

Abstract
A naturally inspired chemical library of 25 molecules was synthesised guided by 3-D dimensionality and natural product likeness factors to explore a new chemical space. The synthesised chemical library, consisting of fused-bridged dodecahydro-2a,6-epoxyazepino[3,4,5-c,d]indole skeletons, followed lead likeness factors in terms of molecular weight, C-sp3 fraction and Clog P. Screening of the 25 compounds against lung cells infected with SARS-CoV-2 led to the identification of 2 hits. Although the chemical library showed cytotoxicity, the two hits (3b, 9e) showed the highest antiviral activity (EC50 values of 3.7 and 1.4 μM, respectively) with an acceptable cytotoxicity difference. Computational analysis based on docking and molecular dynamics simulations against main protein targets in SARS-CoV-2 (main protease Mpro, nucleocapsid phosphoprotein, non-structural protein nsp10–nsp16 complex and RBD/ACE2 complex) were performed. The computational analysis proposed the possible binding targets to be either Mpro or the nsp10–nsp16 complex. Biological assays were performed to confirm this proposition. A cell-based assay for Mpro protease activity using a reverse-nanoluciferase (Rev-Nluc) reporter confirmed that 3b targets Mpro. These results open the way towards further hit-to-lead optimisations.
A naturally inspired chemical library of 25 molecules was synthesised guided by 3-D dimensionality and natural product likeness proved to have antiviral activity against SARS-CoV-2.
Introduction
The late 2019 coronavirus disease outbreak turned out into the global COVID-19 pandemic that is still on-going.1 The causative agent of the COVID-19 disease was identified as the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).2–4 This highly transmissible airborne virus is a positive-sense single-stranded RNA virus, whose genome encodes 29 proteins including four structural proteins.5 Although there are currently different vaccines and treatments available to tackle this pandemic, research efforts should continue to find therapeutic agents against the virus, as well as variants of concern or other zoonotic coronaviruses that might emerge in the future.14–16 There are now several treatments for COVID-19. For example, remdesivir, which has been granted emergency-use authorisation, showed modest benefit to patients with COVID-19.6–9 Paxlovid, the FDA approved oral SARS-CoV-2 main protease Mpro inhibitor, showed benefit for the treatment of COVID-19 patients in severe condition.9b Although the structure of the active site of Mpro is highly conserved among coronaviruses,9c the emergence of drug-resistant variants cannot be excluded. Furthermore, the massive vaccination campaign currently ongoing does not guarantee herd immunity and that vaccinated people with SARS-CoV-2 antibodies will be immune to reinfection.10–13 Therefore, designing small molecule libraries from new unexplored chemical space could open an avenue towards new antiviral drugs. To get better hit rates in drug discovery programs, a beneficial factor is natural product likeness.17 In addition, enhancing the 3-D character and increasing the fraction of sp3 carbons in the designed chemical libraries increase the opportunities to discover hits and consequently potential drug candidates.18,19 Herein, we designed a naturally-inspired high 3-D fused-bridged dodecahydro-2a,6-epoxyazepino[3,4,5-c,d]indole-based chemical library, with up to three points of diversity and six controlled chiral centres, two of them being all-carbon quaternary, accessible in one step. The synthesised 25 compounds were assayed in lung cell-lines infected with SARS-CoV-2 and screened for cytotoxicity leading to the confirmation of two hits promising for further optimisation. Furthermore, to identify the possible target, high-throughput virtual screening was carried out on four main protein targets in SARS-CoV-2: (i) the main protease (Mpro) that is a non-structural cysteine protease and plays a key role in the release of 16 non-structural proteins involved in the virus replication;20 (ii) the nucleocapsid phosphoprotein that packages the viral RNA into a helical ribonucleocapsid (RNP);21 (iii) the non-structural protein nsp10–nsp16 complex;22,30 and (iv) the RBD/ACE2 membrane glycoprotein complex, responsible for the entry into host cells.23 The computational data led to the possible binding of the best two hits 3b and 9e to either Mpro or the nsp10–nsp16 complex. A cellular assay and a biochemical assay for Mpro protease activity confirmed the anti-Mpro activity of 3b, while no activity was validated for 9e. These findings open a promising starting point for the identification of new antiviral lead candidates.
Results and discussion
Chemistry
Tuning the right combination between 3-D dimensionality, natural product likeness, C-sp3 fraction, Clog P and the number of heteroaromatic rings bearing H-donor/acceptor atoms in one molecule is a challenging task. Finding the right blend leads to a better hit rate. Inspired by the synthesis by Shi and his co-workers, compound 1 can be accessed in three steps in a large scale from the commercially available p-anisidine (Scheme 1).24a This hydro-epoxybenzo[c,d]indole skeleton is a natural product-like compound and has a high 3-D character and high C-sp3 fraction along with an optimal Clog P value that is known to favour bioactivity. Modulation of 1 by introducing relevant groups bearing different H-donor/acceptor atoms can increase its biological activity potential. Compound 1 has two double bonds (highlighted in colours in Scheme 1) and a ketone function allowing further chemical modification. Because of the diverse stereoelectronic environment, the two double bonds could be differentiated. The non-conjugated double bond (in magenta) was selectively dihydroxylated with potassium osmate(vi) to give diol 2 (Scheme 1). This intermediate could be transformed into decahydroepoxy-azepino indoles (3a′–c′), which incorporate a constrained morpholine ring present in several bioactive molecules.24b Ring expansion was achieved by oxidation of 2 with sodium periodate to give an intermediate bisaldehyde, which was reacted, without isolation, with different primary amines under reductive amination conditions to give 3a′–c′. Reduction of the ketone with sodium borohydride afforded alcohols 3a–c as single diastereoisomers. These transformations allowed the introduction of a new accessible point of diversity at the nitrogen atom.
Scheme 1. Synthetic routes to the first round of functionalisation. Reagents and conditions: (a) DIAD (1.1 equiv.), PPh3 (1.2 equiv.), 4 Å M.S. in THF, 77%. (b) NMO (1.2 equiv.), K2OsO4·2H2O (0.8 mol%) in THF/H2O 27% over two steps. (c) NaIO4 in H2O (1.4 equiv.), silica in DCM. (d) RNH2 (1.2 equiv.), STAB (3.0 equiv.), AcOH (0.1 equiv.), M.S. 4 Å in THF. (e) NaBH4 (1.1 equiv.) in MeOH. (f) NaH (60% dispersion in mineral oil) in dry THF, 0 °C, 1 h and then 2-chloropyrimidine, r.t., overnight, 67%.

The enone moiety in 1 or 2 was resistant to reductive amination conditions. Indeed, in the preparation of 3a′–c′, no reaction at the ketone function was observed and several attempts to transform 1 into the corresponding secondary amines using primary amines under diverse reductive amination conditions failed to produce the desired secondary amines. Thus, further modulation was conducted by reducing the ketone group of compound 1 to the corresponding alcohol group and then reacted with electrophiles such as 2-chloropyrimidine to afford 4 (Scheme 1).
To introduce other relevant groups bearing different H-donor/acceptor atoms, scaffold 3a′ was hydrogenated in MeOH to give the deprotected secondary amine 5 with concomitant reduction of the double bond (Scheme 2). By-product 6 was also isolated as the result of the reductive amination of the newly formed secondary amine 5 with adventitious formaldehyde, formed in situ from the reaction of MeOH with Pd/C and hydrogen.25 Changing the solvent to ethyl acetate slowed down the reaction and gave 7 resulting from the reduction of the double bond only without removal of the benzyl group (Scheme 2, panel A). Secondary amine 5 allowed the preparation of a diverse set of compounds, showing the reactivity of this position. Reaction with aldehydes under reductive conditions gave tertiary amines and reaction with acyl chlorides under basic conditions, or with carboxylic acids under peptide coupling conditions, gave the corresponding tertiary amides. As reactive functional groups are not desirable, all the intermediate ketones were directly subjected to reduction with NaBH4 to give 8a–g. In parallel, the secondary amine 5 was reacted with triazolopyrimidine, and then the ketone was reduced with NaBH4 to give compound 8h (Scheme 2, panel B).
Scheme 2. Synthetic routes to the second round of functionalisation. Reagents and conditions: (a) H2 (1.0 bar), Pd/C (10 mol%), MeOH (5 and 6) or EtOAc (7). (b) RCOOH (1.1 equiv.), DIPEA (1.5 equiv.), TBTU (2.5 equiv.) in DCM. (c) RCHO (1.2 equiv.), STAB (3.0 equiv.), AcOH (0.1 equiv.), M.S. 4 Å in DCM. (d) N-Methyl-1H-imidazole-1-carboxamide (1.1 equiv.), Et3N (1.1 equiv.) in DCM. (e) RCOCl (1.1 equiv.), Et3N (1.1 equiv.) in DCM. (f) 1-(Methoxymethyl)-4-(1H-1,2,4-triazol-1-yl)pyrimidin-2(1H)-one (1.2 equiv.), Et3N (1.2 equiv.) in MeCN, 70 °C. (g) NaBH4 (1.1 equiv.) in MeOH.

The third round of functionalisation was conducted on 6, 7, and 8a, e, and g (Scheme 3). Reaction of the free alcohol with halogenated heterocycles or alkanes gave compounds 9a–e (Scheme 3, panel A). Similarly, compound 3b reacted with 2-chloropyrimidine to give compound 10. Surprisingly, the dihydroxylation of the left-hand site (LHS) double bond in compound 3b by potassium osmate(vi) was not successful. The isolated product was ketone 3b′, resulting from the oxidation of the allylic alcohol catalysed by osmium salts26 (page S18 in the ESI†).
Scheme 3. Synthetic routes to the third round of functionalisation. Reagents and conditions: (a) NaH (60% dispersion in mineral oil) in dry THF, 0 °C, 1 h and then RCl, r.t., overnight.

Molecular properties
The 3-D dimensionality and molecular properties of the 25 molecules were analysed using the web-free tool LLAMA (Lead-Likeness And Molecular Analysis; https://llama.leeds.ac.uk).27 The chemical library showed an average C-sp3 fraction of 0.5–0.7, average Clog P of 1.0–5.0 and average molecular weight of 400–600, which are all features consistent with lead-likeness factors (Fig. 1, panel A). It also provided a high 3-D character (Fig. 1, panel B), a property that was proven to give better hit rates.19 The natural product likeness was assessed using the Natural Product Likeness Score calculator (NaPLeS) web-free tool and it indicated that all the synthesised compounds fall in the natural product likeness space with an average score of 0.2–1.3 (Fig. 1, panel B).28
Fig. 1. Molecular properties and diversity. Panel A: Molecular weight vs. Clog P (Clog P was calculated using ChemDraw version 15). Panel B: Principal moment of inertia PMI plot. Panel C: Natural product likeness score, where the green line indicates all synthetic products, the orange line the natural products and the red dots the synthesised chemical library.

Biological evaluation
The 25 compounds were tested for inhibition of SARS-CoV-2 replication of the beta variant in the lung cancer A549 cell line (adenocarcinomic human alveolar basal epithelial cells, source ATTC, reference CCL-158) stably transduced with a lentiviral construct bearing the human ACE2 receptor.40 The measurement of viral replication was carried out by quantitative RT-PCR as described in the ESI† Methods section (Table 1). The key scaffolds 1 and its analogue 4 were not active, indicating the importance of the right-hand side (RHS) diversity point. Introducing dihydroxyl groups on compound 1 appeared to be beneficial with an antiviral activity for compound 2 of EC50 = 14 μM. Structure activity relationship (SAR) revealed that the presence of large aromatic groups in the R1 position on the RHS is important for the antiviral activity as compounds 5, 6 and 8f were inactive. It was also found that the presence of the ketone group did not significantly change the viral activity when comparing compounds 3a′ and 8d′ to 3a and 8d, respectively. Substitution in R2 with aromatic groups did not increase the activity; the aliphatic chain with a basic centre gave better activity. In parallel, the cytotoxic activity of the compounds was assessed as the cytotoxicity could interfere with the antiviral activity of the compound (ESI† Methods section). Several hits showed a favourable ratio between the antiviral activity and cytotoxicity. The most active compounds with a favourable antiviral activity to cytotoxicity ratio and do not have electrophilic groups, which could favour covalent binding, are 3b and 9e (Fig. 2). In addition, 3b and 9e remained active against cells infected with the SARS-CoV2 Omicron BA1 variant with an EC50 of 3.5 and 4.4 μM, respectively, with 3b showing no cytotoxicity, while 9e exhibiting cytotoxicity above 20 μM (Table 1 and ESI† Fig. S1).
Anti-SARS-CoV-2 activity on A549-ACE2 cells (RT-qPCR) and cell cytotoxicity of the 22 compounds, expressed as EC50 in μM, the concentration at which 50% inhibition of the maximal signal is observed at 95% confidence interval.

| ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Saturation on the LHS | RT-qPCR Beta EC50 (μM) [95% CI] | Cytotoxicity CC50 (μM) [95% CI] | Ratio cytotoxicity/RT-qPCR |
| 3a′ |
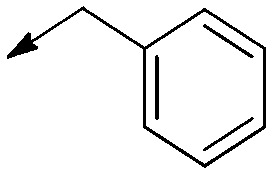
|
— | Double bond | 16 (N/A) | 14 [2.8 to 73] | 0.9 |
| 3a |
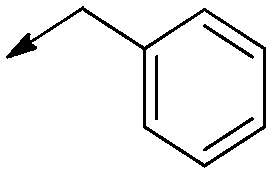
|
H | Double bond | 17 [13 to 23] | >50 | >3 |
| 3b′ |

|
— | Double bond | 13 [7.2 to 24] | 6.6 [2.7 to 16] | 0.5 |
| 3b |

|
H | Double bond | 3.7 [2.0 to 6.9] 3.5 [2.3to 5.6]a | 12 [10 to 15] | 3.3 |
| >50a | ||||||
| 3c |

|
H | Double bond | 8.3 [6.3 to 11] | 13 (N/A) | 1.6 |
| 5 | H | — | Saturated | >50 | >50 | N/A |
| 6 | Me | — | Saturated | >50 | >50 | N/A |
| 7 |
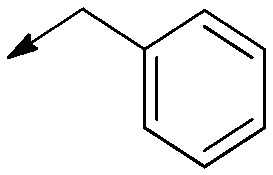
|
— | Saturated | 8.4 [5.5 to 13] | 5.8 [3.0 to 11] | 0.7 |
| 8a |
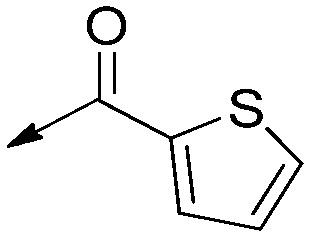
|
H | Saturated | 16 [5.2 to 51] | >50 | >3 |
| 8b |

|
H | Saturated | 14 [8.5 to 22] | 20 [8.2 to 48] | 1.4 |
| 8c |
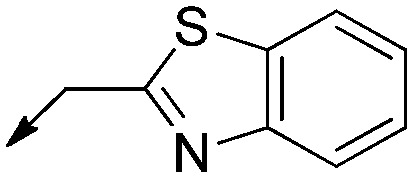
|
H | Saturated | 37 [23 to 59] | >50 | 1.4 |
| 8d′ |
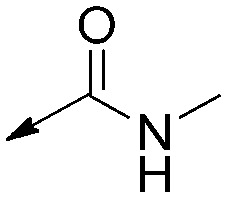
|
— | Saturated | 5.3 [4.0 to 7.1] | 37 N/A | 6.9 |
| 8d |
|
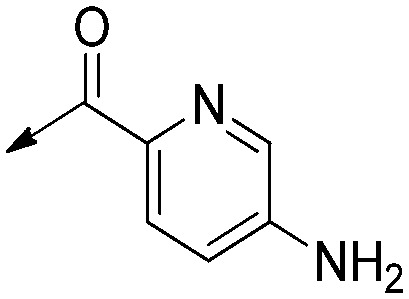
H | Saturated | 6.6 [4.3 to 10] | 12 [6.7 to 21] | 1.6 |
| 8e |

|
H | Saturated | 5.6 N/A | 13 [7.6 to 21] | 2.3 |
| 8f |
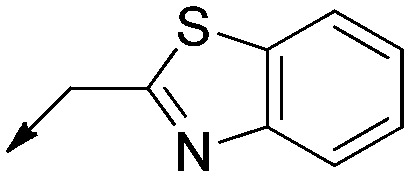
|
H | Saturated | >50 | >50 | N/A |
| 8h |
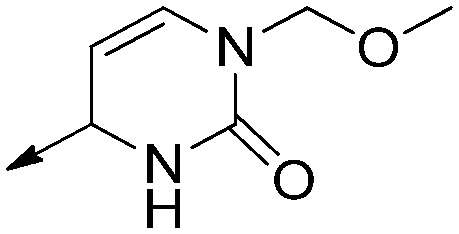
|
H | Saturated | >17 | >50 | N/A |
| 9a |
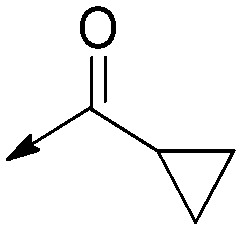
|

|
Saturated | >40 [N/A] | >50 [N/A] | N/A |
| 9b |
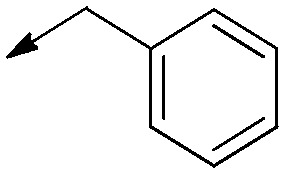
|
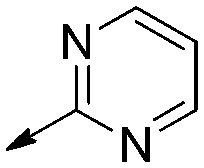
|
Saturated | 6.2 [N/A] | 6.3 [3.7 to 11] | 1.0 |
| 9c | Me |
|
Saturated | 20 [N/A] | 17 [14 to 18] | 0.8 |
| 9d |

|
|
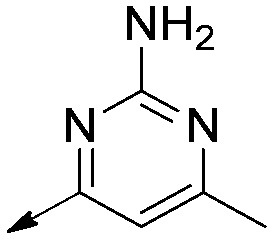
Saturated | >50 | >50 | N/A |
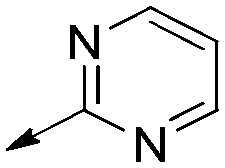
| 9e |
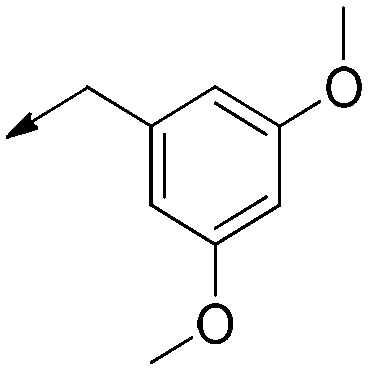
|
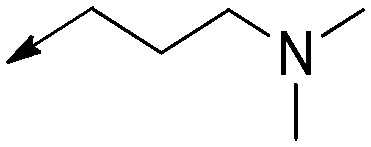
|
Saturated | 1.4 [0.9 to 2.2] 4.4 [2.0 to 9.0]a | 7.7 [5.2 to 12] | 5.7 |
| >20a | ||||||
| 10 |
|

|
Double bond | >50 | — | N/A |
Assays against cells infected with SARS-CoV-2 variant Omicron BA1.
Fig. 2. Anti-SARS-CoV-2 dose response (left) and cytotoxicity (right) curves with respect to the log concentration in μM of the compound. Panel A: Compound 3b. Panel B: Compound 9e. Error bars of triplicates are shown.

In silico studies
To identify the potential target(s) behind the observed activity against SARS-CoV-2 of compounds 3b and 9e, a docking and virtual screening study followed by a molecular dynamics simulation (MD) was conducted against four SARS-CoV-2 proteins. The first selected potential target was the SARS-CoV-2 main protease (Mpro), a non-structural cysteine protease that is one of the key players in the release of 16 non-structural proteins involved in the virus replication.20 The second was the nucleocapsid phosphoprotein that packages the viral RNA into a helical ribonucleocapsid.21 The third selected target was the non-structural nsp10–nsp16 complex that has a 2′-O-methyltransferase (2′-O-MTase) activity involved in the methylation of the RNA cap structure.22,30 Finally, the receptor-binding domain (RBD) of the viral spike protein responsible for the viral entry into host cells was also virtually screened as the fourth plausible target.23 3D structures of the four target proteins were obtained from the Protein Data Bank (PDB) as follows: the SARS-CoV-2 main protease Mpro (PDB ID: 6LU7),31 the 2′-O-methyltransferase nsp10–nsp16 complex (PDB ID: 6W4H),30 the nucleocapsid phosphoprotein RNA binding domain (PDB IDs: 6VYO),29 and the RBD domain/ACE2/B(0)AT1 complex (PDB ID: 6M17).33 These 3D structures were selected based on their high resolution and lack of mutations.
Docking validation and virtual screening
First, we validated the use of the method MOE 2019.01 for the virtual screening study by self-docking co-crystallized ligands in the adopted crystal structures for the nsp10–nsp16 complex (PDB ID: 6W4H)29 and the SARS-CoV-2 main protease Mpro (PDB ID: 6LU7)30 with the respective co-crystallized ligands S-adenosyl-l-methionine (SAM) and the peptide-like inhibitor PRD_00221 (ESI† Fig. S2). The proteins were initially subjected to structure preparation and protonation state fixation, and then the London dG and GBVI/WSA dG scoring functions were used to assess the binding poses and binding interactions. The root mean square deviation (RMSD) of the best scoring poses was calculated using the DockRMSD online server (https://zhanggroup.org/DockRMSD/).31 Since the optimum RMSD for docking validation is conventionally ≤2 Å and the results obtained by MOE 2019.01 were within this range, the adopted docking protocol was considered reliable.
The binding potential of the 25 synthesised compounds was assessed and their potential binding affinity was reported as S-scores (MOE internal scoring function) and compared to those of the co-crystallized ligands, when applicable, for each of the four target proteins (ESI† Table S1). The 2D interactions of the molecules compared to those of the co-crystallized ligands showed that compounds 3b and 9e were among the best virtual hits, which is in line with the antiviral activity profile of these compounds (Table 1). The 2D binding interactions of the two compounds with the four-screened SARS-CoV-2 proteins are shown in Fig. 3.
Fig. 3. 2D interactions of the best 2 hits, compounds 3b and 9e. (A) Binding of co-crystallized ligand (PRD_002214), 3b and 9e with Mpro (PDB ID. 6LU7). (B) Binding of co-crystallized ligand (SAM), 3b and 9e with the nsp10–nsp16 complex (PDB 6W4H). (C) Binding of 3b and 9e with the nucleocapsid phosphoprotein (PDB 6VYO). (D) Binding of 3b and 9e with the RBD/ACE2 complex (PDB 6M17).

Docking studies showed that the peptide-like inhibitor PRD_002214 is covalently bound to Cys145 and forms seven other non-covalent interactions with the active pocket of Mpro (Fig. 3A and ESI† Table S2). Compound 3b forms six interactions: four as a H-bond donor with distances of 3.14 Å, 3.42 Å, 3.61 Å and 3.72 Å, respectively, and with energy scores of −0.6 kcal mol−1, −0.8 kcal mol−1, −0.8 kcal mol−1, and −1.6 kcal mol−1, respectively. It also indicated two pi–H interactions between the ligand's 6-membered ring and Thr25 and Thr26 in the pocket with distances of 4.39 Å and 4.22 Å, respectively, and with energy scores of −1.1 kcal mol−1, and −2.0 kcal mol−1 respectively (Fig. 3A and ESI† Table S2). This suggests that compound 3b might interact with this target protein. In contrast, compound 9e makes only two interactions with Mpro (Fig. 3A and ESI† Table S2). For the nsp10–nsp16 complex, we mapped strong 11 interactions with the co-crystallized ligand SAM (Fig. 3B and ESI† Table S2), while compounds 3b and 9e formed only two interactions, H bonds and pi–H interactions. Concerning the nucleocapsid phosphoprotein, the docking suggests that both compounds 3b and 9e form two interactions only (Fig. 3C). Finally, with the spike protein RBD/ACE2 complex (Fig. 3D), only one H-bond was observed between O62 of compound 3b and Glu165 and one hydrogen bond between O72 of compound 9e and Asp164, suggesting a reduced interaction with this target complex (ESI† Table S1).
Molecular dynamics simulation for compounds 3b and 9e
We then carried out a 100 ns MD simulation of 3b and 9e to assess their binding stability with the target Mpro and nsp10–nsp16 complex (ESI† data and Fig. S3–S6). To measure how much the protein and ligand conformations change along the MD simulation trajectory of the target protein–ligand complex, RMSD values were calculated for the proteins' Cα atomic coordinates and the screened ligands using GROMACS 2021.1.32,343b and Mpro formed a very stable complex reaching stability in the first nanoseconds of the simulation with very low perturbation (less than 1 Å) throughout the whole 100 ns simulation (Fig. 4A). Additionally, the stability of the complex was confirmed by the RMSD of 3b itself, as displayed in Fig. 4B, eliciting perturbations of less than 1 Å throughout the whole simulation. On the other hand, the RMSD of the nsp10–nsp16 complex Cα backbone in its complex with 3b showed small perturbations, while the RMSD of 3b itself gave very high fluctuations after 50 ns.
Fig. 4. Structural dynamics of compound 3b bound to Mpro (black curve) and the nsp10–nsp16 complex (blue curve); RMSD of the Cα backbone of the proteins in nm (A) and RMSD of 3b in nm along the MD trajectory (B).

Then the stability of 9e in complex with Mpro and the nsp10–nsp16 complex was assessed over a 100 ns simulation (Fig. 5). The RMSD of the protein referenced to the backbone was calculated. On the one hand, the complex of 9e bound to Mpro achieved stability at almost 5 ns, showing small perturbations (less than 1 Å) in the region from 65 ns to the end of the simulation (Fig. 5A, black curve). This indicates the stability of the protein upon interaction with 9e. On the other hand, the RMSD of 9e itself showed significant perturbations at the beginning (from 0 ns to 40 ns), representing the ligand jumping away from the binding pocket, and at the end (from 70 ns to 100 ns) (Fig. 5B, black curve). In parallel, the nsp10–nsp16 complex exhibited convergence at 12 ns with small perturbations (∼1 Å), thus indicating the stabilisation of the protein (ESI† Fig. S5). Besides that, the RMSD of 9e complexed to the nsp10–nsp16 complex exhibited a very low RMSD (less than 6 Å) and RMSD perturbations, which reflect the high 9e complex stability as shown in Fig. 5B.
Fig. 5. Structural dynamics of compound 9e bound to Mpro (black curve) and the nsp10–nsp16 complex (blue curve); RMSD of the Cα backbone of the proteins in nm (A) and RMSD of 9e in nm along the MD trajectory (B).

Stable binding of 3b and 9e should reflect the stability of the residues within the binding pocket. The rigidity and flexibility of such residues can be investigated through the root mean square fluctuation (RMSF) of the Cα atoms during the simulation (ESI† Fig. S5).35 The RMSF of the 3b and Mpro complex showed low fluctuations, especially Cys145 and His41 that constitute the catalytic dyad. 9e exhibited fluctuations (∼1.3–3.2 Å) in some residues, with higher fluctuations at residues (185 : 192) than 3b. In parallel, the nsp10–nsp16 complex shows residues within a 5 Å proximity from 3b and 9e, exhibiting lower fluctuations (∼0.4–1.4 Å) (ESI† Fig. S5). The collective results for the in silico studies suggest that the target of 3b is the main protease Mpro, while 9e preferentially interacts with the nsp10–nsp16 complex. To verify the in silico results, compounds 3b and 9e were tested in vitro for inhibition of the 2′-O-methyltransferase activity of the nsp10–nsp16 complex and nsp14 guanine-N7-methyltransferase activity, and in a cellular assay involving Mpro.
Biological target validation
Inhibition of the nsp10–nsp16 complex
The nsp10–nsp16 complex is responsible for catalysing the final step of the coronaviral mRNA capping. Nsp16 is a member of the 2′-O-MTase family, which catalyses the transfer of a methyl group to the RNA substrates from the methyl donor SAM. Nsp16 requires nsp10 for methyltransferase activity and stability. The in vitro activity of the SARS-CoV-2 nsp10–nsp16 complex was assessed by monitoring the transfer of 3H-SAM to the biotinylated N7-meGpppACCCCC RNA substrate (ESI†). The subsequently methylated RNA was captured using scintillation proximity assay (SPA) beads followed by quantifying the level of incorporated 3H-methyl by measuring the radioactivity level (counts per minute [CPM]).41 In dose response experiments (Fig. S7†), commercially available S-adenosyl-l-homocysteine (SAH) showed IC50 values of 3.9 μM and 0.22 μM for the nsp10–nsp16 complex and nsp14 methyltransferase activities, respectively (ESI† Fig. S7 and Table 2). However, 3b and 9e did not show any inhibitory effect on the nsp10–nsp16 2′-O-MTase activity, which did not support the computational analysis prediction for this target. Although 3b showed no inhibitory effect on the methyltransferase activity of nsp14, 9e showed a weak activity against nsp14 at high concentrations (Table 2 and ESI† Fig. S7).
Effect of 3b and 9e on the methyltransferase activities of the nsp10–nsp16 complex and nsp14. S-Adenosyl-l-homocysteine (SAH) was used as control.
| Compound name | IC50nsp10–nsp16 (μM) | Hill slope | IC50nsp14 (μM) | Hill slope |
|---|---|---|---|---|
| SAH | 3.9 | 1.1 | 0.22 | 0.8 |
| 3b | NI | NA | NI | NA |
| 9e | NI | NA | >100 | NA |
Inhibition of SARS-Cov-2 main protease (Mpro)
Mpro is a validated target for treating COVID-19 with the recent approval of the drug Paxlovid.9b Encouraged by the importance of this target and guided by the results obtained from the computational analysis through docking and molecular dynamics simulations, we tested the two hits for their ability to inhibit Mpro in a highly sensitive cell-based luciferase assay that we developed to monitor SARS-CoV-2 main protease activity.36 This gain-of-function assay is based on a reverse-nanoluciferase (Rev-Nluc) reporter in which two nanoluciferase domains are permuted and linked together by a cleavage site recognized by Mpro. Co-expression with the wild-type SARS-CoV-2 Mpro results in cleavage of the reporter and thereby a significant reduction in luciferase activity. The addition of an inhibitor of Mpro results in a dose-dependent restoration in luciferase activity. The assay is run in parallel on a wild-type and a catalytically inactive Mpro, to assess the specificity of the drug (details are provided in the ESI† Methods section). The commercially available GC376 compound was used as a positive control. The dose response curve showed that GC376 reduced Mpro activity, as seen by a gradual increase of nanoluciferase activity measured in the presence of Mpro WT (Fig. 6A), while the luciferase signal measured in the presence of the catalytically inactive mutant remained unchanged. Full inhibition of Mpro was achieved at 10 μM GC376 and the estimated IC50 was in the micromolar range.37–39 Only compound 3b showed a specific activity against Mpro in the Rev-Nluc assay from 10 μM, as indicated by an increase in the luciferase signal, which was not observed with the catalytically inactive Mpro mutant (Fig. 6B). An in vitro enzymatic assay on the purified protein confirmed the weak inhibitory activity of compound 3b (ESI† Fig. S8), while compound 9e was inactive. This observed activity of 3b against SARS-CoV-2 Mpro was consistent with the computational study.
Fig. 6. Cell-based assay luciferase assay to monitor SARS-CoV-2 main protease activity of the wild-type (WT, open symbols) and catalytically inactive C145A mutant (black symbols) of Mpro: (A) dose response curve of the positive control GC376. (B) Dose response curve of compound 3b. The inhibition was assessed using the Rev-Nluc-based assay. The assay was performed as described in the ESI† Methods section. The inhibitor concentrations correspond to 3-fold serial dilutions from 125 to 0.02 μM. The RLUs are shown as the mean ± SD of three independent experiments performed in technical triplicate.

Conclusions
In conclusion, to explore a new chemical space for the design of inhibitors of SARS-CoV-2, a chemical library of 25 fused-bridged dodecahydro-2a,6-epoxyazepino[3,4,5-c,d]indole compounds guided by natural product likeness, 3-D dimensionality and lead likeness was synthesised. The screening in a cellular system of viral infection led to the identification of 2 hits against the SARS-CoV-2 virus, confirming that following lead likeness guidelines and enhancing 3-D dimensionality increases the hit rate in the design of chemical libraries. Although the compounds also showed cytotoxicity, the two hits were confirmed to have antiviral activity against SARS-CoV-2 with an EC50 between 3.7 and 1.4 μM with an acceptable cytotoxicity difference. They also showed anti-viral activity against SARS-CoV-2 variant Omicron BA1, which consolidates their inhibition profile. Computational analysis through docking and molecular dynamics simulations was carried out against four main target proteins of SARS-CoV-2: main protease Mpro, nucleocapsid phosphoprotein, non-structural protein nsp10–nsp16 complex and RBD/ACE2 membrane glycoprotein complex. The analysis identified as possible binding targets either the main protease Mpro or the nsp10–nsp16 complex. A cellular assay using a reverse-nanoluciferase (Rev-Nluc) reporter system confirmed that compound 3b shows some activity against Mpro in cells and a weak activity in an in vitro enzymatic assay with purified Mpro. In contrast, an in vitro inhibition assay of the nsp10–nsp16 complex and nsp14 did not confirm the computational analysis suggesting the methyltransferases as a potential target. The confirmed hits 3b and 9e pave the way to the chemical optimisation of the fused-bridged dodecahydro-2a,6-epoxyazepino[3,4,5-c,d]indole skeleton for the inhibition of SARS-CoV-2. This also confirms the interest in inspired natural product scaffolds with improved drug likeness properties for the identification of novel hits with antiviral activity. Finally, the chemical pathway was designed to allow further modulation of the substituents. Further chemical optimisation is underway to increase antiviral activity and, in particular, to improve the activity against the potential target, the main protease Mpro.
Author contributions
HH designed, synthesised the library and wrote the chemistry, molecular properties and biological activity parts, and revised the manuscript. JC and FA set up and carried out the biological evaluation in cells and participated in the writing and revision of the manuscript. TK, KC and NN set up and carried out the cell-based assay for Mpro protease activity and NN proofread the manuscript. RKA conceptualized the in silico screening. AH and LB carried out the in silico study and primary writing of the relevant sections, and both of them participated equally in the in silico studies part. RKA, AH, and LB revised the results and data analysis and wrote the in silico studies. ED and AD developed and performed the Mpro enzymatic assay. AL, AD, and MV carried out the assay against the methyltransferase activity of nsp10–nsp16 and nsp14. SM resynthesised key compounds and revised the chemistry and biological activity parts. PBA supervised the project, obtained the grants, and wrote and revised the whole manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Fondation de France grant Programme Féderateur de Recherche sur SARS-CoV2 & COVID-19 PRF7 SARS-CoV2 drug pipeline project (to PBA). Vedadi's lab is funded by the US NIH 1U19AI171110-01. The authors thank Dr Benjamin BARDIAUX, Structural Bioinformatics Laboratory, Institute Pasteur, for constructive proofreading of the in silico studies part. The authors thank the Institut Pasteur and Fondation de France grants for the installation and the Drug pipeline against SARS-CoV-2 project. Sylvain Paisant and Marine Ghazarian are acknowledged for providing technical help for the Mpro cell-based assay. TK, KYC and NN were funded by the “URGENCE COVID-19” fundraising campaign of Institut Pasteur and by the Agence Nationale de la Recherche (grants ANR-18-CE18-0026, ANR-18-CE18-0028 and ANR-10-LABX-62-IBEID).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00149g
Notes and references
- Wu F. Zhao S. Yu B. Chen Y.-M. Wang W. Song Z.-G. Hu Y. Tao Z.-W. Tian J.-H. Pei Y.-Y. Yuan M.-L. Zhang Y.-L. Dai F.-H. Liu Y. Wang Q.-M. Zheng J.-J. Xu L. Holmes E. C. Zhang Y.-Z. Nature. 2020;579:265. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. Yang X. L. Wang X. G. Hu B. Zhang L. Zhang W. Si H. R. Zhu Y. Li B. Huang C. L. Chen H. D. Chen J. Luo Y. Guo H. Di Jiang R. Liu M. Q. Chen Y. Shen X. R. Wang X. Zheng X. S. Zhao K. Chen Q. J. Deng F. Liu L. L. Yan B. Zhan F. X. Wang Y. Y. Xiao G. F. Shi Z. L. Nature. 2020;579:270. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q. Guan X. Wu P. Wang X. Zhou L. Tong Y. Ren R. Leung K. S. M. Lau E. H. Y. Wong J. Y. Xing X. Xiang N. Wu Y. Li C. Chen Q. Li D. Liu T. Zhao J. Liu M. Tu W. Chen C. Jin L. Yang R. Wang Q. Zhou S. Wang R. Liu H. Luo Y. Liu Y. Shao G. Li H. Tao Z. Yang Y. Deng Z. Liu B. Ma Z. Zhang Y. Shi G. Lam T. T. Y. Wu J. T. Gao G. F. Cowling B. J. Yang B. Leung G. M. Feng Z. N. Engl. J. Med. 2020;382:1199. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya H. Kokudo N. Global Health Med. 2020;2:53. doi: 10.35772/ghm.2020.01040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Baltimore D. Bacteriol. Rev. 1971;35:235. doi: 10.1128/br.35.3.235-241.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Berman J. J., Group IV Viruses: Single-Stranded (+)Sense RNA, Taxonomic Guide to Infectious Diseases, 2012, pp. 237–246 [Google Scholar]
- Scavone C. Brusco S. Bertini M. Sportiello L. Rafaniello C. Zoccoli A. Berrino L. Racagni G. Rossi F. Capuano A. Br. J. Pharmacol. 2020;177:4813. doi: 10.1111/bph.15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. Cao R. Zhang L. Yang X. Liu J. Xu M. Shi Z. Hu Z. Zhong W. Xiao G. Cell Res. 2020;30:269. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreary E. K. Angus D. C. JAMA, J. Am. Med. Assoc. 2020;324:1041. doi: 10.1001/jama.2020.16337. [DOI] [PubMed] [Google Scholar]
- (a) Spinner C. D. Gottlieb R. L. Criner G. J. López J. R. A. Cattelan A. M. Viladomiu A. S. Ogbuagu O. Malhotra P. Mullane K. M. Castagna A. Chai L. Y. A. Roestenberg M. Tsang O. T. Y. Bernasconi E. le Turnier P. Chang S.-C. SenGupta D. Hyland R. H. Osinusi A. O. Cao H. Blair C. Wang H. Gaggar A. Brainard D. M. McPhail M. J. Bhagani S. Ahn M. Y. Sanyal A. J. Huhn G. Marty F. M. JAMA, J. Am. Med. Assoc. 2020;324:1048. doi: 10.1001/jama.2020.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Owen D. R. Allerton C. M. N. Anderson A. S. Aschenbrenner L. Avery M. Berritt S. Boras B. Cardin R. D. Carlo A. Coffman K. J. Dantonio A. Di L. Eng H. Ferre R. Gajiwala K. S. Gibson S. A. Greasley S. E. Hurst B. L. Kadar E. P. Kalgutkar A. S. Lee J. C. Lee J. Liu W. Mason S. W. Noell S. Novak J. J. Obach R. S. Ogilvie K. Patel N. C. Pettersson M. Rai D. K. Reese M. R. Sammons M. F. Sathish J. G. Singh R. S. P. Steppan C. M. Stewart A. E. Tuttle J. B. Updyke L. Verhoest P. R. Wei L. Yang Q. Zhu Y. Science. 2021;374:1586. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]; (c) Gao X. Qin B. Chen P. Zhu K. Hou P. Wojdyla J. A. Wang M. Cui S. Acta Pharm. Sin. B. 2021;11:237. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S. P. Gupta V. Virus Res. 2020;288:198114. doi: 10.1016/j.virusres.2020.198114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- On behalf of the COVID-19 Commission of Accademia Nazionale dei Lincei, Rome,; Forni G. Mantovani A. Cell Death Differ. 2021;28:626. doi: 10.1038/s41418-020-00720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B. Nielsen T. B. Casadevall A. JAMA Intern. Med. 2021;181:460. doi: 10.1001/jamainternmed.2020.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland G. A. Ovsyannikova I. G. Kennedy R. B. Lancet. 2020;396:1595. doi: 10.1016/S0140-6736(20)32137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T. Manickam M. Namasivayam V. Hayashi Y. Jung S. H. J. Med. Chem. 2016;59:6595. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C. Günther S. Preiser W. van der Werf S. Brodt H.-R. Becker S. Rabenau H. Panning M. Kolesnikova L. Fouchier R. A. M. Berger A. Burguiere A.-M. Cinatl J. Eickmann M. Escriou N. Grywna K. Kramme S. Manuguerra J.-C. Müller S. Rickerts V. Stürmer M. Vieth S. Klenk H.-D. Osterhaus A. D. M. E. Schmitz H. Doerr H. W. N. Engl. J. Med. 2003;348:1967. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Lau S. K. P. Lee P. Tsang A. K. L. Yip C. C. Y. Tse H. Lee R. A. So L.-Y. Lau Y.-L. Chan K.-H. Woo P. C. Y. Yuen K.-Y. J. Virol. 2011;85:11325. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman D. J. Cragg G. M. J. Nat. Prod. 2016;79:629. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Hung A. Ramek A. Wang Y. Kaya T. Wilson J. Clemons P. Young D. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6799. doi: 10.1073/pnas.1015271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway W. R. J. D. Isidro-Llobet A. Spring D. R. Nat. Commun. 2010;1:80. doi: 10.1038/ncomms1081. [DOI] [PubMed] [Google Scholar]
- Zhang L. Lin D. Sun X. Curth U. Drosten C. Sauerhering L. Becker S. Rox K. Hilgenfeld R. Science. 2020;368:409. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. Ye Q. Singh D. Cao Y. Diedrich J. K. Yatas III J. R. Villa E. Clevel D. W. Corbett K. D. Nat. Commun. 2021;12:502. doi: 10.1038/s41467-020-20768-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M. Lugari A. Posthuma C. C. Zevenhoven J. C. Bernard S. Betzi S. Imbert I. Canard B. Guillemot J.-C. Lécine P. J. Biol. Chem. 2014;289:25783. doi: 10.1074/jbc.M114.577353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Petitjean S. J. L. Koehler M. Zhang Q. Dumitru A. C. Chen W. Derclaye S. Vincent S. P. Soumillion P. Alsteens D. Nat. Commun. 2020;11:4541. doi: 10.1038/s41467-020-18319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Cao B. Wei Y. Shi M. Org. Biomol. Chem. 2019;17:3737. doi: 10.1039/C9OB00585D. [DOI] [PubMed] [Google Scholar]; (b) Flagstad T. Min G. Bonnet K. Morgentin R. Roche D. Clausen M. H. Nielsen T. E. Org. Biomol. Chem. 2016;14:4943. doi: 10.1039/C6OB00961A. [DOI] [PubMed] [Google Scholar]
- Xu C.-P. Xiao Z.-H. Zhuo B.-Q. Wang Y.-H. Huang P.-Q. Chem. Commun. 2010;46:7834. doi: 10.1039/C0CC01487G. [DOI] [PubMed] [Google Scholar]
- (a) Fernandes R. A. Bethi V. RSC Adv. 2014;4:40561. doi: 10.1039/C4RA07500E. [DOI] [Google Scholar]; (b) Shibuya M. Tomizawa M. Iwabuchi Y. J. J. Org. Chem. 2008;73:4750. doi: 10.1021/jo800634r. [DOI] [PubMed] [Google Scholar]
- Colomer I. Empson C. J. Craven P. Owen Z. Doveston R. G. Churcher I. Marsden S. P. Nelson A. Chem. Commun. 2016;52:7209. doi: 10.1039/C6CC03244C. [DOI] [PubMed] [Google Scholar]
- Sorokina M. Steinbeck C. J. J. Cheminf. 2019;11:55. doi: 10.1186/s13321-019-0378-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C., Michalska K., Jedrzejczak R., Maltseva N., Endres M., Godzik A., Kim Y. and Joachimiak A., Crystal structure of RNA binding domain of nucleocapsid phosphoprotein from SARS coronavirus 2, to be published
- Rosas-Lemus M. Minasov G. Shuvalova L. Inniss N. L. Kiryukhina O. Brunzelle J. Satchell K. J. F. Sci. Signaling. 2020;13:1202. doi: 10.1126/scisignal.abe1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z. Du X. Xu Y. Deng Y. Liu M. Zhao Y. Zhang B. Li X. Zhang L. Peng C. Duan Y. Yu J. Wang L. Yang K. Liu F. Jiang R. Yang X. You T. Liu X. Yang X. Bai F. Liu H. Liu X. Guddat L. W. Xu W. Xiao G. Qin C. Shi Z. Jiang H. Rao Z. Yang H. Nature. 2020;582:289. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Bell E. W. Zhang J. J. Cheminf. 2019;11:40. [Google Scholar]
- Yan R. Zhang Y. Li Y. Xia L. Guo Y. Zhou Q. Science. 2020;367:1444. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E., Abraham M. J., Hess B. and van der Spoel D., Gromacs 2021, 1 manual, 2021, 10.5281/zenodo.5053220 [DOI] [Google Scholar]
- Ferreira J. C. Fadl S. Villanueva A. J. Rabeh W. M. Front. Chem. 2021;9:491. doi: 10.3389/fchem.2021.692168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. Y. Krischuns T. Varga L. O. Harigua-Souiai E. Paisant S. Zettor A. Chiaravalli J. Delpal A. Courtney D. O'Brien A. Baker S. C. Decroly E. Isel C. Agou F. Jacob Y. Blondel A. Naffakh N. Antiviral Res. 2022;201:105272. doi: 10.1016/j.antiviral.2022.105272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froggatt H. M. Heaton B. E. Heaton N. S. J. Virol. 2020;94:e01265-01220. doi: 10.1128/JVI.01265-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W. Khan M. B. Fischer C. Arutyunova E. Lamer T. Shields J. Saffran H. A. McKay R. T. van Belkum M. J. Joyce M. A. Young H. S. Tyrrell D. L. Vederas J. C. Lemieux M. J. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iketani S. Forouhar F. Liu H. Hong S. J. Lin F.-Y. Nair M. S. Zask A. Huang Y. Xing L. Stockwell B. R. Chavez A. Ho D. D. Nat. Commun. 2021;12:2016. doi: 10.1038/s41467-021-22362-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J. Dufloo J. Hubert M. Monel B. Planas D. Rajah M. M. Planchais C. Porrot F. Guivel-Benhassine F. Van der Werf S. Casartelli N. Mouquet H. Bruel T. Schwartz O. EMBO J. 2020;39:e106267. doi: 10.15252/embj.2020106267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi A. K. Li F. Devkota K. Perveen S. Ghiabi P. Hajian T. Bolotokova A. Vedadi M. SLAS Discovery. 2021;26:757. doi: 10.1177/24725552211008863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.