


Abstract
Most classical integrases of prokaryotic genetic elements specify integration into tRNA or tmRNA genes. Sequences shared between element and host integration sites suggest that crossover can occur at any of three sublocations within a tRNA gene, two with flanking symmetry (anticodon-loop and T-loop tDNA) and the third at the asymmetric 3′ end of the gene. Integrase phylogeny matches this classification: integrase subfamilies use exclusively either the symmetric sublocations or the asymmetric sublocation, although tRNA genes of several different aminoacylation identities may be used within any subfamily. These two familial sublocation preferences imply two modes by which new integration site usage evolves. The tmRNA gene has been adopted as an integration site in both modes, and its distinctive structure imposes some constraints on proposed evolutionary mechanisms.
INTRODUCTION
Genetic elements capable of horizontal transfer and integration into host chromosomes are key agents in the evolution of cellular genomes; bacterial pathogenicity in particular is shaped by integrative elements that can carry with them genes promoting virulence. Integration site specificity is determined primarily by the integrase enzyme typically encoded within elements of several types: temperate bacteriophages, integrative plasmids, pathogenicity islands and conjugative and mobilizable elements. Integrases have also been harnessed for stably inserting foreign genes into bacterial chromosomes in the construction of useful strains. It is therefore important to understand both the mechanism and evolution of integration site usage.
The longest studied integrase, from phage lambda, has the following prototypical properties (1): it (i) catalyzes integration and excision of a genetic element, (ii) has one highly preferred integration site in the host chromosome (attB), (iii) recombines segments of identical sequence within attB and the attP region in the element (2,3), and (iv) belongs to the tyrosine recombinase family. This large protein family, defined by sequence consensus in the C-terminal domain that includes invariant residues involved in catalysis (4,5), contains many members that do not fit the above criteria for a classical integrase; some are involved in resolution of chromosome or plasmid multimers, or shufflon or integron function, and yet other members recombine non-identical sites. Phylogenetic analysis of the tyrosine recombinases has revealed several subfamilies (4), but a robust phylogenetic tree has been difficult to construct, as might be expected for a functionally diverse protein family that often promotes mobility of its own genes.
The lambda integrase is heterobivalent; each molecule can simultaneously bind two types of DNA site, with partitioning among domains: the central domain binds the ‘core’-type site and the N-terminal domain binds the ‘arm’-type site (1). In both attP and attB, an inverted pair of core sites closely surrounds the 7-bp crossover segment where 5′ strands are exchanged. attB consists of little more than this arrangement (6); attP is much larger (>200 bp) because it also contains multiple arm sites, as well as sites for auxiliary proteins that bind and bend DNA. Bivalency allows integrase molecules to couple attB to attP during assembly of a catalytic complex, in which four integrase molecules act on the four DNA strands of the two crossover segments. There seems to be no particular sequence requirement within the crossover segment itself, but activity is severely depressed if the two att sites are not identical there (7). The pattern of core, arm and auxiliary sites in the attP–attB pair differs from that in the pair of hybrid sites attL and attR formed by integration, providing a basis for controlling the directionality (integration versus excision) of recombination.
Less well understood than how established sites are used is how new integration sites arise. Focusing on tRNA genes is useful, as they frequently serve as integration sites (8); indeed this survey shows that they do so in the majority of cases. The uniformity of tRNA genes thus provides a convenient format for comparing most integration sites. Usage of tRNA gene attBs is here sorted into four classes, according to sequence identity between element and host, which are presumed to use three different regions in tDNA as crossover sites. Two of these regions are marked by symmetry, employing tDNA for either the anticodon stem–loop or the T stem–loop. The third region, as originally noted in a study of the attB for phage mv4 (9), is located further 3′ and marked by asymmetry. A full switch to a new attB entails changes in both attP and the integrase gene, whose co-evolution is facilitated by their typically adjacent location in genetic elements. Accordingly, integrase phylogeny is compared with tRNA gene sublocation use, revealing that integrase subfamilies can mix the usage of the two symmetric regions, but that subfamilies using the asymmetric region use it exclusively. This correlation provokes discussion of the evolution of new integration site/integrase combinations.
USE OF tRNA AND tmRNA GENES AS INTEGRATION SITES
tRNA gene attB sites have been characterized at three levels: (i) in vitro analysis of integration at three tRNA gene attBs has identified 7-bp crossover segments where 5′ overhang strands are exchanged (10–12). (ii) Deletion analysis of five tRNA genes has defined minimal segments required for attB function (9–13); these are small (16–30 bp) relative to minimal attPs (>200 bp), and centered at identified crossover segments. (iii) Sequencing of integration site regions for the unintegrated and integrated states defines an uninterrupted block of sequence identity that includes known crossover segments. This identity block is essentially the portion of the tRNA gene that is displaced upon integration yet restored by a viral copy (Fig. 1).
Figure 1.

Integrase action at a tRNA gene. The crossover segment (solid box) where strands exchange is small; in this case it is precisely the 7-bp anticodon-loop tDNA, with anticodon stem tDNA serving as symmetrical integrase core-type sites (filled triangles), and with arm-type sites (open triangles) more distant (14). Blocks of sequence identity (brackets) between reacting DNAs usually extend from the crossover segment to or beyond the downstream end of the gene, and may also extend slightly in the upstream direction. tRNA gene function is retained after integration. For uniformity, the attL and attR designations used in this article refer to tRNA gene orientation as in this diagram and may not match those of previous descriptions of the integrated elements.
Some previously identified attB sites (for Gamma, TPW22 and bIL286) are recognized here as tRNA or tmRNA genes, and the assignment of the phage T12 attB as a tRNA-Ser gene (15) is corrected; it is instead the tmRNA gene (ssrA) of Streptococcus pyogenes. tmRNA is a bacterial RNA with some structural similarity to tRNA (Fig. 2), but a different physiological role (see below, ‘tmRNA gene usage’). Revisiting regions surrounding integrase genes in completed genome projects, endpoints in tRNA genes can be newly proposed for putative cryptic elements in Pseudomonas aeruginosa, Bacillus subtilis, Deinococcus radiodurans, Sinorhizobium meliloti and Thermoplasma acidophilum chromosomes; these elements are given names such as Pae12G (see Table 1) that reflect the host species, size in kilobase pairs and the aminoacylation identity of the tRNA gene used. The B.subtilis element had been annotated previously as φ2 (16) without definition of its endpoints, and Pae12G contains the genome of the filamentous phage Pf1 (17) but with substantial additions on either side that include the integrase gene.
Figure 2.
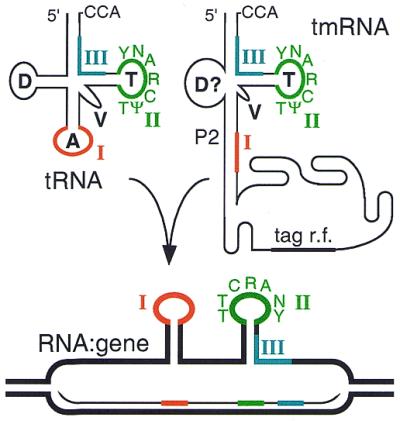
Secondary structures. The 7-nt anticodon and T loops of tRNAs are flanked by the symmetrical sequences that form 5-bp stems. The region corresponding to the tRNA anticodon is instead part of a long stem in tmRNA. For both RNA types, a similar structure might form in a hypothetical hybrid between RNA and gene if an analog of the anticodon stem–loop can fortuitously form in the tmRNA hybrid (gray shading in Fig. 3). T-loop consensus sequence is shown. Three presumed crossover sites used in both types of RNA gene are marked with Roman numerals.
Table 1. tDNA usage by prokaryotic genetic elements.

1Integrase subfamily assignments from the Tyrosine Recombinase Website (www.members.home.net/domespo/trhome.html), adding the new subfamilies NBU (53), SSV (28) and 16-3 (Fig. 3); s, unclassified singleton; ?, integrase sequence unavailable; *, assigned here, integrase absent from website.
2Violet, Proteobacteria; cyan, Gram-positive bacteria.
3tRNA identity in one-letter amino acid code; Z, selenocysteinyl tRNA; X, tmRNA.
4Orientation to int gene: distance in base pairs from discriminator position (see Fig. 3) in attP tDNA to int (negative numbering when int upstream of tDNA); Nterm or Cterm, inside int at N-terminal or C-terminal end; > or <, same or opposite orientation for tRNA fragment and int gene.
5Apparent rho-independent terminator: distance from tDNA discriminator position (see Fig. 3) to last non-T stem nucleotide of terminator; ?, none found within 400 bp; ND, none found within available sequence <200 bp; NA, not applicable, archaeal host or 5′ end duplication.
Examples of tRNA gene usage were considered redundant if the same tRNA gene was involved, and the pairwise distance score of the aligned integrase sequences (see Fig. 4 legend) was smaller than with any other integrase and less than one. In all, 61 unique examples of elements integrating specifically into prokaryotic tRNA or tmRNA genes are available (Table 1; Fig. 3). Five of these cases involve the tmRNA gene. Genes for Glu, Gln, His, Met, Trp and initiator tRNAs are absent from the list; it is not yet clear whether any of these are inherently unsuitable for use by genetic elements. Some integration events appear to damage the displaced portion of the original tRNA gene (Fig. 3, lines 38, 44, 60 and 61), making excision less favorable to the host because it would generate a non-functional gene; such behavior may be an adaptation of the associated integrase.
Figure 4.

Subfamilies of integrases recognizing tDNA or tmDNA. Fitch-Margoliash tree showing percent support (if >50%) for each node, from three phylogenetic analyses (order: Fitch-Margoliash, parsimony, quartet puzzling); tilde, <50% support; x, node absent. attB usage is color-coded: red, class IA; mustard, class IB; green, class II; blue, class III. Brackets mark subfamilies that were recognized previously, and the new 16-3 family; symmetry-preferring subfamilies are in gray and 3′-end-preferring subfamilies are in blue. Aminoacylation identities of attB tRNA genes are shown for subfamilies. *, Node included pSAM2 integrase. Parsimony supported nodes not present on this tree: Oi108 apart from all others (69%); HPI, she and clc (74%); 933I, φR73 and CPS-53 (61%); TPW22, Sfi21 and φ10MC (63%); and the pSE subfamily with φ2 and Dra18R (56%); none of these mix attB class usage. Methods: an alignment of the catalytic segment, corresponding to lambda integrase residues 202–345, of 43 integrases from Figure 3 (and Cre recombinase as an outgroup) was taken from the Tyrosine Recombinase Website, with some manual realignment and addition of 15 integrases absent from the website (final alignment available at sunflower.bio.indiana.edu/~kwilliam/tDNAint). One thousand bootstrap subsamples of this alignment were constructed by SEQBOOT, and trees for the subsamples were found using either FITCH (in a parallelized format implemented by Robert Cruise at Indiana University Information Technology Services) with distances evaluated by Blocks Substitution Matrix 62 or PROTPARS, each with 10 jumblings; majority-rule trees were taken using CONSENSE (65,66). Distance and branch length calculations and quartet puzzling were performed with PUZZLE (67).
Figure 3.

Sequence identity between attPs and attBs in tRNA and tmRNA genes. Genes are aligned according to the secondary structure of the encoded RNA, indicated above for tRNA and below for tmRNA (discriminator position marked by ampersand). attBs are ordered by the gene-internal endpoint of the identity block (underlined) shared with attP, as summarized on the bottom line. The length of continued rightward extension of the identity block is given. Yellow shading marks reported minimal attBs; cyan shading marks crossover segments that have been mapped (all three corresponding precisely to the anticodon loop); gray shading marks symmetry in tmRNA genes resembling anticodon stem–loop tDNA. Lower case marks terminal positions where the gene does not encode the full CCA tail of the mature RNA. Sequence data references not in Table 1: line 4 (16); line 5, E.coli tDNA (24) substitutes for unavailable sequence from natural host; line 17 (58); line 28, M.tuberculosis tDNA (59) substitutes for unavailable sequence from natural host; line 36 (60); lines 37 and 59 (61); line 51, Bacteroides fragilis genome project at The Sanger Centre (www.sanger.ac.uk); line 52 (62). HTML version available at sunflower.bio.indiana.edu/~kwilliam/tDNAint.
To address the question of how frequently integrases of the tyrosine recombinase family use tRNA or tmRNA genes as attB sites, the literature was searched for non-redundant cases where integration specificity was especially well determined (Table 2). Fifty-eight cases were identified, and for 34 of these (59%) the attB is in a tRNA or tmRNA gene. The integrase subfamily with the largest number of unique attBs (LC3, found in Gram-positive hosts) uses tDNA rather infrequently, and if this group is excluded, 75% of the remaining attBs are in tDNAs or tmDNAs. A recent survey of integration sites for pathogenicity islands alone (mostly from proteobacterial hosts) similarly concluded that 75% are in tRNA or tmRNA genes (63). Another indication of the prevalence of tRNA gene attBs comes from Escherichia coli O157:H7, which among currently available genomes has by far the highest known count of apparently intact integrase genes; all are within islands or prophages not present in E.coli MG1655, and 12 of these 20 are flanked by tRNA or tmRNA genes (23). Despite going unnoticed until 1987 (44), tRNA gene usage predominates among prokaryotic elements.
Table 2. tDNA or tmDNA usage frequency among integration sites that are well-determined (with at least three of the attP, attB, attL and attR sites sequenced) and recognized by an integrase of the tyrosine recombinase family.

INTEGRATION SITE SUBLOCATIONS WITHIN tRNA GENES
The sequence-identity block common to an attP–attB pair can be readily determined; it usually extends to, and sometimes well beyond, the 3′ end of the RNA gene (Fig. 1), which can be explained biologically by the need to retain function of the RNA gene after integration. Some of these identity blocks are quite long, implying an event during the evolution of new integration site/integrase combinations in which a segment of the host genome is captured by the attP of the element (64). The end of the identity block internal to the tRNA gene indicates with imprecision the location of strand crossover; identity might continue beyond the true crossover segment by one or a few positions, simply by chance, as a reflection of core site symmetry, or as a remnant from the original host DNA capture event. In Figure 2, tDNA attBs are ordered according to the gene-internal extent of the identity block, which suggests an organization into four classes: in classes IA and IB the identity block encompasses the anticodon loop; in class II it encompasses the T loop without extending into the variable region; in class III it is further 3′ and does not (or occasionally barely does) fully encompass the T loop. Class IB has only one member that provides the single exception to the rule that integrating elements replace the 3′ end of the gene they disrupt; it instead replaces the 5′ end of a tRNA gene (26).
The three tDNA attBs where strand exchange has been examined all fall into class IA and the crossover segments map precisely to the 7 bp encoding the anticodon loop (10,12,33). It has been proposed that this coincidence reflects a preference of the associated integrases for symmetry of flanking segments (a known preference of lambda integrase), which is assured in DNA encoding stem–loop RNA (8,10). The identity block for the class IB attB, although arriving from the 5′ end, also encompasses the anticodon-loop tDNA. The proposal for class II is based on its clear discontinuity from class I and slight discontinuity from the distribution of class III, and moreover on the observation that it fully includes the T loop; class II may therefore reflect integrase preference for flank symmetry centered at the 7-bp T loop, as do classes IA and IB at the anticodon loop.
In contrast, asymmetry was the notable characteristic of the minimal form of the class III attB for phage mv4, suggesting a mode of integrase-attB recognition differing from the symmetry-based lambda model; it was moreover recognized that several additional attB sites are similarly positioned in an asymmetrical setting at the far 3′ end of tRNA genes, quite distant from the anticodon region attBs (9). Crossover segments have not yet been determined for any members of class III, but it can be noted that all their identity blocks contain the same 7-bp stretch corresponding to the last 3 nt of the T stem, abutting 4 nt of the acceptor stem, and that this stretch is also at the center of the minimal class III attB determined for phage mv4. Thus, the study of attP–attB identity blocks tentatively delineates three tDNA attB sublocations, two characterized by their symmetry, and the third by its asymmetry. Determining crossover segments for attBs outside of class IA will be necessary to ascertain whether T-loop tDNA is in fact used by class II, and whether class III is a consistent group using a single precise position in tDNA.
CORRELATION OF INTEGRASE PHYLOGENY WITH attB SUBLOCATION
Although the class II identity block ends appear distinct from those of class III, they might sceptically be viewed as a skewed tail of the latter distribution (bottom line of Fig. 3). The possibility that class II could use the symmetrical T-loop tDNA, which would be impossible for almost all of class III, gives more credence to the distinction. Separate classification receives further support from the phylogeny of the associated integrases. Several subfamilies of related integrases emerge clearly from phylogenetic analysis of the tyrosine recombinase family (4,28,53); the most comprehensive public database of integrase sequence alignments and subfamily assignments is the Tyrosine Recombinase Website (www.members.home.net/domespo/trhome.html). Despite such success, a complete phylogenetic history of the family has not been established. The relationships between subfamilies are mostly ambiguous, and many singleton integrases are too divergent to place in any subfamily. Moreover, only the catalytic domain has been used in these analyses; complete alignment of the other domains, responsible for DNA-binding specificity, has not yet been achieved. Still, if domain shuffling among integrase genes has been infrequent, the phylogeny of the catalytic domain may adequately track relationships among the specificity domains.
An alignment of the catalytic domain sequence from the 58 integrases of Figure 3 for which sequences are available was used for three types of phylogenetic analysis: Fitch-Margoliash, parsimony and quartet puzzling. Figure 4 summarizes the analyses on the framework of the Fitch-Margoliash tree. Most previously described subfamilies were supported by all analyses. A new subfamily emerged clearly, comprising the integrases from φ16-3 and the recently recognized Mlo45V, with some support for inclusion of the D3 integrase. All subfamilies use genes of multiple tRNA identities.
Some subfamilies (P22, CTX, 16-3, SSV) exclusively use one of the attB sublocations characterized by symmetry (classes IA or II), but others (P2 and FRAT) mix the use of these two sublocations. These mixed-use subfamilies contain members known to promote strand exchange precisely at the 7 bp encoding the anticodon loop, strengthening the hypothesis that class II sites similarly exchange strands at the 7 bp encoding the T loop.
The status of the pSE subfamily is of interest because it may include the class IB integrase from Mlo38S, which is unique in replacing the 5′ portion of the tRNA gene into which it integrates. In the current release of the Tyrosine Recombinase Website, the Mlo38S integrase is absent, but its closest known BLAST relative (52% identity, from a segment of Bradyrhizobium japonicum DNA with the same attR as Mlo38S) (26) is included in the pSE subfamily along with the integrases from pSE101, pSE211 and Sco14R. Here, Mlo38S integrase was included in the pSE subfamily by parsimony analysis (along with the pSAM2 integrase), but the four-member pSE subfamily node occurred in only 44% of the Fitch-Margoliash bootstrap trees. The least intensive algorithm, quartet puzzling, which generally provided less support for other subfamilies, did not even support the core of this subfamily, pSE101 together with pSE211. Additional related integrase sequences will be necessary to determine conclusively whether the Mlo38S integrase is part of pSE subfamily; if so, it would be the only integrase subfamily involving hosts from more than one bacterial phylum, and would also link the unusual 5′ tDNA replacement by Mlo38S with the standard 3′ tDNA replacement by the other subfamily members.
Three subfamilies, P4, NBU and LC3, use exclusively the tDNA sublocation marked by asymmetry (class III). A pattern emerging from the phylogenetic analysis is that these latter subfamilies are segregated from those using exclusively the symmetrical sublocations (classes IA, IB or II, or mixtures). Some circularity should be admitted; for example, integrase phylogeny helped to sort the otherwise somewhat ambiguous attB of the element she (Fig. 3, line 38) into class III rather than class II; this in turn was used to tentatively sort Sme19T. What is not circular is that the ordered array of attB/attP identity blocks can be split (Fig. 3, class II/III border) so as to segregate the associated integrase subfamilies into two types. These two subfamily types, symmetry preferring and 3′ end preferring, imply two modes by which new integration site usage arises in tDNA.
Although the trees from the three analyses generally agreed on subfamily assignments, the rest of their branching patterns did not, and no nodes above the subfamily levels received significant levels of support by any analysis; therefore, such nodes in Figure 3 should be discounted and it is premature to discuss order or multiplicity for the emergence of sublocation symmetry preference.
Almost every integrase subfamily within the tyrosine recombinase family contains members using tRNA gene att sites (Table 2). The lambda subfamily can now be included in this group because the island 933M of the E.coli O157:H7 genome appears to use a class IA site in a tRNA gene (Table 1, line 7) and encode an early-branching member of the lambda subfamily (data not shown). The high frequency of tRNA gene use and its dispersion among integrase subfamilies suggests viewing the tDNA fraction of the genome as the true crucible where new site specificity evolves; non-tDNA sites may arise mainly from corruption of original tDNA site usage.
EVOLUTION OF NEW INTEGRATION SITE USAGE
Three general sorts of explanations, not mutually exclusive, can be proposed for how tRNA and tmRNA genes, which comprise <2% of bacterial genomes, come to serve so frequently as integration sites. (i) Drift: sequences similar to an established tDNA attB are most likely to be found in another tRNA gene. (ii) Selection: new site specificity can arise for virtually any chromosomal locus, but most sites do not serve the biology of genetic elements as well as do tRNA genes. (iii) Generic recognition: integrases recognize features that are generic for tRNA genes yet distinctive with respect to non-tDNA. How do the symmetry seeking and 3′ end seeking modes fit with these explanations?
The idea of sequence drift can be dispensed with first: sequences at a particular sublocation within tRNA genes may be so similar that a few mutations in attP can create a match to a new tRNA gene at the same sublocation. The best case in Figure 3 for this argument is the pair of elements pSE211 and pSE101 which function in the same host at class IA attB sites in different tRNA genes, using closely related integrases; the two anticodon stem–loop tDNAs have 16-bp blocks that are identical except for a 3-bp segment within the presumable crossover segment. With an integrase trained on one of these genes, a relatively small change in the attP might allow the element to switch and use the other tRNA gene. This sort of explanation does not seem broadly applicable, because the attB sequences used within integrase subfamilies do not generally appear closely related. For example, the class III region used by the LC3 subfamily varies from purine-rich to pyrimidine-rich among the tRNA genes recognized. However, there may have been pathways of gene switching that are obscured by the incompleteness of the data set.
Although the intact tRNA genes occasionally found within phage genomes are usually thought to improve decoding of phage mRNAs, they have also been proposed to play a role in the evolution of tRNA gene integration site usage through homologous recombination (68), but no specific scenarios have been described.
SELECTION FOR tRNA GENE INTEGRATION SITES
Two factors can be mentioned as favoring tRNA gene sites a priori over protein-coding genes, which typically comprise 85–90% of the bacterial genome. One factor is their reliability (68); combining data of Lynch (69) and Ochman et al. (70) allows the estimate that among bacteria, the sequence divergence rate per base pair for tRNA genes is from 4- to 9-fold (average, 6-fold) lower than for protein-coding genes. The stability of an attB sequence in tDNA may broaden host range or improve long-term survival prospects of a genetic element. A second factor favoring tRNA genes as integration sites is that their small size minimizes the amount of host DNA that must be captured in attP in order to restore the target gene upon integration. From most points within a protein-coding gene or operon, restoration would require the capture of an impractically large host fragment in attP.
Specific proposals have been made for benefits to molecular events in the life cycle of the genetic element from association with a tRNA gene. One is that an element may insert into a gene for a tRNA species decoding a codon that is more abundant in the element than in the host; this proposal has been applied to only one case (63). Another possible benefit could be transcriptional coupling of the integrated element to the tRNA gene, which would allow it to monitor the physiological state of the cell, as tRNA promoters are typically regulated by growth rate (71). However, there is not much regularity in the orientation of prophages to tRNA genes, and in half, or more, of the cases the attP carries with it an apparently strong rho-independent terminator that would act to sever this transcriptional connection (Table 1, columns ‘Orient. int’ and ‘Term.’). The tRNA gene setting might directly affect integrase function or the directionality of recombination in a way that is beneficial for genetic elements. It has been proposed that mature tRNAs occasionally hybridize to their own genes, commencing with the free 3′ CCA tail, and that the hybrid structure might somehow improve integrase action (72). The hypothesis has some problems: CCA tails added after transcription should not initiate hybridization to the 33% of attB tRNA genes that do not encode the tail (Fig. 3), and tDNA attBs are utilized efficiently (in vitro) by integrase in the absence of tRNA.
Whatever selective benefits there may be from tDNA location, coupling them with the typical integrase preference for symmetrical sites (8,10) may suffice to explain the familial use of the classes I and II attB sites. Familial use of the asymmetric class III sites is less obviously explained by selection alone; one possibility is that it may be more difficult to capture long DNA segments than short ones as 3′ gene ends, so that in an integrase subfamily with relaxed symmetry preference, we observe a statistical distribution of capture events that were as short as possible. This hypothesis would predict a heterogeneous pattern of positions for class III crossover segments, which makes their mapping more urgent.
HYPOTHESIS: GENERIC tDNA RECOGNITION
One interpretation of the familial use by integrases of the class III position, despite its lack of symmetry or sequence conservation, is that these integrases can actively recognize that particular sublocation within virtually any tRNA gene during the evolution of new integration site usage. Even the symmetry-preferring integrases could be directed generically to T and anticodon tDNA by some feature that marks tRNA genes and their sublocations.
What might mark tRNA gene sublocations so that integrases could find them? One model for tDNA marking would be the eukaryotic transcription factor IIIC which binds tRNA genes directly and specifically based on their conserved primary sequence features, found primarily at D and T stem–loop tDNA (73). No bacterial protein that acts likewise has been described.
Shape may accompany sequence as a marking principle. Following a proposal of Hou (72), transcription of tRNA genes (or post-transcriptional hybridization of mature tRNAs) could generate distinctive structures that would direct integrase to the DNA. One possibility is inspired by the behavior of RNA polymerase when it transcribes the primer for ColE1 plasmid replication. This RNA forms a special structure as it emerges from the polymerase, which at a certain point prevents the transcription bubble from collapsing behind the advancing polymerase, so that a persistent hybrid forms between transcript and template (74). This example may be a high-frequency form of behavior that occurs at low frequency for tRNA (or any) transcripts. Such template:transcript hybrids would be favored in a more negatively supercoiled domain of DNA, yet short-lived due to susceptibility to ribonuclease H. For a tRNA:tDNA hybrid, the opposite DNA strand would be free to present a distinctive pattern of both conserved primary sequence and secondary structure corresponding to the stem–loops of tRNA (Fig. 2). The strong bias observed in the attP capture of 3′ gene ends rather than 5′ ends might be partly established in the asymmetry of such a hybrid structure.
With such a hypothetical mechanism for reliably finding new tRNA genes as integration sites, benefits to the biology of genetic elements and their hosts arise from another feature of tRNA genes: their abundance in aggregate among genomes. This availability of many different tRNA genes in a host allows combinatorial acquisition of different genetic elements, without interference.
tmRNA GENE USAGE
The tmRNA gene has not been found in more than a single copy in any genome, yet it is used as attB as frequently as any tRNA gene, by members of four different integrase subfamilies at all three tDNA sublocations (Table 1). Its function is not that of a classical tRNA; although it is charged with alanine and the ribosome transfers that moiety to a nascent peptide, tmRNA does not read any codon (75). Rather, it is considered to solve problems arising from ribosomes that have stalled during translation (perhaps at rare codons or at the end of mRNAs that have no stop codon). tmRNA contains a reading frame that is translated, adding a peptide tag to the incomplete protein in the stalled ribosome, after transfer of its alanyl moiety and exchange with the troublesome mRNA. Translation continues to the stop codon in tmRNA, which rescues the stalled ribosome, and the tag targets the protein product for proteolysis. The tmRNA gene shows that prokaryotic genetic elements are not constrained to genes that provide classical tRNA function.
The distinctive structure of tmRNA (76) and its gene constrains hypothetical mechanisms of generic tDNA recognition. One of the arms of the tRNA L-shape, containing the acceptor stem and T stem–loop, probably forms similarly in tmRNA, while the other arm containing the D and anticodon stem–loops would have to differ in tmRNA (Fig. 2). No stem is apparent in the region equivalent to the D stem–loop, and the equivalent of the anticodon stem is elongated into an interrupted stem (P2) of ∼20 bp that is capped by a further-structured giant loop of ∼250 nt. Thus, at the 3′ end of the tmRNA gene, strict analogy to tRNA genes ends upstream of the T stem–loop. The features of the D stem–loop sequence that are conserved among tRNA genes cannot be found in tmRNA genes and should therefore not be included in any mechanism proposed to attract integrases.
One integrase of the P2 subfamily, another member of which is known to use the 7-bp anticodon loop tDNA as its crossover segment (10), exhibits apparent class IA usage of a tmRNA gene (Fig. 3, line 18) despite the absence of an equivalent to anticodon loop tDNA. However, dyad symmetry can still be noted (gray shading in Fig. 3) in the corresponding region of this tmRNA gene, which may satisfy integrase symmetry preference even though not expressed as stem–loop structures in the mature tmRNA. Selection of an imperfect mimic of anticodon stem–loop tDNA due to its position relative to true T stem–loop tDNA would tend to support the hypothesis of generic tDNA recognition. Similar dyads can be proposed for all the tmRNA genes known to serve as attBs, such that they still might form the same RNA–gene hybrid structure proposed for tRNAs (Fig. 2).
CONCLUSIONS
The high frequency at which tRNA genes are adopted as integration sites raises the question of how elements return to this class of genes. The striking outcome of this survey, that integrase subfamilies use characteristic sublocations within tRNA genes, poses a refined question: how do integrase clades direct the return to the same sublocation at many different tRNA genes? Although other explanations are possible, the correlation suggests that integrases may recognize some generic feature or form of tRNA genes before exercising their particular positional preference.
The evolution of new integration site usage may truly be beyond the reach of experimentation; its frequency is low, and it may depend on pre-marking of tRNA genes by events that occur at low frequency. Still, some accessible avenues should be explored further. (i) Crossover segments must be determined for more attBs, especially those of classes IB, II and III, for which there are yet no data; the crossover segments will provide a better basis for attB positional classification than the identity blocks used here. (ii) Investigating mechanisms for integrases that use class III sites could reveal how they apparently function without the symmetry preferences that have been established for lambda and other integrases. (iii) It may become possible to align integrase sequences outside of the catalytic domain, and find mechanistically relevant correlations between tRNA gene sublocation and the integrase domains responsible for DNA specificity. (iv) The binding properties of integrases for tRNA, single-stranded tDNA, or tRNA–tDNA hybrids may prove interesting.
The tRNA gene habit is not obligate among integration systems (a few members of the resolvase family of site-specific recombinases are known to provide integrase function for genetic elements, all at non-tDNA sites), but it is exhibited by elements of both prokaryotes and eukaryotes. Eukaryotic retroelements that target tRNA genes target them (together with other genes for Pol III-transcribed RNAs) collectively, to sites outside the mature RNA-coding sequence, and the elements are found as numerous repeats within a genome (77,78); in the best-studied case, targeting is based on a simple principle in which the genes are marked by binding of a Pol III transcription factor (79). Prokaryotic integrases target particular tRNA genes, one at a time, but may have the ability to move on to new tRNA genes at breakthrough points in their evolutionary history. This system allows genetic elements, often promoting survival of their hosts, to accrue in a combinatorial fashion: a genome will not be flooded by any one element, but rather could harbor a large number of different elements; witness E.coli O157:H7. The prokaryotic and eukaryotic systems are overwhelmingly different (no homology relationships have been detected between their integrases), but may derive similar benefits from their integration specificity. tRNA genes are numerous, reliable, uniform as a class yet distinctive in relation to the rest of the genome and they can be used innocuously.
Integrases may preserve an ancient but still profitable strategy of association; in some views of early evolution, the translational apparatus developed prior to the use of DNA as the genomic material, such that tRNA genes would have been uniform and numerous in the earliest DNA genomes. Continued success of an integration-specificity principle could have led to competition among genetic elements for tRNA genes, driving a subdivision of the niche through segregation of sublocation usage.
NOTE ADDED IN PROOF
Wassarman et al. (80) show that the phage P2 integration site in the E.coli chromosome is at the 3′ end of the gene for a small RNA (of unknown function but conserved among enterobacteria), in the inverted repeat encoding its apparent rho-independent terminator. This is the only example of an attB in a gene encoding something other than tRNA, tmRNA or protein, and points to generality in the correspondence of integration sites with conserved portions at the 3′ ends of transcripts.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by NIH grant GM59881.
REFERENCES
- 1.Azaro M.A. and Landy,A. (2002) λ Integrase and the λ Int family. In Craig,N.L., Craigie,R., Gellert,M. and Lambowitz,A. (eds), Mobile DNA II. ASM Press, Washington DC, 118–148.
- 2.Campbell A.M. (1962) Episomes. Adv. Genet., 11, 101–145. [Google Scholar]
- 3.Weisberg R.A. and Landy,A. (1983) Site-specific recombination in phage lambda. In Hendrix,R.W., Roberts,J.W., Stahl,F.W. and Weisberg,R.A. (eds), Lambda II. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 211–250.
- 4.Esposito D. and Scocca,J.J. (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res., 25, 3605–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nunes-Duby S.E., Kwon,H.J., Tirumalai,R.S., Ellenberger,T. and Landy,A. (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res., 26, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuuchi M. and Mizuuchi,K. (1985) The extent of DNA sequence required for a functional bacterial attachment site of phage lambda. Nucleic Acids Res., 13, 1193–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer C.E., Gardner,J.F. and Gumport,R.I. (1985) Extent of sequence homology required for bacteriophage lambda site-specific recombination. J. Mol. Biol., 181, 187–197. [DOI] [PubMed] [Google Scholar]
- 8.Reiter W.D., Palm,P. and Yeats,S. (1989) Transfer RNA genes frequently serve as integration sites for prokaryotic genetic elements. Nucleic Acids Res., 17, 1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auvray F., Coddeville,M., Ordonez,R.C. and Ritzenthaler,P. (1999) Unusual structure of the attB site of the site-specific recombination system of Lactobacillus delbrueckii bacteriophage mv4. J. Bacteriol., 181, 7385–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hauser M.A. and Scocca,J.J. (1992) Site-specific integration of the Haemophilus influenzae bacteriophage HP1. Identification of the points of recombinational strand exchange and the limits of the host attachment site. J. Biol. Chem., 267, 6859–6864. [PubMed] [Google Scholar]
- 11.Pena C.E., Stoner,J.E. and Hatfull,G.F. (1996) Positions of strand exchange in mycobacteriophage L5 integration and characterization of the attB site. J. Bacteriol., 178, 5533–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith-Mungo L., Chan,I.T. and Landy,A. (1994) Structure of the P22 att site. Conservation and divergence in the lambda motif of recombinogenic complexes. J. Biol. Chem., 269, 20798–20805. [PubMed] [Google Scholar]
- 13.Raynal A., Tuphile,K., Gerbaud,C., Luther,T., Guerineau,M. and Pernodet,J.L. (1998) Structure of the chromosomal insertion site for pSAM2: functional analysis in Escherichia coli. Mol. Microbiol., 28, 333–342. [DOI] [PubMed] [Google Scholar]
- 14.Pena C.E., Kahlenberg,J.M. and Hatfull,G.F. (2000) Assembly and activation of site-specific recombination complexes. Proc. Natl Acad. Sci. USA, 97, 7760–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McShan W.M., Tang,Y.F. and Ferretti,J.J. (1997) Bacteriophage T12 of Streptococcus pyogenes integrates into the gene encoding a serine tRNA. Mol. Microbiol., 23, 719–728. [DOI] [PubMed] [Google Scholar]
- 16.Kunst F., Ogasawara,N., Moszer,I., Albertini,A.M., Alloni,G., Azevedo,V., Bertero,M.G., Bessieres,P., Bolotin,A., Borchert,S. et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- 17.Hill D.F., Short,N.J., Perham,R.N. and Petersen,G.B. (1991) DNA sequence of the filamentous bacteriophage Pf1. J. Mol. Biol., 218, 349–364. [DOI] [PubMed] [Google Scholar]
- 18.Gabriel K., Schmid,H., Schmidt,U. and Rausch,H. (1995) The actinophage RP3 DNA integrates site-specifically into the putative tRNA(Arg)(AGG) gene of Streptomyces rimosus. Nucleic Acids Res., 23, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cianciotto N., Serwold-Davis,T., Groman,N., Ratti,G. and Rappuoli,R. (1990) DNA sequence homology between attB-related sites of Corynebacterium diphtheriae, Corynebacterium ulcerans, Corynebacterium glutamicum and the attP site of gamma-corynephage. FEMS Microbiol. Lett., 54, 299–301. [DOI] [PubMed] [Google Scholar]
- 20.Brasch M.A., Pettis,G.S., Lee,S.C. and Cohen,S.N. (1993) Localization and nucleotide sequences of genes mediating site-specific recombination of the SLP1 element in Streptomyces lividans. J. Bacteriol., 175, 3067–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hochhut B., Jahreis,K., Lengeler,J.W. and Schmid,K. (1997) CTnscr94, a conjugative transposon found in enterobacteria. J. Bacteriol., 179, 2097–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bar-Nir D., Cohen,A. and Goedeke,M.E. (1992) tDNA(ser) sequences are involved in the excision of Streptomyces griseus plasmid pSG1. Gene, 122, 71–76. [DOI] [PubMed] [Google Scholar]
- 23.Perna N.T., Plunkett,G., Burland,V., Mau,B., Glasner,J.D., Rose,D.J., Mayhew,G.F., Evans,P.S., Gregor,J., Kirkpatrick,H.A. et al. (2001) Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature, 409, 529–533. [DOI] [PubMed] [Google Scholar]
- 24.Blattner F.R., Plunkett,G., Bloch,C.A., Perna,N.T., Burland,V., Riley,M., Collado-Vides,J., Glasner,J.D., Rode,C.K., Mayhew,G.F. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- 25.Papp I., Dorgai,L., Papp,P., Jonas,E., Olasz,F. and Orosz,L. (1993) The bacterial attachment site of the temperate Rhizobium phage 16-3 overlaps the 3′ end of a putative proline tRNA gene. Mol. Gen. Genet., 240, 258–264. [DOI] [PubMed] [Google Scholar]
- 26.Zhou S. and Williams,K.P. (2002) An integrative genetic element that reverses the usual target gene orientation. J. Bacteriol., 184, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kropinski A.M. (2000) Sequence of the genome of the temperate, serotype-converting, Pseudomonas aeruginosa bacteriophage D3. J. Bacteriol., 182, 6066–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng X., Holz,I., Zillig,W., Garrett,R.A. and She,Q. (2000) Evolution of the family of pRN plasmids and their integrase-mediated insertion into the chromosome of the crenarchaeon Sulfolobus solfataricus. J. Mol. Biol., 303, 449–454. [DOI] [PubMed] [Google Scholar]
- 29.Muskhelishvili G., Palm,P. and Zillig,W. (1993) SSV1-encoded site-specific recombination system in Sulfolobus shibatae. Mol. Gen. Genet., 237, 334–342. [DOI] [PubMed] [Google Scholar]
- 30.Van Mellaert L., Mei,L., Lammertyn,E., Schacht,S. and Anne,J. (1998) Site-specific integration of bacteriophage VWB genome into Streptomyces venezuelae and construction of a VWB-based integrative vector. Microbiology, 144, 3351–3358. [DOI] [PubMed] [Google Scholar]
- 31.Uchiumi T., Abe,M. and Higashi,S. (1998) Integration of the temperate phage phiU into the putative tRNA gene on the chromosome of its host Rhizobium leguminosarum biovar trifolii. J. Gen. Appl. Microbiol., 44, 93–99. [DOI] [PubMed] [Google Scholar]
- 32.Reed M.R., Shearwin,K.E., Pell,L.M. and Egan,J.B. (1997) The dual role of Apl in prophage induction of coliphage 186. Mol. Microbiol., 23, 669–681. [DOI] [PubMed] [Google Scholar]
- 33.Pena C.E., Stoner,J. and Hatfull,G.F. (1998) Mycobacteriophage D29 integrase-mediated recombination: specificity of mycobacteriophage integration. Gene, 225, 143–151. [DOI] [PubMed] [Google Scholar]
- 34.Brown D.P., Idler,K.B. and Katz,L. (1990) Characterization of the genetic elements required for site-specific integration of plasmid pSE211 in Saccharopolyspora erythraea. J. Bacteriol., 172, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown D.P., Idler,K.B., Backer,D.M., Donadio,S. and Katz,L. (1994) Characterization of the genes and attachment sites for site-specific integration of plasmid pSE101 in Saccharopolyspora erythraea and Streptomyces lividans. Mol. Gen. Genet., 242, 185–193. [DOI] [PubMed] [Google Scholar]
- 36.Lindsey D.F., Mullin,D.A. and Walker,J.R. (1989) Characterization of the cryptic lambdoid prophage DLP12 of Escherichia coli and overlap of the DLP12 integrase gene with the tRNA gene argU. J. Bacteriol., 171, 6197–6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vrijbloed J.W., Madon,J. and Dijkhuizen,L. (1994) A plasmid from the methylotrophic actinomycete Amycolatopsis methanolica capable of site-specific integration. J. Bacteriol., 176, 7087–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiewitz C., Larbig,K., Klockgether,J., Weinel,C. and Tummler,B. (2000) Monitoring genome evolution ex vivo: reversible chromosomal integration of a 106 kb plasmid at two tRNA(Lys) gene loci in sequential Pseudomonas aeruginosa airway isolates. Microbiology, 146, 2365–2373. [DOI] [PubMed] [Google Scholar]
- 39.Freitas-Vieira A., Anes,E. and Moniz-Pereira,J. (1998) The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNA(Ala) gene of Mycobacterium spp. Microbiology, 144, 3397–3406. [DOI] [PubMed] [Google Scholar]
- 40.Hendrix R.W., Smith,M.C., Burns,R.N., Ford,M.E. and Hatfull,G.F. (1999) Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc. Natl Acad. Sci. USA, 96, 2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tojo N., Sanmiya,K., Sugawara,H., Inouye,S. and Komano,T. (1996) Integration of bacteriophage Mx8 into the Myxococcus xanthus chromosome causes a structural alteration at the C-terminal region of the IntP protein. J. Bacteriol., 178, 4004–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z., Xiong,G. and Lutz,F. (1995) Site-specific integration of the phage phi CTX genome into the Pseudomonas aeruginosa chromosome: characterization of the functional integrase gene located close to and upstream of attP. Mol. Gen. Genet., 246, 72–79. [DOI] [PubMed] [Google Scholar]
- 43.Al-Hasani K., Rajakumar,K., Bulach,D., Robins-Browne,R., Adler,B. and Sakellaris,H. (2001) Genetic organization of the she pathogenicity island in Shigella flexneri 2a. Microbial Pathogen., 30, 1–8. [DOI] [PubMed] [Google Scholar]
- 44.Pierson L.S. and Kahn,M.L. (1987) Integration of satellite bacteriophage P4 in Escherichia coli. DNA sequences of the phage and host regions involved in site-specific recombination. J. Mol. Biol., 196, 487–496. [DOI] [PubMed] [Google Scholar]
- 45.Sun J., Inouye,M. and Inouye,S. (1991) Association of a retroelement with a P4-like cryptic prophage (retronphage phi R73) integrated into the selenocystyl tRNA gene of Escherichia coli. J. Bacteriol., 173, 4171–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez M.A., Herrero,M. and Suarez,J.E. (1998) The site-specific recombination system of the Lactobacillus species bacteriophage A2 integrates in gram-positive and gram-negative bacteria. Virology, 250, 185–193. [DOI] [PubMed] [Google Scholar]
- 47.Ravatn R., Studer,S., Zehnder,A.J. and van der Meer,J.R. (1998) Int-B13, an unusual site-specific recombinase of the bacteriophage P4 integrase family, is responsible for chromosomal insertion of the 105-kilobase clc element of Pseudomonas sp. Strain B13. J. Bacteriol., 180, 5505–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sullivan J.T. and Ronson,C.W. (1998) Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc. Natl Acad. Sci. USA, 95, 5145–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chopin A., Bolotin,A., Sorokin,A., Ehrlich,S.D. and Chopin,M.C. (2001) Analysis of six prophages in Lactococcus lactis IL1403: different genetic structure of temperate and virulent phage populations. Nucleic Acids Res., 29, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gindreau E., Torlois,S. and Lonvaud-Funel,A. (1997) Identification and sequence analysis of the region encoding the site-specific integration system from Leuconostoc oenos (Oenococcus oeni) temperate bacteriophage phi 10MC. FEMS Microbiol. Lett., 147, 279–285. [DOI] [PubMed] [Google Scholar]
- 51.Rakin A., Noelting,C., Schropp,P. and Heesemann,J. (2001) Integrative module of the high-pathogenicity island of Yersinia. Mol. Microbiol., 39, 407–416. [DOI] [PubMed] [Google Scholar]
- 52.Shoemaker N.B., Wang,G.R. and Salyers,A.A. (1996) The Bacteroides mobilizable insertion element, NBU1, integrates into the 3′ end of a Leu-tRNA gene and has an integrase that is a member of the lambda integrase family. J. Bacteriol., 178, 3594–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J., Shoemaker,N.B., Wang,G.R. and Salyers,A.A. (2000) Characterization of a Bacteroides mobilizable transposon, NBU2, which carries a functional lincomycin resistance gene. J. Bacteriol., 182, 3559–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petersen A., Josephsen,J. and Johnsen,M.G. (1999) TPW22, a lactococcal temperate phage with a site-specific integrase closely related to Streptococcus thermophilus phage integrases. J. Bacteriol., 181, 7034–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruttin A., Foley,S. and Brussow,H. (1997) The site-specific integration system of the temperate Streptococcus thermophilus bacteriophage phiSfi21. Virology, 237, 148–158. [DOI] [PubMed] [Google Scholar]
- 56.Cheetham B.F., Tattersall,D.B., Bloomfield,G.A., Rood,J.I. and Katz,M.E. (1995) Identification of a gene encoding a bacteriophage-related integrase in a vap region of the Dichelobacter nodosus genome. Gene, 162, 53–58. [DOI] [PubMed] [Google Scholar]
- 57.Karaolis D.K., Johnson,J.A., Bailey,C.C., Boedeker,E.C., Kaper,J.B. and Reeves,P.R. (1998) A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl Acad. Sci. USA, 95, 3134–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White O., Eisen,J.A., Heidelberg,J.F., Hickey,E.K., Peterson,J.D., Dodson,R.J., Haft,D.H., Gwinn,M.L., Nelson,W.C., Richardson,D.L. et al. (1999) Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science, 286, 1571–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cole S.T., Brosch,R., Parkhill,J., Garnier,T., Churcher,C., Harris,D., Gordon,S.V., Eiglmeier,K., Gas,S., Barry,C.E.,3rd et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature, 393, 537–544. [DOI] [PubMed] [Google Scholar]
- 60.Stover C.K., Pham,X.Q., Erwin,A.L., Mizoguchi,S.D., Warrener,P., Hickey,M.J., Brinkman,F.S., Hufnagle,W.O., Kowalik,D.J., Lagrou,M. et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature, 406, 959–964. [DOI] [PubMed] [Google Scholar]
- 61.Galibert F., Finan,T.M., Long,S.R., Puhler,A., Abola,P., Ampe,F., Barloy-Hubler,F., Barnett,M.J., Becker,A., Boistard,P. et al. (2001) The composite genome of the legume symbiont Sinorhizobium meliloti. Science, 293, 668–672. [DOI] [PubMed] [Google Scholar]
- 62.Ruepp A., Graml,W., Santos-Martinez,M.L., Koretke,K.K., Volker,C., Mewes,H.W., Frishman,D., Stocker,S., Lupas,A.N. and Baumeister,W. (2000) The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature, 407, 508–513. [DOI] [PubMed] [Google Scholar]
- 63.Hacker J. and Kaper,J.B. (2000) Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol., 54, 641–679. [DOI] [PubMed] [Google Scholar]
- 64.Campbell A.M. (1992) Chromosomal insertion sites for phages and plasmids. J. Bacteriol., 174, 7495–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Felsenstein J. (1995) PHYLIP (Phylogeny Inference Package), version 3.57c. Distributed by the author. Department of Genetics, University of Washington, Seattle.
- 66.Henikoff S. and Henikoff,J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Strimmer K. and von Haeseler,A. (1996) Quartet puzzling: a quartet maximum likelihood method for reconstructing tree topologies. Mol. Biol. Evol., 13, 964–969. [Google Scholar]
- 68.Cheetham B.F. and Katz,M.E. (1995) A role for bacteriophages in the evolution and transfer of bacterial virulence determinants. Mol. Microbiol., 18, 201–208. [DOI] [PubMed] [Google Scholar]
- 69.Lynch M. (1997) Mutation accumulation in nuclear, organelle and prokaryotic transfer RNA genes. Mol. Biol. Evol., 14, 914–925. [DOI] [PubMed] [Google Scholar]
- 70.Ochman H., Elwyn,S. and Moran,N.A. (1999) Calibrating bacterial evolution. Proc. Natl Acad. Sci. USA, 96, 12638–12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Swenson D.L., Kim,K.J., Six,E.W. and Clegg,S. (1994) The gene fimU affects expression of Salmonella typhimurium type 1 fimbriae and is related to the Escherichia coli tRNA gene argU. Mol. Gen. Genet., 244, 216–218. [DOI] [PubMed] [Google Scholar]
- 72.Hou Y.M. (1999) Transfer RNAs and pathogenicity islands. Trends Biochem. Sci., 24, 295–298. [DOI] [PubMed] [Google Scholar]
- 73.Klemenz R., Stillman,D.J. and Geiduschek,E.P. (1982) Specific interactions of Saccharomyces cerevisiae proteins with a promoter region of eukaryotic tRNA genes. Proc. Natl Acad. Sci. USA, 79, 6191–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Masukata H. and Tomizawa,J. (1990) A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell, 62, 331–338. [DOI] [PubMed] [Google Scholar]
- 75.Karzai A.W., Roche,E.D. and Sauer,R.T. (2000) The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nature Struct. Biol., 7, 449–455. [DOI] [PubMed] [Google Scholar]
- 76.Williams K.P. and Bartel,D.P. (1996) Phylogenetic analysis of tmRNA secondary structure. RNA, 2, 1306–1310. [PMC free article] [PubMed] [Google Scholar]
- 77.Kim J.M., Vanguri,S., Boeke,J.D., Gabriel,A. and Voytas,D.F. (1998) Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res., 8, 464–478. [DOI] [PubMed] [Google Scholar]
- 78.Szafranski K., Glockner,G., Dingermann,T., Dannat,K., Noegel,A.A., Eichinger,L., Rosenthal,A. and Winckler,T. (1999) Non-LTR retrotransposons with unique integration preferences downstream of Dictyostelium discoideum tRNA genes. Mol. Gen. Genet., 262, 772–780. [DOI] [PubMed] [Google Scholar]
- 79.Yieh L., Kassavetis,G., Geiduschek,E.P. and Sandmeyer,S.B. (2000) The Brf and TATA-binding protein subunits of the RNA polymerase III transcription factor IIIB mediate position-specific integration of the gypsy-like element, Ty3. J. Biol. Chem., 275, 29800–29807. [DOI] [PubMed] [Google Scholar]
- 80.Wassarman K.M., Repoila,F., Rosenow,C., Storz,G. and Gottesman,S. (2001) Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev., 15, 1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]