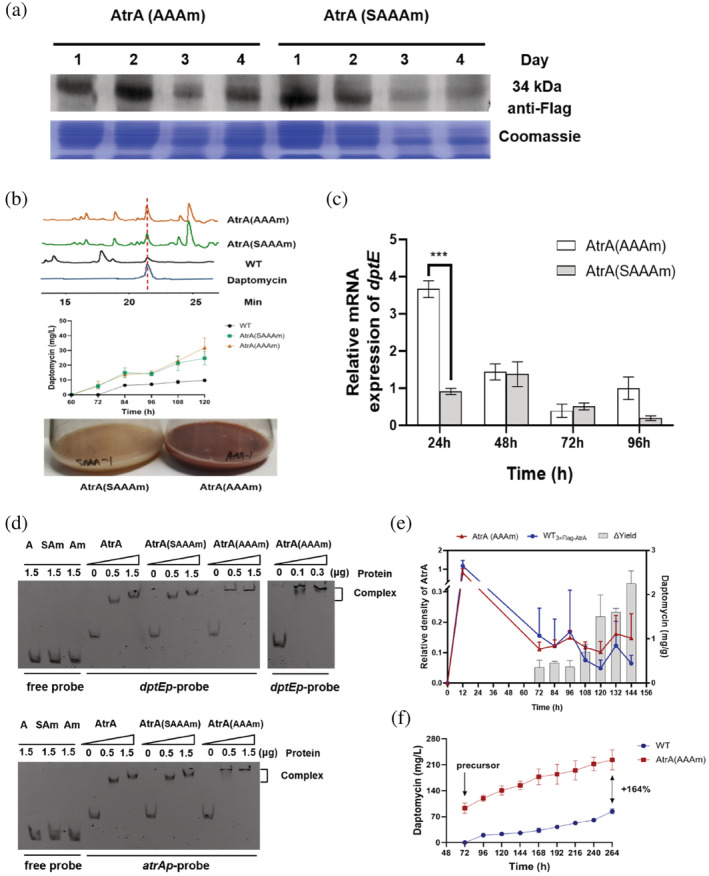
AAA motifs mutation of AtrA is more effective than SAAA motifs mutation. (a) Western blot analysis of the overexpression of mutated AtrA (AAAm/SAAAm) in vivo. Thirty‐four kilodaltons represented the molecular weight of mutated AtrA. (b) The daptomycin fermentation levels of AtrA(AAAm) and AtrA(SAAAm) strains from 60 to 120 h and phenotypic changes of AtrA(AAAm) and AtrA(SAAAm) strains at 72 h. (c) qRT‐PCR was used to detect dptE transcript levels in AtrA(AAAm) and AtrA(SAAAm) strains. The strains were fermented in YEME medium for 24, 48, 72, and 96 h to extract RNA. (d) Electrophoretic mobility shift assay (EMSA) was used to detect the binding ability of AtrA, AtrA(AAAm) and AtrA(SAAAm) to atrAp and dptEp. Proteins were produced using pET‐28a. (e) Relative protein level of AtrA and yield of daptomycin. The relative protein level was calculated by grayscale from Western blot. The daptomycin yield is given in mg per g mycelia dry weight. (f) Daptomycin fermentation levels in 15 L fermenter of WT and AtrA(AAAm) strains. The precursor decanoic acid was fed from 72 to 264 h.
3. DISCUSSION
AtrA, a pleiotropic regulator, is involved in a complex regulatory network in Streptomyces and it is also tightly regulated itself. In this study, we found that AtrA degradation involved the peptidase ClpP in S. roseosporus and the proteasome might play a role in regulating AtrA turnover. The subunits ClpP1 and ClpP2 together constitute ClpP, which is in the same operon as ClpX. ClpX was found to be important for recognition of AtrA. Due to the interchangeable cleavage sites, it is unfeasible to block the degradation by mutating the cleavage sites. Through truncation mutation and incubation assays in vitro, we identified essential AAA motifs for the degradation process. We constructed an AtrA (AAAm) strain to avoid degradation and strengthen the regulatory pathway robustness in vivo. Under the continuous positive regulation of AtrA (AAAm), S. roseosporus entered the secondary metabolism phase earlier and produced more daptomycin. This was particularly evident after cultivation in the 15 L bioreactor.
Furthermore, we found that AAA motifs were present in a large number of proteins. Bioinformatics analysis showed that ArpA, which is in the same regulatory pathway as AtrA, was also likely to be recognized and degraded by ClpXP. Although the AAA motif has a great similarity with the previously discovered recognition sites, there are also noticeable differences in the positions of these motifs. Intriguingly, the first cleavage site of AtrA was very close to the AAA motif. The degradation of AtrA is only a small part of a complex regulatory network and our results provide a blueprint to manipulate this degradation regulation in order to enhance industrial production of valuable natural products.
It is worth noting that the knockout of the proteasome and the ClpP protease had similar effects, suggesting that there is a underlying mutual mechanism of regulation. The degradation system is relatively simple in Streptomyces. And with the development of omics in recent years, taking advantage of multi‐omics to mine protease‐interacting proteins is helpful to construct a complex regulatory network. Based on previous studies on proteases (Bilyk et al., 2020; Busche et al., 2018; Compton et al., 2015; Demir et al., 2019), it can be concluded that AAA+ proteases play an indispensable regulatory role in Streptomyces, including the differentiation process and secondary metabolism. Efforts toward increasing the antibiotic production by knocking out proteases are almost deemed impossible due to the negative effect on the growth. Therefore, it is particularly significant to understand how regulators are recognized by proteases. Particularly, some common recognition sites of the degradation substrates of Lon and FtsH need to be elucidated.
AtrA is highly conserved in Streptomyces, but most studies focused on bacterial differentiation and secondary metabolic regulation (Hirano et al., 2008; Li et al., 2015; Liu et al., 2019; Uguru et al., 2005; Wang et al., 2015). The Western blot results showed that the protein level of endogenous AtrA was higher during primary metabolism, and gradually decreased after shifting to secondary metabolism in S. roseosporus. This is in line with a study that shows that AtrA is involved in primary metabolism regulation in S. avermitilis (Liu et al., 2019). However, the role of AtrA in primary metabolism, and how AtrA participates in the primary metabolic network remains to be revealed.
In summary, we first identified the degradation mechanism of AtrA, an important regulator in S. roseosporus. We further improved the production of daptomycin by circumventing the degradation of this key regulator, which provides an exemplary approach for the subsequent research on the degradation mechanism of other regulators. It also serves for industrial production, proposing a more precise strategy for high quality and high‐yield antibiotics production.
4. MATERIALS AND METHODS
4.1. Bacterial strains, plasmids, and culture conditions
All strains and plasmids used or constructed in this work are listed in Tables S4 and S5.
S. roseosporus L30 (Mao et al., 2017) (China Center for Type Culture Collection [CCTCC] no. M2010136) was used as the wild‐type strain and was grown at 30°C. R5 solid medium was used for sporulation, while MS solid medium was used for conjugation. Liquid tryptic soy broth (TSB) with 5% PEG6000 was used as media for S. roseosporus mycelium growth, and YEME (0.3% malt extract, 0.3% yeast extract, 0.5% tryptone, 4% glucose) was used for daptomycin production. To promote the biosynthesis of daptomycin, starting from 72 h, every 12 h, 0.1% decanoic acid was supplemented to the fermentation broth (YEME). In fed‐batch fermentation, the primary seed medium contained 3% tryptic soy broth (TSB) and 3% maltodextrin (MD), abbreviated as TSB‐MD. The secondary seed medium contained 6% maltodextrin, 2.5% soybean powder, 1.5% glucose, 0.08% (NH4)2Fe(SO4)2·6H2O, 0.1% Yeast extract, 0.5% calcium carbonate, and 0.2% molasses. The fermentation medium contained 7.2% maltodextrin, 1.2% yeast powder, 1.0% glucose, 0.08% (NH4)2Fe(SO4)2·6H2O, 0.72% molasses, and 0.1% defoamer GPE. Starting from 72 h, every 24 h, 0.1% decanoic acid was added to the fermentation medium. All E. coli strains were cultured in LB medium at 37°C.
All the DNA fragments were amplified with the corresponding primers (F + R) and cloned into corresponding plasmids by the ClonExpress II One Step Cloning Kit (Vazyme). All the fragments were amplified with KOD‐Plus‐Neo (Toyobo) using S. roseosporus L30 genomic DNA as the template. E. coli DH5α (Novagen) was the host for plasmid construction, and ET12567/pUZ8002 was used for conjugation to introduce DNA from E. coli to S. roseosporus. The plasmid pKC1139 was used to knock out genes and in situ replacement with corresponding homologous arms. The plasmid pSN7 was used to achieve the overexpression of genes.
4.2. Protein expression and purification
E. coli BL21 (DE3) cells containing plasmid pET28a or pET32a with atrA or mutated atrA were grown at 37°C to OD600 = 0.6, and then induced with 0.1 mM IPTG at 16 °C overnight. The resulting His‐tagged AtrA or mutated AtrA were purified by using Ni‐NTA resin according to the manufacturer's instruction (Novagen). The purified fusion protein was assessed by SDS‐PAGE and the concentration was determined by using the Bradford kit (Sangon Biotech).
4.3. Pull‐down assays in vivo and in vitro
3×Flag‐atrA strain was grown in YEME and 50 mL were collected after 24 h. Cells were resuspended in phosphate buffer (pH = 7.4) (PBS) and sonicated. After centrifugation at 12,000 rpm, the supernatant was transferred and incubated with FLAG affinity gel (Yeasen) at 4 °C. The FLAG affinity gel was washed three times with 1 mL binding buffer (PBS contains 10% glycerol). After centrifugation at 2000 rpm, 500 μL PBS was added, and the interaction proteins were eluted after denaturation. WT strain was used as control. Protein identification and analysis was provided by Jingjie PTM BioLab (Hangzhou, China).
Proteins were expressed in BL21 with the plasmids pET‐28a‐3×Flag‐atrA and pET28a‐clpX, respectively. The purified proteins were mixed and incubated at 30°C for 3 h. The reaction system consisted of 4 μg AtrA, 4 μg ClpX and 3 mM ATP. The elution method is the same as for the pull‐down assay after FLAG affinity gel purification at 4°C. Samples were analyzed by western blot using anti‐His mouse monoclonal antibody.
4.4. Post‐translation modification analysis of AtrA
AtrA was expressed in BL21 with the plasmid pET‐32a‐atrA and incubated with cell lysate of Δprc at 30°C for 1 h. The protein processing and pupylation analysis were provided by Jingjie PTM BioLab (Hangzhou, China).
4.5. HPLC detection of daptomycin
One milliliter of fermentation broth was collected daily, mixed with equal volume of methanol (fermentation medium collected from bioreactor was mixed with seven times the volume of methanol), and then centrifuged to save the supernatant for HPLC (1260 Infnity, Agilent Technologies) on a reverse phase column (Zorbax 300SB‐C18, 5 μm, 4.6 mm × 250 mm, Agilent Technologies) with a fow rate of 1 mL/min. Solution A (H2O containing 0.1% formic acid) and solution B (pure acetonitrile) were used to separate the daptomycin with UV detection set at 215 nm. The method set for HPLC was: 0 min, A:B = 90:10%; 5 min, A:B = 65:35%; 55 min, A:B = 45:55%; 60 min, A:B = 5:95%; 63 min, A:B = 90:10%; and 65 min, A:B = 90:10%. Pure daptomycin was used as a standard.
4.6. Western blot and coomassie staining
Strains were cultured in TSB seed medium, transferred to YEME medium at OD600nm = 0.4. Mycelia were collected and washed three times with PBS (pH 7.4). The PBS was used as the lysis buffer with sonication, the supernatant was collected after centrifugation, and the protein concentration was measured using Bradford kit and microplate reader. Each protein gel lane was loaded with 20 μg protein. After electrophoresis at 100 V for 1.5 h, the protein gel was put in coomassie brilliant blue staining solution for 1 h and then incubated with decolorizing solution (60% deionized water, 30% methanol, and 10% glacial acetic acid) to observe the protein bands in lanes. The protein gel after electrophoresis was transferred to the nitrocellulose membrane for blotting. Then NC membrane was blocked in 5% skimmed milk at room temperature for 1 h TBST (150 mM NaCl, 250 mM Tris–HCl pH = 7.4, 0.5% Tween‐20) was used for washing, and anti‐DYKDDDDK (or anti‐His tag) (1:3000) was used as a primary antibody. Goat anti‐mouse HRP conjugated antibody (1:4000) was used as a secondary antibody. Bands were observed by using chemiluminescence developer.
4.7. Reaction of proteins and cell lysates in vitro
Two micrograms AtrA (or mutated AtrA) was reacted with 2 μg cell lysate at 30°C for 0, 5, 15, and 30 min, respectively. After the reaction, the samples were denatured by heating, separated by SDS‐PAGE, and stained with coomassie blue.
4.8. Electrophoretic mobility shift assay
5′‐FAM‐labeled dptEp and atrAp were amplified from pTA2‐dptEp and pTA2‐atrAp, respectively. The empty probe served as a control. After 200 ng probe and different amounts of proteins (AtrA, AtrA(SAAAm), AtrA(AAAm)) (10 μL system) were incubated at 30°C for 30 min, the reaction mixture was analyzed on a 5% natural polyacrylamide gel with 0.5 × TBE (0.55% H3BO3, 1.08% Tris, 10 mM EDTA pH = 8.0) as the running buffer at 100 V for 1 h. Finally, the results were observed with an imager.
4.9. Construction of bacterial two‐hybrid library
Genes of proteases and the proteasome such as lon, ftsH, prcA, prcB, clpP1, clpP2, and clpX were recombined into pKT25. And atrA was recombined into pUT18. Double plasmids (pKT25‐gene+ pUT18‐gene) were mixed together and transformed into E. coli BTH101, which was grown in LB media with 50 μg/mL kanamycin, 100 μg/mL ampicillin, 15 mM IPTG, and 20 μg/mL X‐gal overnight at 30°C. Double empty plasmids were transformed into E. coli BTH101 and used as negative control.
4.10. RNA preparation and qRT‐PCR
RNA was prepared from mycelia with the RN43‐EASYspin Plus Kit (Aidlab Biotech Co., Ltd. China) according to the manufacturer's protocol. Genomic DNA was removed with RNase‐free DNase I (TaKaRa), and cDNA was prepared with MMLV (TakaRa) as described by the supplier. qRT‐PCR was performed with SYBR Premix Ex Taq II (TaKaRa) for genes hrdB, dptE, clpP1, clpP2, and atrA with primers listed in Table S5. The relative fold changes of gene expression were calculated based on the formula of 2–ΔΔCt .
4.11. Dry weight determination
The strain was cultured in TSB seed medium, transferred to YEME medium at OD600 nm = 0.4. Mycelia were collected at different time points and placed in a metal bath at 60°C for 72 h until the water was completely evaporated. The dry weight was measured with an electronic balance (dry weight = sample tube‐empty tube; n = 3).
AUTHOR CONTRIBUTIONS
Wei‐Feng Xu: Conceptualization (equal); investigation (equal); methodology (equal); visualization (equal); writing – original draft (equal). Chen‐Fan Sun: Formal analysis (equal); investigation (equal); methodology (equal); validation (equal). Wen‐Li Gao: Investigation (equal); methodology (equal); resources (equal). Daniel H. Scharf: Visualization (equal); writing – review and editing (equal). Chen‐Yang Zhu: Investigation (equal); methodology (equal). Qing‐Ting Bu: Writing – original draft (equal); writing – review and editing (equal). Qing‐Wei Zhao: Funding acquisition (equal); resources (equal). Yong‐Quan Li: Conceptualization (equal); funding acquisition (equal); resources (equal); supervision (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Data S1: Supporting Information
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support by National Natural Science Foundation of China (grant number 31730002) and the National Key R&D Program of China (grant number 2019YFA09005400).
Xu W‐F, Sun C‐F, Gao W‐L, Scharf DH, Zhu C‐Y, Bu Q‐T, et al. Degradation mechanism of AtrA mediated by ClpXP and its application in daptomycin production in Streptomyces roseosporus . Protein Science. 2023;32(4):e4617. 10.1002/pro.4617
Review Editor: John Kuriyan
Funding information National Natural Science Foundation of China, Grant/Award Number: 31730002; The National Key R&D Program of China, Grant/Award Number: 2019YFA09005400
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article. The proteomics data have been deposited in the PRIDE database (http://www.ebi.ac.uk/pride) and are available via ProteomeXchange with identifier PXD037377. Username: reviewer pxd037377@ebi.ac.uk. Password: REUhF2dQ.
REFERENCES
- Bilyk B, Kim S, Fazal A, Baker TA, Seipke RF. Regulation of antimycin biosynthesis is controlled by the ClpXP protease. mSphere. 2020;5(2):e00144–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubakri H, Seghezzi N, Duchateau M, Gominet M, Kofroňová O, Benada O, et al. The absence of pupylation (prokaryotic ubiquitin‐like protein modification) affects morphological and physiological differentiation in Streptomyces coelicolor . J Bacteriol. 2015;197(21):3388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche T, Tsolis KC, Koepff J, Rebets Y, Rückert C, Hamed MB, et al. Multi‐omics and targeted approaches to determine the role of cellular proteases in Streptomyces protein secretion. Front Microbiol. 2018;9:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton CL, Fernandopulle MS, Nagari RT, Sello JK. Genetic and proteomic analyses of pupylation in Streptomyces coelicolor . J Bacteriol. 2015;197(17):2747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Crécy‐Lagard V, Servant‐Moisson P, Viala J, Grandvalet C, Mazodier P. Alteration of the synthesis of the Clp ATP‐dependent protease affects morphological and physiological differentiation in Streptomyces . Mol Microbiol. 1999;32(3):505–17. [DOI] [PubMed] [Google Scholar]
- Demir Z, Bayraktar A, Tunca S. One extra copy of lon gene causes a dramatic increase in actinorhodin production by Streptomyces coelicolor A3(2). Curr Microbiol. 2019;76(9):1045–54. [DOI] [PubMed] [Google Scholar]
- Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, et al. Structures of the ATP‐fueled ClpXP proteolytic machine bound to protein substrate. elife. 2020;9:e52774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX‐recognition signals. Mol Cell. 2003;11(3):671–83. [DOI] [PubMed] [Google Scholar]
- Gominet M, Seghezzi N, Mazodier P. Acyl depsipeptide (ADEP) resistance in Streptomyces . Microbiology (Reading). 2011;157(8):2226–34. [DOI] [PubMed] [Google Scholar]
- Guyet A, Benaroudj N, Proux C, Gominet M, Coppée JY, Mazodier P. Identified members of the Streptomyces lividans AdpA regulon involved in differentiation and secondary metabolism. BMC Microbiol. 2014;14:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyet A, Gominet M, Benaroudj N, Mazodier P. Regulation of the clpP1clpP2 operon by the pleiotropic regulator AdpA in Streptomyces lividans . Arch Microbiol. 2013;195(12):831–41. [DOI] [PubMed] [Google Scholar]
- Hirano S, Tanaka K, Ohnishi Y, Horinouchi S. Conditionally positive effect of the TetR‐family transcriptional regulator AtrA on streptomycin production by Streptomyces griseus . Microbiology (Reading). 2008;154(3):905–14. [DOI] [PubMed] [Google Scholar]
- Kahne SC, Darwin KH. Structural determinants of regulated proteolysis in pathogenic bacteria by ClpP and the proteasome. Curr Opin Struct Biol. 2021;67:120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA. A specificity‐enhancing factor for the ClpXP degradation machine. Science. 2000;289(5488):2354–6. [DOI] [PubMed] [Google Scholar]
- Li X, Yu T, He Q, McDowall KJ, Jiang B, Jiang Z, et al. Binding of a biosynthetic intermediate to AtrA modulates the production of lidamycin by Streptomyces globisporus . Mol Microbiol. 2015;96(6):1257–71. [DOI] [PubMed] [Google Scholar]
- Liu L, Cheng Y, Lyu M, Zhao X, Wen Y, Li J, et al. AveI, an AtrA homolog of Streptomyces avermitilis, controls avermectin and oligomycin production, melanogenesis, and morphological differentiation. Appl Microbiol Biotechnol. 2019;103(20):8459–72. [DOI] [PubMed] [Google Scholar]
- Lyu ZY, Bu QT, Fang JL, Zhu CY, Xu WF, Ma L, et al. Improving the yield and quality of daptomycin in Streptomyces roseosporus by multilevel metabolic engineering. Front Microbiol. 2022;13:872397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao XM, Luo S, Li YQ. Negative regulation of daptomycin production by DepR2, an ArsR‐family transcriptional factor. J Ind Microbiol Biotechnol. 2017;44(12):1653–8. [DOI] [PubMed] [Google Scholar]
- Mao XM, Luo S, Zhou RC, Wang F, Yu P, Sun N, et al. Transcriptional regulation of the daptomycin gene cluster in Streptomyces roseosporus by an autoregulator. AtrA J Biol Chem. 2015;290(12):7992–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao XM, Sun N, Wang F, Luo S, Zhou Z, Feng WH, et al. Dual positive feedback regulation of protein degradation of an extra‐cytoplasmic function sigma factor for cell differentiation in Streptomyces coelicolor . J Biol Chem. 2013;288(43):31217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao V, Coëffet‐LeGal MF, Brian P, Brost R, Penn J, Whiting A, et al. Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology (Reading). 2005;151(5):1507–23. [DOI] [PubMed] [Google Scholar]
- Nagy I, Tamura T, Vanderleyden J, Baumeister W, De Mot R. The 20S proteasome of Streptomyces coelicolor . J Bacteriol. 1998;180(20):5448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA+: a class of chaperone‐like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9(1):27–43. [PubMed] [Google Scholar]
- Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin‐like protein involved in the proteasome pathway of mycobacterium tuberculosis . Science. 2008;322(5904):1104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber‐Ban E. Bacterial ubiquitin‐like modifier pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16(6):647–51. [DOI] [PubMed] [Google Scholar]
- Sun CF, Xu WF, Zhao QW, Luo S, Chen XA, Li YQ, et al. Crotonylation of key metabolic enzymes regulates carbon catabolite repression in Streptomyces roseosporus . Commun Biol. 2020;3(1):192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguru GC, Stephens KE, Stead JA, Towle JE, Baumberg S, McDowall KJ. Transcriptional activation of the pathway‐specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor . Mol Microbiol. 2005;58(1):131–50. [DOI] [PubMed] [Google Scholar]
- Wang W, Tian J, Li L, Ge M, Zhu H, Zheng G, et al. Identification of two novel regulatory genes involved in pristinamycin biosynthesis and elucidation of the mechanism for AtrA‐p‐mediated regulation in Streptomyces pristinaespiralis . Appl Microbiol Biotechnol. 2015;99(17):7151–64. [DOI] [PubMed] [Google Scholar]
- Xu WF, Fang JL, Bu QT, Lyu ZY, Zhu CY, Sun CF, et al. A novel strategy of gene screen based on multi‐omics in Streptomyces roseosporus . Appl Microbiol Biotechnol. 2022;106(8):3103–12. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Sun CF, Fu Y, Chen XA, Li YQ, Mao XM. Dual regulation between the two‐component system PhoRP and AdpA regulates antibiotic production in Streptomyces . J Ind Microbiol Biotechnol. 2019;46(5):725–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article. The proteomics data have been deposited in the PRIDE database (http://www.ebi.ac.uk/pride) and are available via ProteomeXchange with identifier PXD037377. Username: reviewer pxd037377@ebi.ac.uk. Password: REUhF2dQ.