

Abstract
Alterations of the proto-oncogene MLL (mixed lineage leukemia) are characteristic for a high proportion of acute leukemias, especially those occurring in infants. The activation of MLL is achieved either by an internal tandem duplication of 5′ MLL exons or by chromosomal translocations that create chimeric proteins with the N-terminus of MLL fused to a variety of different partner proteins. A domain of MLL with significant homology to the eukaryotic DNA methyltransferases (MT domain) has been found to be essential for the transforming potential of the oncogenic MLL derivatives. Here we demonstrate that this domain specifically recognizes DNA with unmethylated CpG sequences. In gel mobility shifts, the presence of CpG was sufficient for binding of recombinant GST–MT protein to DNA. The introduction of 5-methylCpG on one or both DNA strands precluded an efficient interaction. In surface plasmon resonance a KD of ∼3.3 × 10–8 M was determined for the GST–MT/DNA complex formation. Site selection experiments and DNase I footprinting confirmed CpG as the target of the MT domain. Finally, this interaction was corroborated in vivo in reporter assays utilizing the DNA-binding properties of the MT domain in a hybrid MT–VP16 transactivator construct.
INTRODUCTION
The mixed lineage leukemia (MLL) gene at the genomic locus 11q23 is a frequent target of chromosomal aberrations. These events activate the proto-oncogene MLL and are associated with an especially aggressive class of leukemias. 11q23 abnormalities are most prevalent in acute lymphoblastic and acute myeloid leukemias during the infant age and in therapy-related secondary myeloid leukemias after a previous treatment with topoisomerase II inhibitors (reviewed in 1,2).
The unaltered MLL gene codes for a large 3968 amino acid protein with limited but significant homology to Drosophila trithorax (TRX) (3). In the fly TRX is the prototype member of a large genetically defined group of proteins (TRX-G) with a positive influence on transcription. TRX-G members are responsible for epigenetic gene regulation by establishing a transcriptionally active, ‘open’ state of chromatin that can be inherited through cell division. Thus, TRX is necessary for the continued maintenance, but not initiation, of a characteristic temporal and spatial expression pattern of the Hox-cluster genes (HOM-C) (reviewed in 4–6).
A similar function was demonstrated for MLL in mouse knockout studies. While MLL–/– embryos initially were able to establish normal Hox expression domains, they could not maintain Hox gene transcription at later stages and died around day 9.5 of embryonic development (7–9). Support for the role of MLL as a regulator of chromatin architecture comes from various biochemical studies that demonstrate a direct physical interaction of MLL with members of the chromatin remodeling machinery. A highly conserved SET domain at the very C-terminus of MLL (Fig. 1A) mediates the association of MLL with INI-1, a component of the human SWI/SNF chromatin-remodeling complex (10). Another region within the C-terminal portion of MLL was originally identified as a transactivation domain in a heterologous fusion with the GAL4 DNA-binding domain (11,12). Recent reports show that this part of MLL binds to the histone deacetylase and nuclear co-activator CREB-binding protein (CBP) (13).
Figure 1.

Structure of MLL, the MT domain and purification of GST–MT protein. (A) Graphical representation (not to scale) of MLL and its oncogenic derivatives. Conserved domains are indicated as hatched bars, the MT domain is depicted in black. The oncogenic alterations are indicated. AT-hook, AT-hook DNA-binding motifs; PhD, plant homeodomain protein–protein interaction motif; TA, transacting domain interacting with CBP; SET, conserved SET domain associates with chromatin remodeling complexes. (B) Amino acid sequence of the MT domain as used in the experiments as a fusion with GST. The CGxCxxC core is boxed, basic amino acids are shaded. (C) Coomassie-stained SDS–polyacrylamide gel showing purified GST–MT fusion protein. The sizes of the molecular weight standards are indicated to the left. Lane 1, standard; lanes 2 and 3, elution fractions (5 µg protein each) after affinity chromatography.
In the majority of leukemias with altered MLL the complete C-terminus is removed by the chromosomal translocation and replaced in frame by a variety of different fusion partners. The resulting fusion protein acquires novel properties and the leukemogenic potential of this type of MLL fusion has been directly demonstrated in mouse models and in in vitro transformation tests (14–21). Despite the heterogeneity of the fusion partners MLL always contributes the identical N-terminal portion to the various chimeric proteins. A second type of leukemogenic MLL alteration corroborates the importance of the MLL N-terminus. In this case, the same N-terminal moiety of MLL that is retained in the fusion proteins is duplicated in tandem (22–24). This leads to the expression of a MLL protein where the N-terminus is ‘fused’ to itself (Fig. 1A).
In a structure–function analysis (21) two domains within MLL have been identified that are essential for the transforming capacity of an MLL fusion protein. The first one is comprised of three AT-hook motifs that occur close to the start of MLL. AT-hooks have been identified in many different proteins and are known to bind unspecifically to the minor groove of AT-rich DNA that is in a distorted or cruciform conformation (25). A second essential region further downstream marks the C-terminal boundary of the minimum MLL portion that is necessary to produce a functional fusion protein. This 100 amino acid domain displays a homology to the regulatory moiety of the eukaryotic DNA methyltransferase 1 (DNMT1) and has therefore been termed MT-homology or MT domain (26). The MT domain consists of a cysteine-rich core flanked by patches of basic amino acids (hence the alternative name BCB domain; basic–cysteine–basic). The cysteine-rich part is characterized by the presence of two copies of the signature amino acid sequence CGxCxxC. A related CxxC motif was found in proteins connected to processes that involve the recognition of the cytosine methylation status in CpG dinucleotide sequences (DNMT1, methyl-CpG-binding protein, MBD1, and human CpG-binding protein, hCGBP) (27–31). The MT region is part of a larger section of the MLL protein that has been dubbed ‘repression domain’ because it can repress transcription if it is artificially recruited to promoters in a fusion with the GAL4 DNA-binding domain (12).
To clarify the biological activity of this essential domain we investigated the properties of purified MT-domain protein. Here we report that the MT domain specifically recognizes CpG dinucleotide sequences in band shift and competition experiments. Binding is abolished if the cytosine in CpG is methylated on one or both DNA strands. Quantitative measurements with surface plasmon resonance (SPR), site selection and footprinting analysis confirmed this result. As demonstrated by reporter gene studies in transfected cells this interaction also persists in vivo.
MATERIALS AND METHODS
GST-fusion protein expression and purification
For bacterial expression of GST–MT fusion proteins the cDNA coding for the MT domain was amplified by PCR with the MT-specific primers BamMTfw (5′-caagggatcctaaagaaaggacgtcgatcgaggcgg-3′) and EcoMTrev (5′-acacgaattcccaggtttctgactagagtccaccacgttc-3′). The PCR product was cloned into the bacterial expression vector pGEX-3X (Amersham Pharmacia Biotech AB, Uppsala, Sweden) and subsequently electroporated into Escherichia coli BL21 (DE3). Bacteria were grown in TB-medium (terrific broth) to an OD600 of 0.8 and expression was induced with 0.2 mM IPTG for 4 h at 27°C. Protein extracts were obtained by sonication with subsequent centrifugation and purified on a GSTrap™ column (Amersham Pharmacia Biotech AB) according to the manufacturer’s instructions. Protein contents were determined with the Bradford method (Bio-Rad protein concentration determination assay; Bio-Rad, Hercules, CA) and purity was tested by SDS–PAGE.
Gel mobility shift analysis
To analyze the DNA-binding properties of the MT domain the following synthetic double-stranded oligonucleotides were produced by annealing of the corresponding single-stranded precursors: CpG52 (5′-tacccgttggaccggactaacgctagtacccgttggaccggactaacgctag-3′), GpC52 (5′-tagcccttggaggccactaagcctagtagcccttggaggccactaagcctag-3′) and methylated CpG52 [same sequence as CpG52 but nucleotides 5, 13, 21, 31, 39 and 47 substituted by 5-methyl-d(C)]. For complex formation various amounts of GST–MT protein were incubated with 20 000 c.p.m. (Cerenkov radiation) 32P-end-labeled double-stranded oligonucleotides in 1× binding buffer [6 mM Tris–HCl, 6 mM MgCl2, 150 mM NaCl, 1 mM DTT, 10 ng/µl BSA, 10 ng/µl poly(dA•dT), pH 7.9] in a total volume of 12 µl for 15 min at room temperature. Glycerol was added to a final concentration of 10% and the mixtures were analyzed in a native 6% polyacrylamide gel in 0.5× TBE at 4°C for 1.5 h. Bands were visualized by autoradiography. For competition studies complex formation was performed in the same way in the presence of the indicated amounts of unlabeled competitor DNA.
SPR measurement (BIAcore)
The kinetic binding constants of the GST–MT/DNA complex were determined with a BIAcoreX-machine (BIAcore AB, Uppsala, Sweden). The primers CpG52fw (5′-tacccgttggaccggactaacgctagtacccgttggaccggactaacgctag-3′) and GpC52fw (5′-tagcccttggaggccactaagcctagtagcccttggaggccactaagcctag-3′) were 3′-biotinylated with a DIG oligonucleotide 3′-labeling kit (Boehringer Mannheim, Penzberg, Germany) according to the manufacturer’s instructions with the exception that 1 mM Biotin-7-dATP (Gibco/Life Technologies, Karlsruhe, Germany) was used instead of DIG-ddUTP. Double-stranded 3′-biotin-CpG52 or 3′-biotin-GpC52 oligonucleotides were produced by dimerization with the corresponding unlabeled primer. Oligonucleotides were immobilized separately on the flow-cell surfaces of a streptavidin (SA)-coated biosensor chip (BIAcore AB) by injecting twice 5 pmol 3′-biotin-labeled ds-oligonucleotides in a total volume of 50 µl 1× coupling buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% poly-sorbate 20) at a flow rate of 5 µl/min followed by a wash cycle with 1× binding buffer [6 mM Tris–HCl, 6 mM MgCl2, 150 mM NaCl, 1 mM DTT, 10 ng/µl BSA, 100 ng/µl poly(dA•dT), pH 7.9]. Dilutions of GST–MT protein were prepared in 1× binding buffer as indicated and 20 µl of each dilution were injected to both flow cells at a flow rate of 5 µl/min per assay cycle. Bound protein was eluted by injecting 20 µl 1× regeneration buffer (2.5 M NaCl, pH 5.3) followed by a wash cycle with 1× binding buffer between assay cycles. Data were analyzed with the BIAevalution software (BIAcore AB) and the dissociation constant (KD) was calculated from the ratio of the dissociation rate (kd) and association rate (ka).
PCR-based site selection
A pool of double-stranded 74 bp oligonucleotides containing a central randomized 26 bp sequence was produced by annealing equimolar amounts of primers R74poly(N) [5′-caggtcagttcagggcatcctgtc-(N)26-gagggaattcagtgcaactgcagc-3′] and F74 (5′-gctgcagttgcactgaattccctc-3′) and subsequent elongation by Klenow polymerase. Double-stranded DNA was purified and eluted by TAE-agarose gel electrophoresis and DNA contents were determined photometrically. Complex formation was performed by incubating 500 ng GST–MT protein with 100 ng of eluted ds74 bp oligonucleotides in a total volume of 20 µl in 1× binding buffer (see Gel mobility shift analysis) at standard conditions. Complexes were precipitated with GST–agarose beads (Sigma-Aldrich, Deisendorf, Germany) for 30 min at 4°C in a rotator shaker and complexed DNA was amplified in a PCR reaction with specific primers F74 and R74 (5′-caggtcagttcagggcatcctgtc-3′). After three rounds of precipitation and amplification the PCR products were 32P-end-labeled and 250 000 c.p.m. (Cerenkov radiation) were used for a gel shift with 1 µg GST–MT protein at standard conditions. Shifted complexes were purified and eluted by native polyacrylamide gel electrophoresis followed by a second round of PCR amplification and gel shift purification. Retarded fragments were amplified once more by PCR and cloned into the vector pBluescript-SK+ (Stratagene, La Jolla, CA). Randomly picked inserts were sequenced with a ABI Prism 310 sequencing machine (PE Biosystems, Foster City, CA).
DNase I protection assays
A double-stranded 74 bp DNA fragment (5′-caggtcagttcagggcatcctgtcCCGTGCTAGTGCGTCGTACCCGCCGAgagggaattcagtgcaactgcagc-3′) corresponding in sequence to clone 1 obtained in the site selection experiments and labeled at either the top or bottom strand was produced by PCR using either 33P-end-labeled primer R74 or F74 with the corresponding unlabeled counterprimer. After purification and elution from a native polyacrylamide gel, complex formation was performed with 3 or 5 µg GST–MT protein in a total volume of 20 µl at standard conditions. Then, CaCl2 was added to a final concentration of 50 µM and the DNA was digested with 2 U DNase I (Boehringer Mannheim) for 5 min at room temperature. Digestions were stopped by addition of 8 µl DNase I-STOP solution (3.2 M LiCl, 100 mM EDTA, pH 8.0, 2 µg/µl yeast RNA) on ice. Ethanol precipitated DNA was redissolved in 3 µl H2O/3 µl formamide loading dye [96% (v/v) formamide, 4% (v/v) H2O, 0.1% (w/v) bromophenol blue, 0.1% (w/v) xylencyanol FF] and analyzed in an 8% acryalmide/urea [50% (w/v)] sequencing gel. Bands were visualized by autoradiography.
Reporter assays
For testing the DNA-binding capacities of the MT domain in vivo the cDNA coding for the MT domain was amplified by PCR with MT-specific primers MT-1fw (5′-acacgaattcaaagaaaggacgtcgatcgaggcgg-3′) and MT-2rev (5′-tgtggatatcggtttctgactagagtccaccac-3′) and cloned into the eukaryotic expression vector pcDNA3 with the addition of an N-terminal flag epitope tag (Invitrogen, Inchinnan, UK). A fusion of the flag-tagged MT domain with the Herpes simplex transactivator domain VP16 was constructed by the addition of a C-terminal VP16 sequence. For control purposes the MT domain and the MT–VP16 chimera were further fused to the DNA-binding moiety of the GAL4 protein in the expression vector pSG424 (32). Either a plasmid with a luciferase gene driven by the SV40 minimal promoter (pGL3-promoter; Promega, Madison, WI) or a modification thereof (pGL3-CG) with a hexamerized CpG-rich sequence (5′-ccgtgctagtgcgtcgtacccgccga-3′) immediately upstream of the SV40 minimal promoter served as reporter. The CpG-rich sequence was identical to the core insert of clone 1 obtained in the site selection experiments. In addition, a similar plasmid with the hexamer of CpG-rich sequence exchanged for a triplet of GAL4 recognition sites was used for control purposes. To test the effect of methylation, the reporter constructs were methylated in vitro with SssI methylase (New England Biolabs, Beverly, MA) according to the manufacturer’s instructions. Combinations of 0.9 µg expression construct and 0.1 µg reporter construct were cotransfected in triplicates into 293T cells with the FuGENE™ 6 Transfection Reagent (Roche, Penzberg, Germany) according to the manufacturer’s instructions. Luciferase activity was measured with the Luciferase Reporter Assay System (Promega) and a Monolight 2010 luminometer (Analytical Luminiscence Laboratories, San Diego, CA).
RESULTS
The MT domain binds to unmethylated CpG in gel shift analysis
To avoid a possible experimental interference by the DNA-binding activity of the AT-hooks we chose to study the properties of the MT domain as an isolated unit. Therefore, MT protein was produced fusing the cDNA encoding amino acids 1147–1244 of MLL (GenBank accession no. AAA58669) to the gene for glutathione S-transferase in the commercial expression vector pGEX-3X. The corresponding 37 kDa GST–MT fusion protein was purified from E.coli by a one-step glutathione affinity procedure. Approximately 90% pure protein was obtained as judged by SDS–PAGE followed by Coomassie staining (Fig. 1C). All further in vitro experiments have been performed with the GST–MT fusion protein.
The presence of the CxxC motif in the MT domain prompted us to investigate a possible association of the recombinant MT fusion protein with CpG-containing DNA. For this purpose, a double-stranded 52 bp oligonucleotide (CpG52) containing six regularly spaced CpG dinucleotides was synthesized. As a control, a second oligo (GpC52) with the same sequence and overall base composition was used with the exception that all CpG combinations were now inverted to GpC (Fig. 2A). In gel shift experiments the addition of increasing amounts of GST–MT protein produced a retarded band in a dose-dependent fashion with labeled CpG52 oligonucleotide. However, no interaction could be detected with the GpC52 probe (Fig. 2B, left). GST protein alone did not bind to any of the probes tested (data not shown). These results were confirmed in a competition experiment. Whereas the addition of a surplus of unlabeled CpG52 oligo competed efficiently with the binding of GST–MT, a 100-fold excess of GpC52 did not affect the protein–DNA interaction (Fig. 2B, right).
Figure 2.

GST–MT protein binds to unmethylated CpG sequences in DNA. (A) Sequence of the probes used in gel mobility shift analysis. CpG sequences are shaded. (B) Mobility shift experiment (left) and competition experiment (right). For the mobility shift the indicated amounts of GST–MT protein were allowed to bind either to the CpG52 probe or the CpG-free GpC52 probe. Complexes formed were separated in a polyacrylamide gel and visualized by autoradiography. In the competition experiment 400 ng of GST–MT were reacted with labeled CpG52 probe in the presence of the indicated molar excess of unlabeled competitor (either CpG52 or GpC52). (C) Mobility shift experiment with methylated DNA (top). The complex formation of GST–MT protein and methylated DNA was investigated as described for (B). To this purpose either an unmodified probe (CpG52) or a probe with identical sequence but 5mC replacing the cytosine of CpG dinucleotides was used. The label 5-methylCpG52 indicates that the probe was methylated on both strands; hemimethylCpG52 signifies a probe methylated in the top or bottom strand only (as indicated). Competition experiments with methylated DNA (bottom). The complex formation of 400 ng of GST–MT with labeled, unmethylated CpG52 probe was challenged with the indicated excess of unlabeled oligos (left) or homopolymers of DNA (right). The competitor DNA used was either parental CpG52 oligo, CpG52 hemimethylated in the top or bottom strand, completely methylated CpG52 oligo, or homopolymeric DNA consisting of poly-desoxyinosine/desoxycytidine or poly-desoxyadenine/desoxythymidine.
Cytosines in CpG dinucleotides of mammalian DNA are the frequent targets for methylating enzymes that convert the base to 5-methylcytosine (5mC). To determine the influence of cytosine methylation on the DNA-binding capacity of GST–MT, oligos identical in sequence to the CpG52 probe were used but now with 5mC replacing the C of CpG sequences either in both strands (methylated DNA) or in the top or bottom strand only (hemimethylated DNA). In gel shifts this exchange of C to 5mC drastically diminished the affinity of GST–MT for DNA. Compared with the unmethylated CpG52 probe only residual binding at high concentrations of GST–MT remained visible with either fully methylated or hemimethylated CpG52 substrate (Fig. 2C, top). This was supported by the fact that neither fully methylated nor hemimethylated DNA could efficiently compete for the binding of GST–MT to unmethylated CpG52 (Fig. 2C, bottom left).
Initial observations had indicated that the addition of ‘unspecific’ poly(dI•dC) competitor could completely inhibit the association of GST–MT protein with DNA (data not shown). As poly(dI•dC) resembles homopolymeric CpG stretches the influence of poly(dI•dC) on GST–MT binding was studied in a competition test. In this experiment the addition of an excess of poly(dI•dC) effectively competed with the complex formation between GST–MT and the CpG52 oligo whereas the same amount of poly(dA•dT) had no visible consequence (Fig. 2C, bottom right).
Determination of the binding constants in solution by SPR
For quantitative measurements of the specific binding affinity of the GST–MT protein, a kinetic binding study with a SPR biosensor (BIAcore) was carried out. In this experiment the kinetic association and dissociation rates of complexes formed between molecules in solution and substrates pre-bound to a solid surface are determined by measuring the reflecting index change in arbitrary units of response (RU). For this purpose biotinylated CpG52 oligonucleotide was immobilized on the streptavidin sensor chip and variable concentrations of GST–MT protein were applied to the surrounding flow chamber. An example of the corresponding response curves is given in Figure 3. From the obtained initial rates of association and dissociation a KD of 3.28 ± 1.7 × 10–8 M (mean ± SD, n = 8) could be estimated. When repeated with fully methylated CpG52 oligonucleotide a very weak binding of GST–MT was observed at the highest possible protein concentrations (data not shown). Therefore, the affinity constant of methylated DNA could not be determined with accuracy. A rough estimate indicated a KD of >1 mM for the interaction of GST–MT with methylated template.
Figure 3.

Kd determination for the GST–MT/DNA interaction by BIAcore experiments. The CpG52 oligonucleotide probe was immobilized on a plasmon resonance sensor chip and the kinetic association/dissociation constants were determined in a flow cell charged with the given concentrations of GST–MT protein. The curves show the graphical representation of the measurements in a typical experiment. The kinetic values represent the mean ± SD (n = 8).
CpG is the preferred binding site of the MT domain in site selection experiments
Site selection studies were carried out to determine if the MT domain has any binding preferences outside the minimally required CpG sequence. For this purpose, a 74 bp oligonucleotide was synthesized that was composed of 26 random nt in its center flanked by known sequence without any CpG (N74) (Materials and Methods). Two primer molecules complementary to the flanking sequence allowed the amplification of the whole 74 bp DNA by PCR. The single-stranded N74 was converted to double-strand DNA by the Klenow fragment of E.coli DNA polymerase I and one of the flanking primers. The resulting DNA fragment was gel-purified, eluted and incubated in solution with GST–MT protein. Bound DNA was precipitated together with the GST–MT protein by binding to glutathione immobilized on agarose beads. After a wash step, the bound fraction of DNA was amplified by PCR. The gel-purified PCR products were subjected to two more rounds of precipitation/amplification. To increase binding stringency, the selection rounds in solution were followed by two rounds of gel shifts. To this end, the PCR product was end-labeled with [γ-32P]ATP and polynucleotide kinase, reacted with GST–MT in binding buffer and applied to a native polyacrylamide gel. The retarded band was excised, eluted and re-amplified by PCR to serve as the input for the final round of gel selection. Finally, the end products of the selection process were cloned into the pBluescript vector and sequenced. Of 19 clones analyzed all contained from three to seven CpG dinucleotides randomly distributed in the center of the selection oligonucleotide (mean 4.6 ± 1.2 CpG repeats) (Fig. 4). Outside the CpG core there was no consensus sequence discernible and the nucleotides flanking each CpG followed a random distribution. In contrast, none of 19 indiscriminately picked samples from the unselected oligo pool included more than two CpG dinucleotide sequences (mean 0.7 ± 0.7 CpG motifs). These results confirm CpG as apparently the sole binding requirement for the MT domain protein.
Figure 4.
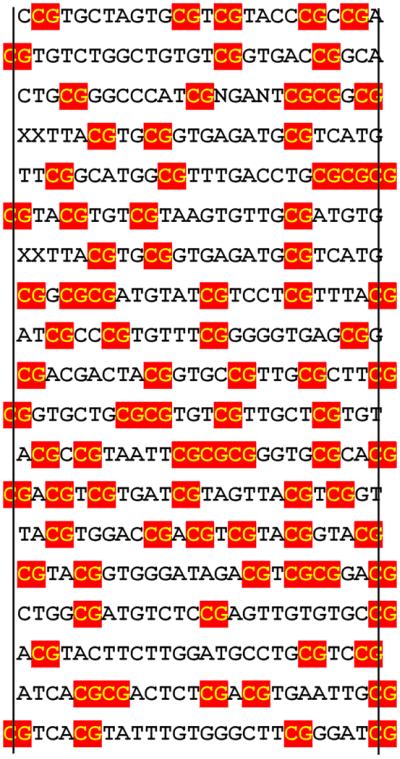
GST–MT prefers CpG sequences in site selection experiments. Site selection experiments (Materials and Methods) were performed with oligonucleotides containing a core of 26 random nt. The sequences of 19 clones with high affinity to GST–MT are shown. The vertical lines indicate the extent of the original randomly designed sequence with flanking nucleotides given if they contribute to form a CpG dinucleotide. CpG sequences are boxed.
GST–MT protects CpG in DNase I footprinting analysis
Next we wanted to determine the extent of DNA that is protected from DNase I digestion by the interaction with the GST–MT protein. As substrate for footprinting a high-affinity binding clone from the site selection experiment (clone 1) was chosen. The corresponding fragment was excised from the respective pBluescript plasmid and the top or bottom strand was labeled specifically by PCR amplification utilizing a 5′ [γ-33P]ATP marked primer. Complexes of end-labeled DNA with GST–MT were subjected to DNase I treatment. The analysis of the reaction products by denaturing PAGE showed three distinct regions protected from DNase I digestion, all centered on CpG dinucleotides (Fig. 5). One area of protection was visible extending from nucleotides 19 to 30 on the top strand. The exact 5′ boundary of this region could not be exactly determined due to the proximity of the labeled 5′ end to the GST–MT-binding site, thus resulting in digestion fragments too small to be separated on the gel. On the bottom strand two more GST–MT footprints were detectable that each covered 5–6 bp (nucleotides 35–40 and 46–51). As the digestion pattern indicated, the binding of GST–MT rendered the DNA outside of the protected center of the molecule increasingly DNase I sensitive with the highest susceptibility for cleavage towards the end of the molecule. Increased band intensity after DNase I treatment could be seen for the top strand starting at nucleotide 40 and extending to the end of the fragment at nucleotide 74. The corresponding pattern for the bottom strand covered the region from nucleotides 1 to 32. A better accessibility for DNase I is indicative of an altered and/or distorted structure of the DNA template in these areas. However, circular permutation experiments did not show any sign of a bend or kink in GST–MT-bound DNA (data not shown).
Figure 5.

GST–MT protects CpG from DNase I digestion. The DNase I footprinting pattern of GST–MT bound to clone 1 (as identified in the site selection experiment) was determined. A corresponding DNA fragment was labeled with [γ-33P]ATP either at the top or the bottom strand, complexed with increasing amounts of GST–MT and subjected to DNase I digestion. The reaction products were separated on a denaturing polyacrylamide gel and visualized by autoradiography. The numbers indicate the respective position of the nucleotides and the vertical bars indicate regions protected by GST–MT. The results are overlaid onto the sequence of the probe in a graphical representation. CpGs in the probe are shaded, protected areas are indicated by boxes and hypersensitive areas are marked by a line on the appropriate DNA strand. The open box at the top strand from nucleotides 20 to 30 signifies that the 5′ extent of the footprint could not be exactly determined due to the proximity of the labeled DNA end to the protected area.
The MT domain associates with CpG-containing DNA in vivo
To prove that the MT domain can bind to CpG also inside a cell, reporter gene assays were performed. The rationale of these studies was that an artificial fusion of the MT domain with the strong transcriptional transactivator VP16 should cause an increased output of reporter gene activity once it is bound to a corresponding promoter. Plasmids were constructed for eukaryotic expression of the MT domain (pMT) and a fusion of MT with the viral transactivator VP16 (pMT–VP16). As controls, similar constructs were used that encoded an additional N-terminal GAL4 DNA-binding domain (pGAL4–VP16, pGAL4–MT–VP16). When transfected into 293T cells all plasmids directed the production of proteins of the correct molecular weight as detected in immunoblots (data not shown). Three different reporter constructs depicted schematically in Figure 6 were applied: (i) a luciferase gene under the control of the minimal SV40 promoter that contains several naturally occurring CpG motifs (pGL3 promoter); (ii) a related construct with six tandem copies of the core sequence of site selection clone 1 preceding the SV40 minimal promoter and therefore increasing the CpG density in the promoter region (pGL3-CG); and (iii) a plasmid with three consensus-binding sites for the GAL4 DNA-binding domain upstream of the SV40 minimal promoter (pGL3-GAL4).
Figure 6.

A fusion of the MT domain with the VP16 transactivator associates with DNA inside living cells. (A) An expression construct coding for MT and a MT–VP16 fusion was cotransfected into 293T cells with a reporter construct containing a luciferase gene under the control of the SV40 minimal promoter (graphical representation under the respective diagram). As control, empty vector was used as indicated. Luciferase activities were determined 48 h after transfection, normalized to protein content and the respective values are given as relative units with the basal SV40 driven luciferase levels set to one unit. The means ± SD of three independent experiments are given. (B) Expression plasmids as in (A) were cotransfected with a reporter containing an increased CpG content (SV40 promoter preceded by six repetitions of the CpG-rich sequence of site selection clone 1). Either unmodified plasmid (left) or DNA pre-methylated at CpG sequences with SssI methylase was used. (C) Control constructs encoding a fusion of the GAL4 DNA-binding domain and either the MT–VP16 chimera or the VP16 transactivator were transfected as in (A). An unmethylated (left) or pre-methylated (right) plasmid with the SV40 promoter preceded by three copies of the GAL4 DNA-binding consensus site served as a reporter.
When cotransfected with pGL3 promoter into 293T cells the MT–VP16 fusion was able to increase the luciferase activity modestly but significantly by ∼2-fold. The transactivation levels were lower as might be expected because of a counteractive repressor activity within the MT domain that was revealed by expression of the MT portion alone (Fig. 6A). Despite the overall weak transactivation potency of the MT–VP16 protein an increase in CpG density of the reporter construct (pGL3-CG) lead to a correspondingly higher transactivation efficiency (Fig. 6B, left). To determine the influence of DNA methylation on the MT–VP16 activity in vivo a pGL3-CG reporter was transfected after pre-methylation of the cytosine residues in CpG sequences by SssI methylase. This pre-treatment reduced the basal promoter activity of the reporter construct to ∼10% compared with the value obtained with unmethylated plasmid. Corresponding to the in vitro-binding results, the transactivation effect of MT–VP16 was completely abolished with this methylated target (Fig. 6B, right). In contrast, when MT–VP16 was directly targeted to the promoter by the GAL4 DNA-binding domain, transactivation could still be seen on methylated as well as native CpG targets (Fig. 6C).
DISCUSSION
In this paper we report experiments identifying the MT homology region of MLL as a DNA-binding domain that specifically recognizes unmethylated CpG sequences. In band shift analysis and SPR studies, recombinant GST–MT protein binds with high affinity to sequences containing CpG. In site selection experiments, CpG is the preferred binding target of GST–MT and in footprinting CpG is protected from DNase I digestion. Moreover, a binding of the MT domain to CpG-containing DNA can be demonstrated also inside living cells. A conversion of cytosine to 5mC eliminates the tight association of MT with DNA in vitro and in in vivo.
These results show that the CxxC domain serves a similar function in different proteins. Except in DNMT1 itself, where the CxxC-containing domain has not been very well characterized with regard to function, two more proteins use CxxC domains for DNA binding. The clearest example is the CpG-binding protein CGBP1 that also contains next to the CxxC motif a transactivator function and has therefore been speculated to be involved in the direct transcriptional activation of promoters with CpG islands (30). In CGBP1, the CxxC region is responsible for binding to DNA and a single CpG is sufficient for this interaction. Methylated sequences are not recognized by CGBP1. A more complicated situation is encountered with the methyl-binding protein MBD1 (27,28). MBD1 includes three CxxC motifs and isoforms are produced by differential splicing that encompass either two or three of these domains. MBD1 binds to methylated as well as unmethylated DNA and it acts as a transcriptional repressor. Despite the presence of three CxxC motifs only one of these seems to be involved in binding to unmethylated DNA and all are dispensable for the association with methylated DNA. A separate methyl-binding domain in MBD1 is responsible for the interaction with methylated sequences. In summary, the CxxC motif seems to be the signature of a special structural fold that confers a CpG-binding activity. However, the overall homology amongst the various CxxC domains is only ∼40% and therefore it cannot be excluded that variable sequences outside of the conserved CGxCxxC core contribute to specific functions in individual proteins.
In our experiments the overexpression of an isolated MT domain had a repressive effect on the transcription of a reporter gene. This fact was noted previously with similar templates containing GAL4 sites and constructs that fused a GAL4 DNA-binding domain to MT (12). The underlying repressive action of MT most likely is one reason why we observed only moderate transactivation levels in the reporter gene assays despite the fusion with the strong VP16 transactivator region. Although an interaction with histone deacetylase has been mapped to the corresponding region in the DNMT1 protein (31), the repressor mechanism of the MT domain is not clear. The MT domain has a high affinity to CpG sequences with a KD in the 10–8 M range. This is comparable with the values obtained in similar experiments, e.g. for the HMG1/DNA interaction with a KD of 4.8 × 10–8 (33). Maybe an isolated MT domain can compete for binding with other DNA-binding proteins, e.g. general transcription factors and in this way cause an unspecific repressive effect.
The homology to fly Trithorax, the knockout studies and the biochemical evidence (3,6,7,10,13) strongly suggest that the function of MLL is to maintain pre-activated promoters in an active transcriptional state by changing the chromatin configuration of the respective DNA. For this purpose MLL needs to recognize active promoters. It is easily visible how a domain that mediates binding to unmethylated CpG islands and prevents the association with DNA that is blocked by methylation could assist in this process. The unique combination of an AT-hook DNA-binding motif and the CpG recognition domain in MLL would target the protein to a specific subset of response elements where AT-rich and/or cruciform DNA neighbors sequence with a high frequency of CpG. The verification of this prediction will have to await the identification of MLL target genes. However, the fact that both the AT-hooks and the MT domain are strictly conserved in the oncogenic MLL proteins strengthens the argument that it is the deregulation of authentic MLL targets that is responsible for the leukemogenic effect. Consequently, a protein that has lost a crucial domain for target finding would be unable to home in on the appropriate promoters and therefore it seems unlikely that such a truncated protein might cause leukemia.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Georg Fey for continous support. This work was supported by grant SFB473/B10 and partially by grants SFB466/C7 and SL27/4-1 from the DFG. R.K.S. is a recipient of a Ria Freifrau-von-Fritsch Stiftung career development award.
REFERENCES
- 1.Dimartino J.F. and Cleary,M.L. (1999) Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br. J. Haematol., 106, 614–626. [DOI] [PubMed] [Google Scholar]
- 2.Look A.T. (1997) Oncogenic transcription factors in the human acute leukemias. Science, 278, 1059–1064. [DOI] [PubMed] [Google Scholar]
- 3.Tkachuk D.C., Kohler,S. and Cleary,M.L. (1992) Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell, 71, 691–700. [DOI] [PubMed] [Google Scholar]
- 4.Pirrotta V. (1997) Chromatin-silencing mechanisms in Drosophila maintain patterns of gene expression. Trends Genet., 13, 314–318. [DOI] [PubMed] [Google Scholar]
- 5.Pirrotta V. (1998) Polycombing the genome: PcG, trxG and chromatin silencing. Cell, 93, 333–336. [DOI] [PubMed] [Google Scholar]
- 6.van Lohuizen M. (1999) The trithorax-group and polycomb-group chromatin modifiers: implications for disease. Curr. Opin. Genet. Dev., 9, 355–361. [DOI] [PubMed] [Google Scholar]
- 7.Yu B.D., Hanson,R.D., Hess,J.L., Horning,S.E. and Korsmeyer,S.J. (1998) MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc. Natl Acad. Sci. USA, 95, 10632–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu B.D., Hess,J.L., Horning,S.E., Brown,G.A. and Korsmeyer,S.J. (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature, 378, 505–508. [DOI] [PubMed] [Google Scholar]
- 9.Hanson R.D., Hess,J.L., Yu,B.D., Ernst,P., van Lohuizen,M., Berns,A., van der Lugt,N.M., Shashikant,C.S., Ruddle,F.H., Seto,M. et al. (1999) Mammalian Trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc. Natl Acad. Sci. USA, 96, 14372–14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rozenblatt-Rosen O., Rozovskaia,T., Burakov,D., Sedkov,Y., Tillib,S., Blechman,J., Nakamura,T., Croce,C.M., Mazo,A. and Canaani,E. (1998) The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc. Natl Acad. Sci. USA, 95, 4152–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasad R., Yano,T., Sorio,C., Nakamura,T., Rallapalli,R., Gu,Y., Leshkowitz,D., Croce,C.M. and Canaani,E. (1995) Domains with transcriptional regulatory activity within the ALL1 and AF4 proteins involved in acute leukemia. Proc. Natl Acad. Sci. USA, 92, 12160–12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeleznik-Le N.J., Harden,A.M. and Rowley,J.D. (1994) 11q23 translocations split the ‘AT-hook’ cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl Acad. Sci. USA, 91, 10610–10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst P., Wang,J., Huang,M., Goodman,R.H. and Korsmeyer,S.J. (2001) Mll and creb bind cooperatively to the nuclear coactivator creb-binding protein. Mol. Cell. Biol., 21, 2249–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiMartino J.F., Miller,T., Ayton,P.M., Landewe,T., Hess,J.L., Cleary,M.L. and Shilatifard,A. (2000) A C-terminal domain of ELL is required and sufficient for immortalization of myeloid progenitors by MLL-ELL. Blood, 96, 3887–3893. [PubMed] [Google Scholar]
- 15.Corral J., Lavenir,I., Impey,H., Warren,A.J., Forster,A., Larson,T.A., Bell,S., McKenzie,A.N., King,G. and Rabbitts,T.H. (1996) An Mll–AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell, 85, 853–861. [DOI] [PubMed] [Google Scholar]
- 16.Dobson C.L., Warren,A.J., Pannell,R., Forster,A., Lavenir,I., Corral,J., Smith,A.J. and Rabbitts,T.H. (1999) The mll–AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J., 18, 3564–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobson C.L., Warren,A.J., Pannell,R., Forster,A. and Rabbitts,T.H. (2000) Tumorigenesis in mice with a fusion of the leukaemia oncogene Mll and the bacterial lacZ gene. EMBO J., 19, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lavau C., Szilvassy,S.J., Slany,R. and Cleary,M.L. (1997) Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J., 16, 4226–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavau C., Luo,R.T., Du,C. and Thirman,M.J. (2000) Retrovirus-mediated gene transfer of MLL-ELL transforms primary myeloid progenitors and causes acute myeloid leukemias in mice. Proc. Natl Acad. Sci. USA, 97, 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavau C., Du,C., Thirman,M. and Zeleznik-Le,N. (2000) Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J., 19, 4655–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slany R.K., Lavau,C. and Cleary,M.L. (1998) The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol. Cell. Biol., 18, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto K., Hamaguchi,H., Nagata,K., Kobayashi,M. and Taniwaki,M. (1997) Tandem duplication of the MLL gene in myelodysplastic syndrome-derived overt leukemia with trisomy 11. Am. J. Hematol., 55, 41–45. [DOI] [PubMed] [Google Scholar]
- 23.Strout M.P., Marcucci,G., Bloomfield,C.D. and Caligiuri,M.A. (1998) The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl Acad. Sci. USA, 95, 2390–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schnittger S., Kinkelin,U., Schoch,C., Heinecke,A., Haase,D., Haferlach,T., Buchner,T., Wormann,B., Hiddemann,W. and Griesinger,F. (2000) Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia, 14, 796–804. [DOI] [PubMed] [Google Scholar]
- 25.Aravind L. and Landsman,D. (1998) AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res., 26, 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Q., Alder,H., Nelson,K.K., Chatterjee,D., Gu,Y., Nakamura,T., Canaani,E., Croce,C.M., Siracusa,L.D. and Buchberg,A.M. (1993) Analysis of the murine All-1 gene reveals conserved domains with human ALL-1 and identifies a motif shared with DNA methyltransferases. Proc. Natl Acad. Sci. USA, 90, 6350–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujita N., Takebayashi,S., Okumura,K., Kudo,S., Chiba,T., Saya,H. and Nakao,M. (1999) Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol. Cell. Biol., 19, 6415–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita N., Shimotake,N., Ohki,I., Chiba,T., Saya,H., Shirakawa,M. and Nakao,M. (2000) Mechanism of transcriptional regulation by methyl-CpG binding protein MBD1. Mol. Cell. Biol., 20, 5107–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cross S.H., Meehan,R.R., Nan,X. and Bird,A. (1997) A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nature Genet., 16, 256–259. [DOI] [PubMed] [Google Scholar]
- 30.Voo K.S., Carlone,D.L., Jacobsen,B.M., Flodin,A. and Skalnik,D.G. (2000) Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax and methyl-CpG binding domain protein 1. Mol. Cell. Biol., 20, 2108–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- 32.Sadowski I. and Ptashne,M. (1989) A vector for expressing GAL4(1–147) fusions in mammalian cells. Nucleic Acids Res., 17, 7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang L., Li,J., Katz,D.S. and Feng,J.S. (2000) Determining the DNA bending angle induced by the non-specific high mobility group (HMG-1) proteins: a novel method. Biochemistry, 39, 3052–3060. [DOI] [PubMed] [Google Scholar]