

Abstract
Immunoglobulin A (IgA) nephropathy (IgAN) is the most common type of primary glomerulonephritis, often progressing to renal failure. IgAN is triggered by IgA deposition in the glomerular mesangium by an undefined mechanism. Here, we show that grouped ddY (gddY) mice, a spontaneous IgAN model, produce serum IgA against mesangial antigens, including βII-spectrin. Most patients with IgAN also have serum anti–βII-spectrin IgA. As in patients with IgAN, IgA+ plasmablasts accumulate in the kidneys of gddY mice. IgA antibodies cloned from the plasmablasts carry substantial V-region mutations and bind to βII-spectrin and the surface of mesangial cells. These IgAs recognize transfected and endogenous βII-spectrin exposed on the surface of embryonic kidney–derived cells. Last, we demonstrate that the cloned IgA can bind selectively to glomerular mesangial regions in situ. The identification of IgA autoantibody and its antigen in IgAN provides key insights into disease onset and redefines IgAN as a tissue-specific autoimmune disease.
The finding of IgA autoantibodies against βII-spectrin on mesangial cells in IgA nephropathy redefines the disease pathogenesis.
INTRODUCTION
Immunoglobulin A (IgA) nephropathy (IgAN) is a common type of primary glomerulonephritis characterized by the deposition of IgA, often with IgG, IgM, and complement C3, in the mesangial region, followed by mesangial cell (MC) proliferation and excessive synthesis of extracellular matrix in the glomeruli of the kidney (1). Approximately 30 to 40% of cases of IgAN progress to kidney failure, necessitating dialysis or kidney transplantation within 10 to 20 years after disease onset (2). IgAN is one of the main causes of chronic kidney diseases and affects 850 million people worldwide. Recently, many cases of IgAN have been newly diagnosed after immunization with the SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) mRNA vaccine, although the underlying mechanism is currently unclear (3–7). Thus, IgAN is now drawing more attention than ever not only in the scientific and clinical fields but also in society in general. Almost half a century has passed since the first report of patients with IgAN by Berger (8), but no specific and causal treatment strategies for the disease have been developed. This is largely due to a poor understanding of the pathogenesis of IgAN, particularly the mechanism by which nephritogenic IgA is deposited in the glomeruli (9–11).
It is known that IgA1 in the sera and the mesangial deposits of patients with IgAN contains undergalactosylated O-linked glycans in its hinge region and is termed galactose-deficient IgA1 (Gd-IgA1) (12, 13). Gd-IgA1 has been proposed to serve as a self-antigen that triggers the generation of autoantibodies (auto-Abs), together forming immune complexes (ICs). Such ICs are trapped in the glomeruli and cause renal injury, which has been considered as the primary cause of IgAN. Thus, IgAN has been classified as a type 3 allergic disease in the Gell and Coombs classification. However, supposing that such ICs would be randomly deposited at various locations in the glomeruli, the mesangium-specific IgA deposition, a hallmark of IgAN, cannot solely be explained by the IC formation of Gd-IgA1. Together with the fact that certain healthy people also have increased serum Gd-IgA1 (13–16), it suggests other reasons for the mesangium-specific IgA deposition.
We have been studying the pathogenesis of IgAN using an animal model of spontaneous IgAN, called grouped ddY (gddY) mice (17–20). The original outbred ddY mice develop IgAN with a low incidence and variable timing of onset. We intercrossed an early-onset group of ddY mice for more than 20 generations to produce gddY mice. Essentially, all individual gddY mice exhibit proteinuria and glomerular IgA deposition by 8 weeks of age, followed by obvious renal failure with a pathology that is similar to human IgAN. Thus, gddY mice are a useful animal model to study disease pathogenesis and for preclinical studies (21). Using gddY mice with a gene edited by the CRISPR-Cas9 system, we recently found that IgA does not form ICs in the blood but is first deposited into the glomerular mesangium before forming ICs with IgG, IgM, and complement C3 in situ (18). In addition, the deposition of IgG, IgM, and C3 and subsequent renal injury were not observed in IgA-deficient gddY mice (18). While it is well known that IgA-IC formation is necessary for the onset of IgAN, these data indicate that the initial IgA deposition in the mesangial region is a prerequisite for IC formation in the glomeruli and therefore for the onset of IgAN. Thus, it is crucial to elucidate the mechanism of IgA deposition in the mesangial region to understand the pathogenesis of IgAN.
In the present study, we found that the sera of gddY mice and human patients with IgAN contain IgA auto-Abs against MC antigens and identified βII-spectrin as a dominant self-antigen. We have demonstrated that βII-spectrin, previously known as an intracellular protein, is exposed on the surface of MCs and directly recognized by the IgA auto-Abs produced by gddY mice. Our unexpected findings indicate that IgAN is an autoimmune disease mediated by auto-Abs targeting tissue self-antigens.
RESULTS
Identification of IgA auto-Abs against glomerular mesangium in gddY mice
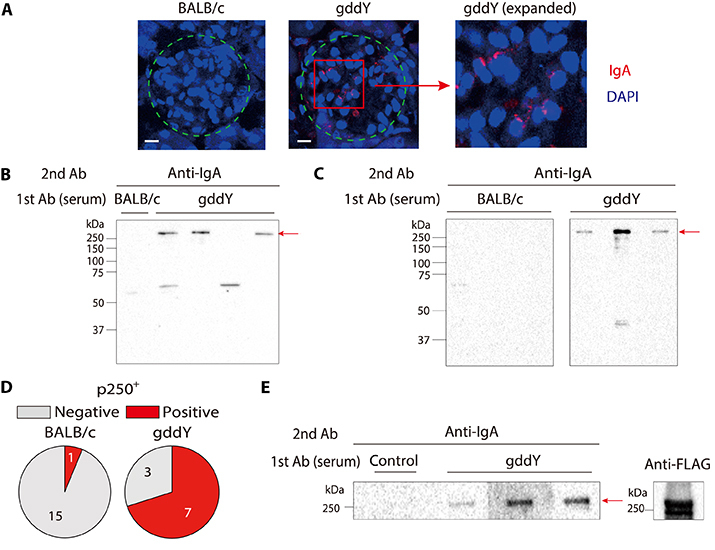
To address whether the serum IgA of gddY mice includes auto-Abs against glomeruli, we stained kidney sections from activation-induced cytidine deaminase (AID)–deficient mice, which lack endogenous IgA, with sera from gddY or BALB/c mice as primary Abs and then with anti-mouse IgA as a secondary Ab. This analysis revealed that the sera of gddY mice, but not control BALB/c mice, contained IgA that bound to the glomeruli (Fig. 1A and fig. S1). Western blot (WB) analysis further demonstrated that serum IgA from all the examined gddY mice detected a few proteins in the lysates of isolated glomeruli (Fig. 1B) and those of primarily cultured MCs (Fig. 1C), with the most prominent one having an apparent molecular weight of >250 kDa (named p250+). The majority of gddY mice (70%), but only a few BALB/c mice (~6%), were found to have serum IgA recognizing the p250+ protein in the MCs (Fig. 1D). These data indicate that gddY mice regularly produce IgA auto-Abs against mesangial antigens, with the p250+ protein being a common target.
Fig. 1. Serum IgA auto-Abs in gddY mice recognizing βII-spectrin in MCs.

(A) Kidney sections from AID-knockout mice were stained with sera from 16-week-old (wo) BALB/c (left) or gddY (center, right) mice followed by PE–anti-IgA Ab (red) and with 4′,6-diamidino-2-phenylindole (DAPI; blue). Dashed circles indicate areas of glomeruli. The scale bars are shown as white lines (10 μm). (B) WB of glomeruli of gddY mice blotted with pooled serum of 12-wo BALB/c or gddY mice (n = 4), followed by anti-IgA Ab. The red arrow indicates the p250+ band. Shown is a representative of three independent experiments. (C) WB of mouse primary MCs blotted with sera of 12-wo BALB/c or 8-wo gddY mice (n = 3), followed by anti-IgA Ab. The red arrow indicates the p250+ band. (D) Reactivity of serum IgA from 8- to 16-wo BALB/c or gddY mice to p250+. The numbers of serum samples are denoted in the pie charts. (E) WB of purified FLAG-FL-Sptbn1 protein probed with serum from a 12-wo ddY mouse (control) or sera from 12-wo gddY mice (n = 3), followed by anti-IgA Ab (left). The presence of the FLAG-FL-Sptbn1 was confirmed by using an anti-FLAG Ab (right). (F) WB of HEK293T cells transfected with mock (−) or FLAG-tagged Sptbn1A, Sptbn1B, or Sptbn1C (+) and probed with pooled sera from four 16-wo BALB/c mice or sera from gddY mice (n = 3), followed by anti-IgA Ab (left). The red arrow indicates Sptbn1C. The expression of each Sptbn1 fragment was confirmed by WB with an anti-FLAG Ab (right). (G) Reactivity of serum IgA of 12- to 16-wo BALB/c (left) and 8- to 16-wo gddY (right) mice to Sptbn1C determined by WB as shown in (F).
βII-spectrin as a common target of gddY mouse IgA auto-Abs
To identify the antigens recognized by the IgA auto-Abs of gddY mice, extracts of mouse kidney glomeruli were immunoprecipitated with IgA purified from the sera of gddY or BALB/c mice. The precipitates were resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and protein bands with sizes similar to those repeatedly detected with the gddY-mouse serum IgA by WB were excised and analyzed by mass spectrometry. From the peptide sequences repeatedly identified in the IgA immunoprecipitate of gddY mice, but not of BALB/c mice, we inferred candidate proteins recognized by the gddY IgA auto-Ab. Candidate proteins were evaluated by complementary DNA (cDNA) cloning and expression. Among these candidates, we identified βII-spectrin [also called spectrin beta chain, non-erythrocytic 1 (Sptbn1)] as a protein recognized by IgA in gddY mice, but not BALB/c mice (Fig. 1E).
βII-spectrin is a 274-kDa protein ubiquitously expressed by all nucleated cells, unlike the other four members of the spectrin β family. βII-spectrin, together with αII-spectrin [also called spectrin alpha chain, non-erythrocytic 1 (Sptan1)], form a polymeric structure underlying the plasma membrane (22). To narrow down the region recognized by gddY IgA, we divided Sptbn1 into three parts (Sptbn1A: 1 to 788 amino acids; Sptbn1B: 789 to 1576 amino acids; Sptbn1C: 1577 to 2364 amino acids), separately expressed each fragment in HEK293T cells, and analyzed binding by WB. Among these fragments, Sptbn1C was most frequently detected by the serum IgA of gddY mice (Fig. 1, F and G). Among 10 other candidate proteins tested using a similar WB approach, only Sptan1 was selectively recognized by the serum IgA of gddY mice, preferentially at its C-terminal one-third (Sptan1C: 1653 to 2478 amino acids), albeit at a much lower frequency (3 of 22 mice) (fig. S2). Thus, we concluded that βII-spectrin is the most representative target antigen of IgA auto-Abs produced by gddY mice.
IgA auto-Abs against βII-spectrin in the sera of patients with IgAN
Next, we examined whether the sera from patients with IgAN also contained IgA auto-Abs against MC components. WB using lysates of primary human MCs revealed that sera of most patients with IgAN (85%) contained IgA commonly recognizing p250+ proteins in addition to some other variable proteins, whereas sera from healthy individuals rarely contained such IgA (Fig. 2, A and B). The similarity of its apparent molecular weight prompted us to test by WB whether the p250+ MC protein is human βII-spectrin (SPTBN1). The purified full-length (FL) SPNBN1 protein encoded by an FL SPTBN1 cDNA was detected by WB using IgA1, a dominant subclass deposited on the glomeruli in patients with IgAN (2), from pooled sera of patients with IgAN, but not from those of healthy individuals (Fig. 2C). WB using independent serum samples from patients with IgAN revealed that approximately 60% of the samples contained anti-SPTBN1 IgA1, whereas no healthy individuals had these antibodies (Fig. 2D). Enzyme-linked immunosorbent assay (ELISA) using SPTBN1 protein purified from the transfected cells as a coating antigen demonstrated that sera of 16 of 45 patients with IgAN contained anti-SPTBN1 IgA, based on a cutoff value of the 95% confidence intervals (CI; mean + 2 SD of healthy individuals) (Fig. 2E), while anti-SPTBN1 IgG could not be detected in the same serum samples (fig. S3). Thus, we have revealed that most patients with IgAN, depending on the assay, have IgA auto-Abs against a specific self-protein, namely, βII-spectrin. Together with the finding that sera of gddY mice also contain IgA auto-Abs against βII-spectrin that binds to glomeruli, these results strongly suggest that IgAN is a tissue-specific autoimmune disease, although this has been little appreciated thus far.
Fig. 2. Presence of IgA auto-Abs against βII-spectrin in the sera of patients with IgAN.

(A) A representative WB of primary human MCs probed with sera from healthy control individuals (HC; n = 4) or patients with IgAN (IgAN; n = 5) followed by anti-human IgA Ab. The red arrow indicates the p250+ band. (B) Reactivity of serum IgA from HC or patients with IgAN to p250+. (C) WB of purified FLAG-tagged FL-SPTBN1 protein probed with purified IgA1 (25 μg/ml) from pooled sera of three HCs or three patients with IgAN, followed by anti-IgA Abs (left). The purified FLAG-tagged FL-SPTBN1 protein developed on an SDS-PAGE gel (5 μg per lane) was stained with Coomassie Brilliant Blue dye (right). (D) Reactivity of serum IgA1 from HC or patients with IgAN to SPTBN1. (E) ELISA determination of anti-SPTBN1 IgA Abs in the sera from HC (n = 15) or patients with IgAN (n = 45). ***P = 0.0007 (Mann-Whitney U test). The dashed lines show the 95% confidence interval(CI) and 99% CI of the control sera. Shown is one of two ELISA experiments with reproducible results. OD, optical density.
Anti–βII-spectrin IgA auto-Abs as a prognostic factor in IgAN
We next compared the clinical characteristics between the groups of IgAN patients with higher concentrations of serum anti-SPTBN1 IgA [optical density (O. D.) > the 99% CI (mean + 3 SD of healthy individuals); “high”] and those with lower concentrations (O. D. < the 95% CI; “low”). We examined several important clinical and pathological parameters, such as the levels of IgA, Gd-IgA1, and complement C3 in the sera; the IgA/C3 ratio; estimated glomerular filtration rate (eGFR); proteinuria; and MEST pathological score (23). Although the levels of proteinuria and MEST pathological scores were comparable between the two groups (fig. S4, A and B), eGFR tended to be low in the “high” group (Fig. 3A). Serum IgA levels were higher in the “high” group than in the “low” group (Fig. 3B), whereas no significant difference was observed in serum Gd-IgA1 (fig. S4C). Notably, the “high” group showed a strikingly higher serum IgA/C3 ratio (Fig. 3, C and D), a well-known independent prognostic factor in IgAN (24–27), suggesting that a higher level of serum anti-SPTBN1 IgA is related to a worse renal outcome in patients with IgAN.
Fig. 3. Increased serum IgA/C3 ratio in IgAN patients with higher anti–βII-spectrin IgA.

(A to D) eGFR (A), serum IgA (B), serum C3 (C), and serum IgA/C3 ratio (D) were compared between the anti-SPTBN1 IgA “low” (Low) and “high” (High) groups of patients with IgAN. Small horizontal lines indicate the mean (black) ± SD (colored) of each group. **P < 0.01 and ****P < 0.0001 (Mann-Whitney U test).
IgA+ PB accumulation in the kidneys of gddY mice and patients with IgAN
Previous studies have suggested that nephritogenic IgA is mainly produced in the bone marrow in IgAN (28), whereas plasma cells (PCs) secreting auto-Abs are often found in the affected kidneys in autoimmune diseases such as lupus (29–32). By flow cytometric analyses, we found that a significantly increased number of IgA+ CD138+ cells accumulated in the kidneys of gddY mice, whereas very few accumulated in the lupus model mice, NZB/W F1, and Faslpr/lpr, or in BALB/c mice (Fig. 4, A and B, and fig. S5A). Most of these IgA+ CD138+ cells in gddY mice expressed the proliferation antigen Ki67 to a level higher than those in the intestine and even to a level comparable to germinal center (GC) B cells in the spleen of immunized mice (fig. S5B), indicating that they are plasmablasts (PBs). IgA+ PBs emerged by 8 weeks of age and further increased with age in gddY mice (Fig. 4C). Immunofluorescence (IF) staining of the kidneys of 8-week-old (wo) gddY mice demonstrated that IgA+ PBs were present in the tubulointerstitial region (fig. S5C). CD138+ B220dull cells in the kidneys of gddY mice were mostly IgA+, whereas those in NZB/W F1 mice were predominantly IgG+ (fig. S5, D and E). Immunohistochemical (IHC) staining of kidney biopsy samples from patients with IgAN also revealed the presence of IgA+ CD138+ cells in the tubulointerstitial region of the kidney (Fig. 4D). The number of CD138+ cells relative to that of glomeruli in the biopsy samples positively correlated with the levels of serum creatinine and proteinuria in patients with IgAN (fig. S6, A and B), as reported similarly for PCs in the case of systemic lupus erythematosus (SLE) (31). Collectively, these data suggest that the accumulation of IgA+ antibody-secreting cells (ASCs) in the kidney is implicated in the pathogenesis of IgAN.
Fig. 4. Accumulation of PBs producing anti–βII-spectrin IgA in gddY mouse kidneys.

(A and B) Leukocytes isolated from the kidneys of the following mice were surface-stained for CD138 and then intracellularly stained for IgA: BALB/c (8 weeks old, n = 6), NZB/W F1 (24 weeks old, n = 6), Faslpr/lpr (24 weeks old, n = 5), and gddY (8 weeks old, n = 8). The numbers in the panels indicate the percentages of IgA+ CD138+ cells among live cells (A). The frequency (left) and the number (right) of IgA+ CD138+ cells in each mouse kidney (B). (C) Frequency of the IgA+ CD138+ cells among live cells in each kidney of gddY mice at the indicated age (n = 8, 8, and 6 for the 4-, 8-, and 16-wo mice, respectively). (D) IHC staining for IgA and CD138 in kidney biopsies from patients with IgAN. The dashed circle indicates a glomerulus. The scale bars are shown as black lines (20 μm). (E) WB of glomerular lysates blotted with the culture supernatants of IgA+ CD138+ cells from gddY mouse kidneys (kidney) or no cells (control) cultured on 40LB/APRIL cells (fig. S7A). (F) WB of HEK293T cells transfected with mock (−) or FLAG-Sptbn1C (+) vectors blotted with supernatants of IgA+ CD138+ cells from the small intestine or kidney of gddY mice cultured as in (E) (left). The arrow indicates exogenous Sptbn1C, as confirmed with an anti-FLAG Ab (right). Data are representative of two or three independent experiments. Small horizontal lines indicate the mean (black) ± SD (colored) of each group (B and C). **P < 0.01, ***P < 0.001, and ****P < 0.0001 (one-way ANOVA with multiple comparison test) (B and C).
Anti–βII-spectrin IgA production from PBs in the gddY mouse kidney
To address whether the IgA+ PBs infiltrating the kidney of gddY mice produced IgA auto-Abs, these cells were sorted and cultured with interleukin-6 (IL-6) on feeder cells expressing exogenous CD40 ligand, a proliferation-inducing ligand (APRIL), and B-cell activating factor (BAFF) to facilitate IgA production in the supernatant (fig. S7A). IF staining and WB demonstrated that the IgA from these PBs bound to glomerular components, particularly the p250+ protein (Fig. 4E and fig. S7B), and specifically to the Sptbn1C fragment (Fig. 4F).
Next, we cloned cDNAs encoding IgA heavy and light chains from single IgA+ PBs isolated from the kidneys of gddY mice and expressed each pair in HEK293T cells to generate a panel of monoclonal recombinant IgA (rIgA) Abs (33). Most of the heavy- and light-chain V-region genes contained substantial numbers of somatic mutations that replaced amino acids (Fig. 5A), and all of the rIgAs carried mutations in either heavy- or light-chain V regions or both (table S1), suggesting their GC origin. GCs can be generated in nonlymphoid tissues, including kidneys, under pathogenic conditions (34, 35), but essentially no GC B cells were detected in the kidney of 8-wo gddY mice, a time when IgA+ PBs had already emerged (fig. S8, A and B), suggesting that the IgA+ PBs had undergone a GC response elsewhere or atypical GC-independent maturation as found in the liver (36).
Fig. 5. Characterization of rIgA generated from IgA+ PBs of gddY mouse kidneys.

(A) Pie charts showing the number of sequences of heavy- or light-chain variable (V) regions carrying the indicated numbers of amino acid mutations per sequence. The V-region genes were cloned from single cells (n = 20) of IgA+ PBs from the kidneys of gddY mice. (B) Flow cytometric analysis of primary cultured mouse MCs stained intracellularly with anti-NP rIgA (#NP) or rIgA #9 followed by anti-IgA Ab (red). Shaded histograms indicate the cells stained with the secondary Ab alone. (C) Anti-FLAG immunoprecipitates from lysates of HEK293T cells transfected with mock (−) or FLAG-Sptbn1C (+) vectors were blotted with rIgA (#NP or #9) followed by anti-IgA Ab (left). The arrow indicates exogenous Sptbn1C, as confirmed with anti-FLAG Abs (right). (D) Reactivity to FL-Sptbn1 of the indicated p-rIgAs at the indicated concentration was evaluated by ELISA and presented as OD450 values. Data are representative of two or three independent experiments.
We screened these monoclonal rIgAs for their intracellular staining of primary-cultured MCs and found that 5 (#9, #11, #13, #18, and #19) of 20 rIgAs could stain MCs to various extents (Fig. 5B and fig. S9, A and B). Two (#9 and #13) of them also bound to Sptbn1C, as shown by WB (Fig. 5C). Since IgAs secreted from the kidney PBs and small intestine ASCs of gddY mice are mainly polymers including dimers (fig. S10A) that presumably bind to antigen with higher avidity than the monomeric IgA, we then made these rIgAs polymeric by expressing them in the HEK293T cells stably transfected with J chain (fig. S10B). The resultant polymeric rIgA (p-rIgA) bound more strongly to the Sptbn1 protein than the corresponding monomeric version (m-rIgA) as assessed by ELISA, as represented by p-rIgA #9 (fig. S10C). Through this strategy, two p-rIgAs (#9 and #11) were verified to bind strongly to the Sptbn1 protein by ELISA (Fig. 5D). These results indicate that PBs accumulating in the kidneys of gddY mice produce IgA Abs that recognize mesangial self-antigens, including βII-spectrin.
βII-spectrin expression on the surface of MCs
Given that the most common target of IgA auto-Abs in gddY mouse serum is βII-spectrin, we addressed how the auto-Abs recognize the protein typically located on the inside of the plasma membrane (37). First, we isolated MCs, endothelial cells (ETs), and podocytes (PDs) from purified glomeruli based on the expression of CD31 and CD73 (38) (Fig. 6A and fig. S11A), confirmed these subset designations by other surface profiles (fig. S11, B and C), and stained these nonpermeabilized cells with p-rIgAs or anti-Sptbn1 Ab. The ex vivo MCs, but not the ETs or PDs, were stained with p-rIgA #9 and p-rIgA #11, with p-rIgA #9 binding far more strongly, but not with the negative controls, p-rIgA #7 and p-rIgA #NP (Fig. 6B). In addition, only the MCs were stained with an Ab against βII-spectrin (Fig. 6C). These data indicate that βII-spectrin is exposed on the surface of MCs and recognized by the monoclonal IgAs originally produced by PBs of gddY mice.
Fig. 6. Binding to βII-spectrin on the MC surface by IgA auto-Abs from gddY mice.

(A) Flow cytometric analysis of mouse glomerular CD45− cells; MCs (CD73+ CD31−), endothelial cells (ETs; CD73− CD31+), and podocytes (PDs; CD73− CD31−). The numbers indicate the percentages of live cells. (B and C) Histograms with MFIs of the cells as defined in (A), after being stained with the indicated p-rIgA Abs followed by anti-IgA Ab (B) or with anti-Sptbn1 Ab (red line) or isotype-matched control Ab (shaded) (C). (D) HEK293T cells were transfected with mock or FLAG-tagged FL-SPTAN1 or FL-SPTBN1 vectors and stained with anti-FLAG Ab on the surface (top) or intracellularly (bottom) 2 days later. The numbers indicate the percentages of dead cell–excluded cells. (E) HEK293T cells transfected with FLAG-tagged FL-Sptbn1 vector and stained 3 days later with anti-FLAG Ab and the indicated p-rIgA Ab (or without the primary Ab), followed by anti-mouse IgA Ab. Histograms with MFIs of FLAG+ or FLAG− (gates shown in the left panel) live cells are shown (center and right). (F and G) IF microscopy. Kidney sections of AID-knockout mice intravenously injected with p-rIgA #7 or p-rIgA #9 (300 μg per mouse) 2 hours previously were stained with anti-IgA (red), anti-WT1 (indicating PDs; green), and DAPI (blue). High-power field presentation of the merged images, with white lines indicating areas of glomeruli, is shown in (G). The scale bars are shown as white lines. Data are one of two independent experiments with similar results.
To confirm the cell-surface expression property of βII-spectrin proteins, we transiently expressed FLAG-tagged human FL-SPTBN1 or FL-SPTAN1 in HEK293T cells. By staining with anti-FLAG Ab, we found that a substantial fraction of the cells expressing SPTBN1 and fewer cells expressing SPTAN1 displayed it on the cell surface (Fig. 6D and fig. S12A). Similarly, mouse FL-Sptbn1 and its N- and C-terminal fragments (Sptbn1A and Sptbn1C), but not the middle fragment (Sptbn1B), could be detected on the surface of the transfected HEK293T cells (fig. S12B). The cell-surface expression was also confirmed with T7-tagged Sptbn1 (fig. S12C) and further confirmed by its marked reduction after treating the cells with accutase, an enzyme with proteolytic and collagenolytic activity (fig. S12D). Notably, the cell-surface expression of exogenous βII-spectrin was barely observed on the mouse fibroblast cell line BALB/c 3T3, the human hepatoma cell line HepG2, or the cervical epithelial carcinoma cell line HeLa (fig. S12, E to G), suggesting that this is a unique property of limited cell types, such as HEK293T cells originating from embryonic kidneys.
HEK293T cells expressing FLAG-tagged FL-Sptbn1 or Sptbn1C on the surface were also positively stained with p-rIgA #9 or p-rIgA #11 Abs, but not with p-rIgA #7 Abs, further confirming that the former Abs bind to the cell-surface βII-spectrin, largely to its C-terminal fragment (Fig. 6E and fig. S12H). In this experiment, we noticed that p-rIgA #9 bound to a fraction of mock-transfected HEK293T cells (fig. S12I), suggesting that HEK293T cells can express their endogenous βII-spectrin on the cell surface. A substantial fraction of untransfected HEK293T cells were stained with the anti-SPTBN1 Ab (fig. S12J). However, HEK293T cells expressing endogenous SPTBN1 completely coexpressed exogenously introduced Sptbn1C or FL-Sptbn1 on their surface (fig. S12K), implying a cell-surface exposure mechanism common to both endogenous and exogenous βII-spectrin.
Last, we examined whether the IgA auto-Abs actually bind to the cell surface of the intact glomerular mesangium in vivo. AID-deficient mice were intravenously injected with the anti–βII-spectrin p-rIgA #9 or the negative control p-rIgA #7, which had respectively been confirmed to bind or not to glomeruli in the kidney sections of AID-deficient mice (fig. S13A). Two hours after the injection, kidney and other tissues were excised, and the deposition of p-rIgA in these tissues was analyzed by IF staining of the sections. The results clearly demonstrated that p-rIgA #9, but not p-rIgA #7, specifically bound to glomeruli in the mesangial region (Fig. 6, F and G) but not anywhere in the other tissues examined (fig. S13B). The binding of p-rIgA #9 was detected until at least 12 hours after injection. These data strongly suggest that p-rIgA #9 reacts in situ with βII-spectrin exposed on the surface of MCs in glomeruli.
Together, these data indicate that βII-spectrin, generally believed to be an intracellular protein, can be expressed on the surface of certain types of cells, including MCs in glomeruli, leading us to propose a unique model of IgAN pathogenesis that is initiated with the recognition of βII-spectrin on MCs by circulating specific IgA auto-Abs.
DISCUSSION
In the pathogenesis of human IgAN, the increase in serum Gd-IgA1 has been believed to be the first hit for its onset since Gd-IgA1 tends to self-aggregate and forms ICs with IgG or IgA Abs against Gd-IgA1, which are likely trapped by glomeruli and induce inflammation (11). However, this scenario would only predict the deposition of the IgA-ICs in the capillary of the glomeruli and cannot easily explain their selective deposition in the mesangial region, a pathological hallmark of IgAN. Our finding that IgA auto-Abs against MC components are produced in IgAN patients and its animal model, gddY mice, has solved this conundrum. We have identified βII-spectrin expressed on the mesangium as one of the self-antigens recognized by IgA auto-Abs. Such anti–βII-spectrin IgA could be detected in the sera of up to 60% of IgAN patients and gddY mice, depending on the assay, but scarcely in the sera of healthy individuals and normal BALB/c mice. The anti–βII-spectrin IgA was shown to bind to the surface of ex vivo MCs and to the mesangial regions in the glomeruli in vivo. Given that IgA can be deposited on the mesangial region before forming ICs with IgG and the complement, IgA auto-Ab binding to the mesangial region should be a trigger in the pathogenesis of IgAN.
Our study revealed that IgA+ PBs accumulating in the tubulointerstitial region of the kidney of gddY mice secrete auto-Abs that bind to the mesangium and βII-spectrin, which was confirmed with rIgA Abs derived from single IgA+ PBs. Thus, the kidney is a possible site for the generation of nephritogenic IgA, in addition to the bone marrow, as previously suggested (28). Although their self-reactivity has yet to be determined, IgA+ ASCs were detected in kidney biopsies from patients with IgAN, and the frequency of ASCs correlated with disease severity, suggesting that such IgA+ ASCs are a source of pathogenic auto-Abs. Similarly, it has been reported that anti-self ASCs often accumulate in the affected tissues in conventional autoimmune diseases such as SLE (30, 31) and rheumatoid arthritis (39).
At present, the mechanisms by which PBs producing IgA auto-Abs are generated and accumulate in the kidneys of gddY mice are unknown. However, the accumulation of somatic hypermutations in the IgA variable region genes cloned from these PBs suggests that they were generated via GCs in a T cell–dependent manner. Although tertiary lymphoid tissues containing GC B cells are known to be generated at inflammation sites in various pathologic conditions, such as autoimmunity, cancer, and allograft rejection (40), as well as in inflamed kidneys of several kidney diseases (34, 41, 42), we could not detect GC B cells in the kidney of 8-wo gddY mice, when IgA+ PBs had already appeared. Therefore, IgA+ PBs are possibly generated in GCs in some lymphoid organs or differentiated from their precursors, namely, memory B cells, which have undergone somatic hypermutation. Another possibility is that such IgA+ PBs are generated in the kidney through a GC-independent process, as recently reported for an antibacterial immune response in the liver (36). In any case, the antigens priming the response that generates such self-reactive B cells remain to be determined. As the “mucosa–bone marrow axis” theory suggests that B cells generating pathogenic IgA are originally primed in the mucosa in IgAN (28), and as IgA+ PCs are mostly generated in mucosal tissues such as the respiratory tract and intestine in response to certain viruses or commensal bacteria (43, 44), it is possible that B cells are primed by commensal bacteria in some mucosal tissues to mature into those cross-reacting with self-antigens and then finally become the IgA+ PBs in the kidney of gddY mice. This scenario also explains the reason why the major isotype of auto-Abs in IgAN is IgA and not IgG.
The data demonstrating the staining of MCs in vitro and in vivo by using the βII-spectrin–reactive rIgA derived from the PBs of gddY mouse kidneys revealed cell-surface expression of βII-spectrin, which typically localizes inside the plasma membrane. The surface expression of βII-spectrin was confirmed in MCs with commercially available βII-spectrin–specific Ab and in HEK293T cells expressing exogenous βII-spectrin. Not only the FL but also the N- and C-terminal fragments of βII-spectrin could be detected on the surface of transfected embryonic kidney–derived HEK293T cells, but not in the other three types of cell lines tested, suggesting that the cell-surface expression of βII-spectrin is not involved in its physiological function but rather a result of aberrant processing. In this regard, Arase (45) proposed that some misfolded self-proteins are exposed on the cell surface in association with MHC class II, which can trigger autoimmune responses. However, the cell-surface expression of βII-spectrin was independent of MHC class II expression, as evident in HEK293T cells. Although the mechanism of the surface expression of βII-spectrin remains unclear, it may be a common feature of renal lineage cells, such as MCs and HEK293T cells, as demonstrated in this study.
Whatever the mechanism of its generation, the βII-spectrin–reactive IgA auto-Ab can be detected in the sera of patients with IgAN at a notable frequency, e.g., ~36% by ELISA, the sensitivity of which may further be increased by narrowing down the antigenic epitope of βII-spectrin. On the other hand, our preliminary examination with a limited number of samples showed that anti–βII-spectrin IgA was not detected in the sera of other kidney diseases, including diabetic kidney disease, focal segmental glomerular sclerosis, and non-IgA proliferative glomerulonephritis. Furthermore, in the sera of IgAN patients, higher levels of anti–βII-spectrin IgA correlated with lower levels of C3 and higher IgA/C3 ratios, the latter of which is known as a predictive factor for poor renal survival (24–27), suggesting the possibility that the anti-SPTBN1 IgA could be a biomarker to predict the renal outcome in IgAN. Although larger cohorts of patients with IgAN and other renal kidney diseases are necessary, these results suggest the possibility that the serum anti–βII-spectrin IgA auto-Abs can be a useful diagnostic and predictive biomarker for IgAN. Our WB analysis using sera from patients with IgAN and gddY mice suggested the presence of other self-antigens besides βII-spectrin that are recognized by the IgA auto-Abs of patients with IgAN. The identification of such self-antigens will enhance the possibility of developing a noninvasive diagnosis strategy using patient sera.
Together, the unexpected identification of IgA auto-Abs and the mesangial autoantigen in IgAN provides strong evidence to redefine IgAN as a tissue-specific autoimmune disease rather than an immune-complex disease (2, 11) and provides deeper insight into the mechanism of the pathogenesis of IgAN. This may lead to a breakthrough in the development of specific and causal therapeutic strategies for IgAN, for example, blocking the deposition of IgA auto-Abs in the mesangium or selective elimination of the auto-Ab–producing PCs or the memory B cells that produce such PCs.
MATERIALS AND METHODS
Mice
The gddY mice were generated by selectively mating individuals within an early-onset group of ddY mice for more than 20 generations (19). Male gddY mice were used in this study. C57BL/6 NCrSlc, BALB/c, ddY, NZB/W F1, and Faslpr/lpr mice were purchased from Sankyo Labo Service Corporation Inc. Only female NZB/W F1 and Faslpr/lpr mice were used in the present study. AID-deficient mice on a C57BL/6 background were provided by Tasuku Honjo (46). Unless otherwise noted, the gddY mice used in this study were 8 weeks old. All mice were maintained in the Tokyo University of Science (TUS) mouse facility under specific pathogen–free conditions. Mouse procedures were performed under protocols approved by the TUS Animal Care and Use Committee.
Human subjects
Sera from healthy individuals and patients with IgAN and renal biopsy specimens from the patients were obtained at Juntendo University Hospital with informed consent from patients and approval of the Research Ethics Committee, Faculty of Medicine, Juntendo University. Laboratory information of patients and healthy volunteers is shown in table S2.
Antibodies
The following Abs were used for IF and IHC staining: anti-mouse IgA Ab-PE (Abcam, 97013) or fluorescein isothiocyanate (FITC; Abcam, 97234), anti-mouse CD138-PE (clone 281-2, BioLegend), anti-human IgA (DAKO, #IR51061), or CD138 (DAKO, M7228). For flow cytometry, the following Abs against mouse antigens were used: B220–APC-Cy7 (clone RA3-6B2, BioLegend), CD138-PE or BV421 (clone 281-2), GL7-PerCP Cy5.5 (clone GL7, BioLegend), Ki67–PE-Cy7 (clone B56, BD), IgG1-FITC (clone A85-1, BD), IgG2a/c-FITC (clone RMG2a-62, BioLegend), IgG2b-FITC (SouthernBiotech, 1092-02), IgG3-FITC (SouthernBiotech, 1102-02), IgA-biotin (clone RMA-1, BioLegend), CD73-biotin (clone TY/11.8, BioLegend), CD140b-APC (clone APB5, BioLegend), CD105–PE-Cy7 (clone MJ7/18, BioLegend), CD45–PerCP-Cy5.5 (clone 30-F11, BioLegend), and CD31-PE (clone 390, BioLegend); other reagents were also used: rabbit polyclonal anti-SPTBN1 (amino acids 2100 to 2150; Abcam, 72239), normal rabbit IgG (an isotype-matched control for the latter; Sigma-Aldrich, 12-370), anti-rabbit IgG-BV421 (clone Poly4064, BioLegend), streptavidin-BV421 (BioLegend, 405226), or APC (BioLegend, 405207).
Histological analyses
For IF microscopy (IFM) in Fig. 1A and figs. S5C and S7B, freshly isolated mouse kidneys were fixed in 4% paraformaldehyde for 6 hours, incubated overnight with 30% sucrose at 4°C, embedded in optimal cutting temperature (OCT) compound (Sakura Finetek), frozen in liquid nitrogen, and stored at −80°C until use. For IFM in Fig. 6 (F and G) and fig. S13, kidneys and the examined tissues were directly embedded in OCT compound, frozen, and stored at −80°C. Frozen specimens were sliced to make 6-μm sections and fixed with ice-cold acetone at −20°C for 4 min. The sections were incubated in 3% bovine serum albumin (BSA)/phosphate-buffered saline (PBS) for 60 min at room temperature (RT) to block nonspecific staining, washed, and stained for 60 min at RT or overnight at 4°C with different combinations of Abs and reagents. Mouse sera were used as the primary Abs at a dilution of 1:10. After washing, slides were mounted with ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). All samples were examined with a confocal microscope (FV3000; Olympus).
Kidney biopsy specimens from patients were fixed in 15% formaldehyde and embedded in paraffin, from which 3-μm sections were prepared. IHC staining was performed after antigen retrieval conducted by heat induction for 40 min (for staining of CD138) or using 0.05% bacterial protease subtilisin A (Sigma-Aldrich; for staining of IgA) for 2 hours at RT. Slides were incubated with primary Abs for 60 min and the EnVision+ System–horseradish peroxidase (HRP)–Labeled Polymer Anti-mouse (DAKO) for 30 min. Staining was visualized with the Liquid DAB+ Substrate Chromogen System (DAKO).
Western blotting
WB was performed as previously reported (47). Briefly, cells were lysed with 1% NP-40 lysis buffer containing protease inhibitors. The lysates were mixed with SDS sample buffer, boiled, and used for SDS-PAGE, followed by immunoblotting using serum and/or Abs. For use as the primary Abs, mouse and human sera were diluted at 1:40 and 1:50, respectively. In some experiments, recombinant FLAG-tagged Sptbn1/SPTBN1 proteins, produced as described below, were used as antigens. In Fig. 2C, human serum IgA1 was purified by using Pierce Jacalin Agarose (Thermo Fisher Scientific) according to the manufacturer’s instructions. Anti-mouse IgA, anti-human IgA, or anti-human IgA1 Abs conjugated with HRP (1:5000) were used as the secondary Abs.
Immunoprecipitation
Magnetically isolated glomeruli from C57BL/6 mouse kidneys were lysed in 1% NP-40 lysis buffer containing protease inhibitors. The lysates were incubated with IgA-immobilized beads (see below), and after washing with 0.5% NP-40 buffer, the precipitates were boiled in sample buffer and subjected to SDS-PAGE. IgA-immobilized beads were generated by coupling purified serum IgA from BALB/c or gddY mice (pooled serum from 10 mice) with Pierce NHS-Activated Magnetic Beads (Thermo Fisher Scientific).
Mass spectrometry analysis
After SDS-PAGE, the gel was stained using a Pierce silver stain for mass spectrometry kit (Thermo Fisher Scientific), from which the regions of interest were excised. After destaining, the gel pieces were dehydrated with 100% acetonitrile for 10 min at RT and dried under vacuum. Then, the gel pieces were reduced and alkylated simultaneously with 10 mM tris (2-carboxyethyl) phosphine hydrochloride (Thermo Fisher Scientific) and 40 mM chloroacetamide (Sigma-Aldrich) in 100 mM tris-HCl (pH 8.5) and incubated at 70°C for 5 min (48). After reduction/alkylation, the gel pieces were washed twice with 50% acetonitrile (ACN) for 10 min each with shaking at RT, dehydrated with 100% ACN for 10 min, and dried. Then, each gel piece was incubated in trypsin/Lys-C (20 μg/liter; Promega) in 100 mM Hepes (pH 8.5) on ice for 30 min and then overnight at 37°C. After digestion, peptides were extracted from the gel pieces three times with 0.1% formic acid in 50% ACN, combined, desalted using an styrene divinylbenzene tip (GL Science, Tokyo), and lyophilized using a centrifugal evaporator. The peptides were identified by nano–liquid chromatography–tandem mass spectrometry (LC-MS/MS) using the Triple TOF 5600+ System operated with Analyst TF 1.7 software and an Eksigent nano LC system (SCIEX, MA). The obtained MS data were interrogated with ProteinPilot 5.0.2 Software (SCIEX) using the UniProt database (2020_04). A confidence cutoff of a 1% false discovery rate was applied for protein identification.
Plasmid construction
Mouse Sptbn1 cDNA was generated from isolated glomeruli of C57BL/6 mice. FL-Sptbn1 cDNA was amplified by using KOD plus Neo DNA polymerases (Toyobo) and subcloned into a pCAT7 neo vector, which provides an N-terminal double T7-tag (49). Sptbn1 cDNA fragments 1A, 1B, and 1C were amplified using KOD Fx Neo or KOD plus Neo DNA polymerases (Toyobo) and subcloned into a p3XFLAG-CMV-10 (Sigma-Aldrich) vector. A FLAG-tagged human FL-SPTBN1 expression vector was provided by Tomozumi Imamichi (50). IgJ chain cDNA was generated from the mRNA of isolated GC B cells of C57BL/6 mice using KOD plus Neo DNA polymerases (Toyobo), fused with a His-tag at the C terminus, and subcloned into the pMXs-ires-GFP (green fluorescent protein) vector (49).
Introduction of expression vectors into cells
The T7 or FLAG-tagged expression vectors were transfected into cell lines by PEI “Max” (Mw 40,000; Polysciences) and analyzed 2 or 3 days later. The pMXs vector encoding the His-tagged J chain (described above) was transfected into Plat-E cells by PEI “Max.” HEK293T cells were transduced with the virus-containing supernatant by spin infection as described previously (47). Two days later, GFP-positive single cells were directly sorted into 96-well plates using a FACSAria II (BD Biosciences) and propagated to produce stable cell lines.
Enzyme-linked immunosorbent assays
Microtiter plates were coated with 50 μl of recombinant Sptbn1 (20 μg/ml) overnight at 4°C. After washing the plates five times with PBS containing 0.05% Tween 20 (wash solution), they were blocked with 100 μl of PBS containing 3% BSA and 0.05% Tween 20. After washing, plates were incubated with 50 μl of diluted patient serum (1:50 for IgA; 1:300 for IgG) or rIgA Abs (4 μg/ml) for 2 hours at RT. After washing, the plates were incubated with 50 μl of goat anti-human IgA1/A2, IgG, or goat anti-mouse IgA Abs conjugated with HRP (1:3000; Abcam) at RT for 60 min. After washing, bound reactants were detected by 2-min incubation with 3,3′,5,5′-tetramethylbenzidine. Absorbance was determined at 450 nm.
Tissue preparation, flow cytometry, and single B cell sorting
Perfused kidneys were cut into 2- to 3-mm pieces, and leukocytes in the kidney were isolated by Percoll gradient centrifugation after digestion with collagenase D (0.6 mg/ml; Roche) and deoxyribonuclease I (100 μg/ml; Roche). Mononuclear cells in the lamina propria of the small intestine from gddY mice or BALB/c mice were isolated as previously described (51). To isolate glomeruli, anesthetized BALB/c mice were perfused through the heart with 8 × 107 Dynabeads M-450 Tosylactivated (Invitrogen) in 40 ml of PBS, which were captured only by the glomeruli. Then, the kidneys were removed, minced into 1- to 2-mm3 pieces, and digested with collagenase D (0.6 mg/ml) in RPMI-1640 (Gibco) containing 10% fetal bovine serum (FBS) at 37°C for 30 min with gentle agitation. Glomeruli were collected magnetically (purity > 98%) and further dispersed with collagenase D (1 mg/ml) to yield single-cell suspensions (38, 52, 53). Dead cells were excluded after staining with Fixable Viability Dye eFluor 506 (eBioscience). To detect intracellular IgA/IgG or Ki67 in PBs/PCs, cells were stained after treatment with a Fixation and Permeabilization solution kit (BD Biosciences) or Foxp3 Staining Buffer Set (eBioscience), respectively. For staining of Ki67, GC B cells were prepared from C57BL/6 mice immunized intraperitoneally with 100 μg of NP-CGG and alum 6 days previously. For rIgA production, mononuclear cells from the kidneys of gddY mice were surface-stained for CD138 and IgA, and single CD138+ IgA+ cells were sorted into 96-well polymerase chain reaction (PCR) plates (Eppendorf) containing 4 μl of lysis solution. The above samples were analyzed using a FACSCalibur, FACSAria II, or FACSCanto II (BD Biosciences). The data were analyzed using FlowJo (Tree Star).
Cell culture
IgA+ PBs from the kidney or small intestine of gddY mice were cultured in “B cell medium” [RPMI-1640 medium (Wako) supplemented with 10% FBS, 10 mM Hepes, 1 mM sodium pyruvate, 5.5 × 10−5 M 2-mercaptoethanol, penicillin (100 U/ml), and streptomycin (100 μg/ml; GIBCO)] with IL-6 (10 ng/ml) on a feeder layer of gamma-irradiated 40LB/APRIL cells [40LB cells (54) transduced with mouse APRIL] at 37°C in 5% CO2 atmosphere. The culture supernatant of the IgA+ PBs was harvested on day 6. Mouse MCs were isolated from the glomeruli of BALB/c mice as described previously (55). The isolated mouse and human MCs were cultured in Dulbecco’s modified Eagle’s medium (high glucose, with l-glutamine and sodium pyruvate) with 20% FBS, 10 mM Hepes, penicillin (100 U/ml), and streptomycin (100 μg/ml). MCs were used between passages 8 and 16.
Generation of recombinant antibodies
rIgA Abs were generated essentially as previously described (33). Briefly, cDNA was synthesized with a kit [SuperPrep II Cell Lysis & RT Kit for qPCR (TOYOBO)] using total RNA extracted from single sorted IgA+ PBs from the kidneys of gddY mice. Variable regions of IgA heavy-chain genes and Igκ or Igλ light-chain genes were amplified by two rounds of nested PCR, although no Igλ variable regions were detected. After sequencing the amplified cDNAs, corresponding germline sequences were assigned by IgBlast (www.ncbi.nlm.nih.gov/igblast/) and VBASE2 (www.vbase2.org/). Then, using the first-round PCR product (0.5 μl) as a template, heavy- and light-chain variable regions from each single cell were amplified with gene-specific V and J primer pairs containing restriction sites (Xho I in the forward primer; Not I in the reverse primer) (tables S1 and S3). After enzyme digestion, the PCR fragments were ligated with the pCAGGS expression vector (56), into which a cDNA-encoding mouse Cα or Cκ had been inserted. The inserted variable region genes were sequenced, and somatic mutations were analyzed (table S1). The resultant vectors were transfected into HEK293T cells or HEK293T cells expressing J chain, and the supernatants containing IgA protein were collected sequentially on days 3 and 6. As a negative control, rIgA Abs specific for the hapten 4-hydroxy-3-nitrophenyl acetyl (NP) were generated by the same protocol with expression vectors producing a VHB1-8hi heavy chain and a λ light chain (57). For in vivo injection, polymerized rIgA was purified with CaptoL (GE Healthcare) according to the manufacturer’s instructions.
Preparation of recombinant βII-spectrin
FLAG-tagged mouse Sptbn1C, FL-Sptbn1, and human FL-SPTBN1 were transiently expressed in HEK293T cells. From the lysates with 1% NP-40, FLAG-tagged proteins were purified using the anti-FLAG M2 Affinity Gel (Sigma-Aldrich), according to the manufacturer’s instructions, eluted using 0.1 M glycine HCl (pH 3.0), and neutralized immediately with tris. Purification was confirmed by Coomassie Brilliant Blue staining.
Statistical analyses
All the statistically analyzed data are derived from biological replicates in this study. Statistical analyses were performed using GraphPad Prism software, version 6.0 (GraphPad Software, La Jolla, CA). Comparisons between two groups in figs. S5E and S8B were analyzed by the Student’s t test, and those in Figs. 2E and 3 and figs. S3 and S4 were analyzed by the Mann-Whitney U test. Comparisons between more than three groups were analyzed by one-way analysis of variance (ANOVA). Spearman regression analysis was used to analyze the correlation between two variables. Differences at P < 0.05 were considered significant.
Acknowledgments
We thank T. Honjo for AID-deficient mice, T. Imamichi for a human SPTBN1 expression vector, T. Kitamura for the retroviral vector system, P. D. Burrows for critical reading of the manuscript, and T. Shibata for research assistance. We thank all members of the Department of Nephrology, Juntendo University Faculty of Medicine and Research Institute for Biomedical Sciences, Tokyo University of Science for useful discussions.
Funding: This work was supported by the Japan Society for the Promotion of Science KAKENHI, Grant-in-Aid for Scientific Research (A) 20H00510 (D.K.).
Author contributions: Conceptualization: Y.N., K.H., Y.S., and D.K. Methodology: Y.N. and K.H. Investigation: Y.N., K.H., M.H., H.I., S.A., Y.F., M.N., H.S., M.K., S.K., and Y.M. Visualization: Y.N. and D.K. Supervision: Y.S. and D.K. Writing—original draft: Y.N. and D.K. Writing—review and editing: Y.N., Y.S., and D.K.
Competing interests: Y.S., Y.N., and D.K. are inventors on patent application (PCT/JP2021/044409) submitted by Juntendo Educational Foundation and Tokyo University of Science Foundation that covers “Autoantibodies related to IgA nephropathy.” All other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Supplementary Materials
This PDF file includes:
Figs. S1 to S13
Tables S1 to S3
Legend for supplementary auxiliary files
Other Supplementary Material for this : manuscript includes the following:
Supplementary auxiliary files
REFERENCES AND NOTES
- 1.R. J. Wyatt, B. A. Julian, IgA nephropathy. N. Engl. J. Med. 368, 2402–2414 (2013). [DOI] [PubMed] [Google Scholar]
- 2.K. N. Lai, S. C. Tang, F. P. Schena, J. Novak, Y. Tomino, A. B. Fogo, R. J. Glassock, IgA nephropathy. Nat. Rev. Dis. Primers. 2, 16001 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Y. Nihei, M. Kishi, H. Suzuki, A. Koizumi, M. Yoshida, S. Hamaguchi, M. Iwasaki, H. Fukuda, H. Takahara, M. Kihara, S. Tomita, Y. Suzuki, IgA nephropathy with gross hematuria following COVID-19 mRNA vaccination. Intern. Med. 61, 1033–1037 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.L. Negrea, B. H. Rovin, Gross hematuria following vaccination for severe acute respiratory syndrome coronavirus 2 in 2 patients with IgA nephropathy. Kidney Int. 99, 1487 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.N. Klomjit, M. P. Alexander, F. C. Fervenza, Z. Zoghby, A. Garg, M. C. Hogan, S. H. Nasr, M. A. Minshar, L. Zand, COVID-19 vaccination and glomerulonephritis. Kidney Int. Rep. 6, 2969–2978 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.K. Matsuzaki, R. Aoki, Y. Nihei, H. Suzuki, M. Kihara, T. Yokoo, N. Kashihara, I. Narita, Y. Suzuki, Gross hematuria after SARS-CoV-2 vaccination: Questionnaire survey in Japan. Clin. Exp. Nephrol. 26, 316–322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.C. Hanna, L. P. Herrera Hernandez, L. Bu, S. Kizilbash, L. Najera, M. N. Rheault, J. Czyzyk, A. M. Kouri, IgA nephropathy presenting as macroscopic hematuria in 2 pediatric patients after receiving the Pfizer COVID-19 vaccine. Kidney Int. 100, 705–706 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.J. Berger, IgA glomerular deposits in renal disease. Transplant. Proc. 1, 939–944 (1969). [PubMed] [Google Scholar]
- 9.B. Knoppova, C. Reily, N. Maillard, D. V. Rizk, Z. Moldoveanu, J. Mestecky, M. Raska, M. B. Renfrow, B. A. Julian, J. Novak, The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front. Immunol. 7, 117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.J. Mestecky, J. Novak, Z. Moldoveanu, M. Raska, IgA nephropathy enigma. Clin. Immunol. 172, 72–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.H. Suzuki, K. Kiryluk, J. Novak, Z. Moldoveanu, A. B. Herr, M. B. Renfrow, R. J. Wyatt, F. Scolari, J. Mestecky, A. G. Gharavi, B. A. Julian, The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 22, 1795–1803 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.J. Yasutake, Y. Suzuki, H. Suzuki, N. Hiura, H. Yanagawa, Y. Makita, E. Kaneko, Y. Tomino, Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol. Dial. Transplant. 30, 1315–1321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Z. Moldoveanu, R. J. Wyatt, J. Y. Lee, M. Tomana, B. A. Julian, J. Mestecky, W.-Q. Huang, S. R. Anreddy, S. Hall, M. C. Hastings, K. K. Lau, W. J. Cook, J. Novak, Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 71, 1148–1154 (2007). [DOI] [PubMed] [Google Scholar]
- 14.S. Bagchi, R. Lingaiah, K. Mani, A. Barwad, G. Singh, V. Balooni, D. Bhowmik, S. K. Agarwal, Significance of serum galactose deficient IgA1 as a potential biomarker for IgA nephropathy: A case control study. PLOS ONE 14, e0214256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Q. Sun, Z. Zhang, H. Zhang, X. Liu, Aberrant IgA1 glycosylation in IgA nephropathy: A systematic review. PLOS ONE 11, e0166700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.H. Yanagawa, H. Suzuki, Y. Suzuki, K. Kiryluk, A. G. Gharavi, K. Matsuoka, Y. Makita, B. A. Julian, J. Novak, Y. Tomino, A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLOS ONE 9, e98081 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.T. Kano, H. Suzuki, Y. Makita, Y. Fukao, Y. Suzuki, Nasal-associated lymphoid tissue is the major induction site for nephritogenic IgA in murine IgA nephropathy. Kidney Int. 100, 364–376 (2021). [DOI] [PubMed] [Google Scholar]
- 18.A. Takahata, S. Arai, E. Hiramoto, K. Kitada, R. Kato, Y. Makita, H. Suzuki, J. Nakata, K. Araki, T. Miyazaki, Y. Suzuki, Crucial role of AIM/CD5L in the development of glomerular inflammation in IgA nephropathy. J. Am. Soc. Nephrol. 31, 2013–2024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.K. Okazaki, Y. Suzuki, M. Otsuji, H. Suzuki, M. Kihara, T. Kajiyama, A. Hashimoto, H. Nishimura, R. Brown, S. Hall, J. Novak, S. Izui, S. Hirose, Y. Tomino, Development of a model of early-onset IgA nephropathy. J. Am. Soc. Nephrol. 23, 1364–1374 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.K. Yamaji, Y. Suzuki, H. Suzuki, K. Satake, S. Horikoshi, J. Novak, Y. Tomino, The kinetics of glomerular deposition of nephritogenic IgA. PLOS ONE 9, e113005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.J. R. Myette, T. Kano, H. Suzuki, S. E. Sloan, K. J. Szretter, B. Ramakrishnan, H. Adari, K. D. Deotale, F. Engler, Z. Shriver, A. M. Wollacott, Y. Suzuki, B. J. G. Pereira, A proliferation inducing ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int. 96, 104–116 (2019). [DOI] [PubMed] [Google Scholar]
- 22.V. Bennett, D. N. Lorenzo, Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr. Top. Membr. 72, 1–37 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Working Group of the International IgA Nephropathy Network and the Renal Pathology Society, D. C. Cattran, R. Coppo, H. T. Cook, J. Feehally, I. S. Roberts, S. Troyanov, C. E. Alpers, A. Amore, J. Barratt, F. Berthoux, S. Bonsib, J. A. Bruijn, V. D'Agati, G. D'Amico, S. Emancipator, F. Emma, F. Ferrario, F. C. Fervenza, S. Florquin, A. Fogo, C. C. Geddes, H.-J. Groene, M. Haas, A. M. Herzenberg, P. A. Hill, R. J. Hogg, S. I. Hsu, J. C. Jennette, K. Joh, B. A. Julian, T. Kawamura, F. M. Lai, C. B. Leung, L.-S. Li, P. K. T. Li, Z.-H. Liu, B. Mackinnon, S. Mezzano, F. P. Schena, Y. Tomino, P. D. Walker, H. Wang, J. J. Weening, N. Yoshikawa, H. Zhang, The Oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int. 76, 534–545 (2009). [DOI] [PubMed] [Google Scholar]
- 24.M. Pan, J. Zhang, Z. Li, L. Jin, Y. Zheng, Z. Zhou, S. Zhen, G. Lu, Increased C4 and decreased C3 levels are associated with a poor prognosis in patients with immunoglobulin A nephropathy: A retrospective study. BMC Nephrol. 18, 231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.J. Zhang, C. Wang, Y. Tang, H. Peng, Z.-C. Ye, C.-C. Li, T.-Q. Lou, Serum immunoglobulin A/C3 ratio predicts progression of immunoglobulin A nephropathy. Nephrology (Carlton) 18, 125–131 (2013). [DOI] [PubMed] [Google Scholar]
- 26.S. J. Kim, H. M. Koo, B. J. Lim, H. J. Oh, D. E. Yoo, D. H. Shin, M. J. Lee, F. M. Doh, J. T. Park, T. H. Yoo, S. W. Kang, K. H. Choi, H. J. Jeong, S. H. Han, Decreased circulating C3 levels and mesangial C3 deposition predict renal outcome in patients with IgA nephropathy. PLOS ONE 7, e40495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.H. Komatsu, S. Fujimoto, S. Hara, Y. Sato, K. Yamada, T. Eto, Relationship between the serum IgA/C3 ratio and the progression of IgA nephropathy. Intern. Med. 43, 1023–1028 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Y. Suzuki, Y. Tomino, The mucosa-bone-marrow axis in IgA nephropathy. Contrib. Nephrol. 157, 70–79 (2007). [DOI] [PubMed] [Google Scholar]
- 29.A. Mueller, C. Brieske, S. Schinke, E. Csernok, W. L. Gross, K. Hasselbacher, J. Voswinkel, K. Holl-Ulrich, Plasma cells within granulomatous inflammation display signs pointing to autoreactivity and destruction in granulomatosis with polyangiitis. Arthritis Res. Ther. 16, R55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.C. Starke, S. Frey, U. Wellmann, V. Urbonaviciute, M. Herrmann, K. Amann, G. Schett, T. Winkler, R. E. Voll, High frequency of autoantibody-secreting cells and long-lived plasma cells within inflamed kidneys of NZB/W F1 lupus mice. Eur. J. Immunol. 41, 2107–2112 (2011). [DOI] [PubMed] [Google Scholar]
- 31.M. Espeli, S. Bökers, G. Giannico, H. A. Dickinson, V. Bardsley, A. B. Fogo, K. G. C. Smith, Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol. 22, 296–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.S. Lacotte, H. Dumortier, M. Décossas, J.-P. Briand, S. Muller, Identification of new pathogenic players in lupus: Autoantibody-secreting cells are present in nephritic kidneys of (NZBxNZW)F1 mice. J. Immunol. 184, 3937–3945 (2010). [DOI] [PubMed] [Google Scholar]
- 33.T. Tiller, C. E. Busse, H. Wardemann, Cloning and expression of murine Ig genes from single B cells. J. Immunol. Methods 350, 183–193 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Y. Sato, A. Oguchi, Y. Fukushima, K. Masuda, N. Toriu, K. Taniguchi, T. Yoshikawa, X. Cui, M. Kondo, T. Hosoi, S. Komidori, Y. Shimizu, H. Fujita, L. Jiang, Y. Kong, T. Yamanashi, J. Seita, T. Yamamoto, S. Toyokuni, Y. Hamazaki, M. Hattori, Y. Yoshikai, P. Boor, J. Floege, H. Kawamoto, Y. Murakawa, N. Minato, M. Yanagita, CD153/CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. J. Clin. Invest. 132, e146071 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Y. Sato, A. Mii, Y. Hamazaki, H. Fujita, H. Nakata, K. Masuda, S. Nishiyama, S. Shibuya, H. Haga, O. Ogawa, A. Shimizu, S. Narumiya, T. Kaisho, M. Arita, M. Yanagisawa, M. Miyasaka, K. Sharma, N. Minato, H. Kawamoto, M. Yanagita, Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight 1, e87680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.N. Trivedi, F. Weisel, S. Smita, S. Joachim, M. Kader, A. Radhakrishnan, C. Clouser, A. M. Rosenfeld, M. Chikina, F. Vigneault, U. Hershberg, N. Ismail, M. J. Shlomchik, Liver is a generative site for the B cell response to ehrlichia muris. Immunity 51, 1088–1101.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.P. Yang, Y. Yang, P. Sun, Y. Tian, F. Gao, C. Wang, T. Zong, M. Li, Y. Zhang, T. Yu, Z. Jiang, βII spectrin (SPTBN1): Biological function and clinical potential in cancer and other diseases. Int. J. Biol. Sci. 17, 32–49 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.F. A. Hatje, U. Wedekind, W. Sachs, D. Loreth, J. Reichelt, F. Demir, C. Kosub, L. Heintz, N. M. Tomas, T. B. Huber, S. Skuza, M. Sachs, S. Zielinski, M. M. Rinschen, C. Meyer-Schwesinger, Tripartite separation of glomerular cell types and proteomes from reporter-free mice. J. Am. Soc. Nephrol. 32, 2175–2193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.C. Masson-Bessière, M. Sebbag, J.-J. Durieux, L. Nogueira, C. Vincent, E. Girbal-Neuhauser, R. Durroux, A. Cantagrel, G. Serre, In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin. Exp. Immunol. 119, 544–552 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.C. Pitzalis, G. W. Jones, M. Bombardieri, S. A. Jones, Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 14, 447–462 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Y. H. Lee, Y. Sato, M. Saito, S. Fukuma, M. Saito, S. Yamamoto, A. Komatsuda, N. Fujiyama, S. Satoh, S. H. Lee, P. Boor, T. Habuchi, J. Floege, M. Yanagita, Advanced tertiary lymphoid tissues in protocol biopsies are associated with progressive graft dysfunction in kidney transplant recipients. J. Am. Soc. Nephrol. 33, 186–200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.T. Miyanaga, K. Mizuguchi, S. Hara, T. Zoshima, D. Inoue, R. Nishioka, I. Mizushima, K. Ito, H. Fuji, K. Yamada, Y. Sato, M. Yanagita, M. Kawano, Tertiary lymphoid tissue in early-stage IgG4-related tubulointerstitial nephritis incidentally detected with a tumor lesion of the ureteropelvic junction: A case report. BMC Nephrol. 22, 34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.J. E. Oh, E. Song, M. Moriyama, P. Wong, S. Zhang, R. Jiang, S. Strohmeier, S. H. Kleinstein, F. Krammer, A. Iwasaki, Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci. Immunol. 6, eabj5129 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.S. Hapfelmeier, M. A. Lawson, E. Slack, J. K. Kirundi, M. Stoel, M. Heikenwalder, J. Cahenzli, Y. Velykoredko, M. L. Balmer, K. Endt, M. B. Geuking, R. Curtiss 3rd, K. D. McCoy, A. J. Macpherson, Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.H. Arase, The major histocompatibility complex: New insights from old molecules into the pathogenesis of autoimmunity. Int. Immunol. 33, 641–645 (2021). [DOI] [PubMed] [Google Scholar]
- 46.M. Wei, R. Shinkura, Y. Doi, M. Maruya, S. Fagarasan, T. Honjo, Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat. Immunol. 12, 264–270 (2011). [DOI] [PubMed] [Google Scholar]
- 47.K. Haniuda, S. Fukao, T. Kodama, H. Hasegawa, D. Kitamura, Autonomous membrane IgE signaling prevents IgE-memory formation. Nat. Immunol. 17, 1109–1117 (2016). [DOI] [PubMed] [Google Scholar]
- 48.J. K. Goodman, C. G. Zampronio, A. M. E. Jones, J. R. Hernandez-Fernaud, Updates of the in-gel digestion method for protein analysis by mass spectrometry. Proteomics 18, e1800236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Y. Imamura, T. Katahira, D. Kitamura, Identification and characterization of a novel BASH N terminus-associated protein, BNAS2. J. Biol. Chem. 279, 26425–26432 (2004). [DOI] [PubMed] [Google Scholar]
- 50.L. Dai, K. B. Lidie, Q. Chen, J. W. Adelsberger, X. Zheng, D. Huang, J. Yang, R. A. Lempicki, T. Rehman, R. L. Dewar, Y. Wang, R. L. Hornung, K. A. Canizales, S. J. Lockett, H. C. Lane, T. Imamichi, IL-27 inhibits HIV-1 infection in human macrophages by down-regulating host factor SPTBN1 during monocyte to macrophage differentiation. J. Exp. Med. 210, 517–534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.H. Luck, S. Khan, J. H. Kim, J. K. Copeland, X. S. Revelo, S. Tsai, M. Chakraborty, K. Cheng, Y. Tao Chan, M. K. Nøhr, X. Clemente-Casares, M. C. Perry, M. Ghazarian, H. Lei, Y. H. Lin, B. Coburn, A. Okrainec, T. Jackson, S. Poutanen, H. Gaisano, J. P. Allard, D. S. Guttman, M. E. Conner, S. Winer, D. A. Winer, Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat. Commun. 10, 3650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.M. Takemoto, N. Asker, H. Gerhardt, A. Lundkvist, B. R. Johansson, Y. Saito, C. Betsholtz, A new method for large scale isolation of kidney glomeruli from mice. Am. J. Pathol. 161, 799–805 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.N. Karaiskos, M. Rahmatollahi, A. Boltengagen, H. Liu, M. Hoehne, M. Rinschen, B. Schermer, T. Benzing, N. Rajewsky, C. Kocks, M. Kann, R.-U. Müller, A single-cell transcriptome atlas of the mouse glomerulus. J. Am. Soc. Nephrol. 29, 2060–2068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.T. Nojima, K. Haniuda, T. Moutai, M. Matsudaira, S. Mizokawa, I. Shiratori, T. Azuma, D. Kitamura, In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat. Commun. 2, 465 (2011). [DOI] [PubMed] [Google Scholar]
- 55.M. Hochane, D. Raison, C. Coquard, O. Imhoff, T. Massfelder, B. Moulin, J.-J. Helwig, M. Barthelmebs, Parathyroid hormone-related protein is a mitogenic and a survival factor of mesangial cells from male mice: Role of intracrine and paracrine pathways. Endocrinology 154, 853–864 (2013). [DOI] [PubMed] [Google Scholar]
- 56.H. Niwa, K. Yamamura, J. Miyazaki, Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 (1991). [DOI] [PubMed] [Google Scholar]
- 57.T.-A. Shih, M. Roederer, M. C. Nussenzweig, Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 3, 399–406 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figs. S1 to S13
Tables S1 to S3
Legend for supplementary auxiliary files
Supplementary auxiliary files