

Abstract
Background
The identification of species or species groups with specific oligo-nucleotides as molecular signatures is becoming increasingly popular for bacterial samples. However, it shows also great promise for other small organisms that are taxonomically difficult to tract.
Results
We have devised here an algorithm that aims to find the optimal probes for any given set of sequences. The program requires only a crude alignment of these sequences as input and is optimized for performance to deal also with very large datasets. The algorithm is designed such that the position of mismatches in the probes influences the selection and makes provision of single nucleotide outloops. Program implementations are available for Linux and Windows.
Background
Identification of species with molecular probes is likely to revolutionize taxonomy, at least for taxa with morphological characters that are difficult to determine otherwise. Among these are the single cell eucaryotes, such as Ciliates and Flagellates, but also many other kinds of small organisms, such as Nematodes, Rotifers, Crustaceans, mites, Annelids or Insect larvae. These organisms constitute the meiofauna in water and soil, which is of profound importance in the ecological network. Efficient ways for monitoring species identity and abundance in the meiofauna should significantly help to understand ecological processes.
Molecular taxonomy with sequence specific oligo-nucleotide probes has been pioneered for bacteria [1,2]. Probes that are specific to particular species or groups of related species can be used in fluorescent in situ hybridization assays to detect the species in complex mixtures or as symbionts of other organisms [3,4]. Alternatively, the microarray technology is increasingly used for this purpose, allowing potentially the parallel screening of many different species. Most of the species-specific sequences that are used so far for this purpose are derived from ribosomal RNA sequences. However, any other sequence is also potentially suitable, as for example mitochondrial D-loop sequences in eucaryotes.
The species-specific probes are usually derived from an alignment of the respective sequences, where conserved and non-conserved regions are directly visible. A program has been developed for ribosomal sequences that helps to build the relevant database, and supports the selection of suitable specific sequences (ARB [5]). In this, a correct alignment is crucial for finding the optimal probes, but alignments are problematical in poorly conserved regions. These, on the other hand, have the highest potential to yield specific probes. Moreover, the current implementation of probe finding calculates only the number of mismatching position to discriminate between the probes, but does not take into account the position of the mismatches within the stretches, which could influence the hybridization behavior. We have therefore devised here a new algorithm that allows working with datasets that need not to be carefully aligned and that takes the position of mismatches along the recognition sequence into account.
The algorithm
The algorithm includes three parts. The first one aims to provide a function that calculates the relative stability of matching oligos in dependence of the number and position of mismatches. The second one provides a strategy for probe finding that scans all possible sequence combinations, but works time efficient. The third part deals with matches caused by single nucleotide outloops of a given sequence.
Stability function
Extensive studies exist for assessing the thermodynamic consequences of internal mismatches in short oligo-nucleotides (see fro example [6,7]). These show that there are no simple rules and that the exact influence on the stability of a hybrid depends on the nature of the mismatch, as well as its flanking nucleotides. For example, mismatches including a G (i.e. G-G, G-T and G-A) tend to be less destabilizing than the other types of mismatches [7], although this can not directly be predicted from steric considerations. Comparable systematic studies on the relative influence of the position of the mismatch within the oligonucleotide do not exist yet, although it is clear that the influence is lower at the ends than in more central positions [7,9]. Preliminary evidence with an oligo-dT stretch harboring A mismatches along the sequence suggests that the position dependence could be a continuous function [8]. We have therefore decided to use an ad hoc approach for the stability calculation that is mainly designed to discriminate against sequences with more central mismatch positions.
We model the relative stability of mismatched oligos as follows. The position of the mismatch can be considered to be a "weak point". The location of the "weak point" is expressed as a probability function that takes into account the differential contribution of central versus terminal positions. The probability that the "weak point" is at position x is defined by p1. Under the experimental conditions of melting, the presence of the "weak point" is true, meaning that [sum(p1) for all x] = 1.
We assume a Gauss distribution as the respective probability function, with the maximum in the middle of the duplex and the integral value along the duplex length set to 1 (Equation 1).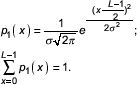
Equation 1. "Weak point" location probability. L – duplex length, σ – distribution parameter, x – duplex position.
Note that the function in Equation 1 refers to discrete positions within the sequence, while the Gauss distribution is continuous and the integration from -∞ to +∞ is set to yield 1. The parameter σ is therefore chosen such that the discrete sum approaches 1 at any intended precision. In the program discussed below the accuracy of the sum value is 0.999.
Although the preliminary experimental evidence [8] suggests that the destabilization function can be approximated with the Gauss distribution, the program implementation allows also to use a flat distribution, i.e. where a position-independent effect on the melting is assumed as an alternative, to compare the outputs of the two different assumptions.
For assessing the relative amount of destabilization caused by a certain mismatch, we assume that the mismatch disturbs the surrounding base pairs from (y-n) to (y+n) positions, n can be called a border parameter that will need to be experimentally verified in the future. Because n can currently only be guessed, it is set as a program variable with a default value of 5. n might also depend on the nature of the mismatch, i.e. some types of mismatches might influence the surrounding bases less than the others. We therefore implemented further program variables that allow to define a different n depending on the nature of the mismatch (i.e. it is possible to set a particular n value for each possible type of mismatch).
The overall relative stability of a given duplex is then expressed as a probability function. It is expressed as the sum of products of the individual position probabilities p1 (determined by the stability function) and p2 (determined by the border parameter). The value of p2 it the probability of "melting", conditioned that the "weak point" is disturbed. (Equation 2).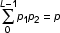
Equation 2. L – the length of the duplex, p1 – the "weak point" location probability, p2 – the "melting" probability due to the disturbance of the "weak point".
p2 is a conditional probability of "melting" with p2 = 1 if the "weak point" is disturbed (in the region y ± n) and p2 = 0 at non-affected positions. This allows transforming Equation 2 into Equation 3.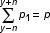
Equation 3. y – the mismatch position, n – the border parameter
p1 can then be substituted by the function in Equation 1, to yield Equation 4.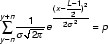
Equation 4: x – the duplex position, y – the mismatch position, n – the border parameter, σ-distribution parameter
In the case of several mismatches, the summing is done along all the respective mismatch regions. If the mismatches occur next to each other, their disturbed regions simply overlap and the summing is performed across the respective region.
Probe finding
The probe finding strategy is devised in a way (i) to avoid the need for exact alignments, (ii) to check probe specificity along the whole available sequence and (iii) to optimize performance. The workflow is depicted in Figure 1. It starts with a database in which each organism is represented by a single continuous sequence, such as a defined region of the 18S or 28S ribosomal genes. From this it takes first the sequences of the In-group organism(s) for which specific probes should be found and cuts these into short pieces of the specified oligo-nucleotide length (set as a program variable), following an approach proposed by Bavykin et al [11]. This is accomplished by a sliding window scheme with 1-nucleotide shifts across the whole length of the sequence(s). Two separate lists are created in this way. The first list is simply a straight list of all possible fragments from all In-group organisms. The second one consists of an array of lists for each of the In-group organisms (the two lists are identical if only one In-group organism is chosen). All duplicate oligos from the first list are then removed and each of the remaining oligos is checked whether it matches with each of the In-group organisms in the second list. A match is positive, when the relative melting probability is within the range of 0–25%, employing the function of Equation 4. Thus, this first calculation simply ensures that all candidate probes match with all In-group organisms. This calculation would be largely dispensable, if only a single In-group organism is chosen.
Figure 1.

Scheme of the probe finding algorithm. Details are explained in the text.
The next step is to subtract all oligos that match in any of the Out-group organisms. To avoid the comparison of all candidate oligos against all Out-group sequences, we identify first a group of sequences that is closely related to the In-group. For this one requires a rough alignment of all sequences, to calculate percentage similarity between them. Note that this serves only to identify a subgroup of sequences for speeding up the calculations, i.e. mistakes in the alignment are of no concern. The similarity calculator in the program extracts this related group of sequences by a simple percentage identity calculation across the given alignment. All sequences that are at least 90% similar to the In-group are used as Related-group. This percentage can be set as a program variable and should be set such that the Related-group does not become more than 5–10% of all sequences.
The sequences of the Related-group are again converted into a fragment list as above, duplicates are removed and all candidate oligos are matched with this list. Now only those oligos are retained, which have a melting probability of at least 75% (the exact percentage values are program variables). The majority of oligos is removed in this step. The remaining candidate oligos are then matched against the remaining sequences in the Out-group with the same cut-off criterion.
This stepwise selection scheme allows to significantly speed up the calculations even for very large datasets, but still ensures that all oligo-nucleotides of the desired length were directly or indirectly matched against all possible other oligos in the database.
Single nucleotide loops
Structure analysis with experimental oligo-nucleotides has shown that in a pair of hybridized oligos, one nucleotide can loop out, without interfering much with the stability of the hybridized pair [12]. This implies that one base of one strand of a duplex can loop out from the duplex and the rest of the strand can shift one position. This is depicted in Figure 2. A standard linear scanning algorithm would recognize the situation at the left as one with 11 mismatches, i.e. would suggest it as a specific probe. However, if the single nucleotide loop is taken into account, the match would be perfect and the probe would have to be considered as unspecific. Our scanning algorithm takes this problem into account by re-checking all candidate probes after the completion of the filtering steps. It does this by sequentially removing one nucleotide from the candidate probe and shifting the remainder by one position. The melting probability of the new oligo is then calculated and checked. The removed nucleotide is then reinserted and the cycle is repeated for the next position. The same procedure is done for the target sequence, so that outloops are considered to be possible on both strands of the duplex. Note that outloops of two nucleotides are considered to destabilize the helix too much to warrant a separate analogous calculation.
Figure 2.

Scheme of the single-nucleotide outloop problem; asterisks represent mismatches, columns represent matches.
Parallel computation
A parallel program version allows probe finding to be done in parallel on several processors. Essentially the same algorithm is used in the parallel version of the program, whereby the parallelism is introduced in the matching steps. Each process takes its own part of the database and performs the matching as well as the stability calculations. The results are then gathered by the root process and superimposed.
Program implementation
The algorithm is implemented in a program called PROBE. The program consists of three modules that can be used independently. The first module finds the probes based on the given task (specificity group, length of probes, source database).
The second one is the analytic module, which can be used if it is impossible to design a probe for a given organism group. This module depicts the situation with the given In-group and enables to find the closest group for which the task can be accomplished. The use of the analytic mode comes into play when PROBE fails to identify a set of probes for the given organism group. Such a failure can have two reasons – either there is no probe, which identifies all organisms in the specificity group, or there is another organism outside the specificity group, which is also identified by all candidate probes suitable for the specificity group.
For the first case, the specificity group must be broken down into several subgroups and the probes must be identified for these subgroups separately. For the second case, the organism that is very similar to the specificity group should be added to the specificity group and this may then have to be broken down into smaller subgroups.
The analytic module creates a table with the organisms of the specificity group as well as the most related organisms. This table depicts then the matching or non-matching patterns for each of the possible probes, allowing a simple visual inspection of the best specificity groups. The output can be viewed and modified with spreadsheet programs such as Excel.
The third module provides a report for the identified probe, including the mismatches in the duplexes within the specificity group, the best match out of the group and some other information.
The program is written in standard C++ in a platform independent manner. Therefore, the program can be easily compiled for Linux and Windows without any modifications. The program binary files for Linux and Windows are available from the http://biochip.genetik.uni-koeln.de/probe as freeware accompanied with all its source files, and a manual that describes further details.
Results
As an example of the performance of the program we have used the full SSU database (RDP, release 8.1) [13] containing approximately 16.000 sequences to find a specific oligo-nucleotide probe with a length on 20 nt for Thermotoga maritima. The search was done on a Pentium III (800 MHz, 512 MB RAM) PC and took about 1.5 hours without outlooping and 16 hours with outlooping, indicating that the most time intensive step is the outlooping subroutine. The parallel version running on a cluster with 24 nodes (with the slowest node being a Pentium II – 400 MHz with 256 MB RAM) took 2 hours for the same full task.
Figure 3 depicts the output from the check module, which allows comparing the oligos and their specificity that were found in this particular comparison. It shows that ARB suggests two oligos that are rejected by PROBE either because of mismatches occurring only at the ends, or under the outloop routine. Both programs find one oligo with acceptable high specificity.
Figure 3.

Comparison of specific oligos suggested by ARB and PROBE for Thermotoga maritima, in comparison to the whole SSU database. A) Oligo suggested by ARB, but found to have lower than 70% melting probability in two other species. This was therefore rejected by PROBE because of insufficient specificity. B) Oligo suggested by ARB, but found to have lower than 70% melting probability when outlooping is considered. This was therefore also rejected by PROBE because of insufficient specificity. C) Oligo suggested by both programs, whereby the best outgroup matches have a higher than 70% melting probability.
Discussion
The algorithm presented here does not take into account the effect of relative GC content and stacking interactions of neighboring bases on the melting temperature of the oligo-nucleotides. Accordingly, the oligo-nucleotides suggested by the program can differ significantly in melting temperature. However, as this can easily be adjusted after the selection is made, we have not included a subroutine that takes GC content into account during the primary search, because this would slow down the calculations. Furthermore, we expect that GC content differences may be of less importance for the applications envisioned here, because they can be largely compensated by the choice of experimental conditions, such as buffers that compensate stability differences [13].
A more general problem is our way of calculating the relative stability factor. This does currently not take the nucleotide composition into account either. The reason is that there are too few experimental data as yet, that would allow to unequivocally include this in the calculations. The current experimental data sets focus on the types of mismatches in particular contexts, but not systematically on position specific effects [7,15]. Moreover, they deal with relatively short model oligos only (up to 12 nt). However, the probes used for species identification are longer and the different effects can currently not be accurately assessed from experimental data for such longer probes. In our equation, it is mainly the border parameter n that would be affected by base composition and nearest neighbor interactions and we have therefore left this as a variable that can be set according to experimental results. In principle, it seems possible that n differs for different sequence compositions, i.e. GC-rich stretches have a smaller n than AT-rich ones. Thus, if one chooses a low n, one would risk that GC-rich oligos are suggested as specific probes that still show cross hybridization. However, it seems that these can easily be eliminated after the selection is made. Still, if experimental data indicate that this is a major problem, the program could easily accommodate such new insights.
Finally, the stability function proposed in Equation 1 could possibly also have other shapes than Gaussian. Again this is a factor that needs further experiments. If it turns out that other functions are more appropriate, one can include this as additional options into the program. At the present we offer the extreme, namely a flat function, as an alternative option.
Conclusion
We have designed a versatile algorithm for finding optimal species- and group-specific probes for molecular taxonomy that is sufficiently open to implement further experimental insights into the nature of the stability of mismatched oligo-nucleotides.
Contributor Information
Alexander E Pozhitkov, Email: alex.pozhitkov@uni-koeln.de.
Diethard Tautz, Email: tautz@uni-koeln.de.
Acknowledgements
We are grateful to Dr. Lysov from the Engelgardt Institute of Molecular Biology, Russian Academy of Sciences for the supporting A.P. in the initial phase of the project. We thank Prof. Speckenmeyer at the Institute of Informatics, University of Cologne for providing access to their LINUX cluster and Jens Rühmkorf for his help with installing the parallel version. This project was supported by a grant from the Ministerium für Schule Wissenschaft und Forschung des Landes Nordrhein-Westfalen.
References
- Guschin D, Mobarry B, Proudnikov D, Stahl D, Brittman, Mirzabekov A. Oligonucleotide microchips as genosensors for determinative and environmental studies in microbiology. Appl Environ Microbiol. 1997;63:2397–2402. doi: 10.1128/aem.63.6.2397-2402.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann R, Ludwig W. Ribosomal RNA-targeted nucleic acid probes for studies in microbial ecology. FEMS Microbiol Rev. 2000;24:555–565. doi: 10.1016/S0168-6445(00)00044-9. [DOI] [PubMed] [Google Scholar]
- Amann R, Ludwig W, Schleifer K. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E DeLong, G Wickham, N Pace. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science. 1989;243:1360–1363. doi: 10.1126/science.2466341. [DOI] [PubMed] [Google Scholar]
- The ARB project. http://www.arb-home.de/
- Allawi H, SantaLucia J Jr. Thermodynamics and NMR of Internal G•T Mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- Peyret N, Senevirante P, Allawi H, SantaLucia J Jr. Nearest-Neighbor Thermodynamics and NMR of DNA Sequences with Internal A•A, C•C, G•G and T•T Mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- Fodor S, Liphutz R, Xiaohua Huang. The Robert A Welch Foundation 37th Conference on Chemical Research 40 Years of the DNA Double Helix. Houston, Texas. 1993.
- Leijon M, Gräslund A. Effects of sequence and length on imino proton exchange and base pair opening kinetics in DNA oligonucleotide duplexes. Nucleic Acids Res. 1992;20:5339–5343. doi: 10.1093/nar/20.20.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DJ, Kozlowski SA, Ikuta S, Itakura K. Deoxyadenosine-deoxycytidine pairing in the d(C-G-C-G-A-A-T-T-C-A-C-G) duplex: conformation and dynamics at and adjacent to the dA X dC mismatch site. Biochemistry. 1984;23:3218–26. doi: 10.1021/bi00309a016. [DOI] [PubMed] [Google Scholar]
- Bavykin S, Mikhaylovich V, Zakharyev V, Lysov Yu, Kelly J, Flax J, Jackman J, Stahl D, Mitzabekov A. Discrimination of Bacillus anthractis and closely related organisms by analysis of 16S and 23S rRNA with oligonucleotide microchips. Appl Environ Microbiol. 2002. in press .
- Kalnik M, Norman D, Li B, Swann P, Patel D. Conformational transitions in thymidine bulge-containing deoxytridecanucleotide duplexes. Role of flanking sequence and temperature in modulating the equilibrium between looped out and stacked thymidine bulge states. J Biol Chem. 1990;265:636–647. [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Lilburn TG, Parker CT Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior W Jr, von Hippel P. Alteration of the relative stability of dA-dT and dG-dC base pairs in DNA. Proc Natl Acad Sci USA. 1973;70:298–302. doi: 10.1073/pnas.70.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doktycz M, Morris M, Dormady S, Beattie K, Jacobson B. Optical melting of 128 octamer DNA duplexes. Effects of base pair location and nearest neighbors on thermal stability. Journal of Biological Chemistry. 1995;270:8439–8445. doi: 10.1074/jbc.270.15.8439. [DOI] [PubMed] [Google Scholar]