

Abstract
G protein-coupled receptors (GPCRs), the largest family of signaling receptors, as well as important drug targets, are known to activate extracellular-signal-regulated kinase (ERK), a master regulator of cell proliferation and survival1. However, the precise mechanisms underlying GPCR-mediated ERK activation are not clearly understood2–4. Here we investigated how spatially organized β2-adrenergic receptor (β2AR) signaling controls ERK. Using subcellularly targeted ERK activity biosensors5, we show that β2AR signaling induces ERK activity at endosomes, but not at the plasma membrane. This pool of ERK activity depends on active, endosome-localized Gαs and requires ligand-stimulated β2AR endocytosis. We further identify an endosomally localized non-canonical signaling axis consisting of Gαs, Raf and mitogen-activated protein kinase kinase (MEK), resulting in endosomal ERK activity that propagates into the nucleus. Selective inhibition of endosomal β2AR and Gαs signaling blunted nuclear ERK activity, c-Myc gene expression and cell proliferation. These results uncover a non-canonical mechanism for the spatial regulation of ERK via GPCR signaling and identify a functionally important endosomal signaling axis.
Ligand-stimulated G protein-coupled receptors (GPCRs) activate heterotrimeric G proteins to initiate signaling, thereby regulating a variety of pivotal physiological processes. Extracellular-signal-regulated kinase (ERK), a master regulator of cell proliferation and survival, is among the most important downstream components of GPCR signaling. However, the molecular mechanism by which GPCRs activate ERK signaling remains poorly understood and is a topic of intensive investigation2–4,6. Critical to a better mechanistic understanding of GPCR-mediated ERK signaling is the ability to add missing spatial information, as both ERK and GPCR signaling are spatially compartmentalized7,8. In particular, emerging evidence has suggested that GPCRs signal from not only the plasma membrane but also early endosomes following clathrin- and β-arrestin (β-arr)-mediated endocytosis, thus allowing for spatiotemporal control of downstream signaling9–11. However, how GPCR-mediated ERK signaling is spatially regulated and how these two spatiotemporally segregated pools of GPCRs are mechanistically coupled to ERK are not fully understood. Here, we show non-canonical regulation of ERK at endosomes by localized β-adrenergic receptors and Gαs. Following ligand-stimulated receptor endocytosis, endosome-localized active Gαs functionally recruits ERK signaling components such as Raf and MEK to stimulate endosomal and subsequently nuclear ERK activity, thereby controlling gene expression and cell proliferation.
GPCR-stimulated ERK activity is spatially compartmentalized
Activation of GPCRs, such as β2-adrenergic receptors (β2ARs), is known to stimulate ERK activity12,13, but the molecular mechanisms are not clearly understood2–4,6,14–16. Although it has been suggested that an ERK-activating complex is formed at the plasma membrane and then internalized to endosomes, thereby impacting the cytosolic and nuclear distribution of ERK activity17,18, spatial regulation of GPCR-mediated ERK activity has not been systematically examined. Given the established ability of genetically encoded kinase activity reporters to selectively detect local kinase activities on membrane compartments5,19–23, we utilized an enhanced FRET-based ERK Activity Reporter (EKAR4) targeted to different subcellular locations to examine GPCR-controlled subcellular ERK activities. EKAR4 provides a sensitive readout of ERK activity with kinetics consistent with other ERK substrates24,25. In addition to plasma membrane-, cytosol- and nuclear-targeted EKAR45, we generated endosome-localized EKAR4 (endoEKAR4) by appending tandem FYVE domains26 to the C-terminus of EKAR4. Subcellularly targeted EKAR4 variants showed proper localization to the plasma membrane, cytosol, nucleus and endosomes and were sensitive to EGF stimulation in HEK293T cells (Extended Data Fig.1).
We then co-expressed these EKAR4 variants together with β2AR fused to a SNAP tag (SNAP-β2AR) in HEK293T cells. Live-cell labeling with BG-JF646 allowed monitoring of β2AR expression and trafficking (Extended Data Fig.2a and Supplementary Movie1). As shown in Fig.1a, epinephrine stimulation yielded an 11.8±0.75% (mean±SEM) increase in the Y/C emission ratio in the cytosol of cytoEKAR4-expressing cells, showing β-adrenergic-stimulated ERK activity in the cytosol. Although previous studies implied GPCR-stimulated ERK signaling at the plasma membrane17,18, we showed that epinephrine failed to generate any significant responses in pmEKAR4-expressing cells compared to mock treatment (P=0.2283, Fig.1b), despite the fact that epidermal growth factor (EGF) elicited a strong response from pmEKAR4 (Extended Data Fig.2b). Epinephrine stimulation robustly elevated plasma membrane cAMP production and PKA activity (Extended Data Fig.2c), potentially suppressing local ERK activity via PKA-ERK crosstalk27. However, co-expression of a PKA inhibitor failed to enhance pmEKAR4 responses (Extended Data Fig.2d and e). Furthermore, cells expressing a truncated β2AR mutant (β2AR-341T) that localizes to the plasma membrane without internalization28 showed no significant increase in epinephrine-induced ERK phosphorylation compared with control cells, further indicating a lack of plasma membrane ERK activity (Extended Data Fig.2f and g and Supplementary Fig.1). In contrast, epinephrine increased ERK activity at endosomes visualized with endoEKAR4, showing an 11.2±0.70% increase in the Y/C emission ratio (Fig.1c). Immunostaining with an anti-phospho-ERK antibody showed that epinephrine stimulation increased colocalization between the phospho-ERK signal and an endosomal marker (Extended Data Fig.2h and i), further supporting this finding. Taken together, these data suggest that GPCR-stimulated ERK activity may in fact originate at endosomes, and not at the plasma membrane.
Fig. 1. GPCR-stimulated ERK activity is spatially compartmentalized.
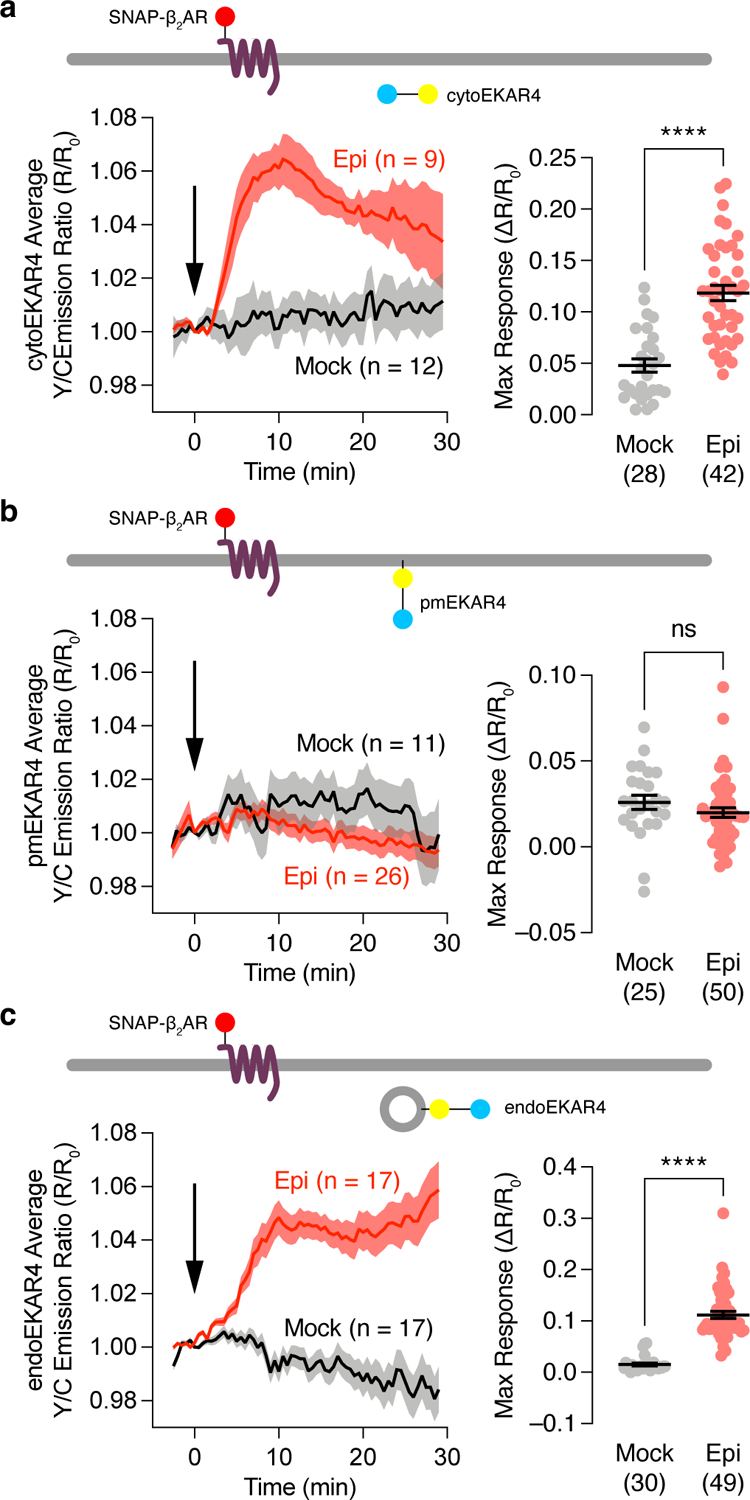
Epinephrine-stimulated responses from EKAR4 targeted to the cytosol, plasma membrane or endosomes. (a-c) Left: Representative yellow/cyan (Y/C) emission ratio time courses from HEK293T cells co-transfected with cytoEKAR4 (a), pmEKAR4 (b), or endoEKAR4 (c) plus SNAP-b2AR and treated with mock (HBSS) or 10 μM epinephrine (Epi) stimulation. Right: Maximum responses from cytoEKAR4 (a) pmEKAR4 (b) or endoEKAR4 (c). ns, not significant, ****P < 0.0001; unpaired two-tailed Student t-test without (a, b) or with (c) Welch’s correction.
GPCR-mediated endosomal ERK activity is regulated by GPCR endocytosis and endosomal Gαs activity
Endosomes have been suggested to serve as a GPCR-signaling platform where internalized receptors, G proteins and adenylyl cyclases orchestrate specific cAMP signaling that promotes PKA nuclear translocation29 and regulates transcription10. Moreover, agonist-stimulated GPCR endocytosis has been linked to ERK activation14,17,18, although its exact role is not fully understood15,30–32. We hypothesized that β-adrenergic receptor-stimulated ERK signaling originates specifically at endosomes through this pool of internalized, agonist-bound receptors. We first tested the role of β-arrestins and agonist-promoted receptor endocytosis. Preincubating cells with barbadin33, a selective blocker of adaptor protein 2 (AP2)-β-arr interactions and inhibitor of clathrin-mediated GPCR endocytosis (Fig.2a), or Dyngo4a, which blocks clathrin-mediated endocytosis (Extended Data Fig.3a and b), diminished epinephrine-stimulated endosomal ERK activity (−: 11.2±0.70%; +Barbadin: 3.40±0.58%; P=1.58×10−12, +Dyngo4a: 4.32±0.91%; P=3.86×10−6). Furthermore, small interfering RNA (siRNA)-mediated knockdown of β-arr1/2 prevented receptor internalization (Extended Data Fig.3) and abolished epinephrine-stimulated endosomal ERK activity, which was rescued by heterologous β-arr2 expression (siCon: 4.16±0.61%; siβ-arr1/2: 1.52±0.26%; siβ-arr1/2+β-arr2: 13.8±0.17%; P=1.81×10−5, Fig.2b and Extended Fig.3c), consistent with what we observed in β-arr1/2 double-KO cells2 (Extended Data Fig.3d). These data suggest that agonist-promoted and arrestin-mediated receptor internalization is required for β-adrenergic-stimulated endosomal ERK activity.
Fig. 2. GPCR-mediated endosomal ERK activity is regulated by GPCR endocytosis and endosomal Gαs activity.
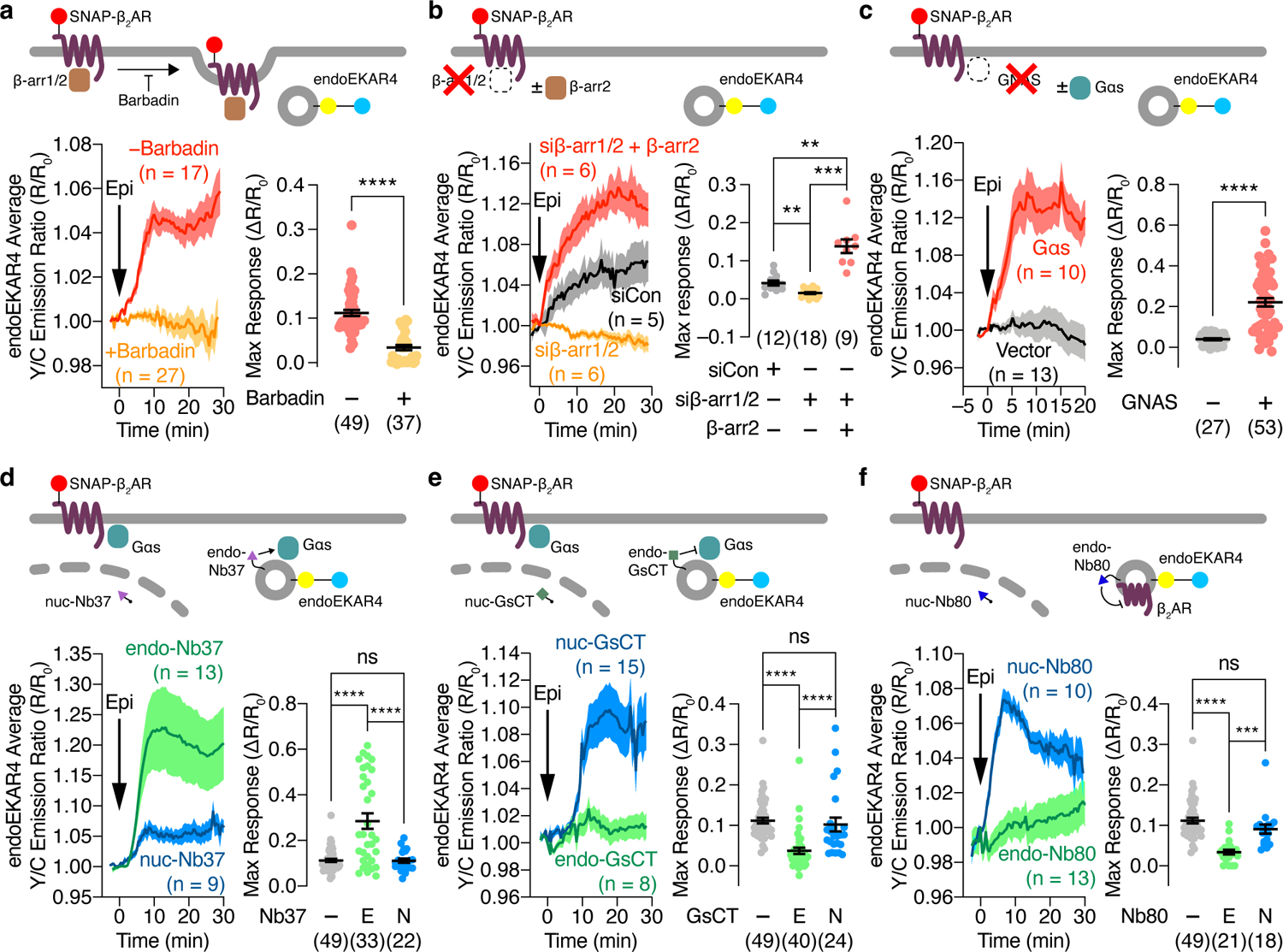
(a-f) Representative time courses showing the 10 μM epinephrine (Epi)-stimulated yellow/cyan (Y/C) emission ratio changes in cells co-expressing endoEKAR4 plus SNAP-β2AR (left) and maximum endoEKAR4 responses (right). (a) HEK293T cells additionally preincubated with (+) or without (−) 100 μM barbadin. (b) HEK293T cells additionally transfected with control siRNA or with siRNA targeting β-arr1/2 plus vector or β-arr2. (c) GNAS KO HEK293A cells additionally transfected with vector or Gαs. (d-f) HEK293T cells additionally transfected with (d) endo- or nuc-NB37, (e) endo- or nuc-GsCT, or (f) endo- or nuc-Nb80. ns, not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001; Mann-Whitney U-test (a), Welch ANOVA followed by Dunnett’s T3 multiple comparison test (b), unpaired two-tailed Student t-test (c), or ordinary one-way ANOVA (d-f). Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM. Negative control curves and data points shown in a and d-f are reproduced from Fig. 1c.
Given the importance of Gαs in β2AR signaling2,3, we then tested the involvement of Gαs in regulating endosomal ERK activity. In GNAS-KO cells2 expressing SNAP-β2AR, epinephrine induced minimal changes in the endoEKAR4 Y/C emission ratio (3.97±0.52%), whereas overexpressing Gαs rescued the β2AR-stimulated endoEKAR4 response (22.1±2.1%; P=2.85×10−8, Fig.2c). Given the recently identified role of Gαi in regulating ERK6, we expressed pertussis toxin S1 subunit (PTX), a Gαi inhibitor (Extended Data Fig.3e), and monitored epinephrine-stimulated endoEKAR4 responses. PTX expression did not alter the response of endoEKAR4 versus cells transfected with empty vector (Extended Data Fig.3f). These data suggest that β2AR-stimulated endosomal-ERK activity specifically requires active Gαs.
Next, we tested whether β2AR-stimulated endosomal ERK activity requires endosomally localized β2AR signaling components. We found that Gαs translocated off the plasma membrane upon epinephrine stimulation and partially co-localized with endosomes, in a β-arr1/2-independent manner (Extended Data Fig.3g–i), consistent with previous studies34,35. To selectively perturb this endosomal Gαs pool, we employed a strategy for location-specific perturbation of signaling activities23,36 and sought to selectively activate endosomal Gαs using Nb37, a nanobody that stabilizes the open conformation of Gαs, promotes nucleotide exchange and thus promotes the active state37. We attached a C-terminal tandem FYVE domain to generate endosome-targeted Nb37 (endo-Nb37) (Extended Data Fig.4a), and utilized nuclear-targeted Nb37 (nuc-Nb37) as a negative control. Epinephrine stimulated higher endosomal ERK activity in cells expressing endo-Nb37 (28.5±3.4%) than in cells expressing nuc-Nb37 (11.1±0.93%; P=7.11×10−7) (Fig.2d), which showed comparable responses to cells lacking Nb37 (11.2±0.70%, Fig.1c, P=0.970). Cells expressing endo-Nb37 also showed significantly increased phospho-ERK immunostaining, in addition to enhanced phospho-ERK colocalization with an endosome marker, upon epinephrine stimulation (Extended Data Fig.4d–f). We next assessed the effects of endosome-specific Gαs inhibition using an endosome-targeted Gαs peptide inhibitor, GsCT38 (endo-GsCT, Extended Data Fig.4b). Compared with control cells, expression of endo-GsCT diminished the epinephrine-stimulated endoEKAR4 response (3.70±0.77%, P=5.93×10−8), whereas nuc-GsCT expression had no effect (10.2±1.7%, P=0.507) (Fig.2e). Furthermore, we tested whether inhibiting endosome-localized β2AR affects endosomal ERK activity by using Nb80, a nanobody that binds and stabilizes the β2AR active conformation and, more importantly, blocks Gas binding and coupling37,39. We reasoned that targeting Nb80 to endosomes would leave arrestin-mediated endocytosis unaffected and only prevent Gαs coupling subsequent to internalization, whereas in the absence of targeting, the nanobody would prevent both arrestin and Gαs from binding to β2AR. We therefore generated endosome-targeted Nb80 (endo-Nb80) (Extended Data Fig.4c). As shown in Fig.2f, overexpressing endo-Nb80 abolished epinephrine-stimulated endoEKAR4 responses (3.39±0.59%, P=4.68×10−9), whereas nuclear-targeted Nb80 had no effect (9.09±1.1%, P=0.0883). Taken together, these data suggest that epinephrine-stimulated endosomal ERK activity requires active, endosomally localized β2AR and Gαs.
Gαs recruits Raf1 and MEK1 to regulate endosomal GPCR-mediated ERK activity
Canonical ERK activation involves a well-defined signaling cascade involving Raf-mediated phosphorylation and activation of the MAPKK, MEK, resulting in the phosphorylation and activation of ERK7. To identify regulators of β2AR-stimulated endosomal ERK activity, we first measured endoEKAR4 responses while perturbing MAPK signaling components via pharmacological inhibition of either Raf (SB-590885) or MEK (U0126). As shown in Fig.3a, epinephrine-stimulated endoEKAR4 responses were significantly decreased upon Raf or MEK inhibition (NT: 28.5±3.4%, MEKi: 2.30±0.27%, Rafi: 5.68±0.54%), suggesting that the Raf/MEK axis is required for β-adrenergic receptor-stimulated endosomal ERK activity.
Fig. 3. Gαs interacts with Raf1 and MEK1 to regulate endosomal GPCR-mediated ERK activity.
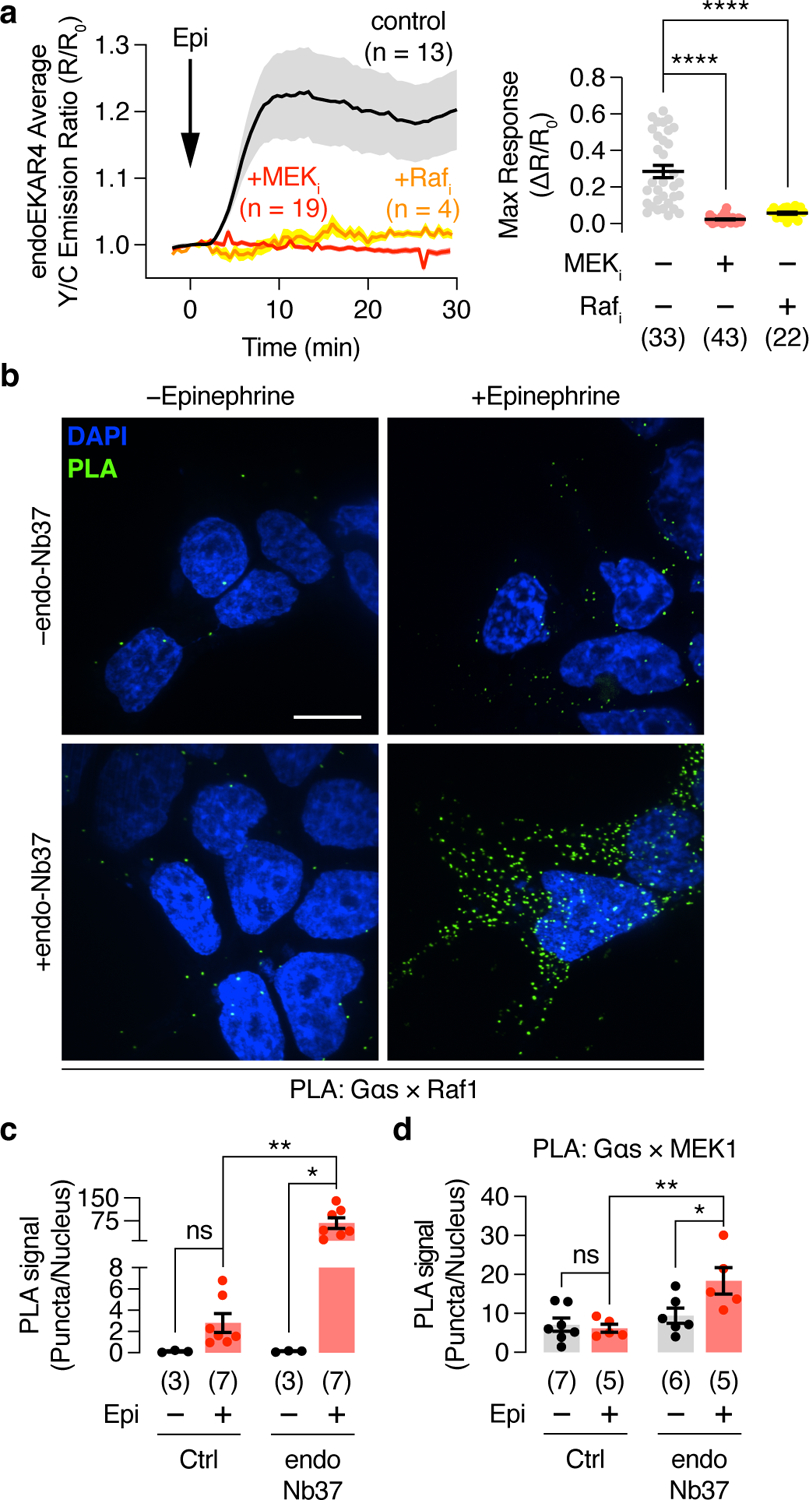
(a) Left: Representative traces showing 10 μM epinephrine (Epi)-stimulated yellow/cyan (Y/C) emission ratio changes from HEK293T cells co-expressing endoEKAR4, SNAP-β2AR, and endo-Nb37 in the presence or absence of 10 μM U0126 (+MEKi) or 10 μM SB-590885 (+Rafi). Solid lines indicate the mean, and shaded areas show SEM. Right: Maximum endoEKAR4 responses. (b) Representative Proximily Ligation Assay (PLA) images for the Gαs × Raf1 association under the indicated conditions. (c) Bar graph of PLA quantification. (d) Quantification of PLA particle density for Gαs × MEK1. Bar graphs and scatter plots depict the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001; ordinary one-way ANOVA followed by Holm-Šídák’s multiple-comparison test (a), two-way ANOVA followed by Turkey’s multiple-comparison test (c, d). Negative control curve and data points in (a) are reproduced from Fig. 2e.
Using the proximity ligation assay (PLA), we also detected the formation of complexes containing Gαs and Raf1 in epinephrine-treated cells (Fig.3b and c). On average, 2.80±0.88 PLA puncta per nucleus were observed when measuring the Gαs and Raf1 association, which is significantly (P=0.00117) above the background signal observed with Gαs antibody plus IgG control (Extended Data Fig.5). Furthermore, co-expression of endo-Nb37 produced a dramatic, 24.2±6.9-fold increase in the Gαs-Raf1 PLA signal in epinephrine-treated cells compared with those lacking endo-Nb37 (Fig.3b and c). Endo-Nb37 co-expression similarly produced a significant increase in the PLA signal detected using antibodies against Gαs and MEK1 (Fig.3d and Extended Data Fig.5). Taken together, our results strongly suggest that endosome-localized, active-Gαs mediates β2AR stimulation of ERK activity by recruiting Raf1 and MEK1 into a signaling complex.
Endosomal Gαs regulates nuclear ERK activity and function
The Raf-MEK-ERK pathway regulates a variety of cellular processes, many of which involve the accumulation of nuclear ERK activity40. Thus, we utilized nucEKAR4 to test whether β2AR-initiated ERK activity can propagate into the nucleus (Fig.4a). Epinephrine stimulation yielded a 9.52±1.02% increase in the Y/C emission ratio in the nuclei of nucEKAR4-expressing cells, indicating that β2AR stimulation leads to the accumulation of nuclear ERK activity (Fig.4b). This nuclear ERK activity is dependent on receptor endocytosis, as endocytosis inhibition and β-arr1/2 knockdown markedly decreased the epinephrine-stimulated nucEKAR4 response (Extended Data Fig.6a and b). Co-expression of endo-Nb37 to selectively activate endosomal- Gαs strongly enhanced the epinephrine-stimulated nucEKAR4 response (26.2±2.97%, Fig.4c), whereas selective inhibition of endosomal Gαs using endo-GsCT completely abolished epinephrine-stimulated nucEKAR4 response (2.65±0.53%, P=5.97×10−9, Fig.4d). Endosome-specific inhibition of β2AR coupling to Gs via endo-Nb80 overexpression also abolished the epinephrine-stimulated nucEKAR4 response (1.21±0.17%, P=3.97×10−17, Fig.4e). The epinephrine-stimulated cytoEKAR4 response was similarly blocked by barbadin pretreatment or endo-GsCT co-expression, further supporting signal propagation from endosomes to the cytosol/nucleus, whereas no response was observed with pmEKAR4 even in the presence of these inhibitors (Extended Data Fig.6c and d). Of note, HEK293T and H9c2 cells expressing endogenous β2AR displayed this same pattern of compartmentalized β2AR-stimulated ERK activity, which was also dependent on endocytosis (Extended Data Fig.7). These findings were further recapitulated in HEK293T cells overexpressing other Gs-coupled GPCRs, prostaglandin EP4 receptor (EP4), dopamine receptor D1 (D1DR) and vasopressin V2 receptor (V2R) (Extended Data Fig.8), suggesting that our model of GPCR-stimulated ERK signaling originating from endosomes in an endocytosis-dependent manner extends beyond β2AR.
Fig. 4. Endosomal Gαs regulates nuclear ERK activity and function.
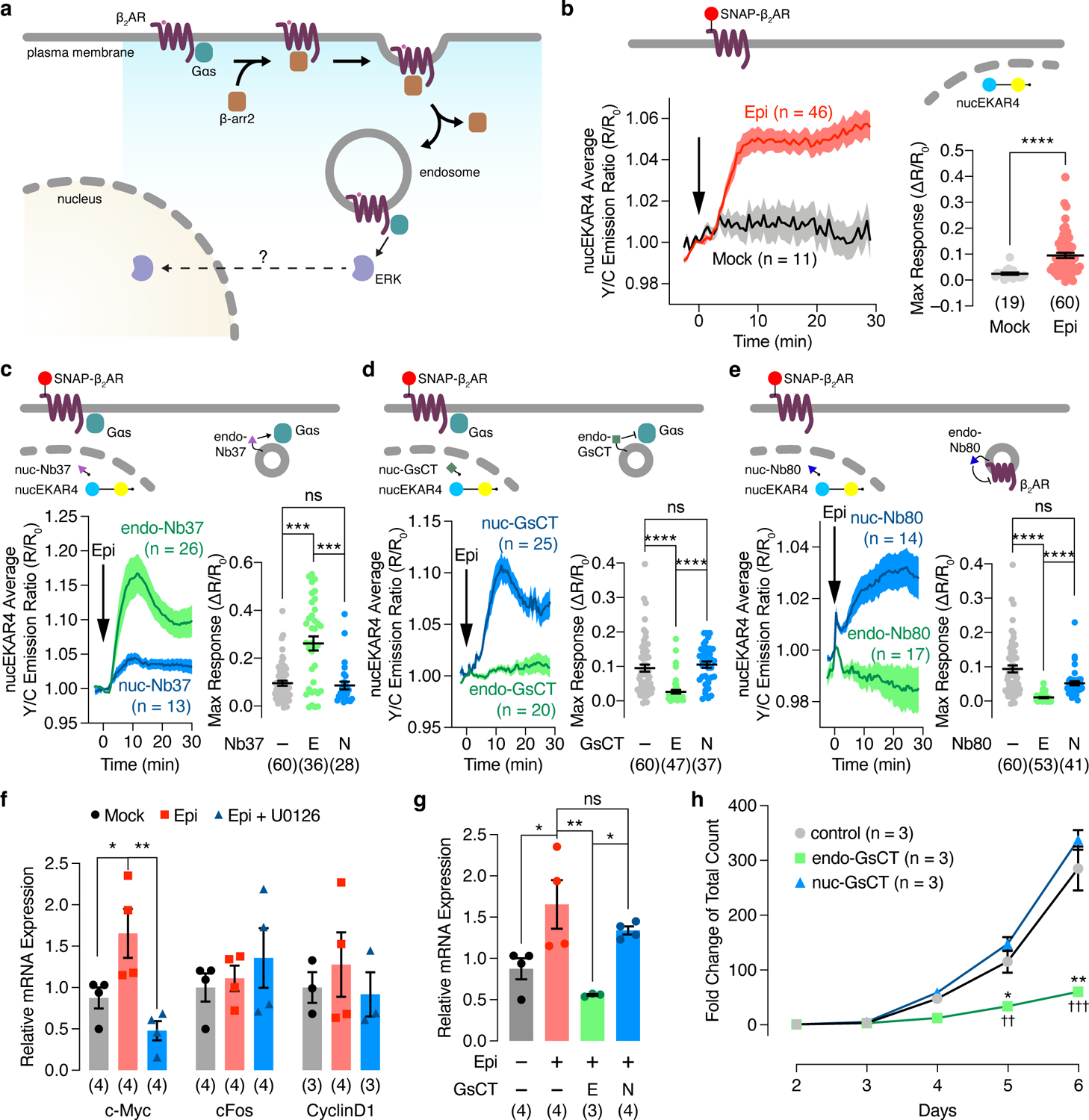
(a) Model illustrating the potential regulation of nuclear ERK activity by endosomal Gas activity. (b-e) Left: Representative yellow/cyan (Y/C) emission ratio traces from HEK293T cells co-expressing nucEKAR4 plus (b) SNAP-β2AR only, or additionally transfected with (c) endo- or nuc-Nb37, (d) endo- or nuc-GsCT, (e) endo- or nuc-Nb80, or in GNAS KO HEK293A cells co-expressing SNAP-β2AR plus endo (i) or nuc (j) EKAR4 and additionally expressing vector, Gαs-L or Gαs-S with or without 10 μM epinephrine (Epi) stimulation. Right: Maximum nucEKAR4 responses. (f, g) qPCR analysis of (f) c-Myc, c-fos and cyclin D1 mRNA in Epi-stimulated HEK293T cells with or without U0126 preincubation or (g) c-Myc mRNA in HEK293T cells with or without Epi stimulation and transfected with vector (−), endo-GsCT (E), or nuc-GsCT (N). Numbers in parenthesis represent independent experiments. (h) Growth curves of HEK293T cells stably expressing mCherry, mCherry-endo-GsCT, or mCherry-nuc-GsCT. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; Mann-Whitney U-test (b), Kruskal-Wallis test followed by Dunn’s multiple-comparison test (c-e), ordinary one-way ANOVA followed by Tukey’s multiple-comparison test (f, g). *P < 0.05, **P < 0.01 vs. control and ††P < 0.01, †††P < 0.001 vs. nuc-GsCT; ordinary one-way ANOVA followed by Tukey’s multiple-comparison test (h). Solid lines in b-e indicate the mean, and shaded areas show SEM. Horizonal lines in b-e depict the mean ± SEM. Bars in f and g and data points in h represent the mean ± SEM.
Because ERK is known to regulate gene expression7,41, we next tested the effect of endosomal Gαs-initiated nuclear ERK signaling on the transcription of ERK target genes by selectively blocking endosomal Gαs activity using endo-GsCT. In contrast to ERK-dependent increases in the expression of c-Myc, c-Fos and cyclin D1 upon EGF stimulation, epinephrine stimulation only induced c-Myc expression (Fig.4f and Extended Data Fig.9). Moreover, the epinephrine-stimulated induction of c-Myc mRNA expression was diminished by endo-GsCT expression (Fig.4g), indicating a functional role for endosomal Gαs-ERK signaling in controlling a select repertoire of ERK target genes. In agreement with endosome-initiated effects on ERK target gene transcription, endosomal Gαs-mediated ERK signaling regulates cell proliferation. Strikingly, HEK293T cells stably expressing endo-GsCT exhibited decreased cell proliferation compared with cells stably expressing nuc-GsCT, which showed comparable proliferation to mCherry-expressing HEK293T cells (Fig.4h).
We recently reported that a splicing variant of Gαs, Gαs long (Gαs-L), selectively upregulates ERK in myelodysplastic syndrome (MDS), a blood cell differentiation-impaired disease. To assess the differential effects of Gαs splice variants on β2AR-stimulated ERK activity, we expressed Gαs-L or Gαs short (Gαs-S) in SNAP-β2AR-expressing GNAS-KO HEK293A cells and monitored endo- or nucEKAR4 responses. Epinephrine stimulated dramatic increases in the endo- and nucEKAR4 Y/C emission ratios in Gαs-L-expressing cells (endoEKAR4: 19.9±0.19%, n=55, nucEKAR4: 13.7±0.16%, n=49), whereas only minimal emission ratio changes were observed in Gαs-S-expressing cells (endoEKAR4: 1.83±0.66%, n = 31, nucEKAR4: 5.54±0.68%, n=34), suggesting that only the long, but not short, splice variant of Gαs is capable of controlling endosomal- and nuclear-ERK activity (Extended Data Fig.9b and c). Taken together, our data reveal that β2AR regulation of nuclear ERK is dependent on internalized receptor and endosomal Gαs activity, in the same fashion as endosomal ERK activity, and imply that ERK signals propagate from endosomes to the nucleus. Our results thus define a non-canonical Gαs-ERK axis at endosomes that plays a spatiotemporally distinct role in regulating specific cell functions by controlling the expression of a select subset of ERK-mediated target genes.
Discussion
In the canonical view of GPCR activation, signaling cascades are initiated at the plasma membrane, such as through the activation of adenylyl cyclase, phospholipase C and the regulation of ion channels42. However, the lack of response from the PM-targeted EKAR (Figure 1b), which selectively measures ERK activity at the plasma membrane, indicates that there is essentially no ERK signaling occurring from this location. Taken together our data provide strong evidence that GPCR-mediated ERK signaling, in the context of Gs-coupled receptors, originates at the endosome. This GPCR-stimulated endosomal ERK signaling requires endocytosis and endosomally localized active receptor and Gαs. Given that arrestin is essential for ligand-mediated endocytosis, our findings are consistent with the well-documented critical contributions of both G protein and arrestin to GPCR-regulated ERK signaling. In our model, endocytosed GPCRs recruit and activate Gαs at endosomes, which in turn recruits Raf1 and MEK1 to initiate ERK signaling. Our data suggest that this model may be applicable to a variety of GPCRs, including both Class A and Class B receptors. Future studies will examine how endosomal lipid compositions, which could lead to distinct GPCR conformations43, and/or different concentrations of ERK signaling components at the plasma membrane versus the endosome membrane44 play a role in endosome-specific ERK activation machinery.
Although we cannot exclude the possibility that β-arr1/2, which was shown to interact with Raf1 among 330 different interaction partners45, plays a role in helping organize β2AR-stimulated ERK activation15,17,18,30–32, our results strongly support a role for Gαs in scaffolding β2AR-stimulated endosomal ERK activation. The PLA signal indicating the epinephrine-induced association between Gαs and Raf1 was increased by 24.2±6.9-fold in the presence of endo-Nb37, which enhances endosomal Gαs activity. In addition, selective perturbations of endosomal Gαs directly impact nuclear ERK activity, suggesting endosomal Gαs is critically involved in orchestrating β2AR-stimulated ERK activity that originates at endosomes and propagates into the nucleus.
GPCRs play a significant role in regulating transcription, and recent studies have suggested that PKA-mediated regulation of CREB-dependent gene transcription is controlled by cAMP signals originating from endosomes in a GPCR- and G protein-dependent manner, rather than from the plasma membrane10,29. This mechanism of localized signaling control makes good geographic sense, given the close placement of endosomes near the nucleus. Our current study highlights similar spatial advantages in GPCR-mediated ERK activation, where signals originate from endosomes and progress to the nucleus to regulate transcription. Interestingly, we found that the endosomal Gαs-ERK pathway selectively controls the induction of c-Myc, an important ERK target gene, whereas EGF additionally induces c-fos and cyclin D1 gene expression. A comparison of the EKAR responses reveals that epinephrine-induced ERK activity is weaker and exhibits slower kinetics compared with EGF stimulation (t1/2 for Epi: 7.98±0.59 min, n=41 cells; EGF: 3.30±0.24 min, n=31; P=5.83×10−9, Mann-Whitney U-test). Thus, endosomal GPCR-ERK signaling may utilize distinct spatiotemporal dynamics to exert specific transcriptional control.
Given the importance of both ERK and c-Myc in cancer, the functional impact of this endosomal Gαs-ERK signaling axis on c-Myc expression and cell proliferation observed in our study not only indicates the importance of this non-canonical pathway in normal physiology, but also opens a new window into the role of GPCRs in cancer. Indeed, many types of cancers have been shown to contain hotspot mutations and copy number changes in the GNAS gene46, which have also been shown to critically contribute to tumorigenesis47. Although current studies have focused on the Gαs-cAMP-PKA signaling pathway in cancers48, we recently reported that mutations in splicing factors U2AF1 and SRSF2 that are associated with MDS, selectively increase expression of Gαs-L and drive abnormal signaling through ERK but not the classical cAMP/PKA axis49. Indeed, our data suggest that Gαs-L, but not Gαs-S, is capable of organizing endosomal ERK activity, which could play a critical role in driving pathological signaling through signal propagation. Given the therapeutic potential of targeting oncogenic GPCRs46,50, the spatiotemporal activity architecture orchestrated by non-canonical endosomal GPCR-Gαs-ERK signaling promises to influence diverse GPCR biology, cancerous dysregulation and therapeutic vulnerability.
Methods
1.1. Plasmids and siRNAs
To construct endoEKAR4, the tandem FYVE domain was PCR-amplified from mKate2-P2A-APEX2-2xFYVE_hrs (gift of Rob Parton; Addgene plasmid #67663) using the forward primer 5’- GTATGAATGAATTGTACAAAGAATTCCGAATTGAATTCGAAAGTGAT and the reverse primer 5’- TGAACTATAGAATAGGGCCCTCTAGATTATGCCTTCTTGTTCAGCTG. The PCR fragments were inserted into EcoRI/XbaI-digested cytoEKAR4 using Gibson assembly.
Negative-control T/A mutants of endoEKAR4 and nucEKAR4 were generated via site-directed mutagenesis from endoEKAR4 and nucEKAR using the forward primer 5’- CCAGATGTCCCTAGAGCTCCAGTGGATAAA and the reverse primer 5’- CTTTGCTTTATCCACTGGAGATCTAGGGACATC.
To construct SNAP-tagged β2AR-T2A-Nb37/Nb80 and SNAP-tagged β2AR-T2A-GαsL/S constructs, SNAP-tagged β2AR was PCR-amplified to add a C-terminal T2A sequence using the forward primer 5’- TTAAGCTTGGTACCGAGCTCGGATCCATGAAGACGATCATCGCCCTG and the reverse primer 5’- ACGTCACCGCATGTTAGCAGACTTCCTCTGCCCTCCCCTCCGGGCCCCAGCAGTGAGTCATTTGTACT. Nb37 was PCR-amplified using the forward primer 5’- CTGCTAACATGCGGTGACGTCGAGGAGAATCCTGGCCCAACCGGTATGGCCCAGGTGCAGCTGCAG and the reverse primer 5’-AGGCTGATCAGCGGGTTTAAACCTAGGCTTCAGGTTCGTGATG. Nb80 was PCR-amplified using the forward primer 5’- CGAGGAGAATCCTGGCCCAACCGGTATGGAGTCTGGGGGAGGCTTG and the reverse primer 5’-AGGCTGATCAGCGGGTTTAAACCTAGGCTTCAGGTTCGTGATG. GαsL/S was PCR-amplified using the forward primer 5’- GACGTCGAGGAGAATCCTGGCCCAACCGGTATGGGCTGCCTCGGGAACAGT for both L and S and the reverse primer for L 5’- GCACAGTCGAGGCTGATCAGCGGGTTTAAACTTAGAGCAGCTCGTACTGACG and the reverse primer for S 5’- CACAGTCGAGGCTGATCAGCGGGTTTAAACTTATAGCAGCTCGTACTGACG. SNAP-β2AR-T2A was then combined with Nb37, Nb80 or Gαs using Gibson assembly.
To construct SNAP-β2AR-T2A-Nb37-FYVE and SNAP-β2AR-T2A-Nb37-NLS, SNAP-β2ART2A-Nb37 was linearized via AgeI and PmeI digestion. Nb37 was PCR-amplified using the forward primer 5’- CTGCTAACATGCGGTGACGTCGAGGAGAATCCTGGCCCAACCGGTATGGCCCAGGTGCAGCTGCAG and the reverse primer 5’- GAATTCTCCACCAGCACTACCAGCACTATCGGCTTCAGGTTCGTGATGGTG. The tandem FYVE domain was PCR-amplified using the forward primer 5’- GAAGCCGATAGTGCTGGTAGTGCTGGTGGAGAATTCCGAATTGAATTCGAA and the reverse primer 5’- TCGAGGCTGATCAGCGGGTTTAAACAGAATAGGGCCCTCTAGATTATGC. The Nb37 and 2xFYVE PCR fragments were combined with AgeI/PmeI-digested SNAP-β2AR-T2A using Gibson assembly. To construct SNAP-β2AR-T2A-Nb37-NLS, Nb37 was PCR-amplified using the forward primer 5’- CTGCTAACATGCGGTGACGTCGAGGAGAATCCTGGCCCAACCGGTATGGCCCAGGTGCAGCTGCAG and the reverse primer 5’- TCCACCAGCACTACCAGCACTATCGGCTTCAGGTTCGTGATGGTG, and an NLS was PCR amplified by using the forward primer 5’- AGCCGATAGTGCTGGTAGTGCTGGTGGAGAATTCCGAATTGAATTCGAACCCAAAAAGAAGAGAAAGGTG and the reverse primer 5’- CAACAGATGGCTGGCAACTAGAAGGCACAGTCGAGGCTGATCAGCGGGTTTAAACTGCGTCTTCCACCTT. The Nb37 and NLS PCR fragments were inserted into AgeI/PmeI-digested SNAP-β2AR-T2A using Gibson assembly.
To construct FYVE-mCherry-GsCT and H2A-mCherry-GsCT, 2xFYVE, H2A, mCherry and GsCT were PCR-amplified using the forward primer 5’- GACTCACTATAGGGAGACCCAAGCTTGCGGCCGCCACC and the reverse primer 5’- TCCTCGCCCTTGCTCACCATGCTCACCATGGGATCTGC for 2xFYVE; the forward primer 5’- CTATAGGGAGACCCAAGCTTGCGGCCGCCACCATGTCTGG and the reverse primer 5’- TCGCCCTTGCTCACCATGGGATCCGCTTTGCTTTTGGCT for H2A; the forward primer 5’- TGAGCATGGTGAGCAAGGGCGAGGAG and the reverse primer 5’- CGAGCAAATTCTGGGAATTCCTTGTACAGCTCGTCCATGCC for mCherry; and the forward primer 5’- ACGAGCTGTACAAGGAATTCCCAGAATTTGCTCGCTACACT and the reverse primer 5’- TGACACTATAGAATAGGGCCCTTAGAGCAGCTCGTACTGACG for GsCT. PCR products were inserted into HindIII/ApaI-digested pcDNA3 using Gibson assembly.
To construct SNAP-β2AR 341T, SNAP-b2AR was PCR-amplifed using the forward primer 5’- CTAGTCTAGAATGGGGCAACCCGGGAACGGC and the reverse primer 5’- AGCTTTGTTTAAACCTATTAGCACAGAAGCTCCTGGAAGGC. PCR products were inserted into XbaI/PmeI-digested SNAP-β2AR using T4 ligase.
siRNAs targeting ARRB1 (siARRB1 s1623) and ARRB2 (siARRB2 s1625) were purchased from Thermo Fisher. Both siRNAs target intron regions, enabling gene replacement experiments using β-arr2 cDNA. siControl was purchased from Qiagen (siScramble 105250726).
1.2. Cell culture and transfection
GNAS KO HEK293A and β-arr1/2 dKO HEK293S cells (generously provided by Asuka Inoue, Tohoku University), HEK293T and H9c2 cells were cultured in Dulbecco’s modified Eagle medium (DMEM, GIBCO) containing 1 g L−1 glucose and supplemented with 10% (v/v) fetal bovine serum (FBS, Sigma) and 1% (v/v) penicillin–streptomycin (Pen-Strep, Sigma-Aldrich). All cells were maintained in a 37°C incubator with a humidified 5% CO2 atmosphere. GNAS KO HEK293A, β-arr1/2 dKO HEK293S and HEK293T cells were plated onto sterile, poly-D-lysine coated 35-mm glass-bottom dishes for imaging or coverslips for proximity ligation assays and grown to 50%–70% confluence for transfection. GNAS KO HEK293A, β-arr1/2 dKO HEK293S and HEK293T cells were transfected using Polyjet (SignaGen) and H9c2 were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions and grown for an additional 16–24 h before imaging. Transfection conditions are described in Supplementary table 1.
1.3. Fluorescence imaging and FRET biosensor analysis
Cells were washed twice with Hank’s balanced salt solution (HBSS) and incubated at 37°C for 30 min prior to imaging. Cells were subsequently imaged in HBSS in the dark at 37°C. For SNAP-tagged protein labeling, cells were preincubated in HBSS containing 0.3 μM JF646-SNAP-tag ligand for 3 min and then washed 3 times with HBSS and incubated at 37°C for 30 min prior to imaging. Epinephrine (Epi; Sigma-Aldrich), Epidermal growth factor (EGF; Sigma-Aldrich), Barbadin (Axon medchem), U0126 (Sigma-Aldrich), Dyngo4a (abcam), PGE2 (Sigma-Aldrich), Dopamine (DA; Sigma-Aldrich), DESMO (Sigma-Aldrich), Lysophosphatidic acid (LPA; Sigma-Aldrich) and SB-590885 (Sigma-Aldrich) were added as indicated. Epifluorescence imaging was performed on a Zeiss AxioObserver Z7 microscope (Carl Zeiss) equipped with a 40x/1.3 NA objective and a Photometrics Prime 95B sCMOS camera (Photometrics), controlled by METAFLUOR 7.7 software (Molecular Devices). The following excitation/emission filter combinations were used: CFP – EX420/20, 455lp dichroic mirror, EM473/24; YFP– EX495/10, 515lp dichroic mirror, EM535/25; CYFRET– EX420/20, 455lp dichroic mirror, EM535/25; mCherry– EX572/35, 594rdc dichroic mirror, EM645/75; JF646– EX640/30, 660lp dichroic mirror, EM700/75. All filter sets were alternated using an LEP MAC6000 control module (Ludl Electronic Products Ltd). Exposure times were 50 ms (YFP channel) and 500 ms (all other channels), and images were acquired every 30 s. Cells to be analyzed were chosen based on the proper localization of biosensor fluorescence. Fluorescence intensities were background-corrected by subtracting the background fluorescence intensity of a cell-free region from the emission intensities of biosensor-expressing cells. Yellow/cyan emission ratios for EKAR4 and AKAR4 and cyan/yellow emission ratios for Epac2-camps and ICUE4 were then calculated at each time point. The time-course data were normalized by dividing the ratio at each time point by the basal value at time zero (R/R0), which was defined as the last time point immediately preceding drug treatment. Maximum ratio changes (ΔR/R0) were calculated as (Rmax−R0)/R0, where Rmax is the maximum ratio value recorded after drug treatment.
1.4. Stable cell line generation
For lentivirus packaging, HEK293T cells were co-transfected with lentiviral vector (containing sequences encoding mCherry, mCherry-endoGsCT or mCherry-nucGsCT) + psPAX2 + pMD2.G using PolyJet transfection reagent (SignaGen Laboratories, MD, USA) according to manufacturer’s instructions. The supernatants were collected after 48 h and then concentrated using a Lenti-X concentrator (Takara Bio USA, Inc.) according to the manufacturer’s instructions. For infection, HEK293T cells were seeded, and concentrated solutions containing lentiviral particles were added into the cell culture medium. After 48 h, cells were passed in fresh growth medium and were treated with puromycin (2 μg/mL) to select transduced cells. Cells were maintained in selection medium for one week, and samples were collected for proliferation assays.
1.5. Immunocytochemistry
Cells were seeded on cover glass and fixed with 4% paraformaldehyde for 20 min followed by permeabilization using 0.1% Triton X-100 for 20 min in a 37°C incubator with a humidified 5% CO2 atmosphere. The cells were then blocked with 5% bovine serum albumin for 1 h in a 37°C incubator with a humidified 5% CO2 atmosphere followed by incubation with primary antibody (mouse anti-pERK (1:500), Cat#9106, Cell Signaling Technologies; mouse anti-Raf (1:500), Cat#ab50858, Abcam; rabbit anti-EEA1 (1:500), Cat#3288, Cell Signaling Technologies) for 1 h at RT or overnight at 4°C.
The cells were incubated with secondary antibody (atto488-conjugated goat anti-mouse IgG, Cat#62197, Sigma-Aldrich; Alexa Flour 555-conjugated goat anti-rabbit IgG, Cat#A21428, Invitrogen/ThermoFisher; 1 hr at 37°C) and mounted on slides using ProLong glass antifade Mountant with NucBlue (Invitrogen/ThermoFisher, P36982) and stored at −20°C before imaging. Samples were imaged on a Nikon Eclipse Ti2 equipped with a Yokogawa CSU-W1 SoRa confocal scanner unit, Nano-Drive positioning stage, Sola light engine, 405 nm (DAPI), 488 nm (Atto488), and 561 nm (Alexa Flour 555) laser lines, and a Photometrics Prime 95B camera. Super-resolution images were generated by microlensing the emission pinhole using the Yokogawa CSU-W1 SoRa confocal scanner unit, controlled by OMERO, and colocalization analysis was performed using the ImageJ coloc2 plugin.
1.6. Proximity Ligation Assay (PLA)
PLA experiments were done using the Duolink® in situ detection reagent green kit (Cat#DUO92008, Sigma-Aldrich) following the provided protocol. Briefly, cells were fixed and permeabilized as above before incubation with primary antibodies (rabbit anti-Gαs, #ab235959, Abcam; mouse anti-Raf1, Cat#ab50858, Abcam; mouse anti-MEK1, Cat #ab139343, Abcam; GenScript USA; normal mouse IgG, Cat #12–371, Millipore-Sigma) for 1 h at RT, followed by incubation with the provided oligonucleotide-conjugated secondary antibodies for 30 min at 37°C with washes after each step. Ligation and amplification were performed by incubating cells with the provided ligase, polymerase and detection solutions. PLA experiments performed using anti-normal mouse IgG were used as negative controls. Representative PLA images were acquired on a Nikon Eclipse Ti2 as described above, with illumination at 405 nm for DAPI and 488 nm for PLA puncta. Images for PLA puncta quantification were acquired on a Zeiss AxioObserver Z1 microscope (Carl Zeiss) equipped with a 40x/1.3NA objective and a Photometrics Evolve 512 EMCCD (Photometrics) controlled by METAFLUOR 7.7 software (Molecular Devices). The DAPI channel was acquired using a 405DF40 excitation filter, T470lp dichroic mirror, and 475DF40 emission filter. PLA signal was acquired using a 480DF30 excitation filter, 505dcxr dichroic mirror, and 535DF45 emission filter. All filter sets were alternated by a Lambda 10–2 filter-changer (Sutter Instruments). PLA fluorescence images were analyzed using FIJI. Raw fluorescence images were corrected by background subtraction and converted to gray-scale. Images of PLA puncta and cell nuclei were then adjusted for brightness and converted to binary mode. PLA puncta and nuclei were counted using the analyze particle plugin in FIJI based on the size criteria 0.3–10 pixel2 for PLA puncta and 30-infinite pixel2 for nuclei.
1.7. Cell proliferation assay
HEK293T cells stably expressing vector, endoGsCT or nucGsCT were seeded in 6-wells plates at 150,000 cells/well. Total mCherry expressing cell numbers were counted using a flow cytometer (Fortessa, BD) on the indicated days.
1.8. Quantitative PCR
HEK293T cells were transiently transfected with plasmid DNA for 24 h and treated with the indicated stimuli for 6 h. Total RNA was then isolated from HEK293T cells using TRIzol reagent, and 500 ng of RNA was reverse-transcribed into cDNA using PrimeScript RT master mix (TAKARA). Quantitative real-time PCR analysis was carried out using iTaq universal SYBR green supermix (Bio-Rad). Each sample measurement was performed in triplicate. The following sense and antisense primers were used. c-Myc: sense, TGAGGAGACACCGCCCAC, antisense, CAACATCGATTTCTTCCTCATCTTC; c-fos: sense, CGTCTCCAGTGCCAACTTCA, antisense, GGTCCGGACTGGTCGAGAT; cyclin D1: sense, CCGTCCATGCGGAAGATC, antisense, GAAGACCTCCTCCTCGCACT. Relative mRNA levels were quantified using the comparative Ct method after normalization to GAPDH.
1.9. Immunoblotting
Cells were washed with ice-cold PBS followed by lysis in RIPA buffer containing protease inhibitor cocktail, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, and 25 nM calyculin A. Total cell lysates were centrifuged at 4°C for 15 min. Total protein was separated via 4–15% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with TBS containing 0.1% Tween-20 and 5% bovine serum albumin and then incubated with primary antibodies overnight at 4°C (mouse anti-phospho-p44/42 MAPK, 1:1000, Cat#9106, Cell signaling technology, rabbit anti-ERK1/2 antibody 1:1000, Cat#17942, Abcam). After incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies (horseradish peroxidase-labeled goat anti-rabbit, Cat#PI31460, or anti-mouse, Cat#PI31430 secondary antibodies, 1:10000, Pierce), the membranes were developed using horseradish peroxidase-based chemiluminescent substrate (Cat#34579 and 34076, Thermo Fisher Scientific).
1.10. Statistics and reproducibility
All experiments were independently repeated as noted in the figure legends and Supplementary table 2. Statistical analyses were performed using Graphpad Prism 9 (Graphpad Software). Values are reported as the mean ± SEM. Pairwise comparisons were performed using either the Mann-Whitney U-test or Student’s t test, as indicated in the figure legends. Comparisons involving three or more groups were performed using the Kruskal-Wallis test or ordinary one-way ANOVA, followed by the indicated multiple comparison test. Statistical significance was set a P < 0.05. Cells to be analyzed were chosen based on the proper localization of biosensor fluorescence, as well as proper receptor internalization, reflecting correct cell functionality in response to agonist stimulation, and cells displaying clearly unusual behavior (e.g., apoptotic blebbing) were excluded from analysis.
Extended Data
Extended Data Fig. 1. Characterization of subcellularly targeted EKAR4.
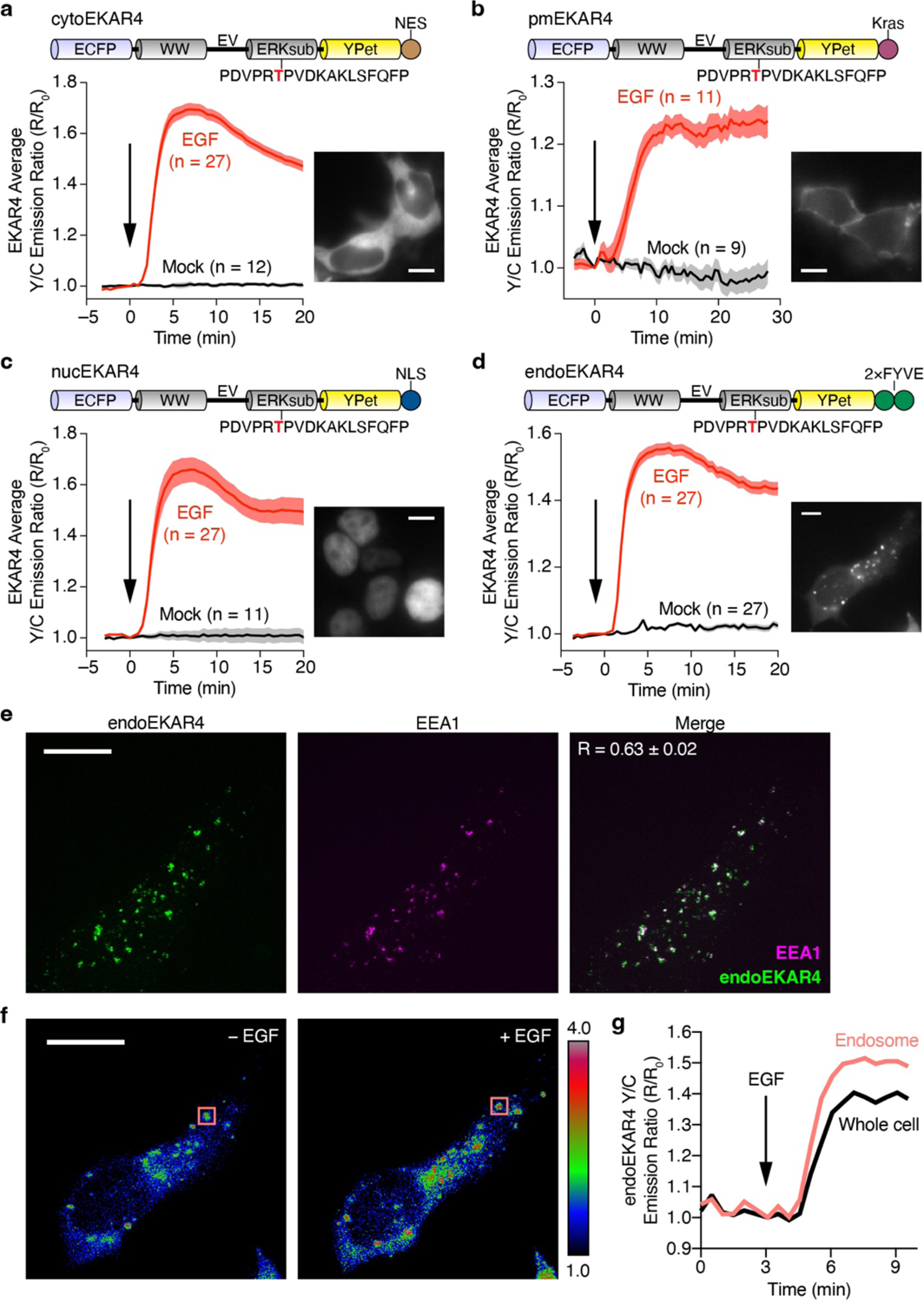
(a-d) Representative yellow/cyan (Y/C) emission ratio time courses from HEK293T cells expressing (a) cytoEKAR4 (EGF: n = 27; Mock: n = 12), (b) pmEKAR4 (EGF: n = 11; Mock: n = 9), (c) nucEKAR4 (EGF: n = 27; Mock: n = 11), or (d) endoEKAR4 (n = 27 each) and stimulated with or without 100 ng/mL EGF. Domain schematics (top) and epifluorescence images showing biosensor localization (inset) are also shown. (e) Maximum-intensity-projected super-resolution fluorescence images of HEK293T cells expressing endoEKAR (left) and stained for EEA1 (middle, endosomes; goat anti-rabbit Alexa Fluor 647 × rabbit anti-EEA1) and merged (right) image. (f) Representative ratio images (pseudocolored) before and after EGF treatment. Pseudocolor images where warmer colors represent higher emission ratios show that emission ratio changes are confined to endosomes. (g) Representative yellow/cyan (Y/C) emission ratio time courses comparing the responses from a single endosome (boxed region in f) or whole HEK293T cell expressing endoEKAR4 and stimulated with 100 ng/mL EGF, indicating that endoEKAR4 responses specifically come from endosomes. Solid lines in time courses indicate the mean, and shaded areas show SEM. Scale bars, 10 μm.
Extended Data Fig. 2. Compartmentalized GPCR signaling.
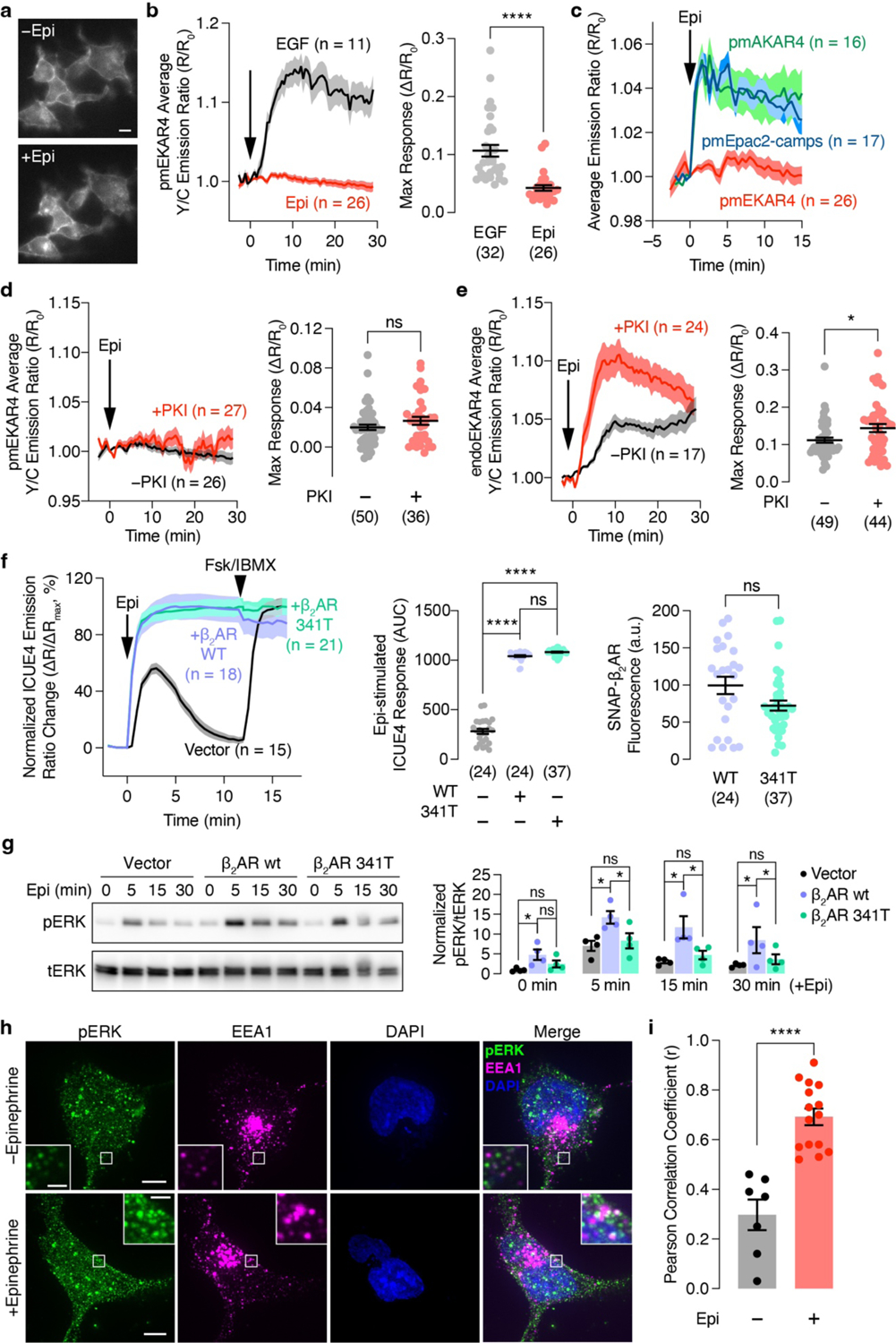
(a) Live-cell epifluorescence images of HEK293T cells expressing SNAP-β2AR and labeled with JF646 before (top) and 30 min after (bottom) 10 μM epinephrine (Epi) stimulation. (b) Left: Representative yellow/cyan (Y/C) emission ratio time course from HEK293T cells co-expressing pmEKAR4 plus SNAP-β2AR stimulated with EGF or Epi. Right: Maximum pmEKAR4 responses (n = 32 and 26 for EGF and Epi, respectively). (c) Representative time courses showing average Epi-promoted emission ratio changes from plasma membrane targeted PKA activity biosensor AKAR4 (pmAKAR4; n = 16), cAMP biosensor Epac2-camps (pmEpac2-camps; n = 17), and EKAR4 (pmEKAR4; n = 26). (d and e) Left: Representative time courses showing the 10 μM Epi-stimulated yellow/cyan (Y/C) emission ratio changes in HEK293T cells co-expressing SNAP-β2AR, PKI or vector and (d) pmEKAR4 or (e) endoEKAR4. Right: Maximum EKAR4 responses. (f) Left: Representative time courses showing the normalized cyan/yellow (C/Y) emission ratio changes in cells co-expressing another cAMP biosensor ICUE4 plus vector, SNAP-β2AR or SNAP-β2AR341T upon stimulation with 10 μM epinephrine (Epi) followed by 50μM Fsk and 100 μM IBMX (Fsk/IBMX). Middle: Total epinephrine-stimulated ICUE4 responses. Right: Expression level of the indicated receptors. (g) Western blot analysis of phospho-ERK (pERK) and total ERK (tERK) levels in HEK293T cells expressing vector, SNAP-β2AR or SNAP-β2AR 341T following stimulation with Epi for 0, 5, 15, 30 min. Representative blot (left) and quantification of normalized pERK/tERK levels at each time point (right). Full blots are shown in Supplementary Fig. 2. (h) Maximum-intensity-projected super-resolution fluorescence images of HEK293T cells expressing SNAP-β2AR with (lower) or without (upper) epinephrine stimulation and stained for phospho-ERK (pERK; goat anti-mouse Atto488 × mouse anti-pERK), EEA1 (endosomes; goat anti-rabbit Alexa Fluor 647 × rabbit anti-EEA1), and DAPI (nuclei). (i) Histogram of the Pearson correlation coefficient for EEA1 and pERK colocalization in HEK293T cells expressing SNAP-β2AR with or without Epi stimulation (n = 7 and 15 for − and + and Epi, respectively). ns, not significant, *P < 0.05, ****P < 0.0001; Mann-Whitney U-test (b), unpaired two-tailed Student t-test without (d, e, and i) or with (f right) Welch’s correction, ordinary one-way ANOVA followed by Holm-Šídák’s multiple-comparison test (f middle and g). Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM. Scale bars, 10 μm. Negative control curves in (d and e) are reproduced from Fig. 1a and b.
Extended Data Fig. 3. GPCR-mediated endosomal ERK activity is regulated by GPCR endocytosis and endosomal Gαs activity.
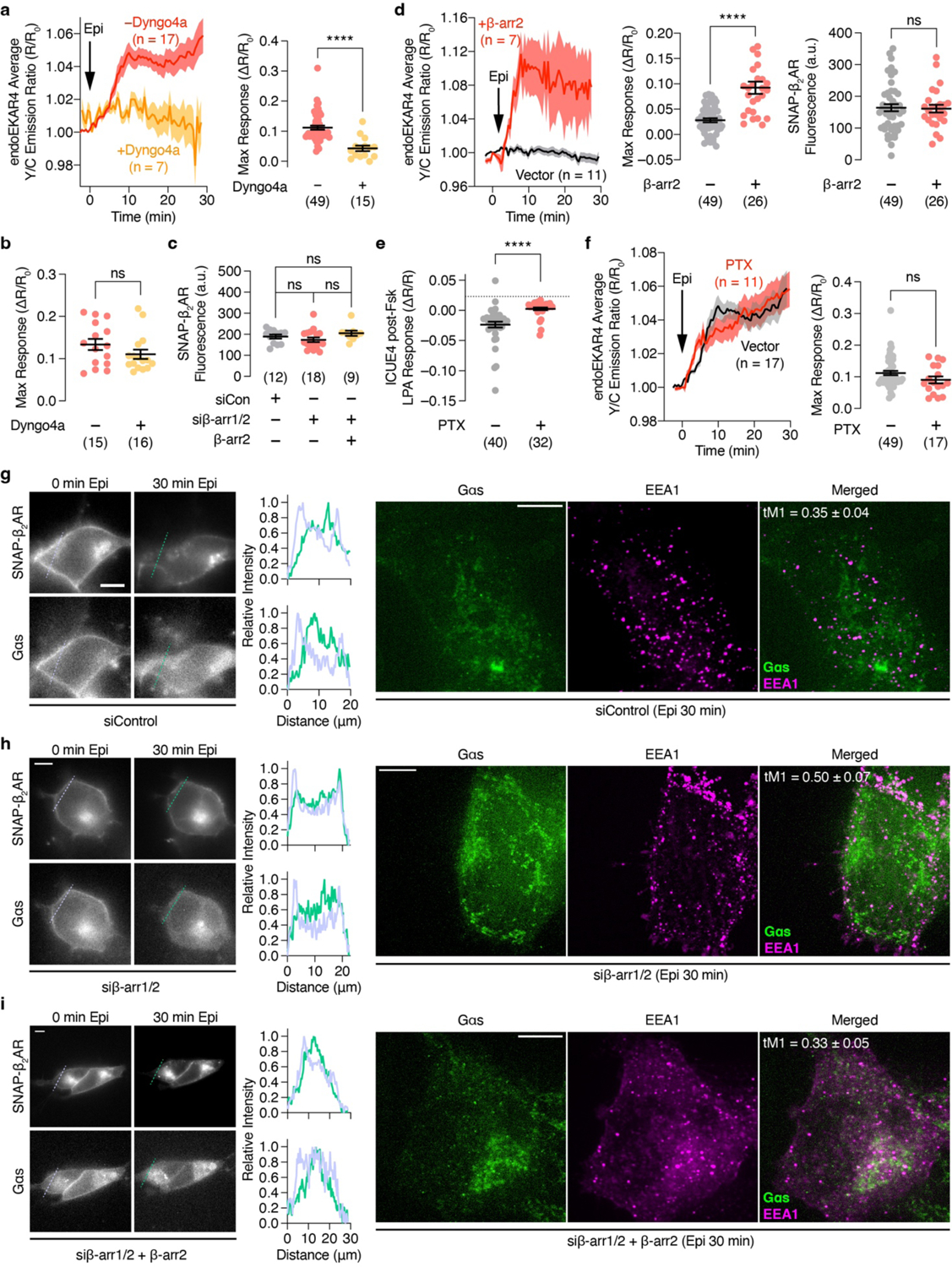
(a, d and f) Left: Representative time courses showing the 10 μM epinephrine (Epi)-stimulated yellow/cyan (Y/C) emission ratio changes in cells co-expressing endoEKAR4 and SNAP-β2AR (a) with or without preincubation of 5 μM of Dyngo4a in HEK293T cells or (d) β-arr1/2 dKO HEK293S cells additionally transfected with vector or β-arr2 or (f) HEK293T cells additionally transfected with vector or PTX. Right (a, f) or middle (d): Maximum EKAR4 responses. Right (d): Intensity of JF646-labeled SNAP-β2AR. (b) Maximum response of pmEKAR4 upon EGF stimulation with or without Dyngo4a pretreatment. (c) Intensity of JF646-labeled SNAP-β2AR; corresponds to Fig. 2b. (e) Maximum decrease of ICUE4 cyan/yellow emission ratio (ΔR/R) upon stimulation with 500 nM LPA in HEK293T cells preincubated with 50 μM Fsk and expressing vector or PTX. ns, not significant, ****P < 0.0001; unpaired two-tailed Student’s t test without (a, d right) or with (d middle) Welch’s correction, Kruskal-Wallis test followed by Dunn’s multiple-comparison test (c), Mann-Whitney U-test (b, e, f). Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM. (g-i) Epifluorescence (left) and maximum-intensity-projected super-resolution fluorescence images (right) of HEK293T cells transfected with (g) control siRNA, (h) siRNAs targeting β-arrestin1/2, and (i) siRNAs targeting β-arrestin1/2 plus β-arrestin2-mCherry cDNA. Individual epifluorescence channel images show (upper) JF646-SNAP-β2AR or (lower) Gαs-mEos3.2 before (left) and after (right) Epi stimulation. Line intensity profile plots corresponding to the dashed lines on the images are shown to the right of the images (light blue: 0 min; green: 30 min after Epi). Individual maximum-intensity-projected super-resolution fluorescence channel images of (left) Gαs, (middle) EEA1 (endosomes; goat anti-rabbit Alexa Fluor 568 × rabbit anti-EEA1) and (right) merged channels. Numbers represent average Manders’ tM1 overlap coefficient from 10 (g), 4 (h) and 8 (i) different cells after 30 min Epi stimulation. Scale bars, 10 μm (epifluorescence images) and 5 μm (super-resolution images). Negative control curve and data points in (a and f) are reproduced from Fig. 1c.
Extended Data Fig. 4. Localization of endosome-targeted nanobodies and GsCT and the effect on ERK.
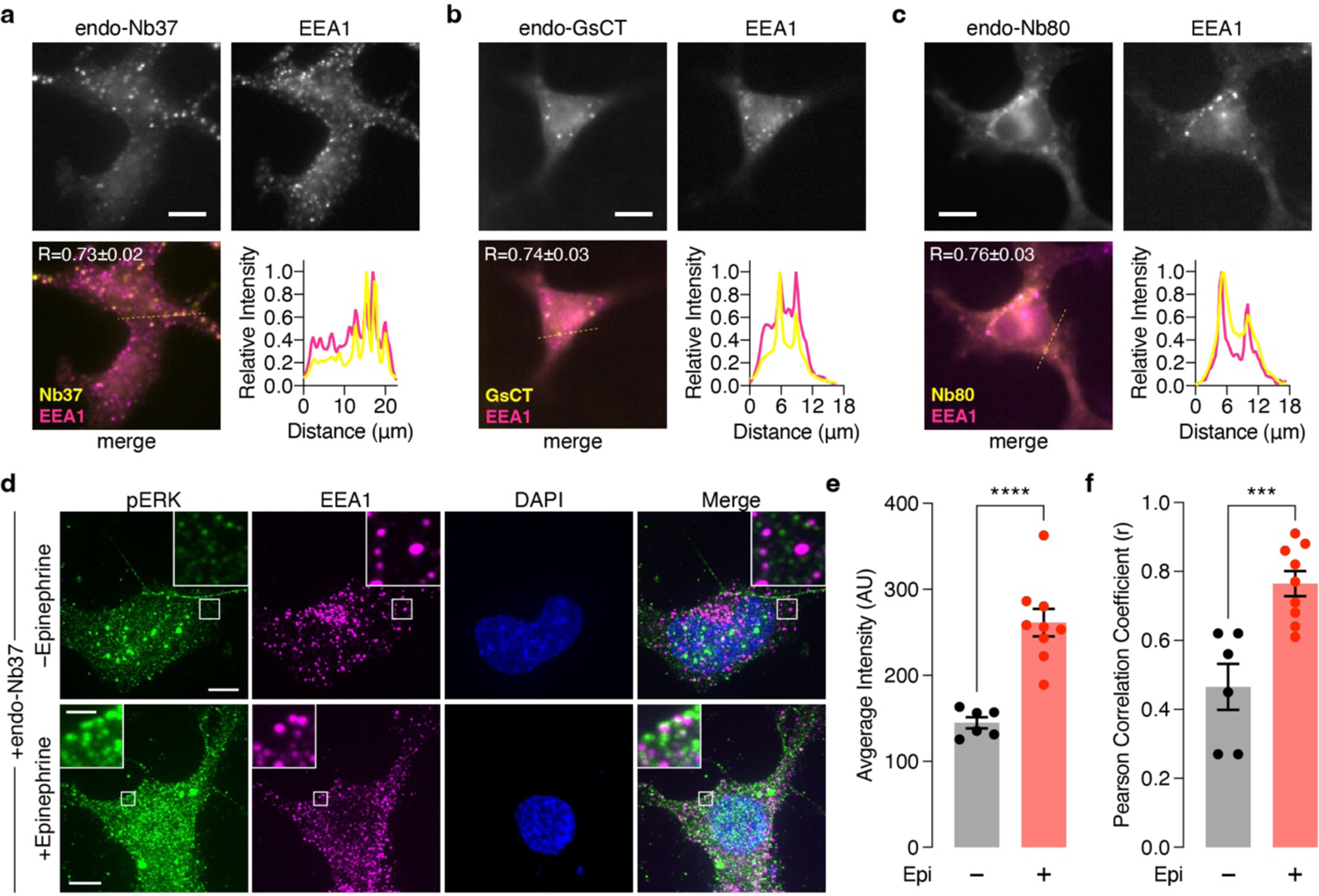
(a-c) Epifluorescence microscopy analysis of (a) endo-Nb37, (b) endo-GsCT, and (c) endo-Nb80 colocalization with the endosomal marker EEA1 in HEK293T cells. Individual channel images of (upper left) mCherry fluorescence from endo-Nb37, endo-GsCT, and endo-Nb80 and (upper right) Alexa Fluor 647 fluorescence (goat anti-rabbit Alexa Fluor 647 × rabbit anti-EEA1), as well as (lower left) merged channels are shown. Line intensity profile plots corresponding to the dashed yellow lines on the merge images are shown to the lower right. (d) Maximum-intensity-projected super-resolution fluorescence images of HEK293T cells co-expressing SNAP-β2AR plus endo-Nb37 with (lower) or without (upper) epinephrine stimulation and stained for phospho-ERK (pERK; goat anti-mouse Atto488 × mouse anti-pERK), EEA1 (endosomes; goat anti-rabbit Alexa Fluor 647 × rabbit anti-EEA1), and DAPI (nuclei). (e, f) Histograms showing (e) the total intensity from the 488 nm (pERK) channel and (f) the Pearson correlation coefficient for EEA1 and pERK colocalization in HEK293T cells co-expressing SNAP-β2AR plus endo-Nb37 with (+) or without (−) epinephrine (Epi) stimulation (n = 6 and 9 for − and + Epi, respectively). Scale bars, 10 μm. ***P < 0.001, ****P < 0.0001; unpaired two-tailed Student’s t-test. Lines in scatter plots represent mean ± SEM.
Extended Data Fig. 5. Gαs interacts with Raf1 and MEK1 to regulate endosomal GPCR-mediated ERK activity.
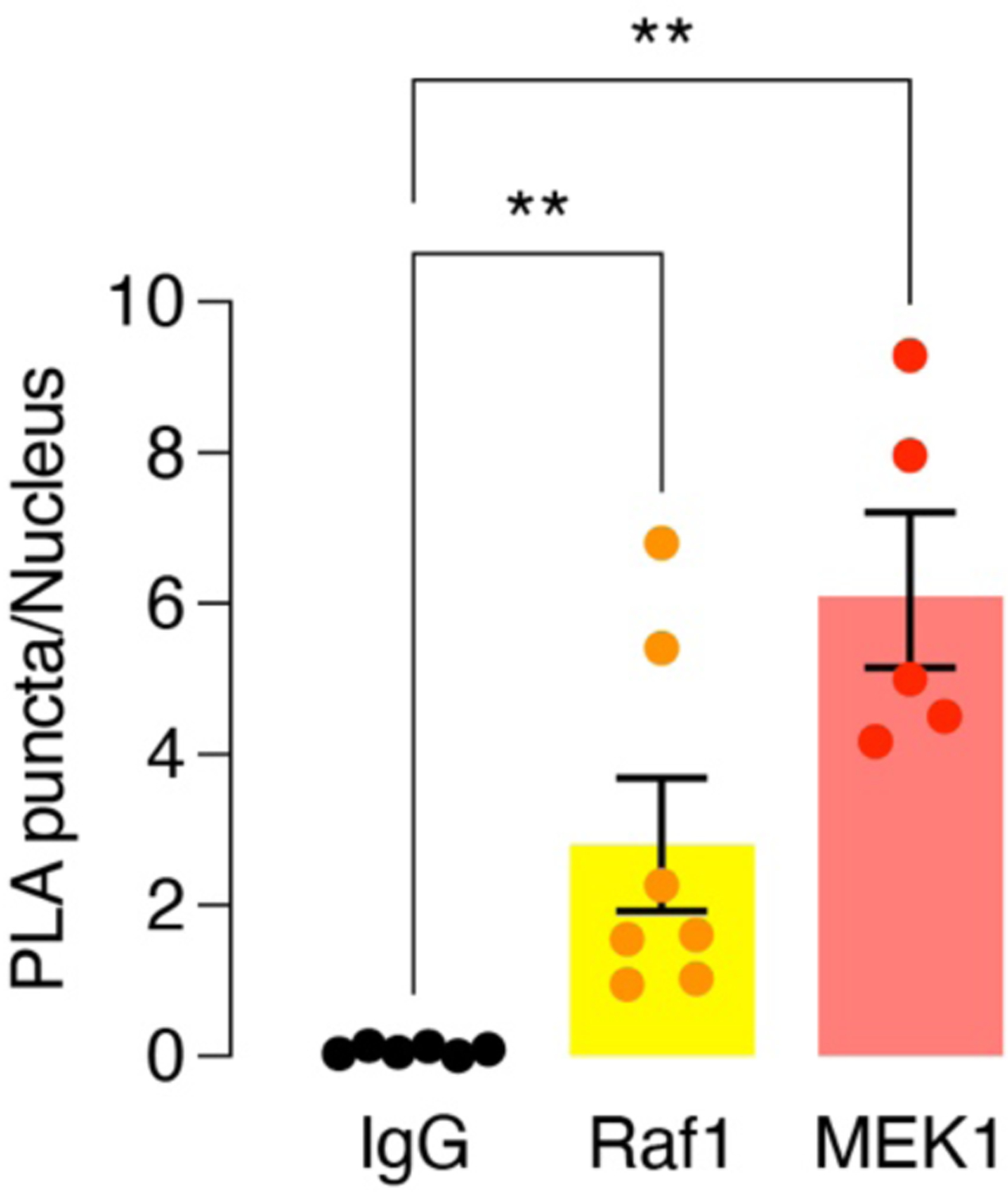
Quantification of PLA particle density for SNAP-β2AR-expressing HEK293T cells incubated with rabbit anti-Gαs plus normal mouse IgG, mouse anti-Raf1 or mouse anti-MEK1 (n = 6, 7, and 5 different randomly selected fields of view for IgG, Raf1 and MEK1, respectively) following stimulation with 10 μm epinephrine. Bars correspond to the mean ± SEM. **P < 0.01; Mann-Whitney U-test.
Extended Data Fig. 6. Endosomal Gαs regulates nuclear ERK activity and function.
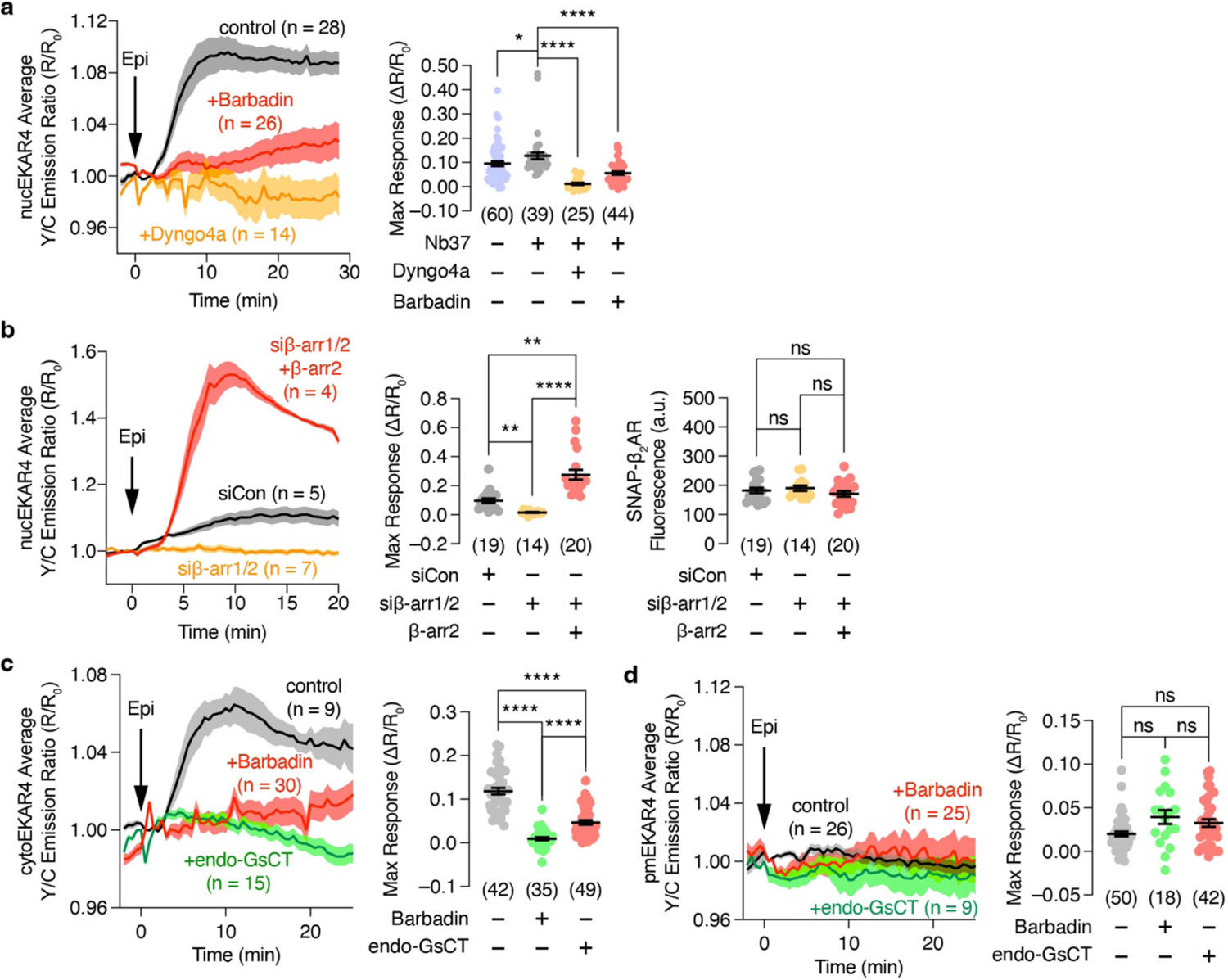
(a, b) Left: Representative 10 μM epinephrine (Epi)-stimulated yellow/cyan (Y/C) emission ratio changes from HEK293T cells co-expressing nucEKAR4, SNAP-β2AR and (a) Nb37 in the presence or absence of 100 μM barbadin or 5 μM of Dyngo4a preincubation or (b) additionally transfected with control siRNA or siRNA targeting β-arr1 and 2 plus vector or β-arr2. (a) Right, (b) middle: Maximum nucEKAR4 responses under the indicated conditions. (b) Right: SNAP-β2AR fluorescence intensity under the indicated conditions. (c and d) Left: Representative time courses showing the 10 μM Epi-stimulated yellow/cyan (Y/C) emission ratio changes of (c) cytoEKAR4 or (d) pmEKAR4 in HEK293T cells co-expressing SNAP-β2AR with or without endoGsCT or preincubation with 100 μM barbadin. Right: Maximum EKAR4 responses. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; Kruskal-Wallis test followed by Dunn’s multiple-comparison test (a, b middle), ordinary one-way ANOVA followed by Holm-Šidák’s multiple-comparison test (b right), Welch ANOVA followed by Dunnett’s T3 multiple-comparison test (c, d). Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM. Negative control curve and data points in (c and d) are reproduced from Fig. 1a and c, respectively.
Extended Data Fig. 7. Endogenous β-adrenergic-stimulated ERK activation.
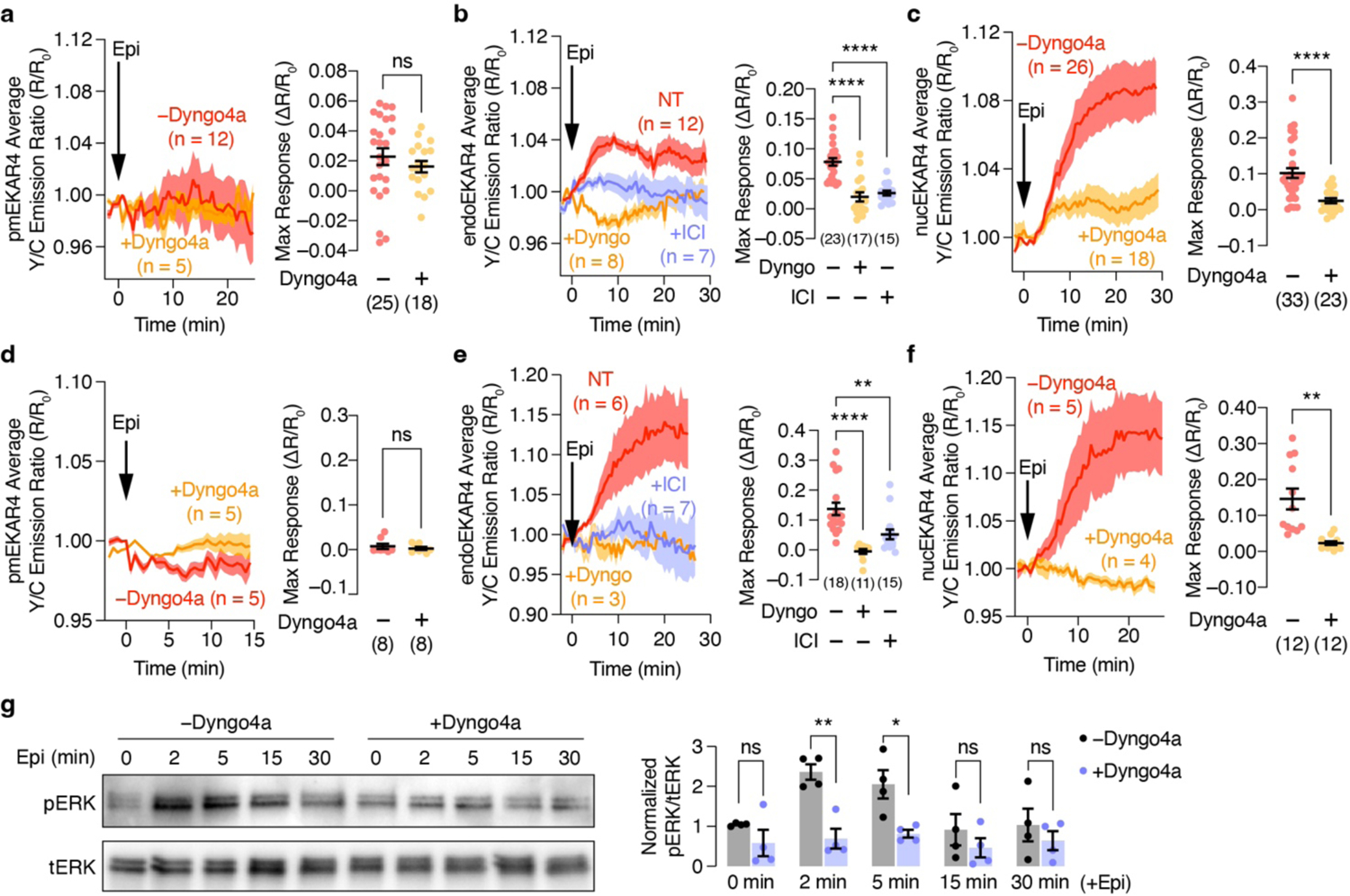
Left: Representative 10 μM epinephrine (Epi)-stimulated yellow/cyan (Y/C) emission ratio changes from (a-c) HEK293T cells or (d-f) H9c2 cells expressing (a, d) pmEKAR4, (b, e) endoEKAR4, or (c, f) nucEKAR4 with or without 5 μM Dyngo4a (b, e) with ICI-118,551 preincubation. Right: Maximum EKAR4 responses under the indicated conditions. Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM. (g) Western blot analysis of phospho-ERK (pERK) and total ERK (tERK) levels in HEK293T cells stimulated with Epi for 0, 2, 5, 15, and 30 min with or without Dyngo4a preincubation. Representative blot (left) and quantification of normalized pERK/tERK levels at each time point (right). Full blots are shown in Supplementary Fig. 1. ns, not significant, **P < 0.01, ***P < 0.001, ****P < 0.0001; Mann-Whitney U-test (a) or unpaired two-tailed Student’s t-test without (c, d) or with (f,g) Welch’s correction, or ordinary one-way ANOVA followed by Holm-Šídák’s multiple-comparison test (b, e).
Extended Data Fig. 8. Compartmentalized ERK activation by the stimulation of multiple types of GPCRs.
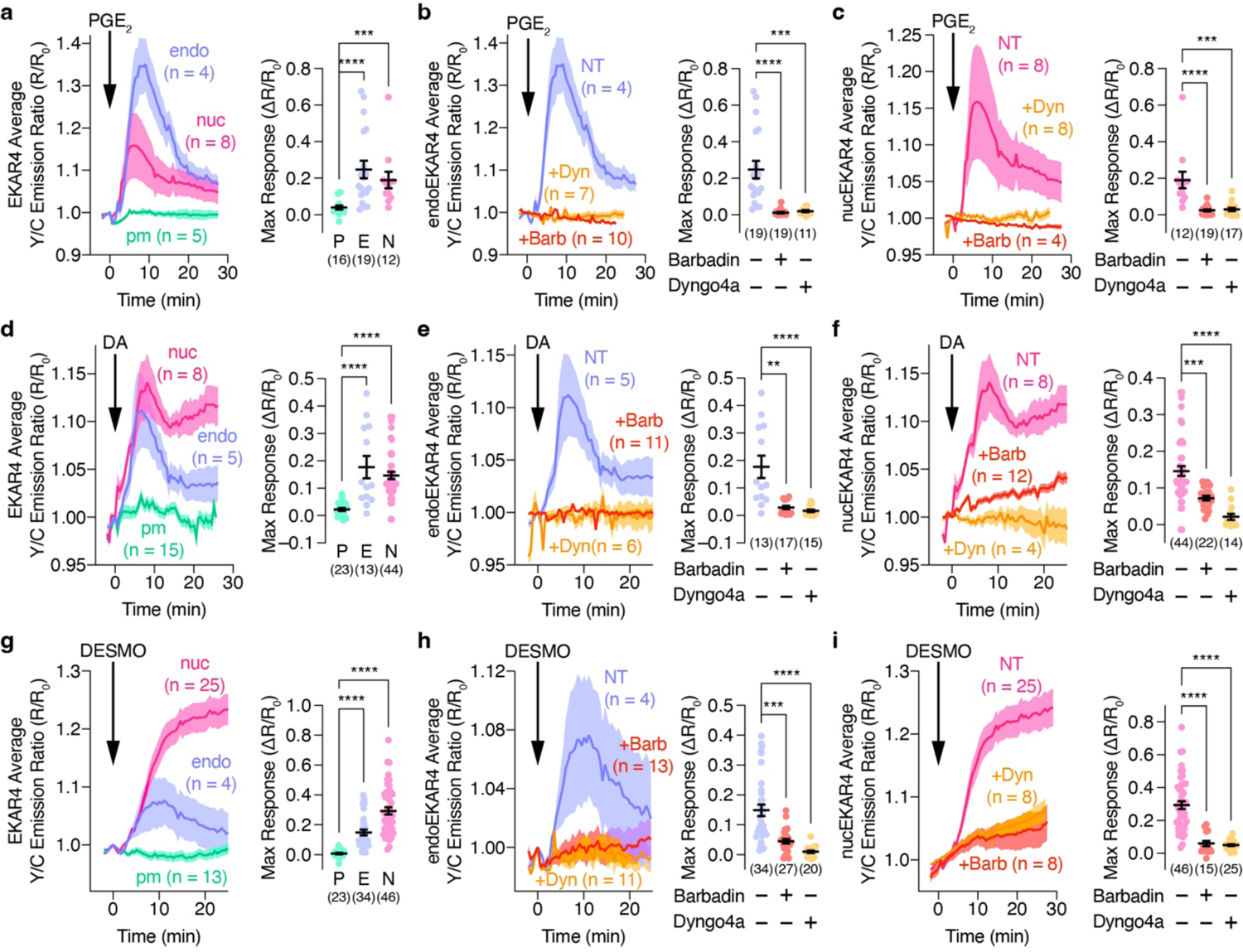
Left: Representative yellow/cyan (Y/C) emission ratio changes of HEK293T cells co-expressing (a-c) EP4, (d-f) D1DR, or (g-i) V2R plus (a, d, g) diffusible, (b, e, h) endo- or (c, f, i) nucEKAR4 upon stimulation with 10 μM (a-c) PGE2, (d-f) 100 μM DA or (g-i) 100 nM DESMO with or without preincubation with 5 μM Dyngo4a or 100 μM barbadin. Right: Maximum EKAR4 responses under the indicated conditions. **P < 0.01, ***P < 0.001, ****P < 0.0001; Kruskal-Wallis test followed by Dunn’s multiple-comparison test. Solid lines in time courses indicate the mean, and shaded areas show SEM. Lines in scatter plots represent mean ± SEM.
Extended Data Fig. 9. Growth factor-induced ERK target gene expression.
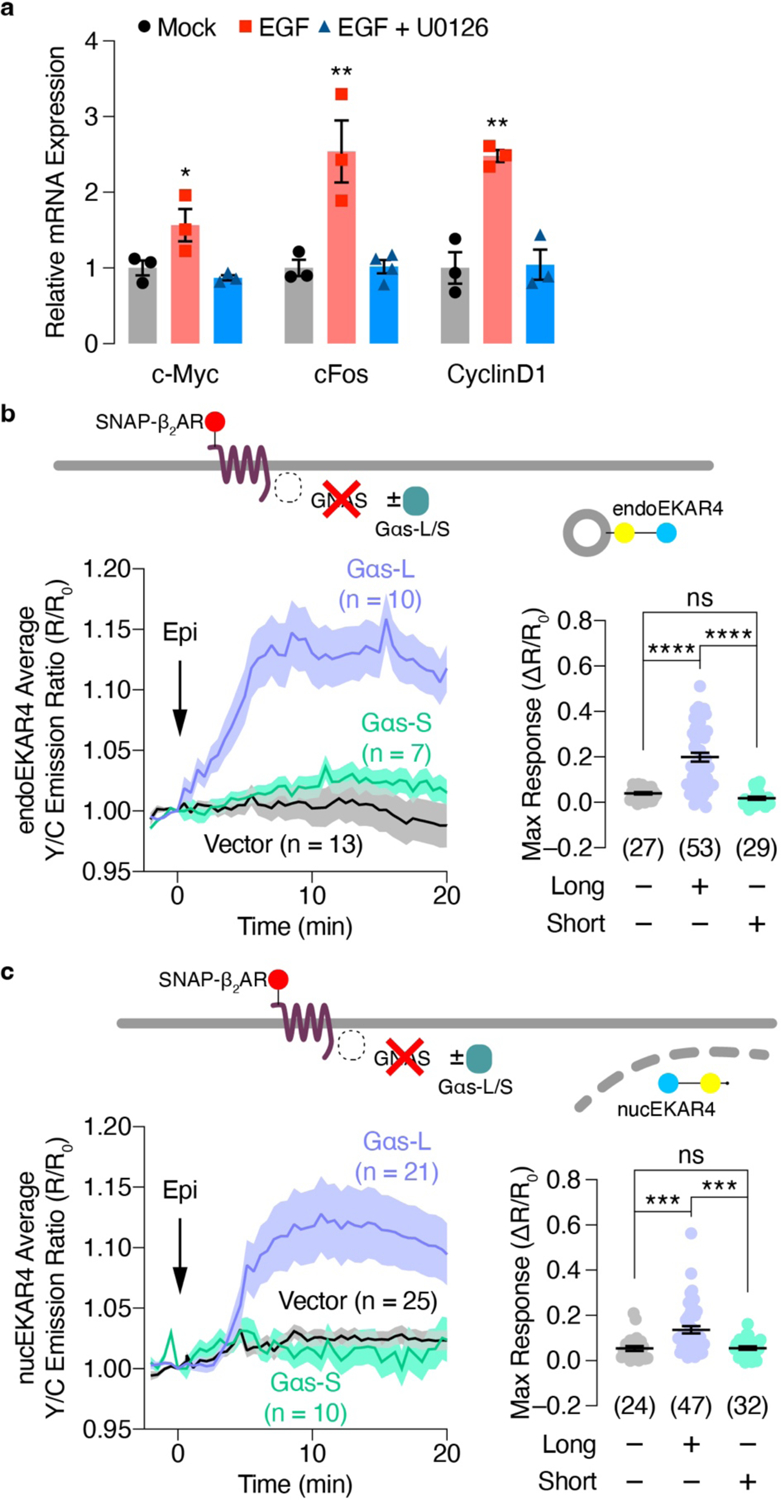
(a) qPCR analysis of c-Myc, c-fos and cyclin D1 mRNA in EGF-treated HEK293T cells with or without U0126 preincubation. (n = 3 independent experiments). *P < 0.05, **P < 0.01, ****P < 0.0001 vs. Mock or EGF + U0126; ordinary one-way ANOVA followed by Tukey’s multiple-comparison test. (b, c) Left: Representative yellow/cyan (Y/C) emission ratio traces from GNAS KO HEK293 cells co-expressing (b) endo or (c) nucEKAR4 and additionally expressing vector, Gαs-L or Gαs-S with or without 10 μM epinephrine (Epi) stimulation. Right: Maximum EKAR4 responses. ns, not significant, ***P < 0.001, ****P < 0.0001; Kruskal-Wallis test followed by Dunn’s multiple-comparison test (b, c). Solid lines in b and c indicate the mean, and shaded areas show SEM. Horizonal lines in b and c depict the mean ± SEM. Negative control and Gαs-L curves and data points in (b) are reproduced from Fig. 2c.
Supplementary Material
Acknowledgements
The authors are grateful to Asuka Inoue for kindly providing the GNAS KO HEK293A and β-arr1/2 dKO cell lines, Dr. Daniel Mayer for comments on the manuscript, the members of the Nikon Imaging Center at UC San Diego for help with imaging experiments, the members of the Human Embryonic Stem Cell Core Facility for assistance of the proliferation assay, and Dr. Luke D. Lavis and HHMI Janelia Research Campus and for providing the JF646-SNAP ligand. This work was supported by the NIH (R35 CA197622 and R01 DK073368 to J.Z.) and the University of California Tobacco-Related Disease Research Program (T32FT4691 to Y.K.).
Footnotes
Competing Interests
The authors declare no competing financial interests.
Supplementary Information is available for this paper.
Data availability
Source data are provided with this paper.
References
- 1.Smith JS, Lefkowitz RJ & Rajagopal S Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17, 243–260, doi: 10.1038/nrd.2017.229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Hayre M et al. Genetic evidence that beta-arrestins are dispensable for the initiation of beta2-adrenergic receptor signaling to ERK. Sci Signal 10, doi: 10.1126/scisignal.aal3395 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundmann M et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 9, 341, doi: 10.1038/s41467-017-02661-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luttrell LM et al. Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 11, doi: 10.1126/scisignal.aat7650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keyes J et al. Signaling diversity enabled by Rap1-regulated plasma membrane ERK with distinct temporal dynamics. Elife 9, doi: 10.7554/eLife.57410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith JS et al. Noncanonical scaffolding of Galphai and beta-arrestin by G protein-coupled receptors. Science 371, doi: 10.1126/science.aay1833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavoie H, Gagnon J & Therrien M ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol 21, 607–632, doi: 10.1038/s41580-020-0255-7 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Eichel K & von Zastrow M Subcellular Organization of GPCR Signaling. Trends Pharmacol Sci 39, 200–208, doi: 10.1016/j.tips.2017.11.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irannejad R et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538, doi: 10.1038/nature12000 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsvetanova NG & von Zastrow M Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10, 1061–1065, doi: 10.1038/nchembio.1665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kotowski SJ, Hopf FW, Seif T, Bonci A & von Zastrow M Endocytosis promotes rapid dopaminergic signaling. Neuron 71, 278–290, doi: 10.1016/j.neuron.2011.05.036 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Rooij J et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477, doi: 10.1038/24884 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Kolch W et al. Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature 364, 249–252, doi: 10.1038/364249a0 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Daaka Y et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem 273, 685–688, doi: 10.1074/jbc.273.2.685 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Pierce KL, Maudsley S, Daaka Y, Luttrell LM & Lefkowitz RJ Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci U S A 97, 1489–1494, doi: 10.1073/pnas.97.4.1489 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurevich VV & Gurevich EV Arrestin-mediated signaling: Is there a controversy? World J Biol Chem 9, 25–35, doi: 10.4331/wjbc.v9.i3.25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luttrell LM et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A 98, 2449–2454, doi: 10.1073/pnas.041604898 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tohgo A et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278, 6258–6267, doi: 10.1074/jbc.M212231200 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Schmitt DL et al. Spatial regulation of AMPK signaling revealed by a sensitive kinase activity reporter. doi: 10.1101/2021.10.11.463987 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M, Sun T, Zhong Y, Zhou X & Zhang J A Highly Sensitive Fluorescent Akt Biosensor Reveals Lysosome-Selective Regulation of Lipid Second Messengers and Kinase Activity. ACS Cent Sci 7, 2009–2020, doi: 10.1021/acscentsci.1c00919 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X et al. Dynamic Visualization of mTORC1 Activity in Living Cells. Cell Rep 10, 1767–1777, doi: 10.1016/j.celrep.2015.02.031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen MD & Zhang J Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun 348, 716–721, doi: 10.1016/j.bbrc.2006.07.136 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Miyamoto T et al. Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell Rep 11, 657–670, doi: 10.1016/j.celrep.2015.03.057 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaidyanathan H et al. ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc Natl Acad Sci U S A 104, 19837–19842, doi: 10.1073/pnas.0704514104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boned Del Rio I et al. SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc Natl Acad Sci U S A 116, 13330–13339, doi: 10.1073/pnas.1902658116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ariotti N et al. Modular Detection of GFP-Labeled Proteins for Rapid Screening by Electron Microscopy in Cells and Organisms. Dev Cell 35, 513–525, doi: 10.1016/j.devcel.2015.10.016 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Gerits N, Kostenko S, Shiryaev A, Johannessen M & Moens U Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal 20, 1592–1607, doi: 10.1016/j.cellsig.2008.02.022 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Eichel K et al. Catalytic activation of beta-arrestin by GPCRs. Nature 557, 381–386, doi: 10.1038/s41586-018-0079-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng GE, Pessino V, Huang B & von Zastrow M Spatial decoding of endosomal cAMP signals by a metastable cytoplasmic PKA network. Nat Chem Biol 17, 558–566, doi: 10.1038/s41589-021-00747-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luttrell LM et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661, doi: 10.1126/science.283.5402.655 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Pierce KL, Luttrell LM & Lefkowitz RJ New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene 20, 1532–1539, doi: 10.1038/sj.onc.1204184 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Luttrell LM ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol 30, 117–126, doi: 10.1677/jme.0.0300117 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Beautrait A et al. A new inhibitor of the beta-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat Commun 8, 15054, doi: 10.1038/ncomms15054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin BR & Lambert NA Activated G Protein Galphas Samples Multiple Endomembrane Compartments. J Biol Chem 291, 20295–20302, doi: 10.1074/jbc.M116.729731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazar AM et al. G protein-regulated endocytic trafficking of adenylyl cyclase type 9. Elife 9, doi: 10.7554/eLife.58039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X et al. Location-specific inhibition of Akt reveals regulation of mTORC1 activity in the nucleus. Nat Commun 11, 6088, doi: 10.1038/s41467-020-19937-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rasmussen SG et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180, doi: 10.1038/nature09648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feldman DS et al. Selective inhibition of heterotrimeric Gs signaling. Targeting the receptor-G protein interface using a peptide minigene encoding the Galpha(s) carboxyl terminus. J Biol Chem 277, 28631–28640, doi: 10.1074/jbc.M204753200 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Irannejad R et al. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 13, 799–806, doi: 10.1038/nchembio.2389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plotnikov A, Zehorai E, Procaccia S & Seger R The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 1813, 1619–1633, doi: 10.1016/j.bbamcr.2010.12.012 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Zhang W & Liu HT MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 12, 9–18, doi: 10.1038/sj.cr.7290105 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Weis WI & Kobilka BK The Molecular Basis of G Protein-Coupled Receptor Activation. Annu Rev Biochem 87, 897–919, doi: 10.1146/annurev-biochem-060614-033910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones AJY, Gabriel F, Tandale A & Nietlispach D Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 25, doi: 10.3390/molecules25204729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arumugam S & Kaur A The Lipids of the Early Endosomes: Making Multimodality Work. Chembiochem 18, 1053–1060, doi: 10.1002/cbic.201700046 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Xiao K et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A 104, 12011–12016, doi: 10.1073/pnas.0704849104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu V et al. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J Biol Chem 294, 11062–11086, doi: 10.1074/jbc.REV119.005601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patra KC et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol 20, 811–822, doi: 10.1038/s41556-018-0122-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arang N & Gutkind JS G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett 594, 4201–4232, doi: 10.1002/1873-3468.14017 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler EC et al. Integrative RNA-omics discovers GNAS alternative splicing as a phenotypic driver of splicing factor-mutant neoplasms. Cancer Discov, doi: 10.1158/2159-8290.CD-21-0508 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Insel PA et al. GPCRomics: GPCR Expression in Cancer Cells and Tumors Identifies New, Potential Biomarkers and Therapeutic Targets. Front Pharmacol 9, 431, doi: 10.3389/fphar.2018.00431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data are provided with this paper.