

Abstract
Although modern clinical practices such as cesarean sections and perinatal antibiotics have improved infant survival, treatment with broad-spectrum antibiotics alters intestinal microbiota and causes dysbiosis. Infants exposed to perinatal antibiotics have an increased likelihood of life-threatening infections, including pneumonia. Here, we investigated how the gut microbiota sculpt pulmonary immune responses, promoting recovery and resolution of infection in newborn rhesus macaques. Early-life antibiotic exposure interrupted the maturation of intestinal commensal bacteria and disrupted the developmental trajectory of the pulmonary immune system, as assessed by single-cell proteomic and transcriptomic analyses. Early-life antibiotic exposure rendered newborn macaques more susceptible to bacterial pneumonia, concurrent with increases in neutrophil senescence and hyperinflammation, broad inflammatory cytokine signaling, and macrophage dysfunction. This pathogenic reprogramming of pulmonary immunity was further reflected by a hyperinflammatory signature in all pulmonary immune cell subsets coupled with a global loss of tissue-protective, homeostatic pathways in the lungs of dysbiotic newborns. Fecal microbiota transfer was associated with partial correction of the broad immune maladaptations and protection against severe pneumonia. These data demonstrate the importance of intestinal microbiota in programming pulmonary immunity and support the idea that gut microbiota promote the balance between pathways driving tissue repair and inflammatory responses associated with clinical recovery from infection in infants. Our results highlight a potential role for microbial transfer for immune support in these at-risk infants.
INTRODUCTION
Distinct immune responses adapted for early postnatal life also render newborns more vulnerable to infections (1). Incomplete understanding of immune development early in life contributes to our inability to reduce neonatal morbidity caused by respiratory infections, which kill more newborns than any other cause (2). Whereas a series of coordinated events control the development of the newborn immune system (3), few are as important as the interactions of immune cells with successive waves of commensal bacteria that colonize the newborn’s intestine after birth (4, 5). Evolving microbial signals play a critical role in the maturation of neutrophils, CD4+ T cells, monocytes, and innate lymphoid cells (ILCs) in the peripheral blood and the intestine (6, 7). Nevertheless, the role of intestinal commensal bacteria in functional programming of effector immune cells at extraintestinal mucosal sites, such as the lungs, is unclear (8).
Nearly half of vaginally delivered infants are exposed to antibiotics given to their mother to prevent group B Streptococcus sepsis (9–11). Some of these infants continue to receive empiric antibiotics, such as ampicillin and gentamicin, for neonatal sepsis (12). The use of vancomycin, although still uncommon, has increased because of the increased incidence of coagulase-negative Staphylococcus sepsis, especially in sick and preterm neonates (13). Although current clinical practices are essential for reducing infant mortality, they may extract a substantial biological cost in long-term disruption of the gut microbiota. Infants treated with antibiotics experience greater morbidity in response to sepsis and pneumonia compared to milder disease and spontaneous resolution in infants not exposed to antibiotics (14, 15).
In this study, we defined how gut microbiota inform immune responses that mediate recovery and resolution rather than severe disease. We used a rhesus macaque model because it more closely resembles a human infant lung in structure, developmental stage, physiology, and mucosal immune mechanisms (16) compared to murine lungs (17). In addition, similar to humans, nonhuman primates develop lobar pneumonia and demonstrate a heterogeneous clinical response (18). Their larger size and similarity to human newborns enabled integration of clinical signs, longitudinal assessment of disease progression, and the ability to distinguish mild, self-resolving pneumonia from severe, often fatal pneumonia.
Here, we defined a tiered immunologic development program, anchored by stepwise engagement of effector cells to limit tissue damage and promote recovery, which was experimentally disrupted by early-life antibiotic exposure. Remodeling of the pulmonary immune response was anchored by the appearance of a population of senescent and hyperinflammatory neutrophils and dysregulated antigen presentation in macrophages. This was all coupled with global loss of tissue-protective, homeostatic pathways in dysbiotic newborns. Fecal microbiota transfer was associated with correction of broad immune maladaptations and protection against severe pneumonia in dysbiotic newborn macaques. Our data suggest that gut microbiota reinforce the balance between regenerative pathways driving tissue homeostasis and inflammatory responses, thereby limiting pathogenesis and promoting clinical recovery. Our results also highlight a potential role for microbiota transfer as immune support in these at-risk infants.
RESULTS
Antibiotic exposure during the first postnatal week delays microbiota maturation and reconfigures the peripheral immune system before infection
We treated a cohort of vaginally delivered, nursery raised rhesus macaques (table S1) with either a cocktail of antimicrobials or with saline (n = 4 in each experimental group) from postnatal day 1 (PN1) to PN7 (Fig. 1A) and profiled the fecal bacterial communities daily. Stools in saline-treated newborn macaques, referred to hereafter as control newborns, were dominated by facultative aerobic Enterobacteriaceae during the first week of life before changing to strict anaerobes, principally Bacteroides and Clostridium in the second week of life (table S2), similar to the pattern seen in human neonates (19, 20). Maternal microbiota, a critical driver of fecal microbial colonization (21–23) and immune development in the newborn (21, 24), were not different between the experimental groups (table S3).
Fig. 1. Antibiotic exposure during the first week of life is associated with delayed microbiota maturation and reconfiguration of the peripheral immune system.
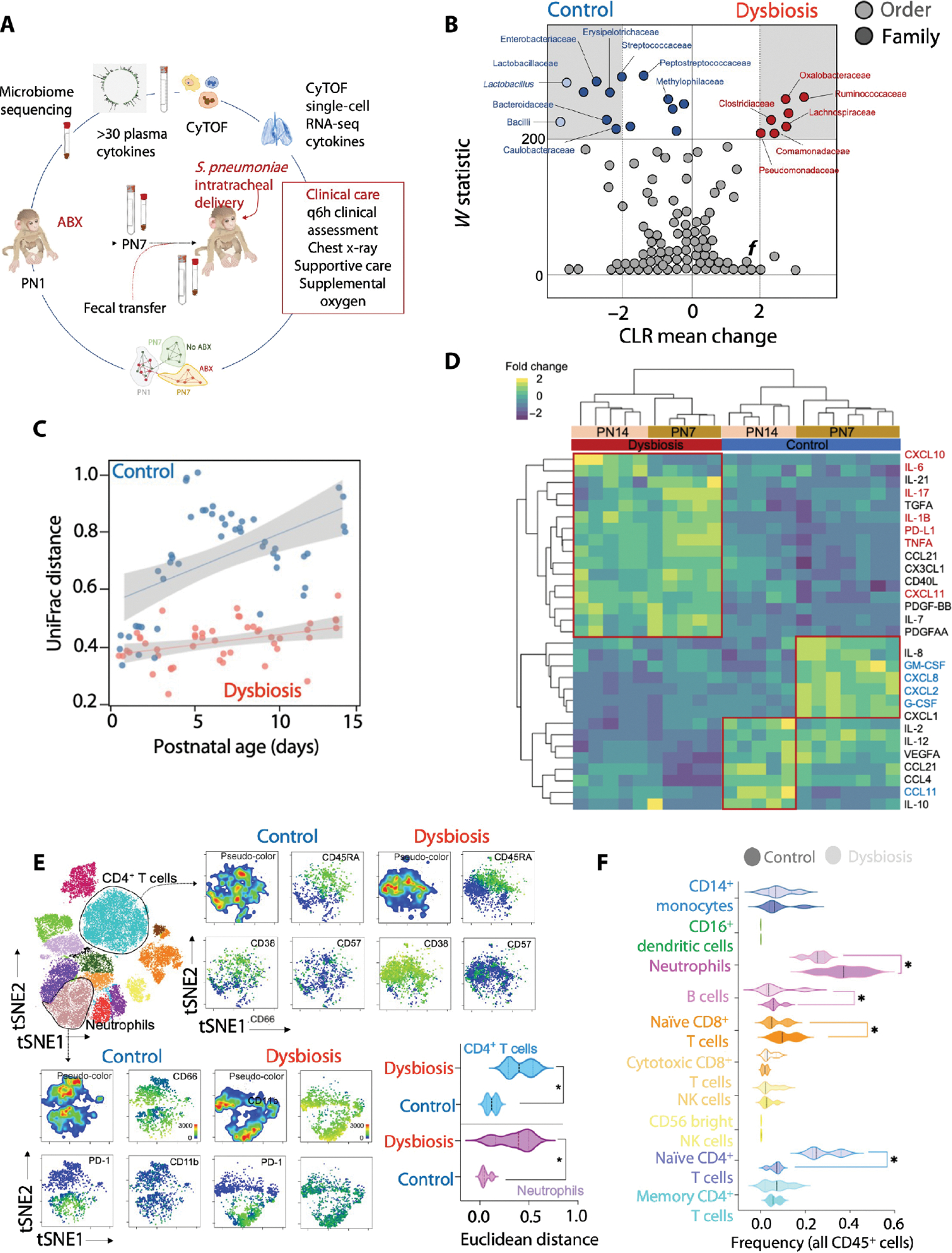
(A) Cohort of vaginally delivered, nursery-raised rhesus macaques was treated with a cocktail of antimicrobials (ABX) from postnatal day 1 (PN1) to PN7 (dysbiosis) or with saline (control) (n = 4 in each experimental group). (B) Differentially abundant taxa [FDR q ≤ 0.05, center log transformation (CLR) > 2] between control and dysbiosis groups are presented in gray margins. The center log transformation mean difference represents compositional differences in microbial communities. (C) β-Diversity (unweighted UniFrac) of fecal bacterial communities at the indicated day of life in control and dysbiotic newborn macaques. Linear fit is shown, and margins represent 95% confidence limits. (D) Abundance of plasma proteins in control and dysbiotic newborn macaques at 7 or 14 days of life, normalized against all subjects and scaled by row. k-means clustering was used to arrange subjects and plasma protein abundance. (E) Unsupervised analysis of cytometry by time of flight (CyTOF) data for CD4+ helper T cells or neutrophils. t-SNE (t-distributed stochastic neighbor embedding) projection of indicated functional markers in CD4+ helper T cells (top) or neutrophils (bottom) in peripheral blood of control and dysbiotic newborn macaques at 7 days of life. Pairwise Euclidean distances between CD4+ helper T cells (top) or neutrophils (bottom) in peripheral blood of control and dysbiotic newborn macaques at 7 days of life (n = 8; four in each experimental group; *P < 0.05, Student’s t test). Solid lines, median; dotted lines, quartiles. (F) Frequencies of the indicated immune cell types in peripheral blood of control and dysbiotic newborn macaques at 7 days of life (n = 8; four in each experimental group; P < 0.05, one-way ANOVA with Tukey’s correction for multiple comparisons). Solid lines, median; dotted lines, quartiles.
Antibiotic use diminished the abundance of Enterobacteriaceae, reduced phylogenetic diversity, and delayed the maturation of intestinal microbiota (Fig. 1, B and C, and table S2). Antibiotic use also created a maladapted immune state, highlighted by reduced amounts of hematopoietic cytokines, for example, granulocyte colony-stimulating factor (G-CSF) and interleukin-2 (IL-2) (25–27), and cytokines critical for mobilization of monocytes from bone marrow, such as C-X3-C motif chemokine ligand 1 (CX3CL1) and C-X-C motif chemokine ligand 8 (CXCL8) (28, 29), as assessed by a nonhuman primate multiplex cytokine kit (Fig. 1D and table S4). These immune changes occurred contemporaneously with changes in the stool microbiota (Fig. 1D and tables S2 and S4 to S6).
Peripheral blood neutrophils in antibiotic-exposed macaques, hereafter referred to as dysbiotic newborns, were phenotypically distinct, marked by increased expression of CD11b, a marker for neutrophil activation (30), and programmed cell death protein 1 (PD-1), a marker associated with senescence and exhaustion (Fig. 1E) (31, 32). The frequency and absolute number of neutrophils were decreased in the dysbiotic newborns (Fig. 1F; fig. S1, A to D; and table S5). Peripheral blood T cells in dysbiotic newborns were also phenotypically distinct with higher expression of CD45RA, CD38, and CD57, which are markers associated with T cell differentiation (33) and senescence (Fig. 1E) (34–36).
Gene networks associated with “cellular stress,” “ubiquitination,” and “apoptosis” were enriched in the transcriptomic analysis of peripheral blood from dysbiotic newborns (fig. S1E and table S7). Collectively, these data demonstrated that disruption of commensal microbes by early-life antibiotic use was associated with a maladapted state marked by both a robust, proinflammatory bias and neutrophil and T cell activation.
Early-life antibiotic exposure is associated with clinically severe pneumonia
After discontinuing antibiotics for 1 week, we challenged the newborn macaques in each experimental group with Streptococcus pneumoniae (serotype 19F), a common and often fatal respiratory pathogen in human infants (37). Vital signs, including heart rate, respiratory effort, blood pressure, urine output, oxygen saturation, and overall clinical condition of these newborn macaques, were monitored every 6 hours. In addition, chest radiographs were obtained daily. The pediatric early warning score (PEWS) (38), an extensively validated clinical decision-making and severity-scoring tool, was used to guide therapy, including intravenous fluids and supplemental oxygen (table S8).
The clinical status of dysbiotic newborns deteriorated, evidenced by higher PEWS and radiographic signs of lung consolidation (Fig. 2, A and B; fig. S2, A and B; and table S8). The progression of symptoms was also more rapid in the dysbiotic newborns (Fig. 2C), and all dysbiotic newborns received supportive treatment by 60 hours (Fig. 2D). At this time point, dysbiotic newborns had more severe disease (PEWS = 8 ± 1.5) than control newborns (PEWS = 5 ± 0.6) despite similar pathogen burden (fig. S2, C and D). These data recapitulate the rapid progression and increased morbidity seen in dysbiotic human infants (39).
Fig. 2. Antibiotic exposure during the first week of life is associated with increased susceptibility to pneumonia.

(A) Pediatric early warning score (PEWS) after infection with Streptococcus pneumoniae in control (blue) or dysbiotic (red) newborn macaques. (B) Representative chest radiographs obtained at euthanasia in control and dysbiotic newborn macaques. Arrows indicate areas of consolidation. (C) Progression of PEWS after infection with S. pneumoniae in control (blue) or dysbiotic (red) newborn macaques. Lines represent the best-fit curve by the smoothed spline of the longitudinal distribution of PEWS from the start of infection. Broken lines represent time (after infection) to PEWS > 8, a predetermined threshold to initiate supportive therapy. (D) Kaplan-Meier plot of the fraction of control and dysbiotic newborn macaques requiring supportive therapy at indicated times after infection (n = 8; four in each experimental group; *P < 0.05, Mantel-Cox log-rank test).
Last, we sought to determine whether a unique immune signature in dysbiotic newborns before infections could predict disease severity and clinical outcomes after challenge with S. pneumoniae. We found that reduced serum concentrations of hematopoietic cytokines—G-CSF, IL-2, CX3CL1, and CXCL8—before infection were associated with lower PEWS and less severe illness after challenge with S. pneumoniae (fig. S2E). In contrast, increased serum levels of proinflammatory cytokines, such as IL-1β, IL-6, IL-17, IL-21, and tumor necrosis factor–α (TNF-α), before infection were associated with higher PEWS and more severe illness after challenge with S. pneumoniae (fig. S2E). These data highlight the relevance of our newborn macaque model in profiling immune responses in target tissues, such as the lungs, which is often impossible in human newborns.
Early-life antibiotic exposure remodels pulmonary immune responses to S. pneumoniae infection
The host inflammatory response to pneumonia is highly compartmentalized to the lungs (40), so systemic immune cell or cytokine responses alone may not fully explain the differential clinical response to pneumonia in dysbiotic newborns. We therefore used a combination of high-parameter (>20 markers) cytometry by time of flight (CyTOF) and single-cell RNA sequencing (scRNA-seq) to profile the innate and adaptive immune cell responses in the lungs of control or dysbiotic newborns at 60 hours after infection with S. pneumoniae. The frequency of neutrophils measured by CyTOF (fig. S3, A to E) decreased significantly (P < 0.01) in the lungs of dysbiotic newborns compared to control newborns after infection with S. pneumoniae (fig. S3F). In contrast, the frequency of CD4+ T cells (fig. S3, A to E) increased significantly (P < 0.01) in the lungs of dysbiotic newborns after infection with S. pneumoniae (fig. S3F).
Cell clusters identified by scRNA-seq of pulmonary immune cells after infection with S. pneumoniae in control or dysbiotic newborns were annotated using signature genes from published scRNA-seq atlases (Fig. 3, A and B, and table S9) (41, 42). In dysbiotic newborns, the proportion of neutrophils identified by scRNA-seq decreased, whereas the proportion of T cells increased, consistent with the CyTOF findings (table S9).
Fig. 3. Antibiotic exposure for the first week remodels the pulmonary immune transcriptome during response to respiratory pathogens.
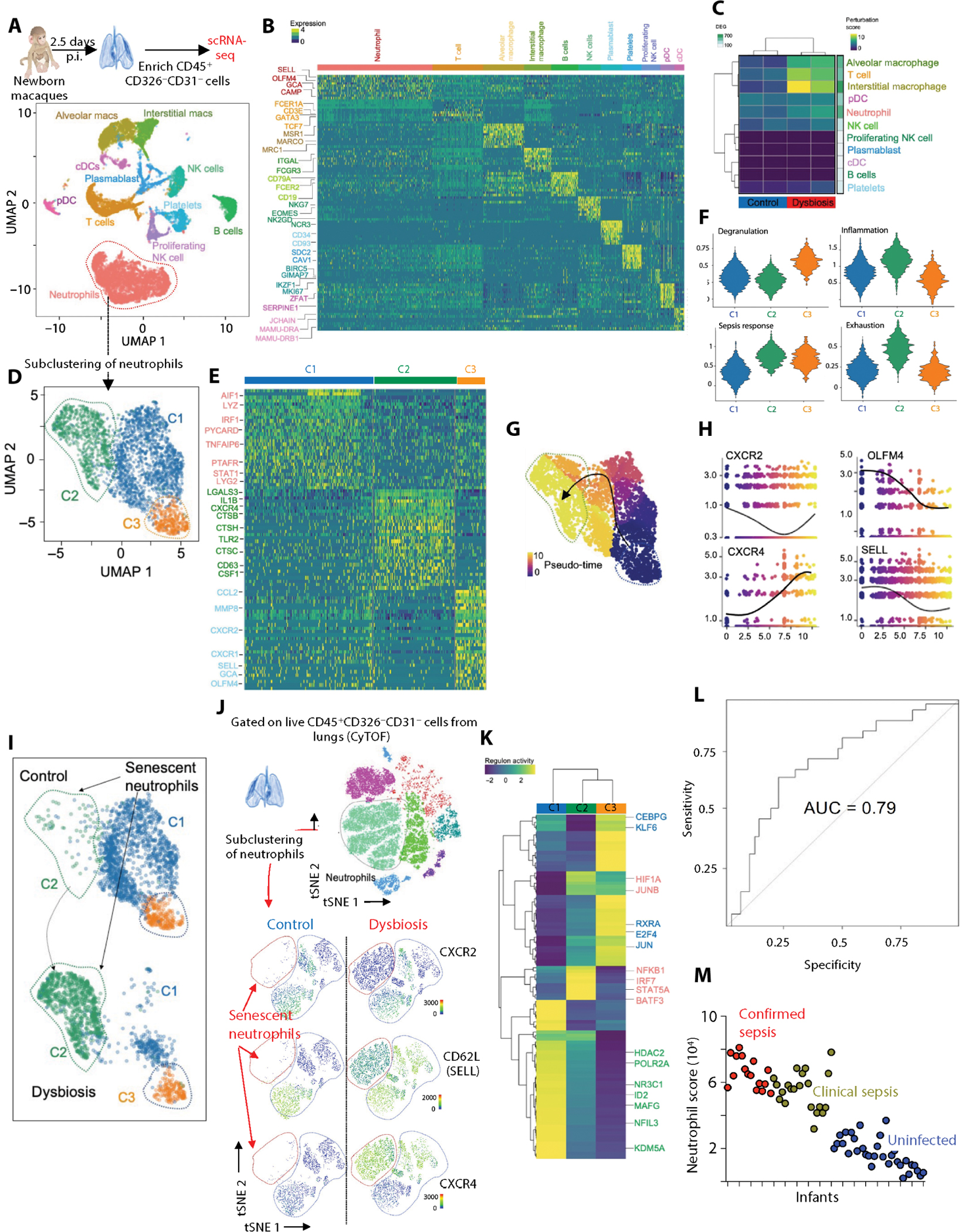
(A) Lung from control and dysbiotic newborn macaques (n = 4; two in each group) was obtained at 60 hours after infection. p.i., post infection. Lung samples were dissociated into cell suspensions, enriched for immune cells (EPCAM−CD31−CD45+), and used for single-cell RNA sequencing (scRNA-seq). Uniform manifold approximation and projection (UMAP) embedding of all samples (n = 13,377 cells) colored by cell clusters was performed on scRNA-seq data of these pulmonary immune cells. pDC, plasmacytoid dendritic cells; cDC, classical dendritic cells. (B) Row-scaled expression of the highest differentially expressed genes (DEGs) in each cluster (Bonferroni-adjusted P < 0.05). pDC, plasmacytoid dendritic cells; cDC, classical dendritic cells. (C) Cellular perturbation scores in indicated samples. The number of DEGs between dysbiotic and control newborn macaques for each cell type is indicated on the right. (D) UMAP embedding of neutrophils (n = 4768) extracted from a larger dataset of lung immune cells and reclustered into three distinct clusters. (E) Row-scaled expression of the highest DEGs in each cluster (Bonferroni-adjusted P < 0.05). (F) Module scores for each cluster of neutrophils showing enrichment of genes related to degranulation, inflammation, sepsis, and exhaustion. (G) UMAP embedding of neutrophils colored by pseudo-time with overlaid trajectory and (H) scatterplots showing expression of selected cluster-defining genes across pseudo-time. (I) UMAP embedding of neutrophils colored by cluster in control and dysbiotic newborn macaques indicating the emergence of the senescent C2 cluster in dysbiotic newborns. (J) CyTOF. t-SNE embedding of neutrophils extracted from a larger dataset of lung immune cells (top). Expression of key phenotypic markers (CXCR2, CD62L, and CXCR4) is shown (bottom). Neutrophil cluster coexpressing CXCR4 and CD62L is absent in control newborn macaques. (K) Row-scaled regulon activity for neutrophil clusters. k-means clustering was used to arrange clusters and regulons (n = 4; two in each experimental group; Benjamini and Hochberg–adjusted P < 0.01). (L) Receiver operating characteristic curve depicting sensitivity and specificity of sepsis diagnosis using the gene signature of senescent, hyperinflammatory neutrophils (HIF1A, CXCR4, CD274, LTF, and S100A8) in an independent cohort of 69 at-risk infants. AUC, area under the curve. (M) Scatterplot representing the ranked senescent neutrophil signature score (aggregated expression of HIF1A, CXCR4, CD274, LTF, and S100A8; see Materials and Methods) for each infant sample in bulk transcriptomic dataset (see Materials and Methods), colored by clinical diagnosis.
Immune pathways dominated by inflammation, such as IL-1β activation, and dysfunction, such as apoptosis and cellular stress, were seen in all pulmonary immune cells of dysbiotic newborns (fig. S4A and table S10). Canonical immune programs anchored by cell migration, differentiation, activation, and tissue repair were also broadly disrupted in these same cells after infection (fig. S4A). Neutrophils and alveolar macrophages (AMs) were the principal sources of inflammatory gene programs after infection with S. pneumoniae (fig. S4B), consistent with their critical roles in lung defense against pneumonia. Neutrophils and AMs also exhibited transcriptomic remodeling (Fig. 3C) and differentially expressed transcripts related to inflammation and tissue repair (fig. S4A) in dysbiotic newborn macaques after infection.
Consistent with transcriptomic changes, cytokines associated with inflammation, such as CXCL8, CXCL10, and transforming growth factor–β (TGFβ), and activation, such as IL-6, IL-8, and TNF-α, were increased in the bronchial lavage fluid of dysbiotic newborns, whereas cytokines associated with tissue repair, such as vascular endothelial growth factor (VEGF), IL-10, and platelet-derived growth factor (PDGF), were decreased (fig. S4C and table S11). A similar reconfiguration of immune responses was associated with rapid disease progression, worse symptoms, and increased mortality in humans in both bacterial and viral [influenza and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)] pneumonia (43–45).
Paired scRNA-seq and CyTOF analyses of matched blood and lung samples reveal compartmentalized production of chemokines and activation of neutrophils in the lungs
Immune cells, including neutrophils in peripheral blood and lung, were phenotypically distinct (fig. S5, A and B, and table S12). Compared to neutrophils from the lungs, neutrophils in the peripheral blood expressed higher amounts of transcripts for chemokine and homing/egress receptors, such as CX3CR1, C-C motif chemokine receptor 2 (CCR2), and SELL (encoding CD62L) (fig. S5, C and D). In contrast, pulmonary neutrophils expressed higher amounts of proinflammatory mediators, including chemokines for recruitment of monocytes and other neutrophils, such as C-C motif chemokine ligand 2 (CCL2), CCL3, and CCL4. They also express higher amounts of complement components, such as complement C1q B chain (C1QB) and C1QC; antimicrobial peptides, such as S100A8 and S100A9; and matrix metalloproteinases (MMPs), such as MMP8 and MMP9, which are implicated in tissue damage in acute respiratory distress syndrome (ARDS) (fig. S5, C and D) (46–48).
Consistent with these scRNA-seq findings, cytokines associated with neutrophil and monocyte migration, such as CCL2, CCL4, and CCL11; activation, such as IL-1β, IL-6, IL-8, IL-17, and TNF-α; and inflammation, such as CXCL8, CXCL10, and TGFβ, were uniformly abundant in the lungs (fig. S5E). The frequency of activated (CXCR2lo, CXCR4hi, and CD62Llo) neutrophils was significantly higher in the lungs compared to the peripheral blood (P < 0.05; fig. S5F). These data support the idea of compartmentalized activation of neutrophils, which may further explain the increased morbidity of dysbiotic newborns.
Senescent and hyperinflammatory neutrophils dominate pulmonary immune responses to S. pneumoniae infection in dysbiotic newborns
We identified three transcriptionally distinct neutrophil populations in the lungs after infection with S. pneumoniae (Fig. 3, D to F; fig. S6A; and table S13). Neutrophil heterogeneity is influenced by developmentally encoded cell programs and by environment and pathogen-specific factors (49). To identify distinct neutrophil populations, we used module scores (42), reflecting the average expression of all genes related to neutrophil development, maturation, and activation using published gene signatures from granulocytes during homeostasis and in the setting of sepsis (Fig. 3F and table S14) (50). Cluster 1 represented mature neutrophils defined by the expression of genes associated with pathogen response and cytokine signaling, reflecting neutrophil heterogeneity caused by pathogen exposure (fig. S6, A and B). Cluster 2 represented hyperinflammatory, stressed neutrophils characterized by high expression of CXCR4 and CD63 and reduced expression of CXCR2 and SELL (fig. S6, A and B) (51). These cells also had increased gene transcripts associated with glycolysis, a pathway associated with hyperinflammatory responses (fig. S6, B and C) (52). Cluster 3 represented immature neutrophils identified on the basis of the expression of gene transcripts related to neutrophil maturation (secretory vesicles, lysozymes, and phagocytosis) (fig. S6, A and B). The developmental relationship among these three clusters was predicted by the cellular trajectory analysis (Fig. 3, G and H) (53). Pseudo-time analysis of granule proteins, olfactomedin 4 (OLFM4) and SELL, and trafficking receptors, CXCR2 and CXCR4, known to be involved in neutrophil maturation, suggests a continuum of differentiation from immature neutrophils (cluster 3) to mature neutrophils (cluster 1) to senescent, hyperinflammatory neutrophils (cluster 2) (Fig. 3H).
Early-life antibiotic exposure strongly influenced the activation state of the lung neutrophil compartment after infection with S. pneumoniae. Pseudo-bulk RNA-seq analysis of these pulmonary neutrophils identified distinct signatures associated with dysbiosis (fig. S6C). Senescent, hyperinflammatory neutrophils were unique to the lungs of dysbiotic newborns, whereas mature neutrophils were absent in the lungs of dysbiotic newborns (Fig. 3I). CyTOF also showed consistent remodeling of the pulmonary neutrophil pool with the emergence of stressed, senescent (CXCR2lo, CXCR4hi, and CD62Llo) neutrophils in the lungs of dysbiotic newborns after infection with S. pneumoniae (Fig. 3J).
There was broad induction of NFκB, enrichment of phagocytosis and degranulation gene sets, and increased expression of epigenetic regulators associated with inflammatory neutrophils, including protein-arginine deiminase 4 (PADI4), which is required for neutrophil extracellular trap formation (NETosis) (54, 55), and CD274 [encoding for programmed cell death ligand 1 (PD-L1)], a marker of cell exhaustion in pulmonary neutrophils from dysbiotic newborns (table S15). We noted consistent changes in neutrophil-activating and chemotactic cytokines in the bronchial washings of dysbiotic newborns after infection with S. pneumoniae (fig. S6D). A similar reconfiguration of immune responses was associated with rapid disease progression, worse symptoms, and increased mortality in humans in both bacterial and viral (influenza and SARS-CoV-2) pneumonia (43–45).
Predicted regulatory networks active in pulmonary neutrophils from dysbiotic newborns after infection with S. pneumoniae include KDMA5 and NFIL3
Immature neutrophils (cluster 3) were enriched for regulatory networks anchored by the transcription factors (TFs) CCAAT/enhancer binding protein γ (C/EBPγ) and Kruppel-like factor 6 (KLF6), which are essential for neutrophil development (56–61), whereas mature neutrophils (cluster 1) were enriched for regulatory networks anchored by defense response–associated TFs (62–64), such as NFKB, interferon (IFN) regulatory factor 7 (IRF7), signal transducer and activator of transcription 5A (STAT5A), basic leucine zipper activating TF–like TF 3 (BATF3), and hypoxia-inducible factor 1A (HIF1A) (Fig. 3K, fig. S6E, and table S16). In contrast, the senescent, hyperinflammatory neutrophils (cluster 2) were enriched for regulatory networks anchored by epigenome-modifying enzymes, lysine demethylase 5A (KDM5A), histone deacetylase 2 (HDAC2), and nuclear factor IL-3 (NFIL3), a component of the circadian clock (Fig. 3K, fig. S6E, and table S16). NFIL3 and KDM5A are implicated in the microbiota-driven regulation of both nonhematopoietic cells, such as the intestinal epithelium, and hematopoietic cells, such as ILCs and natural killer (NK) cells, by chromatin histone modification (65, 66). Disruption of NFIL3 and KDM5A regulatory pathways may explain neutrophil dysfunction in dysbiotic newborns and highlights the need for experimental validation of their role in neutrophil homeostasis.
Gene scores of hyperinflammatory neutrophils from dysbiotic newborn macaques predict disease severity in an independent human cohort
Dysfunctional neutrophils contribute to pulmonary damage in experimental models of acute lung injury (67). We therefore hypothesized that the senescent, hyperinflammatory neutrophils from the dysbiotic newborn macaques would predict severe pneumonia outcomes. We identified the five most differentially expressed genes between senescent, hyperinflammatory neutrophils and all other cells in our dataset: HIF1A, CXCR4, CD274, lactotransferrin (LTF), and S100A8. We then used publicly available whole-blood bulk transcriptomic datasets of infants and children with severe sepsis and pneumonia as a reference for scoring with this identified neutrophil gene signature (68). We scored each sample in these datasets by the aggregated expression of these five genes. Our senescent, hyperinflammatory neutrophil gene score predicted sepsis diagnosis (area under the curve (AUC) = 0.79) in a cohort of at-risk infants (Fig. 3, L and M, and tables S17 and S18). Our data suggest that gene signatures from senescent, hyperinflammatory neutrophils might be potential prognostic indicators in human infants with sepsis and pneumonia.
Macrophages with dysfunctional features are a hallmark of the remodeled pulmonary immune response in the dysbiotic macaques
To interrogate macrophage plasticity, which is informed by developmental programs and tissue- and stress-specific signals, we used MacSpectrum (69), an analytical tool to stratify macrophage maturation and activation. The dominant population enriched for gene transcripts associated with antigen processing and presentation represents terminally differentiated mature AM (cluster 1); the population enriched for transcripts associated with inflammation, purinergic-inflammasome signaling, and IL-1 receptor activation represents polarized, inflammatory AMs (cluster 2); and immature AMs were represented by the expression of gene transcripts associated with DNA replication and cell division (cluster 3) (Fig. 4A; fig. S7, A to D; and table S19). Cellular trajectory analysis further identified a relationship between differentiation and activation as the AMs developed from immature (cluster 3) to mature (cluster 1) to polarized inflammatory AMs (cluster 2) (Fig. 4, B and C).
Fig. 4. Antibiotic exposure during the first week remodels the pulmonary macrophages and helper T cell compartments.
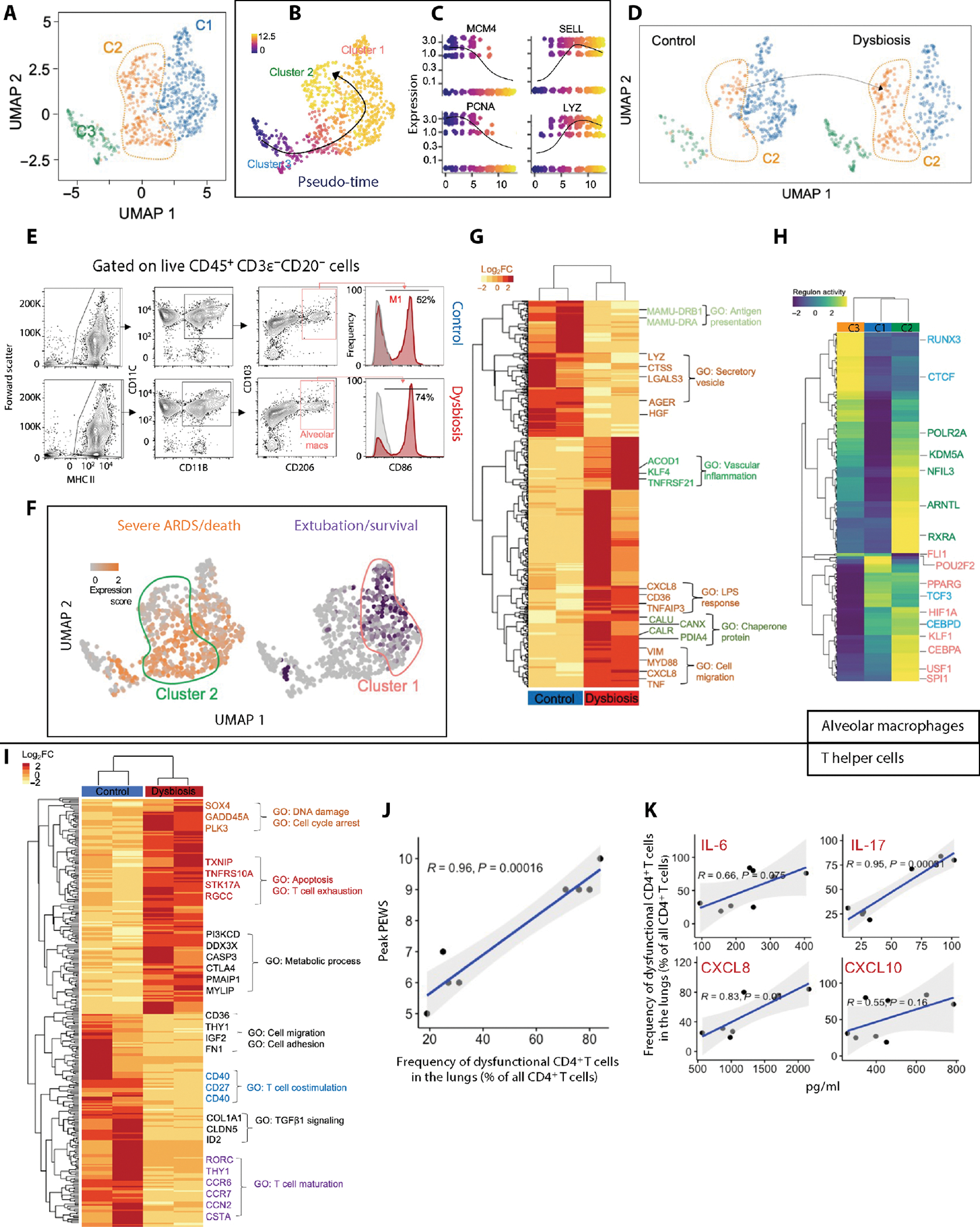
(A) UMAP embedding of AMs extracted from a larger dataset of lung immune cells colored by clusters. (B) UMAP embedding of AMs colored by pseudo-time with overlaid trajectory and (C) scatterplots showing expression of selected cluster-defining genes across pseudo-time. (D) UMAP embedding of AMs split by control and dysbiotic macaques showing an emergence of a unique dysbiotic cluster (cluster 2). (E) Flow cytometry. Bivariate contour plots showing gating strategy to identify macrophage subsets and histograms showing coexpression of activation markers, CD86 and CD206, on AMs from control (top) and dysbiotic (bottom) newborn macaques. Numbers indicate the relative frequencies of M1-activated macrophage subset (n = 8; four in each experimental group). (F) UMAP embedding of AMs colored by average expression of genes associated with severe ARDS/death (orange) and survival/extubation (purple). These gene signatures were derived from public gene expression datasets from monocytes in pediatric bacterial sepsis subjects (see Materials and Methods). (G) Row-scaled expression of DEGs in AMs from control and dysbiotic newborn macaques (n = 4; two in each experimental group), normalized against all subjects. k-means clustering was used to arrange subjects and transcripts (n = 4; two in each experimental group; Benjamini and Hochberg–adjusted P < 0.01, log2 fold change (FC) > 2, Wald’s test]. GO, Gene Ontology. (H) Row-scaled regulon activity for AM clusters. k-means clustering was used to arrange clusters and regulons (n = 4; two in each experimental group; Benjamini and Hochberg–adjusted P < 0.01). (I) Row-scaled expression of DEGs in the pulmonary T cells from control and dysbiotic newborn macaques (n = 4; two in each experimental group), normalized against all subjects. k-means clustering was used to arrange subjects and transcripts (n = 4; two in each experimental group; Benjamini and Hochberg–adjusted P < 0.01, log2 fold change > 2, Wald’s test). (J) Pearson correlation between peak PEWS and the frequency of dysfunctional (LAG-3+) CD4+ T cells (percentage of all CD4+ T cells from lungs). Correlation coefficient (R) and significance with the associated P value is indicated. (K) Pearson correlation between indicated cytokines from bronchial washings (pg/ml) and the frequency of dysfunctional CD4+ T cells (% of all CD4+ T cells from lungs). Correlation coefficient (R2) and significance with the associated P value is indicated.
Early-life antibiotic exposure influenced the development and activation state of AMs after infection with S. pneumoniae. Mature AMs (cluster 1), known to maintain noninflammatory states by promoting tolerance and facilitating tissue repair (70), were decreased, whereas the frequency of polarized, inflammatory AMs (cluster 2) was increased in dysbiotic newborn macaques (Fig. 4D). In addition, the frequency of M1-activated AMs [identified as live major histocompatibility complex II–positive (MHC class II+), CD11C+, and CD86+ cells] was increased in dysbiotic newborns after infection with S. pneumoniae (Fig. 4E) and correlated with disease severity (fig. S7E).
We hypothesized that dysfunctional inflammatory macrophages were associated with severe pneumonia in dysbiotic macaques. Using published gene signatures from AMs in ARDS in humans (50), we found that gene transcripts predicting recovery were enriched in cluster 1, whereas gene transcripts predicting severe ARDS/death were enriched in cluster 2 (Fig. 4F and table S20). The expression of genes related to tissue damage and vascular inflammation was differentially enriched in dysbiotic macaques (Fig. 4G and table S21).
Adenosine triphosphate (ATP) released from damaged epithelium and endothelium activates an ATP-driven purinergic-inflammasome signaling pathway and is associated with fatal pneumonia and severe ARDS (71). We found increased ATP in the bronchial lavage fluid of dysbiotic macaques, although this did not reach statistical significance (P = 0.06) (fig. S7F). Last, we noted decreased expression of tolerance-promoting MHC class II genes in AMs of dysbiotic newborns (Fig. 4G).
Conserved gene regulatory networks are associated with remodeled pulmonary neutrophil and macrophage compartments in dysbiotic newborns
Regulatory networks anchored by peroxisome proliferator–activated receptor γ (PPARγ), C/EBPα, PU.1 (encoded by SPI1) (72), and upstream stimulatory factor 1 (USF1), TFs critical in macrophage development (73), were enriched in all mature AMs, whereas HIF1A and early growth response (EGR), critical drivers of transcriptional programs underlying macrophage activation and terminal polarization (74), were enriched in stressed and hyperinflammatory macrophages (Fig. 4H and table S22).
A regulatory network anchored by aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL) [encoded by brain and muscle ARNT-like 1 (BMAL1)], a component of the circadian clock, was overrepresented in the stressed and hyperinflammatory macrophages (cluster 2) that were expanded in dysbiotic newborns (Fig. 4H and fig. S7G). Diurnal oscillations of intestinal microbiota are thought to drive the programming of host immune responses via ARNTL (75) and other components of the circadian clock. In addition, retinoid X receptor alpha (RXRα) and NFIL3 were overrepresented in regulatory networks for cluster 2 (Fig. 4H). These data, coupled with similar observations in neutrophils (Fig. 3K), suggested a role for shared regulatory networks anchored by circadian clock components in the transcriptional remodeling of pulmonary neutrophil and AM compartments in dysbiotic newborns.
Antibiotic exposure also remodeled the interstitial macrophage (IM) pool after infection with S. pneumoniae. Analogous to AMs, we identified three unique populations of mature IMs marked by expression of genes related to macrophage migration, phagocytosis, tolerance promotion, and wound repair (cluster 1); activated IMs identified by their expression of genes related to antigen presentation, IL-1 receptor activation, and CD4+ T cell differentiation (cluster 2); and a population marked by expression of genes associated with endoplasmic reticulum stress, exhaustion, and apoptotic clearing, identified as exhausted and stressed IM (cluster 3) (fig. S7, H and I). In addition, antibiotic exposure was associated with contraction of pro-repair IMs and expansion of stressed, exhausted proinflammatory IMs (fig. S7, J and K, and table S23). These data collectively suggested the loss of prophagocytic, tolerance-promoting, and antigen-presenting programs, which facilitate protective functions of lung macrophages and expansion of ATP-purinergic inflammasome signaling and proinflammatory programs, and are associated with lung damage and increased pneumonia-related morbidity in dysbiotic macaques.
Dysbiotic newborns display ineffective pulmonary T helper cell responses after infection with S. pneumoniae
Consistent with transcriptomic changes seen in other pulmonary cell types, gene transcripts associated with tissue repair and growth factor signaling, T cell fitness, and effector differentiation were decreased, whereas transcripts for genes in the aerobic glycolysis pathway, linked to T cell exhaustion (76), were increased in T cells of dysbiotic newborns (Fig. 4I and table S24). Pulmonary CD4+ T cells from dysbiotic newborns coexpressed T cell immunoglobulin and mucin-domain containing-3 (TIM-3) and lymphocyte activating gene-3 (LAG-3), which are markers linked to T cell exhaustion (fig. S8A) (31, 77). The frequency of dysfunctional CD4+ T cells (expressing LAG-3) correlated with disease severity (Fig. 4J). Furthermore, cytokines associated with T cell differentiation and effector responses, such as IL-6, IL-17, CXCL8, and CXCL10, were increased in the bronchial lavage fluid of dysbiotic macaques (Fig. 4K). Our data highlighted T cell responses marked by a failure to limit “cytotoxicity” and potentially increased tendency toward “exhaustion” in dysbiotic newborns, which is consistent with recent studies highlighting a critical role of exhausted T cells in severe coronavirus disease 2019 (COVID-19) (78).
Early-life antibiotic exposure increases stressed, inflammatory NK cells in lungs of dysbiotic newborns after infection with S. pneumoniae
Optimal NK cell functions promote infection control by serving as a rheostat in regulating T cell responses with excessive activation contributing to immunopathology (79). Consistent with these observations, we found that most NK cells in the lungs of dysbiotic newborns coexpressed activation marker CD69 and produced granzyme B and IFN-γ (fig. S9A).
Similar remodeling of the pulmonary NK pool was supported by scRNA-seq data. Expression of cytotoxic effector and activation markers, such as NK cell granule protein 7 (NKG7), CD38, and CD52 (44, 80, 81), and proliferation markers, such as minichromosome maintenance (MCM), proliferating cell nuclear antigen (PCNA), and eukaryotic translation initiation factor 4A1 (EIF4A1), identified CD56+ NK cells (cluster 2) and proliferating NK cells (cluster 3), respectively, whereas a cluster expressing transcripts associated with cell survival, cellular stress, and inflammation represented cytotoxic and stressed NK cells (cluster 1) (fig. S9, B and C). The numbers of cytotoxic NK cells expanded in dysbiotic newborns (fig. S9D). The transcriptomic analysis further revealed an increased abundance of transcripts associated with T cell activation (82), inflammation, and exhaustion in dysbiotic newborns (fig. S9E and table S25). These data, coupled with reports implicating dysfunctional cytotoxic NK cell responses in severe COVID-19 (44), suggest that defects in NK cell cytotoxicity may be associated with adverse outcomes in dysbiotic newborns.
Miscommunication between innate and adaptive immune cells uncouples inflammatory and pro-repair pathways and contributes to increased pneumonia severity in dysbiotic newborns
To identify altered cell-cell communication in the lungs of dysbiotic newborns after infection, we used the analytical tool CellChat (83) to infer cell signaling interactions based on ligand-receptor transcriptional abundance. Globally, signaling pathways related to chemotactic localization, such as CXCL, CCL, and selectin P ligand (SELPLG), and tissue homeostasis and repair (84, 85), such as NOTCH and semaphorin 4 (SEMA4), were dominant in control newborns, whereas signaling pathways related to inflammation, such as CD86 and RESISTIN (86, 87), and cell exhaustion, such as PD-L1 and PD-L2, were dominant in dysbiotic newborns (Fig. 5, A and B), potentially suggesting a global rewiring of immune cell-to-cell communication network in dysbiotic newborn macaques (Fig. 5C and fig. S10, A to C).
Fig. 5. Changes in communication circuits between neutrophils and macrophages underlie a dysfunctional remodeling of the pulmonary myeloid compartment.
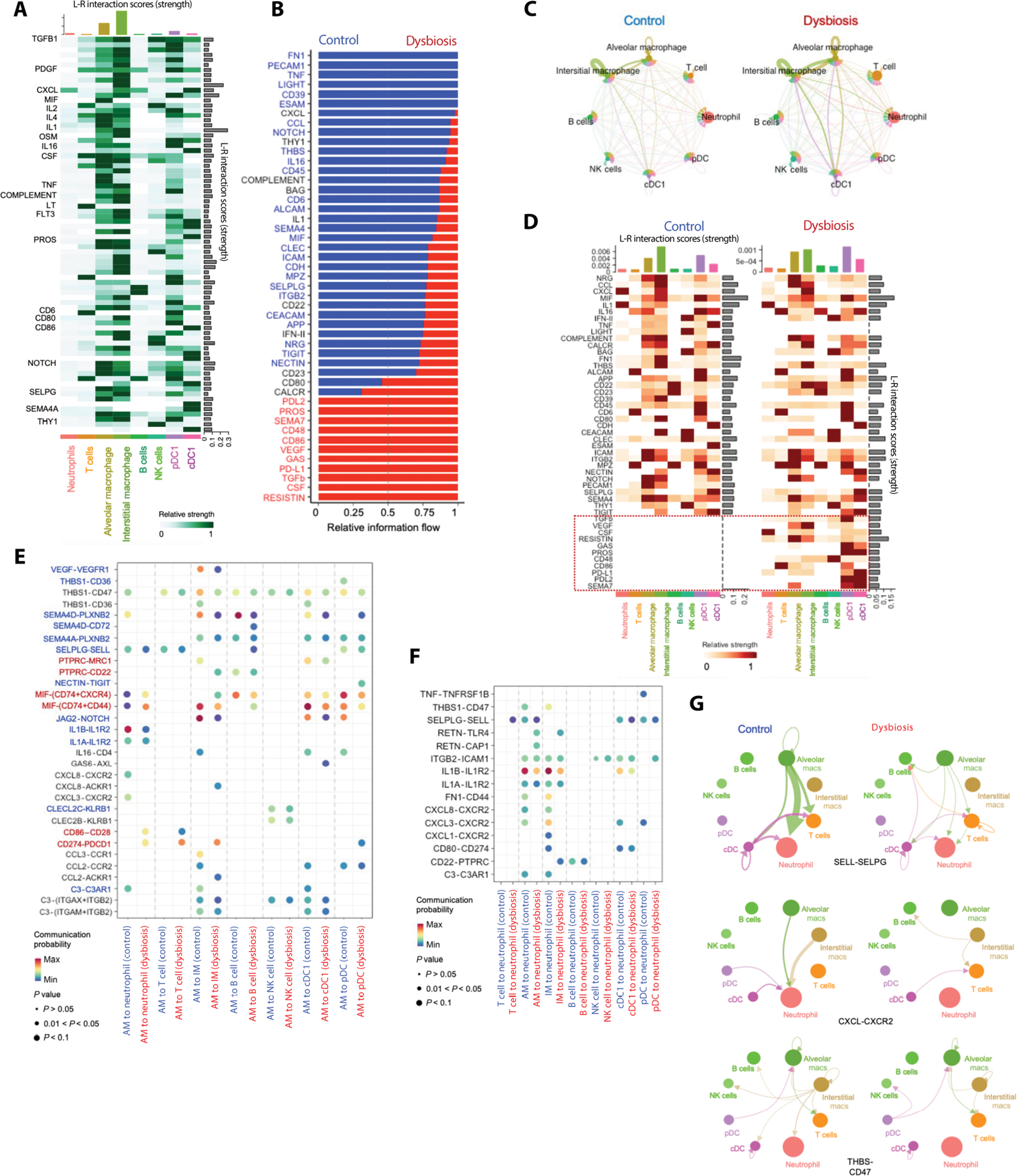
(A) Cellcell communication network between different pulmonary immune cells. Bar graphs at the top indicate ligand-receptor interaction scores (strength) for each indicated cell type. Bar graphs on the right show the ligand-receptor interaction scores (strength) of each ligand-receptor interaction. The network is dominated by pathways related to inflammation, chemotaxis, and tissue repair, as indicated by selected signal transcripts (on the left). (B) Cell-cell communication pathways ranked by overall information flow in control or dysbiotic newborn macaques. Cell-cell communications enriched in control exposed newborn macaques (in blue text) are dominated by pathways related to tissue homeostasis. Pathways related to chemotaxis (in black text) are equally enriched in control or dysbiotic newborn macaques. Cell-cell communications increased in dysbiotic newborn macaques (in red text) are dominated by pathways related to inflammation. (C) Circle plot showing differential number of interactions in the cellcell communication network between control (left) and dysbiotic (right) newborn macaques. Macrophages and dendritic cells are the hubs (senders), whereas neutrophils and T cells are targets (receivers) of cell-cell communication networks. (D) Communication pathways related to cell exhaustion and cell activation (boxed) are abundant in dysbiotic compared to control macaques. Bar graphs at the top indicate ligand-receptor interaction scores (strength) for each indicated cell type. Bar graphs on the right show the ligand-receptor interaction scores (strength) of each ligand-receptor interaction. (E) Dot plot of outgoing signaling patterns from AM (sender) to other immune cells in control (blue) or dysbiotic macaques (red). Dot color reflects communication probabilities and dot size represents computed P values. Empty space means that the communication probability is zero (P values calculated from one-sided permutation test). (F) Dot plot of incoming signaling patterns to neutrophils (receiver) from other immune cells in control (blue) or dysbiotic macaques (red) (P values computed from one-sided permutation test). (G) Autocrine and paracrine signaling pathways related to neutrophil migration (CXCL-CXCR2 and THBS1-CD47) and neutrophil activation (SELPLG-SELL) in control or dysbiotic macaques. Circle sizes are proportional to the number of cells in each cell group, and edge width represents the strength of cell-cell communication.
Dysfunctional macrophages were the central hub of misdirected cell-cell communications (Fig. 5, C to E, and fig. S10, A and B). In contrast, neutrophils and, to a lesser extent, T cells were predicted to be targets of such miscommunications, which included immune-inhibitory interactions, such as CD274(PD-L1)–programmed cell death protein 1 (PDCD1), macrophage migration inhibitory factor (MIF)–CD74/CD44/CXCR4, CD86-CD28, and protein tyrosine phosphatase receptor type C (PTPRC)–CD22 in dysbiotic newborns (Fig. 5, D and F, and fig. S10, C to F). In control newborns, communication circuits between macrophages and neutrophils or T cells were dominated by pathways related to interleukin and chemokine signaling, such as IL-1A/B-IL-1R2 and CXCL1/3/8-CXCR2; immune costimulation and complement activation, such as C3-C3AR1; and tissue repair, such as NOTCH, SEMA4, and THY1 (Fig. 5D and fig. S10, G to I) (88, 89). These data identified a potential mismatch between inflammatory and pro-repair pathways anchored by dysfunctional macrophages in dysbiotic newborns.
To identify the communication circuits informing the pathogenic remodeling of the pulmonary neutrophil pool, we interrogated specific signaling circuits between AMs and neutrophils. AM-derived paracrine signals related to neutrophil migration, such as CXCL-CXCR2 and thrombospondin 1 (THBS1)–CD47; neutrophil extravasation, such as integrin-subunit β2 (ITGB2)–intracellular adhesion molecule 1 (ICAM1); and neutrophil activation, such as SELPLG-SELL, were enriched in control newborns. In contrast, pathways related to programmed cell death [CD80-CD274(PD-L1)] were dominant in dysbiotic newborns (Fig. 5, E to G, and fig. S10, C to E). PD-L1 negatively regulates the activation of T cell receptors and mediates lymphocyte apoptosis. Severe and often fatal sepsis is marked by increased neutrophil PD-L1 expression (88), suggesting a role for PD-L1 in pathogenic responses.
Collectively, these findings highlight the miscommunication between innate and adaptive immune cells in dysbiotic newborns, contributing to a hyperinflammatory signature in neutrophils and relative immune paralysis in the T cells (fig. S10, J and K). This pattern supports the concept that the uncoupling of inflammatory and pro-repair pathways contributes to the increased pneumonia severity in dysbiotic newborns.
Fecal transfer is associated with favorable changes in pulmonary immune cell responses and improved host resistance to pneumonia in dysbiotic newborns
Although no specific bacterial taxa have been consistently associated with pulmonary host resistance to pathogens, fecal microbiota transplantation, which transfers the entire gut microbiota from one host to another, has demonstrated improved clinical outcomes in immunotherapy trials (90, 91). We performed fecal transfer (FT) of the fecal contents of control newborns to dysbiotic newborns on PN8. Dysbiotic newborns who received the FT, referred hereafter as FT recipient newborns, were challenged with S. pneumoniae on PN14 (6 days after FT). FT recipients had lower PEWS after infection, less rapid progression of symptoms, and reduced need for supportive therapy (Fig. 6, A to D, and fig. S11A). FT recipients also showed a bacterial burden closer to control (fig. S11B). Although all recipients demonstrated clinical benefit, the benefit was variable (Fig. 6, D and E).
Fig. 6. Fecal transfer was associated with favorable changes in pulmonary immune cell responses and improved host resistance to pneumonia in dysbiotic macaques.
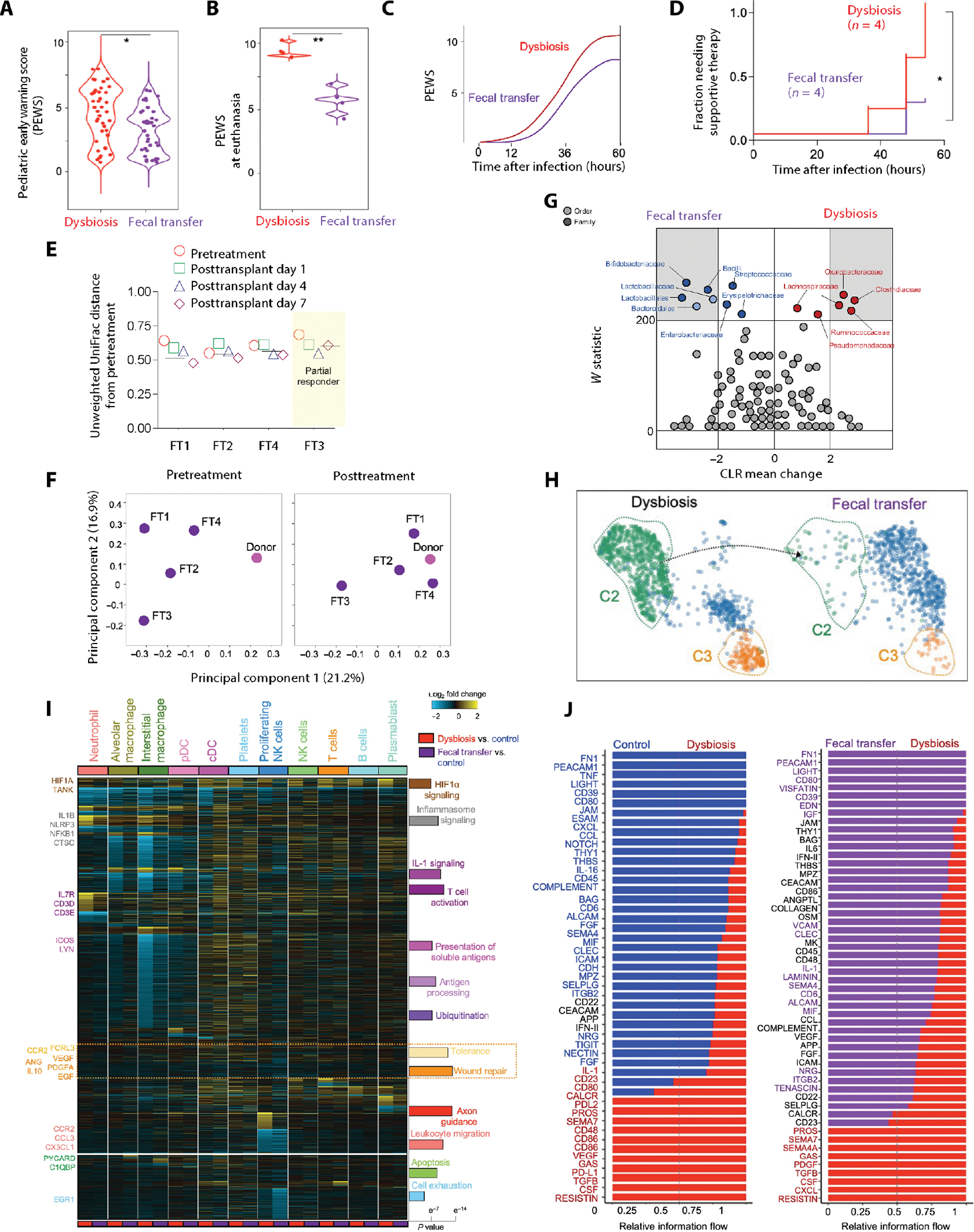
(A) ABX-exposed (dysbiosis) or fecal transfer (FT) recipient newborn macaques were challenged with S. pneumoniae (serotype 19F) on PN14. PEWS was determined every 6 hours (n = 8; four in each experimental group; the P value is indicated, Student’s t test). (B) PEWS at euthanasia (n = 8; four in each experimental group; the P value is indicated, Student’s t test). (C) Progression of PEWS after infection in dysbiotic or FT recipient newborn macaques. Broken lines (C) represent time (after infection) to PEWS > 8, a predetermined threshold to initiate supportive therapy. (D) Kaplan-Meier plot of the fraction of dysbiotic or FT recipient newborn macaques requiring supportive therapy at indicated times after infection (n = 8; four in each experimental group; *P < 0.05, Mantel-Cox log-rank test). (E) Plot of unweighted UniFrac distances for each FT recipient from the corresponding pretreatment sample at 1, 4, or 7 days after transfer. 0 represents identical microbiota compositions, and 1 represents completely dissimilar compositions. The horizontal line represents the average posttreatment distance. (F) Principal component analysis (PCA) of fecal bacterial communities of the donor (pink) and the recipients (purple) before (pretreatment) or 7 days after FT (posttreatment), based on β-diversity (unweighted UniFrac). The distance between samples on the plot represents their dissimilarity. (G) Relative abundance of specific taxa in the recipients before (pretreatment) or 7 days after FT (posttreatment). Differentially abundant taxa (FDR q ≤ 0.05, center log transformation > 2) are presented in gray margins. The center log transformation mean difference represents compositional differences in microbial communities. (H) UMAP embedding of pulmonary neutrophils colored by cluster in dysbiotic and FT recipient newborn macaques, showing the reappearance of cluster 1. (I) Heatmap of DEGs in all pairwise cell type comparisons (fold > 1.2 and eBayes t test P < 0.05, FDR corrected) in dysbiosis or FT versus control (cellHarmony). Bar plot denotes the Fisher’s exact P values (FDR corrected) of GO terms adjacent to the enriched cellHarmony DEG cluster. (J) Cell-cell communication pathways ranked by overall information flow in dysbiotic (red text) or FT recipient newborn macaques (purple text). Miscommunication in pathways related to inflammation, immune costimulation, and cell exhaustion is reversed in FT recipient newborn macaques. Cell-cell communication associated with tissue repair and cell migration remains uncorrected after FT.
Gut microbiota composition of the FT recipients (after treatment) differed from their baseline (pre-FT) (Fig. 6F and tables S26 and S27). After FT, gut microbiota had a higher abundance of Bifidobacterium bifidum, a favorable modulator of immune responses in humans (Fig. 6G) (7, 20). Although our study lacked the power to establish a clear association between specific bacterial taxa and clinical response to pneumonia, our results suggest that FT could be feasible in dysbiotic newborns.
Next, we evaluated the immunological effects of FT after challenge with S. pneumoniae. The pulmonary neutrophil pool expanded after FT, although the numbers remained lower than control newborns (fig. S12, A and B). FT partially corrected the remodeling of the neutrophil pool observed in dysbiotic newborns (Fig. 6H). FT was associated with changes in the transcriptome of neutrophils compared to dysbiotic macaques. Transcripts of genes related to leukocyte apoptosis, such as cathepsin C (CTSC), TNF type 1–associated DEATH domain (TRADD), PYD and CARD domain containing (PYCARD), and C1QBP; respiratory burst, such as myeloperoxidase (MPO), neutrophil cytosolic factor (NCF1), and NCF2; and cellular stress and ubiquitination, such as HIF1A, EGR1, NOP53, and TRAFF family member–associated nuclear factor κB (NFκB) activator (TANK), which were previously increased in neutrophils of dysbiotic macaques, were decreased in FT recipient newborn macaques (Fig. 6I, fig. S12C, and table S28).
Expression of genes related to inflammasome or IL-1 signaling, such as nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin-domain containing (NLRP3), IL1B, IL10RA, and NFKB1, decreased in AMs of FT recipient macaques compared to dysbiotic macaques, whereas gene transcripts related to macrophage migration, such as CCR2, CCL3, and CX3CL1; phagocytosis, such as C-type lectin domain containing (CLEC7A), CD47, and C3; molecules promoting tolerance, such as CCR2, adenosine deaminase (ADA), IL10, and Fc-receptor like 3 (FCRL3); and wound repair, such as angiogenin (ANG), IL10, and VEGFA, remained unchanged in FT recipients (Fig. 6I, fig. S12D, and table S29). Transcripts associated with T cell activation and differentiation, such as IL7R, CCR7, CD3D, and CD3E, and antigen processing and presentation, such as CCR4, inducible T cell co-stimulator (ICOS), and LYN, which were severely decreased in T cells of dysbiotic newborns, partially increased after FT (Fig. 6I, fig. S12E, and table S30). Transcripts associated with tissue repair and growth, such as VEGF, PDGFA, and EGF, did not recover (Fig. 6I and table S30).
Miscommunication between the innate and adaptive immune cells was reversed after FT, although signaling pathways associated with tissue repair, such as NOTCH, IL-10, and SEMA4, and chemotaxis, such as CXCL, remained dysregulated in FT recipients (Fig. 6J and fig. S12F). These data suggested that, although FT reversed cell stress and apoptosis and mitigated proinflammatory signatures in the pulmonary immune cells, it did not fully reverse the loss of innate immune cell migration, differentiation, activation, and tissue repair signatures.
DISCUSSION
We leveraged a nonhuman primate model and systems immunology approach to examine the effect of antibiotic-induced dysbiosis on the clinical and immunologic progression of neonatal pneumonia, the most common infectious cause of infant mortality (2). Early-life antibiotic exposure was associated with an overrepresentation of Enterococcus and Clostridium, approximating the gut microbial communities in a large cohort of vaginally delivered infants exposed to perinatal antibiotics (92, 93) and increased morbidity to S. pneumoniae, recapitulating epidemiological observations (93). Early-life antibiotic exposure was associated with a remodeled pulmonary neutrophil compartment and the presence of senescent neutrophils with exhausted and hyperinflammatory signatures. We speculate that extended neutrophil life span and the resulting exhaustion, coupled with failure to remove senescent and exhausted neutrophils from the infected lungs, caused severe tissue damage because of the release of proteases, cationic peptides, and cytokines, which were increased in dysbiotic newborn macaques. An extended neutrophil life span was observed in patients with sepsis (94) and ARDS (95) and was associated with disease progression and poor prognosis.
Dysbiotic newborn macaques also displayed strong inflammatory patterns, terminally polarized macrophages, and a trend toward increased ATP amounts. Expanded ATP–purinergic inflammasome signaling is associated with ARDS, fibrosis, and worse clinical outcomes in viral pneumonia, including severe COVID-19 (96, 97). We speculate that, in the absence of tolerance-promoting and antigen-presenting function, the proinflammatory program in macrophages is pathogenically turned on in dysbiotic newborn macaques, contributing to excessive lung damage and increased infection-related morbidity. These data suggest that therapeutic approaches targeting hyperinflammation, neutrophil clearance from inflamed tissues, or induction of neutrophil apoptosis may potentially improve clinical outcomes in infected, dysbiotic infants.
Intestinal microbiota undergoes diurnal compositional and functional oscillations (98), which, in turn, contribute to the homeostatic programming of host immune cells via clock genes. Antibiotics disrupt these diurnal oscillations of the microbiota and lead to desynchronization of the circadian clock network both locally in the intestine and systemically (98). Circadian oscillations, anchored by BMAL1 and NFIL3 in the sympathetic neurons, regulate the egress of neutrophils from the bone marrow into the periphery via the CXCL12-dependent mechanism (99). Trafficking of neutrophils from blood to infected tissues is constrained by BMAL1-dependent regulation of CXCR4 and CCL2 expression (100, 101), and neutrophil aging and clearance of senescent neutrophils are regulated by circadian rhythms (51, 102). Last, NFIL3-driven circadian rhythms limit macrophage heterogeneity and amplify the pathogenic inflammatory responses in the macrophages (103). These observations, coupled with our present data identifying these TFs in regulatory networks controlling immune cells in dysbiotic newborns, suggest that disrupted circadian oscillations underpin both the disrupted neutrophil maturation and macrophage dysfunction.
The present study identifies dysfunctional macrophages as a hub of misdirected cell-cell communications. Macrophages and dendritic cells sense respiratory pathogens and initiate immune responses, resulting in rapid recruitment of target cells, such as neutrophils, through the secretion of first-order cytokines, such as CXCR2/CXCL1, CXCL8, IL-1A, and IL-1B/IL-R2. Our study suggests that neutrophils transform the first-order cytokine signals into second-order cytokines that enhance the trafficking and extravasation [oncostatin M (OSM)– leukemia inhibitory factor receptor (LIFR)], immune coactivation (complement C3-C3AR1), and effector function, such as phagocytosis [PTPRC–mannose receptor C-type 1 (MRC1)], to eliminate pathogens. At the same time, reciprocal interactions limit macrophage activation (MIF-CD74/CXCR2 and CD83–platelet and endothelial adhesion molecule 1 (PECAM1)] and promote tissue repair factors (CSF1-CSFR1, TGFBR3-TGFB1, SEMA4–neuropilin (NRP)]. This stepwise, tiered response was disrupted in dysbiotic macaques because of dysfunctional macrophages. Immune-inhibitory interactions, such as CD274(PD-L1)-PDCD1, MIF-CD74/CD44/CXCR4, CD86-CD28, and PTPRC-CD22, which promote neutrophil and lymphocyte dysfunction, dominated immune responses in dysbiotic newborns. Similarly, macrophage-anchored signaling pathways related to neutrophil migration, such as CXCL-CXCR2 and THBS1-CD47; neutrophil extravasation, such as ITGB2-ICAM1; and neutrophil activation, such as SELPLG-SELL, were disrupted in dysbiotic newborns. These changes in the communication circuits may explain the shared hyperinflammatory signature in all immune cells and global loss of tissue-protective, homeostatic pathways.
We show that FT was associated with restored immune responses and protection of the dysbiotic newborn against pneumonia. Our findings show that FT shifted the composition of the recipient’s microbiota toward Gammaproteobacteria and B. bifidum, taxa favoring beneficial immune responses in humans. FT mitigated certain aspects of immune dysfunction seen in dysbiotic newborns. Although broad dysregulation in signaling pathways related to inflammation, complement activation, and cell exhaustion was corrected in FT-recipients, miscommunication in pathways related to tissue repair and chemotaxis persisted.
Several scenarios may explain this partial restoration. Distinct taxa drive the maturation of individual immune cells, so a lack of response may reflect the absence of favorable taxa in FT or failure of such taxa to engraft. A more logical explanation is that resolution of pneumonia requires a tiered response with the sequential engagement of innate immune cells and first-order cytokines, which activate other innate and adaptive immune cells via second-order cytokines. FT may not completely mitigate the disruption of such first- and second-order effectors, most likely because of persistent macrophage dysfunction, contributing to suboptimal benefit. Nevertheless, our study provides proof-of-concept evidence for the ability of FT to improve clinical outcomes in at-risk dysbiotic newborns.
Our study has some limitations. The necessity of frequent clinical examination, sample collection, and invasive procedures precluded us from using dam-reared infants. Therefore, infants in our study received a diet consisting of formula, not breast milk. As infants grow, feeding practices play an increasing role in determining the composition of the infant gut microbiota (104–106); however, delivery mode and perinatal antibiotic use have a stronger influence on the composition of the microbial community immediately after birth (107). Nevertheless, further studies are needed to delineate the relative contribution of infant diet and microbial metabolites (108) on pulmonary immune maturation during infancy.
In conclusion, these findings reinforce the concept that the gut microbiota play a crucial role in programming the balance between tissue inflammatory and regenerative pathways in response to infection and highlight the role of a healthy gut microbiome in limiting pathogens and promoting clinical recovery.
MATERIALS AND METHODS
Study design
The overall goal of this study was to use a nonhuman primate model to investigate the immune response to respiratory pathogens within the lungs. Our overall hypothesis was that disruption of early-life microbiota would lead to dysfunctional pulmonary immune responses and worsened outcomes to bacterial pneumonia. The Institutional Animal Care and Use Committees (IACUCs) at Cincinnati Children’s Hospital Medical Center and University of California at Davis approved all the animal studies (IACUC2016–19222), which were carried out in Association for the Assessment and Accreditation of Laboratory Animal Care–accredited facilities at the California National Primate Research Center (CNPRC). Because there were no previously published studies in newborn primates, published data in newborn mice were used to estimate that four animals in each group would be sufficient to detect a 20% difference in morbidity with 80% power and an α of 0.05. Twelve vaginally delivered Indian-origin rhesus macaque infants (Macaca mulatta) (table S1) were used in this study, which was conducted per National Institutes of Health (NIH)’s Guide for the Care and Use of Laboratory Animals. Infant macaques were separated from their dams (for demographic characteristics of macaque dams, see table S31), raised in a nursery from the day of birth, and exposed to a normal light cycle (lights on for 12 hours starting at 08:00). The infant macaques had a diet consisting of Enfamil Lipil with iron for the study duration. The necessity of frequent clinical examination, sample collection, and invasive procedures precluded us from using dam-reared infants in our study. For PN1 to PN7, infant macaques were randomized to one of three treatment groups: saline (n = 4; control); a cocktail of vancomycin, gentamicin, and ampicillin (n = 4; dysbiotic); or the same cocktail of antibiotics and a FT at PN8 (n = 4; FT; see the “Fecal transplant” section below). This antibiotic regimen targeted both Gram-positive and Gram-negative intestinal bacteria and mimicked antibiotic exposure in human infants.
At PN7, antibiotics were discontinued to allow 1 week of washout before subsequent infection on PN14. Clinical assessments (both parameters for PEWS and other clinical parameters) were made by the veterinary staff caring for the rhesus macaque neonates, and these veterinary staff were blinded to the intervention. PEWS values were calculated as previously described (38). Briefly, clinical parameters, such as heart rate and respiratory rate, are given scores on the basis of predetermined ranges and are combined for an overall PEWS, with a higher PEWS reflecting higher clinical severity of the disease. The veterinary staff provided supportive care to the rhesus macaque newborns based on a predetermined PEWS threshold, which consisted of intravenous fluids if PEWS = 8 and intravenous fluids plus oxygen supplementation if PEWS ≥ 9.
Infectious studies
We grew S. pneumoniae serotype 19A (American Type Culture Collection, 700674) with gentle aeration (37°C, 200 rpm) in tryptic soy broth to log-phase growth. Neonatal macaques were inoculated with S. pneumoniae (106 colony-forming units) via the intratracheal route on PN14. See Supplementary Materials and Methods for full description.
Fecal transplant
FTs were performed in four newborn macaques who had received the cocktail of vancomycin, gentamicin, and ampicillin from PN1 to PN7. This group was separate from the other group of newborn macaques that just received the cocktail of antibiotics (the dysbiotic newborns). See Supplementary Materials and Methods for full description.
Microbiota analysis
Fecal samples were collected daily (PN1 to PN14) from saline-treated (control) or antibiotic-treated (dysbiotic infants) infants and from dams at the time of delivery. Infant fecal samples were collected daily (PN10 to PN14) in FT recipient infants. Fecal samples were frozen (−80°C) immediately after collection. All samples were analyzed together to prevent batch effects. See Supplementary Materials and Methods for full description.
Cytometry by time of flight
Frozen whole-blood leukocytes or CD326−CD31−CD45+ lung cells were thawed and plated at 1 × 106 cells per well in a 96-well U-bottom plate. All samples were analyzed together to prevent batch effects. See Supplementary Materials and Methods for full description.
Flow cytometry
Pulmonary CD4+ T cells, AMs, and pulmonary NK cells were thawed and further characterized using markers for functional status, as seen and noted in Fig. 4 and fig. S9. See table S32 for the specific antibodies used. Stained cells were acquired on a custom four-laser BD LSRFortessa flow cytometer (Becton Dickinson) with FACSDiva software and analyzed with FlowJo Software (TreeStar).
Single-cell RNA sequencing
Frozen CD326−CD31−CD45+ cells from digested lungs or frozen peripheral blood mononuclear cells were thawed and resuspended in 50 μl at a concentration of 1000 cells/μl. After excluding dead cells, ~20,000 cells (~3 to 4000 cells per animal, barcoded to identify individual animals) were then loaded into one channel of the Chromium system using the v3 Single Cell Reagent Kit (10x Genomics) at the Cincinnati Children’s Hospital Medical Center DNA Sequencing and Genotyping Core. After capture and lysis, complementary DNA (cDNA) was synthesized and amplified as per the manufacturer’s protocol (10x Genomics). The amplified cDNA was used to construct Illumina sequencing libraries that were each sequenced using an Illumina HiSeq 4000. See Supplementary Materials and Methods for full details on the analyses done.
Alignment and quality control
Raw sequencing data were aligned to the rhesus macaque reference Mmul_10 with Cell Ranger 1.3 (10x Genomics), generating expression count matrix files (see table S33 for dataset metrics). Cells that had fewer than 750 unique molecular identifiers (UMIs) or greater than 15,000 UMIs, as well as cells that contained greater than 20% of reads from mitochondrial genes or ribosomal RNA genes (RNA18S5 or RNA28S5) or hemoglobin genes, were considered low quality and were removed from further analysis. Putative multiplets were removed with DoubletFinder (version 2.0). Genes that were expressed in fewer than 10 cells were removed from the final count matrix.
Data analysis
The Seurat package (version 3.1.0; https://satijalab.org/seurat/) was used to identify common cell types across different experimental conditions, differential expression analysis, and most visualizations. Percentages of mitochondrial genes, ribosomal genes, and hemoglobin genes were regressed during data scaling to remove unwanted variation due to cell quality using the SCTransform() function in Seurat. Principal components analysis was performed using the 3000 most highly variable genes, and the first 20 principal components (PCs) were used to perform uniform manifold approximation and projection to embed the dataset into two dimensions. Next, the first 20 PCs were used to construct a shared nearest neighbor (SNN) graph [FindNeighbors ()], and this SNN was used to cluster the dataset [FindClusters ()]. Manual annotation of cellular identity was performed by finding differentially expressed genes for each cluster using Seurat’s implementation of the Wilcoxon rank sum test [FindMarkers()] and comparing those markers to known cell type–specific genes from published studies (41, 42). Global differential gene expression profiles between all cell types were identified and organized with the software cellHarmony (109), using these Seurat clusters [fold change > 1.2, empirical Bayes t test P < 0.05, false discovery rate (FDR) corrected].
Sepsis diagnosis prediction using senescent neutrophil differentially expressed genes
To test whether the gene signature of senescent neutrophils could be used to predict sepsis diagnosis, we first developed a five-gene signature of the senescent neutrophils by identifying the most differentially enriched genes in cluster 2 neutrophils in our transcriptomic dataset relative to all other cells. Next, we downloaded normalized transcript counts from a publicly available whole-blood bulk transcriptomic dataset (GSE696686) (68). We then scored each sample in this dataset by the expression of the five genes enriched in our senescent neutrophil cluster (HIF1A, CXCR4, CD274, LTF, and S100A8). Last, we used these gene signature scores as a predictor variable and disease/diagnosis metadata reported by Wynn et al. (68) as the response variable to construct a receiver operating characteristic curve to quantify and visualize the sensitivity and specificity of the prediction.
Statistical analysis
All data met the assumptions of the statistical tests used. Statistical tests used for microbiome or single-cell analyses are described in relevant sections. For comparing the differences between groups, we used either unpaired two-tailed Student’s t test, analysis of variance (ANOVA), or Wilcoxon signed-rank test. We used Pearson correlation coefficient to measure correlation between different variables. We used the Kaplan-Meier log-rank test to compare morbidity between groups (all in GraphPad Prism 8.0). P values are indicated as follows: *P ≤ 0.05 or **P ≤ 0.01.
Supplementary Material
Acknowledgments:
We thank the Cincinnati Children’s Hospital Research Foundation’s DNA Sequencing Core and Flow Cytometry Laboratories (supported by AR47363, DK78392, and DK90971 from the NIH). We are grateful to the staff at the CNPRC, especially P. Sosa, J. Kendrick, and S. Davis for help in animal care. We thank J. Whitsett, S. Way, A. Walton, A. Jobe, and S. Pasare for helpful comments.
Funding:
These studies were supported by HD084686, HL155611, ES029234, and HL142708 (NIH) (to H.D.); the Francis Family Foundation (to H.D.); ES029234 and AG053498 (NIH) (to C.C.); HD028827 (NIH) (to L.P.); HL140178 (NIH) (to W.Z.); HL149366 and AI152100 (NIH) (to I.L.); CA226802 and HL148865 (NIH) (to N.S.); U01HL134745 and U01HL122642 (NIH) (to Y.X. and M.G.); HD89939, EB029863, GM128452, and HD89939 (NIH) (to J.L.W.); AI138553 and HL142485 (NIH) (to L.M.); AI157626, AI150554, 143554, and OD27094 (NIH) (to D.H.O.); and P51 OD11107 (NIH) (to CNPRC).
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (accession number GSE176408). Processed data can be further explored at the Lung MAP atlas portal at https://research.cchmc.org/pbge/lunggens/lungExternal/Deshmukh_Macaque_2021.html. The reagents and metadata included in this manuscript are not covered by any material transfer agreement (MTA) and can be obtained from the corresponding author upon request. Scripts used for data analysis are available from GitHub at https://github.com/Deshmukh-Lab/2021_Stevens_Macaque (DOI: 10.5281/zenodo.6569295).
REFERENCES AND NOTES
- 1.Dowling DJ, Levy O, Ontogeny of early life immunity. Trends Immunol. 35, 299–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, Lawn JE, Cousens S, Mathers C, Black RE, Global, regional, and national causes of under-5 mortality in 2000–15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 388, 3027–3035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacGillivray DM, Kollmann TR, The role of environmental factors in modulating immune responses in early life. Front. Immunol. 5, 434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cash HL, Whitham CV, Behrendt CL, Hooper LV, Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elinav E, Strowig T, Henao-Mejia J, Flavell RA, Regulation of the antimicrobial response by NLR proteins. Immunity 34, 665–679 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS, The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 20, 524–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rooks MG, Garrett WS, Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wypych TP, Wickramasinghe LC, Marsland BJ, The influence of the microbiome on respiratory health. Nat. Immunol. 20, 1279–1290 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Spaetgens R, DeBella K, Ma D, Robertson S, Mucenski M, Davies HD, Perinatal antibiotic usage and changes in colonization and resistance rates of group B streptococcus and other pathogens. Obstet. Gynecol. 100, 525–533 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Lin FY, Weisman LE, Azimi P, Young AE, Chang K, Cielo M, Moyer P, Troendle JF, Schneerson R, Robbins JB, Assessment of intrapartum antibiotic prophylaxis for the prevention of early-onset group B Streptococcal disease. Pediatr. Infect. Dis. J. 30, 759–763 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knight M, Chiocchia V, Partlett C, Rivero-Arias O, Hua X, Hinshaw K, Tuffnell D, Linsell L, Juszczak E; ANODE Collaborative group, Prophylactic antibiotics in the prevention of infection after operative vaginal delivery (ANODE): A multicentre randomised controlled trial. Lancet 393, 2395–2403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, Bizzarro MJ, Goldberg RN, Frantz III ID, Hale EC, Shankaran S, Kennedy K, Carlo WA, Watterberg KL, Bell EF, Walsh MC, Schibler K, Laptook AR, Shane AL, Schrag SJ, Das A, Higgins RD; Eunice Kennedy Shriver National Institute of Child and Human Development Neonatal Research Network, Early onset neonatal sepsis: The burden of group B Streptococcal and E. coli disease continues. Pediatrics 127, 817–826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boghossian NS, Page GP, Bell EF, Stoll BJ, Murray JC, Cotten CM, Shankaran S, Walsh MC, Laptook AR, Newman NS, Hale EC, McDonald SA, Das A, Higgins RD; Eunice Kennedy Shriver National Institute of Child and Human Development Neonatal Research Network, Late-onset sepsis in very low birth weight infants from singleton and multiple-gestation births. J. Pediatr. 162, 1120–1124, 1124 e1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuppala VS, Meinzen-Derr J, Morrow AL, Schibler KR, Prolonged initial empirical antibiotic treatment is associated with adverse outcomes in premature infants. J. Pediatr. 159, 720–725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arboleya S, Sanchez B, Milani C, Duranti S, Solis G, Fernandez N, de los Reyes-Gavilan CG, Ventura M, Margolles A, Gueimonde M, Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J. Pediatr. 166, 538–544 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Miller LA, Royer CM, Pinkerton KE, Schelegle ES, Nonhuman primate models of respiratory disease: Past, present, and future. ILAR J. 58, 269–280 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao L, Reese TA, Making mouse models that reflect human immune responses. Trends Immunol. 38, 181–193 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Philipp MT, Purcell JE, Martin DS, Buck WR, Plauche GB, Ribka EP, DeNoel P, Hermand P, Leiva LE, Bagby GJ, Nelson S, Experimental infection of rhesus macaques with Streptococcus pneumoniae: A possible model for vaccine assessment. J. Med. Primatol. 35, 113–122 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO, Development of the human infant intestinal microbiota. PLOS Biol. 5, e177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R, Current understanding of the human microbiome. Nat. Med. 24, 392–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macpherson AJ, de Aguero MG, Ganal-Vonarburg SC, How nutrition and the maternal microbiota shape the neonatal immune system. Nat. Rev. Immunol. 17, 508–517 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Perez PF, Dore J, Leclerc M, Levenez F, Benyacoub J, Serrant P, Segura-Roggero I, Schiffrin EJ, Donnet-Hughes A, Bacterial imprinting of the neonatal immune system: Lessons from maternal cells? Pediatrics 119, e724–e732 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J, The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, McCoy KD, Macpherson AJ, The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, Woloszynek JR, Greenbaum AM, Link DC, G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 28, 1851–1860 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metcalf D, Hematopoietic cytokines. Blood 111, 485–491 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O’Shea MA, Fauci AS, The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J. Immunol. 181, 6738–6746 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z, Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Soehnlein O, Drechsler M, Doring Y, Lievens D, Hartwig H, Kemmerich K, Ortega-Gomez A, Mandl M, Vijayan S, Projahn D, Garlichs CD, Koenen RR, Hristov M, Lutgens E, Zernecke A, Weber C, Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 5, 471–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weirich E, Rabin RL, Maldonado Y, Benitz W, Modler S, Herzenberg LA, Herzenberg LA, Neutrophil CD11b expression as a diagnostic marker for early-onset neonatal infection. J. Pediatr. 132, 445–451 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R, Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. U.S.A. 107, 14733–14738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharpe AH, Pauken KE, The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Johannisson A, Festin R, Phenotype transition of CD4+ T cells from CD45RA to CD45R0 is accompanied by cell activation and proliferation. Cytometry 19, 343–352 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kared H, Martelli S, Ng TP, Pender SL, Larbi A, CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol. Immunother. 65, 441–452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morandi F, Airoldi I, Marimpietri D, Bracci C, Faini AC, Gramignoli R, CD38, a receptor with multifunctional activities: From modulatory functions on regulatory cell subsets and extracellular vesicles, to a target for therapeutic strategies. Cell 8, 1527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostapchuk M, Roberts DM, Haddy R, Community-acquired pneumonia in infants and children. Am. Fam. Physician 70, 899–908 (2004). [PubMed] [Google Scholar]
- 38.Duncan H, Hutchison J, Parshuram CS, The Pediatric Early Warning System score: A severity of illness score to predict urgent medical need in hospitalized children. J. Crit. Care 21, 271–278 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Rello J, Ausina V, Ricart M, Castella J, Prats G, Impact of previous antimicrobial therapy on the etiology and outcome of ventilator-associated pneumonia. Chest 104, 1230–1235 (1993). [DOI] [PubMed] [Google Scholar]
- 40.Boutten A, Dehoux MS, Seta N, Ostinelli J, Venembre P, Crestani B, Dombret MC, Durand G, Aubier M, Compartmentalized IL-8 and elastase release within the human lung in unilateral pneumonia. Am. J. Respir. Crit. Care Med. 153, 336–342 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA, A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck III WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandes M, Klauschen F, Kuchen S, Germain RN, A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martinez-Colon GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, Simpson LJ, Grant P, Subramanian A, Rogers AJ, Blish CA, A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T, Zhang X, Liu Z, Yao T, Zheng D, Gan J, Yu S, Li L, Chen P, Sun J, Single-cell RNA sequencing reveals the sustained immune cell dysfunction in the pathogenesis of sepsis secondary to bacterial pneumonia. Genomics 113, 1219–1233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbins RA, Russ WD, Rasmussen JK, Clayton MM, Activation of the complement system in the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 135, 651–658 (1987). [DOI] [PubMed] [Google Scholar]
- 47.Davey A, McAuley DF, O’Kane CM, Matrix metalloproteinases in acute lung injury: Mediators of injury and drivers of repair. Eur. Respir. J. 38, 959–970 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Chen L, Long X, Xu Q, Tan J, Wang G, Cao Y, Wei J, Luo H, Zhu H, Huang L, Meng F, Huang L, Wang N, Zhou X, Zhao L, Chen X, Mao Z, Chen C, Li Z, Sun Z, Zhao J, Wang D, Huang G, Wang W, Zhou J, Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 17, 992–994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng LG, Ostuni R, Hidalgo A, Heterogeneity of neutrophils. Nat. Rev. Immunol. 19, 255–265 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, Hung DT, Levy BD, Baron RM, Blainey PC, Goldberg MB, Hacohen N, An immune-cell signature of bacterial sepsis. Nat. Med. 26, 333–340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M, Frenette PS, Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Neill LA, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan MA, Palaniyar N, Transcriptional firing helps to drive NETosis. Sci. Rep. 7, 41749 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagareddy PR, Sreejit G, Abo-Aly M, Jaggers RM, Chelvarajan L, Johnson J, Pernes G, Athmanathan B, Abdel-Latif A, Murphy AJ, NETosis is required for S100A8/A9-induced granulopoiesis after myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 40, 2805–2807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner J, Crossley M, Basic Krüppel-like factor functions within a network of interacting haematopoietic transcription factors. Int. J. Biochem. Cell Biol. 31, 1169–1174 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Matsumoto N, Kubo A, Liu H, Akita K, Laub F, Ramirez F, Keller G, Friedman SL, Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood 107, 1357–1365 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cloutier A, Guindi C, Larivee P, Dubois CM, Amrani A, McDonald PP, Inflammatory cytokine production by human neutrophils involves C/EBP transcription factors. J. Immunol. 182, 563–571 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Cao Z, Sun X, Icli B, Wara AK, Feinberg MW, Role of Kruppel-like factors in leukocyte development, function, and disease. Blood 116, 4404–4414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Humbert M, Halter V, Shan D, Laedrach J, Leibundgut EO, Baerlocher GM, Tobler A, Fey MF, Tschan MP, Deregulated expression of Kruppel-like factors in acute myeloid leukemia. Leuk. Res. 35, 909–913 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Lawrence SM, Corriden R, Nizet V, The ontogeny of a neutrophil: Mechanisms of granulopoiesis and homeostasis. Microbiol. Mol. Biol. Rev. 82, e00057–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oeckinghaus A, Hayden MS, Ghosh S, Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12, 695–708 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Liao W, Lin JX, Leonard WJ, IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 23, 598–604 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun T, Rojas OL, Li C, Ward LA, Philpott DJ, Gommerman JL, Intestinal Batf3-dependent dendritic cells are required for optimal antiviral T-cell responses in adult and neonatal mice. Mucosal Immunol. 10, 775–788 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV, The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 357, 912–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orozco-Solis R, Aguilar-Arnal L, Circadian regulation of immunity through epigenetic mechanisms. Front. Cell. Infect. Microbiol. 10, 96 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT, Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wynn JL, Guthrie SO, Wong HR, Lahni P, Ungaro R, Lopez MC, Baker HV, Moldawer LL, Postnatal age is a critical determinant of the neonatal host response to sepsis. Mol. Med. 21, 496–504 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li C, Menoret A, Farragher C, Ouyang Z, Bonin C, Holvoet P, Vella AT, Zhou B, Single cell transcriptomics based-MacSpectrum reveals novel macrophage activation signatures in diseases. JCI Insight 5, e126453 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allard B, Panariti A, Martin JG, Alveolar macrophages in the resolution of inflammation, tissue repair, and tolerance to infection. Front. Immunol. 9, 1777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grazioli S, Dunn-Siegrist I, Pauchard LA, Blot M, Charles PE, Pugin J, Mitochondrial alarmins are tissue mediators of ventilator-induced lung injury and ARDS. PLOS ONE 14, e0225468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Langlais D, Barreiro LB, Gros P, The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 213, 585–603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, Fonseca GJ, Troutman TD, Spann NJ, Strid T, Sakai M, Yu M, Hu R, Fang R, Metzler D, Ren B, Glass CK, Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–1809.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Corcoran SE, O’Neill LA, HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest. 126, 3699–3707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thaiss CA, Levy M, Korem T, Dohnalova L, Shapiro H, Jaitin DA, David E, Winter DR, Gury-BenAri M, Tatirovsky E, Tuganbaev T, Federici S, Zmora N, Zeevi D, Dori-Bachash M, Pevsner-Fischer M, Kartvelishvily E, Brandis A, Harmelin A, Shibolet O, Halpern Z, Honda K, Amit I, Segal E, Elinav E, Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 167, 1495–1510.e12 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Salmond RJ, mTOR regulation of glycolytic metabolism in T cells. Front. Cell Dev. Biol. 6, 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Enyindah-Asonye G, Nwankwo A, Rahman MA, Hunegnaw R, Hogge C, Helmold Hait S, Ko EJ, Hoang T, Robert-Guroff M, Overexpression of CD6 and PD-1 identifies dysfunctional CD8+ T-cells during chronic SIV infection of Rhesus Macaques. Front. Immunol. 10, 3005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Z, John Wherry E, T cell responses in patients with COVID-19. Nat. Rev. Immunol. 20, 529–536 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Waggoner SN, Cornberg M, Selin LK, Welsh RM, Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Malarkannan S, NKG7 makes a better killer. Nat. Immunol. 21, 1139–1140 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Smith SL, Kennedy PR, Stacey KB, Worboys JD, Yarwood A, Seo S, Solloa EH, Mistretta B, Chatterjee SS, Gunaratne P, Allette K, Wang YC, Smith ML, Sebra R, Mace EM, Horowitz A, Thomson W, Martin P, Eyre S, Davis DM, Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 4, 1388–1406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, Whibley N, Tucci A, Chen X, Lindeman I, Emerton G, Krausgruber T, Shields J, Haniffa M, Powrie F, Teichmann SA, Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q, Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chigurupati S, Arumugam TV, Son TG, Lathia JD, Jameel S, Mughal MR, Tang SC, Jo DG, Camandola S, Giunta M, Rakova I, McDonnell N, Miele L, Mattson MP, Poosala S, Involvement of notch signaling in wound healing. PLOS ONE 2, e1167 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xia J, Swiercz JM, Banon-Rodriguez I, Matkovic I, Federico G, Sun T, Franz T, Brakebusch CH, Kumanogoh A, Friedel RH, Martin-Belmonte F, Grone HJ, Offermanns S, Worzfeld T, Semaphorin-plexin signaling controls mitotic spindle orientation during epithelial morphogenesis and repair. Dev. Cell 33, 299–313 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Nolan A, Kobayashi H, Naveed B, Kelly A, Hoshino Y, Hoshino S, Karulf MR, Rom WN, Weiden MD, Gold JA, Differential role for CD80 and CD86 in the regulation of the innate immune response in murine polymicrobial sepsis. PLOS ONE 4, e6600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A, Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 174, 5789–5795 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS, Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J. Leukoc. Biol. 100, 1239–1254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, de Macedo MP, Cotechini T, Kumar T, Chen WS, Reddy SM, Sloane RS, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Sanchez EMR, Zhang Y, Le Chatelier E, Zitvogel L, Pons N, Austin-Breneman JL, Haydu LE, Burton EM, Gardner JM, Sirmans E, Hu J, Lazar AJ, Tsujikawa T, Diab A, Tawbi H, Glitza IC, Hwu WJ, Patel SP, Woodman SE, Amaria RN, Davies MA, Gershenwald JE, Hwu P, Lee JE, Zhang J, Coussens LM, Cooper ZA, Futreal PA, Daniel CR, Ajami NJ, Petrosino JF, Tetzlaff MT, Sharma P, Allison JP, Jenq RR, Wargo JA, Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF, The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L, Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, Barratt MJ, VanArendonk LG, Zhang Q, Province MA, Petri WA Jr., Ahmed T, Gordon JI, Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 510, 417–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tapiainen T, Koivusaari P, Brinkac L, Lorenzi HA, Salo J, Renko M, Pruikkonen H, Pokka T, Li W, Nelson K, Pirttila AM, Tejesvi MV, Impact of intrapartum and postnatal antibiotics on the gut microbiome and emergence of antimicrobial resistance in infants. Sci. Rep. 9, 10635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O, Ertel W, Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood 90, 3356–3363 (1997). [PubMed] [Google Scholar]
- 95.Matute-Bello G, Liles WC, Radella II F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR, Neutrophil apoptosis in the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 156, 1969–1977 (1997). [DOI] [PubMed] [Google Scholar]
- 96.Aggarwal NR, King LS, D’Alessio FR, Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L709–L725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schulte-Schrepping J, Reusch N, Paclik D, Bassler K, Schlickeiser S, Zhang B, Kramer B, Krammer T, Brumhard S, Bonaguro L, De Domenico E, Wendisch D, Grasshoff M, Kapellos TS, Beckstette M, Pecht T, Saglam A, Dietrich O, Mei HE, Schulz AR, Conrad C, Kunkel D, Vafadarnejad E, Xu CJ, Horne A, Herbert M, Drews A, Thibeault C, Pfeiffer M, Hippenstiel S, Hocke A, Muller-Redetzky H, Heim KM, Machleidt F, Uhrig A, de Jarcy LB, Jurgens L, Stegemann M, Glosenkamp CR, Volk HD, Goffinet C, Landthaler M, Wyler E, Georg P, Schneider M, Dang-Heine C, Neuwinger N, Kappert K, Tauber R, Corman V, Raabe J, Kaiser KM, Vinh MT, Rieke G, Meisel C, Ulas T, Becker M, Geffers R, Witzenrath M, Drosten C, Suttorp N, von Kalle C, Kurth F, Handler K, Schultze JL, Aschenbrenner AC, Li Y, Nattermann J, Sawitzki B, Saliba AE, Sander LE; Deutsche COVID-19 OMICS Initiative (DeCOI), Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thaiss CA, Zeevi D, Levy M, Zilberman-Schapira G, Suez J, Tengeler AC, Abramson L, Katz MN, Korem T, Zmora N, Kuperman Y, Biton I, Gilad S, Harmelin A, Shapiro H, Halpern Z, Segal E, Elinav E, Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159, 514–529 (2014). [DOI] [PubMed] [Google Scholar]
- 99.Mendez-Ferrer S, Lucas D, Battista M, Frenette PS, Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A, Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 341, 1483–1488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, Huerga-Encabo H, Silvestre-Roig C, Rossaint J, Cossio I, Lechuga-Vieco AV, Garcia-Prieto J, Gomez-Parrizas M, Quintana JA, Ballesteros I, Martin-Salamanca S, Aroca-Crevillen A, Chong SZ, Evrard M, Balabanian K, Lopez J, Bidzhekov K, Bachelerie F, Abad-Santos F, Munoz-Calleja C, Zarbock A, Soehnlein O, Weber C, Ng LG, Lopez-Rodriguez C, Sancho D, Moro MA, Ibanez B, Hidalgo A, A neutrophil timer coordinates immune defense and vascular protection. Immunity 50, 390–402.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, Huerga-Encabo H, Silvestre-Roig C, Rossaint J, Cossio I, Lechuga-Vieco AV, Garcia-Prieto J, Gomez-Parrizas M, Quintana JA, Ballesteros I, Martin-Salamanca S, Aroca-Crevillen A, Chong SZ, Evrard M, Balabanian K, Lopez J, Bidzhekov K, Bachelerie F, Abad-Santos F, Munoz-Calleja C, Zarbock A, Soehnlein O, Weber C, Ng LG, Lopez-Rodriguez C, Sancho D, Moro MA, Ibanez B, Hidalgo A, A neutrophil timer coordinates immune defense and vascular protection. Immunity 51, 966–967 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Allen NC, Philip NH, Hui L, Zhou X, Franklin RA, Kong Y, Medzhitov R, Desynchronization of the molecular clock contributes to the heterogeneity of the inflammatory response. Sci. Signal. 12, eaau1851 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, Lieber AD, Wu F, Perez-Perez GI, Chen Y, Schweizer W, Zheng X, Contreras M, Dominguez-Bello MG, Blaser MJ, Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 8, 343ra382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Forbes JD, Azad MB, Vehling L, Tun HM, Konya TB, Guttman DS, Field CJ, Lefebvre D, Sears MR, Becker AB, Mandhane PJ, Turvey SE, Moraes TJ, Subbarao P, Scott JA, Kozyrskyj AL; Canadian Healthy Infant Longitudinal Development (CHILD) Study Investigators, Association of exposure to formula in the hospital and subsequent infant feeding practices with gut microbiota and risk of overweight in the first year of life. JAMA Pediatr. 172, e181161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ardeshir A, Narayan NR, Mendez-Lagares G, Lu D, Rauch M, Huang Y, Van Rompay KK, Lynch SV, Hartigan-O’Connor DJ, Breast-fed and bottle-fed infant rhesus macaques develop distinct gut microbiotas and immune systems. Sci. Transl. Med. 6, 252ra120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mitchell CM, Mazzoni C, Hogstrom L, Bryant A, Bergerat A, Cher A, Pochan S, Herman P, Carrigan M, Sharp K, Huttenhower C, Lander ES, Vlamakis H, Xavier RJ, Yassour M, Delivery mode affects stability of early infant gut microbiota. Cell Rep. Med. 1, 100156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, Esaulova E, Artyomov MN, Morales DJ, Holtzman MJ, Boon ACM, Lenschow DJ, Stappenbeck TS, The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 357, 498–502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.DePasquale EAK, Schnell D, Dexheimer P, Ferchen K, Hay S, Chetal K, Valiente-Alandí Í, Blaxall BC, Grimes HL, Salomonis N, cellHarmony: Cell-level matching and holistic comparison of single-cell transcriptomes. Nucleic Acids Res. 47, e138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (accession number GSE176408). Processed data can be further explored at the Lung MAP atlas portal at https://research.cchmc.org/pbge/lunggens/lungExternal/Deshmukh_Macaque_2021.html. The reagents and metadata included in this manuscript are not covered by any material transfer agreement (MTA) and can be obtained from the corresponding author upon request. Scripts used for data analysis are available from GitHub at https://github.com/Deshmukh-Lab/2021_Stevens_Macaque (DOI: 10.5281/zenodo.6569295).