

Abstract
Current anticancer agents suffer from narrow therapeutic indexes and suboptimal therapeutic combinations stemming from mixtures of drugs with dissimilar physical properties. Nanomedicine platforms for drug delivery could address these challenges but it remains unclear whether synergistic free drug ratios translate to nanocarriers and whether nanocarriers with multiple drugs outperform mixtures of single-drug nanocarriers at the same dose. Here, we report a bottlebrush prodrug (BPD) platform to answer these questions in the context of multiple myeloma (MM) therapy. We show that a bortezomib-based BPD monotherapy slows tumor progression in vivo and that mixtures of bortezomib, pomalidomide, and dexamethasone BPDs exhibit in vitro synergistic, additive, or antagonistic patterns distinct from the free drug counterparts. BPDs carrying a statistical mixture of 3 drugs in a synergistic ratio outperform the free drug combination at the same ratio and a mixture of single-drug BPDs in the same ratio. Our results address unanswered questions in the field of nanomedicine, offering design principles for combination nanomedicines and strategies for improving current front-line mono- and combination therapies for MM.
Graphical Abstract
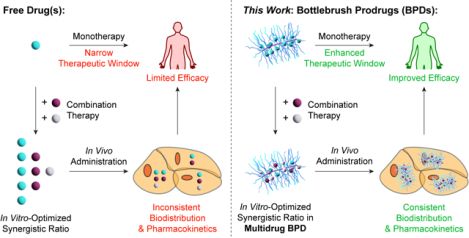
Controlling the tissue exposure of drugs remains the most persistent challenge of modern cancer therapies and the ‘holy grail’ of drug delivery.1–4 By exploiting features such as size, shape, composition, and release kinetics, nanocarriers can enhance the therapeutic indexes (TIs) of drugs by increasing their exposure in diseased sites and/or avoiding major sites of toxicity.1,3,4 The development of nanomedicine combination therapies represents a frontier of modern cancer treatment.5–10 While recent pioneering advancements in cancer biology have greatly improved the ability to identify and predict synthetic lethalities of drug combinations, the clinical translation of such combinations suffers from fundamental barriers.5–9 For instance, due to the distinct physical properties of dissimilar drugs, combinations of those drugs that are synergistic in vitro may not accumulate in target tissues/cells in vivo.6–10 Due to this disconnect, many clinical combination therapies are derived empirically based on the maximum tolerated dose (MTD) of each component drug rather than rational synergies.6–8 Combination therapies present an exciting nanomedicine opportunity wherein multiple drugs that are pharmacologically different may be delivered to the same tissue/cell in precise ratios to empower their synergistic mechanisms. For example, Vyxeos (CPX-351), a clinically successful liposomal formulation of 5:1 cytarabine:daunorubicin, maintained a synergistic drug ratio (from 5:1 to 9:1) in the blood compartment over 24 h post-injection while the free drugs exhibited a 1923:1 ratio 15 min post-injection.11,12
While strategies for incorporating mixtures of structurally dissimilar drugs through encapsulation, chemical conjugation, and/or self-assembly have been studied extensively, nanocarriers that simultaneously achieve controlled drug ratios, multi-drug release kinetics, and/or sequential release for 2 or more drugs remain rare.5–9,11–20 Moreover, due to differences in cell uptake pathways, rates of cellular internalization, and/or drug release kinetics, multidrug-nanocarriers could exhibit synergistic ratios that are distinct from their free drug counterparts, necessitating the identification of optimal ratios in the nanocarrier context. Given that most nanocarriers rely on supramolecular interactions between the drugs, the vehicle, and/or surfactant(s), which depend on the physical properties of the drugs, exchanging one drug for another may result in changes to the physical properties of the final nanocarrier. It is, thus, difficult to make multiple single-drug nanocarriers and multi-drug nanocarriers with varying drug ratios but otherwise identical properties. Hence, in combination nanocarriers employed to date, the synergistic drug ratios exemplified for the free drugs are typically translated to the nanocarriers directly, without consideration for the possibility that these ratios may no longer be optimal.11,12 Moreover, it remains unknown whether multidrug nanocarriers have fundamental advantages over mixtures of single drug nanocarriers.
Here, we introduce a polymer-based nanocarrier design that allows us to address these questions in the context of the second most common hematologic malignancy in the United States, which remains incurable in most patients21—multiple myeloma (MM). Our approach leverages “bottlebrush prodrugs” (BPDs) comprising the clinically relevant three-drug combination of a proteasome inhibitor (PI) bortezomib (Btz), an immunomodulatory drug (IMiD) pomalidomide (Pom) and a corticosteroid dexamethasone (Dex). This drug combination is able to overcome resistance to the front-line and standard-of-care regimen of Lenalidomide (Len)/Btz/Dex as Pom allows for higher target binding affinity when compared to Len.22–25 In spite of the empirical derivation of this combination in the clinic, it offers prolonged progression-free survival in lenalidomide (Len)-refractory patients (17.8 vs. 9.5 months) as well as in non-Len refractory patients (22 months vs. 12 months); moreover, it improves overall survival rates in both settings (85.9% vs. 50.8%, and 95.37% vs. 60.0%, respectively).26 Nevertheless, the combination suffers from significant drawbacks that primarily arise from off-tissue toxicities, poor stability, and the development of Btz.27 While several examples of nanoparticle Btz formulations have been reported as monotherapies,28–32 so far they have shown only minor improvements over free Btz in terms of efficacy.33,34 By contrast, combination nanomedicines for MM are exceptionally rare, and nanocarriers incorporating the clinical combination of Btz, Pom and Dex have not been reported.33–39 Moreover, no examples of >2 drug combination therapies with systematically optimized synergistic ratios have been demonstrated in any disease context. Here, we show that (1) synergies between free drugs identified in vitro do not necessarily translate to BPDs; and (2) BPDs bearing a statistical mixture of drugs in a synergistic ratio are more effective than a mixture of 3 different physically equivalent single-drug BPDs administered at the same ratio. The latter finding is explained mathematically using a Monte-Carlo simulation approach.
Bottlebrush Prodrug (BPD) Design and Synthesis
Our BPD manufacturing involves the synthesis of macromonomer prodrugs of Btz, Pom, and Dex. For Btz, racemic 1,2-tertiary diol azide linker 1 was synthesized from tetraethylene glycol and 2,3-dimethyl-2-butene (see Supplementary Information for full synthetic details). Azido-boronic ester Btz-N3 was formed from 1 and Btz in 70% yield and was subsequently coupled to alkyne 2 through copper-catalyzed alkyne-azide cycloaddition “click” chemistry, affording Btz-M (Fig. 1A).40 Following a similar workflow but with different linkers tailored to the inherent functionality of each API, Pom-M and Dex-M were prepared (Fig. 1A). The structures of each macromonomer and its precursors were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and by either high-resolution mass spectrometry (HR-MS) or matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-ToF MS) where appropriate (Fig. S1–S17).
Figure 1. Synthesis and characterization of BPDs.
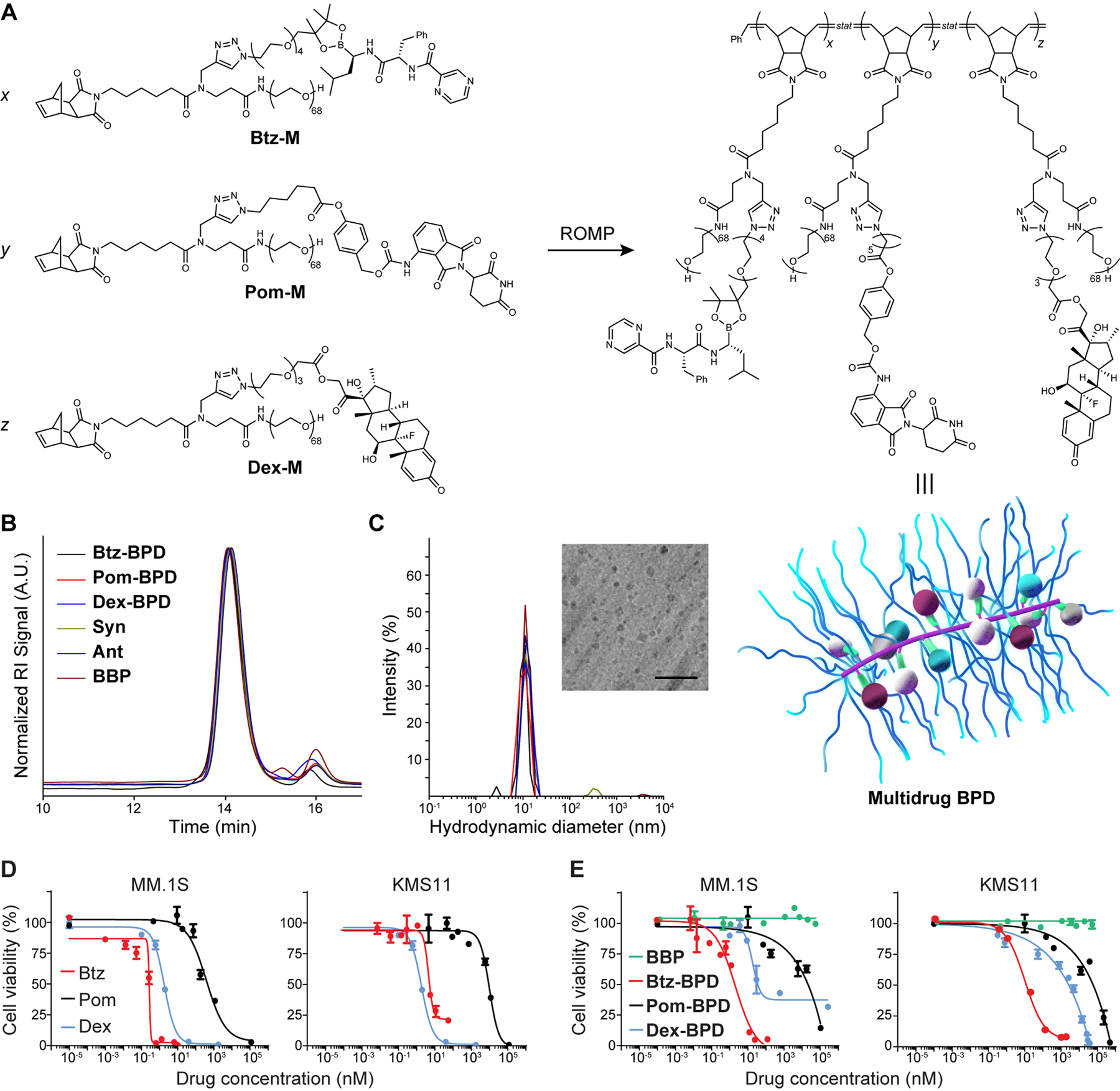
(A) Chemical structures of prodrug macromonomers used in this work. These macromonomers, or mixtures thereof, were subjected to ROMP via exposure to Grubbs 3rd-generation bis-pyridyl complex to produce the corresponding BPDs. Schematic of multidrug BPD is provided (not drawn to scale). Maroon, blue and white spheres = different drugs arranged randomly along the BPD backbone; green = cleavable linkers that activate to release the drugs; purple = BPD backbone; blue strands = poly(ethylene glycol) (PEG) shrouds the drugs and backbone, providing similar physical properties for BPDs regardless of drug identity. (B) Size exclusion chromatography traces of BPDs. The minor peak at 16 min elution time corresponds to residual macromonomers. (C) Hydrodynamic diameters (Dh) of BDPs as determined by dynamic light scattering (DLS). Inset: Cryogenic electronic microscopy image of 3-drug BPD “Syn” (scale bar = 50 nm). (D) Free drugs and (E) 1-drug BPDs were evaluated in MM.1S and KMS11 cell lines (BBP refers to a drug-free bottlebrush polymer). Cell viability (n = 3 biologically independent samples) was evaluated by the CellTiter Glo assay at 48 h after incubation with varying concentrations. Data are presented as mean ± SEM.
Btz-M, Pom-M, and Dex-M were polymerized by ring-opening metathesis polymerization (ROMP) to afford single-drug BPDs Btz-BPD, Pom-BPD, and Dex-BPD, respectively, with number-average degrees of polymerization of 10 (Fig. 1A). Multidrug BPDs with varying ratios of Btz : Pom : Dex were synthesized by copolymerization of these macromonomers in various feed ratios (Fig. S18–S20 and Table S1). A drug-free control polymer (BBP) was synthesized for comparison.41 For in vivo studies, a cyanine5.5 (Cy5.5) dye was incorporated into each BPD.42 Gel permeation chromatography (GPC; Fig. 1B) and dynamic light scattering (DLS; Fig. 1C) revealed efficient macromonomer-to-BPD conversions and hydrodynamic diameters (Dh) of ~10–15 nm, respectively. All samples, regardless of payload compositions (i.e., mono-, multidrug, or no drug), displayed consistent sizes (Table S1). Cryogenic electron microscopy revealed ellipsoidal structures with dimensions of ~10 nm and average aspect ratios of 1.1 (Table S1, Fig. S21). Release of Btz from Btz-BPD in pH 7.4 PBS was much slower (Fig. S22) than was observed for Btz-M (Fig. S17), suggesting that the BPD architecture stabilizes the boronic ester linker from rapid hydrolysis. Nevertheless, exposure to glucose and ATP as well as acidic buffer—established triggers for boronic ester cleavage in the tumor microenvironment43–45—led to significantly enhanced Btz release (e.g., 25.9 ± 2.2% in 1 h at pH 4 or 34.6 ± 2.5% in 1 h at 100 mM glucose, Figure S23). We note that alkyl boronic esters are typically unstable in water at neutral pH; placement of the boronic ester along the relatively hydrophobic BPD backbone shields it from immediate hydrolysis.43–47
In vitro and in vivo characterization of single-drug BPDs
The potency of each single-drug BPD was examined in vitro, using cell viability assays (Cell TiterGlo, Promega) performed after 48 h incubation and with 2 different multiple myeloma cell lines (MM.1S and KMS11). In all cases, the BPDs exhibited half-maximal inhibitory concentrations (IC50) comparable to their free drug counterparts. For instance, in MM.1S cells Btz-BPD was slightly less potent than free Btz (IC50 = 13.1 ± 0.9 nM versus 2.8 ± 0.4 nM, respectively, Fig. 1D and 1E), which could be attributable to differences in cell uptake (transmembrane diffusion versus cellular endocytosis for free Btz and Btz-BPD, respectively) or the slowed release of Btz from Btz-BPD. Dex-BPD was similarly less potent than free Dex (IC50 = 70.9 ± 1.9 nM versus 17 ± 2.8 nM, respectively, Fig. 1D and 1E). Pom-BPD displayed a similar IC50 compared to free Pom (IC50 = 354.2 ± 4.9 nM versus 308.6 ± 3.5 nM, respectively, Fig. 1D and 1E). The BBP was not toxic at any dose level, suggesting that the observed toxicities for the BPDs are due to drug release.
As Btz-associated toxicities remain a hurdle in clinical MM therapy, we first assessed Btz-BPD as a monotherapy in vivo. Gross toxicity was assessed in healthy BALB/c mice (n = 5 animals per group) for free Btz (at 0.75, 1, and 1.25 mg/kg doses administered twice a week for 4 weeks via s.c. injection) and Btz-BPD (at 5, 10, and 18.75 mg/kg doses administered twice a week for 4 weeks via i.v. injection) (Fig. 2A). For Btz-BPD, the 5, 10, and 18.75 mg/kg groups correspond to 0.47, 0.95, and 1.78 mg/kg of Btz, respectively. The drug-free polymer was not examined here as it was previously shown to be well-tolerated at doses up to 2 g/kg.42 Moreover, since subcutaneous administration of Btz is used in the clinic, and displays improved safety with similar efficacy compared to intravenous administration, we use it here for the fairest possible comparison to Btz-BPD.47,48 For Btz, the 0.75 mg/kg dose was observed to be safe, which is consistent with previous reports (Fig. 2A).28 Higher doses induced toxicities as reflected by decreased survival rates and dramatic losses in body weight. By contrast, Btz-BPD was tolerated at all doses with no evidence of mortality or substantial weight loss (Fig. 2B). Toxicology studies were performed in BALB/c mice (twice a week over a 2-week period; 4 injections/mouse) using the same test compounds. Metabolic profiles (Fig. 2C), complete blood counts (Fig. 2D) and white blood cell differential counts (Fig. 2E) were obtained 13 days after the last injected dose of either Btz (0.75 mg/kg via s.c. injection) or Btz-BPD (18.75 mg/kg via i.v. injection). Animals in the Btz-BPD group did not display any changes with respect to the aforementioned parameters (two-tailed student t-test, P > 0.05). The safety of Pom-BPD was evaluated following similar protocols in the CRBNI391V mouse model known to be sensitive to IMiD toxicity49 (Fig. S24–S26; see supplemental information). We did not test the MTD of Dex-BPD alone due to its low in vitro toxicity and the role of Dex as a mitigator of toxicity in clinical therapy.
Figure 2. Safety assessments of Btz-BPD.
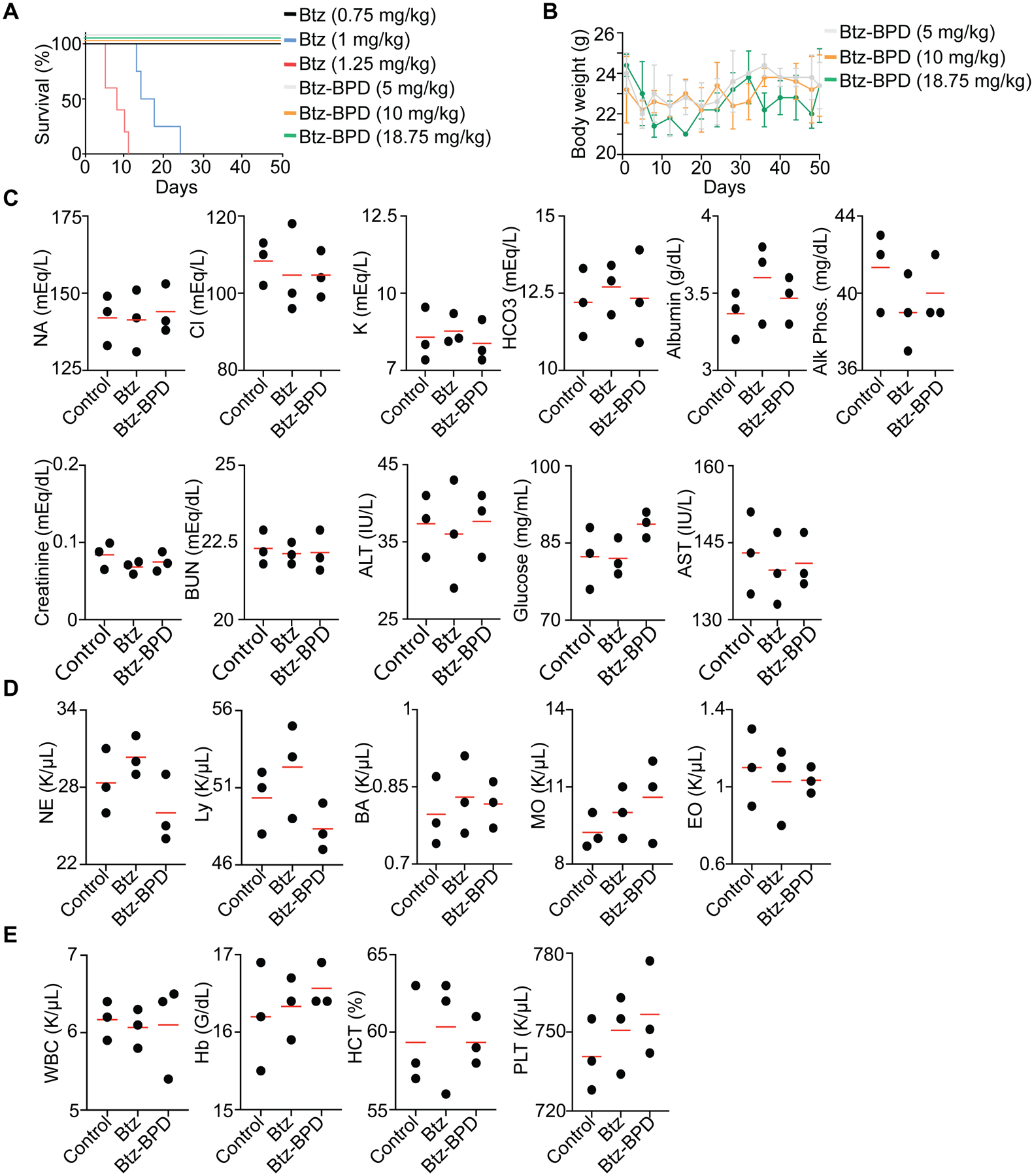
Healthy BALB/c mice were adminstered either PBS, Btz, or Btz-BPD twice a week for 4 weeks. (A) Kaplan-Meier survival curves for mice treated with each agent (n = 5 mice per group). (B) Body weight measurements of BALB/c mice administered Btz-BPD (i.v.) at various doses (n = 5 mice per group). Data are presented as mean ± SEM. (C) Basic metabolic profiles, (D) complete blood counts, and (E) white blood cell differential counts for healthy BALB/c mice (n = 3 mice per group) that were administered each treatment (twice per week for 2 weeks) followed by 2 weeks of rest (i.e., no injection) prior to blood draw and analysis.
The accumulation of Btz-BPD in subcutaneous MM tumors (KMS11) was next evaluated. Fluorescence microscopy revealed significant intratumoral accumulation within 1 h of administration (Fig. 3A). Additional subcutaneous KMS11 tumor-bearing mice (n = 5 per group) were treated with either PBS, Btz (0.75 mg/kg via s.c. injection), Btz-BPD at a mass-equivalent dose of Btz (0.75 mg/kg via i.v. injection, or ‘low dose’), or with Btz-BPD at its highest tested dose level (18.75 mg/kg via i.v. injection, or ‘high dose’). We note that the “low dose” corresponds to 0.071 mg/kg of Btz—more than 10-fold lower that the free drug dose. Groups of mice were treated twice a week for 4 weeks (Fig. 2A and 2B); tumor volumes and body weights were monitored. The study endpoint was reached when a tumor measured >2 cm in longest axis or the animal experienced >20% body weight loss.
Figure 3. Btz-BPD provides significant therapeutic enhancements over Btz in subcutaneous (A-D) and aggressive orthotopic models (E-H) of MM.
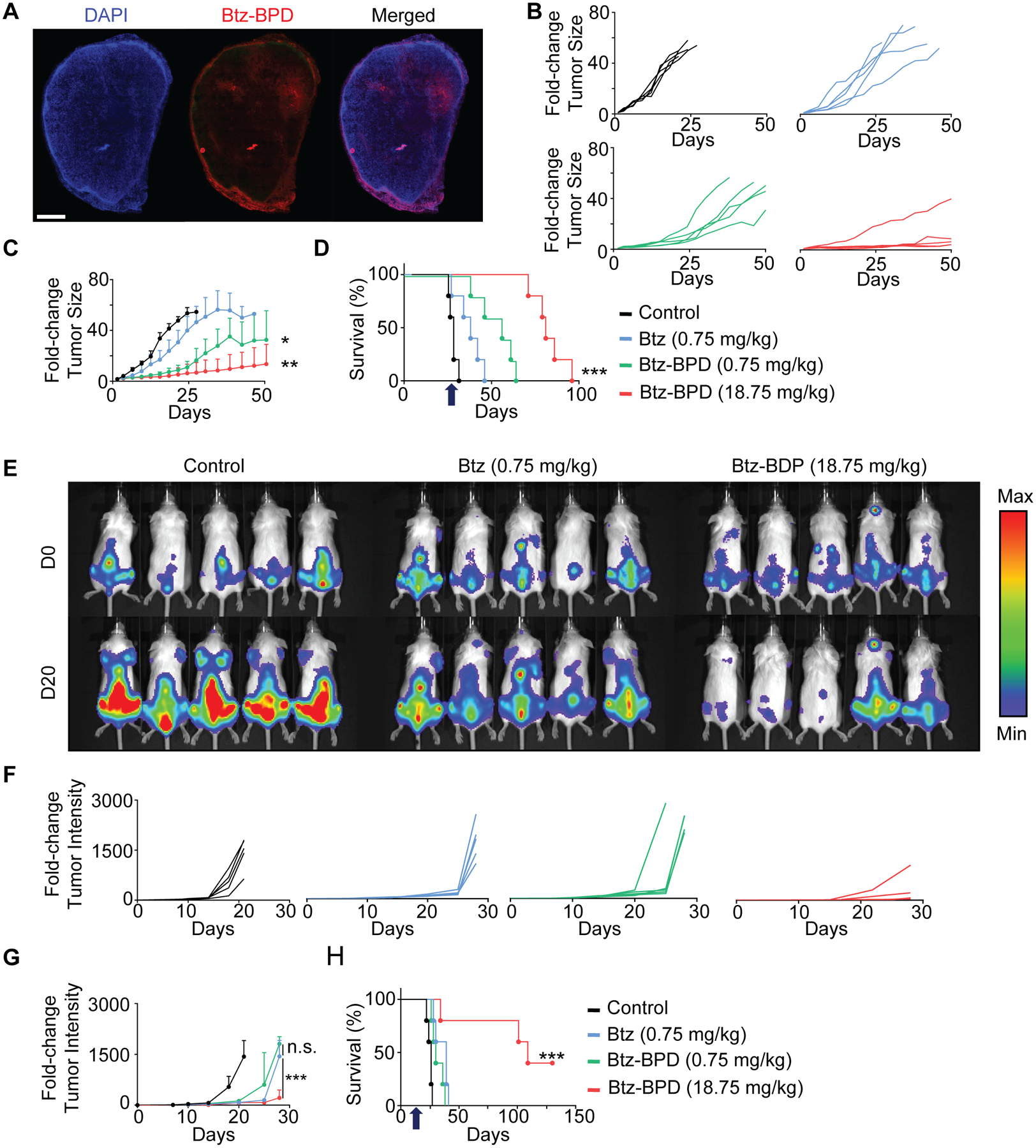
(A) Evaluation of tumor accumulation and penetration of Cy5.5-labelled Btz-BPD at 1 h post-administration (i.v.) as assessed by fluorescence microscopy of harvested tumor upon animal euthanasia (scale bar = 200 μm); representative micrograph is shown, and similar results were acquired in 3 independent biological samples. For efficacy evaluation, KMS11 subcutaneous tumor-bearing mice were injected with PBS, Btz, or Btz-BPD, starting when their tumors reached 5 mm in the largest axis. (B) Spider plots of tumor growth and (C) average tumor size (± SEM) over the course of the study (n = 5 mice per group). Statistical analysis was performed by using a two-tailed t-test between the Btz and Btz-BPD groups. P = 0.0025— Btz-BPD (18.75 mg/kg) vs. Btz (0.75 mg/kg), P = 0.0325— Btz-BPD (0.75 mg/kg) vs. Btz (0.75 mg/kg). (D) Kaplan-Meier survival curves, revealing significant enhancements in therapeutic outcomes for animals treated with Btz-BPD vs. Btz at equivalent doses and with further improvements based on increased Btz-BPD dose level. Arrow indicates the last administered dose. Statistical analysis was performed by using a Log-Rank test, P < 0.0002. (E) Bioluminescence imaging of MM.1SLUC+/GFP+ cells after intravenous dissemination and as a function of the time (day 0 vs. day 20) after administration of PBS (control), Btz (0.75 mg/kg), or Btz-BDP (18.75 mg//kg). (F) Individual spider plots and (G) average tumor size (± SEM) over the course of the study (n = 5 mice per group). Statistical analysis was performed by using a two-tailed t-test between the Btz-BPD and Btz groups. P = 0.0002— Btz-BPD (18.75 mg/kg) vs. Btz-BPD (0.75 mg/kg), P = 0.0525— Btz-BPD (0.75 mg/kg) vs. Btz (0.75 mg/kg). (H) Kaplan-Meier survival curves confirm significant enhancements in the therapeutic outcomes for animals treated at the high dose of Btz-BPD (18.75 mg/kg) as when compared to those treated at the MTD of Btz (0.75 mg/kg). Arrow indicates the last administered dose. Statistical analysis was performed by using a Log-Rank test, P = 0.0002. For statistical tests, ns: non-significant, *: P<0.05, **: P<0.01, ***: P<0.001.
Btz displayed modest activity in this aggressive MM model when compared to the control (mean survival time of 42 ± 6 days vs. 22 ± 5 days for the control group) (Fig. 3B–D). Btz-BPD outperformed free Btz at the “low dose” (e.g., mean survival time of 61 ± 9 days vs. 42 ± 6 days). Moreover, the “high dose” further prolonged survival (mean survival 84 ± 13 days, P < 0.0002 as compared to Btz and other groups). The enhanced activity of Btz-BPD is attributable to its tumor accumulation and Btz release (Fig. 3A).43–47
Next, we evaluated Btz-BPD in a more challenging, orthotopic model of MM, which primarily develops in the bone marrow compartment. Tumors were induced via i.v. injection of luciferase-expressing MM.1SLuc+/GFP+ cells; tumor progression was quantified by bioluminescence imaging (Fig. 3E). Mice were removed from the study when they exhibited hind limb paralysis or a loss of >20% body weight. Mice (n = 5 per group) were treated with the same doses described above at 4 different time points (e.g., day 1, 5, 8, and 12 after tumor cell inoculation). Statistically insignificant efficacy (P = 0.0525) was not observed for Btz and Btz-BPD when administered at the “low dose” (Fig. 3E–G). On the other hand, the “high dose” of Btz-BPD offered significant improvements in tumor growth inhibition and survival (with a mean duration of 108 ± 11 days as compared to 24 ± 4 days for the control group for instance, P = 0.0002). Complete responses were observed in 40% of the animals (2 of 5) while none were seen for either free Btz or PBS (Fig. 3F). Thus, Btz-BPD is more effective as a single-agent PI therapy than Btz. Lastly, we note that Pom-BPD gave similar trends as a monotherapy (Fig. S27).
The serum distribution and PI activity of Btz and Btz-BPD were assessed to explain their differences in efficacy and MTD. Proteasomes are present in micromolar concentrations in red blood cells (RBCs); binding of PIs to RBC proteasomes limits bioavailability and contributes to hematologic toxicity.50,51 Stable boronic ester prodrugs may overcome this limitation. To test this hypothesis, Btz-BPD was incubated in human blood for various times. The plasma and cell fractions were separated, and the amount of Btz-BPD in each fraction was quantified (Table S2). The concentration of Btz-BPD in plasma was 7–10-fold greater than in RBCs at all time points, which represents a >100-fold reversal compared to free Btz, as reported previously.50,51 PI activity was next assessed. The IC50 values (concentrations of PI at which the proteasome is 50% active) for Btz and Btz-BPD were 11.83 nM and 80.50 nM, respectively (Figure S28). Thus, even when exposed directly to its target, Btz-BPD is relatively stable, which would shift its exposure away from RBCs and thereby improve bioavailability in vivo.
In vitro characterization of combination nanomedicines
Next, we investigated potential synergies between Btz, Pom, and Dex as free drugs and Btz-BPD, Pom-BPD, and Dex-BPD as single-drug BPDs in vitro using a full-factorial design approach in MM.1S and KMS-11 cell lines (Fig. S29). Synergistic, additive, or antagonistic relationships were determined using the Chou-Talalay method (Fig. S29 and S30). Notably, free drugs and BPDs displayed distinct combination indexes (CIs) (Fig. S30), suggesting that direct translation of free drug ratios to nanocarriers would be detrimental in this system. Additionally, the addition of Dex gives improvements in the efficacies of both Btz and Pom, and cell death is mostly driven by the concentration of Btz (Fig. S30).
CI maps of the 3 free drugs and of the 3 single-agent BPDs were generated using the Loewe additivity method (Fig. 4A), holding the concentration of Dex constant (2 nM for free drugs and 20 nM for Dex-BPD). Leveraging the Bliss independence model, which predicts the toxicity of additive drug combinations, we identified a ratio of 0.2:9.46:0.34 Btz:Pom:Dex as synergistic (it is more toxic than the Bliss model prediction) and a ratio of 0.02:9.98:0.01 as antagonistic (as it is less toxic than the Bliss model prediction) for the single-drug BPDs. Two new 3-drug BPDs “Syn” and “Ant” were synthesized bearing these average synergistic and antagonistic drug ratios, respectively, by copolymerization of Btz-MM, Pom-MM, and Dex-MM (Fig. 1A–D and Table S1). Syn and Ant were incubated with 4 different MM cell lines (MM.1S, KMS11, U266, and KMS18); Syn exhibited greater toxicity and Ant lower toxicity than the Bliss model prediction (Fig. 4B), which confirms the synergistic and antagonistic nature of these 3-drug BPDs.
Figure 4. 3-drug BPD combination index (CI) studies.
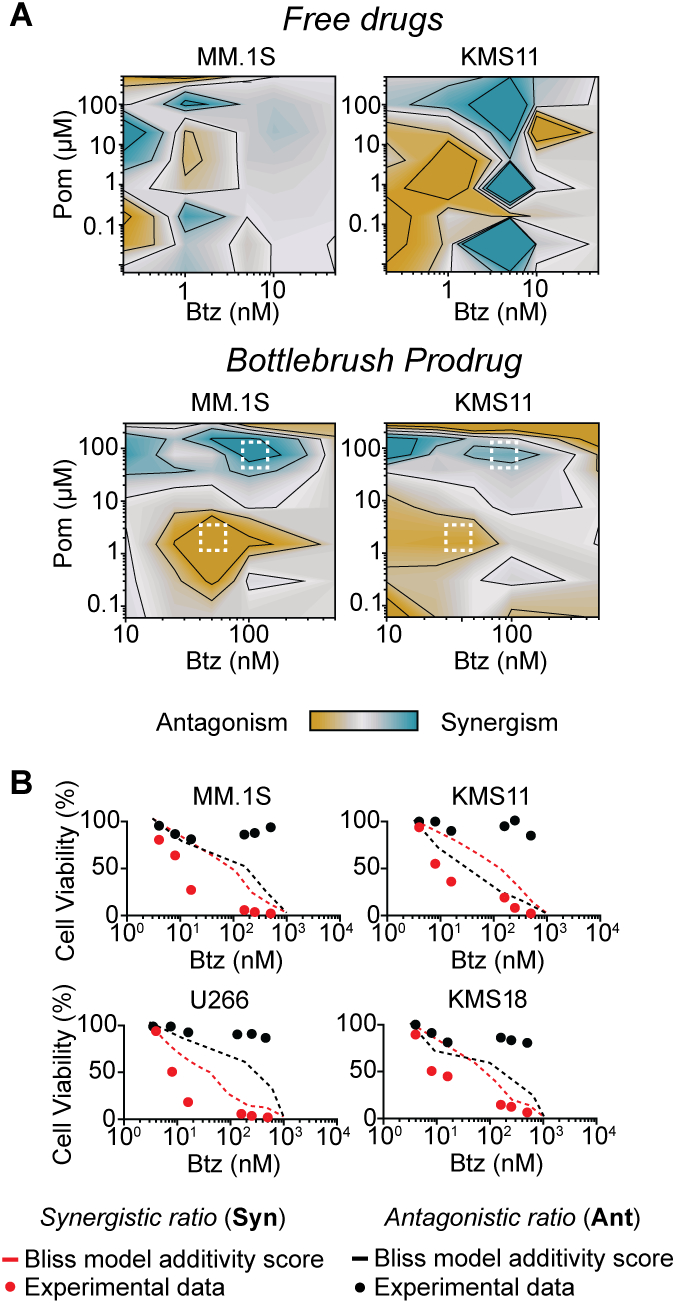
(A) CI maps obtained by using the Chou-Talalay method with a fixed dose of Dexamethasone (Dex, 2 nM) and by varying the concentrations of Btz and Pomalidomide (Pom) used in free drugs combinations (top); a fixed dose of Dex-BPD (20 nM) and varying Btz-BPD and Pom-BPD were employed for BPD combinations (bottom) after 48h of treatment. (B) Ratio validation using viability assays of 3-drug BPDs at Syn and Ant were performed in 4 MM cell lines (KMS11, MM.1S, U266, and KMS18), confirming that the selected ratios were synergistic and antagonistic in comparison to the Bliss model (for additive drug activity).
In vivo evaluation combination nanomedicines.
We propose that 3-drug BPDs should outperform mixtures of single-drug BPDs at the same synergistic ratio in vivo. To rationalize this proposal, the variance from the target drug ratio as a function of the number of BPD molecules for 3-drug BPDs (assuming random copolymerization) and mixtures of 1-drug BPDs was modeled (Fig. S31, see Supplementary Information for details). For small BPD sample sizes (<10k BPD molecules), the statistical mixture is more likely to reflect the target ratio. For example, if one randomly selects 1000 BPD molecules, the sample will be ~80% reflective of the target ratio for the 3-drug BPD and only ~20% reflective of the target ratio for the 1-drug BPD mixture (Fig. S31). Other reports have suggested that cells take up ~102 nanoparticles per vesicle regardless of nanoparticle dose,52,53 which could amplify this effect.
Using the same MM models as above (n = 5), Syn was administered at two doses: 5.3 mg/kg (SynLD, 0.01 mg/kg Btz, 0.38 mg/kg Pom, 0.02 mg/kg Dex), and 25.0 mg/kg (SynHD, 0.05 mg/kg Btz, 1.64 mg/kg Pom, 0.08 mg/kg Dex; Ant was administered at 50 mg/kg (0.01 mg/kg Btz, 3.48 mg/kg Pom, 0.01 mg/kg Dex); a mixture of single-drug BPDs (1D-BPD) was administered at 5.3 mg/kg (0.1 mg/kg Btz-BPD, 5 mg/kg Pom-BPD, 0.2 mg/kg Dex-BPD), corresponding to the same dose as SynLD. The free drugs (FD) were administered in a total mass that matched the mass of SynLD (0.1 mg/kg Btz, 5 mg/kg Pom, 0.2 mg/kg Dex) (Fig. 5). Lower doses of PI were used for these combination therapy studies compared to the monotherapy studies above to more easily differentiate between the study groups. Bioluminescence imaging for the MM.1S model was done on day 0 (study initiation) and day 20, (a known cutoff date for control mice in this model).28
Figure 5. Improved therapeutic efficacy of synergistic 3-Drug BPD in MM mouse models (n = 5 mice per treatment group).
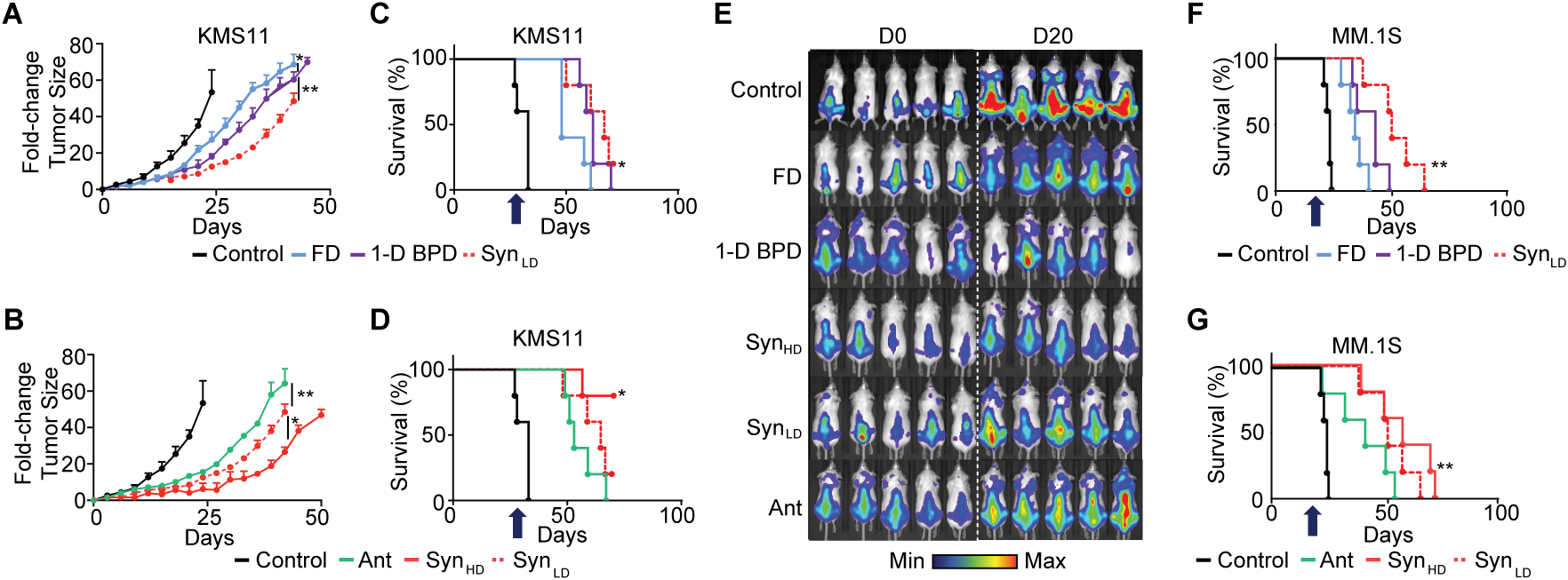
(A) Tumor fold-change based on the delivery methods for the synergistic drug ratio in subcutaneous KMS11 mouse model. FD – free drug combination; 1-D BPD: mixture of 1 drug-loaded BPDs (synergistic ratio); SynLD: low dose Syn (synergistic ratio, 3-drug BPD). Data are presented as mean ± SEM. Statistical analysis was performed by using a two-tailed t-test to compare the different groups at a fixed time points for the tumor fold-changes. 1-D BPD vs. FD: P = 0.045; 1-D BPD vs. SynLD: P = 0.007. (B) Tumor fold-change comparing the therapeutic outcomes in the context of synergistic and antagonistic drug delivery profiles with 3-drug BPD in subcutaneous KMS11 mouse model. Ant: antagonistic ratio; SynLD: low dose Syn (synergistic ratio); SynHD: high dose Syn (synergistic ratio). Data are presented as mean ± SEM. Statistical analysis was performed by using a two-tailed t-test to compare the different groups at a fixed time points for the tumor fold-changes. Ant vs. SynLD: P = 0.0075; SynLD vs. SynHD: P = 0.045. (C) Associated Kaplan-Meier curves comparing the therapeutic outcomes based on the delivery methods for the synergistic drug ratio in subcutaneous KMS11 mouse model and (D) the synergistic and antagonistic drug delivery profiles with 3-drug BPD. Statistical analysis was performed by using a Log-Rank test, with P = 0.045 (Panel C) and P = 0.0325 (Panel D). (E) Bioluminescence imaging of orthotopic MM.1S mouse models at day 0 and day 20 during treatment. (F) Kaplan-Meier curves comparing the therapeutic outcomes based on the delivery methods for the synergistic drug ratio in orthotopic MM.1SGFP+/LUC+ mouse model and (G) the synergistic and antagonistic drug delivery profiles with 3-drug BPD. Statistical analysis was performed by using a Log-Rank test, with P = 0.0025 (Panel F) and P = 0.025 (Panel G). For statistical tests, ns: non-significant, *: P<0.05, **: P<0.01, ***: P<0.001. Arrow indicates the last administered dose. We note that panel A-D, and F-G display different study groups within the same experiment, which share the same controls and SynLD group, separated for visualization purposes to support the comparisons at hand. Mean survival times were as the following: FD (47 ± 6 days for KMS11 model, and 41 ± 9 days for MM.1S model), 1-D BPD (53 ± 4 days for KMS11 model, and 48 ± 4 days for MM.1S model), SynLD (61 ± 9 days for KMS11 model, and 53 ± 14 days for MM.1S model), SynHD (unavailable for KMS11 model as >50% of mice survived until the end of the study, and 62 ± 8 days for MM.1S model), Ant (52 ± 6 days for KMS11 model, and 46 ± 5 days for MM.1S model).
In support of our modeling, SynLD outperformed 1D-BPD and FD (Fig. 5) in slowing tumor progression (Fig. 5A and 5E) and increasing survival (Fig. 5C and 5F). Moreover, SynHD provided further enhancements in efficacy compared to SynLD (Fig. 5A), while still using less drug than FD. On the other hand, Ant displayed inferior efficacy compared to SynLD despite having the same Btz dose and a 10-fold higher dose of Pom (Fig. 5D and 5G), suggesting that the synergistic ratio is preferred over a ‘more is better’ approach.6–8 Interestingly, Ant outperformed FD despite having a smaller amount of drug, which may be due to improved delivery of the drugs to tumor cells via the BPD.
Conclusions
We introduce a nanomedicine strategy that offers a promising new PI-based treatment for MM and potentially other cancers, as well as rapid translation of 3-drug synergies determined in vitro to in vivo.6–8 First, a PI-based monotherapy (Btz-BPD) is introduced that offers significantly improved efficacy compared to the standard PI Btz while displaying no detectable toxicities in two in vivo models of MM. Then, by manufacturing single-drug BPDs of Btz, Pom, and Dex, we observe that BPDs display synergistic, additive, or antagonistic patterns distinct from their free drugs, showing that synergies should be measured in the nanocarrier context. Finally, 3-drug BPDs are shown to outperform a mixture of 3 single-drug BPDs and free drugs in vivo, which is modeled quantitatively. Overall, this work offers potentially translatable therapies for MM and offers new mechanistic insights into optimizing and manufacturing combination nanomedicines in other disease contexts.
This approach also raises regulatory questions that will be important as the field of combination therapeutics moves forward. For example, could nanocarriers bearing a statistical mixture of drugs generally classify as single entities for regulatory purposes? If so, such an approach may be advantageous compared to mixtures of nanocarriers that each would need independent evaluation. Additionally, while it was shown here that synergy identified for Btz, Dex, and Pom holds in 4 different cell lines, it is uncertain that this ratio would be optimal for all cell lines and patients given the heterogeneity of multiple myeloma. A future clinical workflow could involve (1) biopsy to isolate a patient’s cancer cells, (2) CI screening to determine if synergy is maintained in those cells or if an alternative synergistic ratio exists. (3) If the former, then existing BPDs could be administered; if the latter, BPDs with patient-specific ratios could be generated on-demand. The latter would be facilitated if components of BPD combination therapies, such as prodrug macromonomers or single-drug BPDs, could undergo translational steps as one package.54,55 Altogether, these questions and directions for the field of combination nanomedicine are fascinating.
Methods
Representative Procedure for Combination BPD with Pom:Btz:Dex Ratio of 9.5:0.2:0.3 (Syn).
To a vial containing a stir bar, Pom-M (34.3 mg, 8.7 μmol, 9.5 eq) was added. To another 3 separate vials, a solution of Btz-M (20 mg/mL in THF), a solution of Dex-M (20 mg/mL in THF), and a solution of 3rd generation Grubbs catalyst (G3-Cat, 0.02 M in THF) were freshly prepared. THF (38.7 μL) was then added to the vial containing Pom-M, followed by the addition of the Btz-M (37.9 μL, 0.19 μmol, 0.2 eq) and the Dex-M (61.7 μL, 0.31 μmol, 0.3 eq) solution. To the macromonomer mixture, G3-Cat solution (46.1 μL, 0.92 μmol, 1.0 eq) was added, affording the desired total DP of 10, a Pom:Btz:Dex ratio of 9.5:0.2:0.3, and to a total macromonomer concentration of 0.05 M. The reaction mixture was allowed to stir for 3 h at room temperature. To quench the polymerization, a drop of ethyl vinyl ether was then added. The reaction mixture was transferred to 8 kDa molecular weight cutoff dialysis tubing in 3 mL nanopure water; and, the solution was dialyzed against H2O (500 mL × 3, solvent exchange every 6 h). The dialyzed solution of Syn was then concentrated as desired via centrifugation with a filter tube. Alternatively, Syn was also acquired by lyophilization, or precipitation in diethyl ether.
Cell lines.
MM.1S (CRL-2974, ATCC) and U266 (TIB-196, ATCC) cells were obtained from ATCC (Manassas, VA, USA). KMS11 (JCRB1179, JCRB) and KMS18 (CVCL-A637, JCRB) cells were obtained from the JCRB Cell Bank. All cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (ThermoFisher Scientific) supplemented with 10% FBS (VWR), 1% Penicillin/Streptomycin (ThermoFisher Scientific) and 1% glutamine (ThermoFisher Scientific). MM.1SLuc+/GFP+ cells were generated by retroviral transduction and authenticated by short tandem repeat DNA profiling. All cell lines were confirmed to be mycoplasma-free by using the MycoAlert Mycoplasma kit (Lonza). Cell lines were housed in 37 °C incubators under 5% CO2.
Animal usage.
All experiments involving animals were reviewed and approved by the Dana-Farber Cancer Institute’s Committee for Animal Care. The maximum tumor size/burden permitted by the committee was not exceeded in these studies. For the free drug comparison, Btz injection was administered subcutaneously (as intravenous toxicity otherwise governed this route); Dex and Pom were injected intravenously. All BPDs were injected intravenously.
Supplementary Material
Acknowledgements
We thank the NIH-NCI (1R01CA220468-01 for J.A.J., P.P.G., and R01CA205954 for I.M.G), the Leukemia and Lymphoma Society, and the National Science Foundation (Graduate Research Fellowship for H.V.-T.N.) for support of this research. This work was further supported in part by the Koch Institute Core Grant P30-CA14051 from the NCI. A.D. acknowledges support from the International Myeloma Foundation, the Fondation Française pour la Recherche contre le Myélome et les Gammapathies (FFRMG), and Inserm Cancer. A.D., J.A.J, and I.M.G. acknowledge support from the Stand Up to Cancer Dream Team Multiple Myeloma grant. P.P.G. acknowledges the generous support of the Charles W. and Jennifer C. Johnson Clinical Investigator Fund as well as the Kathryn Fox Samway Foundation.
Footnotes
Competing Interests Statement
A.D., H.V.-T.N., Y.J., I.M.G., P.P.G., and J.A.J. are named inventors on a patent application (US Patent Application No. 16/825,269) filed jointly by the Massachusetts Institute of Technology and the Dana-Farber Cancer Institute on the bortezomib macromolecular proteasome inhibitors described in this work. H.V.-T.N., Y.J., and J.A.J. are co-founders and shareholders of Window Therapeutics, which seeks to translate this technology to clinical cancer therapies. The remaining authors declare no competing interests.
Data Availability
All data supporting the findings of this study are available within the article and its Supplementary Information and can also be obtained from the corresponding authors upon reasonable request.
References
- 1.Tibbitt MW; Dahlman JE; Langer R Emerging Frontiers in Drug Delivery. J. Am. Chem. Soc 2016, 138, 704–717. [DOI] [PubMed] [Google Scholar]
- 2.Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi J; Xiao Z; Kamaly N; Farokhzad OC Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc. Chem. Res 2011, 44, 1123–1134. [DOI] [PubMed] [Google Scholar]
- 4.Kakkar A; Traverso G; Farokhzad OC; Weissleider R; Langer R Evolution of Macromolecular Complexity in Drug Delivery Systems. Nat. Rev. Chem 2017, 1, 0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma L; Kohli M; Smith A Nanoparticles for Combination Drug Therapy. ACS Nano 2013, 7, 9518–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mignani S; Bryszewska M; Klajnert-Maculewicz B; Zablocka M; Majoral J-P Advances in Combination Therapies Based on Nanoparticles for Efficacious Cancer Treatment: An Analytical Report. Biomacromolecules 2015, 16, 1–27. [DOI] [PubMed] [Google Scholar]
- 7.Zhang RX; Wong HL; Xue HY; Eoh JY; Wu XY Nanomedicine of Synergistic Drug Combinations for Cancer Therapy – Strategies and Perspectives. J. Control. Release 2016, 240, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Q; Sun W; Wang C; Gu Z Recent Advances of Cocktail Chemotherapy by Combination Drug Delivery Systems. Adv. Drug Deliv. Rev 2016, 98, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shim G; Kim M-G; Kim D; Park JY; Oh Y-K Nanoformulation-Based Sequential Combination Cancer Therapy. Adv. Drug Deliv. Rev 2017, 115, 57–81. [DOI] [PubMed] [Google Scholar]
- 10.Jia J; Zhu F; Ma X; Cao ZW; Li YX; Chen YZ Mechanisms of Drug Combinations: Interaction and Network Perspectives. Nat. Rev. Drug Discov 2009, 8, 111–128. [DOI] [PubMed] [Google Scholar]
- 11.Tardi P; Johnstone S; Harasym N; Xie S; Harasym T; Zisman N In Vivo Maintenance of Synergistic Cytarabine:Daunorubicin Ratios Greatly Enhances Therapeutic Efficacy. Leuk. Res 2009, 33, 129–139. [DOI] [PubMed] [Google Scholar]
- 12.Batist G; Gelmon KA; Chi KN; Miller WH; Chia SK; Mayer LD Safety, Pharmacokinetics, and Efficacy of CPX-1 Liposome Injection in Patients with Advanced Solid Tumors. Clin. Cancer Res 2009, 15, 692–700. [DOI] [PubMed] [Google Scholar]
- 13.Lehar J; Krueger AS; Avery W; Heilbut AM; Johansen LM; Price ER; Rickles RJ; Short GF III; Staunton JE; Jin X; Lee MS; Zimmermann GR; Borisy AA Synergistic Drug Combinations Tend to Improve Therapeutically Relevant Selectivity. Nat. Biotechnol 2009, 27, 659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolishetti N; Dhar S; Valencia PM; Lin LQ; Karnik R; Lippard SJ; Langer R; Farokhzad OC Engineering of Self-Assembled Nanoparticle Platform for Precisely Controlled Combination Drug Therapy. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 17939–17944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng ZJ; Morton SW; Ben-Akiva E; Dreaden EC; Shopsowitz KE; Hammond PT Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment. ACS Nano 2013, 7, 9571–9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aryal S; Hu C-MJ; Zhang L Polymeric Nanoparticles with Precise Ratiometric Control over Drug Loading for Combination Therapy. Mol. Pharmaceutics 2011, 8, 1401–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lammers T; Subr V; Ulbrich K; Peschke P; Huber PE; Hennink WE; Storm G Simultaneous Delivery of Doxorubicin and Gemcitabine to Tumors in vivo Using Prototypic Polymeric Drug Carriers. Biomaterials 2009, 30, 3466–3475. [DOI] [PubMed] [Google Scholar]
- 18.Wang H; Wu J; Xie K; Fang T; Chen C; Xie H; Zhou L; Zheng S Precise Engineering of Prodrug Cocktails into Single Polymeric Nanoparticles for Combination Cancer Therapy: Extended and Sequentially Controllable Drug Release. ACS Appl. Mater. Interfaces 2017, 9, 10567–10576. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L; Su H; Liu Y; Pang N; Li J; Qi X-R Enhancing Solid Tumor Therapy with Sequential Delivery of Dexamethasone and Docetaxel Engineered in a Single Carrier to Overcome Stromal Resistance to Drug Delivery. J. Control. Release 2019, 294, 1–16. [DOI] [PubMed] [Google Scholar]
- 20.Cai L; Xu G; Shi C; Guo D; Wang X; Luo J Telodendrimer Nanocarrier for Co-Delivery of Paclitaxel and Cisplatin: A Synergistic Combination Nanotherapy for Ovarian Cancer Treatment. Biomaterials 2015, 37, 456–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howlader N; Noone AM; Krapcho M; Miller D; Bishop K; Altekruse SF; Kosary CL; Yu M; Ruhl J; Tatalovich Z; Mariotto A; Lewis DR; Chen HS; Feuer EJ; Cronin KA SEER Cancer Statistics Review, 1975–2013, National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/archive/csr/1975_2013/, based on November 2015 SEER data submission, posted to the SEER web site, April 2016. [Google Scholar]
- 22.Attal M; Lauwers-Cances V; Hulin C; Leleu X; Caillot D; Escoffre M; Arnulf B; Macro M; Belhadj K; Garderet L; Roussel M; Payen C; Mathiot C; Fermand JP; Meuleman N; Rollet S; Maglio ME; Zeytoonjian AA; Weller EA; Munshi N; Anderson KC; Richardson PG; Facon T; Avet-Loiseau H; Harousseau JL; Moreau P; Study IFM Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med 2017, 376, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nooka AK; Kaufman JL; Muppidi S; Langston A; Heffner LT; Gleason C; Casbourne D; Saxe D; Boise LH; Lonial S Consolidation and Maintenance Therapy with Lenalidomide, Bortezomib and Dexamethasone (RVD) in High-Risk Myeloma Patients. Leukemia 2014, 28, 690–693. [DOI] [PubMed] [Google Scholar]
- 24.Richardson PG; Oriol A; Beksac M; Liberati AM; Galli M Schjesvold F; Lindsay J; Weisel K; White D; Facon T; San Miguel J; Sunami K; O’Gorman P; Sonneveld P; Robak P; Semochkin S; Schey S; Yu X; Doerr T; Bensmaine A; Biyukov T; Peluso T; Zaki M; Anderson K; Dimopoulos M OPTIMISMM Trial Investigators. Pomalidomide, Bortezomib, and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): a Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 781–794. [DOI] [PubMed] [Google Scholar]
- 25.Chanan-Khan AA; Swaika A; Paulus A; Kumar SK; Mikhael JR; Rajkumar SV; Dispenzieri A; Lacy MQ Pomalidomide: The New Immunomodulatory Agent for The Treatment of Multiple Myeloma. Blood Cancer J. 2013, 3, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimopoulos M; Weisel K; Moreau P; Anderson LD Jr.; White D; San-Miguel J; Sonneveld P; Engelhardt M; Jenner M; Corso A; Dürig J; Pavic M; Salomo M; Casal E; Srinivasan S; Yu X; Nguyen TV; Biyukov T; Peluso T; Richardson P Pomalidomide, Bortezomib, and Dexamethasone for Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): Outcomes by Prior Treatment at First Relapse. Leukemia. 2020, In Press. DOI: 10.1038/s41375-020-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.San Miguel J; Blade J; Boccadoro M; Cavenagh J; Glasmacher A; Jagannath S; Lonial S; Orlowski RZ; Sonneveld P; Ludwig H A Practical Update on the Use of Bortezomib in the Management of Multiple Myeloma. Oncologist 2006, 11, 51–61. [DOI] [PubMed] [Google Scholar]
- 28.Swami A; Reagan MR; Basto P; Mishima Y; Kamaly N; Glavey S; Zhang S; Moschetta M; Seevaratnam D; Zhang Y; Liu J; Memarzadeh M; Wu J; Manier S; Shi J; Bertrand N; Lu ZN; Nagano K; Baron R; Sacco A; Roccaro AM; Farokhzad OC; Ghobrial IM Engineered Nanomedicine for Myeloma and Bone Microenvironment Targeting. Proc. Natl. Acad. Sci. U. S. A 2014, 111,10287–10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashley JD; Stefanick JF; Schroeder VA; Suckow MA; Kiziltepe T; Bilgicer B Liposomal Bortezomib Nanoparticles via Boronic Ester Prodrug Formulation for Improved Therapeutic Efficacy in vivo. J. Med. Chem 2014, 57, 5282–5292. [DOI] [PubMed] [Google Scholar]
- 30.Xu W; Ding J; Li L; Xiao C; Zhuang X; Chen X Acid-Labile Boronate-Bridged Dextran-Bortezomib Conjugate with Up-Regulated Hypoxic Tumor Suppression. Chem. Commun 2015, 51, 6812–6815. [DOI] [PubMed] [Google Scholar]
- 31.Lu X; Chai Z; Lu L; Ruan H; Wang R; Zhan C; Xie C; Pan J; Liu M; Wang H; Lu W Bortezomib Dendrimer Prodrug‐Based Nanoparticle System. Adv. Funct. Mater 2019, 29, 1807941. [Google Scholar]
- 32.Zhu J; Huo Q; Xu M; Yang F; Li Y; Shi H; Niu Y; Liu Y Bortezomib-Catechol Conjugated Prodrug Micelles: Combining Bone Targeting and Aryl Boronate-Based pH-Responsive Drug Release for Cancer Bone-Metastasis Therapy. Nanoscale 2018, 10, 18387–18397. [DOI] [PubMed] [Google Scholar]
- 33.Detappe A; Bustoros M; Mouhieddine TH; Ghoroghchian PP, Advancements in Nanomedicine for Multiple Myeloma. Trends. Mol. Med 2018, 24, 560–574. [DOI] [PubMed] [Google Scholar]
- 34.Mu C-F; Shen J; Liang J; Zheng H-S; Xiong Y; Wei Y-H; Li F Targeted Drug Delivery for Tumor Therapy Inside the Bone Marrow. Biomaterials 2018, 155, 191–202. [DOI] [PubMed] [Google Scholar]
- 35.Zhong W; Zhang X; Zhao M; Wu J; Lin D Advancements in Nanotechnology for The Diagnosis and Treatment of Multiple Myeloma. Biomater. Sci 2020, 8, 4692–4711. [DOI] [PubMed] [Google Scholar]
- 36.Ashley JD; Quinlan CJ; Schroeder VA; Suckow MA; Pizzuti VJ; Kiziltepe T; Bilgicer B Dual Carfilzomib and Doxorubicin–Loaded Liposomal Nanoparticles for Synergistic Efficacy in Multiple Myeloma. Mol. Cancer Ther 2016, 15, 1452–1459. [DOI] [PubMed] [Google Scholar]
- 37.Soodgupta D; Pan D; Cui G; Senpan A; Yang X; Lu L; Weilbaecher KN; Prochownik EV; Lanza GM; Tomasson MH Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol. Cancer Ther 2015, 14, 1286–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deshantri AK; Fens MHAM; Ruiter RWJ; Metselaar JM; Storm G; Mandhane SN; Graat GHM; Lentjes EGW; Yuan H; Bruijn J. D. d.; Mutis T; Martens ACM; Groen RWJ; Schiffelers RM Complete Tumor Regression by Liposomal Bortezomib in a Humanized Mouse Model of Multiple Myeloma. Hemasphere 2020, 4, e463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deshantri AK; Fens MH; Ruiter RWJ; Metselaar JM; Storm G; Bloois L. v.; Varela-Moreira A; Mandhane SN; Mutis T; Martens ACM; Groen RWJ; Schiffelers RM Liposomal Dexamethasone Inhibits Tumor Growth in an Advanced Human-Mouse Hybrid Model of Multiple Myeloma. J. Control. Release 2019, 296, 232–240. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen HV-T; Gallagher NM; Vohidov F; Jiang Y; Kawamoto K; Zhang H; Park JV; Huang Z; Ottaviani MF; Rajca A; Johnson JA Scalable synthesis of multivalent macromonomers for ROMP. ACS Macro Lett. 2018, 7, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J; Burts AO; Li Y; Zhukhovitskiy AZ; Ottaviani MF; Turro NJ; Johnson JA “Brush-first” method for the parallel synthesis of photocleavable, nitroxide-labeled PEG star polymers. J. Am. Chem. Soc 2012, 134, 16337–16344. [DOI] [PubMed] [Google Scholar]
- 42.Sowers MA; McCombs JR; Wang Y; Paletta JT; Morton SW; Dreaden EC; Boska MD; Ottaviani MF; Hammond PT; Rajca A; Johnson JA Redox-responsive branched-bottlebrush polymers for in vivo MRI and fluorescence imaging. Nat. Commun 2014, 5, 5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stubelius A; Lee S; Almutairi A The Chemistry of Boronic Acids in Nanomaterials for Drug Delivery. Acc. Chem. Res 2019, 52, 3108–3119. [DOI] [PubMed] [Google Scholar]
- 44.Antonio JPM; Russo R; Carvalho CP; Cal PMSD; Gois PMP Boronic Acids as Building Blocks for the Construction of Therapeutically Useful Bioconjugates. Chem. Soc. Rev 2019, 48, 3513–3536. [DOI] [PubMed] [Google Scholar]
- 45.Brooks WLA; Sumerlin BS Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine. Chem. Rev 2016, 116, 1375–1397. [DOI] [PubMed] [Google Scholar]
- 46.Graham BJ; Windsor IW; Gold B; Raines RT Boronic Acid with High Oxidative Stability and Utility in Biological Contexts. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2013691118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Millenium Pharmaceuticals, Inc. Approval Package for: Application Number 21–602/S-015 (Velcade). Center for Drug Evaluation and Research 2008. [Google Scholar]
- 48.Merz M; Salwender H; Haenel M; Mai EK; Bertsch U; Kunz C; Hielscher T; Blau IW; Scheid C; Hose D; Seckinger A; Jauch A; Hillengass J; Raab MS; Schurich B; Munder M; Schmidt-Wolf IG; Gerecke C; Lindemann HW; Zeis M; Weisel K; Duerig J; Goldschmidt H Subcutaneous Versus Intravenous Bortezomib in Two Different Induction Therapies for Newly Diagnosed Multiple Myeloma: an Interim Analysis from the Prospective GMMG-MM5 Trial. Haematologica, 2015, 100, 964–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fink EC; McConkey M; Adams DN; Haldar SD; Kennedy JA; Guirguis AA; Udeshi ND; Mani DR; Chen M; Liddicoat B; Svinkina T; Nguyen AT; Carr SA; Ebert BL CrbnI391V is Sufficient to Confer in vivo Sensitivity to Thalidomide and its Derivatives in Mice. Blood 2018, 132, 1535–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hemeryck A; Geerts R; Monbaliu J; Hassler S; Verhaeghe T; Diels L; Verluyten W; van Beijsterveldt L; Mamidi RNVS; Janssen C; De Coster R Tissue Distribution and Depletion Kinetics of Bortezomib and Bortezomib-Related Radioactivity in Male Rats After Single and Repeated Intravenous Injection of 14 C-Bortezomib. Cancer Chemother. Pharmacol 2007, 60, 777–787. [DOI] [PubMed] [Google Scholar]
- 51.Sanchorawala V; Palladini G; Kukreti V; Zonder JA; Cohen AD; Seldin DC; Dispenzieri A; Jaccard A; Schonland SO; Berg D; Yang H; Gupta N; Hui A-M; Comenzo RL; Merlini G A Phase 1/2 Study of the Oral Proteasome Inhibitor Ixazomib in Relapsed or Refractory AL Amyloidosis. Blood 2017, 130, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Summers HD; Rees P; Holton MD; Brown MR; Chappell SC; Smith PJ; Errington RJ Statistical Analysis of Nanoparticle Dosing in a Dynamic Cellular System. Nat. Nanotechnol 2011, 6, 170–174. [DOI] [PubMed] [Google Scholar]
- 53.Rees P; Wills JW; Brown MR; Barnes CM; Summers HD The Origin of Heterogeneous Nanoparticle Uptake by Cells. Nat. Commun 2019, 10, 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lancet JE; Uy GL; Cortes JE; Newell LF; Lin TL; Ritchie EK; Stuart RK; Strickland SA; Hogge D; Solomon SR; Stone RM; Bixby DL; Kolitz JE; Schiller GJ; Wieduwilt MJ; Ryan DH; Hoering A; Banerjee K; Chiarella M; Louie AC; Medeiros BC CPX-351 (Cytarabine and Daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol 2018, 36, 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell MJ; Billingsley MM; Haley RM; Wechsler ME; Peppas NA; Langer R Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug. Discov 2021, 20, 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the article and its Supplementary Information and can also be obtained from the corresponding authors upon reasonable request.