

Abstract
Nucleosides play central roles in all facets of life, from metabolism to cellular signaling. Due to their physiochemical properties, nucleosides are lipid bilayer impermeable and thus rely on dedicated transport systems to cross biological membranes. In humans, two unrelated protein families mediate nucleoside membrane transport: the concentrative and equilibrative nucleoside transporter families. The objective of this review is to provide a broad outlook on the current status of nucleoside transport research. We will discuss the role played by nucleoside transporters in human health and disease, with emphasis placed on recent structural advancements that have revealed detailed molecular principles of these important cellular transport systems and exploitable pharmacological features.
Graphical Abstract
1. Introduction
Nucleosides play central roles in human metabolism, physiology and pharmacology. As nucleotide precursors, nucleoside pools are required for nucleic acid synthesis and cellular metabolism. Additionally, adenosine signals through specified cell surface receptors with heavy involvement in processes ranging from cellular response to hypoxic stress to neurotransmission and neuromodulation. Owing to the hydrophilic nature of these small biomolecules, nucleosides rely on dedicated transport systems to cross the kinetic barrier of the lipid bilayer. Therefore, nucleoside transport systems play key roles in the regulation of many aspects of nucleoside-related biology. Furthermore, nucleoside transport systems contribute significantly to nucleoside-derived antiviral and anticancer drug absorption, delivery, metabolism and excretion (ADME). Nucleoside transport systems are mediated by choreographed actions of the integral membrane proteins knows as nucleoside transporters (NTs). Due to the physiological and therapeutic importance of NTs, there have been numerous studies of the role of NTs in nucleoside-related physiology and drug efficacy. We wish to note detailed aspects of cellular nucleoside transport and its relation to human physiology and pharmacology have been excellently reviewed elsewhere1–7. Therefore, the goal of this review is to provide a comprehensive, detailed overview of the molecular and mechanistic features of NTs and their exploitable pharmacological features. Emphasis will be placed on recent experimental NT structures that have not only aided in our understanding of the mechanism of nucleoside transport across the membrane by these proteins, but also have paved the way for future therapeutic interventions in nucleoside biology.
1.1. Nucleosides in human health and disease
Comprised of a ribose sugar linked to a nitrogenous base through a β-N-glycosidic linkage, nucleosides are hydrophilic biomolecules that are either sourced from diet or de novo biosynthesized in humans. As nucleosides can be readily converted to nucleotides via phosphorylation8, 9, nucleoside pools are required for maintenance of cellular energy stores, DNA/RNA synthesis and cellular signaling. Conversely, nucleosides can be generated from nucleotide precursors through the actions of various enzymes9, 10. The purine nucleoside adenosine is of particular importance to cell signaling, as it serves as a ligand for cell-surface adenosine G-protein coupled receptors (GPCRs). Basal concentrations of adenosine are typically in the nanomolar range, though certain physiological and pathophysiological states can trigger extracellular adenosine concentrations to reach millimolar levels11. Under hypoxic/ischemic conditions, this occurs through the release of ATP into the extracellular matrix by pannexins, followed by subsequent hydrolysis of adenine nucleotides into adenosine by ectonucleotidases3, leading to the accumulation of adenosine that ultimately stimulates signaling through adenosine GPCRs11–14. Downstream of adenosine signaling in these contexts are pathways that lead to vasodilation, angiogenesis and metabolic regulation, making adenosine a protective agent to tissues undergoing hypoxic stress12. Adenosine receptor signaling is also a key component in neurotransmission and neuromodulation, involved in sleep or wakefulness, alcohol intoxication and use disorders, and implicated in numerous neurological and addictive behavioral disorders11, 13, 15–24.
Due to their role as precursors in nucleic acid synthesis, intervention of cellular and viral replication through use of exogenous nucleoside-analogs (Figure 1) has proven a viable therapeutic strategy in the treatment of cancers and viral infections25–28. Nucleoside-analog antineoplastic agents exhibit antimetabolite activities, achieved by simple competition of the endogenous nucleoside-pool, resulting in early chain termination, lesion incorporation into the genome, or aberrant DNA methylation, all of which result in general cytotoxicity29. The modest selectivity of the nucleoside-analog antineoplastic agents comes from the fact that cellular replication levels are significantly higher in cancel cells than normal cells. Nucleoside-analog antiviral drugs work in a similar manner, inhibiting viral replication via inhibition of viral polymerases, chain termination or introduction of mutations into the viral genome. Notably, studies have shown that many antiviral drugs can be rationally modified to specifically inhibit viral DNA polymerases, viral RNA dependent RNA polymerases or viral reverse transcriptases, not endogenous human polymerases, therefore exhibiting suitable target specificities26, 27. Some common nucleoside analog drugs include gemcitabine – a chemotherapeutic agent used in the treatment of pancreatic cancer, cytarabine – a therapeutic used in treatment of leukemia, ribavirin – a therapeutic used to treat hepatitis C, and azidothymidine (AZT) – a therapeutic used in the treatment of human immunodeficiency virus (HIV) infection. Of recent relevance to global public health, several nucleoside-analog antivirals have shown initial promise in the treatment of novel coronavirus disease 2019 (COVID-19)30–32.
Figure 1.
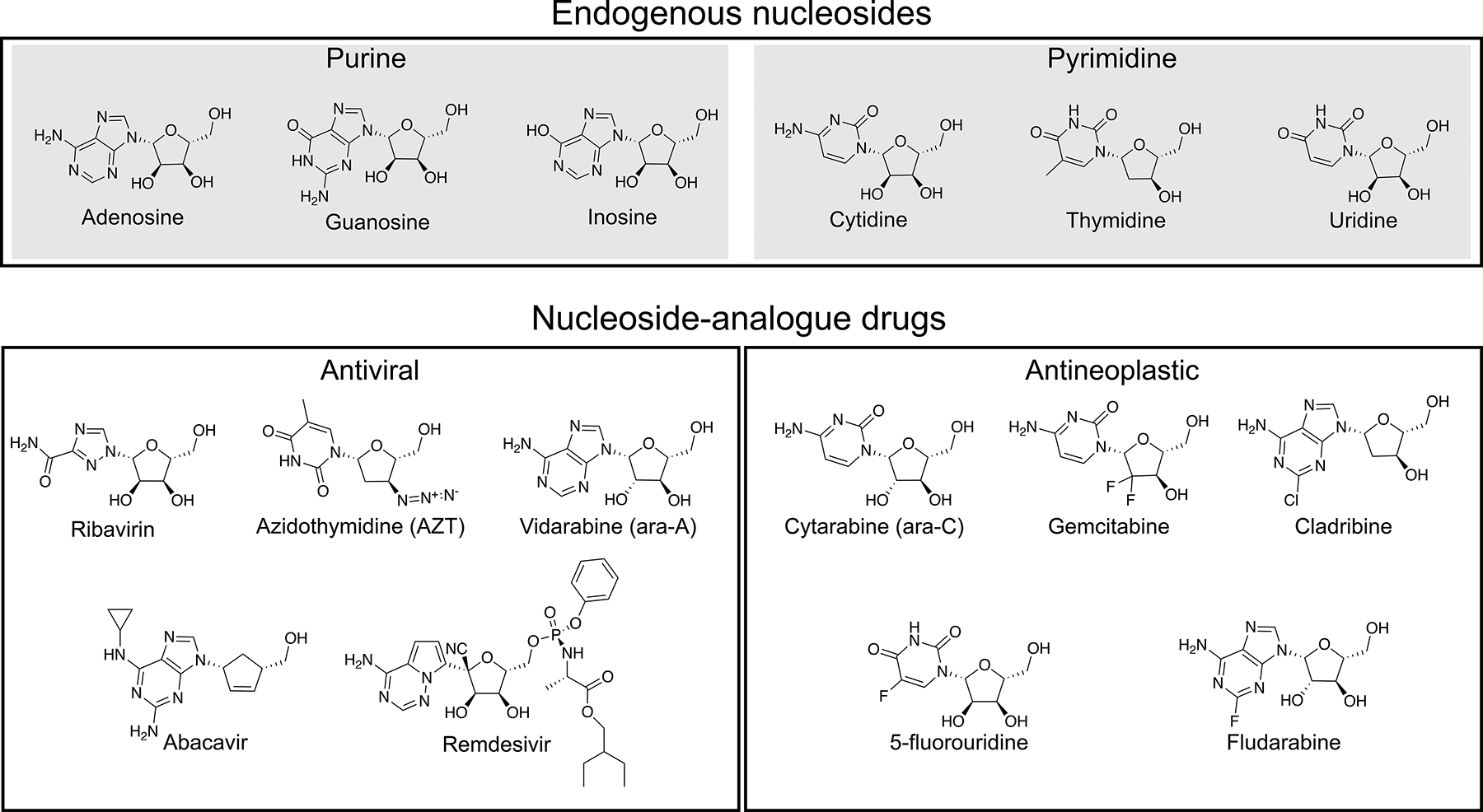
Select endogenous nucleosides (top) and nucleoside/nucleotide antiviral analog drugs (bottom).
1.2. Cellular nucleoside transport systems
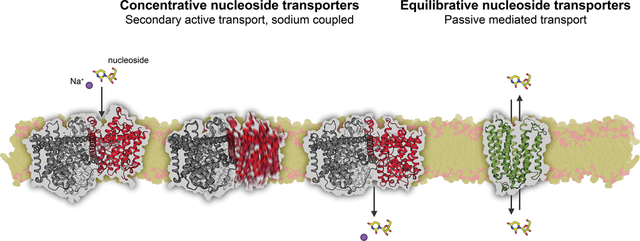
In humans, nucleoside and nucleoside-analog drug cellular uptake and excretion is mediated by two genetically distinct protein families: sodium-dependent concentrative nucleoside transporters (CNTs, gene family solute-carrier 28; SLC28) and sodium-independent equilibrative nucleoside transporters (ENTs, gene family solute-carrier 29; SLC29)4. While ENTs are in most cases facilitative uniporters, CNTs are secondary active symporters that transport nucleoside against its concentration gradient by thermodynamic coupling with electrochemical gradient of cation. The human CNT family consists of three isoforms (hCNT1-3) and the human ENT family consists of four isoforms (hENT1-4), with each isoform exhibiting varying tissue distributions and functional properties (to be discussed later in this review, also reviewed in4).
Human evolution has taken advantage of the distinctive functional features of these two transport protein families, with many tissue types exhibiting strategic spatial expression of CNTs and ENTs. For example, in intestine epithelia CNTs are expressed at apical membranes, acting as a “pump” to drive the uptake of solute from lumen to tissue, while ENTs are expressed at basolateral membranes to allow for trans-tissue diffusion of solute33. Such a system drives efficient vectorial transport of nucleosides and nucleoside-analog drugs from lumen to blood, controlling nucleoside metabolite and nucleoside-analog drug intestinal absorption. Therefore nucleoside transporters are key determinants of drug oral bioavailability in many cases34 (also reviewed in35).
1.3. Nucleoside transporters in efficacy and disposition of nucleoside-analog therapeutics
Nucleoside transporters mediate cellular uptake and efflux of numerous nucleoside-analog anticancer and antiviral therapeutics, making them key regulators of drug ADME profiles36, 37. The anticancer agent gemcitabine is arguably the most extensively established case of transporter involvement in drug disposition properties. Numerous studies report expression levels and polymorphisms in certain CNTs and ENTs are valid markers to predict cellular efficacy, cellular cytotoxicity, along with patient outcomes to gemcitabine treatment in cancers38–50. Additionally, similar involvement of nucleoside transporters have been reported for the anticancer agents cytarabine (Ara-C)50, decitabine and azacitidine51, and for the antiviral nucleoside-analog drug ribavirin52–57. For example, multiple studies have shown that human ENT1 is the major transporter for the antiviral ribavirin and single nucleotide polymorphisms (SNPs) in human ENT1 affect ribavirin treatment response against chronic hepatitis C viral infection significantly53, 54, 57. ENTs and CNTs have also been shown to transport the antiviral drug azidothymidine (AZT)58–60.
It was thought that other nucleoside-analog antiviral prodrugs, such as remdesivir, are hydrophobic and independently membrane permeable, and they are hydrolyzed to directly form an active monophosphate intracellularly27. However, animal pharmacokinetics studies on remdesivir, sofosbuvir and a N4-hydroxycytidine prodrug have shown that these prodrugs are metabolized into nucleoside form in the blood after systemic or oral administration61–65, suggesting that nucleoside transporters likely play important roles in their target availability, systemic distribution and drug excretion/clearance. It has yet to be established what roles nucleoside transporters would play in the drug ADME of these prodrug antiviral therapeutics, which would significantly impact development of improved nucleoside-analog antivirals against COVID-19.
1.4. Nucleoside transporter inhibitors
As nucleoside transport processes have numerous physiological implications, inhibitors targeting specific CNTs or ENTs hold high therapeutic potential. Due to the wide tissue distributions of human ENTs and their major role in adenosine transport, ENT inhibitors block adenosine reuptake (adenosine reuptake inhibitors, termed AdoRIs) and thus significantly potentiate purine signaling. As adenosine-GPCR agonists exhibit undesired side-effects due to poor tissue selectivity15, 66, prolonged adenosine signaling achieved through its cellular reuptake inhibition would, in theory, have preferential effects on tissue microenvironments experiencing heightened extracellular adenosine accumulation. For these reasons interest has been shown in use of AdoRIs as therapeutic interventions in adenosine biology, for treatment of neurological disorders, hypertension, heart disease and renal disorders, and several of which are currently used in the clinic13, 67–69. AdoRIs targeting human ENTs are broad and chemically diverse, which we will discuss in detail in a later section of this review. Although CNT inhibitors have potential to be used for therapeutic intervention, only a class of sub-micromolar inhibitors against human CNTs have been reported to date70. Development of CNT and ENT isoform-specific inhibitors would greatly aid in the functional dissection of nucleoside transport, allowing for further insight into this cellular transport system in the context of human physiology and nucleoside-analog drug pharmacology.
2. Concentrative nucleoside transporters
2.1. Functional properties
Early studies determined cellular nucleoside transport consisted of multiple components, with each component identifiable by three general functional properties: transport dependence on extracellular sodium, transport sensitivity to S-(4-nitrobenzyl)-6-thioinosine (NBMPR), and permeant specificities1. Sodium-independent nucleoside transport components were later determined to be mediated by the ENT family (to be expanded on later in this review), whereas the sodium-dependent components were later discovered to be mediated by the CNT family. Early nomenclature of the CNT family stratified the three sodium-dependent components of cellular nucleoside transport by permeant specificity: concentrative/NBMPR-insensitive/transports-thymidine (cit), concentrative/NBMPR-insensitive/transports-formycin (cif) and concentrative/NBMPR-insensitivie/transports-both-purines-and-pyrimidines (cib)4.
2.1.1. Isoform features and transporter regulation
In a series of papers by Young and colleagues, the molecular identification of individual members of the human CNT family yielded three distinct isoforms with apparent pairwise sequence identities in the range of 40–60%, with each isoform exhibiting an apparent 13 transmembrane domain (TM) topology based on sequence hydropathy analysis59, 71, 72. These studies unambiguously assigned the previously designated cit, cif and cib cellular nucleoside transport components to human CNT isoforms 1–3, respectively, and also showed the three CNT isoforms exhibit diverse tissue distributions and high tissue abundance in epithelia59, 71, 72 (reviewed further in4).
A more detailed understanding of permeant specificities through human CNTs was advanced by the initial molecular identification of individual human CNT isoforms and subsequent work. It is now well established while all human CNTs transport uridine, human CNT1 preferentially transports pyrimidines, human CNT2 transports purines, and human CNT3 exhibits broad nucleoside selectivity transporting both purines and pyrimidines. It is not clear if human CNTs transport nucleobases, as transport of nucleobase by any CNT has yet to be reported. Later transport studies suggested the importance of ribose hydroxyl groups for nucleoside recognition and transport by all human CNT isoforms73. Transporter interactions with other moieties of solute were determined to be different between isoforms, providing an initial molecular basis for solute preference exhibited by different CNT isoforms73. Another feature of solute specificity exhibited by all three human CNT isoforms is the strong preference for North ribose ring (3’-endo) conformations, determined by transport studies conducted with nucleoside analogs featuring sterically constrained ribose moieties74.
Concerning cation-cotransport properties, human CNT1 and CNT2 exhibit a strict 1:1 sodium: nucleoside coupling stoichiometry58, 75, however human CNT3 features more complex coupling criteria. It has been shown that human CNT3 can cotransport with 2:1 sodium: nucleoside stoichiometry but proton can be utilized for transport in the absence of sodium with 1:1 proton: nucleoside stoichiometry76. Proton-coupling is also observed in some fungal and bacterial concentrative nucleoside transporters that are sodium-independent, such as Candida albicans CNT (CaCNT) and Escherichia coli NupC77, 78. These differences in solute specificities and substrate coupling ratios present between each human CNT isoform may allow for fine-tuning of concentrative cellular nucleoside uptake in response to specific physiological environments.
Regulation of human CNT expression also likely serves to control cellular nucleoside uptake. It has been reported glucocorticoid treatment leads to increased CNT expression in rat intestinal epithelia79, and starvation or nucleotide-deficient diet leads to increased CNT1 expression in rat jejunum80, leading Pastor-Anglada et. al. to suggest maturation of epithelial cells and nutritional regulation as two possible factors regulating CNT expression81. Furthermore, human CNT3 expression in HL-60 cells is stimulated by phorbol myristate acetate (PMA), and does contain an upstream PMA response element71, demonstrating transcriptional control of a human CNT.
2.2. Structural advancements
As stated above, the first molecular cloning of individual human CNT isoforms predicted a 13-TM transporter membrane topology based on sequence, however following accessibility studies suggested mammalian CNTs contained 13 or 15-TM82–84, with an intracellular N-terminus and extracellular C-terminal. Work on rat CNT1 determined N-linked glycosylation sites are located within the extracellular C-terminal domain82. It was originally postulated the first 3-TM were dispensable, as bacterial CNT homologs lack these putative N-terminal TMs. Subsequent work confirmed this hypothesis, as truncations of the first putative 3-TM of human and rat CNT1 resulted in wild-type like transporter function, while providing insight into the overall topology of the transporter82. It is important to note while the CNT N-terminal domain is functionally dispensable, it likely plays important roles in mediating protein-protein interactions, controlling transporter trafficking and may contribute to transporter stability85, 86. Although these initial studies were important advancements to the field of nucleoside research, it remained a challenge to accurately predict the transporter architecture. To gain a detailed understanding of the molecular requirements for nucleoside recognition, ion-coupling and the mechanism of nucleoside membrane translocation exhibited by CNTs, experimental transporter structures were in dire need.
2.2.1. Crystal structure of a sodium-dependent nucleoside transporter from Vibrio cholerae
A 2.4-Å resolution crystal structure of a sodium-dependent CNT from Vibrio cholerae (vcCNT) was reported by Johnson et. al., which marked the first structure determination of any nucleoside transporter87. Being that vcCNT exhibits an overall 39% sequence identity to human CNT3, with higher conservation apparent in the nucleoside/sodium sites (~91% seq. identity), this study presented structural data with high relevance to human CNTs.
2.2.1.1. Novel Transporter architecture and the sodium binding site of vcCNT
vcCNT forms a homotrimer, with each protomer comprised of 8 TM (TM1-TM8), 3 interfacial helices (IH1-IH3), 2 re-entrant helix-turn-helix hairpins (HP1 & HP2) and short extracellular helices (EH) (Figure 2A). The CNT protein fold is novel and can be divided into two structural domains: the scaffold domain and transport domain. Comprised of TM1-3, IH1, EH and TM6, the scaffold domain is located at the exterior of a CNT protomer, appearing to contribute to structural integrity of the homotrimer as it forms extensive oligomeric contacts (Figure 2B). Located inside the scaffold domain are two different structural elements that comprise the transport domain - TM4, TM5, IH2 and HP1 form a single group that exhibits apparent internal two-fold symmetry with a structural element formed by TM7, TM8, IH3 and HP2. This observation of apparent internal symmetry, in addition to the presence of conserved positions previously implicated in human CNT function, led to the designation of these structural elements as the transport domain. The high-resolution crystal structure of vcCNT also unambiguously resolved the atomic coordinates of uridine and sodium in complex with the transporter, revealing the CNT substrate binding and ion coordination sites within the transport domain. Located between HP1, HP2, TM4 and TM7 of the transport domain and facing the trimer axis, uridine forms multiple interactions with transporter, many of which were previously reported by functional studies of human CNTs83, 88–90 (Figure 2B). Approximately 8 Å from substrate nucleoside is an octahedrally coordinated sodium ion, which was designated as the sodium binding site in vcCNT based on its coordination distances and geometry (Figure 2B). Because subsequent higher-resolution (2.1-Å resolution) structure of vcCNT in complex with uridine did not reveal any additional sodium site91, we suggest that vcCNT adopts 1:1 sodium: nucleoside stoichiometry. The domain architecture of vcCNT is vaguely similar to that of the bacterial excitatory amino acid transporter homolog GltPh92–96, which also features distinct transport and scaffold domains. Further, the GltPh transport domain contains two helix-turn-helix hairpins related by internal two-fold symmetry, similar to vcCNT. However, several stark structural differences are apparent, including highly distinct locations of coupled ions and different scaffold domain architectures.
Figure 2.
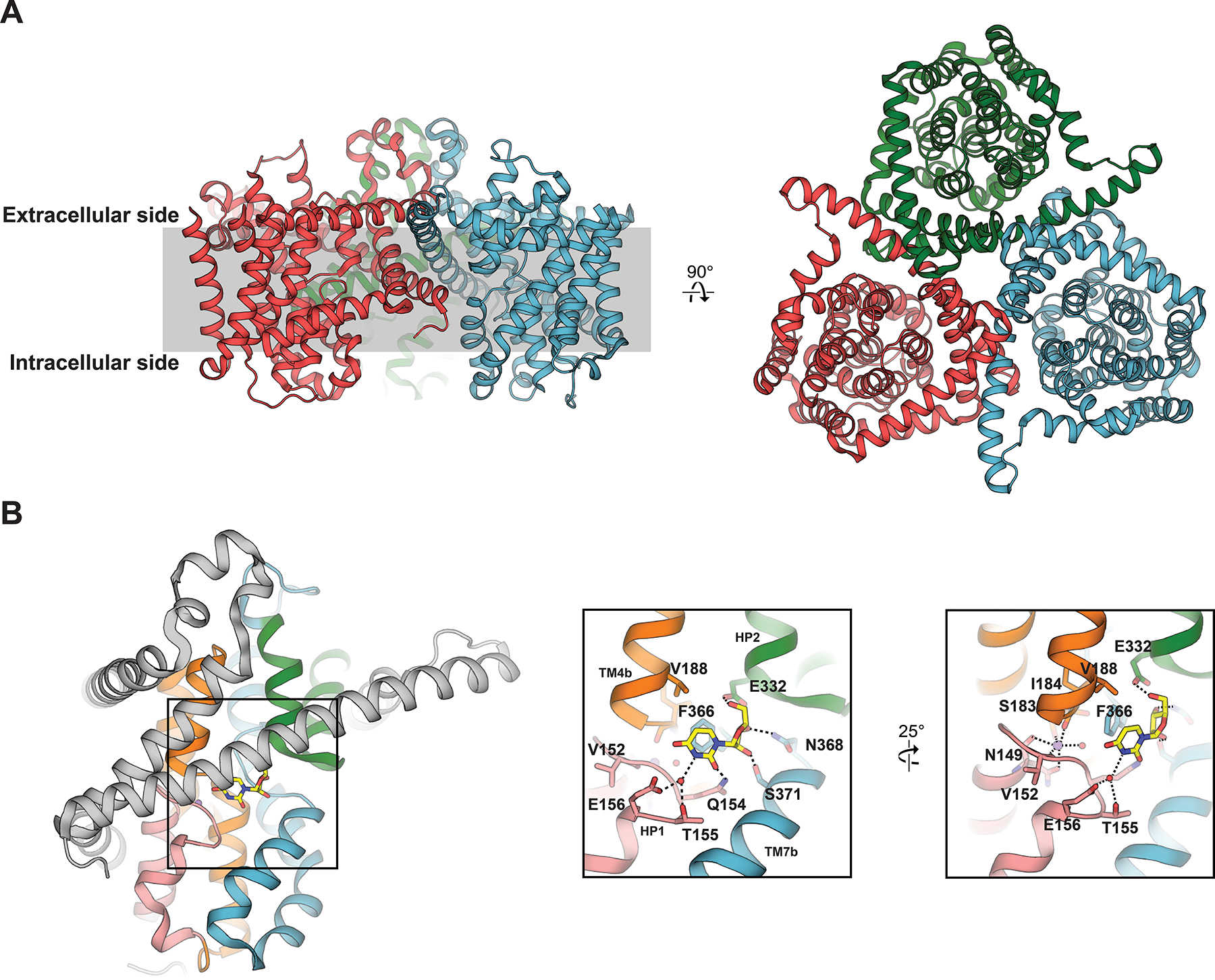
A) Overview of the vcCNT trimeric structure (PDB ID 3TIJ). B) Detailed interactions of substrate nucleoside (yellow sticks) and coordinated coupled sodium ion (purple sphere) with vcCNT. Scaffold domain depicted in grey, HP1 in salmon, HP2 in green, TM4/5 in orange and TM7/8 in blue.
2.2.1.2. Mechanistic implications
The vcCNT structure adopts a conformation in which the substrate uridine and sodium are facing the intracellular side of the membrane. Interaction of TM3 and TM6 of the scaffold domain with TM4 and HP2 of the transport domain form a thick seal, completely preventing sodium and uridine access to the extracellular side. Although the sodium and uridine sites are solvent accessible to the intracellular side, HP1 appears it would sterically prevent sodium and uridine release, representing an inward-facing occluded state in the context of the alternating access model of membrane transport97, 98.
A finding from this structural study with clear mechanistic implication on sodium-dependent nucleoside transport: the sodium ion did not directly interact with substrate uridine, which is distinct from the sodium-dependent amino acid transporter LeuT. In LeuT, a sodium ion is coordinated by the carboxylate moiety of leucine in LeuT, suggesting simultaneous binding of sodium ion and leucine by the transporter99. Johnson et. al. proposed that in vcCNT sodium serves to promote nucleoside binding – as residues interacting with uracil base are located on HP1, stabilization of HP1 by sodium would elicit or stabilize a conformation posed to productively bind nucleoside. The proposed role of sodium in stabilizing the nucleoside binding site was consistent with the follow up equilibrium binding studies that mutation of sodium coordinating residues result in decreased affinity for nucleoside100. These observations, in combination with structural studies of the bacterial excitatory amino acid transporter homolog GltPh92–96, led Johnson et. al. to propose a transport mechanism for CNT. In order to transition to additional conformational states, the transport domain would likely rearrange relative to the scaffold domain, which would allow nucleoside and sodium sites alternating access to each side of the membrane (discussed in detail in Section 2.2.3). The proposed role of sodium in the affinity control of nucleoside binding by vcCNT, together with the alternate access model, provides an initial glimpse into the sodium-dependent transport mechanism by vcCNT. When CNT is in an outward facing conformation where the substrate binding sites in the transport domain faces the extracellular side, the high concentration of sodium in the extracellular solution will increase sodium occupancy in the transporter, which in turn increases nucleoside binding. On the contrary, when CNT is in an inward facing conformation, the low concentration of sodium in the intercellular side lowers sodium occupancy of the transporter, which in turn decreases nucleoside binding, driving nucleoside release. This proposed role of affinity control by sodium is in line with the reported kinetic studies on hCNTs which revealed sodium binds to the transporter before nucleoside58. Our proposed role of sodium in controlling affinity for substrate could be widely extended to other types of sodium-coupled transporters, as not all LeuT fold transporters feature a coupled-sodium directly interacting with substrate101, 102.
2.2.2. Structural basis of substrate specificity
As vcCNT was determined to be a suitable tool to structural interrogate sodium-dependent nucleoside transport87, various nucleosides and nucleoside analogs were later assessed for binding to vcCNT in parallel with co-crystal structure determination (Figure 3A), in order to interrogate general principles of nucleoside recognition and transport by CNTs91.
Figure 3.
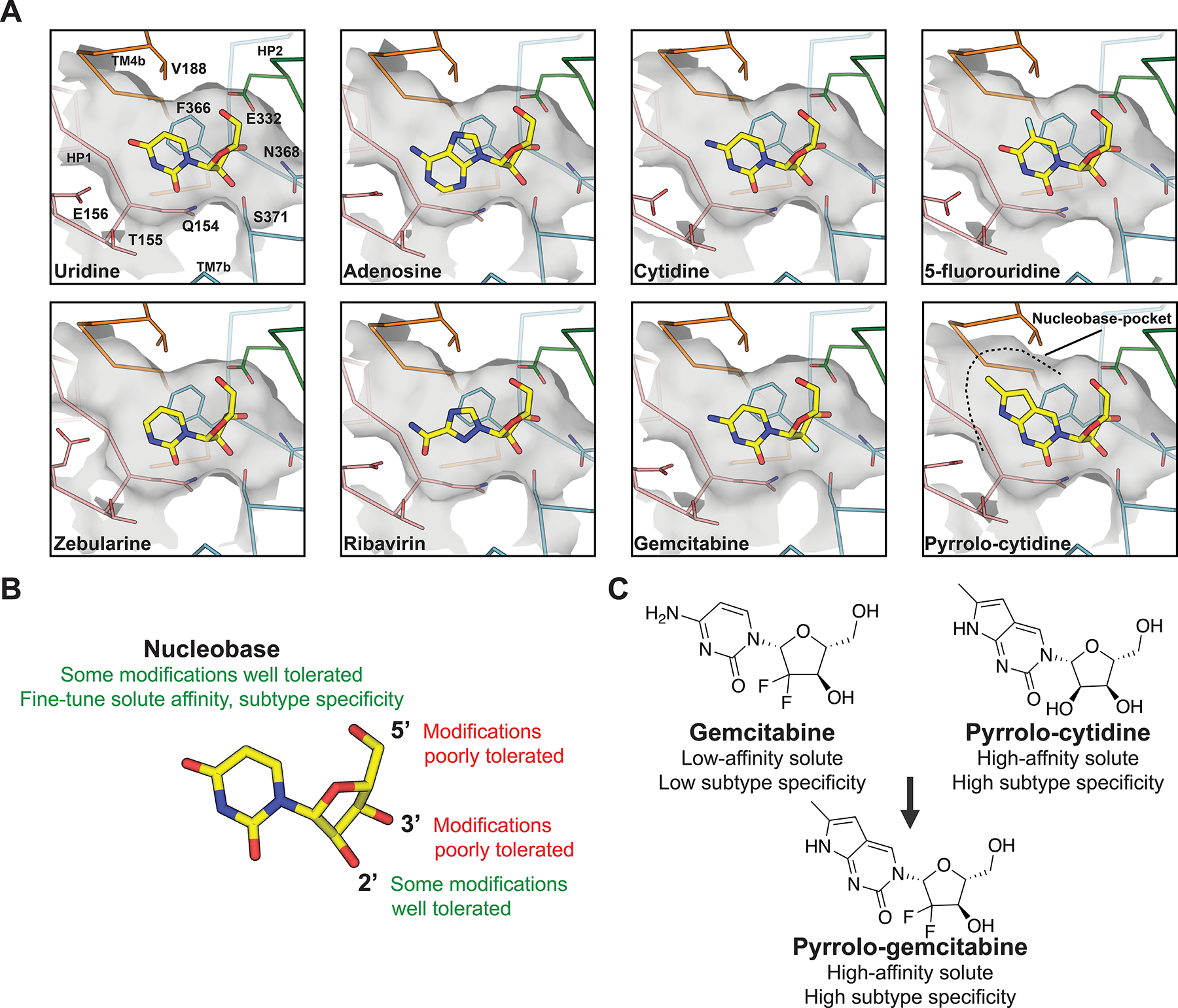
A) Detailed interactions of nucleosides and nucleoside analog drugs with vcCNT (PDB IDs, top left to bottom right: 4PD6, 4PD9, 4PDA, 4PB2, 4PD7, 4PB1, 4PD5, 4PD8). B) Summary of chemical determinants of solute recognition by vcCNT C) Rational chemical modifications that resulted in a chemotherapeutic drug with enhanced transport properties.
2.2.2.1. Nucleobase interactions
Compared to uridine nucleoside, substitutions at the N3 and C4 positions of the uracil base were found to decrease nucleoside affinities. Furthermore, adenosine and guanosine-analog ribavirin also exhibited lower affinities to transporter. This is explained by the crystallographic data, in which uracil base makes important interactions with Q154, T155 and E156 of HP1, many of which are disrupted in the cocrystal structures of nucleoside analogs featuring said substitutions (Figure 3A). However, uridine-analogs featuring substituents on the C5 position of the uracil base exhibited differential affinities, where marked decreases in nucleoside affinities correlate with increasing atomic radii and decreasing in electronegativity of the C5 substituent. The atomic radii trend is an unlikely explanation for this effect, as crystallographic data shows C5-substituents of varying sizes could be accommodated in the nucleoside binding site, therefore substituent electronegativity is the driving factor. Located ~3.5Å from the uracil base is the highly conserved F366 of TM7, which is required for nucleoside binding. This position participates in offset pi-pi stacking interaction with nucleobase, along with CH-pi interactions with ribose. Electron withdrawing groups at the C5 position of the uracil ring would strengthen the offset the pi-pi stacking interaction, which is reflected by the increased affinities of 5-flourouridine and 5-chlorouridine for vcCNT (KD~14–16 μM) compared to that of uridine (KD ~36 μM).
2.2.2.2. Ribose interactions
As stated previously in this review, early functional studies highlighted the importance of the hydroxyl groups of ribose for nucleoside recognition by CNT73. Crystallographic data on vcCNT supports this87, 91, as conserved residues N368 of TM7, and E332 of HP2, are in close proximity and interact with the 3’-OH of nucleoside. Additionally, S371 of TM7 and E332 of HP2 form interactions with 2’ and 5’-OH groups, respectively. Consistent with previous functional studies and the crystallographic data, 3’- and 5’- deoxyuridine analogs exhibit large decreases in binding affinities to transporter, whereas a 2’-deoxyuridine analog did not exhibit appreciable binding loss. However, other perturbations at the epimeric 2’ position opposite to -OH (such as in the anticancer drugs gemcitabine and cytarabine) result in more dramatic decreases in nucleoside binding affinities – these observations are explained by the crystallographic data, as substituents at this position disrupt the CH-pi interaction between ribose and residue F366 (Figures 2B and 3A).
2.2.2.3. Molecular features of nucleoside recognition by CNT are exploitable for rational drug modification
Since many anticancer and antiviral agents feature substituents on the ribose moiety of nucleoside, they exhibit low-affinity transport through human CNTs due to the strict requirements for ribose recognition by CNTs. Pyrrolo-cytidine (MePrPmR) is a human CNT inhibitor70 that also exhibits high-affinity binding to vcCNT. A co-crystal structure revealed the base of the pyrrolo group of pyrrolo-cytidine uniquely occupies a pocket within vcCNT formed by TM4 and TM6, which may explain the high-affinity exhibited by this nucleoside analog (Figure 3A). Johnson et. al replaced the cytidine ring of the anticancer drug gemcitabine with the nucleobase of pyrrolo-cytidine to compensate for the low affinity of gemcitabine to human CNTs (Figure 3C). The new nucleoside-analog, pyrrolo-gemcitabine, exhibited a higher affinity to the bacterial CNT compared to gemcitabine. Interestingly, pyrrolo-gemcitabine exhibited subtype-selective transport for human CNT1 - whereas human CNT3 was inhibited by the nucleoside analog, transport by hCNT1 was enhanced compared to that of gemcitabine. Modelling suggests the pocket of hCNT3 is similar to vcCNT so that it is snuggly fit with the pyrrolo-cytidine base, but in human CNT1 the pocket is not compatible with pyrrolo-cytidine binding91. Therefore, increase in affinity by occupying this pocket in human CNT3 is likely the basis for apparent transport inhibition rather than solute permeation. These studies strongly suggest that by modifying nucleobase, the affinity of nucleoside analog drug for CNTs can be tuned to maximize its transportability and isoform specificity by CNTs.
Taken together, the structural and functional data presented on vcCNT by Johnson et. al. reveal that nucleoside recognition by CNTs is mediated by both nucleobase and ribose specific interactions: HP1 and TM4 serve to interact with nucleobase, whereas HP2 and TM7 form interactions with ribose. The residue F366 is central to nucleoside recognition by the transporter, as it forms pi-pi stacking interactions with nucleobase in addition to CH-pi interactions with ribose. Structural features of nucleoside recognition by vcCNT were found to be exploitable for modelling of human CNTs, as rational modification of gemcitabine not only resulted in a nucleoside-analog with higher apparent affinity to the bacterial transporter, but also yielded a permeant with high subtype specificity for transport by certain human CNT isoforms. This proves that it is possible to use structural data in the rational modification of drugs, so they exhibit more desirable transport properties through human nucleoside transporters.
2.2.3. Multiple conformations elucidate novel insights into the elevator transport mechanism
In its simplest form, the generalized mechanism of membrane transport involves solute access to either side of the cell membrane in alternating fashion97, 98, 103. In such a model, transporter gates are elements which serve to fully occlude solute within the transporter substrate cavity, preceding protein conformational transitions between inward and outward facing end states. In the case of ion-coupled cotransporters, solute occlusion by orchestrated transporter gate openings and closures are also necessary to prevent non-specific ion leak. More specific mechanisms of membrane transport are categorized by structural features of conformational change between end states and include the rocking-bundle, rocker-switch and elevator models103. The elevator mechanism is an emerging model where the substrate-binding transport domain moves a large distance across the membrane while the scaffold domain remains static in the membrane103. As the CNT protein fold is comprised of a scaffold domain that mediates oligomeric contacts, and a transport domain that largely contains the substrate binding site, it was initially predicted to utilize an elevator-mechanism for membrane transport87. Many initial studies concerning the elevator mechanism were from the glutamate transporter homolog GltPh92–96. The elevator model was inferred as a single rigid-body transition of the transport domain, mainly because structures were available for two end states103. However, biophysical studies suggested that intermediate states exist104, 105. This has posed interesting questions for CNT transport mechanism. Does the elevator motion occur in a single concerted step or multiple steps? How do interactions between the transport and scaffold domain change while preventing non-specific ion leak during the elevator motion? Does the transport domain move as a rigid body, or does it undergo conformational changes during the elevator motion?
To interrogate the transport mechanism exhibited by CNT, structural elucidation of multiple conformations along the transport cycle are required. As vcCNT was refractory to crystallization in conformations other than the substrate-bound inward-facing occluded state, Hirschi et. al. identified a sodium-dependent bacterial nucleoside transporter from Neisseria wadsworthii (CNTNW) that exhibited high biochemical stability in the absence of nucleoside and sodium100. In addition to obtaining crystal structures of inward-facing occluded CNTNW in complex with sodium and uridine, multiple conformational states were captured with crystallography by utilizing mutations that abrogated sodium and nucleoside binding (CNTNWN149S,F336A, CNTNWN149S,E332A and CNTNWN149L). These structural studies, combined with biochemical studies, provide structural and mechanistic insights into the conformational pathway of the elevator transition of CNT and provides novel insights into the elevator mechanism.
2.2.3.1. Outward-facing and intermediate states of CNTNW
The crystal structures of both CNTNWN149S,F336A and CNTNWN149S,E332A were determined in the absence of nucleoside and sodium100. These structures feature two protomers (A and B) in similar conformations to the previously observed inward-facing occluded state (nucleoside substrate bound). However, the third protomer (C) adopts a distinct conformation. Crystal packing analyses shows that the transport domains of the protomers A and B are involved in packing interactions while that of the protomer C is unhindered for movement. While TM3 and TM6 of the scaffold domain in protomer C exhibited no significant differences when compared to the inward-facing state, the entire transport domain is displaced substantially towards the extracellular side of the membrane in a rigid-body manner. This conformational change results in a ~12 Å displacement of the nucleoside binding site across TM6 towards the extracellular environment, allowing for solvent access of extracellular solution to the nucleoside-binding site (Figure 4). The interface between the scaffold domain (TM3 and TM6) and components of the transport domain (HP1 and TM7) seal off access to the intracellular side, therefore Hirschi et. al. proposed this observed conformation represents the outward-facing state. Intriguingly, by utilizing the CNTNWN149L construct, additional conformers of protomer C were observed with crystallography, all of which feature the transport domain in a position somewhere between inward and outward facing states, thus representing intermediate transporter states. Moreover, X-ray data collected from different CNTNWN149L crystals revealed three distinct intermediate conformations – intermediate-1, intermediate-2 and intermediate-3, named based on their position in the conformational path from inward to outward facing states (Figure 4). In all three of these intermediate states, the nucleoside binding site is located directly behind TM6 of the scaffold domain and fully sealed from both extracellular and intracellular solutions. Adding biochemical relevance to these structural observations, Hirschi et. al. performed structure-guided double cysteine crosslinking to show the observed inward, intermediate and outward-facing transporter conformers exist in membranes and proteoliposomes. Importantly, while cross-linked mutants reconstituted in proteoliposomes are nearly inactive, when reduced by DTT, the mutants restore transport activity substantially, strongly suggesting that the intermediate and outward conformations are physiologically relevant states, not the states in off-pathways.
Figure 4.
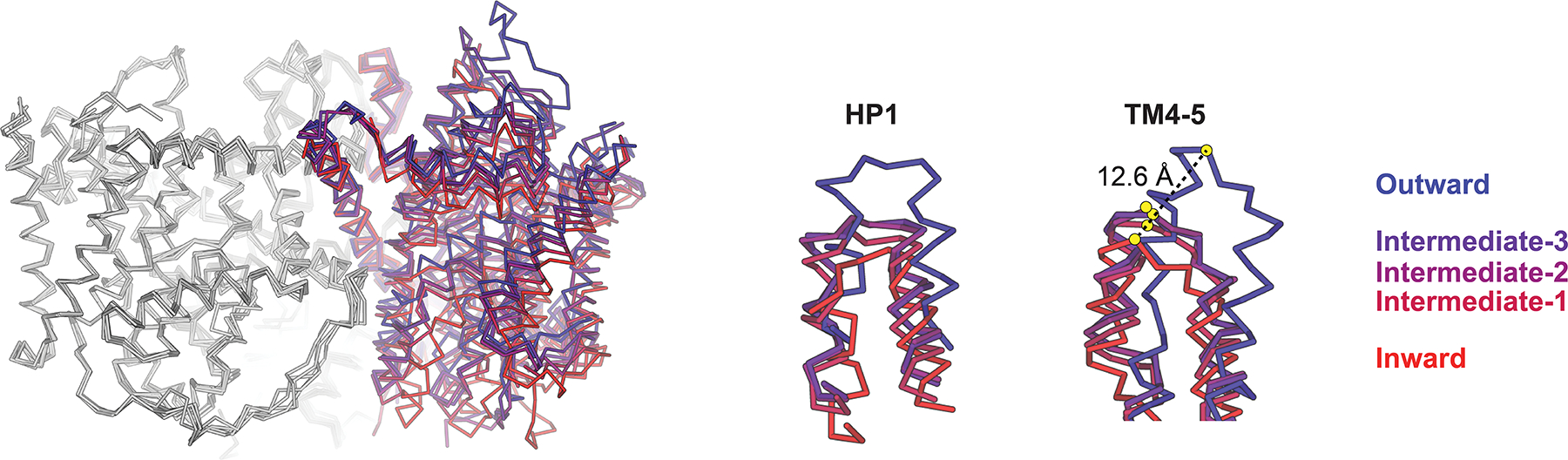
Structural superposition of crystal structures of CNTNW in different conformational states (structures aligned with reference to protomers A and B, protomers A and B colored in grey, color key for protomer C at right). Large displacements in HP1 and TM4-5 due to conformational transitions are highlighted at right (PDB IDs: 5L26, 5L27, 5L24, 5U9W, 5L2A for inward, intermediate-1, intermediate-2, intermediate-3 and outward, respectively).
2.2.3.2. Conformational changes within the transport domain
In addition to the large rigid body like movements of the transport domain, local conformational rearrangements of HP1 and TM4 underlie the transition from inward to outward facing end states of CNTNW. Our structural snapshots represent the structural transition from the inward-occluded (substrate-bound) to outward-open state. Notably, the nucleoside binding site within the transport domain in these intermediate states are collapsed and thus cannot accommodate nucleosides – however this nucleoside binding pocket is restored in both inward and outward states. These structures illustrate that in the return path of substrate-free CNT in the transport cycle, the transporter domain appears to undergo a conformational change to a “collapsed” state during transport, which could be necessary prevent transport of unwanted molecules (e.g. nucleoside or ions or some non-specific small molecules).
HP1 appears to function as the intracellular gate, as it interacts with substrate nucleoside in the inward-facing occluded state in both vcCNT and CNTNW structures. In CNTNWN149S,F336A and CNTNWN149L structures, HP1 of protomers A and B adopt a variety of conformations in which HP1 is moved away from the central nucleoside binding site to varying degrees. This would allow productive nucleoside release, thus representing inward-facing open states. Additionally, Hirschi et. al. noted the presence of an additionally conformation in which HP1 is partially unwound and “tucked in” towards the center of the nucleoside binding site. This represents an inward-facing pre-translocation state, a rearrangement of HP1 required to transition from inward-apo to intermediate-apo transporter states. Such a state prevents steric clash of HP1 with the scaffold domain during such transitions. This is clear in protomer C of intermediate CNTNW structures – HP1 and TM4 move closer together during these conformational transitions, making the transport domain more compact. In the outward-facing transporter state, TM4 is displaced upward substantially allowing the nucleoside binding site solvent accessibility to the extracellular solution (Figure 5). Therefore, TM4 likely operates as the extracellular gate. This “fixed barrier elevator with two gates” mechanism of transport exhibited by CNT is distinguished from other transporters that utilize elevator mechanisms106, and has yet to be observed in other transporter families.
Figure 5.

Conformational changes in the transport domain mediates collapse of the nucleoside binding pocket during the elevator-like transitions. Surface representation of each transporter conformer shown, with the nucleoside pocket circled in red (PDB IDs: 5L26, 5L27, 5L24, 5U9W, 5L2A for inward, intermediate-1, intermediate-2, intermediate-3 and outward, respectively. Protomer C depicted for each conformer, except for inward pre-translocation which is depicted from chain A of PDB ID 5U9W).
In summary, structures of CNTNW in the inward occluded (substrate-bound), inward open, inward-pre-translocation, intermediate, and outward open states reveal the conformational trajectory from inward substrate-bound to outward open states. Notably the new intermediate states with the transport domain halfway across the membrane provides that elevator motion by CNTNW is composed of multiple steps including the intermediate steps. Possible reasons for the existence of these intermediate states are : 1) to provide an energetic barrier between two end states, as without the barrier switching between the end states would lead to sodium leakage, an idea suggested by smFRET studies of GltPh104, 105, and 2) to guide the path of large-scale motions of the transport domain, to prevent the non-specific leakage through the interface between the scaffold and the transport domains. Consistent with this idea, mutation of a glutamate residue on the domain interface in the intermediate state leads to nucleoside-uncoupled sodium leak currents in human CNT1 and CNT389, 90. It is remained to be seen whether these intermediate states exist in other transporters that exploit elevator transport mechanisms.
2.2.4. Structural mechanism of CNT mediated nucleoside transport
Taken together, structural studies performed on bacterial CNTs to date have uncovered important molecular principles of the mechanism of sodium-dependent cellular nucleoside transport. A key feature of sodium coupling in CNT is the fact there is no direct interaction between cation and solute. Observations of varying structural states highlight the likely role of sodium in affinity control of solute nucleoside – occupancy of sodium on an exofacial transporter site would enhance nucleoside affinity, whereas sodium release from an endofacial transporter site reduces nucleoside affinity, thus promoting release of solute into the intracellular environment. Indeed, in sodium-free conditions CNTNW exhibits a wide range of HP1 conformations in protomers which adopt the inward-facing state (Figure 6), providing direct structural evidence for sodium release driving HP1 opening, followed by nucleoside release to the intracellular environment.
Figure 6.
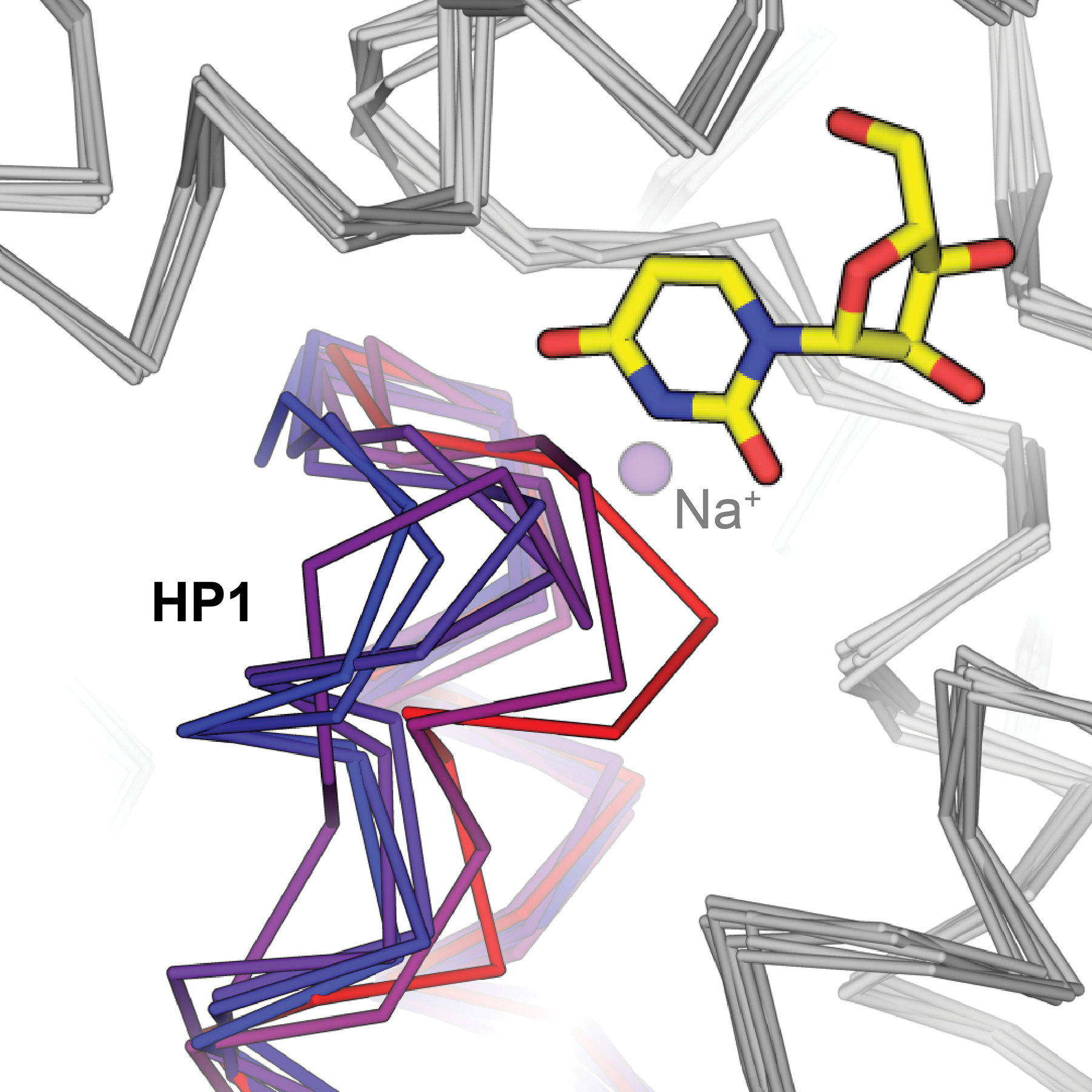
HP1 of CNTNW adopts a variety of conformations in the absence of sodium. Inward-facing sodium- and substrate-bound CNTNW HP1 depicted in red, with substrate uridine shown as sticks and sodium as a purple sphere (PDB ID 5L26). HP1 conformers from protomers A and/or B from sodium free conditions shown in shades of violet to blue (PDB IDs 5L27, 5L24, 5UW9, 5L2A).
Intermediate states of the transporter on its return path from solute release to the intracellular solution back to outward-facing conformations showed the nucleoside binding pocket collapses, forming tight seals between the transport domain and scaffold domain. We propose this substrate-free return path to an outward facing transporter state serves to prevent hysteresis and non-specific transport events. Therefore, our transport mechanism illustrates the conformational dynamics of the CNT transport mechanism: starting from inward facing occluded states (solutes bound), to solute sodium and nucleoside release, and then back to the outward facing state posed for sodium and nucleoside recognition through compact intermediate states (Figure 7). The mechanism of CNT from outward-facing substrate bound to inward-facing occluded is currently not known. Additionally, it remains to be seen if the elevator mechanism of CNT is applicable to other transporters that exhibit elevator motions associated with transport.
Figure 7.
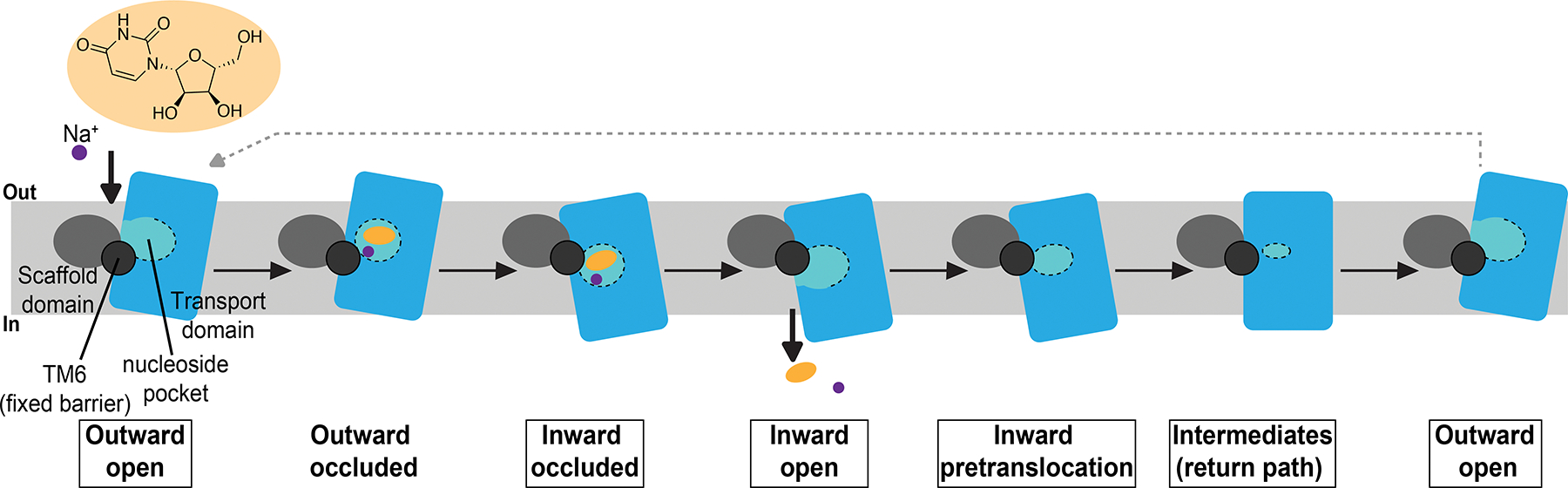
Current working model for the mechanism of nucleoside transport mediated by CNTs. Structural states in which experimental structures are available are boxed. Scaffold domain depicted in grey, transport domain in blue. The nucleoside binding pocket (light blue) is demarcated with dotted line.
3. Equilibrative nucleoside transporters
3.1. Functional properties
Use of the mercaptopurine NBMPR has allowed for numerous functional and genetic advancements in the nucleoside transporter field, which includes the identification, cloning and initial characterization of individual human ENT isoforms107–115. The ENT family was primarily designated as equilibrium/NBMPR-sensitive (es) and equilibrium/NBMPR-insensitive (ei) components of sodium-independent cellular nucleoside transport4, 115.
3.1.1. Isoform features and transporter regulation
Human ENT1 was the first sodium-independent nucleoside transporter to be cloned109. Responsible for es nucleoside transport, human ENT1 is potently inhibited by NBMPR with the half maximal inhibitory concentration (IC50) in the sub- to low-nanomolar range109, 115. Human ENT2 was cloned next and determined to be the major contributor to ei sodium-independent nucleoside transport, as it is far less sensitive to NBMPR (micromolar or weaker inhibition)116. Human ENT3 was later identified as a primarily organellar transporter which exhibited virtually no sensitivity to NBMPR117, 118. Human ENT4 was the final human isoform to be identified and cloned – this ENT isoform is also completely insensitive to NBMPR119–121.
All human ENT isoforms feature an apparent 11-TM topology with sequence identities ranging from 25 to 50%, with human ENT4 exhibiting large sequence divergence from the other human ENTs. One shared feature of solute recognition among all human ENT isoforms is the preference for South (C2’-endo) ribose ring conformation for nucleoside substrate, which contrasts the preference for North (C3’-endo) ribose conformation exhibited by CNTs74. Human ENT 1–3 exhibit broad tissue distributions and can transport both purines and pyrimidines, therefore exhibiting much broader substrate selectivity compared to CNTs. Additionally, human ENT1 and ENT2 have been shown to mediate low-affinity transport of nucleobases108, 122. The permeant specificity exhibited by human ENT4 is distinct from human ENT 1–3, as the only nucleoside it can transport is adenosine at acidic pH119. Human ENT4 is also known as the plasma membrane monoamine transporter (PMAT) as it can also permeate monoamines, such as serotonin121. Additionally, human ENT4 exhibits distinct tissue distributions compared to the other human ENT isoforms, as it was found to be enriched in heart and brain tissue119, 121.
It is well understood human ENT1 and ENT2 are facilitative nucleoside carriers, however human ENT3 and ENT4 have been shown to exhibit pH-dependent transport of nucleosides117, 119. Putative titratable residues responsible for the pH-dependence of nucleoside transport for human ENT3 and ENT4 have been reported119, 123, 124. Electrophysiological studies on human ENT3 failed to detect proton transport during adenosine transport, and thus suggested that proton is not a co-transporting ion but regulates adenosine transport in human ENT3123,, 125. Although further studies are necessary to validate the conclusion of the role of pH in hENT3, the allosteric role of pH in adenosine transport in hENT3 is intriguing. The pH sensing exhibited by human ENT3 may reflect the environment in which the transporters is posed to operate – in acidic organellar compartments117. Because acidic pH is critical for maintaining lysosomal function, it is possible that hENT3 has evolved to operate only in the acidic pH to synchronize the metabolic needs of the lysosome. Adenosine transport by human ENT4 has been reported to be pH-dependent, however monoamine transport does not involve cotransport of ions or exhibit pH-dependence119 and was found to be membrane potential dependent121. Although it has yet to be established if pH-dependent adenosine permeation requires ion co-transport, pH-dependent purine transport mediated by human ENT4 may be physiologically relevant in the context of ischemia of the brain and heart119, 126.
Being that human ENT1 is most widely expressed and well-studied, its transcriptional regulation is best understood out of all ENT isoforms. In general, human ENT1 expression is coordinated in response to cell cycle and nutrient availability (reviewed in127). In line with its important role in purinergic signaling, equilibrative nucleoside transporter expression has been reported to respond to stress. Specifically, it was determined human ENT1 expression during hypoxia is repressed by HIF-1α and the human ENT1 promoter contains two putative HIF-1α recognition sites128. Additionally, activation of the JNK-cJun pathway has been reported to negatively regulate ENT1 expression in rat129. It is important to note sequence elements for other regulatory factors are also present within the human ENT1 promoter130.
3.1.2. Genetic disease and genome-wide associations in the human ENT family
Currently, intracellularly localized human ENT3 is the only nucleoside transporter identified to be associated with human genetic disease. Rare mutations in the slc29a3 gene have been identified as the cause of recessive autosomal disorders including H-syndrome131, pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome132, Faisalabad histiocytosis, familial Rosai-Dorfman disease133 and dysosteosclerosis134. These slc29a3 associated disorders are autoinflammatory in nature, with lysosomal and mitochondrial instability as a likely driver of inflammation135. In further support of this disease etiology, human ENT3 has been shown to play roles in bone physiology136 immune function, macrophage homeostasis137 and T-cell survival138, through maintenance of organellar integrity and function. Although some mutations of slc29a3 that lead to such disorders are nonsense or frameshift139, 140, many are missense that diminish transporter stability, localization and transport activity141. Further, many of these mutations map to conserved positions of potential structural and functional importance in the recently determined human ENT1 crystal structure142 (to be discussed later in this review).
Human ENT1 is a key controller of adenosine signaling, and as adenosine is implicated in alcoholism19–24, human ENT1 has been shown to regulate ethanol preference and sensitivity143–146. A human polymorphism in slc29a1, 647T/C, is significantly associated with alcohol dependence with withdrawal seizures145. This polymorphism results in the I216T mutation on TM6 of human ENT1, and is a common variant prevalent in the human population147. A current working hypothesis suggests human ENT1 is inhibited by ethanol, and that the I216T variant alters the transporters sensitivity to ethanol145 (to be discussed later in this review). It is important to note other polymorphisms of the ENT family are prevalent in the human population and are discussed further by Schaller and Lauschke147.
3.1.3. ENT inhibitors
As mentioned in the introduction, human ENTs are sensitive to inhibition by a chemically diverse class of compounds termed adenosine-reuptake inhibitors (AdoRIs, Figure 8). In general, human ENT1 exhibits the highest sensitivity to many AdoRIs. Beyond the hallmark AdoRI NBMPR, other high-affinity inhibitors include dipyridamole – an FDA approved coronary vasodilator148, 149, dilazep – a low nanomolar hENT1 inhibitor used as a therapeutic in renal IgA nephropathy69, 150, draflazine/soluflazine (and derivatives) – highly potent AdoRIs with varying subtype and species selectivities151, and rapadocin – a recently discovered novel rapamycin-derivative which exhibits highly potent and subtype-specific inhibition of human ENT1 in a FKBP12-dependent manner68. Adding to this list are other putative transporter inhibitors likely targeting human ENTs. T1-11 is an adenosine-mimetic chemically similar to NBMPR with lower apparent toxicity, which inhibits adenosine transport in the low micromolar range and has shown promise as an adenosinergic modulator in a mouse model of Huntington’s disease67. Ticagrelor is a P2Y12 receptor inverse agonist and FDA approved antiplatelet drug which was shown to inhibit human ENT1152, 153 – however, the extent to which inhibition of human ENT1 by ticagrelor contributes to its pharmacological properties remains unclear154, 155. It is also important to note human ENTs may be inhibited by ethanol6, 143, 156–158, certain JNK inhibitors, serine/threonine kinase inhibitors3, 159, calcium channel antagonists160, and cannabinoids161 (Figure 8) – however it has yet to be established if these compounds directly act on human ENTs, or act on upstream pathways.
Figure 8.
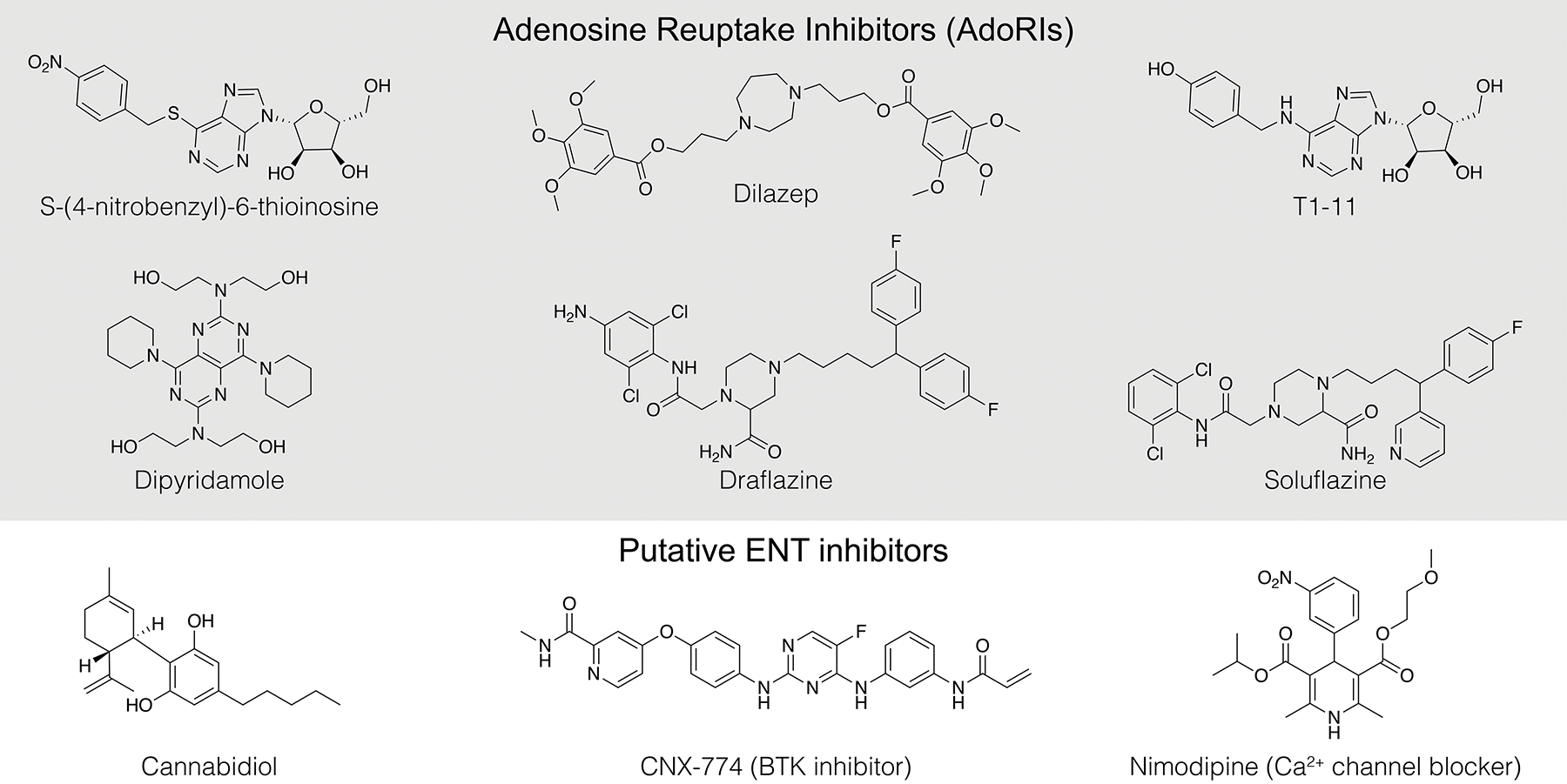
Chemical structures of select Adenosine Reuptake Inhibitors (AdoRIs) and other putative ENT inhibitors.
3.2. Structural advancements
As stated earlier, upon the molecular cloning of individual human ENT isoforms and subsequent topology studies, evidence for an 11-TM protein fold emerged109, 112, 116–121. Featuring a cytosolic facing N-terminal and extracellular facing C-terminal, the ENT fold also contains two large disordered loops which are located between TM1-2 and TM6-7. The TM1-2 loop contains N-linked glycosylation sites and has been implicated in membrane trafficking and protein folding112, 162. The TM6-7 loop contains multiple phosphorylation sites and has been shown to be important in the regulation of transporter function163, 164. Both the TM1-2 and TM6-7 loops have been shown to be dispensable in human ENT1, without affecting general functional properties of transport and inhibition165. Studies using chimeric constructs elucidated the role of various TM regions in sensitivity to NBMPR and other AdoRIs, along with substrate recognition107. Mutagenesis and functional characterization of transporter interaction with substrate and inhibitors were conducted, shedding considerable light onto the molecular determinants of such interactions110, 111, 166–169. Functional studies utilizing the Leishmania donovani nucleoside transporters, members of the ENT family, also contributed greatly to early efforts to functionally interrogate ENTs170, 171.
Sequence analysis of the ENT family provides evidence they share a common ancestor with the ubiquitous Major Facilitator Superfamily (MFS) of transporters172, 173 – however, extremely low sequence identities (<15%) are apparent between members of the ENT family and MFS members. Using this as a framework for initial structural studies, experimental structures of MFS members E. coli lactose permease (LacY)174 and fucose permease (FucP)175 were used as a templates for ab initio homolog modeling. These studies resulted in the construction of a structural models of L. donovani nucleoside transporter 1.1 (LdNT1.1) in inward and outward facing conformations, which combined with mutagenesis highlighted general features of the ENT fold176, 177. Furthermore, ENT structure predictions have been performed with de novo prediction pipelines7, 178–180.
These initial structural studies were important advancements to the field of ENT research. However, constructed homology models of ENTs are generally of low fidelity, due to low sequence homology to template or the challenges of membrane protein structure prediction using de novo methods. To comprehensively characterize nucleoside recognition, inhibition by AdoRIs and the transport mechanism exhibited by human ENTs, experimental transporter structures were determined142.
3.2.1. Crystal structures of human ENT1 in complex with adenosine reuptake inhibitors
Work on bacterial CNTs highlighted the feasibility of utilizing biochemically stable prokaryotic orthologs for structural studies87, 91, 100. Human ENTs lack any significant evolutionary relation to prokaryotic transporters and purified human transporter exhibits biochemical instability180–182. Furthermore, the requirement of milligram quantities of purified transporter underlies any crystallographic effort. To circumvent these issues, Wright and Lee engineered a functional variant of human ENT1 that is biochemically stable and crystallizable142. This enabled structure determination of the human transporter in complex with the adenosine reuptake inhibitors dilazep and NBMPR, marking the first available experimental structures of any ENT family member.
3.2.1.1. Transporter architecture and evolutionary insights
Crystal structures of human ENT1 confirmed the transporter adopts a 11-TM topology112, with the N-terminal in the cytoplasmic side and C-terminal in the extracellular side. The structure can be divided into two domains referred to as the N-domain and C-domain, where the first 6 TMs comprise the N-domain and the final 5 TMs comprise the C-domain (Figure 9). These two domains exhibit pseudo two-fold symmetry, a structural arrangement similar to that exhibited by MFS transporters183. Superposition of human ENT1 with various MFS transporter structures reveals structural homology between the two protein folds. The ENT fold exhibits several notable differences from the MFS fold, including slight displacement of TM9 resulting from the lack of the canonical TM12. This results in a lower apparent internal two-fold symmetry between the N- and C- domains within the ENT fold compared to the level of internal two-fold symmetry apparent in MFS142. We speculate that the lower internal two-fold symmetry provides a gating mechanism that deviates from that of MFS (rocker switch – see 3.2.1.3).
Figure 9.
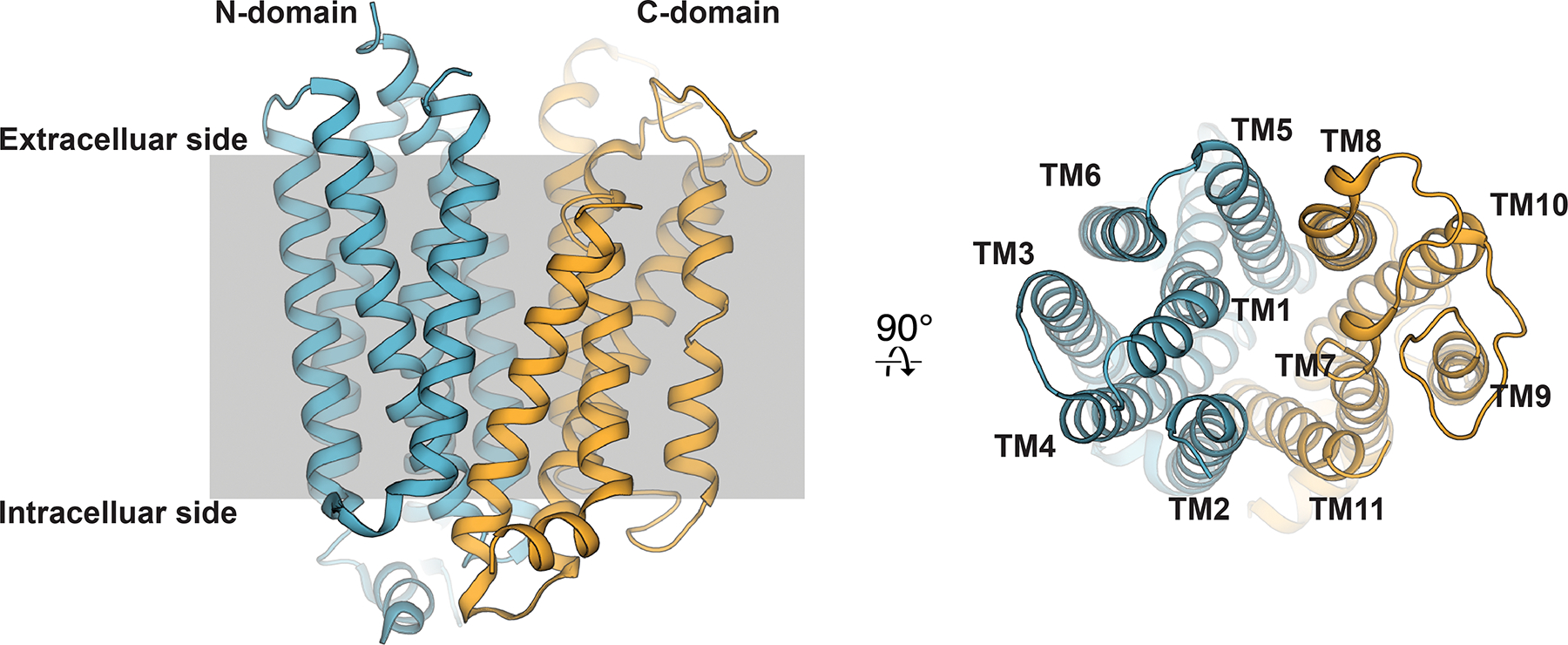
High-resolution crystal structure of the dilazep human ENT1 co-crystal structure (PDB ID 6OB7). The first 6 transmembrane helices colored blue (N-domain), and last 5 transmembrane helices colored orange (C-domain).
The 11-TM topology of human ENT1 and its structural similarity with MFS has posed an interesting question regarding the design principle of functional transporter from the evolutionary standpoint. Based on the semi-SWEET and SWEET transporter structures184–186, it was suggested that each lobe of MFS can be further broken into a 3+3 TM assembly, suggesting the 3 TM bundles in the MFS fold (TMS 1–3, 4–6, 7–9, 10–12) are the minimal building block with which functional transporter can be built. Consistent with the nomenclature presented by Nordlund and colleagues, these 3 TM bundles consists of a sequential arrangement of A-, B- and C- helices183. Currently available structural data concerning alternating access through MFS and semi-SWEET indicates interactions between A- and B- helices contribute most extensively to transporter gating183. These structural observations led to the hypothesis SWEET and semi-SWEET families may represent evolutionary predecessors to MFS186, consistent with the hypothesis that MFS may have evolved from gene duplication events of triple-helix bundles187, 188. The human ENT1 structure highlights the achievability of a functional transporter fold without following the strict 12 TM topology of MFS or the 3 TM building block strategy from SWEET transporters. It is possible that the C-helices (TMs 3, 6, 9 or 12) are less critical for transporter function, and that the ENT family simply lost a single C-helix TM during evolution. These studies pose an interesting question as to what the minimal structural requirements are to build a functional transporter.
Located in the center of the transporter between the N- and C-domains is a hydrophilic cavity lined by conserved residues previously implicated in transporter function, substrate recognition and inhibitor binding110, 111, 166–169. This central cavity is sealed from the intracellular side by extensive hydrophobic contacts formed by TMs 4, 5, 10 and 11. Due to the large protein volume apparent at this intracellular region of the transporter, we term this structural feature the “thick-gate.” At the extracellular side of the transporter TMs 1 and 7 are located in close proximity at the top of this central cavity but are still far enough apart to allow solvent accessibility from the extracellular solution. We term this structural feature the transporter “thin-gate”, as the barrier does not exhibit extensive protein volume between the extracellular side and central transporter cavity but would occlude nucleoside release from the central cavity (Figure 10A). Therefore, these human ENT1 structures represent the transporter in the outward-facing occluded state.
Figure 10.
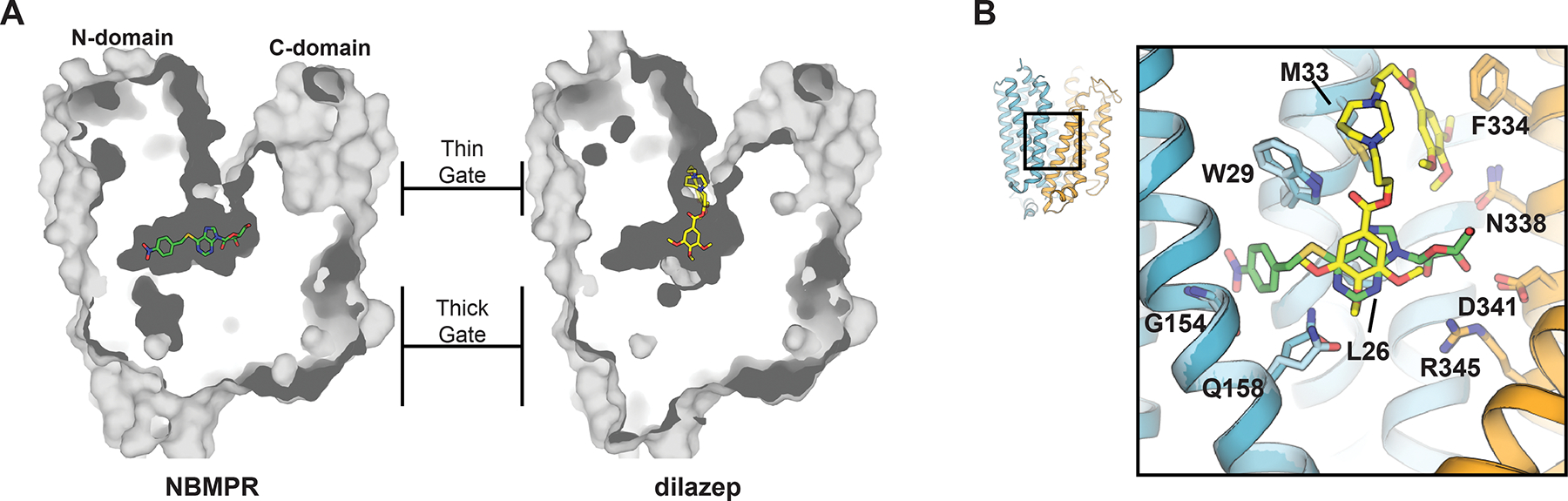
A) Dilazep and NBMPR binding sites in human ENT1 (dilazep depicted in yellow and NBMPR green, PDB IDs 6OB6 and 6OB7, respectively). B) Detailed depiction of inhibitor-transporter interactions. Protein portion of N-domain colored blue and C-domain colored orange (lighter shades of either color used for the NBMPR human ENT1 cocrystal structure). Interacting residues depicted as sticks.
3.2.1.2. Adenosine reuptake inhibitor binding sites
A large portion of the adenosine analog NBMPR occupies the central cavity of human ENT1, where its thioinosine nucleoside moiety forms interactions with several residues previously implicated in nucleoside recognition110, 111, 166–169 (Figure 10B). The p-nitrobenzyl moiety of NBMPR sits in a cavity within the N-domain of human ENT1 (this cavity is also present in the dilazep structure), connected and adjacent to the transporter central cavity. The N-domain cavity is lined by TM1, TM3, and TM4 and contains G154 at the proximal side – a residue responsible for the species/subtype specificity exhibited by NBMPR169 and I216 at the distal side – a human polymorphism at this position is associated with ethanol addiction and withdrawal in humans145, 147 (discussed in section 3.1.2 and to be discussed in 3.2.1.4). Dilazep is chemically dissimilar to nucleoside and is comprised of two trimethoxybenzoic acid groups and a central diazepane ring. One of the trimethoxybenzoic acid groups occupies the central transporter cavity, while the diazepane ring and other trimethoxybenzoic acid group protrude upward towards the extracellular solution and form contacts with the extracellular thin gate (Figure 10A). Therefore, NBMPR and dilazep both feature occupancy of the transporter central cavity, which is proposed to be the nucleoside binding site. However, specific chemical moieties of NBMPR and dilazep also occupy their own distinct sites on human ENT1 – one site being a cavity within the N-domain (NBMPR), another site being the extracellular thin gate (dilazep). These structural observations provide a possible explanation for the high avidity exhibited by these transporter inhibitors to human ENT1. In addition to the favorable interactions observed within the nucleoside-binding site, there are substantial interactions observed in each of the allosteric sites. Therefore, the energetic contributions to inhibitor binding are likely, to some degree, an additive combination of interactions observed within the nucleoside binding site and allosteric site.
3.2.1.3. Mechanistic implications
Structural mechanisms of MFS transporters have been thoroughly characterized183, 189–191, and fall under the rocker-switch category for membrane transport103. This model for membrane transport involves the sequential formation and disruption of transporter protein barriers, or gates, at either side of the membrane. Conformational changes involving movement of the two symmetric halves of the transporter relative to the membrane normal occur between gate-formation and gate-disruption steps. These transitions allows the solute within the central cavity, located between two symmetric halves of the transporter, alternating access to either side of the membrane192 (summarized in Figure 11). Due to the overall similarities of the ENT and MFS folds, their transport mechanisms are likely analogous. In the structure of dilazep bound human ENT1, inhibition of transporter function is likely achieved by steric block of extracellular thin gate occlusion – this inhibitory mechanism has been reported for other transporter inhibitors193–196. This highlights the mechanistic requirement of gate closure by rearrangements of TM1 and TM7, purposed to fully seal the central cavity from extracellular solution. Based on the current working model for membrane transport through MFS transporters, this step would precede transporter exchange to inward-facing conformational states. In the structure of NBMPR bound human ENT1, the transporter adopts a conformation similar to the dilazep-bound structure. However, no NBMPR moiety contacts the human ENT1 extracellular thin gate, ruling out steric hindrance of extracellular gate occlusion as a plausible mechanism for transport inhibition exhibited by NBMPR. Further, it is well accepted NBMPR recognizes an exofacial transporter site114, 197. Therefore, inhibition appears to be solely mediated by the occupancy of the human ENT1 N-domain cavity and results in the transporter being trapped in an outward-facing state. Since the N-domain cavity is lined by TM1, TM3 and TM4, we propose that these structural elements undergo key conformational rearrangements that occur after extracellular gate closure to drive transporter to inward-facing states. The trapping of the transporter in the outward facing state by occupancy of a site adjacent to the substrate binding cavity is distinct from steric block of gate closure and is therefore a unique mechanism of transporter inhibition.
Figure 11.
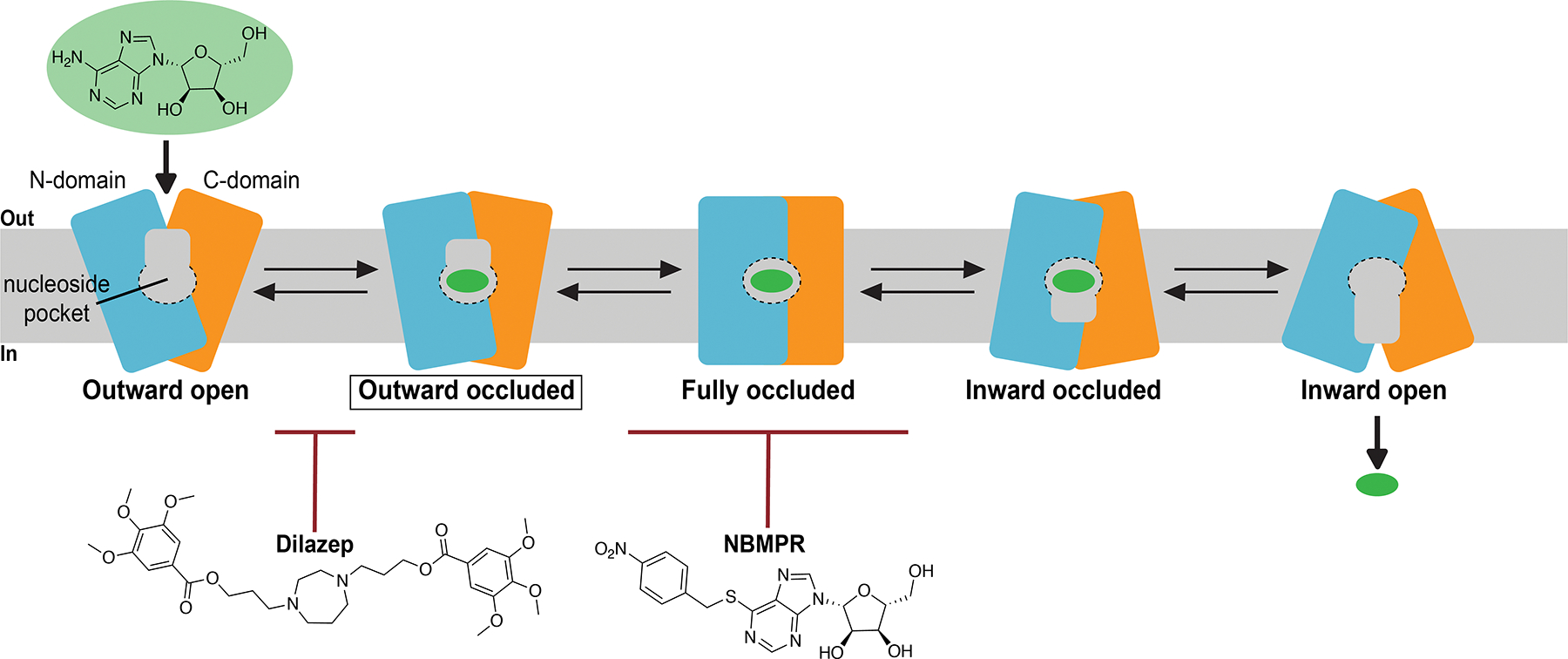
Current working model for the mechanism of nucleoside transport mediated by human ENT1. Currently, only the outward occluded transporter conformer has been structurally elucidated (boxed in diagram). Adenosine reuptake inhibitors dilazep and NBMPR exhibit distinct mechanisms of inhibition by perturbing two different conformational transitions in the transport cycle.
3.2.1.4. Insights into nucleoside recognition by hENT1 and its comparison with vcCNT
Since NBMPR is an adenosine-mimetic, we can postulate structural features of purine recognition in human ENT1 using the NBMPR-bound human ENT1 structure and compare with the adenosine-bound vcCNT structure (Figure 12). Intriguingly, although these two transporter families both recognize and transport nucleosides, they exhibit highly distinct structural architectures. Further, they appear to recognize nucleoside within their substrate binding cavities in different ways. For example, critical for nucleoside recognition through CNTs is a F366, a residue located in the middle of the nucleoside binding site – this residue not only forms pi-pi stacking interactions with base, but also mediates favorable CH-pi interactions with ribose. In human ENT1, the purine ring of NBMPR appears to be recognized through hydrophobic interactions mediated by L26 and L442, along with polar interaction with Q158. However, some commonalities do appear to exist between the two transporter families, such as the presence of interactions specified for 2’ and 3’ OH, which are mediated by polar/charged transporter residues.
Figure 12.
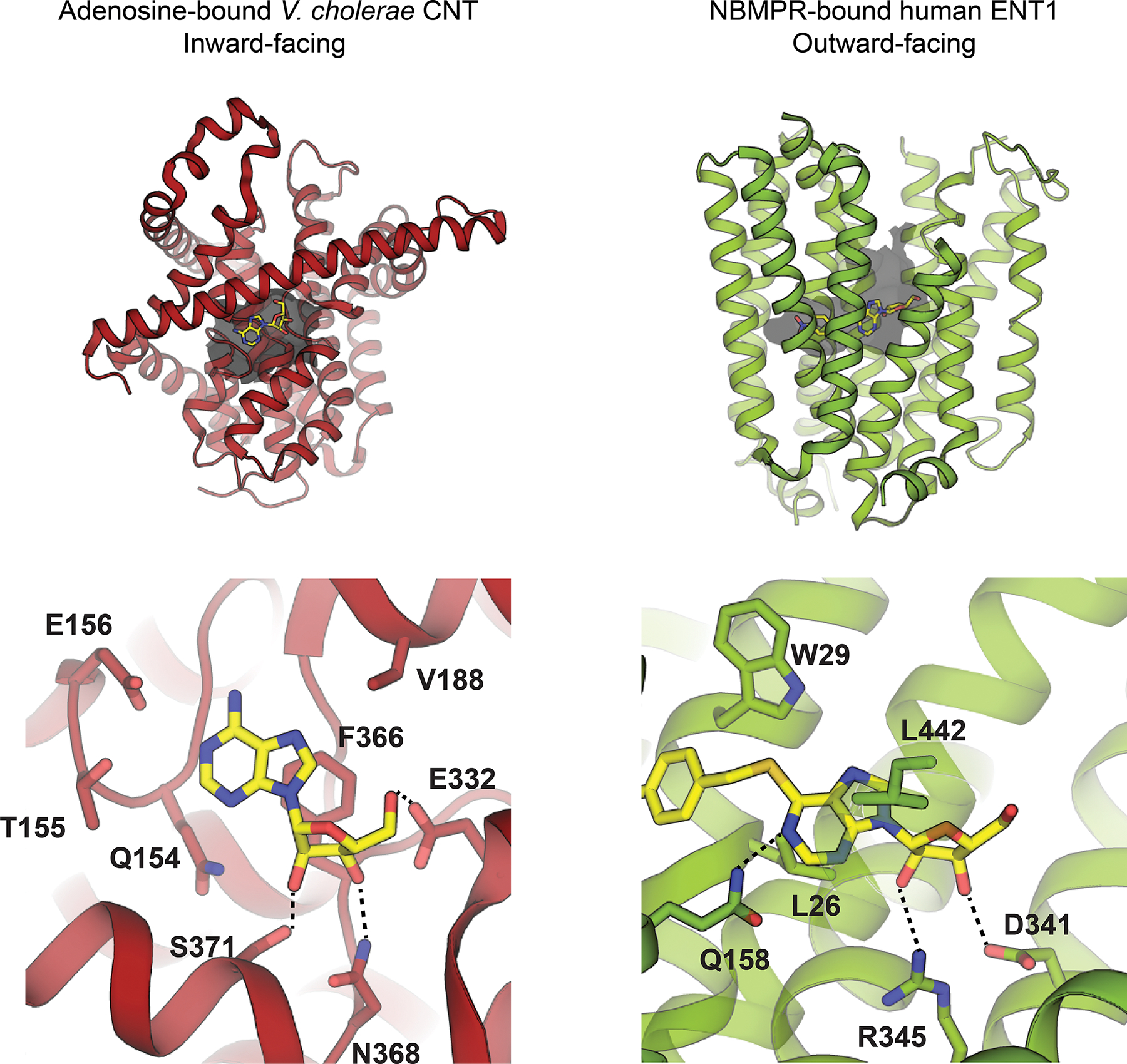
Comparison of inward-facing adenosine-bound V. cholerae CNT (left, PDB ID 4PD9) with outward-facing NBMPR-bound human ENT1 (right, PDB ID 6OB6).
3.2.1.5. Structural basis of slc29a3 genetic disorders
Rare mutations in human slc29a3 are associated with a wide spectrum of autosomal recessive genetic disorders that are often autoinflammatory in nature. The availability of an ENT experimental structure allows for preliminary understanding of the molecular basis of slc29a3 genetic disorders – disease associated mutations were mapped to their corresponding conserved positions on the human ENT1 crystal structure (Figure 13). The mutation M116R132 has previously been shown to disrupt protein trafficking and results in human ENT3 retention in the ER/Golgi141. Three additional disease mutations (G427S, G437R and T449R)132, 133, 198, 199 impaired human ENT3 activity but appeared to traffic normally in the same study141. G437R and T449R map to the TM10-11 loop, which contains one of the putative pH-sensitive residues in human ENT3123,, 125 (human ENT3 residue E447, conserved position in human ENT1 is E428). Other mutations identified from human patients include R134H/C198, S203P134, R363Q200 and R386Q201. While the mutation R386Q likely affects protein stability or turnover, examination of the locations corresponding to R134H/C, S203P and R363Q in the available ENT crystal structure are of particular interest. Human ENT3 position R134 is highly conserved across the entire ENT family and when mapped on the human ENT1 structure, would interact with the conserved position corresponding to the pH-sensing residue E447 in human ENT3123, 125. R363Q has clear implications in disrupting nucleoside binding in the central cavity of the transporter as this corresponding position in the human ENT1 crystal structure (R345) interacts with the ribose ring of the adenosine mimetic NBMPR142. Human ENT3 disease mutant S203P, a polymorphism associated with dysosteosclerosis134, is located in close proximity to this conserved position. In summary, while various mutations or truncations of slc29a3 associated with rare genetic disease result in changes in protein fold, stability or expression, many mutations result in perturbations of functionally important positions that would diminish transport activities or alter functional properties of nucleoside transport mediated by human ENT3.
Figure 13.
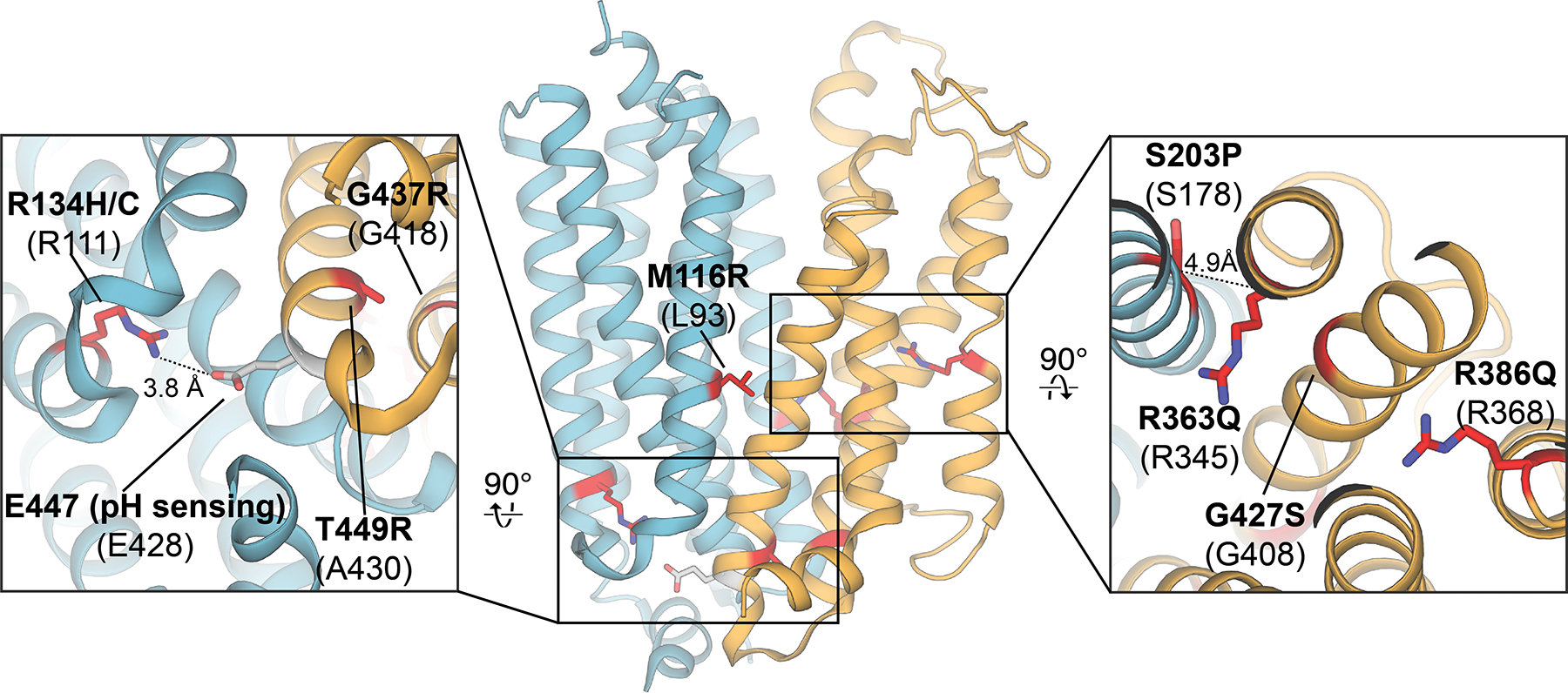
Mapping of slc29a3 mutations associated with a broad spectrum of genetic disorders onto their corresponding conserved positions in the human ENT1 crystal structure (PDB ID 6OB7; mutation sites highlighted in red, pH sensing residue highlighted in grey). Mutations in human ENT3 are labeled in bold, and the corresponding conserved positions in human ENT1 are listed below in parentheses.
3.2.1.6. Potential role of human ENT1 in ethanol related physiology and disease
As discussed in Section 3.1.2, a common variant in slc29a1 (I216T) may be associated with alcohol dependence with withdrawal seizures145. Located on TM6 of human ENT1, I216 faces the distal end of the cavity within the transporter N-domain in which the p-nitrobenzyl ring of NBMPR occupies in the co-crystal structure. This cavity is still present in the dilazep co-crystal structure, despite the absence of ligand occupancy at this site. Due to the high-resolution of the dilazep co-crystal structure, several well-ordered waters were resolved at the proximal end of this cavity142. It has been reported that the I216T mutation in human ENT1 results in a ~2 fold decrease in NBMPR affinity145, which is consistent with this mutation changing the electrostatic properties of this otherwise apolar site (Figure 14, surface electrostatic potential map calculated with the Adaptive Poisson-Boltzmann Solver202). Therefore, ethanol could possibly occupy the distal end of this cavity and achieve transport inhibition in a similar mechanism to that observed for NBMPR, where the I216T mutation would result in an altered binding affinity of ethanol at this site. Consistent with this hypothesis, acute ethanol exposure has been shown to inhibit nucleoside transport, and in some cases potentiate adenosine signaling156–158. However convincing these observations, the molecular basis for ethanol inhibition of human ENT1, along with the role played by I216T in altering human ENT1 sensitivity to ethanol, has yet to be rigorously investigated with biochemical and structural studies.
Figure 14.

Human ENT1 residue I216 lines the distal end of the cavity within the transporter N-domain. Surface electrostatic potential map calculated for the dilazep human ENT1 cocrystal structure (PDB ID 6OB7) using the Adaptive Poisson-Boltzmann Solver. Well-ordered waters observed in the proximal end of the N-domain cavity in the high-resolution structure are depicted as red spheres.
4. Future advancements
Although representative experimental structures are now available for both the CNT and ENT protein families, our understanding of their transport mechanisms are still limited. The transport mechanism exhibited by CNT is most extensively characterized100, however the outward-facing occluded state (substrate bound) remains to be determined. Moreover, despite their high sequence similarities, would human CNTs exhibit any differences in solute recognition and transport mechanism compared to that established for bacterial sodium-dependent nucleoside transporters? For example, would human CNTs employ a multi-step elevator motion including the intermediate states that are observed in CNTNW? Where is the second sodium site in human CNT3? A polymorphism in human CNT3 (C602R) disrupts the observed 2:1 coupling stoichiometry203, 204, indicating it might be in proximity to this second sodium site - would this second sodium also contribute to nucleoside substrate affinity control that we observe in bacterial CNTs? What is the structural basis of proton involvement in transport mediated by human CNT3? What are the timescales of conformational transitions during the transport cycle, and could they be measured with biophysical approaches? Additional functional, structural and biophysical studies focused on all three human CNT isoforms are needed to address these remaining questions.
Our understanding of the transport mechanism by ENTs is at an even earlier stage with many remaining questions – what are the structural features that underlie nucleoside transport? Although many nucleoside drugs bind to hENT1, why do some drugs act as permeants while others act as inhibitors? Facilitative solute transport characterizes the ENT family, but isoforms 3 and 4 utilize pH-dependent transport mechanisms. What is the structural and mechanistic basis for these pH-dependent transport mechanisms, and what specific purposes do they serve in human physiology? Understanding the structural basis of solute recognition as well as transport dynamics would help answer these important questions. This would require structural data on human ENT1 conformations beyond the previously reported outward-facing occluded state142 and in complex with additional ligands. Structures of ENT isoforms 2–4 would also be needed to understand the molecular basis of functional discrepancies between the different ENT isoforms. Further functional and biophysical approaches would also be needed to interrogate transport dynamics exhibited by ENTs.
The availability of the current structural data on bacterial CNTs and human ENT1 sets the stage for the advancement of rational drug design. Nucleoside-analog antiviral or anticancer drugs exhibiting improved ADME properties and transporter inhibitors with desirable pharmacological activities stand to be gained from utilizing this available structural information. Future efforts would certainly impact the effectiveness of therapeutics purposed to intervene in nucleoside biology.
Acknowledgments
This work was supported by the National Institutes of Health, United States (R01 GM137421 to S.-Y.L.) and the American Heart Association (AHA 20PRE35210058 to N.J.W.). We thank Jiyong Hong for the long-term collaboration on the CNT and ENT studies. We thank the former Lee lab members Zachary Johnson and Marscha Hirschi for their contribution to structural and functional studies of CNT.
Biographies
Biographies
Nicholas Wright earned a B.S. in biochemistry from Roanoke College in 2016. He is currently an American Heart Association predoctoral fellow pursuing Ph.D. training at Duke University in the Department of Biochemistry, under the supervision of Dr. Seok-Yong Lee. His research has been focused on the structure and mechanism of membrane transport proteins.
Seok-Yong Lee, Ph.D., is a professor of Biochemistry at Duke University School of Medicine. Dr. Lee completed his doctoral work in 2003 at the University of California at Berkeley under the supervision of Dr. David Wemmer. He then pursued postdoctoral studies with Dr. Roderick MacKinnon at Rockefeller University, where he carried out structural and biophysical studies of potassium channels. In 2009, Dr. Lee started his independent career at Duke University and built a research program focusing on the structural biology of ion channels, transporters and membrane-embedded enzymes. He has been recognized for his contributions to the field of membrane protein structural biology by multiple awards including the NIH Director’s New Innovator Award, NIGMS award, the SER-CAT Outstanding Science Award, and McKnight Scholar Award.
Footnotes
Author Information The authors declare no competing financial interests.
Note added during review: During the revision of this paper a cryo-electron microscopy structure of apo human CNT3ins (human CNT3 splice variant) was reported205. This study revealed there is a high level of structural similarity is apparent between the bacterial CNT and human CNT folds. Additional features resolved in the human CNT3 structure are the 3 additional N-terminal transmembrane helices, which are not present in prokaryotic homologs.
References
- 1.Baldwin SA; Mackey JR; Cass CE; Young JD, Nucleoside transporters: Molecular biology and implications for therapeutic development. Mol Med Today 1999, 5, 216–224. [DOI] [PubMed] [Google Scholar]
- 2.King AE; Ackley MA; Cass CE; Young JD; Baldwin SA, Nucleoside transporters: From scavengers to novel therapeutic targets. Trends Pharmacol Sci 2006, 27, 416–425. [DOI] [PubMed] [Google Scholar]
- 3.Parkinson FE; Damaraju VL; Graham K; Yao SYM; Baldwin SA; Cass CE; Young JD, Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr Top Med Chem 2011, 11, 948–972. [DOI] [PubMed] [Google Scholar]
- 4.Young JD; Yao SYM; Baldwin JM; Cass CE; Baldwin SA, The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Molecular Aspects of Medicine 2013, 34, 529–547. [DOI] [PubMed] [Google Scholar]
- 5.Young JD; Yao SYM; Sun L; Cass CE; Baldwin SA, Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 2008, 38, 995–1021. [DOI] [PubMed] [Google Scholar]
- 6.Boswell-Casteel RC; Hays FA, Equilibrative nucleoside transporters—a review. Nucleosides, Nucleotides and Nucleic Acids 2017, 36, 7–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehan S; Shahid S; Salminen TA; Jaakola VP; Paavilainen VO, Current progress on equilibrative nucleoside transporter function and inhibitor design. SLAS Discov 2019, 24, 953–968. [DOI] [PubMed] [Google Scholar]
- 8.Van Rompay AR; Johansson M; Karlsson A, Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther 2000, 87, 189–198. [DOI] [PubMed] [Google Scholar]
- 9.Yegutkin GG, Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit Rev Biochem Mol Biol 2014, 49, 473–497. [DOI] [PubMed] [Google Scholar]
- 10.Robson SC; Sevigny J; Zimmermann H, The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal 2006, 2, 409–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H; Xia Y, Beneficial and detrimental role of adenosine signaling in diseases and therapy. J Appl Physiol (1985) 2015, 119, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borea PA; Gessi S; Merighi S; Varani K, Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends in Pharmacological Sciences 2016, 37, 419–434. [DOI] [PubMed] [Google Scholar]
- 13.Layland J; Carrick D; Lee M; Oldroyd K; Berry C, Adenosine: Physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv 2014, 7, 581–591. [DOI] [PubMed] [Google Scholar]
- 14.Mubagwa K; Flameng W, Adenosine, adenosine receptors and myocardial protection: An updated overview. Cardiovasc. Res. 2001, 52, 25–39. [DOI] [PubMed] [Google Scholar]
- 15.Chen JF; Eltzschig HK; Fredholm BB, Adenosine receptors as drug targets--what are the challenges? Nat Rev Drug Discov 2013, 12, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boison D; Chen JF; Fredholm BB, Adenosine signaling and function in glial cells. Cell Death & Differentiation 2010, 17, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hack SP; Christie MJ, Adaptations in adenosine signaling in drug dependence: Therapeutic implications. Crit Rev Neurobiol 2003, 15, 235–274. [DOI] [PubMed] [Google Scholar]
- 18.Bjorness TE; Greene RW, Adenosine and sleep. Curr Neuropharmacol 2009, 7, 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruby CL; O’Connor KM; Ayers-Ringler J; Choi DS, Adenosine and glutamate in neuroglial interaction: Implications for circadian disorders and alcoholism. Adv Neurobiol 2014, 11, 103–119. [DOI] [PubMed] [Google Scholar]
- 20.Nam HW; Bruner RC; Choi DS, Adenosine signaling in striatal circuits and alcohol use disorders. Mol Cells 2013, 36, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nam HW; McIver SR; Hinton DJ; Thakkar MM; Sari Y; Parkinson FE; Haydon PG; Choi DS, Adenosine and glutamate signaling in neuron-glial interactions: Implications in alcoholism and sleep disorders. Alcohol Clin Exp Res 2012, 36, 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asatryan L; Nam HW; Lee MR; Thakkar MM; Saeed Dar M; Davies DL; Choi DS, Implication of the purinergic system in alcohol use disorders. Alcohol Clin Exp Res 2011, 35, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruby CL; Adams CA; Knight EJ; Nam HW; Choi DS, An essential role for adenosine signaling in alcohol abuse. Curr Drug Abuse Rev 2010, 3, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindberg D; Shan D; Ayers-Ringler J; Oliveros A; Benitez J; Prieto M; McCullumsmith R; Choi DS, Purinergic signaling and energy homeostasis in psychiatric disorders. Curr Mol Med 2015, 15, 275–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galmarini CM; Mackey JR; Dumontet C, Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol 2002, 3, 415–424. [DOI] [PubMed] [Google Scholar]
- 26.Seley-Radtke KL; Yates MK, The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res 2018, 154, 66–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yates MK; Seley-Radtke KL, The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part ii: Complex modifications to the nucleoside scaffold. Antiviral Res 2019, 162, 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jordheim LP; Durantel D; Zoulim F; Dumontet C, Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 2013, 12, 447–464. [DOI] [PubMed] [Google Scholar]
- 29.Tsesmetzis N; Paulin CBJ; Rudd SG; Herold N, Nucleobase and nucleoside analogues: Resistance and re-sensitisation at the level of pharmacokinetics, pharmacodynamics and metabolism. Cancers (Basel) 2018, 10, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang M; Cao R; Zhang L; Yang X; Liu J; Xu M; Shi Z; Hu Z; Zhong W; Xiao G, Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020, 30, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheahan TP; Sims AC; Zhou S; Graham RL; Pruijssers AJ; Agostini ML; Leist SR; Schäfer A; Dinnon KH; Stevens LJ; Chappell JD; Lu X; Hughes TM; George AS; Hill CS; Montgomery SA; Brown AJ; Bluemling GR; Natchus MG; Saindane M; Kolykhalov AA; Painter G; Harcourt J; Tamin A; Thornburg NJ; Swanstrom R; Denison MR; Baric RS, An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Science Translational Medicine 2020, 12, eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grein J; Ohmagari N; Shin D; Diaz G; Asperges E; Castagna A; Feldt T; Green G; Green ML; Lescure FX; Nicastri E; Oda R; Yo K; Quiros-Roldan E; Studemeister A; Redinski J; Ahmed S; Bernett J; Chelliah D; Chen D; Chihara S; Cohen SH; Cunningham J; D’Arminio Monforte A; Ismail S; Kato H; Lapadula G; L’Her E; Maeno T; Majumder S; Massari M; Mora-Rillo M; Mutoh Y; Nguyen D; Verweij E; Zoufaly A; Osinusi AO; DeZure A; Zhao Y; Zhong L; Chokkalingam A; Elboudwarej E; Telep L; Timbs L; Henne I; Sellers S; Cao H; Tan SK; Winterbourne L; Desai P; Mera R; Gaggar A; Myers RP; Brainard DM; Childs R; Flanigan T, Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 2020, 382, 2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Govindarajan R; Bakken AH; Hudkins KL; Lai Y; Casado FJ; Pastor-Anglada M; Tse CM; Hayashi J; Unadkat JD, In situ hybridization and immunolocalization of concentrative and equilibrative nucleoside transporters in the human intestine, liver, kidneys, and placenta. Am J Physiol Regul Integr Comp Physiol 2007, 293, R1809–R1822. [DOI] [PubMed] [Google Scholar]
- 34.Damaraju VL; Sawyer MB; Mackey JR; Young JD; Cass CE, Human nucleoside transporters: Biomarkers for response to nucleoside drugs. Nucleosides, nucleotides & nucleic acids 2009, 28, 450–463. [DOI] [PubMed] [Google Scholar]
- 35.Pastor-Anglada M; Urtasun N; Perez-Torras S, Intestinal nucleoside transporters: Function, expression, and regulation. Compr Physiol 2018, 8, 1003–1017. [DOI] [PubMed] [Google Scholar]
- 36.Giacomini KM; Huang SM, Transporters in drug development and clinical pharmacology. Clinical pharmacology and therapeutics 2013, 94, 3–9. [DOI] [PubMed] [Google Scholar]
- 37.Koczor CA; Torres RA; Lewis W, The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin Drug Metab Toxicol 2012, 8, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenhalf W; Ghaneh P; Neoptolemos JP; Palmer DH; Cox TF; Lamb RF; Garner E; Campbell F; Mackey JR; Costello E; Moore MJ; Valle JW; McDonald AC; Carter R; Tebbutt NC; Goldstein D; Shannon J; Dervenis C; Glimelius B; Deakin M; Charnley RM; Lacaine F; Scarfe AG; Middleton MR; Anthoney A; Halloran CM; Mayerle J; Oláh A; Jackson R; Rawcliffe CL; Scarpa A; Bassi C; Büchler MW, Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. JNCI: Journal of the National Cancer Institute 2014, 106. [DOI] [PubMed] [Google Scholar]
- 39.Skrypek N; Duchene B; Hebbar M; Leteurtre E; van Seuningen I; Jonckheere N, The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the concentrative nucleoside transporter family. Oncogene 2013, 32, 1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhutia YD; Hung SW; Patel B; Lovin D; Govindarajan R, CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer research 2011, 71, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voutsadakis IA, Molecular predictors of gemcitabine response in pancreatic cancer. World J Gastrointest Oncol 2011, 3, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagmann W; Jesnowski R; Löhr JM, Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2010, 12, 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka M; Javle M; Dong X; Eng C; Abbruzzese JL; Li D, Gemcitabine metabolic and transporter gene polymorphisms are associated with drug toxicity and efficacy in patients with locally advanced pancreatic cancer. Cancer 2010, 116, 5325–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okazaki T; Javle M; Tanaka M; Abbruzzese JL; Li D, Single nucleotide polymorphisms of gemcitabine metabolic genes and pancreatic cancer survival and drug toxicity. Clin. Cancer Res. 2010, 16, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marechal R; Mackey JR; Lai R; Demetter P; Peeters M; Polus M; Cass CE; Young J; Salmon I; Deviere J; Van Laethem JL, Human equilibrative nucleoside transporter 1 and human concentrative nucleoside transporter 3 predict survival after adjuvant gemcitabine therapy in resected pancreatic adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2009, 15, 2913–2919. [DOI] [PubMed] [Google Scholar]
- 46.Farrell JJ; Elsaleh H; Garcia M; Lai R; Ammar A; Regine WF; Abrams R; Benson AB; Macdonald J; Cass CE; Dicker AP; Mackey JR, Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 2009, 136, 187–195. [DOI] [PubMed] [Google Scholar]
- 47.Marcé S; Molina-Arcas M; Villamor N; Casado FJ; Campo E; Pastor-Anglada M; Colomer D, Expression of human equilibrative nucleoside transporter 1 (hENT1) and its correlation with gemcitabine uptake and cytotoxicity in mantle cell lymphoma. Haematologica 2006, 91, 895–902. [PubMed] [Google Scholar]
- 48.Spratlin J; Sangha R; Glubrecht D; Dabbagh L; Young JD; Dumontet C; Cass C; Lai R; Mackey JR, The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2004, 10, 6956–6961. [DOI] [PubMed] [Google Scholar]
- 49.Mackey JR; Mani RS; Selner M; Mowles D; Young JD; Belt JA; Crawford CR; Cass CE, Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer research 1998, 58, 4349–4357. [PubMed] [Google Scholar]
- 50.Girardi E; César-Razquin A; Lindinger S; Papakostas K; Konecka J; Hemmerich J; Kickinger S; Kartnig F; Gürtl B; Klavins K; Sedlyarov V; Ingles-Prieto A; Fiume G; Koren A; Lardeau C-H; Kumaran Kandasamy R; Kubicek S; Ecker GF; Superti-Furga G, A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nature Chemical Biology 2020, 16, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Damaraju VL; Mowles D; Yao S; Ng A; Young JD; Cass CE; Tong Z, Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides, nucleotides & nucleic acids 2012, 31, 236–255. [DOI] [PubMed] [Google Scholar]
- 52.Rau M; Stickel F; Russmann S; Manser CN; Becker PP; Weisskopf M; Schmitt J; Dill MT; Dufour JF; Moradpour D; Semela D; Mullhaupt B; Geier A; Swiss Hepatitis CCSG, Impact of genetic SLC28 transporter and itpa variants on ribavirin serum level, hemoglobin drop and therapeutic response in patients with hcv infection. Journal of hepatology 2013, 58, 669–675. [DOI] [PubMed] [Google Scholar]
- 53.Iikura M; Furihata T; Mizuguchi M; Nagai M; Ikeda M; Kato N; Tsubota A; Chiba K, ENT1, a ribavirin transporter, plays a pivotal role in antiviral efficacy of ribavirin in a hepatitis C virus replication cell system. Antimicrob Agents Chemother 2012, 56, 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsubota A; Shimada N; Yoshizawa K; Furihata T; Agata R; Yumoto Y; Abe H; Ika M; Namiki Y; Chiba K; Fujise K; Tada N; Aizawa Y, Contribution of ribavirin transporter gene polymorphism to treatment response in peginterferon plus ribavirin therapy for HCV genotype 1b patients. Liver Int 2012, 32, 826–836. [DOI] [PubMed] [Google Scholar]
- 55.Doehring A; Hofmann WP; Schlecker C; Zeuzem S; Sarrazin C; Berg T; Muller T; Herrmann E; Geisslinger G; Lotsch J, Role of nucleoside transporters SLC28A2/3 and SLC29A1/2 genetics in ribavirin therapy: Protection against anemia in patients with chronic hepatitis C. Pharmacogenetics and genomics 2011, 21, 289–296. [DOI] [PubMed] [Google Scholar]
- 56.Morello J; Cuenca L; Soriano V; Medrano J; Madejon A; Vispo E; Barreiro P; Labarga P; Jimenez-Nacher I; Rodriguez-Novoa S, Influence of a single nucleotide polymorphism at the main ribavirin transporter gene on the rapid virological response to pegylated interferon-ribavirin therapy in patients with chronic hepatitis C virus infection. J Infect Dis 2010, 202, 1185–1191. [DOI] [PubMed] [Google Scholar]
- 57.Endres CJ; Moss AM; Ke B; Govindarajan R; Choi DS; Messing RO; Unadkat JD, The role of the equilibrative nucleoside transporter 1 (ENT1) in transport and metabolism of ribavirin by human and wild-type or Ent1-/- mouse erythrocytes. J Pharmacol Exp Ther 2009, 329, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith KM; Ng AM; Yao SY; Labedz KA; Knaus EE; Wiebe LI; Cass CE; Baldwin SA; Chen XZ; Karpinski E; Young JD, Electrophysiological characterization of a recombinant human Na+-coupled nucleoside transporter (hCNT1) produced in Xenopus oocytes. J Physiol 2004, 558, 807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritzel MW; Yao SY; Huang MY; Elliott JF; Cass CE; Young JD, Molecular cloning and functional expression of cDNAs encoding a human Na+-nucleoside cotransporter (hCNT1). Am J Physiol 1997, 272, C707–C714. [DOI] [PubMed] [Google Scholar]
- 60.Yao SY; Ng AM; Sundaram M; Cass CE; Baldwin SA; Young JD, Transport of antiviral 3’-deoxy-nucleoside drugs by recombinant human and rat equilibrative, nitrobenzylthioinosine (NBMPR)-insensitive (ENT2) nucleoside transporter proteins produced in Xenopus oocytes. Mol Membr Biol 2001, 18, 161–167. [DOI] [PubMed] [Google Scholar]
- 61.Agostini ML; Andres EL; Sims AC; Graham RL; Sheahan TP; Lu X; Smith EC; Case JB; Feng JY; Jordan R; Ray AS; Cihlar T; Siegel D; Mackman RL; Clarke MO; Baric RS; Denison MR, Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, e00221–00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren TK; Jordan R; Lo MK; Ray AS; Mackman RL; Soloveva V; Siegel D; Perron M; Bannister R; Hui HC; Larson N; Strickley R; Wells J; Stuthman KS; Van Tongeren SA; Garza NL; Donnelly G; Shurtleff AC; Retterer CJ; Gharaibeh D; Zamani R; Kenny T; Eaton BP; Grimes E; Welch LS; Gomba L; Wilhelmsen CL; Nichols DK; Nuss JE; Nagle ER; Kugelman JR; Palacios G; Doerffler E; Neville S; Carra E; Clarke MO; Zhang L; Lew W; Ross B; Wang Q; Chun K; Wolfe L; Babusis D; Park Y; Stray KM; Trancheva I; Feng JY; Barauskas O; Xu Y; Wong P; Braun MR; Flint M; McMullan LK; Chen SS; Fearns R; Swaminathan S; Mayers DL; Spiropoulou CF; Lee WA; Nichol ST; Cihlar T; Bavari S, Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denning J; Cornpropst M; Flach SD; Berrey MM; Symonds WT, Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob Agents Chemother 2013, 57, 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawitz E; Rodriguez-Torres M; Denning JM; Albanis E; Cornpropst M; Berrey MM; Symonds WT, Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, following multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob Agents Chemother 2013, 57, 1209–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toots M; Yoon JJ; Cox RM; Hart M; Sticher ZM; Makhsous N; Plesker R; Barrena AH; Reddy PG; Mitchell DG; Shean RC; Bluemling GR; Kolykhalov AA; Greninger AL; Natchus MG; Painter GR; Plemper RK, Characterization of orally efficacious influenza drug with high resistance barrier in ferrets and human airway epithelia. Sci Transl Med 2019, 11, eaax5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao ZG; Jacobson KA, Emerging adenosine receptor agonists: An update. Expert Opin Emerg Drugs 2011, 16, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang NK; Lin JH; Lin JT; Lin CI; Liu EM; Lin CJ; Chen WP; Shen YC; Chen HM; Chen JB; Lai HL; Yang CW; Chiang MC; Wu YS; Chang C; Chen JF; Fang JM; Lin YL; Chern Y, A new drug design targeting the adenosinergic system for Huntington’s disease. PLoS One 2011, 6, e20934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Z; Hong SY; Wang J; Rehan S; Liu W; Peng H; Das M; Li W; Bhat S; Peiffer B; Ullman BR; Tse CM; Tarmakova Z; Schiene-Fischer C; Fischer G; Coe I; Paavilainen VO; Sun Z; Liu JO, Rapamycin-inspired macrocycles with new target specificity. Nat Chem 2019, 11, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshida H; Kanatsu K; Muso E; Kuwahara T; Sekita K; Ono T; Konishi N; Kanda C; Minakata T; Morimoto K, Effects of an anti-platelet drug (dilazep) in IgA nephropathy: Comparison of clinical effects with renal biopsy findings. Nihon Jinzo Gakkai Shi 1994, 36, 339–344. [PubMed] [Google Scholar]
- 70.Damaraju VL; Smith KM; Mowles D; Nowak I; Karpinski E; Young JD; Robins MJ; Cass CE, Interaction of fused-pyrimidine nucleoside analogs with human concentrative nucleoside transporters: High-affinity inhibitors of human concentrative nucleoside transporter 1. Biochem Pharmacol 2011, 81, 82–90. [DOI] [PubMed] [Google Scholar]
- 71.Ritzel MW; Ng AM; Yao SY; Graham K; Loewen SK; Smith KM; Ritzel RG; Mowles DA; Carpenter P; Chen XZ; Karpinski E; Hyde RJ; Baldwin SA; Cass CE; Young JD, Molecular identification and characterization of novel human and mouse concentrative Na+-nucleoside cotransporter proteins (hCNT3 and mCNT3) broadly selective for purine and pyrimidine nucleosides (system cib). J Biol Chem 2001, 276, 2914–2927. [DOI] [PubMed] [Google Scholar]
- 72.Ritzel MW; Yao SY; Ng AM; Mackey JR; Cass CE; Young JD, Molecular cloning, functional expression and chromosomal localization of a cdna encoding a human Na+/nucleoside cotransporter (hCNT2) selective for purine nucleosides and uridine. Mol Membr Biol 1998, 15, 203–211. [DOI] [PubMed] [Google Scholar]
- 73.Zhang J; Smith KM; Tackaberry T; Visser F; Robins MJ; Nielsen LP; Nowak I; Karpinski E; Baldwin SA; Young JD; Cass CE, Uridine binding and transportability determinants of human concentrative nucleoside transporters. Molecular pharmacology 2005, 68, 830–839. [DOI] [PubMed] [Google Scholar]
- 74.Damaraju VL; Mowles D; Smith KM; Yao SY; Young JD; Marquez VE; Cass CE, Influence of sugar ring conformation on the transportability of nucleosides by human nucleoside transporters. Chembiochem 2011, 12, 2774–2778. [DOI] [PubMed] [Google Scholar]
- 75.Smith KM; Slugoski MD; Cass CE; Baldwin SA; Karpinski E; Young JD, Cation coupling properties of human concentrative nucleoside transporters hCNT1, hCNT2 and hCNT3. Mol Membr Biol 2007, 24, 53–64. [DOI] [PubMed] [Google Scholar]
- 76.Smith KM; Slugoski MD; Loewen SK; Ng AM; Yao SY; Chen XZ; Karpinski E; Cass CE; Baldwin SA; Young JD, The broadly selective human Na+/nucleoside cotransporter (hCNT3) exhibits novel cation-coupled nucleoside transport characteristics. J Biol Chem 2005, 280, 25436–25449. [DOI] [PubMed] [Google Scholar]
- 77.Loewen SK; Ng AM; Mohabir NN; Baldwin SA; Cass CE; Young JD, Functional characterization of a H+/nucleoside co-transporter (caCNT) from Candida albicans, a fungal member of the concentrative nucleoside transporter (CNT) family of membrane proteins. Yeast 2003, 20, 661–675. [DOI] [PubMed] [Google Scholar]
- 78.Loewen SK; Yao SY; Slugoski MD; Mohabir NN; Turner RJ; Mackey JR; Weiner JH; Gallagher MP; Henderson PJ; Baldwin SA; Cass CE; Young JD, Transport of physiological nucleosides and anti-viral and anti-neoplastic nucleoside drugs by recombinant escherichia coli nucleoside-H(+) cotransporter (NupC) produced in xenopus laevis oocytes. Mol Membr Biol 2004, 21, 1–10. [DOI] [PubMed] [Google Scholar]
- 79.Aymerich I; Pastor-Anglada M; Casado FJ, Long term endocrine regulation of nucleoside transporters in rat intestinal epithelial cells. J Gen Physiol 2004, 124, 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valdes R; Ortega MA; Casado FJ; Felipe A; Gil A; Sanchez-Pozo A; Pastor-Anglada M, Nutritional regulation of nucleoside transporter expression in rat small intestine. Gastroenterology 2000, 119, 1623–1630. [DOI] [PubMed] [Google Scholar]
- 81.Pastor-Anglada M; Cano-Soldado P; Errasti-Murugarren E; Casado FJ, SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica 2008, 38, 972–994. [DOI] [PubMed] [Google Scholar]
- 82.Hamilton SR; Yao SY; Ingram JC; Hadden DA; Ritzel MW; Gallagher MP; Henderson PJ; Cass CE; Young JD; Baldwin SA, Subcellular distribution and membrane topology of the mammalian concentrative Na+-nucleoside cotransporter rCNT1. J Biol Chem 2001, 276, 27981–27988. [DOI] [PubMed] [Google Scholar]
- 83.Slugoski MD; Ng AM; Yao SY; Lin CC; Mulinta R; Cass CE; Baldwin SA; Young JD, Substituted cysteine accessibility method analysis of human concentrative nucleoside transporter hCNT3 reveals a novel discontinuous region of functional importance within the CNT family motif (G/A)XKX3NEFVA(Y/M/F). J Biol Chem 2009, 284, 17281–17292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang J; Tackaberry T; Ritzel MW; Raborn T; Barron G; Baldwin SA; Young JD; Cass CE, Cysteine-accessibility analysis of transmembrane domains 11–13 of human concentrative nucleoside transporter 3. The Biochemical journal 2006, 394, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernandez-Calotti P; Casulleras O; Antolin M; Guarner F; Pastor-Anglada M, Galectin-4 interacts with the drug transporter human concentrative nucleoside transporter 3 to regulate its function. FASEB J 2016, 30, 544–554. [DOI] [PubMed] [Google Scholar]
- 86.Errasti-Murugarren E; Casado FJ; Pastor-Anglada M, Different N-terminal motifs determine plasma membrane targeting of the human concentrative nucleoside transporter 3 in polarized and nonpolarized cells. Molecular pharmacology 2010, 78, 795–803. [DOI] [PubMed] [Google Scholar]
- 87.Johnson ZL; Cheong C-G; Lee S-Y, Crystal structure of a concentrative nucleoside transporter from vibrio cholerae at 2.4 å. Nature 2012, 483, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loewen SK; Ng AM; Yao SY; Cass CE; Baldwin SA; Young JD, Identification of amino acid residues responsible for the pyrimidine and purine nucleoside specificities of human concentrative Na(+) nucleoside cotransporters hCNT1 and hCNT2. J Biol Chem 1999, 274, 24475–24484. [DOI] [PubMed] [Google Scholar]
- 89.Slugoski MD; Smith KM; Ng AM; Yao SY; Karpinski E; Cass CE; Baldwin SA; Young JD, Conserved glutamate residues glu-343 and glu-519 provide mechanistic insights into cation/nucleoside cotransport by human concentrative nucleoside transporter hCNT3. J Biol Chem 2009, 284, 17266–17280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yao SY; Ng AM; Slugoski MD; Smith KM; Mulinta R; Karpinski E; Cass CE; Baldwin SA; Young JD, Conserved glutamate residues are critically involved in Na+/nucleoside cotransport by human concentrative nucleoside transporter 1 (hCNT1). J Biol Chem 2007, 282, 30607–30617. [DOI] [PubMed] [Google Scholar]
- 91.Johnson ZL; Lee J-H; Lee K; Lee M; Kwon D-Y; Hong J; Lee S-Y, Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. eLife 2014, 3, e03604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reyes N; Oh S; Boudker O, Binding thermodynamics of a glutamate transporter homolog. Nat Struct Mol Biol 2013, 20, 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Verdon G; Boudker O, Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat Struct Mol Biol 2012, 19, 355–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reyes N; Ginter C; Boudker O, Transport mechanism of a bacterial homologue of glutamate transporters. Nature 2009, 462, 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boudker O; Ryan RM; Yernool D; Shimamoto K; Gouaux E, Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 2007, 445, 387–393. [DOI] [PubMed] [Google Scholar]
- 96.Yernool D; Boudker O; Jin Y; Gouaux E, Structure of a glutamate transporter homologue from pyrococcus horikoshii. Nature 2004, 431, 811–818. [DOI] [PubMed] [Google Scholar]
- 97.Krishnamurthy H; Piscitelli CL; Gouaux E, Unlocking the molecular secrets of sodium-coupled transporters. Nature 2009, 459, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jardetzky O, Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970. [DOI] [PubMed] [Google Scholar]
- 99.Yamashita A; Singh SK; Kawate T; Jin Y; Gouaux E, Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 2005, 437, 215–223. [DOI] [PubMed] [Google Scholar]
- 100.Hirschi M; Johnson ZL; Lee S-Y, Visualizing multistep elevator-like transitions of a nucleoside transporter. Nature 2017, 545, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Faham S; Watanabe A; Besserer GM; Cascio D; Specht A; Hirayama BA; Wright EM; Abramson J, The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 2008, 321, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weyand S; Shimamura T; Yajima S; Suzuki S; Mirza O; Krusong K; Carpenter EP; Rutherford NG; Hadden JM; O’Reilly J; Ma P; Saidijam M; Patching SG; Hope RJ; Norbertczak HT; Roach PC; Iwata S; Henderson PJ; Cameron AD, Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 2008, 322, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Drew D; Boudker O, Shared molecular mechanisms of membrane transporters. Annu Rev Biochem 2016, 85, 543–572. [DOI] [PubMed] [Google Scholar]
- 104.Erkens GB; Hanelt I; Goudsmits JM; Slotboom DJ; van Oijen AM, Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature 2013, 502, 119–123. [DOI] [PubMed] [Google Scholar]
- 105.Hanelt I; Jensen S; Wunnicke D; Slotboom DJ, Low affinity and slow Na+ binding precedes high affinity aspartate binding in the secondary-active transporter GltPh. J Biol Chem 2015, 290, 15962–15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garaeva AA; Slotboom DJ, Elevator-type mechanisms of membrane transport. Biochem Soc Trans 2020, 48, 1227–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sundaram M; Yao SYM; Ng AML; Cass CE; Baldwin SA; Young JD, Equilibrative nucleoside transporters: Mapping regions of interaction for the substrate analogue nitrobenzylthioinosine (NBMPR) using rat chimeric proteins. Biochemistry 2001, 40, 8146–8151. [DOI] [PubMed] [Google Scholar]
- 108.Ward JL; Sherali A; Mo ZP; Tse CM, Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. ENT2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem 2000, 275, 8375–8381. [DOI] [PubMed] [Google Scholar]
- 109.Griffiths M; Beaumont N; Yao SY; Sundaram M; Boumah CE; Davies A; Kwong FY; Coe I; Cass CE; Young JD; Baldwin SA, Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997, 3, 89–93. [DOI] [PubMed] [Google Scholar]
- 110.Endres CJ; Unadkat JD, Residues met89 and ser160 in the human equilibrative nucleoside transporter 1 affect its affinity for adenosine, guanosine, s6-(4-nitrobenzyl)-mercaptopurine riboside, and dipyridamole. Molecular pharmacology 2005, 67, 837–844. [DOI] [PubMed] [Google Scholar]
- 111.Endres CJ; Sengupta DJ; Unadkat JD, Mutation of leucine-92 selectively reduces the apparent affinity of inosine, guanosine, NBMPR [s6-(4-nitrobenzyl)-mercaptopurine riboside] and dilazep for the human equilibrative nucleoside transporter, hENT1. Biochemical Journal 2004, 380, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sundaram M; Yao SYM; Ingram JC; Berry ZA; Abidi F; Cass CE; Baldwin SA; Young JD, Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. Journal of Biological Chemistry 2001, 276, 45270–45275. [DOI] [PubMed] [Google Scholar]
- 113.Vickers MF; Mani RS; Sundaram M; Hogue DL; Young JD; Baldwin SA; Cass CE, Functional production and reconstitution of the human equilibrative nucleoside transporter (hENT1) in Saccharomyces cerevisiae. Interaction of inhibitors of nucleoside transport with recombinant hENT1 and a glycosylation-defective derivative (hENT1/N48Q. The Biochemical journal 1999, 339 ( Pt 1), 21–32. [PMC free article] [PubMed] [Google Scholar]
- 114.Agbanyo FR; Cass CE; Paterson AR, External location of sites on pig erythrocyte membranes that bind nitrobenzylthioinosine. Molecular pharmacology 1988, 33, 332–337. [PubMed] [Google Scholar]
- 115.Jarvis SM; Young JD, Nucleoside transport in rat erythrocytes: Two components with differences in sensitivity to inhibition by nitrobenzylthioinosine and p-chloromercuriphenyl sulfonate. J Membr Biol 1986, 93, 1–10. [DOI] [PubMed] [Google Scholar]
- 116.Crawford CR; Patel DH; Naeve C; Belt JA, Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. J Biol Chem 1998, 273, 5288–5293. [DOI] [PubMed] [Google Scholar]
- 117.Baldwin SA; Yao SY; Hyde RJ; Ng AM; Foppolo S; Barnes K; Ritzel MW; Cass CE; Young JD, Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem 2005, 280, 15880–15887. [DOI] [PubMed] [Google Scholar]
- 118.Hyde RJ; Cass CE; Young JD; Baldwin SA, The ENT family of eukaryote nucleoside and nucleobase transporters: Recent advances in the investigation of structure/function relationships and the identification of novel isoforms. Mol Membr Biol 2001, 18, 53–63. [PubMed] [Google Scholar]
- 119.Barnes K; Dobrzynski H; Foppolo S; Beal PR; Ismat F; Scullion ER; Sun L; Tellez J; Ritzel MW; Claycomb WC; Cass CE; Young JD; Billeter-Clark R; Boyett MR; Baldwin SA, Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circ Res 2006, 99, 510–519. [DOI] [PubMed] [Google Scholar]
- 120.Acimovic Y; Coe IR, Molecular evolution of the equilibrative nucleoside transporter family: Identification of novel family members in prokaryotes and eukaryotes. Mol Biol Evol 2002, 19, 2199–2210. [DOI] [PubMed] [Google Scholar]
- 121.Engel K; Zhou M; Wang J, Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem 2004, 279, 50042–50049. [DOI] [PubMed] [Google Scholar]
- 122.Yao SYM; Ng AML; Cass CE; Baldwin SA; Young JD, Nucleobase transport by human equilibrative nucleoside transporter 1 (hENT1). Journal of Biological Chemistry 2011, 286, 32552–32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rahman MF; Askwith C; Govindarajan R, Molecular determinants of acidic pH-dependent transport of human equilibrative nucleoside transporter 3. J Biol Chem 2017, 292, 14775–14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhou M; Xia L; Engel K; Wang J, Molecular determinants of substrate selectivity of a novel organic cation transporter (PMAT) in the SLC29 family. J Biol Chem 2007, 282, 3188–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Singh A; Govindarajan R, ENT3 utilizes a pH sensing mechanism for transport. Channels (Austin) 2018, 12, 78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang C; Leung GP, Equilibrative nucleoside transporters 1 and 4: Which one is a better target for cardioprotection against ischemia-reperfusion injury? J Cardiovasc Pharmacol 2015, 65, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Baldwin SA; Beal PR; Yao SY; King AE; Cass CE; Young JD, The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 2004, 447, 735–743. [DOI] [PubMed] [Google Scholar]
- 128.Eltzschig HK; Abdulla P; Hoffman E; Hamilton KE; Daniels D; Schonfeld C; Loffler M; Reyes G; Duszenko M; Karhausen J; Robinson A; Westerman KA; Coe IR; Colgan SP, HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 2005, 202, 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leisewitz AV; Zimmerman EI; Huang M; Jones SZ; Yang J; Graves LM, Regulation of ENT1 expression and ENT1-dependent nucleoside transport by c-Jun N-terminal kinase. Biochem Biophys Res Commun 2011, 404, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Abdulla P; Coe IR, Characterization and functional analysis of the promoter for the human equilibrative nucleoside transporter gene, hENT1. Nucleosides, nucleotides & nucleic acids 2007, 26, 99–110. [DOI] [PubMed] [Google Scholar]
- 131.Bhatti S; Jamil A; Siddiqui SH; Yaqoob U; Virk LN; Bhatti A, The h syndrome: A genodermatosis. Cureus 2018, e2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cliffe ST; Kramer JM; Hussain K; Robben JH; de Jong EK; de Brouwer AP; Nibbeling E; Kamsteeg EJ; Wong M; Prendiville J; James C; Padidela R; Becknell C; van Bokhoven H; Deen PM; Hennekam RC; Lindeman R; Schenck A; Roscioli T; Buckley MF, SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum Mol Genet 2009, 18, 2257–2265. [DOI] [PubMed] [Google Scholar]
- 133.Morgan NV; Morris MR; Cangul H; Gleeson D; Straatman-Iwanowska A; Davies N; Keenan S; Pasha S; Rahman F; Gentle D; Vreeswijk MP; Devilee P; Knowles MA; Ceylaner S; Trembath RC; Dalence C; Kismet E; Koseoglu V; Rossbach HC; Gissen P; Tannahill D; Maher ER, Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet 2010, 6, e1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Campeau PM; Lu JT; Sule G; Jiang MM; Bae Y; Madan S; Hogler W; Shaw NJ; Mumm S; Gibbs RA; Whyte MP; Lee BH, Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis. Hum Mol Genet 2012, 21, 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cagdas D; Surucu N; Tan C; Kayaoglu B; Ozgul RK; Akkaya-Ulum YZ; Aydinoglu AT; Aytac S; Gumruk F; Balci-Hayta B; Balci-Peynircioglu B; Ozen S; Gursel M; Tezcan I, Autoinflammation in addition to combined immunodeficiency: SLC29A3 gene defect. Mol Immunol 2020, 121, 28–37. [DOI] [PubMed] [Google Scholar]
- 136.Nair S; Strohecker AM; Persaud AK; Bissa B; Muruganandan S; McElroy C; Pathak R; Williams M; Raj R; Kaddoumi A; Sparreboom A; Beedle AM; Govindarajan R, Adult stem cell deficits drive SLC29A3 disorders in mice. Nat Commun 2019, 10, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hsu CL; Lin W; Seshasayee D; Chen YH; Ding X; Lin Z; Suto E; Huang Z; Lee WP; Park H; Xu M; Sun M; Rangell L; Lutman JL; Ulufatu S; Stefanich E; Chalouni C; Sagolla M; Diehl L; Fielder P; Dean B; Balazs M; Martin F, Equilibrative nucleoside transporter 3 deficiency perturbs lysosome function and macrophage homeostasis. Science 2012, 335, 89–92. [DOI] [PubMed] [Google Scholar]
- 138.Wei CW; Lee CY; Lee DJ; Chu CF; Wang JC; Wang TC; Jane WN; Chang ZF; Leu CM; Dzhagalov IL; Hsu CL, Equilibrative nucleoside transporter 3 regulates t cell homeostasis by coordinating lysosomal function with nucleoside availability. Cell Rep 2018, 23, 2330–2341. [DOI] [PubMed] [Google Scholar]
- 139.Bakhchane A; Kindil Z; Charoute H; Benchikhi K; Khadir K; Nadifi S; Baline K; Roky R; Barakat A, Compound heterozygous SLC29A3 mutation causes h syndrome in a moroccan patient: A case report. Curr Res Transl Med 2016, 64, 65–68. [DOI] [PubMed] [Google Scholar]
- 140.Noavar S; Behroozi S; Tatarcheh T; Parvini F; Foroutan M; Fahimi H, A novel homozygous frame-shift mutation in the SLC29A3 gene: A new case report and review of literature. BMC Med Genet 2019, 20, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kang N; Jun AH; Bhutia YD; Kannan N; Unadkat JD; Govindarajan R, Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J Biol Chem 2010, 285, 28343–28352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wright NJ; Lee S-Y, Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nature Structural & Molecular Biology 2019, 26, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Choi DS; Cascini MG; Mailliard W; Young H; Paredes P; McMahon T; Diamond I; Bonci A; Messing RO, The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat Neurosci 2004, 7, 855–861. [DOI] [PubMed] [Google Scholar]
- 144.Nam HW; Hinton DJ; Kang NY; Kim T; Lee MR; Oliveros A; Adams C; Ruby CL; Choi DS, Adenosine transporter ent1 regulates the acquisition of goal-directed behavior and ethanol drinking through A2A receptor in the dorsomedial striatum. J Neurosci 2013, 33, 4329–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim JH; Karpyak VM; Biernacka JM; Nam HW; Lee MR; Preuss UW; Zill P; Yoon G; Colby C; Mrazek DA; Choi DS, Functional role of the polymorphic 647 T/C variant of ENT1 (SLC29A1) and its association with alcohol withdrawal seizures. PLoS One 2011, 6, e16331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen J; Rinaldo L; Lim SJ; Young H; Messing RO; Choi DS, The type 1 equilibrative nucleoside transporter regulates anxiety-like behavior in mice. Genes Brain Behav 2007, 6, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schaller L; Lauschke VM, The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet 2019, 138, 1359–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Leonardi-Bee J; Bath PM; Bousser MG; Davalos A; Diener HC; Guiraud-Chaumeil B; Sivenius J; Yatsu F; Dewey ME; Dipyridamole in Stroke C, Dipyridamole for preventing recurrent ischemic stroke and other vascular events: A meta-analysis of individual patient data from randomized controlled trials. Stroke 2005, 36, 162–168. [DOI] [PubMed] [Google Scholar]
- 149.Fitzgerald G, Drug therapy: Dipyridamole. The New England Journal of Medicine 1987, 316, 1247–1257. [DOI] [PubMed] [Google Scholar]
- 150.Marzilli M; Trivelia MG; Levantesi D; Pelosi G; Vacche MD; Taddei L; rsquo; Abbate A, Effect of dilazep on coronary and systemic circulations. Pharmacology 1985, 31, 82–87. [DOI] [PubMed] [Google Scholar]
- 151.Hammond JR, Interaction of a series of draflazine analogues with equilibrative nucleoside transporters: Species differences and transporter subtype selectivity. Naunyn Schmiedebergs Arch Pharmacol 2000, 361, 373–382. [DOI] [PubMed] [Google Scholar]
- 152.Armstrong D; Summers C; Ewart L; Nylander S; Sidaway JE; van Giezen JJJ, Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. Journal of Cardiovascular Pharmacology and Therapeutics 2014, 19, 209–219. [DOI] [PubMed] [Google Scholar]
- 153.Aungraheeta R; Conibear A; Butler M; Kelly E; Nylander S; Mumford A; Mundell SJ, Inverse agonism at the P2Y12 receptor and ENT1 transporter blockade contribute to platelet inhibition by ticagrelor. Blood 2016, 128, 2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Parker WAE; Eriksson N; Becker RC; Voora D; Akerblom A; Himmelmann A; James SK; Wallentin L; Storey RF; Investigators P, Equilibrative nucleoside transporter 1 gene polymorphisms and clinical outcomes following acute coronary syndromes: Findings from the PLATelet inhibition and patient Outcomes (PLATO) study. Platelets 2019, 30, 579–588. [DOI] [PubMed] [Google Scholar]
- 155.van den Berg TN; El Messaoudi S; Rongen GA; van den Broek PH; Bilos A; Donders AR; Gomes ME; Riksen NP, Ticagrelor does not inhibit adenosine transport at relevant concentrations: A randomized cross-over study in healthy subjects in vivo. PLoS One 2015, 10, e0137560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nagy LE; Diamond I; Casso DJ; Franklin C; Gordon AS, Ethanol increases extracellular adenosine by inhibiting adenosine uptake via the nucleoside transporter. J Biol Chem 1990, 265, 1946–1951. [PubMed] [Google Scholar]
- 157.Ramadan A; Naydenova Z; Stevanovic K; Rose JB; Coe IR, The adenosine transporter, ENT1, in cardiomyocytes is sensitive to inhibition by ethanol in a kinase-dependent manner: Implications for ethanol-dependent cardioprotection and nucleoside analog drug cytotoxicity. Purinergic Signal 2014, 10, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Allen-Gipson DS; Jarrell JC; Bailey KL; Robinson JE; Kharbanda KK; Sisson JH; Wyatt TA, Ethanol blocks adenosine uptake via inhibiting the nucleoside transport system in bronchial epithelial cells. Alcohol Clin Exp Res 2009, 33, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Abt ER; Rosser EW; Durst MA; Lok V; Poddar S; Le TM; Cho A; Kim W; Wei L; Song J; Capri JR; Xu S; Wu N; Slavik R; Jung ME; Damoiseaux R; Czernin J; Donahue TR; Lavie A; Radu CG, Metabolic modifier screen reveals secondary targets of protein kinase inhibitors within nucleotide metabolism. Cell Chem Biol 2020, 27, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li RW; Tse CM; Man RY; Vanhoutte PM; Leung GP, Inhibition of human equilibrative nucleoside transporters by dihydropyridine-type calcium channel antagonists. Eur J Pharmacol 2007, 568, 75–82. [DOI] [PubMed] [Google Scholar]
- 161.Carrier EJ; Auchampach JA; Hillard CJ, Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proc Natl Acad Sci U S A 2006, 103, 7895–7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bicket A; Coe IR, N-linked glycosylation of N48 is required for equilibrative nucleoside transporter 1 (ENT1) function. Bioscience Reports 2016, 36, e00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hughes SJ; Cravetchi X; Vilas G; Hammond JR, Adenosine a1 receptor activation modulates human equilibrative nucleoside transporter 1 (hENT1) activity via PKC-mediated phosphorylation of serine-281. Cell. Signal. 2015, 27, 1008–1018. [DOI] [PubMed] [Google Scholar]
- 164.Reyes G; Nivillac NMI; Karim MZ; Desouza L; Siu KWM; Coe IR, The equilibrative nucleoside transporter (ENT1) can be phosphorylated at multiple sites by PKC and PKA. Mol. Membr. Biol. 2011, 28, 412–426. [DOI] [PubMed] [Google Scholar]
- 165.Aseervatham J; Tran L; Machaca K; Boudker O, The role of flexible loops in folding, trafficking and activity of equilibrative nucleoside transporters. PloS One 2015, 10, e0136779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Visser F; Sun L; Damaraju V; Tackaberry T; Peng Y; Robins MJ; Baldwin SA; Young JD; Cass CE, Residues 334 and 338 in transmembrane segment 8 of human equilibrative nucleoside transporter 1 are important determinants of inhibitor sensitivity, protein folding, and catalytic turnover. Journal of Biological Chemistry 2007, 282, 14148–14157. [DOI] [PubMed] [Google Scholar]
- 167.Visser F; Baldwin SA; Isaac RE; Young JD; Cass CE, Identification and mutational analysis of amino acid residues involved in dipyridamole interactions with human and Caenorhabditis elegans equilibrative nucleoside transporters. Journal of Biological Chemistry 2005, 280, 11025–11034. [DOI] [PubMed] [Google Scholar]
- 168.Visser F; Zhang J; Raborn RT; Baldwin SA; Young JD; Cass CE, Residue 33 of human equilibrative nucleoside transporter 2 is a functionally important component of both the dipyridamole and nucleoside binding sites. Molecular pharmacology 2005, 67, 1291–1298. [DOI] [PubMed] [Google Scholar]
- 169.SenGupta DJ; Unadkat JD, Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochemical Pharmacology 2004, 67, 453–458. [DOI] [PubMed] [Google Scholar]
- 170.Arastu-Kapur S; Arendt CS; Purnat T; Carter NS; Ullman B, Second-site suppression of a nonfunctional mutation within the Leishmania donovani inosine-guanosine transporter. Journal of Biological Chemistry 2005, 280, 2213–2219. [DOI] [PubMed] [Google Scholar]
- 171.Arastu-Kapur S; Ford E; Ullman B; Carter NS, Functional analysis of an inosine-guanosine transporter from Leishmania donovani. The role of conserved residues, aspartate 389 and arginine 393. J Biol Chem 2003, 278, 33327–33333. [DOI] [PubMed] [Google Scholar]
- 172.Damaraju VL; Damaraju S; Young JD; Baldwin SA; Mackey J; Sawyer MB; Cass CE, Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [DOI] [PubMed] [Google Scholar]
- 173.Wang SC; Davejan P; Hendargo KJ; Javadi-Razaz I; Chou A; Yee DC; Ghazi F; Lam KJK; Conn AM; Madrigal A; Medrano-Soto A; Saier MH Jr., Expansion of the major facilitator superfamily (MFS) to include novel transporters as well as transmembrane-acting enzymes. Biochim Biophys Acta Biomembr 2020, 183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Abramson J; Smirnova I; Kasho V; Verner G; Kaback HR; Iwata S, Structure and mechanism of the lactose permease of escherichia coli. Science 2003, 301, 610–615. [DOI] [PubMed] [Google Scholar]
- 175.Dang S; Sun L; Huang Y; Lu F; Liu Y; Gong H; Wang J; Yan N, Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734–738. [DOI] [PubMed] [Google Scholar]
- 176.Valdés R; Elferich J; Shinde U; Landfear SM, Identification of the intracellular gate for a member of the equilibrative nucleoside transporter (ENT) family. J. Biol. Chem. 2014, 289, 8799–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Valdés R; Arastu-Kapur S; Landfear SM; Shinde U, An ab initio structural model of a nucleoside permease predicts functionally important residues. J. Biol. Chem. 2009, 284, 19067–19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Roy A; Kucukural A; Zhang Y, I-tasser: A unified platform for automated protein structure and function prediction. Nat Protoc 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kallberg M; Wang H; Wang S; Peng J; Wang Z; Lu H; Xu J, Template-based protein structure modeling using the RaptorX web server. Nat Protoc 2012, 7, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Huang W; Zeng X; Shi Y; Liu M, Functional characterization of human equilibrative nucleoside transporter 1. Protein Cell 2017, 8, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rehan S; Jaakola V-P, Expression, purification and functional characterization of human equilibrative nucleoside transporter subtype-1 (hENT1) protein from Sf9 insect cells. Protein Expr. Purif. 2015, 114, 99–107. [DOI] [PubMed] [Google Scholar]
- 182.Boswell-Casteel RC; Johnson JM; Roe-Žurž Z; Duggan KD; Schmitz H; Hays FA, Expression and purification of human and Saccharomyces cerevisiae equilibrative nucleoside transporters. Protein Expr. Purif. 2018, 142, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Quistgaard EM; Löw C; Guettou F; Nordlund P, Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. [DOI] [PubMed] [Google Scholar]
- 184.Lee Y; Nishizawa T; Yamashita K; Ishitani R; Nureki O, Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nat Commun 2015, 6, 6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jaehme M; Guskov A; Slotboom DJ, Crystal structure of the vitamin B3 transporter PnuC, a full-length SWEET homolog. Nat Struct Mol Biol 2014, 21, 1013–1015. [DOI] [PubMed] [Google Scholar]
- 186.Xu Y; Tao Y; Cheung LS; Fan C; Chen LQ; Xu S; Perry K; Frommer WB; Feng L, Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 2014, 515, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pao SS; Paulsen IT; Saier MH Jr., Major facilitator superfamily. Microbiol Mol Biol Rev 1998, 62, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Madej MG; Dang S; Yan N; Kaback HR, Evolutionary mix-and-match with MFS transporters. Proc Natl Acad Sci U S A 2013, 110, 5870–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Deng D; Sun P; Yan C; Ke M; Jiang X; Xiong L; Ren W; Hirata K; Yamamoto M; Fan S; Yan N, Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. [DOI] [PubMed] [Google Scholar]
- 190.Nomura N; Verdon G; Kang HJ; Shimamura T; Nomura Y; Sonoda Y; Hussien SA; Qureshi AA; Coincon M; Sato Y; Abe H; Nakada-Nakura Y; Hino T; Arakawa T; Kusano-Arai O; Iwanari H; Murata T; Kobayashi T; Hamakubo T; Kasahara M; Iwata S; Drew D, Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 2015, 526, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Qureshi AA; Suades A; Matsuoka R; Brock J; McComas SE; Nji E; Orellana L; Claesson M; Delemotte L; Drew D, The molecular basis for sugar import in malaria parasites. Nature 2020, 578, 321–325. [DOI] [PubMed] [Google Scholar]
- 192.Mitchell P, A general theory of membrane transport from studies of bacteria. Nature 1957, 180, 134–136. [DOI] [PubMed] [Google Scholar]
- 193.Deng D; Xu C; Sun P; Wu J; Yan C; Hu M; Yan N, Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [DOI] [PubMed] [Google Scholar]
- 194.Singh SK; Piscitelli CL; Yamashita A; Gouaux E, A competitive inhibitor traps LeuT in an open-to-out conformation. Science 2008, 322, 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Coleman JA; Green EM; Gouaux E, X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Penmatsa A; Wang KH; Gouaux E, X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yao SYM; Ng AML; Cass CE; Young JD, Role of cysteine 416 in N-ethylmaleimide sensitivity of human equilibrative nucleoside transporter 1 (hENT1). The Biochemical journal 2018, 475, 3293–3309. [DOI] [PubMed] [Google Scholar]
- 198.Al-Haggar M; Salem N; Wahba Y; Ahmad N; Jonard L; Abdel-Hady D; El-Hawary A; El-Sharkawy A; Eid AR; El-Hawary A, Novel homozygous SLC29A3 mutations among two unrelated Egyptian families with spectral features of H-syndrome. Pediatr Diabetes 2015, 16, 305–316. [DOI] [PubMed] [Google Scholar]
- 199.Molho-Pessach V; Lerer I; Abeliovich D; Agha Z; Abu Libdeh A; Broshtilova V; Elpeleg O; Zlotogorski A, The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet 2008, 83, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Melki I; Lambot K; Jonard L; Couloigner V; Quartier P; Neven B; Bader-Meunier B, Mutation in the SLC29A3 gene: A new cause of a monogenic, autoinflammatory condition. Pediatrics 2013, 131, e1308–1313. [DOI] [PubMed] [Google Scholar]
- 201.Farooq M; Moustafa RM; Fujimoto A; Fujikawa H; Abbas O; Kibbi AG; Kurban M; Shimomura Y, Identification of two novel mutations in SLC29A3 encoding an equilibrative nucleoside transporter (hENT3) in two distinct syrian families with H syndrome: Expression studies of SLC29A3 (hENT3) in human skin. Dermatology 2012, 224, 277–284. [DOI] [PubMed] [Google Scholar]
- 202.Jurrus E; Engel D; Star K; Monson K; Brandi J; Felberg LE; Brookes DH; Wilson L; Chen J; Liles K; Chun M; Li P; Gohara DW; Dolinsky T; Konecny R; Koes DR; Nielsen JE; Head-Gordon T; Geng W; Krasny R; Wei GW; Holst MJ; McCammon JA; Baker NA, Improvements to the APBS biomolecular solvation software suite. Protein Sci 2018, 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Arimany-Nardi C; Claudio-Montero A; Viel-Oliva A; Schmidtke P; Estarellas C; Barril X; Bidon-Chanal A; Pastor-Anglada M, Identification and characterization of a secondary sodium-binding site and the main selectivity determinants in the human concentrative nucleoside transporter 3. Mol Pharm 2017, 14, 1980–1987. [DOI] [PubMed] [Google Scholar]
- 204.Errasti-Murugarren E; Molina-Arcas M; Casado FJ; Pastor-Anglada M, The human concentrative nucleoside transporter-3 C602R variant shows impaired sorting to lipid rafts and altered specificity for nucleoside-derived drugs. Molecular pharmacology 2010, 78, 157–165. [DOI] [PubMed] [Google Scholar]
- 205.Zhou Y; Liao L; Wang C; Li J; Chi P; Xiao Q; Liu Q; Guo L; Sun L; Deng D, Cryo-EM structure of the human concentrative nucleoside transporter CNT3. PLoS Biol 2020, 18, e3000790. [DOI] [PMC free article] [PubMed] [Google Scholar]