

Abstract
Wiskott–Aldrich syndrome (WAS) is a rare immunodeficiency X-linked genetic disorder. It is often featured with a clinical triad of thrombocytopenia with low mean platelet volume, eczematoid dermatitis and recurrent infections. The clinical manifestation of WAS, depending on the underlying variant, shows wide heterogeneity. We present a case of a 10-month-old boy who came in with a history of recurrent fever, skin lesions since birth and episodes of bloody diarrhoea. He had severe anaemia and thrombocytopenia (with normal mean platelet volume). Genetic analysis revealed the patient to be hemizygous for a pathogenic WAS gene splice variant (NM_000377.2:c.360+1G>A). He was managed with supportive treatment and regular follow up, but died 4 months later. As it is a rare genetic disease, the diagnosis of WAS can easily be missed, especially in settings with scarce healthcare resources that do not have easy access to genetic testing. Thus, a high index of suspicion is needed when a male child presents with recurrent infections and bleeding tendencies.
Plain language summary
Management challenges of a rare genetic disorder in a resource-limited country: a case report of Wiskott–Aldrich syndrome in Tanzania
Wiskott–Aldrich syndrome (WAS) is a rare inherited disease that mainly affects boys. Patients will typically present with low levels of a single line of little particles of cells that clot the blood called platelets, whole-body skin rashes and recurrent infections. Nevertheless, the clinical presentation can vary between individuals. We present a case of a 10-month-old boy who came in with a history of recurrent fever, skin rash since birth and episodes of bloody diarrhoea. He had very low levels of red blood cells and platelets. Genetic analysis confirmed the patient to have WAS. He was managed with supportive treatment, followed up on a regular clinic but unfortunately died 4 months later. Being a rare genetic disease, the diagnosis of WAS can easily be missed, especially in regions with scarce healthcare resources that do not have easy access to genetic testing. Thus, doctors should suspect WAS in boys presenting with recurrent infections and bleeding problems.
Keywords: case report, eczema, thrombocytopenia, Wiskott–Aldrich syndrome
Introduction
Wiskott–Aldrich syndrome (WAS) is a rare immunodeficiency X-linked genetic disorder often featured with a clinical triad of thrombocytopenia (with low mean platelet volume), eczematoid dermatitis and recurrent infections.1 The syndrome was first described more than 80 years ago. Incidence is estimated at 1–10 per 1 million cases per live birth.2–4 The culprit gene product, Wiskott–Aldrich syndrome protein (WASP), is a protein encoded on the short arm of the X chromosome, found in the cytoplasm of nonerythroid haematopoietic cells. Variants can lead to absence, reduced or increased expression of the WASP protein, manifesting as classical WAS, X-linked thrombocytopenia (XLT) and X-linked neutropenia (XLN) phenotypes, respectively. The exact function of the WASP protein has not been completely elucidated, however, several studies have singled it out as an important component in signalling pathways and arrangement of actin filaments in the cytoskeleton.5,6
The clinical manifestation of WAS, depending on the underlying variant, shows wide heterogeneity, and a high index of suspicion is required to establish clinical diagnosis. In addition to the classical triad of micro-thrombocytopenia, eczema and immunodeficiency, patients with WAS may present with an autoimmune disease with a propensity for developing lymphoreticular neoplasms, such as lymphoma, leukaemia and myelodysplasia.7,8
Clinical symptoms and signs usually commence early in life and are often missed or managed as a different condition. The majority of patients are usually diagnosed in their second year of life.8 Advanced techniques, which most of the time are not readily available in resource-limited settings, such as sequence analysis for the WAS gene, are required to confirm laboratory diagnosis of WAS.9
The current standard-of-care for infants with WAS is early haematopoietic stem cell transplantation (HSCT), preferably from human leukocyte antigen (HLA)-identical siblings with a reported survival rate of more than 80%.10In the absence of HSCT, supportive care alone aimed at managing complications such as bleeding and infections remains the mainstay of management with poor outcomes.11
WAS has a wide range of phenotypical presentation and fatal outcomes. We present this case report to share the challenges in the diagnosis and management of WAS in a setting of limited healthcare resources.
Case description
A 10-month-old boy, a resident of Dar es Salaam, Tanzania, was referred to our hospital with fever, recurrent skin lesions and bloody diarrhoea. The fever was low grade, associated with excessive night sweats and subsided with paracetamol. No report of any respiratory symptoms or convulsions was noted. The onset of the skin lesions was gradual, noticed first behind the ears as dark painless spots, before spreading to the rest of the body. Later on, the skin lesions became itchy, gradually increasing in size and bled occasionally. The symptoms were followed by three episodes of bloody diarrhoea which only lasted for 1 day. The patient had no history of bleeding from the gums or nose, and he did not report any joint pain or swelling. He had normal urine colour and micturition habits.
The patient was born with a body weight of 3.5 kg, via a caesarean section following a prolonged obstructed labour with an Apgar score of 4 at 0 min and 7 at 5 min. He was discharged home with his mother on day 7 in a good clinical condition. At the age of 2 weeks, he was admitted for the first time due to a high-grade fever and generalised skin lesions. He was treated as a case of sepsis and eczema with delayed improvement before being discharged. At 6 months of age, he was re-admitted with a similar presentation and management.
The child had attained normal developmental milestones for his age. He was the second child in his family. His sister was well, without a history of similar illness. There was no history of a similar condition from his uncles and cousins on his mother’s side.
During presentation, physical examination revealed ecchymosis and eczematoid lesions over the scalp, neck, chest and back, together with the upper and lower limbs, with bleeding in some lesions (Figure 1(a) and (b)). The largest lesion was 2 × 3 cm. The patient was febrile (38.2°C) with moderate pallor, not icteric and there were no palpable lymph nodes. Other system examination findings were normal.
Figure 1.

Ecchymosis and eczematoid lesions with bleeding in some lesions.
Figure 2.
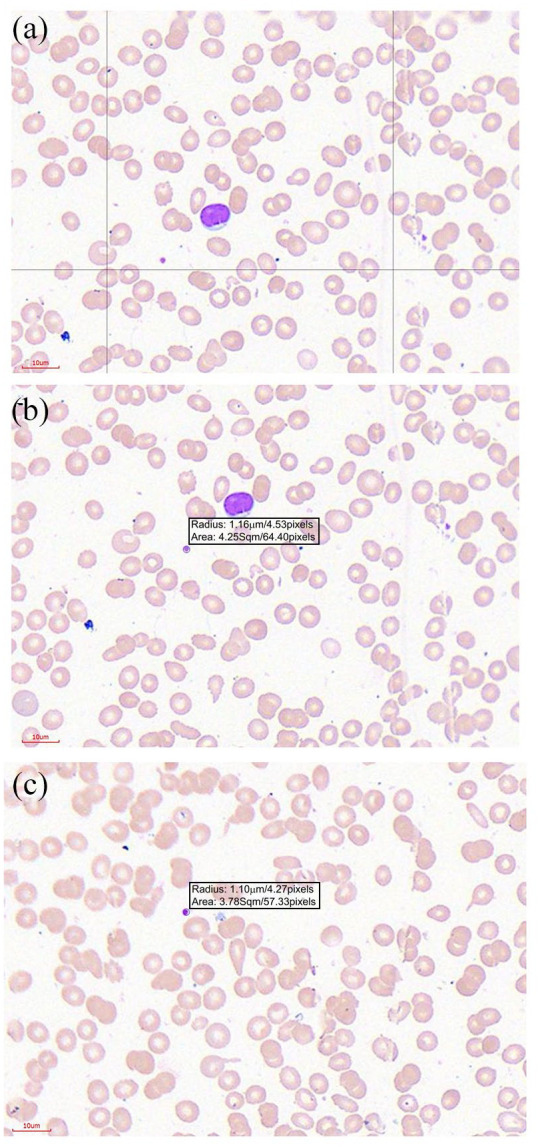
Light microscopy of peripheral blood smear. (a) The slide shows the blood smear with reduced erythrocytes (most are microcytic) with very few normal staining platelets per field. (b) A field section with a platelet measuring a radius of 1.16 µm/diameter of 2.32 µm. (c) A field section with a normal staining platelet measuring a radius of 1.10 µm/diameter of 2.2 µm.
The blood count at presentation showed leucopenia, microcytic anaemia and thrombocytopenia with normal mean platelet volume (MPV). (Table 1) The peripheral blood smear showed microcytic hypochromic red cells consistent with iron deficiency anaemia and a pancytopenia (Figure 2). The platelet diameter was quantified by computer-assisted image analysis using the Motic Easyscan Pro and the Motic DSAassistant software (Motic, Hong Kong, China). The mean platelet diameter was 2.28 µm (Figure 2(b) and (c)). A mean platelet diameter of <2.6 µm is considered to be consistent with the presence of hereditary thrombocytopenias with normal or reduced size.12 Platelet immunofluorescence studies were not available. Bone marrow aspiration cytology showed erythroid hyperplasia with a myeloid–erythroid ratio of 1:1. Megakaryocyte series was not increased and no abnormal cell infiltration was observed. C-reactive protein and lactate dehydrogenase were raised. Prothrombin time and activated partial thromboplastin time were within normal range. The immunological profile revealed high immunoglobulin (Ig) IgE and IgA and normal IgG and IgM (Table 2).
Table 1.
Patient’s complete blood count trend.
| Test | Reference range | Baseline | Follow-up counts | ||
|---|---|---|---|---|---|
| Week 4 | Week 8 | Week 12 | |||
| White blood count, total, (per L) | 4–16 × 109 | 3.18 × 109 | 3.45 × 109 | 5.43 × 109 | 3.9 × 109 |
| Neutrophil, (per L) | 1–7 × 109 | 0.05 × 109 | 1.26 × 109 | 0.89 × 109 | 1.35 × 109 |
| Lymphocyte, (per L) | 3.5–11 × 109 | 0.89 × 109 | 0.75 × 109 | 3.26 × 109 | 1.33 × 109 |
| Monocyte, (per L) | 0.2–1.0 × 109 | 2.22 × 109 | 1.21 × 109 | 0.87 × 109 | 1.47 × 109 |
| Basophil, (per L) | 0.0–0.11 × 109 | 0.02 × 109 | 0.04 × 109 | 0.15 × 109 | 0.04 × 109 |
| Eosinophil, (per L) | 0.1–1.0 × 109 | 0.00 × 109 | 0.19 × 109 | 0.26 × 109 | 0.01 × 109 |
| Haemoglobin | 11.1–14.1 | 6.63 | 10.1 | 11.1 | 12.4 |
| MCV (fl) | 72–84 | 63 | 82 | 79.8 | 75.8 |
| MCH (pg) | 25–29 | 20 | 26.7 | 25.6 | 24 |
| MCHC (g/L) | 320–360 | 320 | 325 | 320 | 316 |
| Platelets (per L) | 200–550 × 109 | 19 × 109 | 14.5 × 109 | 49.9 × 109 | 9.68 × 109 |
| MPV (fl) | 8.0 - 15.0 | 13.5 | - | 8.2 | - |
MCH, mean corpuscular haemoglobin; MCHC, mean corpuscular haemoglobin concentration; MCV, mean corpuscular volume; MPV, mean platelet volume.
Table 2.
Other blood test results at presentation.
| Test | Result | Reference range |
|---|---|---|
| C-reactive protein | 3095 mg/L | 0–50 mg/L |
| Lactate dehydrogenase | 279 U/L | 60–100 U/L |
| Coagulation tests | ||
| Prothrombin time | 13 s | 11–13.5 s |
| Activated partial thromboplastin time | 32 s | 30–40 s |
| Immunological profile | ||
| IgE | 10,000 IU/ml | 0–100 IU/ml |
| IgA | 1.57 g/L | 0–0.83 g/L |
| IgG | 2.07 g/L | 2.31–14.11 g/L |
| IgM | 0.24 g/L | 0–1.45 g/L |
Ig, immunoglobulin.
Urea and creatinine levels were within the normal range. Tissue biopsy of the skin lesions revealed multifocal inflammatory cells infiltrate, predominantly lymphocytes. Serology tests for syphilis, cytomegalovirus (CMV) and HIV were all negative. Blood culture could not be performed due to laboratory diagnostic limitations that were present during the time of patient admission.
Thrombocytopenia, early childhood onset and history of recurrent eczematoid skin lesions, recurrent infections, and the biological sex of our patient led us to suspect WAS. Even though our patient had normal-sized platelets, his presentation was consistent with Ochs et al.11 disease classification score of 4. The diagnosis was confirmed with genetic testing abroad. Using Sanger sequencing, a likely pathogenic variant (NM_000377.2:c.360+1G>A) was detected. This variant disrupts one of the two invariant nucleotides of the splice donor site and is predicted to lead to aberrant splicing.13 It has previously been detected in individuals/families with WAS.14,15 Two other splicing variants have also been reported at nucleotide c.360+1, c.360+1G>C and c.360+1G>T, both associated with WAS.16,17 This variant is therefore predicted to be pathogenic.
We were not able to perform genetic analysis of the patient’s mother, therefore, we could not establish, whether this variant had occurred de novo or was inherited in an X-linked fashion.
The child was started on intravenous (IV) paracetamol 150 mg, 6 h for 3 days, then switched to oral administration. He was also initiated on IV meropenem 200 mg, 8 h for 10 days. The skin was managed with topical steroid application, with daily wound care. He also received a total of 450 ml of packed red blood cells (PRBC) and 250 ml of platelet concentrates (non-irradiated as equipment for irradiation was not readily available) during the time of admission. The child’s condition improved significantly and he was later discharged home in a stable condition. He was discharged on prophylactic antibiotics (oral azithromycin 200 mg, 3 days per week and topical mupirocin cream to apply two times daily) and topical steroids (hydrocortisone cream to apply two times daily), and his mother was advised to attend regular paediatrics clinic for follow up and monitoring. He attended monthly clinics in the 3 months following discharge. Haemoglobin and other levels remained relatively within range; however, the white cell line remained low with severe thrombocytopenia throughout. Follow-up MPV after 2 months was within the normal range (Table 1) In between clinic visits, he was placed on iron supplements (ferrous sulphate 30 mg, twice daily), a topical steroid (hydrocortisone cream to apply two times daily) and prophylactic antibiotics (oral azithromycin 200 mg, 3 days per week and topical mupirocin cream to apply two times daily). At 4 months after the first discharge, he was re-admitted with severe anaemia, purpura, eczema and bleeding from wounds and nose. He was managed with PRBC and platelet concentrates replacement. He was also started on IV tranexamic acid 250 mg, 8 h, IV methylprednisolone 250 mg, once daily, and IV pantoprazole 20 mg, once daily. The patient’s clinical condition improved and he was discharged home after 2 weeks with topical corticosteroids, iron supplements and prophylactic antibiotics. At 3 weeks after discharge, the patient died shortly after presenting to the emergency department with a cough and severe difficulty in breathing. The cause of death was thought to be severe pneumonia and sepsis. An autopsy report could not be obtained. The lack of HSCT services in the country and region and socio-economic constraints limited the HSCT option for the patient.
Discussion and conclusion
Our patient presented with some classic WAS features of bleeding, eczema and thrombocytopenia. However, he had normal-sized platelets. He had an altered immunoglobulin profile (raised IgE and IgA), but IgG and IgM were within the normal range. Clinical features pointing to WAS are often present at birth or early on in life. In this report, the patient presented with symptoms at 2 weeks of age.
The common manifestation is usually bleeding, presenting as petechiae, bruises, epistaxis and bloody diarrhoea. Severe forms of bleeding may occur and may involve intracranium.
Thrombocytopenia with reduced platelet volume is a persistent feature in WAS gene variants, except for patients presenting with the XLN phenotype.18 Platelet counts are usually between 20 × 109/L and 50 × 109 /L, however, levels may drop to below 10 × 109/L. The mean platelet volume is usually below the normal lower limit of 7.1 fl and often ranges between 3.8 fl and 5.0 fl.19 Nevertheless, few reports have documented rare cases of WAS with normal-sized thrombocytes and even macrothrombocytopenia.20,21 Remold-O’Donnell et al. reported a similar variant to that found in our patient, that is, G>A transition at the [+1] position at the 5′ end of intron 3, in a 3-year-old boy who had severe thrombocytopenia of 10 × 109/L.15 However, in this case, the platelet size was not recorded. Our report, therefore, is the first to report the platelet size in a patient with a c.360+1G>A variant.
Recurrent infections due to immunodeficiency, mostly bacterial in origin, are common. Fungal infections are found in rare cases with severe immune deficiency. Commonly reported organisms are Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae. Varicella and CMV are also commonly observed viruses. Patients usually present with otitis media, bacterial pneumonia, sinusitis, meningitis, sepsis, colitis and skin infections.22 The severity of the immune deficiency and infections in patients with WAS depends largely on the variant and its effect on the levels of protein expression and function.23
Eczema, resembling atopic dermatitis, affects nearly half of patients with WAS in the first 12 months of life. The lesions are often superinfected and, in some instances, there is bleeding into the lesions as was the case with our patient.24
A wide range of autoantibodies has been observed both in classic WAS and in XLT. Coombs test-positive autoimmune haemolytic anaemia is the commonest autoimmune feature. Other autoimmune presentations include neutropenia, vasculitis, renal disease, inflammatory bowel disease and Henoch–Schönlein-like purpura.25
WAS affects the function of both T and B lymphocytes and generally lymphocyte counts decrease with age. Antibody activity and responses vary across the types of antigen, and while there may be sufficient response to some antigens, reduced or total loss of response has been observed with a set of different antigens, in the same individual. IgG and IgM levels are usually within the normal range or slightly reduced, whereas IgA and IgE are elevated.19,26,27 While WAS impairs chemotactic responses of polymorphonuclear cells, the phagocytic activity of neutrophils is mostly normal.19
Ochs et al.11 developed a scoring system that helps to differentiate distinct WAS variant clinical phenotypes based on clinical findings. A score of 1 or 2 is consistent with XLT while a score of 3–4 points to classic WAS. A score of 5 defines patients with either XLT or WAS who develop autoimmunity and/or malignancies. A patient’s score may change with time as the disease manifestation progresses.
WAS gene sequence analysis is used to establish the presence and type of variant. Our patient was hemizygous for (NM_000377.2:c.360+1G>A). One study15 that reported a similar variant identified a mixture of small (∙1.9 kb) and normal-sized transcripts, however lacked detectable WASP suggesting a problem with splicing. Indeed, the absence of WASP in cases with this variant is postulated to be due to defective WASP homology one domain resulting from skipping of exon 3 in the 5′ donor site, resulting in small (∙1.9 kb) RNA.6,28 WASP homology one domain is involved in the formation of the secondary or tertiary structure of WASP.6 Unfortunately, we could not measure RNA size or WASP levels in our patient.
Flow cytometry with anti-WASP antibodies can be used to screen lymphocytes for quantitative WASP defects, although it may not be able to identify mutated or nonfunctional protein using this method.29 Due to the lack of anti-WAS antibodies in our settings, this was not possible.
The survival rate in patients with WAS (without HSCT) is disappointingly low.8 Some studies have reported median survival of only 20 years in patients with WAS.11 However, patients with XLT have been shown to have higher overall survival.30 HSCT is currently the only available definitive treatment for WAS. For WAS, the younger the patient the better the outcome of HSCT. The HSCT outcomes have improved over the years, across all donor types. The overall survival rate of over 80% has been demonstrated with HLA-identical sibling donors.30 Matched unrelated donor HSCT has also shown good outcomes in cases without HLA-identical sibling donors. Haplo-identical HSCT has poor outcomes with reports of survival rates of approximately 50% in large studies.30,31 New treatments are emerging and currently, studies and trials of gene therapy are underway.30
For patients who are awaiting HSCT and for those for whom HSCT is not an option as with our patient, supportive care is the mainstay.11 Supportive care encompasses paying close attention to prevent and promptly treat infections. Splenectomy may be an option for some patients to increase circulating platelet counts. However, the decision should be approached with care, as it may expose patients to a significant risk of septicaemia, and patients will require indefinite penicillin (or equivalent) prophylaxis. Splenectomy is also not recommended if there are plans for HSCT.30,32 For our patient, splenectomy was not a favourable option as he lives in a malaria-endemic region.33
Regular or persistent viral infections may warrant antiviral prophylaxis.30 Evidence supporting the use of immunosuppressants and plasmapheresis for the treatment of autoimmune cytopenia is limited.34 IV immunoglobulin (IVIG) therapy is used to treat severe antibody deficiency, especially for prevention of severe infections. In addition, corticosteroids and rituximab (anti-CD 20) are utilised. For the treatment of thrombocytopenia, irradiated blood products (PRBC and platelets) screened for CMV are essential to maintain adequate red cell mass and platelet levels, especially in bleeding patients. Plasmapheresis, IVIG therapy, blood products and rituximab are all expensive treatments that are limited in our setting, therefore, the mainstay of management is with corticosteroids.
As it is a rare genetic disease, the diagnosis of WAS can easily be missed, especially in resource-limited healthcare settings. A high index of suspicion is needed when a boy presents with early onset of recurrent infections and bleeding tendencies. In the absence of HSCT, treatment remains supportive with very poor outcome.
Acknowledgments
We extend our sincere gratitude to the patient and his family for allowing us to report this case of rare disease. We also thank all the attending staff of the Paediatrics and Haematology departments at Muhimbili National Hospital and Muhimbili University of Health and Allied Sciences. We would also like to send our appreciation to Professor Lucio Luzzatto for his support and critical review of this report.
Footnotes
Author contributions: WFM: conceptualization; data curation; formal analysis; investigation; methodology; project administration; validation; writing original draft; writing–review and editing.
HI: data curation; formal analysis; methodology; writing original draft; writing–review and editing. CAK: formal analysis; methodology; writing–review and editing.
AN: data curation; formal analysis; writing–review and editing.
AS: formal analysis; investigation; resources; supervision; validation; writing–review and editing.
The authors declare that there is no conflict of interest.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
Consent to publication: Written informed consent for publication of their clinical details and clinical images was obtained from the parent of the patient. A copy of the consent form is available for review by the editor.
Ethics Statement: Ethical approval to develop this case report was granted by the Muhimbili National Hospital Ethical Review Board with certificate reference number: MNH/IRB-CR/2019/001.
ORCID iD: William Frank Mawalla  https://orcid.org/0000-0002-0016-7551
https://orcid.org/0000-0002-0016-7551
Contributor Information
William Frank Mawalla, Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Science (MUHAS), P.O. Box 65001, Upanga, Dar es Salaam, Tanzania.
Hamisa Iddy, Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences. Dar es Salaam, Tanzania.
Christine Aloyce Kindole, Department of Paediatrics and Child Health, Muhimbili National Hospital, Dar es Salaam, Tanzania.
Ahlam Nasser, Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences. Dar es Salaam, Tanzania.
Anna Schuh, Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences. Dar es Salaam, Tanzania; Oxford Molecular Diagnostic Centre, Department of Oncology, University of Oxford, John Radcliffe Hospital, Oxford, UK.
References
- 1. Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics 1954; 13; 133–139. [PubMed] [Google Scholar]
- 2. Bosticardo M, Marangoni F, Aiuti A, et al. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood 2009; 113: 6288–6295. [DOI] [PubMed] [Google Scholar]
- 3. Ryser O, Morell A, Hitzig WH. Primary immunodeficiencies in Switzerland: first report of the national registry in adults and children. J Clin Immunol 1988; 8: 479–485. [DOI] [PubMed] [Google Scholar]
- 4. Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol 2005; 25: 73–77. [DOI] [PubMed] [Google Scholar]
- 5. Bouma G, Burns SO, Thrasher AJ. Wiskott–Aldrich syndrome: immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology 2009; 214: 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Symons M, Derry J, Karlak B, et al. Wiskott- Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 1996; 84: 723–734. [DOI] [PubMed] [Google Scholar]
- 7. Dupuis-Girod S, Medioni J, Haddad E. Autoimmunity in Wiskott-Aldrich syndrome risk factors, clinical features, and outcome in a single center cohort of 55 patients. Pediatrics 2003; 111: 622–627. [DOI] [PubMed] [Google Scholar]
- 8. Sullivan KE, Mullen CA, Blaese RM, et al. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994; 125: 876–885. [DOI] [PubMed] [Google Scholar]
- 9. José MB, María LL, Rocío B, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018; 103: 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi R. Outcome in patients with Wiskott-Aldrich syndrome following stem cell transplantation: an analysis of 57 patients in Japan. Br J Haematol 2006; 135: 362–366. [DOI] [PubMed] [Google Scholar]
- 11. Ochs HD, Filipovich AH, Veys P, et al. WiskottAldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transpl 2009; 15: 84–90. [DOI] [PubMed] [Google Scholar]
- 12. Greinacher A, Pecci A, Kunishima S, et al. Diagnosis of inherited platelet disorders on a blood smear: a tool to facilitate worldwide diagnosis of platelet disorders. J Thromb Haemost 2017; 15: 1511–1521. [DOI] [PubMed] [Google Scholar]
- 13. Interactive-Biosoftware. Alamut Visual, https://www.interactive-biosoftware.com/alamut-visual/ (2020, accessed 25 May 2020).
- 14. The human gene mutation database. Cardiff: Cardiff University, http://www.hgmd.cf.ac.uk/ac/index.php (2017, accessed 25 May 2020). [Google Scholar]
- 15. Remold-O’Donnell E, Cooley J, Shcherbina A, et al. Variable expression of WASP in B cell lines of Wiskott-Aldrich syndrome patients. J Immunol 1997; 158: 4021–4025. [PubMed] [Google Scholar]
- 16. Liu DW, Zhang ZY, Zhao Q, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia in China: clinical characteristic and genotype-phenotype correlation. Pediatr Blood Cancer 2015; 62: 1601–1608. [DOI] [PubMed] [Google Scholar]
- 17. Schindelhauer S, Weiss M, Hellebrand H, et al. Wiskott-Aldrich syndrome: no strict genotype-phenotype correlations but clustering of missense mutations in the amino-terminal part of the WASP gene product. Hum Genet 1996; 98: 68–76. [DOI] [PubMed] [Google Scholar]
- 18. Devriendt K, Kim AS, Mathijs G. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 2001; 27: 313–317. [DOI] [PubMed] [Google Scholar]
- 19. Ochs HD, Slichter SJ, Harker LA. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood 1980; 55: 243–252. [PubMed] [Google Scholar]
- 20. Mantadakis E, Sawalle-Belohradsky J, Tzanoudaki M. X-linked thrombocytopenia in three males with normal sized platelets due to novel WAS gene mutations. Pediatr Blood Cancer 2014; 61: 2305–2306. [DOI] [PubMed] [Google Scholar]
- 21. Bastida JM, Del Rey M, Revilla N. Wiskott-Aldrich syndrome in a child presenting with macrothrombocytopenia. Platelets 2017; 28: 417–420. [DOI] [PubMed] [Google Scholar]
- 22. Mullen CA, Anderson KD, Blaese RM. Splenectomy and/or bone marrow transplantation in the management of the Wiskott-Aldrich syndrome: long-term follow-up of 62 cases. Blood 1993; 82: 2961–2966. [PubMed] [Google Scholar]
- 23. Jin Y, Mazza C, Christie JR. Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood 2004; 104: 4010–4019. [DOI] [PubMed] [Google Scholar]
- 24. Presa JG, de Carvalho VO, Morrisey LR, et al. Cutaneous manifestations in patients with Wiskott-Aldrich syndrome submitted to haematopoietic stem cell transplantation. Arch Dis Child 2013; 98: 304–307. [DOI] [PubMed] [Google Scholar]
- 25. Crestani E, Volpi S, Candotti F. Broad spectrum of autoantibodies in patients with Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol 2015; 136: 1401–1404.e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blaese RM, Strober W, Brown RS, et al. The Wiskott-Aldrich syndrome. A disorder with a possible defect in antigen processing or recognition. Lancet 1968; 1: 1056–1061. [DOI] [PubMed] [Google Scholar]
- 27. Ozcan E, Notarangelo LD, Geha RS. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol 2008; 122: 1054–1062; quiz 1063–1064. [DOI] [PubMed] [Google Scholar]
- 28. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 1992; 90: 41–54. [DOI] [PubMed] [Google Scholar]
- 29. Chiang SCC, Vergamini SM, Husami A. Screening for Wiskott-Aldrich syndrome by flow cytometry. J Allergy Clin Immunol 2018; 142: 333–335.e8. [DOI] [PubMed] [Google Scholar]
- 30. Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet 2014; 7: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin CR, Kim MO, Li D. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transpl 2012; 47: 1428–1435. [DOI] [PubMed] [Google Scholar]
- 32. Albert MH, Bittner TC, Nonoyama S. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood 2010; 115: 3231–3238. [DOI] [PubMed] [Google Scholar]
- 33. Mahlaoui N, Pellier I, Mignot C. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood 2013; 121: 1510–1516. [DOI] [PubMed] [Google Scholar]
- 34. Bach O, Baier M, Pullwitt A. Falciparum malaria after splenectomy: a prospective controlled study of 33 previously splenectomized Malawian adults. Trans R Soc Trop Med Hyg 2005; 99: 861–867. [DOI] [PubMed] [Google Scholar]