

Abstract
Human Exo1 is a member of the RAD2 nuclease family with roles in replication, repair and recombination. Despite sharing significant amino acid sequence homology, the RAD2 proteins exhibit disparate nuclease properties and biological functions. In order to identify elements that dictate substrate selectivity within the RAD2 family, we sought to identify residues key to Exo1 nuclease activity and to characterize the molecular details of the human Exo1–DNA interaction. Site-specific mutagenesis studies demonstrate that amino acids D78, D173 and D225 are critical for Exo1 nuclease function. In addition, we show that the chemical nature of the 5′-terminus has a major impact on Exo1 nuclease efficiency, with a 5′-phosphate group stimulating degradation 10-fold and a 5′-biotin inhibiting degradation 10-fold (relative to a 5′-hydroxyl moiety). An abasic lesion located within a substrate DNA strand impedes Exo1 nucleolytic degradation, and a 5′-terminal abasic residue reduces nuclease efficiency 2-fold. Hydroxyl radical footprinting indicates that Exo1 binds predominantly along the minor groove of flap DNA, downstream of the junction. As will be discussed, our results favor the notion that the single-stranded DNA structure is pinched by the helical arch of the protein and not threaded through this key recognition loop. Furthermore, our studies indicate that significant, presumably biologically relevant, differences exist between the active site dynamics of Exo1 and Fen1.
INTRODUCTION
Nucleases function in many cellular processes, including DNA repair, recombination, replication and apoptosis. These enzymes are either non-specific in nature (i.e. bind generically to DNA) or specific for a unique DNA sequence, chemistry or structure. Defects in nuclease function have been associated with complex phenotypes, such as genetic instability and elevated cancer risk. The RAD2 family of nucleases is conserved from bacteriophage to human (1,2) and consists of a range of nuclease activities that operate in several biological pathways. The RAD2 family members maintain a conserved nuclease core, which is comprised of the ‘N’ (N-terminal) and ‘I’ (internal) domains. The eukaryotic proteins have been divided into three classes based on amino acid sequence identity, positioning of the N and I domains and substrate specificity.
RAD2 Class I consists of the XPG-like proteins that operate in nucleotide excision repair. XPG incises the target strand 3′ to the bubble-like, damage-containing structure formed as an intermediate during the repair process (3). Mutations in the human XPG gene are associated with the disorder xeroderma pigmentosum, characterized by hypersensitivity to sunlight and an increased likelihood of developing skin cancer (4). Little is known about how these proteins recognize target substrates or catalyze incision.
The RAD2 Class II family is composed of the Fen1-like proteins. These enzymes exhibit a flap endonuclease activity for bifurcated DNA structures produced by polymerase strand displacement or as intermediates during DNA recombination (5,6). The 5′-nuclease activity of Fen1 is also responsible for the excision of Okazaki RNA fragments, which are used as primers for lagging strand DNA synthesis during replication (7,8). FEN1 knockouts in yeast (Δrad27) exhibit a temperature-sensitive phenotype (9–11) and an increased expansion or contraction of simple repetitive DNA sequences (12) due to inadequate processing of replication intermediates. Mutations in the FEN1 gene of Saccharomyces cervisiae are also associated with increased sensitivity to the alkylating agent methylmethane sulfonate, but not to γ-rays, suggesting a prominent role in base excision repair (9).
RAD2 Class III consists of the Exo1-like enzymes reported in yeast, fly and mammals (13–20). Exo1 was first identified in Schizosaccharomyces pombe as a meiotically induced 5′→3′ exonuclease (21). The S.cerevisiae Exo1 protein was subsequently identified as an interacting partner of the mismatch recognition protein Msh2 (14). Human Exo1 has been shown to likewise interact with components of the mismatch recognition complex (18,22–24). Notably, yeast genetic studies have found that exo1 mutant cells exhibit a mutator phenotype consistent with a mismatch repair (MMR) defect (12,13). Moreover, germline variation has been observed in the human EXO1 gene of patients with hereditary nonpolyposis colorectal cancer (HNPCC) (as well as several atypical HNPCC forms), a dysplasia commonly associated with defects in MMR (25).
The nuclease activity of S.cerevisiae Exo1 has also been shown to be required for the in vitro recombination of linear DNA molecules with overlapping homology (26). Consistently, exo1 yeast mutants display a reduction in recombination of tandem, homologous repeats (15), indicating a prominent role in single-stranded annealing. Last, EXO1 transcription is highest in cell types or at developmental phases associated with meiotic recombination in yeast, fly and mice (16,20,21), suggesting that Exo1 contributes to meiotic recombination. Consistent with this notion, Δexo1 yeast mutants exhibit reduced reciprocal crossing-over, resulting in meiotic chromosome non-disjunction and spore death (27,28). In combination, these data suggest that Exo1 operates as a 5′-nuclease (i.e. either a 5′-flap endonuclease or a 5′→3′ exonuclease) during stages of homologous recombination and MMR.
The structures of several RAD2 family members, including Fen1 from Methanococcus jannashii (29) and Pyrococcus furiosus (30), T4 RNase H (31,32) and T5 exonuclease (33), have been solved. These structures reveal a similar core architecture, consisting of a conserved region with a β-sheet and two α-helices forming the base of the active site pocket. An additional notable feature of these proteins is the flexible loop (or helical arch), which is just large enough (8 × 25 Å) to accommodate a single-stranded DNA molecule. Based on this structural consideration, site-specific mutation effects and the fact that the loop region is lined with basic residues, it was proposed that the RAD2 enzymes translocate the target flap strand using a threading mechanism, in which the single strand is fully encircled by the loop domain (29,30,33). Consistent with this model, the 5′-end of the flap must be ‘free’ for efficient Fen1 cleavage, as bubble structures, complementary primers or proteins present at the 5′-terminus inhibit nuclease degradation (34,35). Moreover, the discoveries that a 5′-strepavidin–biotin complex prevents Fen1 activity and that Fen1 can be ‘trapped’ on a 5′-flap-containing DNA substrate upon annealing a complementary primer (34) further supported the threading mechanism. However, the observation that Fen1 is able to traverse and excise flap segments that contain large covalent DNA adducts and 11-nt branches argues against this mode of recognition (36). Thus, an alternative model has been proposed in which, like the thumb subdomain of reverse transcriptases (37), the Fen1 loop structure (helical arch) closes down upon the target strand, allowing the DNA to slide through the active site, while never completely encircling the single strand. Assuming this model, the previous 5′-end inhibitory effects may have resulted from interference with initial DNA recognition and not the prevention of DNA threading.
In vitro biochemical assays have revealed that Exo1 exhibits a 5′→3′ exonuclease activity and that the RAD2 nuclease domain of the human protein maintains a robust 5′-flap-specific endonuclease activity similar to that of Fen1 (17,38). In addition, Exo1, like Fen1 (7), can degrade the RNA strand of an RNA–DNA hybrid (its RNase H activity; 39). Yet despite these apparent biochemical similarities, the EXO1 and FEN1 yeast mutants display distinct phenotypic characteristics (see above), indicating important differences in their cellular functions. To gain further insight into the biochemical properties of Exo1, particularly in relation to Fen1, and to specifically address the issue of targeted recognition, we characterized wild-type and several site-specific Exo1 mutants for DNA binding and excision activities using a variety of DNA substrates and in vitro assays.
MATERIALS AND METHODS
Construction and purification of human Exo1 mutant proteins
To generate the human Exo1 mutants, site-directed mutagenesis was performed using an overlapping PCR method (40). Briefly, a site-specific mutant primer (D78A, 5′-CTC GTATTT GCT GGA TGT ACT-3′; D173A, 5′-AAA GCT AGG AGA GCC GAG TCC TCT G-3′; D225A, 5′-GAT GAC AGG TAG GCA CAA CCT GA-3′) was used in combination with primer 5′-GGC ACC ATG GGG ATA CAG GGA T-3′ to generate PCR fragment 1. PCR fragment 2 was generated using primers 5′-GGC ACC ATG GGG ATA CAG GGA T-3′ and 5′-CGG GAT CCT CAC TTC AAT TGT GGG GCA TC-3′. These two overlapping PCR products were then mixed at a ratio of 10:1 (fragment 1:fragment 2) and PCR fragment 3 was produced using primers 5′-GGC ACC ATG GGG ATA CAG GGA T-3′ and 5′-CGG GAT CCT CAC TTC AAT TGT GGG GCA TC-3′. Fragment 3 was subcloned into the NcoI and BamHI sites of pET-11d (Novagen, Madison, WI) following appropriate restriction enzyme digestion to create Hex1-N2-D78A, Hex1-N2-D173A and Hex1-N2-D225A. These plasmids were transformed into Escherichia coli BL21 (λDE3) and the Hex1-N2 RAD2 nuclease domain was purified as previously described (38). All mutants showed a similar expression level, except D78A, which was expressed at 50-fold less in terms of total protein quantity per cell. Different temperatures or alternative expression constructs did not improve D78A protein production. The concentration of all proteins was measured by the BCA colorimetric assay (Pierce, Rockford, IL) using BSA as the standard according to the manufacturer’s instructions, and confirmed by SDS–PAGE analysis as shown in Figure 1.
Figure 1.
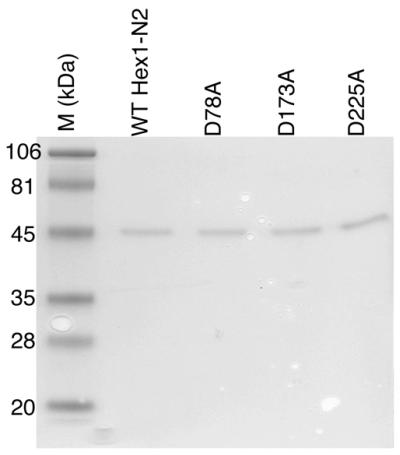
Wild-type (WT) and mutant Hex1-N2 proteins. Proteins (1 µg) were separated by 10% SDS–PAGE and stained with Coomassie brilliant blue R250M. Molecular weight standards (M) are shown in kDa.
Preparation of DNA substrates
For purification of DNA oligonucleotides, DNAs were electrophoresed on a 20% denaturing polyacrylamide gel, visualized by UV shadowing, and recovered from the gel as previously described (38). For 3′-labeling, oligonucleotides were incubated with terminal deoxynucleotidyl transferase (NEB, Beverly, MA) and [α-32P]dCTP as instructed by the manufacturer. 5′-End-labeling was carried out using T4 polynucleotide kinase and [γ-32P]ATP. Excess labeled dCTP or ATP was removed using a Microspin G-25 column and the labeled strand was then annealed to an equal molar concentration of the complementary oligonucleotide as described (41). DNA substrates used in these studies were reported previously (38); sequence information is provided in the relevant table. 5′-Biotin-labeled DNA was synthesized by Life Technology. Abasic DNA was from Genesis and 5′-phosphate substrates were obtained by T4 polynucleotide kinase reaction using unlabeled ATP.
Nuclease assays
Nuclease assays were performed using 0.05 ng protein (Hex1-N2 or hFen1) and 1 pmol end-labeled DNA substrate (38) in a final volume of 10 µl containing 20 mM HEPES pH 7.5, 50 mM KCl, 0.5 mM DTT, 5 mM MgCl2, 0.05% Triton X-100, 100 µg/ml BSA and 5% glycerol, unless otherwise noted. Reactions were carried out at 37°C for 20 min and stopped immediately by adding 4 µl of formamide loading buffer and heating at 90°C for 5 min. An aliquot was subsequently analyzed on a 16% polyacrylamide–8 M urea denaturing gel to determine the percent conversion of substrate to mononucleotide or 10 nt product (38).
Electrophoretic mobility shift assay (EMSA)
A 41mer continuous flap oligonucleotide (see Results) was 5′-labeled and annealed as above. To measure apparent Kd (i.e. the protein concentration needed to shift 50% of the available DNA substrate), wild-type or mutant Hex1-N2 protein (at the concentrations indicated) was incubated with 1 pmol DNA substrate for 5 min on ice in 10 µl of binding buffer (20 mM HEPES–KOH pH 7.5, 50 mM KCl, 50 µg/ml BSA, 0.025% Triton X-100, 10% glycerol and 4 mM EDTA). Binding reactions (8 µl) were then separated on a 5% non-denaturing gel. Band visualization and quantification of bound and unbound DNA was achieved using a Molecular Dynamics (Sunnyvale, CA) STORM 860 Phosphorimager and Molecular Dynamics ImageQuant v.2.10 software.
Hydroxyl radical footprinting protection assay
The 41mer continuous flap DNA was labeled at the 5′-end, annealed and purified as above. Binary protein–DNA complexes were formed by incubating 1 pmol of the labeled DNA substrate with various amounts of either Hex1-N2 or Hex1-N2 D225A mutant protein in 10 µl of binding buffer (see above) for 5 min on ice. An aliquot of 40 µl of 0.32% H2O2 and 5 mM sodium ascorbate solution was then added, followed immediately by the addition of 2 or 4 µl of 50 mM Fe(EDTA)2. The cleavage reaction was continued for 30 s and was stopped with 50 µl of 0.1 M thiourea (42). Samples were then processed and footprinting signals were visualized as described (43). The locations of protected bases were mapped relative to various chemical sequencing ladders, which were generated as described (44).
RESULTS
Functional characterization of residues in the hExo1 active site
Based on previous Fen1 studies (45), residues D78, D173 and D225 of Exo1, which were predicted to be critical for substrate binding and/or cleavage, were mutated to alanine (see alignment available online as Supplementary Material). Given the past difficulties in expressing and purifying the full-length hExo1 protein (17,39) and the in vitro functional similarities of the RAD2 nuclease domain (Hex1-N2) to partially purified full-length material (38), these site-specific mutants were characterized as nuclease fragments (Fig. 1). We then measured the ability of the resulting mutant proteins to degrade 5′-32P-end-labeled blunt end duplex DNAs (observed as the release of mononucleotides; Fig. 2A) and flap DNA substrates (observed as the release of 10-nt single-stranded fragments; Fig. 2B). Each of the mutants exhibited a 100- or 1000-fold reduced specific activity relative to the wild-type in degrading duplex blunt end or 5′-flap DNAs, respectively (Table 1). A mutation at residue T169, a position previously suggested to be involved in the catalytic reaction (45), only resulted in a 3-fold reduction in enzymatic activity, indicating a non-essential role for this amino acid in recognition or catalysis (data not shown).
Figure 2.

Exo- and endonuclease activities of wild-type (Wt) and mutant Exo1 proteins. (A) An aliquot of 1 pmol double-stranded (D.S.) blunt end DNA substrate was incubated with 0.01, 0.1 and 0.5 ng of the indicated protein and the nuclease reactions were analyzed on a polyacrylamide denaturing gel (see Materials and Methods). The positions of the substrate and mononucleotide (1 nt) are shown. The far left lane indicates the no protein control. (B) Endonuclease activities of wild-type (Wt) and mutant Hex1-N2 proteins were measured using a 5′-flap DNA substrate. The position of the flap junction incision product (10 nt) is shown. The substrates used in these assays and the specific activities of these proteins are given in Table 1.
Table 1. Specific activities of wild-type and mutant Hex1-N2 proteins for double-stranded (D.S.) blunt end and 5′-flap DNA substrates.
| Specific activity (D.S.) (pmol min–1 mg–1) | Fold reduction | Specific activity (5′-flap) (pmol min–1 mg–1) | Fold reduction | |
|---|---|---|---|---|
| Hex1-N2 | (0.97 ± 0.3) × 105 | (0.89 ± 0.2) × 105 | ||
| D78A | (0.47 ± 0.2) × 103 | 206 | (0.86 × 0.1) × 102 | 1035 |
| D173A | (1.74 ± 0.4) × 103 | 56 | (1.33 ± 0.1) × 102 | 669 |
| D225A | (1.00 ± 0.1) × 103 | 97 | (2.34 ± 0.3) × 102 | 380 |
The averages and standard deviations of three independent nuclease reactions are shown, as well as the fold reduction in activity compared to wild-type Hex1-N2. The oligonucleotides used to create the double-stranded DNA substrate were *5′-TAG AGG ATC CCC GCT AGC GGG TAC CGA GCT CGA ATT CAC TGG-3′ and 5′-CCA GTG AAT TCG AGC TCG GTA CCC GCT AGC GGG GAT CCT CTA-3′, and to create the 5′-flap DNA were *5′-ATT GGT TAT TTA CCG AGC TCG AAT TCA CTG G-3′, 5′-TAG AGG ATC CCC GCT AGC GGG-3′ and 5′-CCA GTG AAT TCG AGC TCG GTA CCC GCT AGC GGG GAT CCT CTA-3′. The asterisk indicates the oligonucleotide that was 32P-labeled. See Materials and Methods for details.
We subsequently measured the ability of the mutant proteins to form stable protein–DNA complexes with either double-stranded blunt end or 5′-flap DNA (see legend to Table 1 for substrate descriptions). In these experiments we observed no obvious protein–DNA complex with blunt end duplex DNAs, yet observed multiple complexes with our initial 5′-flap substrates (data not shown). Varying the pH (pH 6–8), salt concentration (0–200 mM KCl or NaCl) or EDTA concentration (which inhibits nuclease degradation) had no beneficial effect on these outcomes. No binding was observed under conditions hat permit incision and non-productive metals, such as cobalt, calcium and iron, did not improve complex formation (data not shown).
The multiple complexes detected with the 5′-flap structures and the lack of complex formation with the blunt end substrates suggested that Exo1 may bind the flap region and subsequently the blunt ends cooperatively. Thus, in order to obtain a single resolved complex, we designed a continuous 5′-flap DNA substrate without a double-stranded blunt end (Fig. 3A). Hex1-N2, as well as the mutant proteins, was found to form a single, stable DNA–protein complex with this DNA (Fig. 3B). Importantly, Hex1-N2 exhibited a flap endonuclease activity of 1.0 ± 0.2 × 105 pmol min-1 mg-1 on this substrate, similar to what has been observed for other flap DNAs (38). A more comprehensive binding analysis revealed that the mutant protein D225A binds with better than wild-type affinity (by 5-fold), while the D173A mutant exhibits wild-type binding affinity and D78A demonstrates a 5-fold lower affinity (Fig. 3).
Figure 3.

Binding affinities of wild-type (Wt) and mutant Exo1 proteins. (A) Depiction of the continuous 5 nt 5′-flap substrate (5′-GAG TTG TAC CGA GTT CTC GGTACC CGCTAG CTT GCT AGC GG-3′) used in binding experiments. (B) Representative EMSA. An aliquot of 1 pmol of DNA substrate was incubated with different amounts of protein (indicated) and binding reactions were analyzed on a 5% non-denaturing gel (see Materials and Methods). The positions of the protein–DNA complex (DNA-Protein) and unbound DNA (Free DNA) are shown. (C) Binding plot of the Exo1 proteins. Percent binding was determined as [(bound value)/(bound value + unbound value)] × 100 by phosphorimager quantification. Proteins (ng) are indicated. (D) Apparent Kd of Exo1 proteins. Apparent Kd values were determined as the protein concentration that results in 50% of labeled DNA substrate being bound. Values shown are the average and standard deviation of three different experiments.
Effects of DNA modifications on the nuclease activities of hExo1
To determine the effect of 5′-end chemistry on the nuclease potency of Exo1, flap or blunt end DNAs containing either a 5′-hydroxyl, 5′-phosphate or 5′-biotin were generated. Nuclease assays, first using blunt end DNA substrates, revealed that a 5′-biotin moiety inhibits Exo1 degradative activity by at least 10-fold, relative to DNAs harboring a 5′-hydroxyl group (Fig. 4A and Table 2). Conversely, a 5′-phosphate group was found to stimulate the blunt end exonuclease activity of Exo1 ∼10-fold. Subsequent studies using flap-containing DNA substrates found similar inhibitory and stimulatory effects on Exo1 nuclease activity for biotin and phosphate, respectively (Fig. 4B and Table 2).
Figure 4.

Effect of 5′ modifications on the nuclease activities of Exo1 and Fen1. (A) 5′ Modification effects on degradation of blunt end double-stranded DNA substrates by Exo1 (Hex1-N2). As a 5′→3′ exonuclease, Exo1 (at 0.1 and 1 ng) has been shown to degrade 3′-labeled substrates down to 6 nt, before dissociating (38); a single mononucleotide (1 nt) is released with 5′-labeled DNAs. (B) 5′ Modification effects on degradation of 5′-flap substrates by Exo1 and Fen1. Exo1 (1 ng) generates the expected 6- and 10-nt products with 3′- and 5′-labeled DNAs, respectively. hFen1 (1 ng), as primarily a 5′-flap endonuclease, produces 21- and 10-nt products with 3′- and 5′-labeled DNAs, respectively. 5′-Biotin- (b), 5′-hydroxyl (OH) or 5′-phosphate (P) oligonucleotides (32mer) were 3′-end labeled and then annealed to the appropriate complementary oligonucleotide to form blunt end or 5′-flap DNAs (see Materials and Methods). The nucleotide sequences of the oligonucleotides used in these experiments are given in Table 1. Specific degradation activities of Hex1-N2 and hFen1 are shown in Table 2. A 5′-32P-labeled DNA substrate (shown on the right in both panels) was used as a comparative control, with the asterisk denoting the position of the radiolabel.
Table 2. Effects of 5′-end modifications on the exo/endonuclease activities of the Hex1-N2 and hFen1 enzymes.
| 5′-End modification | Double-stranded | 5′-Flap | |||||
|---|---|---|---|---|---|---|---|
| Hex1-N2 | hFen1 | Hex1-N2 | hFen1 | ||||
| pmol min–1 mg–1 | RA | pmol min–1 mg–1 | pmol min–1 mg–1 | RA | pmol min–1 mg–1 | RA | |
| Hydroxyl (OH) | (1.0 ± 0.3) × 104 | 1 | n.d. | (1.0 ± 0.1) × 104 | 1 | (1.2 ± 0.1) × 105 | 1 |
| Phosphate | (1.3 ± 0.4) × 105 | 13 | n.d. | (0.6 ± 0.2) × 105 | 6 | (2.0 ± 0.4) × 105 | 1.7 |
| Biotin | (1.0 ± 0.3) × 103 | 0.1 | n.d. | (0.8 ± 0.2) × 103 | 0.1 | (1.4 ± 0.3) × 105 | 1.2 |
The averages and standard deviations of three independent experiments are shown, as well as relative activity (RA) compared to substrates containing a 5′-OH group. Substrates are as described in Table 1. See Materials and Methods for details. n.d., not determined.
For comparison, we also examined the effects of these 5′-moieties on the flap endonuclease activity of purified human Fen1 protein (38). These experiments revealed that the nuclease potency of hFen1 was not affected by a 5′-biotin group, whereas a 5′-phosphate group was found to stimulate its activity 1.7-fold (similar to that seen by Wu et al.; 46). This pattern of inhibition and stimulation of Fen1 is noticeably distinct from what was observed with the nuclease domain of hExo1 (Fig. 4 and Table 2).
To determine whether the nuclease activity of hExo1 is affected by the presence of a common DNA lesion/intermediate, i.e. an internal abasic site or a 5′-abasic residue (47), corresponding DNA substrates were created. With DNAs containing a centrally located abasic site analog tetrahydrofuran (F), the hExo1 nuclease domain was able to degrade up to one to two bases before the abasic site, before pausing. At high enzyme concentrations, bypass of the lesion was detected (Fig. 5, 41F). When the abasic site was positioned at the 5′-terminus within a nick (mimicking a base excision repair intermediate; 47), hExo1 was able to excise past this modification (Fig. 5, 41IF), but at an efficiency ≥2-fold less than the comparable 5′-OH nicked double-stranded DNA substrate (data not shown).
Figure 5.

Effect of an internal abasic site or a nick containing a 5′-abasic residue on the nuclease activity of Exo1. (A) Schematic of the duplex DNA substrates used. F, the tetrahydrofuran residue, is a synthetic abasic site analog (51). (Top) The intact abasic site-containing duplex (41F). (Bottom) The single-strand nick 5′-abasic residue-containing duplex (41IF). The asterisk indicates the location at which these DNAs were 32P-labeled for the experiments in (B). (B) Hex1-N2 degradation of internal and 5′-F-containing substrates. An aliquot of 1 pmol of substrate was incubated with 0, 0.01 or 0.1 ng wild-type Hex1-N2 protein for 20 min at 37°C and the reactions were analyzed on 20% polyacrylamide–8 M urea denaturing gels (see Materials and Methods). The arrows indicate the sizes of the prominent DNA products (22 and 6 nt) and the location of the abasic residue (F).
Defining the molecular interface of hExo1 with substrate DNA
As shown above, the hExo1 nuclease domain is capable of forming stable protein–DNA complexes with continuous 5′-flap substrates. To determine the molecular nature of this interaction, we employed the chemical footprinting reagent hydroxyl radical (OH·) (42). As shown in Figure 6, hExo1 protects five bases on the strand containing the single-stranded 5′-flap and eight bases on the complementary strand. The Exo1-protected region on the flap strand begins three bases 3′ of the flap junction. Notably, the footprinted regions are downstream of the disjoint and offset by 2–3 bp in the 3′ direction (Fig. 6B). OH· footprinting experiments using hFen1 protein, under either Exo1 binding conditions (see Materials and Methods) or the binding conditions of Shen et al. (45), produced no obvious protein-protected region. This result may suggest that hExo1 has a longer (more stable) complex half-life than hFen1 with DNA substrates containing a short 5′-flap, or that our conditions are not optimal for stable Fen1 binding.
Figure 6.

The DNA footprint of Exo1 bound to the continuous 5′-flap substrate. (A) Hydroxyl radical footprinting reactions with Hex1-N2 and D225A mutant protein bound to 5′-flap DNA. A representative phosphorimager scan of three independent experiments is shown. The first lane (far left) is the no cleavage control. The – lanes are the no protein controls. Wild-type Hex1-N2 was used at 0.956 and 1.912 µg and the D225A Hex1-N2 mutant at 0.21 and 0.63 µg (from left to right). The A+G chemical sequencing ladder is shown, as are relevant nucleotide positions in the continuous 5′-flap substrate of (B). (B) Summary of the hydroxyl radical footprinting data. The filled vertical bars above or below each base indicate sites of protection from cleavage. The height indicates the relative strength of footprinting protection. The arrow indicates the site of endonuclease cleavage, between positions T5 and G6. While the data in this panel are the footprint of wild-type Hex1-N2, an essentially identical footprint was observed with the D225A Hex1-N2 mutant [see (A)].
DISCUSSION
Human Exo1 exhibits amino acid sequence similarity, as well as overlapping substrate specificities, with members of the RAD2 nuclease family, most notably human Fen1. However, there are important differences not only in the biological contributions of these proteins, but also in the nuclease potency of these enzymes for various nucleic acid substrates. In particular, Exo1 has been found to exhibit a much more robust 5′→3′ exonuclease activity than Fen1 (38 and references within), which displays a preference for unannealed 5′-tails or flap structures (1). In order to gain insight into the active site dynamics of Exo1, we designed several site-specific mutants (Fig. 7, left) and examined the biochemical activities of these protein derivatives.
Figure 7.

Summary model of the Exo1–DNA interface. (Left) The active site residues of hExo1 characterized herein. A hExo1 active site model (yellow protein structure diagram) was achieved by structural and multiple sequence alignments [Ceslovas Venclovas, Lawrence Livermore National Laboratory; see Nucleic Acids Research, 26 (16), cover]. Residues D78, D173 and D225 of hExo1 are shown as space-filled atoms. Also shown is the Methanococcus jannaschii Fen1 structure (blue ribbon diagram; 29) and the two Mn2+ ions in its active site (green, space-filled). The model of hExo1 and the MjFen1 structure were aligned using the LGA program (52). (Right) The DNA protection sites of hExo1 on a 5′-flap substrate based on the footprinting experiments (Fig. 6). The protected sites are indicated by space-filled atoms for the corresponding sugars. Because DNA hydrolysis requires direct contact with Mn2+ ions, the DNA likely binds with the branch point (arrow) between the active site Mn2+ ions shown in the image on the left. The figures were was created with vmd (53) using STRIDE (54) and raster 3D (55).
As anticipated, mutagenesis at conserved residue positions in both hFen1 and hExo1 produced generally similar biochemical outcomes. For instance, the D86A mutant of Fen1 (45) and the equivalent D78A Exo1 mutant were both found to exhibit major reductions in incision capacity, as well as mild 5–10-fold reductions in DNA-binding affinity. In addition, the D181A mutant of hFen1 (45), like the D173A mutant of Exo1, was nucleolytically inactive, while maintaining wild-type (or better) DNA-binding capacity. Recent biophysical evidence (48), coupled with earlier crystallography results, strongly suggests that this amino acid in hFen1 is involved in direct metal binding. Yet despite the similarities in active site composition, there were noticeable differences in the outcomes of certain Exo1 and Fen1 mutations. In particular, whereas the D233A Fen1 mutant was shown to have reduced DNA-binding activity (as determined by a competition assay with the wild-type enzyme; 45), we found that the equivalent D225A Exo1 mutant exhibited a 5-fold increase in DNA-binding affinity. These results demonstrate not only the functional importance of these conserved acidic residues in Exo1 and Fen1 (and the RAD2 family in general), but also suggest that the human proteins, while displaying generally similar properties, possess distinct active site dynamics that may influence divalent metal coordination, substrate specificity and their mode of action.
As seen with hFen1 (36,46) and the S.pombe Fen1 equivalent (Rad2p; 49), we observed complex (although differing from Fen1) effects of 5′-end modifications on Exo1 nuclease activity. In particular, 5′-phosphate residues significantly enhanced the 5′-nuclease activity of Exo1, while 5′-biotin groups inhibited this activity. These results suggest that the 5′-end may be the first site of DNA interaction by Exo1 and that the nature of the 5′-terminus may determine the biological affinity (and thus in vivo targets) of the RAD2 proteins.
Hydroxyl radical footprinting studies show that Exo1 protects a region on both DNA strands downstream of the disjoint and offset 2–3 bp in the 3′ direction. Assuming that the double-stranded portion of the 5′-flap is B-form DNA, this pattern of protection (i.e. offset by 2–3 bp in the 3′ direction) is consistent with the interaction taking place in the minor groove of the substrate (Fig. 7, right); binding of hExo1 in the major groove would have generated a footprint that is offset in the 5′ direction (42). Moreover, the fact that the hExo1 nuclease domain most noticeably protects the double-stranded region 3′ to the flap junction and not the single-stranded flap itself, appears to argue that this protein does not encircle the target DNA strand and thus employ a threading mechanism. Thus, as suggested for Fen1 (36), we propose that hExo1 first interacts with a free 5′-end and, where a single-stranded tail exists, pinches the flap structure with its helical arch domain, allowing the protein to slide down the single-stranded segment until it reaches the double-stranded junction, where the enzyme forms a stable complex prior to endonuclease cleavage. The loop domain likely makes subtle (i.e. less protective) contacts with the target flap region to orient the enzyme for eventual catalysis. Recent ethylation interference studies using the 5′-nuclease domain of E.coli DNA polymerase I suggest that its single-stranded tail interactions are primarily with the phosphate backbone (50).
In the context of RAD2 nuclease recognition, it is important to note that there are significant differences between our hExo1 footprint and the footprint previously reported for hFen1 (35). Barnes et al. (35), using a low resolution micrococcal nuclease footprinting assay, reported that RTH1/Fen1 protects a 10–25-nt region on a 73-nt-long 5′-flap and not the duplex region. We, unfortunately, for reasons presently unclear, were unable (under multiple binding conditions, at either 0 or 37°C) to obtain a high resolution footprint with hFen1 on the 10-nt continuous 5′-flap DNA substrate. Yet, we emphasize that our footprinting pattern is consistent with the ethylation interference studies of Xu et al. (50), who reported that the exonuclease domain of E.coli DNA polymerase I binds one face of the DNA helix downstream of the cleavage site.
In closing, despite the similarities between human Fen1 and Exo1, there is emerging evidence that these two family members have important differences that presumably give rise to their disparate biochemical activities, and thus biological functions. Comparative structural studies aimed at determining the molecular details of appropriate protein–DNA complexes should shed light on these enigmatic issues.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Masood Hadi, Binghui Shen and Michael P. Thelen for insightful comments during the course of these studies. This work was performed under the auspices of the US Department of Energy by the University of California, Lawrence Livermore National Laboratory under contract no. W-7405-Eng-48 and supported by NIH grant CA79056 to D.M.W. III.
REFERENCES
- 1.Lieber M.R. (1997) The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays, 19, 233–240. [DOI] [PubMed] [Google Scholar]
- 2.Ceska T.A. and Sayers,J.R. (1998) Structure-specific DNA cleavage by 5′ nucleases. Trends Biochem. Sci., 23, 331–336. [DOI] [PubMed] [Google Scholar]
- 3.Wood R.D. (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie, 81, 39–44. [DOI] [PubMed] [Google Scholar]
- 4.de Bore J. and Hoeijmakers,J.H. (2000) Nucleotide excision repair and human syndromes. Carcinogenesis, 21, 453–460. [DOI] [PubMed] [Google Scholar]
- 5.Harrington J.J. and Lieber,M.R. (1994) Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev., 8, 1344–1355. [DOI] [PubMed] [Google Scholar]
- 6.Harrington J.J. and Lieber,M.R. (1994) The characterization of a mammalian DNA structure-specific endonuclease. EMBO J., 13, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waga S., Bauer,G. and Stillman,B. (1994) Reconstitution of complete SV40 DNA replication with purified replication factors. J. Biol. Chem., 269, 10923–10934. [PubMed] [Google Scholar]
- 8.Turchi J.J., Huang,L., Murante,R.S., Kim,Y. and Bambara,R.A. (1994) Enzymatic completion of mammalian lagging-strand DNA replication. Proc. Natl Acad. Sci. USA, 91, 9803–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reagan M.S., Pittenger,C., Siede,W. and Friedberg,E.C. (1995) Characterization of a mutant strain of Saccharomyces cerevisiae with a deletion of the RAD27 gene, a structural homolog of the RAD2 nucleotide excision repair gene. J. Bacteriol., 177, 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sommers C.H., Miller,E.J., Dujon,B., Prakash,S. and Prakash,L. (1995) Conditional lethality of null mutations in RTH1 that encodes the yeast counterpart of a mammalian 5′- to 3′-exonuclease required for lagging strand DNA synthesis in reconstituted systems. J. Biol. Chem., 270, 4193–4196. [DOI] [PubMed] [Google Scholar]
- 11.Vallen E.A. and Cross,F.R. (1995) Mutations in RAD27 define a potential link between G1 cyclins and DNA replication. Mol. Cell. Biol., 15, 4291–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tishkoff D.X., Filosi,N., Gaida,G.M. and Kolodner,R.D. (1997) A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell, 88, 253–263. [DOI] [PubMed] [Google Scholar]
- 13.Szankasi P. and Smith,G.R. (1995) A role for exonuclease I from S. pombe in mutation avoidance and mismatch correction. Science, 267, 1166–1169. [DOI] [PubMed] [Google Scholar]
- 14.Tishkoff D.X., Boerger,A.L., Bertrand,P., Filosi,N., Gaida,G.M., Kane,M.F. and Kolodner,R.D. (1997) Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl Acad. Sci. USA, 94, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiorentini P., Huang,K.N., Tishkoff,D.X., Kolodner,R.D. and Symington,L.S. (1997) Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro.Mol. Cell. Biol., 17, 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Digilio F.A., Pannuti,A., Lucchesi,J.C., Furia,M. and Polito,L.C. (1996) Tosca: a Drosophila gene encoding a nuclease specifically expressed in the female germline. Dev. Biol., 178, 90–100. [DOI] [PubMed] [Google Scholar]
- 17.Wilson D.M., Carney,J.P., Coleman,M.A., Adamson,A.W., Christensen,M. and Lamerdin,J.E. (1998) Hex1: a new human Rad2 nuclease family member with homology to yeast exonuclease 1. Nucleic Acids Res., 26, 3762–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmutte C., Marinescu,R.C., Sadoff,M.M., Guerrette,S., Overhauser,J. and Fishel,R. (1998) Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res., 58, 4537–4542. [PubMed] [Google Scholar]
- 19.Tishkoff D.X., Amin,N.S., Viars,C.S., Arden,K.C. and Kolodner,R.D. (1998) Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer Res., 58, 5027–5031. [PubMed] [Google Scholar]
- 20.Lee B.I., Shannon,M., Stubbs,L. and Wilson,D.M. (1999) Expression specificity of the mouse exonuclease 1 (mExo1) gene. Nucleic Acids Res., 27, 4114–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szankasi P. and Smith,G.R. (1992) A DNA exonuclease induced during meiosis of Schizosaccharomyces pombe. J. Biol. Chem., 267, 3014–3023. [PubMed] [Google Scholar]
- 22.Rasmussen L.J., Rasmussen,M., Lee,B.I., Rasmussen,A.K., Wilson,D.M., Nielsen,F.C. and Bisgaard,H.C. (2000) Identification of factors interacting with hMSH2 in the fetal liver utilizing the yeast two-hybrid system. In vivo interaction through the C-terminal domains of hExo1 and hMSH2 and comparative expression analysis. Mutat. Res., 460, 41–52. [DOI] [PubMed] [Google Scholar]
- 23.Jager A.C., Rasmussen,M., Bisgaard,H.C., Singh,K.K., Nielsen,F.C. and Rasmussen,L.J. (2001) HNPCC mutations in the human DNA mismatch repair gene hMLH1 influence assembly of hMutLalpha and hMLH1-hEXO1 complexes. Oncogene, 20, 3590–3595. [DOI] [PubMed] [Google Scholar]
- 24.Schmutte C., Sadoff,M.M., Shim,K.S., Acharya,S. and Fishel,R. (2001) The interaction of DNA mismatch repair proteins with human exonuclease I. J. Biol. Chem., 26, 26. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y., Berends,M.J., Post,J.G., Mensink,R.G., Verlind,E., Van Der Sluis,T., Kempinga,C., Sijmons,R.H., van der Zee,A.G., Hollema,H., Kleibeuker,J.H., Buys,C.H. and Hofstra,R.M. (2001) Germline mutations of EXO1 gene in patients with hereditary nonpolyposis colorectal cancer (HNPCC) and atypical HNPCC forms. Gastroenterology, 120, 1580–1587. [DOI] [PubMed] [Google Scholar]
- 26.Huang K.N. and Symington,L.S. (1993) A 5′-3′ exonuclease from Saccharomyces cerevisiae is required for in vitro recombination between linear DNA molecules with overlapping homology. Mol. Cell. Biol., 13, 3125–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khazanehdari K.A. and Borts,R.H. (2000) EXO1 and MSH4 differentially affect crossing-over and segregation. Chromosoma, 109, 94–102. [DOI] [PubMed] [Google Scholar]
- 28.Tsubouchi H. and Ogawa,H. (2000) Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell, 11, 2221–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang K.Y., Baek,K., Kim,H.Y. and Cho,Y. (1998) The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nature Struct. Biol., 5, 707–713. [DOI] [PubMed] [Google Scholar]
- 30.Hosfield D.J., Mol,C.D., Shen,B. and Tainer,J.A. (1998) Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell, 95, 135–146. [DOI] [PubMed] [Google Scholar]
- 31.Mueser T.C., Nossal,N.G. and Hyde,C.C. (1996) Structure of bacteriophage T4 RNase H, a 5′ to 3′ RNA-DNA and DNA-DNA exonuclease with sequence similarity to the RAD2 family of eukaryotic proteins. Cell, 85, 1101–1112. [DOI] [PubMed] [Google Scholar]
- 32.Bhagwat M., Meara,D. and Nossal,N.G. (1997) Identification of residues of T4 RNase H required for catalysis and DNA binding. J. Biol. Chem., 272, 28531–28538. [DOI] [PubMed] [Google Scholar]
- 33.Ceska T.A., Sayers,J.R., Stier,G. and Suck,D. (1996) A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature, 382, 90–93. [DOI] [PubMed] [Google Scholar]
- 34.Murante R.S., Rust,L. and Bambara,R.A. (1995) Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J. Biol. Chem., 270, 30377–30383. [DOI] [PubMed] [Google Scholar]
- 35.Barnes C.J., Wahl,A.F., Shen,B., Park,M.S. and Bambara,R.A. (1996) Mechanism of tracking and cleavage of adduct-damaged DNA substrates by the mammalian 5′- to 3′-exonuclease/endonuclease RAD2 homologue 1 or flap endonuclease 1. J. Biol. Chem., 271, 29624–29631. [DOI] [PubMed] [Google Scholar]
- 36.Bornarth C.J., Ranalli,T.A., Henricksen,L.A., Wahl,A.F. and Bambara,R.A. (1999) Effect of flap modifications on human FEN1 cleavage. Biochemistry, 38, 13347–13354. [DOI] [PubMed] [Google Scholar]
- 37.Patel P.H., Jacobo-Molina,A., Ding,J., Tantillo,C., Clark,A.D., Raag,R., Nanni,R.G., Hughes,S.H. and Arnold,E. (1995) Insights into DNA polymerization mechanisms from structure and function analysis of HIV-1 reverse transcriptase. Biochemistry, 34, 5351–5363. [DOI] [PubMed] [Google Scholar]
- 38.Lee B.I. and Wilson,D.M. (1999) The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J. Biol. Chem., 274, 37763–37769. [DOI] [PubMed] [Google Scholar]
- 39.Qiu J., Qian,Y., Chen,V., Guan,M.X. and Shen,B. (1999) Human exonuclease 1 functionally complements its yeast homologues in DNA recombination, RNA primer removal and mutation avoidance. J. Biol. Chem., 274, 17893–17900. [DOI] [PubMed] [Google Scholar]
- 40.Ausubel F.M. (ed.) (1997) Current Protocols in Molecular Biology. John Wiley & Sons, New York, NY.
- 41.Hadi M.Z., Coleman,M.A., Fidelis,K., Mohrenweiser,H.W. and Wilson,I.D. (2000) Functional characterization of Ape1 variants identified in the human population. Nucleic Acids Res., 28, 3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon W.J., Hayes,J.J., Levin,J.R., Weidner,M.F., Dombroski,B.A. and Tullius,T.D. (1991) Hydroxyl radical footprinting. Methods Enzymol., 208, 380–413. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen L.H., Barsky,D., Erzberger,J.P. and Wilson,D.M. (2000) Mapping the protein-DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J. Mol. Biol., 298, 447–459. [DOI] [PubMed] [Google Scholar]
- 44.Kassavetis G.A., Riggs,D.L., Negri,R., Nguyen,L.H. and Geiduschek,E.P. (1989) Transcription factor IIIB generates extended DNA interactions in RNA polymerase III transcription complexes on tRNA genes. Mol. Cell. Biol., 9, 2551–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen B., Nolan,J.P., Sklar,L.A. and Park,M.S. (1997) Functional analysis of point mutations in human flap endonuclease-1 active site. Nucleic Acids Res., 25, 3332–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu X., Li,J., Li,X., Hsieh,C.L., Burgers,P.M. and Lieber,M.R. (1996) Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res., 24, 2036–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson D.M.,III and Barsky,D. (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res., 485, 283–307. [DOI] [PubMed] [Google Scholar]
- 48.Kim C.Y., Park,M.S. and Dyer,R.B. (2001) Human flap endonuclease-1: conformational change upon binding to the flap DNA substrate and location of the Mg2+ binding site. Biochemistry, 40, 3208–3214. [DOI] [PubMed] [Google Scholar]
- 49.Alleva J.L. and Doetsch,P.W. (2000) The nature of the 5′-terminus is a major determinant for DNA processing by Schizosaccharomyces pombe Rad2p, a FEN-1 family nuclease. Nucleic Acids Res., 28, 2893–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y., Potapova,O., Leschziner,A.E., Grindley,N.D. and Joyce,C.M. (2001) Contacts between the 5′ nuclease of DNA polymerase I and its DNA substrate. J. Biol. Chem., 276, 30167–30177. [DOI] [PubMed] [Google Scholar]
- 51.Wilson D.M. III, Takeshita,M., Grollman,A.P. and Demple,B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem., 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- 52.Zemla A., Venclovas,C., Moult,J. and Fidelis,K. (1999) Processing and analysis of CASP3 protein structure predictions. Proteins, suppl. 3, 22–29. [DOI] [PubMed] [Google Scholar]
- 53.Humphrey W., Dalke,A. and Schulten,K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics, 14, 27–28, 33–38. [DOI] [PubMed] [Google Scholar]
- 54.Frishman D. and Argos,P. (1995) Knowledge-based protein secondary structure assignment. Proteins, 23, 566–579. [DOI] [PubMed] [Google Scholar]
- 55.Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- 56.Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 24, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.