

Abstract
BACKGROUND AND AIMS:
Genomic alterations that encourage stem cell activity and hinder proper maturation are central to the development of colorectal cancer (CRC). Key molecular mediators that promote these malignant properties require further elucidation to galvanize translational advances. We therefore aimed to characterize a key factor that blocks intestinal differentiation, define its transcriptional and epigenetic program, and provide preclinical evidence for therapeutic targeting in CRC.
METHODS:
Intestinal tissue from transgenic mice and patients were analyzed by means of histopathology and immunostaining. Human CRC cells and neoplastic murine organoids were genetically manipulated for functional studies. Gene expression profiling was obtained through RNA sequencing. Histone modifications and transcription factor binding were determined with the use of chromatin immunoprecipitation sequencing.
RESULTS:
We demonstrate that SRY-box transcription factor 9 (SOX9) promotes CRC by activating a stem cell–like program that hinders intestinal differentiation. Intestinal adenomas and colorectal adenocarcinomas from mouse models and patients demonstrate ectopic and elevated expression of SOX9. Functional experiments indicate a requirement for SOX9 in human CRC cell lines and engineered neoplastic organoids. Disrupting SOX9 activity impairs primary CRC tumor growth by inducing intestinal differentiation. By binding to genome wide enhancers, SOX9 directly activates genes associated with Paneth and stem cell activity, including prominin 1 (PROM1). SOX9 up-regulates PROM1 via a Wnt-responsive intronic enhancer. A pentaspan transmembrane protein, PROM1 uses its first intracellular domain to support stem cell signaling, at least in part through SOX9, reinforcing a PROM1-SOX9 positive feedback loop.
CONCLUSIONS:
These studies establish SOX9 as a central regulator of an enhancer-driven stem cell–like program and carry important implications for developing therapeutics directed at overcoming differentiation defects in CRC.
Keywords: SOX9, PROM1, Differentiation Block, Colorectal Cancer
Graphical Abstract
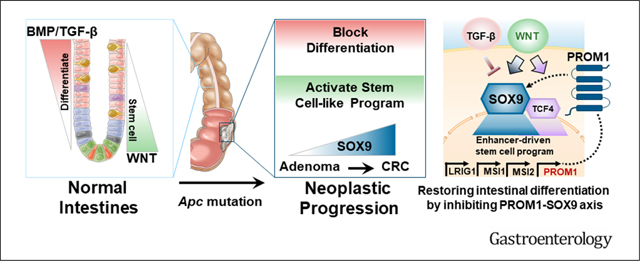
Intestinal stem cells and their immediate progeny replicate frequently in the crypt to sustain the most rapidly renewing epithelium in the human body. Wingless/integrated (Wnt) signaling is critical for maintaining stem cell reservoirs and crypt homeostasis. As the immediate progeny push beyond the crypt into the villus, they enter postmitotic differentiation, giving rise to mature intestinal cells that provide absorptive, barrier, and endocrine functions. Broadly, Wnt activity hinders and transforming growth factor (TGF) β/bone morphogenetic protein (BMP) signaling supports differentiation of progenitors into mature enterocytes, establishing a crypt-villus gradient.1 Disrupting the balance between stem cell and differentiation programs is a defining property of colorectal cancer (CRC), which remains the third most common and second most deadly malignancy worldwide.2
The near-universal initiating event in sporadic CRC involves genomic alterations that activate Wnt signaling, most often through loss-of-function APC mutations.3–5 Aberrant Wnt activation leads to stem cell expansion, shifting the homeostasis between the crypt and villus.6 As these early colonic lesions evolve, they often develop insensitivity to pro-differentiation cues by selecting TGF-β/BMP pathway alterations.7 As such, genomic alterations that hinder intestinal differentiation, either by activating stem cell–like pathways or disrupting differentiation pathways, are central to CRC development. However, we have yet to uncover critical regulators of aberrantly active stem cell–like programs or to convert this understanding into effective treatment for CRC.
SRY-box transcription factor 9 (SOX9) is a key developmental transcription factor that guides cell fate decisions during developmental and adult homeostasis in diverse tissues, including cartilage,8 testis,9 skin,10 and breast.11 In the intestines, bi-allelic genetic inactivation of SOX9 led to impaired Paneth cell differentiation in genetically engineered mouse models.12,13 However, the role of SOX9 in CRC remains unclear as there is evidence for oncogenic and tumor suppressor functions.14–24 In the present study, we identify SOX9 as a key mediator of a differentiation block, delineate functional components of an enhancer-driven stem cell–like program, and provide rationale for therapeutics designed to induce intestinal differentiation in CRC.
Materials and Methods
See the Supplementary Methods for descriptions of cell culture, lentivirus packing, and transduction, cell proliferation assays, RNA isolation, quantitative polymerase chain reaction, RNA sequencing, immunoblot, histopathology, immunofluorescence and immunohistochemistry, reporter assay, genetic dependencies, chromatin immunoprecipitation (ChIP-seq) analysis and visualization, and statistical analysis.
Animal Studies
All mouse experiments were approved by the Institutional Animal Care and Use Committee of the Dana-Farber Cancer Institute (11–009). The Lgr5-EGFP-Ires-CreERT2 mouse strain was described earlier.25 To inactivate Apc in intestinal tissue, we crossed Apcflox/flox mice26 (a generous gift from Christine Perret) with Lgr5-EGFP-Ires-CreERT2. These mice were further crossed with R26-LSL-tdTomato mice27 (Jackson Laboratory) for conditional tdTomato labeling. To activate conditional alleles, experimental mice (6–8 weeks) were injected intraperitoneally with a single dose of tamoxifen (50 μL of 10 mg/mL in sunflower oil). Mice were euthanized at the experimental end point and intestines were harvested for histopathologic and immunohistochemical analyses as well as organoid generation.
For tumor xenografts, 1.5 × 106 ApcKOKrasG12D colon organoid cells or 1 × 106 HT-29 CRC cells were injected into flanks of athymic Ncr-nu/nu mice. Tumor measurements were made with the use of a caliper, and tumor volumes were calculated using the following formula: volume = length × width2 × 0.5. Tumors were harvested, fixed in 10% formalin overnight, and paraffin embedded for histologic analysis. Fresh tissue was also collected for RNA isolation, protein collection, and flash-frozen for long-term storage.
Human Tissue Histopathology and Analysis
Human neoplastic colorectal samples without any previous chemotherapeutic or radiation therapy were obtained from the Department of Pathology of Chongqing Medical University, under approval (protocol 2020–444) by the Internal Review Board of the First Affiliated Hospital of Chongqing Medical University, Chongqing, China. Endoscopic resections of advanced adenomas (n = 17), surgical resections of paired adenoma/adenocarcinoma from patients with familial adenomatous polyposis (n = 4), and surgical resections of sporadic carcinoma (n = 3) and adenomas (n = 3) were selected (Supplementary Table 1).
SOX9 expression was evaluated by an experienced gastrointestinal pathologist without access to clinical data (Y.Y.). For the semi-quantitative assessment, histoscore (H-score) was calculated based on the intensity and percentage of nuclear SOX9 expression. The intensity of staining was classified as 0 to 3 (0: negative; 1: weak; 2: intermediate; 3: strong) and the positive rate was scored from 0 to 100. In each case, a H-score with a potential range of 0–300 was calculated as follows: H-score = (1 × % weakly stained cells) + (2 × % moderately stained cells) + (3 × % strongly stained cells).
Intestinal Organoid Culture
Colonic glands were isolated, treated with EDTA, and then resuspended in 30–50 μL of Matrigel (BD Bioscience) and plated in 24-well plates. Wnt/R-spondin/Noggin (WRN) containing DMEM/F12 with HEPES (Sigma-Aldrich) containing 20% fetal bovine serum, 1% penicillin/streptomycin, and 50 ng/mL recombinant mouse epidermal growth factor (Life Technologies) was used for culturing ApcKO colon organoids. For the first 2–3 days after seeding, the media were also supplemented with 10 mmol/L ROCK inhibitor Y-27632 (Sigma Aldrich) and 10 mmol/L SB431542 (Sigma Aldrich), an inhibitor for the TGF-β type I receptor, to avoid anoikis. For passage, colon organoids were dispersed by trypsin-EDTA and transferred to fresh Matrigel. Passage was performed every 3–4 days with a 1:3–5 split ratio. For normal human colon organoid culture, the previous media were supplemented with antibiotics 100 μg/mL Primocin (Invivogen), 100 μg/mL Normocin (Invivogen), serum-free supplements 1× B27 (Thermo Fisher [Gibco]) and 1 × N2 (Thermo Fisher [Gibco]), chemical supplements 10 mmol/L nicotinamide (Sigma) and 500 mmol/L N-acetylcysteine (Sigma), hormone 50 mmol/L [Leu15]-Gastrin (Sigma), growth factor 100 μg/mL fibroblast growth factor 10 (recombinant human; Thermo Fisher), and 500 nmol/L A-83–01 (Sigma), an inhibitor of TGF-β receptors ALK4, 5, and 7.
Generation of Stable Cell Lines
All genetically manipulated CRC cell lines and colon organoids were generated with the use of the protocol described by Shalem et al.28 In brief, short guide (sg) RNAs targeting SOX9 were designed and cloned into Lenti-CRISPR v2.29 Short hairpin (sh) RNAs against SOX9/PROM1 were cloned into PLKO.1, TET-PLKO, and TET-Cellecta vectors. To generate SOX9 knockout (KO) or knockdown (KD) and PROM1 KD stable cells, CRC cells were selected with 1.5 μg/mL puromycin and colon organoids were selected with the use of 3 μg/mL puromycin at 24 hours after infection. To generate stable cells constitutively expressing SOX9 or PROM1, PLX304 vectors containing indicated complementary (c) DNA were constructed and 15 μg/mL blasticidin selection was started 24 hours after lentiviral infection. For V5-tagged inducible expression of SOX9, PLIX403 vectors were used and 15 μg/mL blasticidin selection was started 24 hours after infection. For complete oligos and cloning, see Supplementary Table 2.
Chromatin Immunoprecipitation Followed by DNA Sequencing
HT115 cells were washed with phosphate-buffered saline solution and cross-linked with 1% formaldehyde for 10 minutes for histone 3 lysine 27 acetylation (H3K27ac) immunoprecipitation or cross-linked with 2 agents starting with 2 mmol/L DSG (Pierce) for 45 minutes at room temperature followed by 1 mL of 1% formaldehyde for 10 minutes for the V5 Tag Antibody. Cross-linked cell lines were quenched with 0.125 mol/L glycine for 5 minutes at room temperature. Cross-linked material was resuspended in 1% sodium dodecyl sulfate (50 mmol/L Tris-HCl pH 8, 10 mmol/L EDTA) and sonicated for 5 minutes with the use of a Covaris E220 instrument (5% duty cycle, 140 peak incident power, 200 cycles per burst, 1 mL AFA Fiber Millitubes). Soluble chromatin (5 μg) was immunoprecipitated with 10 μg H3K27ac (Diagenode C15410196 antibody) or 40 μg chromatin with 10 μg V5 Tag Antibody (Invitrogen Cat# R96025 Lot# 1949337). ChIP-seq libraries were constructed using the Accel-NGS 2S DNA library kit from Swift Biosciences. Fragments of the desired size were enriched with the use of AMPure XP beads (Beckman Coulter); 36-bp paired-end reads were sequenced on a Nextseq instrument (Illumina).
Results
SOX9 Is Overexpressed in Intestinal Adenoma Mouse Models and Human CRC
Sporadic CRC development is most often initiated by inappropriate Wnt activation, typically through mutations in the pathway negative regulator APC. To evaluate Sox9 expression during cancer initiation, we developed a mouse model that conditionally deletes Apc26 in Lgr5eGFP-expressing intestinal stem cells25 (Lgr5-ApcKO) (Figure 1A). Compared with ApcWT control, ApcKO lesions displayed transcriptional activation of Wnt pathway components (ie, Axin2, Myc), consistent with known biology, and Sox9 (Figure 1A). Sox9 activation in intestinal adenomas was confirmed by analyzing gene expression data from a previously published mouse model of inducible Apc KD with or without mutant K-rasG12D activation30 (Figure 1B). These data suggest that Apc inactivation leads to elevated expression of Sox9 during intestinal cancer initiation.
Figure 1.
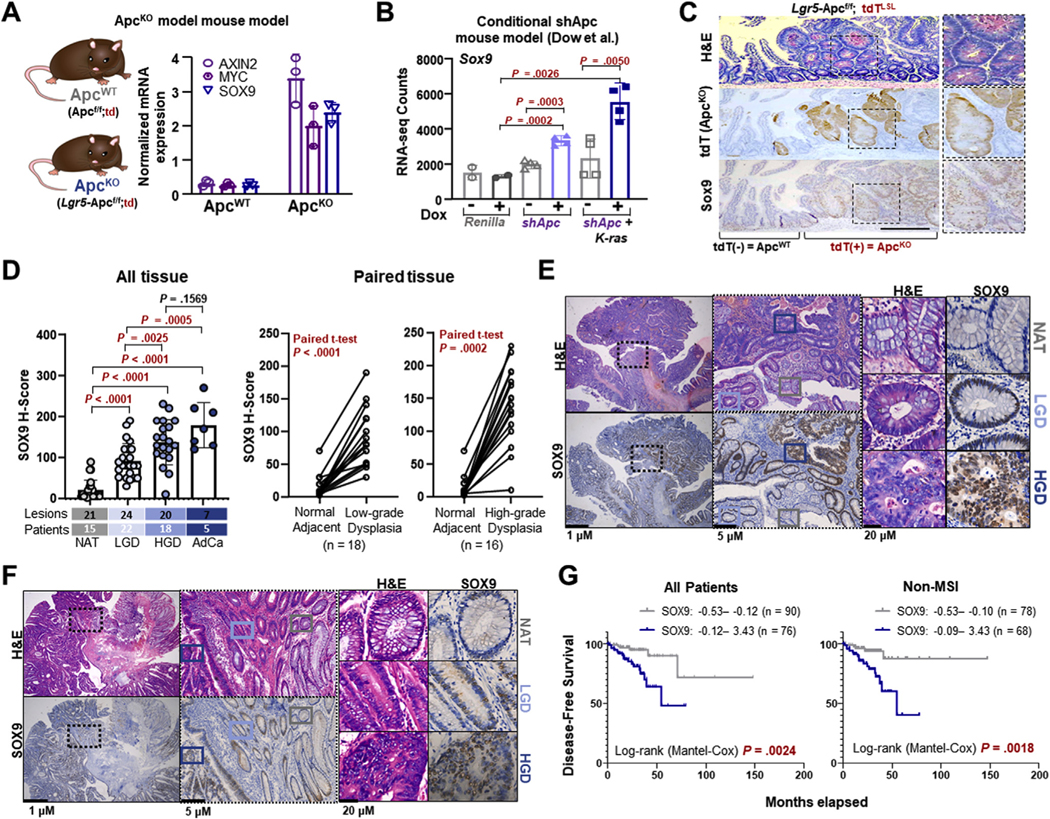
SOX9 is overexpressed in mouse models and human tissue of intestinal adenomas and colorectal cancer (CRC). (A) mRNA expression of Wnt target genes in ApcWT and ApcKO intestines according to quantitative reverse-transcription polymerase chain reaction (qRT-PCR); mean ± SD. (B) Sox9 mRNA expression in intestinal lesions from Apc shRNA mice ± K-ras mutation ± doxycycline (Dox)30; mean ± SD Student’s t test P values. (C) Representative histopathology of intestinal lesions (hematoxylin and eosin [H&E], td-RFP immunohistochemistry [IHC], Sox9 IHC) from Lgr5-Apcf/f;td mice treated with tamoxifen 28 days before harvesting tissue; scale bar = 100 μm. (D) SOX9 IHC in human specimens. Immunostaining was quantified as H-score (left). Paired lesions were analyzed (right). AdCa, adenocarcinoma; HGD, high-grade dysplasia; LGD, low-grade dysplasia; NAT, normal adjacent tissue. (E, F) Representative histopathology (H&E, SOX9 IHC) of human adenomas on the same specimen slide; scale bars = 1 μm, 5 μm, and 20 μm. (G) Kaplan-Meier curves indicating disease-free survival of patients with CRC (left) and non–microsatellite instability (MSI) CRC (right) with indicated expression of SOX9.
We next analyzed protein expression of Sox9 in intestinal adenomas of Lgr5-ApcKO mice. We took advantage of tomato red (tdT) labeling,27 which marked ApcKO cells in our mouse model. Compared with the crypt-restricted Sox9 expression in normal villi, tdT+ ApcKO lesions demonstrated ectopic and elevated expression of Sox9 (Figure 1C and Supplementary Figure 1A). To validate and extend these findings, we obtained 17 endoscopic resection samples of advanced colorectal adenomas (Supplementary Table 1) that contained normal adjacent tissue (NAT), low-grade dysplasia (LGD), and/or high-grade dysplasia (HGD) on the same specimen slide, controlling for intra-patient molecular heterogeneity that can be observed when pursing multi-region sampling.31 Adenocarcinomas (AdCas) with corresponding adenomas from patients with familial adenomatous polyposis and sporadic disease were included as well. SOX9 gradually increased in expression from paired NAT, LGD, and HGD to AdCa (Figure 1D to F and Supplementary Figure 1B to D), with the greatest relative increase in this progression between NAT and LGD. While heterogeneous SOX9 expression was observed in AdCa, levels were consistently higher than paired NAT and dysplasia. Collectively, these data indicate that SOX9 is overexpressed in 2 mouse models of intestinal adenomas and human adenoma to CRC progression, which may indicate an important role in cancer initiation.
To examine whether SOX9 expression is associated with clinical outcome, we analyzed patients with CRC from the pan-cancer cohort of The Cancer Genome Atlas. With the use of different expression cutoffs, our analyses robustly show that elevated expression of SOX9 is associated with decreased survival, which was consistent with previous work.32 Importantly, elevated SOX9 expression was associated with poor outcomes independently from MSI status (Figure 1G and Supplementary Figure 1E).
Human CRC Is Dependent on SOX9
To investigate whether SOX9 is required for proliferation in human CRC, we first analyzed large genome-scale functional cell line datasets.33 SOX9 KD and KO by means of RNA interference (n = 41) and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (n = 33), respectively, led to proliferation defects in CRC cell lines (Figure 2A and Supplementary Figure 2A); cell lines with higher SOX9 expression or a requirement for β-catenin were more dependent on SOX9 (Figure 2A and B). To confirm these results, we suppressed SOX9 by means of stable shRNA expression and observed proliferation defects in several CRC cell lines surveyed during adherent or ultra-low-attachment culture (Supplementary Figure 2B to D and Supplementary Table 3). Focusing on CRC cell lines most sensitive to SOX9 depletion, we validated that constitutive or inducible SOX9 KD with the use of multiple shRNAs impaired HT-115, HT-29, and COLO-205 proliferation (Figure 2C and D; Supplementary Figure 2E to G). Consistent with these phenotypes, SOX9 KD was associated with reduced expression of Hippo/Yes-associated protein (YAP) downstream target TAZ (Supplementary Figure 2F), which is a marker of survival and proliferation in the intestines34,35 and driver of CRC tumorigenesis.36 Reexpression of SOX9 cDNA rescued ultra-low-attachment colony-forming defects and restored TAZ expression in COLO-205 cells stably expressing a 3′UTR-targeting SOX9 shRNA (Supplementary Figure 2G). Overexpression of SOX9 alone did not improve adherent growth or ultra-low-attachment colony formation in CRC cells, presumably owing to high endogenous SOX9 expression levels in CRC (Supplementary Figure 2G to I). Constitutive or inducible SOX9 KD partially reduced soft-agar colony formation in vitro (Figure 2E and Supplementary Figure 2I) and impaired primary tumor growth in vivo (Figure 2F and Supplementary Figure 2K).
Figure 2.

SOX9 is required for CRC proliferation and primary tumor growth. (A, B) Dependency for SOX9 CRISPR knockout (KO) plotted against SOX9 mRNA expression (left) and CTNNB1 CRISPR dependency (right); blue regression line; Pearson’s correlation P value. (C) SOX9 and β-actin expression in HT-115 control and SOX9 KD according to immunoblot. Adherent proliferation by CellTiterGlo. Area of ultra-low-attachment colonies; mean SD; Student’s t test P values. (D) Immunoblot (SOX9, vinculin) and colony formation of Dox-inducible HT-29 non-targeting control (NTC), short hairpin (sh) RNA#1 or shRNA#5 ± 0.5 μg/mL Dox. (E) Soft-agar colony formation assay of COLO-205 control and SOX9 shRNA; mean ± SD of 3 cell culture replicates and representative phase-contrast images; Student’s t test P values. (F) Primary xenograft tumor growth of HT-29 control or SOX9 shRNA; mean ± SD (n = 5); Student’s t test P value (*P < 0.05). (G) Proportions of wild-type (WT)/heterozygous (Het), Het, and homozygous SOX9 inactivation in single-cell clones in 3 CRC Cas9/SOX9 short guide (sg) RNAs. Proliferation by CellTiterGlo. (H) LS180 nucleofected with Cas9/SOX9 sgRNA complex, then amplicon sequenced to quantify in-frame and frameshift indels. Abbreviations as in Figure 1.
We next attempted to eliminate SOX9 in CRC cell lines with the use of CRISPR/Cas9. Among the CRC cell lines tested, only COLO-205 was able to tolerate SOX9 KO, which resulted in compromised proliferation (Supplementary Figure 3A). We further analyzed single cell clones from 3 cell lines stably expressing an inactivating SOX9 sgRNA and Cas9. HT-115 and HT-29 clones did not demonstrate homozygous deletions, whereas a minority of COLO-205 clones harbored bi-allelic SOX9 inactivation (Figure 2G). As anticipated, COLO-205 clones with bi-allelic SOX9 deletion grew poorly in vitro (Figure 2G) and lost the ability to form primary tumor xenografts in vivo (0/10 vs 10/10).
SOX9 is mutated in approximately 10% of CRC.14,37 Although the functional significance of these mutations is poorly understood, we have evidence that the majority are heterozygous alterations (Duronio et al, unpublished). We therefore asked whether the LS180 CRC cell line, which harbors a heterozygous SOX9 mutation, is dependent on the remaining wild-type (WT) SOX9 allele. Genomic analysis of LS180 cells constitutively expressing an inactivating SOX9 sgRNA/Cas9 demonstrated that the majority of indels are in-frame (Figure 2H and Supplementary Figure 3B), likely preserving the function of WT SOX9. Collectively, these results suggest that human CRC requires SOX9.
SOX9 Blocks Intestinal Differentiation in Human CRC
Blocking differentiation is a key mechanism by which cancers survive, persist, and grow, especially in regenerative tissue such as the hematopoietic system and intestinal epithelium. SOX9 is known to regulate cell fate decisions in different tissue contexts.10,11 We therefore hypothesized that SOX9 dependency in human CRC may relate to an ability to regulate intestinal differentiation. Keratin-20 (Krt20) is a tissue-specific marker of intestinal differentiation,30 as shown by its villus-restricted expression in the normal mouse intestines (Figure 3A). In a mutually exclusive pattern, Sox9 is expressed in crypts of murine intestines (Figure 3A), suggesting that Sox9 may restrict intestinal differentiation.
Figure 3.

SOX9 blocks intestinal differentiation in human CRC. (A) Sox9 and Krt20 IHC in normal mouse small intestines; scale bar = 100 μm. (B) Immunoblot (V5-tagged, KRT20 GAPDH) of indicated LS180 ± 0.5 μg/mL Dox. (C) SOX9, KRT20, MUC2, and CDX2 mRNA expression in indicated LS180 ± 0.5 μg/mL DOX according to RT-PCR; mean ± SD; Student’s t test: ***P < 0.005, ****P < 0.001. (D) Immunoblot (SOX9, KRT20, GAPDH) of indicated HT-115 ± 0.25 μg/μL Dox. (E) Immunoblot (E-cadherin, vimentin, GAPDH) in indicated COLO-205 Cas9/SOX9 sgRNA single-cell clones; phase-contrast images; scale bar = 100 μm. Abbreviations as in Figures 1 and 2.
To investigate this hypothesis, we manipulated SOX9 in human CRC cell lines. Inducible overexpression of SOX9 reduced expression of absorptive and secretory intestinal differentiation markers KRT20, mucin 2 (MUC2), and CDX2 in LS180 CRC cells, whereas overexpression of a truncated, transcriptionally hypomorphic form of SOX9 (SOX9ΔC) had diminished to no effect (Figure 3B and C). Constitutive SOX9 overexpression also led to diminished expression of general epithelial differentiation marker E-cadherin in COLO-205, a CRC cell line without KRT20 expression (Supplementary Figure 4A). These data suggest that SOX9 negatively regulates intestinal differentiation in human CRC.
In contrast, inducible SOX9 KD promoted intestinal differentiation in HT-115 CRC cells as indicated by KRT20 induction (Figure 3D). Constitutive or inducible SOX9 KD in COLO-205 induced CDH1/E-cadherin expression proportional to shRNA strength (Supplementary Figure 4B to D). Inducible SOX9 KD led to junctional localization of E-cadherin by immunofluorescence (Supplementary Figure 4E). Notably, the few COLO-205 clones that tolerated bi-allelic SOX9 KO displayed a pronounced “cobblestone” morphology that corresponded to marked E-cadherin expression induction (Figure 3E), phenocopying SOX9 KD. These data indicate that SOX9 disruption promotes intestinal differentiation and likely explains the resultant impairment in human CRC growth.
SOX9 KD Impairs Proliferation and Induces Differentiation in Neoplastic Mouse Organoids
Organoids capture intestinal stem cell behavior and differentiation dynamics,38 serving as a well suited platform to investigate SOX9 function in CRC. We first examined whether SOX9 KD promotes intestinal differentiation in normal human colon organoids. Gene expression profiling by means of RNA sequencing revealed that SOX9 suppression induces a broad intestinal differentiation program and reduces expression of a subset of stem cell markers (Figure 4A). We next used ApcKO-KrasG12D neoplastic colon organoids derived from genetically engineered mice39 to investigate the impact of Sox9 KD on proliferation and differentiation. Constitutive Sox9 KD reduced growth of ApcKO-KrasG12D colon organoids by 2- to 3-fold, as indicated by in vitro culture assays and Ki67 immunohistochemistry (Figure 4B and C; Supplementary Figure 5A). The growth defects were associated with induction of intestinal differentiation, as shown by Krt20 immunofluorescence (Supplementary Figure 5A). Sox9 KD reduced mRNA expression of Wnt/stem cell markers and induced Krt20 expression (Figure 4D). These results were validated with inducible Sox9 KD in ApcKO and ApcKO-KrasG12D colon organoids (Supplementary Figure 5B and C).
Figure 4.
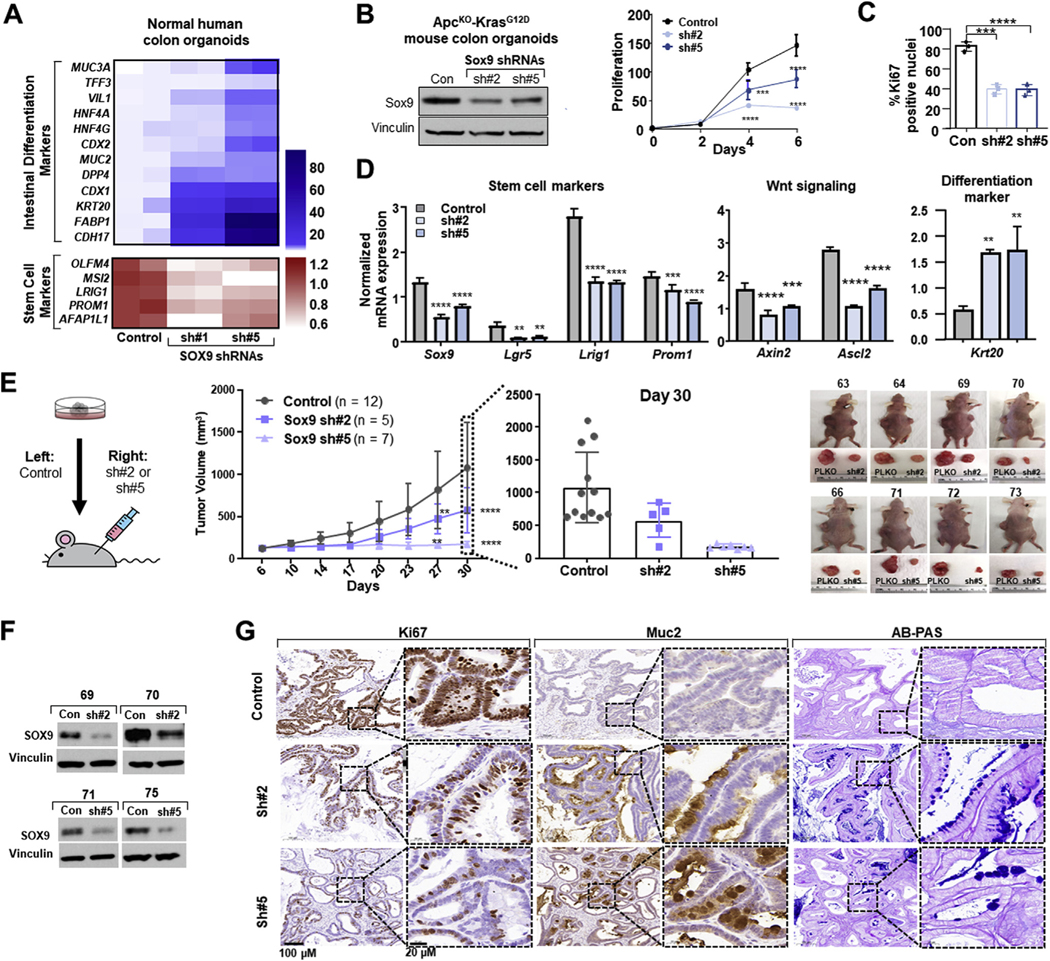
SOX9 knockdown promotes intestinal differentiation in organoid models of CRC. (A) mRNA expression heat map of intestinal differentiation (blue) and stem cell (red) markers in control and indicated SOX9 shRNA human colon organoids. (B) Immunoblot (Sox9 and vinculin) of mouse control or indicated Sox9 shRNA ApcKO KrasG12D colon organoids. Proliferation by CellTiterGlo; mean ± SD; Student’s t test: ***P < 0.005, ****P < 0.001. (C) Ki67 IHC quantification of indicated fixed mouse ApcKO KrasG12D colon organoids; mean ± SD; Student;s t test ***P < 0.005, ****P < 0.001. (D) Sox9, Lgr5, Lrig1, Prom1, Axin2, Ascl2, Krt20 mRNA expression in ApcKO KrasG12D colon organoids according to qRT-PCR; mean ± SD; Student’s t test: **P < 0.01, ***P < 0.005, ****P < 0.001. (E) ApcKO KrasG12D Sox9 xenograft schematic. Primary tumor xenograft growth curve and day 30 quantification. Representative images of xenograft tumors. (F) Immunoblot (Sox9 and vinculin) of xenograft tumors. (G) Ki67 and Muc2 immunohistochemistry and Alcian blue–periodic acid Schiff (AB-PAS) staining of xenograft tumors; scale bars = 100 μm and 20 μm. Abbreviations as in Figures 1 and 2.
We next evaluated the ability of Sox9 KD to affect ApcKO-KrasG12D colon organoid growth and differentiation in vivo with the use of primary tumor xenograft assays. Compared with control samples injected into the contralateral flanks of nude mice, Sox9 KD organoids displayed significantly impaired primary tumor growth (Figure 4E). Histopathologic analysis confirmed reduced proliferative capacity by Ki67 immunohistochemistry and demonstrated a clear induction of intestinal differentiation by Muc2 immunohistochemistry and Alcian blue staining in Sox9 KD xenografts (Figure 4G). We collected protein from tumors at the experimental end point to validate Sox9 suppression. Although the majority of Sox9 KD tumors demonstrated reduced Sox9 expression (Figure 4F), there was a subset of outliers that escaped shRNA-mediated Sox9 KD and demonstrated increased xenograft growth (Supplementary Figure 6). These results demonstrated that Sox9 KD leads to intestinal differentiation and decreased tumor growth in neoplastic murine organoid models.
SOX9 Activates a Stem and Paneth Cell Transcriptional Program by Binding Genome-Wide Enhancers
To determine the mechanism by which SOX9 blocks differentiation in human CRC, we performed gene expression profiling of LS180 CRC cells engineered to conditionally overexpress green fluorescent protein (GFP) control, WT SOX9, or SOX9ΔC with the use of RNA sequencing (Figure 5A). Principal component analysis of the top 500 differentially expressed genes indicated that SOX9 induces a distinct transcriptional state (Supplementary Figure 7A). Gene ontology analysis demonstrated that Ras signaling, lysozyme and pluripotency are the top 3 gene-sets upregulated by SOX9 (Supplementary Figure 7B), which was consistent with individual gene expression patterns (Figure 5A). Furthermore, genes up-regulated by SOX9 were enriched for an Lgr5 intestinal stem cell signature40 according to gene set enrichment analysis (Supplementary Figure 7C). In contrast, differentiation genes associated with both absorptive and secretory cell lineages were among significantly down-regulated genes (Figure 5A). These data indicate that SOX9 may block differentiation by promoting a stem cell–like and Paneth cell transcriptional program.
Figure 5.

SOX9 activates an enhancer-driven stem and Paneth cell transcriptional program. (A) Gene expression heat map of indicated LS180 CRC ± Dox. Selected up-regulated (red) and down-regulated (blue) gene sets displayed. (B) Gene ontology (GO Biological Process) of SOX9-bound genes. False discovery rate (FDR) q-value after Bonferroni correction displayed. (C) SOX9-binding distribution among intergenic, intronic, promoter, and exonic regions. (D) Transcription factor–binding similarity (Giggle score64) of publicly available vs our V5-SOX9 chromatin immunoprecipitation sequencing (ChIP-seq) data. (E) Integrative Genomics Viewer (IGV) gene track at indicated gene loci of H3K27ac (red) and V5-ChIP (blue) in indicated HT-115; signal track for TCF4 (purple) from published endogenous LS180 ChIP-seq data.65 GFP, green fluorescent protein; other abbreviations as in Figure 1.
To investigate the direct transcriptional program mediated by SOX9 in CRC, we next performed genome-wide V5 and H3K27Ac chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) in CRC cells conditionally expressing V5-tagged GFP, WT SOX9, and SOX9ΔC. Indeed, genes directly bound and activated by SOX9 were significantly enriched in gene sets associated with stem cell activity (Figure 5B); 94% of SOX9 binding occurred at intergenic and intronic regions of the genome (Figure 5C), and more than a fifth of genome-wide regions occupied by SOX9 are typical enhancers (~374 sites). Super-enhancers are composed of a high density of individual enhancers and often regulate tissue-specific genes involved in cellular identity.41,42 We observed that SOX9-binding sites were more prevalent in super-enhancers than typical enhancers. Of the H3K27Ac-marked super-enhancers, 26% (33/128) were bound by SOX9, which was greater than the 15% (374/2455) of typical enhancers occupied by SOX9. These results are consistent with a potential role for SOX9 as a transcriptional regulator of stem cell–like activity by engaging enhancers.
We next investigated whether other transcription factors bind to regions occupied by SOX9, reasoning that these mediators may co-regulate stem cell–like behavior. Motif analysis inferred that binding sites for CDX2, an intestinal lineage transcription factor,43,44 and TCF7L2/TCF4, an essential Wnt signaling co-factor,5,6,45 are enriched at regions bound by SOX9 (Supplementary Figure 7D). Supporting this result, publicly available genome-wide transcription factor binding data indicated a strong overlap among TCF4- and SOX9-bound regions, with most of these studies using CRC cell lines (Figure 5D). Consistent with gene ontology analysis, SOX9 occupied intronic enhancers of several stem cell genes and Paneth cell gene Lyz (Figure 5E and Supplementary Figure 7E and F). LRIG1 and PROM1 are 2 stem cell genes co-occupied by SOX9 and TCF4 at intronic enhancers (Figure 5E). These results suggest that the Wnt effector TCF4 may collaborate with SOX9 at specific enhancers to regulate a stem cell–like program in CRC.
To determine whether these relationships are found in patient samples, we queried The Cancer Genome Atlas transcriptional profiles from samples with high and low expression of SOX9. Several genes associated with stem cell activity displayed elevated expression in CRC samples with high SOX9 expression (Supplementary Figure 7G). These data indicate that SOX9 mediates a stem cell–like transcriptional program by binding to genome-wide enhancers in CRC.
To examine whether SOX9 is regulated by stem cell and differentiation cues, we evaluated the impact of the Wnt and BMP/TGF-β signaling pathways on SOX9 expression, respectively. Wnt signaling activates stem cell transcriptional programs in the enteric crypt, whereas BMP/TGF-β activity promotes post-mitotic differentiation in the villus.1 Disrupting the Wnt pathway by means of the chemical inhibitor ICG-001 or dominant-negative form of TCF4 (dnTCF4) reduced SOX9 expression in CRC cells (Supplementary Figure 8A), corroborating previous results that SOX9 is positively regulated by Wnt signaling.20,46 In contrast, TGF-β signaling negatively regulated SOX9 expression: Recombinant TGF-β treatment reduced SOX9 expression whereas TGF-β inhibitor exposure increased SOX9 expression in CRC cells (Supplementary Figure 8B). These results position SOX9 as a central effector that can integrate stem cell cues mediated by Wnt activation and differentiation signals from TGF-β signaling.
SOX9 Directly Activates PROM1 Via a Wnt-Responsive Intronic Enhancer
To identify members of the SOX9-mediated stem cell–like program in CRC, we performed an integrative analysis of RNA sequencing, V5-SOX9 ChIP-seq, and H3K27Ac ChIP-seq data from our previous experiments. Among the genes implicated by all 3 data-sets (Figure 6A), PROM1 (Prominin-1; CD133) was the most attractive candidate based on 1) its association with intestinal stem cell biology47 and 2) its elevated expression in poor-prognosis human CRCs.48,49 We therefore proceeded to examine whether PROM1 is broadly activated by SOX9 in relevant experimental systems. SOX9 KD reduced PROM1 expression in normal human organoids and neoplastic murine organoids (Figure 6B and Supplementary Figure 9A to C). Moreover, PROM1 expression corresponded with the genomic status of SOX9 in human CRC tumors (Figure 6C). SOX9 overexpression led to a modest but consistent increase in mRNA and protein expression of PROM1 in HT-115 CRC cells; proportional to the severity of truncation, overexpression of various SOX9ΔC proteins had little to no impact on PROM1 activation (Supplementary Figure 9D and E). Inducible overexpression of WT SOX9 but not SOX9ΔC led to a 6-fold induction in PROM1 mRNA levels in LS180 CRC cells, which corresponded with protein levels according to immunoblot (Figure 6D, Supplementary Figure 9F).
Figure 6.

SOX9 directly activates PROM1 via a Wnt-responsive intronic enhancer. (A) Volcano plot displaying log2 fold change of differentially expressed genes (SOX/GFP) along x-axis (FDR <0.05). Blue points mark genes significantly associated with SOX9-binding or differential H3K27ac. (B) Prom1 mRNA expression in indicated ApcKO KrasG12D colon organoids according to qRT-PCR; mean ± SD; Student’s t test: ****P < 0.001. (C) PROM1 mRNA expression in human CRC with WT, Het, or homozygous (Homo) SOX9 mutations. TCGA, The Cancer Genome Atlas. (D) mRNA and protein expression of PROM1 in indicated cell lines ± 0.5 μg/mL Dox. (E) IGV gene track of PROM1 intron1; signal tracks of H3K27ac (red) and V5-ChIP (blue) in indicated conditions; peak scale displayed in top right. (F) PROM1 enhancer reporter assay: phase-contrast and fluorescence images and quantification of HEK293T transiently transfected with indicated plasmids and/or treated with WNT3A. Abbreviations as in Figures 1, 2, and 5.
To investigate the precise mechanism of PROM1 activation, we analyzed V5-SOX9 and H3K27Ac ChIP-seq data. Although WT SOX9 and SOX9ΔC both bound to an enhancer in intron 1 of PROM1, only WT SOX9 led to greater H3K27Ac deposition at the locus (Figure 6E). To further study this regulatory element, we cloned a 573-bp sequence encompassing the PROM1 intronic enhancer into a GFP reporter construct by means of Gibson assembly. WT SOX9 but not SOX9ΔC stimulated reporter activity by more than 2-fold (Figure 6F). Moreover, activation of the WNT pathway with the use of recombinant WNT3A stimulated reporter activity by more than 3-fold, whereas genetic inhibition with the use of dnTCF4 blocked WNT3A-mediated reporter induction (Figure 6F). Together, these data indicate that SOX9 directly activates PROM1 using a Wnt/TCF4-responsive intronic enhancer.
A PROM1-SOX9 Positive Feedback Loop Blocks Differentiation in CRC
PROM1 is a marker for intestinal stem cell activity50 and tumorigenic capacity.51 However, whether PROM1 is functionally required for human CRC has not been fully addressed.52 To investigate whether PROM1 is required for CRC growth, we conditionally suppressed PROM1 with the use of multiple shRNAs (Supplementary Figure 10 A and B). Phenocopying SOX9 suppression, PROM1 KD led to proliferation defects in HT-115 and LS180 cells in vitro and impaired primary tumor growth of HT-29 in vivo (Figure 7A to C and Supplementary Figure 10C and D). Inducible PROM1 KD also stimulated intestinal differentiation in HT-115 and LS180 cells, as shown by KRT20 induction (Figure 7D and E; Supplementary Figure 10E). In contrast, overexpression of PROM1 blocked intestinal differentiation in LS180 cells, albeit to a lesser extent than SOX9 (Figure 7F). These data suggest that, like SOX9, PROM1 blocks differentiation in CRC, perhaps by activating stem cell signaling.
Figure 7.

PROM1 blocks intestinal differentiation via its first intracellular domain. (A, B) Proliferation and colony formation of indicated control and PROM1 shRNA HT-115 and LS180 ± 0.25 μg/mL Dox; mean ± SD; Student’s t test: ***P < 0.005. (C) Primary tumor xenograft growth curve and weight of NTC or indicated PROM1-knockdown HT-29 cell line; Student’s t test P values. (D) Immunoblot (PROM1, KRT20, and GAPDH) in indicated HT115 cell lines ± 0.5 μg/mL Dox. (E) Immunoblot (KRT20 and vinculin) in indicated LS180 cell lines ± 0.5 μg/mL Dox. (F) Immunoblot (KRT20, PROM1, SOX9, and vinculin) in indicated control or LS180 overexpression cells. (G) Immunoblot (KRT20, PROM1, SOX9, GAPDH, and vinculin) in indicated HT-115 and LS180 cells. (H) Immunoblot (KRT20, PROM1, SOX9, AXIN2, GAPDH, and vinculin) in inducible PROM1 shRNA HT-115 and LS180 cells with indicated Dox treatment. (I) KRT20 mRNA and KRT20, SOX9, vinculin protein expression in NTC or PROM1 shRNA LS180 cells expressing GFP or WT-SOX9 ± 0.5 μg/μL Dox; mean ± SD; Student’s t test: ****P < 0.001. (J) KRT20 mRNA and protein expression in indicated LS180 overexpression. (K) Schematic summarizing the SOX9-PROM1 reinforcing feedback loop. Abbreviations as in Figures 1, 2, and 5.
PROM1 is a glycosylated pentaspan apical transmembrane protein. We next wondered whether a molecular mediator connects its membrane localization to its ability to block intestinal differentiation. We noticed that constitutive and inducible PROM1 suppression reduced SOX9 expression (Figure 7G and H), suggesting that PROM1 may also function upstream and potentially block differentiation through SOX9 activity. Consistent with this hypothesis, the extent of SOX9 repression by individual PROM1 shRNAs correlated with the magnitude of intestinal differentiation induction, as shown by KRT20 expression in HT-115 and LS180 cells (Figure 7G and Supplementary Figure 10F). A time course of PROM1 expression restoration following doxycycline withdrawal also showed a gradual return of SOX9 levels that corresponded to KRT20 repression (Figure 7H). To definitively test whether SOX9 functions downstream of PROM1, we asked whether exogenous SOX9 expression could prevent intestinal differentiation induced by PROM1 KD. SOX9 overexpression effectively blocked intestinal differentiation mediated by PROM1 KD (Figure 7I and Supplementary Figure 10G and H). These data indicate that PROM1 obstructs intestinal differentiation through SOX9, establishing a positive feedback loop.
PROM1 has 3 intracellular domains: The first intracellular loop engages HDAC6 to stabilize β-catenin53 and the 59–amino acid C-terminal domain participates in PI3K/Akt signaling.54,55 Consistent with its regulation of β-catenin, inducible PROM1 KD reduced levels of canonical Wnt-target AXIN2 (Figure 7H). Because the Wnt pathway positively regulates SOX9 activity (Supplementary Figure 8A),46 we suspected that PROM1 regulates SOX9 and stem cell activity through its ability to stabilize β-catenin via its first intracellular domain. Consistent with this model, overexpression of a truncated PROM1 protein without its C-terminal intracellular domain (PROM1ΔC) retained the ability to block intestinal differentiation as indicated by KRT20 expression, suggesting that the C-terminal domain is expendable for this function (Figure 7F). In contrast, overexpression of a truncated PROM1 protein lacking the first intracellular domain (PROM11−129) lost its ability to suppress intestinal differentiation (Figure 7J). Notably, overexpression of a truncated PROM1 protein that preserves the first intracellular domain (PROM11−178) regained the ability to suppress intestinal differentiation (Figure 7J). These findings support a reinforcing feedback loop whereby PROM1 and SOX9 positively regulate each other to activate a stem cell–like transcriptional program that counteracts intestinal differentiation in CRC (Figure 7K).
Discussion
There is conflicting evidence as to the function of SOX9 in CRC. Sox9 inactivation in mouse intestines led to hyperplasia,12 suggesting a tumor suppressor role that is supported by genomic14 and functional17–19 studies in human CRC. In contrast, there is also evidence that SOX9 promotes CRC in mouse models20,21 and human CRC studies.22–24 The present study positions SOX9 as a key early mediator of CRC with the use of a combination of mouse models, human tissue analyses, and functional experiments in cancer cell lines and organoids. Although there is evidence that SOX9 can act as a transcriptional repressor,46 our data indicate that SOX9 behaves primarily as a transcriptional activator, binding to enhancers that up-regulate genes associated with Paneth and stem cell function. In addition to supporting previous studies12,13 that demonstrated a requirement for Sox9 in Paneth cell fate, we extend the role of SOX9 to a direct activator of stem cell–like signaling. SOX9 may therefore regulate a crypt-restricted gene expression program without distinguishing a stem cell or a Paneth cell fate, which likely requires additional factors. Suppressing SOX9 reliably induced absorptive and secretory lineage differentiation in human CRC and neoplastic murine organoids. The extent to which intestinal differentiation was induced by SOX9 KD was variable, with stronger phenotypes observed in normal human colon organoids and ApcKO-KrasG12D murine organoids compared with human CRC cell lines. These data indicate that advanced cancers with a greater burden of defects that prohibit differentiation may be more difficult to reprogram, and therefore treat, than early lesions, such as well differentiated adenomas.
Wnt and TGF-β are powerful developmental pathways that orchestrate intestinal stem cell and differentiation cues, respectively.1 In CRC, oncogenes tend to support stem cell programs whereas tumor suppressor pathways endorse differentiated cell fates. For example, the TGF-β–SMAD4 pathway, known to be a tumor suppressor in gastrointestinal tissue,56 ensures a differentiated enterocyte cell identity through hepatocyte nuclear factors 4α and 4γ activity.57 As such, the evolution of CRC can be viewed as a progressive deviation from proper cellular differentiation and serial acquisition of aberrant stem cell–like behavior. Reprogramming CRC to obey differentiation cues and regulate stem cell signaling is therefore a promising therapeutic approach, as has been demonstrated in other cancer types.58 Expressing functional APC can restore Wnt pathway regulation, promote cellular differentiation, and inhibit cancer functions.30,59,60 Furthermore, inhibiting Lgr5+ stem cells can impair CRC tumor growth and metastasis.61,62 Translation into effective therapy, however, has proven difficult as Wnt pathway inhibitors have not advanced in clinical testing.63 By defining critical mediators of the stem cell–like program co-opted by CRC, we may uncover new therapeutic strategies designed to induce differentiation. Drug compounds that inhibit stem cell–like activity mediated by PROM1 and SOX9 have the potential to restore intestinal differentiation without evoking toxicities associated with disrupting the pleotropic effects of Wnt signaling.
Aberrant activation of stem cell–like programs and their ability to block proper differentiation are central to the pathogenesis of CRC. We provide evidence that human CRC is dependent on an enhancer-driven stem cell–like program supported by a PROM1-SOX9 positive feedback loop. Blocking the PROM1-SOX9 axis restores intestinal differentiation and impairs CRC growth, providing rationale for the development of therapeutic agents that disrupt this pathway.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Aberrant stem cell activity that prevents intestinal differentiation is a hallmark of colorectal cancer (CRC). The molecular mediators that facilitate differentiation blocks in CRC are poorly defined.
NEW FINDINGS
SOX9 is a functional mediator that blocks differentiation by activating an enhancer-driven stem cell–like program in CRC. Disrupting the PROM1-SOX9 positive feedback circuit promotes intestinal differentiation and impedes tumor growth.
LIMITATIONS
The functional studies are carried out in human CRC cell lines and neoplastic murine organoids. Validation in a genetically engineered mouse model would strengthen the claim.
IMPACT
These results define key molecular players of a reinforcing transcriptional circuit that blocks intestinal differentiation and provide rationale for designing therapeutics that overcome differentiation blocks in CRC.
Acknowledgments
The authors thank Ramesh Shivdasani, Manav Korpal, Adam Sperling, William G. Kaelin Jr, Shridar Ganesan, Mathew Hemming, David Liu, Doug Micalizzi, and Ankur Nagaraja for insightful discussions; Klothilda Lim for ChIP-seq technical assistance; Aniket Gad and Lay-Hong Ang for immunohistochemistry assistance; Jatin Roper and Omer Yilmaz for normal colon organoid; Yi-Jang Lin and Kevin Haigis for murine ApcKOKrasG12D organoids; Dana-Farber/Harvard Cancer Center for the use of the Specialized Histopathology Core Laboratory, which provided histology and immunohistochemistry service; Harvard Digestive Disease Center and National Institutes of Health grant P30DK034854 for core services, resources, technology, and expertise. Dana-Farber/Harvard Cancer Center is supported in part by a National Cancer Institute Cancer Center Support Grant (NIH 5 P30 CA06516).
Funding
This work was funded by grants from the National Cancer Institute (P01 CA098101) to Adam J. Bass, and a Perry Fellowship, Claudia Barr Award, and Karen Grunebaum Foundation Award to Nilay S. Sethi.
Adam J. Bass receives funding from Merck, Bayer and Novartis, and is an advisor to Earli and Helix Nano and a cofounder of Signet Therapeutics. Sandor Spisak and Nilay S. Sethi are inventors on an unpublished US Provisional Patent application that is partly based on this work.
Abbreviations used in this paper:
- APC/Apc
adenomatous polyposis coli (human/mouse)
- BMP
bone morphogenic protein
- ChIP
chromatin immunoprecipitation
- CRC
colorectal cancer
- GFP
green fluorescent protein
- H3K27Ac
histone 3 lysine 27 acetylation
- KD
knockdown
- KO
knockout
- KRT20/Krt20
keratin 20 (human/mouse)
- Lgr5
Lgr5CreERT2-T2A-eGFP(mouse)
- MUC2/Muc2
mucin 2 (human/mouse)
- PROM1/Prom1
prominin 1 (human/mouse)
- sgRNA
short guide RNA
- shRNA
short hairpin RNA
- SOX9/Sox9
SRY-box transcription factor 9 (human/mouse)
- TGF-β
transforming growth factor beta
- WT
wild-type
Footnotes
Data Transparency
All RNA sequencing and ChIP sequencing data and generated reagents (eg, organoids, cell lines, and/or plasmids) will be shared either by depositing in public domains or through material transfer agreements in compliance with our institution.
Conflicts of interest
The other authors declare no conflicts.
CRedit Authorship Contributions
Xiaoyan Liang, M.D., Ph.D. (Formal analysis: Lead; Investigation: Lead; Validation: Lead; Writing – original draft: Supporting; Writing – review & editing: Equal); Gina N Duronio, B.S. (Formal analysis: Supporting; Investigation: Supporting; Validation: Supporting; Writing – original draft: Supporting; Writing – review & editing: Equal); Yaying Yang, M.D. (Formal analysis: Lead; Histopathology: Lead); Pratyusha Bala, Ph.D. (Investigation: Supporting; Validation: Supporting); Prajna Hebbar, n/a (Investigation: Supporting); Sandor Spisak, Ph.D. (Investigation: Supporting); Pranshu Sahgal, Ph.D. (Writing – review & editing: Supporting); Harshabad Singh, M.D. (Investigation: Supporting; Writing – review & editing: Supporting); Yanxi Zhang, Ph.D. (Investigation: Supporting); Yingtian Xie, Ph.D. (Investigation: Supporting); Paloma Cejas, Ph.D. (Investigation: Supporting); Henry W Long, Ph.D. (Investigation: Supporting); Adam J Bass, M.D. (Conceptualization: Supporting; Funding acquisition: Supporting; Investigation: Supporting; Supervision: Supporting; Writing – review & editing: Supporting); Nilay S Sethi, M.D., Ph.D. (Conceptualization: Lead; Formal analysis: Supporting; Funding acquisition: Lead; Investigation: Lead; Supervision: Lead; Validation: Supporting; Writing – original draft: Lead; Writing – review & editing: Equal).
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2021.09.044.
References
- 1.Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol 2004;20:695–723. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996;87:159–170. [DOI] [PubMed] [Google Scholar]
- 5.Morin PJ, Sparks AB, Korinek V, et al. Activation of β-catenin–Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997;275:1787–1790. [DOI] [PubMed] [Google Scholar]
- 6.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin–Tcf complex in APC−/− colon carcinoma. Science 1997; 275:1784–1787. [DOI] [PubMed] [Google Scholar]
- 7.Jung B, Staudacher JJ, Beauchamp D. Transforming growth factor beta superfamily signaling in development of colorectal cancer. Gastroenterology 2017;152: 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyama H, Kim JE, Nakashima K, et al. Osteo-chon-droprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A 2005; 102:14665–14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jakob S, Lovell-Badge R. Sex determination and the control of Sox9 expression in mammals. FEBS J 2011; 278:1002–1009. [DOI] [PubMed] [Google Scholar]
- 10.Kadaja M, Keyes BE, Lin M, et al. SOX9: a stem cell transcriptional regulator of secreted niche signaling factors. Genes Dev 2014;28:328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo W, Keckesova Z, Donaher JL, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012;148:1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bastide P, Darido C, Pannequin J, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol 2007; 178:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori-Akiyama Y, van den Born M, van Es JH, et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 2007; 133:539–546. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas N.Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu B, Fang Y, Xu J, et al. Analysis of SOX9 expression in colorectal cancer. Am J Clin Pathol 2008;130:897–904. [DOI] [PubMed] [Google Scholar]
- 16.Prevostel C, Blache P. The dose-dependent effect of SOX9 and its incidence in colorectal cancer. Eur J Cancer 2017;86:150–157. [DOI] [PubMed] [Google Scholar]
- 17.Darido C, Buchert M, Pannequin J, et al. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res 2008;68:4258–4268. [DOI] [PubMed] [Google Scholar]
- 18.Prevostel C, Rammah-Bouazza C, Trauchessec H, et al. SOX9 is an atypical intestinal tumor suppressor controlling the oncogenic Wnt/β-catenin signaling. Oncotarget 2016;7:82228–82243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blache P, Canterel-Thouennon L, Busson M, et al. A Short SOX9 peptide mimics SOX9 tumor suppressor activity and is sufficient to inhibit colon cancer cell growth. Mol Cancer Ther 2019;18:1386–1395. [DOI] [PubMed] [Google Scholar]
- 20.Feng Y, Sentani K, Wiese A, et al. Sox9 induction, ectopic Paneth cells, and mitotic spindle axis defects in mouse colon adenomatous epithelium arising from conditional biallelic Apc inactivation. Am J Pathol 2013; 183:493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiramatsu Y, Fukuda A, Ogawa S, et al. Arid1a is essential for intestinal stem cells through Sox9 regulation. Proc Natl Acad Sci U S A 2019;116:1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcker Espersen ML, Linnemann D, Christensen IJ, et al. SOX9 expression predicts relapse of stage II colon cancer patients. Hum Pathol 2016;52:38–46. [DOI] [PubMed] [Google Scholar]
- 23.Carrasco-Garcia E, Lopez L, Aldaz P, et al. SOX9-regulated cell plasticity in colorectal metastasis is attenuated by rapamycin. Sci Rep 2016;6:32350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matheu A, Collado M, Wise C, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res 2012;72:1301–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–1007. [DOI] [PubMed] [Google Scholar]
- 26.Colnot S, Niwa-Kawakita M, Hamard G, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest 2004;84:1619–1630. [DOI] [PubMed] [Google Scholar]
- 27.Madisen L, Zwingman TA, Sunkin SM, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010; 13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dow LE, O’Rourke KP, Simon J, et al. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015; 161:1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pectasides E, Stachler MD, Derks S, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov 2018;8:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Javier BM, Yaeger R, Wang L, et al. Recurrent, truncating SOX9 mutations are associated with SOX9 overexpression, KRAS mutation, and TP53 wild type status in colorectal carcinoma. Oncotarget 2016;7:50875–50882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghandi M, Huang FW, Jane-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moroishi T, Park HW, Qin B, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 2015;29:1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taniguchi K, Wu LW, Grivennikov SI, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015;519:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 2015; 15:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 2018;33:721–735.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–265. [DOI] [PubMed] [Google Scholar]
- 39.Haigis KM, Kendall KR, Wang Y, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet 2008;40:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent “+4” cell markers. EMBO J 2012; 31:3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009; 457:854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.James R, Erler T, Kazenwadel J. Structure of the murine homeobox gene cdx-2. Expression in embryonic and adult intestinal epithelium. J Biol Chem 1994; 269:15229–15237. [PubMed] [Google Scholar]
- 44.Suh E, Chen L, Taylor J, et al. A homeodomain protein related to caudal regulates intestine-specific gene transcription. Mol Cell Biol 1994;14:7340–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 1998;19:379–383. [DOI] [PubMed] [Google Scholar]
- 46.Blache P, van de Wetering M, Duluc I, et al. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 2004;166:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snippert HJ, van Es JH, van den Born M, et al. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 2009; 136:2187–2194.e1. [DOI] [PubMed] [Google Scholar]
- 48.Spelt L, Sasor A, Ansari D, et al. The prognostic role of cancer stem cell markers for long-term outcome after resection of colonic liver metastases. Anticancer Res 2018;38:313–320. [DOI] [PubMed] [Google Scholar]
- 49.Espersen ML, Olsen J, Linnemann D, et al. Clinical implications of intestinal stem cell markers in colorectal cancer. Clin Colorectal Cancer 2015;14:63–71. [DOI] [PubMed] [Google Scholar]
- 50.Hou NY, Yang K, Chen T, et al. CD133+ CD44+ subgroups may be human small intestinal stem cells. Mol Biol Rep 2011;38:997–1004. [DOI] [PubMed] [Google Scholar]
- 51.Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009;457:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shmelkov SV, Butler JM, Hooper AT, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133 metastatic colon cancer cells initiate tumors. J Clin Invest 2008;118:2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mak AB, Nixon AM, Kittanakom S, et al. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep 2012; 2:951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimozato O, Waraya M, Nakashima K, et al. Receptortype protein tyrosine phosphatase κ directly dephosphorylates CD133 and regulates downstream AKT activation. Oncogene 2015;34:1949–1960. [DOI] [PubMed] [Google Scholar]
- 55.Wei Y, Jiang Y, Zou F, et al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci U S A 2013;110:6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350–353. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Toke NH, Luo S, et al. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat Genet 2019;51:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Thé H.Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–127. [DOI] [PubMed] [Google Scholar]
- 59.Faux MC, Ross JL, Meeker C, et al. Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J Cell Sci 2004;117:427–439. [DOI] [PubMed] [Google Scholar]
- 60.Groden J, Joslyn G, Samowitz W, et al. Response of colon cancer cell lines to the introduction of APC, a colon-specific tumor suppressor gene. Cancer Res 1995;55:1531–1539. [PubMed] [Google Scholar]
- 61.de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017;543:676–680. [DOI] [PubMed] [Google Scholar]
- 62.Shimokawa M, Ohta Y, Nishikori S, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017;545:187–192. [DOI] [PubMed] [Google Scholar]
- 63.Cheng X, Xu X, Chen D, et al. Therapeutic potential of targeting the Wnt/β-catenin signaling pathway in colorectal cancer. Biomed Pharmacother 2019;110: 473–481. [DOI] [PubMed] [Google Scholar]
- 64.Layer RM, Pedersen BS, DiSera T, et al. GIGGLE: a search engine for large-scale integrated genome analysis. Nat Methods 2018;15:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol 2012;26:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.