

Abstract
A key challenge in studying organisms and diseases is to detect rare molecular programs and rare cell populations (RCPs) that drive development, differentiation, and transformation. Molecular features such as genes and proteins defining RCPs are often unknown and difficult to detect from unenriched single-cell data, using conventional dimensionality reduction and clustering-based approaches. Here, we propose an unsupervised approach, SCMER (Single-Cell Manifold presERving feature selection), which selects a compact set of molecular features with definitive meanings that preserve the manifold of the data. We applied SCMER in the context of hematopoiesis, lymphogenesis, tumorigenesis, and drug resistance and response. We found that SCMER can identify non-redundant features that sensitively delineate both common cell lineages and rare cellular states. SCMER can be used for discovering molecular features in a high dimensional dataset, designing targeted, cost-effective assays for clinical applications, and facilitating multi-modality integration.
2. Introduction
A tissue in a living organism often consists of millions to billions of cells. While the terminally differentiated cells with relatively distinct molecular profiles can be readily distinguished via single-cell RNA sequencing (scRNA-seq) at current sampling depth, many cells involved in development, differentiation, and transformation remain difficult to detect1,2. For example, a fraction of tumor cells in renal cell carcinomas can go through sarcomatoid transformation driven by epithelial to mesenchymal transformation (EMT)3,4; tumor cells in pancreatic ductal adenocarcinomas can transiently express stemness features (e.g., SOX2) at its invasion fronts5–7. These cells can be relatively rare in the sampled populations, transiently expressing certain molecular features and thereby may not form distinct clusters in high dimensional feature spaces8,9.
To detect characteristic features (e.g., genes, proteins) in a single-cell dataset, studies8,10–13 often employ unsupervised clustering followed by one-cluster-vs-all differential expression (DE) analysis, the optimal way for two-group hypothesis testing. These approaches can detect major cell types governed by lineage features that dominate data variance, but are insensitive to rare but unique features that have relatively small variance and manifest as level gradients within cell-type clusters (a.k.a. cell states)14. They are also clumsy at detecting features affecting multiple clusters, e.g., transcription factors (TFs) regulating multiple cell types15, as that involves comparison of an exponentially growing number of cluster combinations. To detect features associated with continuous developmental processes, many studies perform trajectory inference16 followed by correlation/regression analysis to identify correlated features (e.g., Monocle 217). The selection of features depends on trajectories, which could be challenging to infer accurately for complex processes. A detailed comparison was performed by RankCorr12 across various methods such as statistical tests, logistic regression, MAST10, scVI11, and COMET13.
Most existing approaches regard features as independent variables without exploring their interactions18. As a result, they tend to identify redundant features (e.g., CD3D, CD3E, and CD3G for T cells). Some recent work such as scHOT18 and SCMarker19 started to exploit correlational patterns among co- or anti-expressing genes. However, they do not model complex interactions of more than two genes. SCMarker cannot characterize continuous cell states, and scHOT relies on the accuracy of trajectory inference.
To enhance sensitivity in detecting rare features and RCPs, many studies20,21 had to slice and dice data spaces in empirical, multifaceted ways8 or perform iterative gating22 and re-clustering at variable resolutions, which may lead to biased, irreproducible results. For example, GiniClust223 selects a set of features to decide the major clusters and another set of features to discover RCPs (Supplementary Note 1). EDGE24 slices feature space randomly to attempt to find RCPs. CellSIUS25 refines clustering by examining gene sets upregulated in RCPs.
Increasing the number and variety of molecular features and improving the fidelity of the measurements can help discover RCPs26. However, they unavoidably increase the already high cost of experiments. To make assays cost-effective towards clinical applications, it is important to select a compact actionable set of molecular features that unbiasedly represent molecular diversity in high dimensional data. This ability is important for designing and manufacturing customized assays, e.g., 10x targeted gene expression, MissionBio Tapestri and NanoString GeoMx, which perform multi-omics measurements of hundreds of selected DNA, RNA, and proteins.
To address these fundamental challenges, we developed SCMER (Single-Cell Manifold presERving feature selection), which selects an optimal set of features such as genes or proteins from a single-cell dataset. Similar to t-Distributed Stochastic Neighbor Embedding (t-SNE)27 and Manifold Approximation and Projection (UMAP)28, we hypothesize that a manifold defined by pairwise cell similarity scores sufficiently represents the complexity of the data, encoding both global relationship between cell groups and local relationship within cell groups29. By preserving such a manifold while performing feature selection, the most salient features that unbiasedly represent the original molecular diversity will be selected.
SCMER does not require clusters or trajectories, and thereby circumvents the associated biases. It detects diverse features that delineate common and rare cell types, continuously changing cell states, and multicellular programs15 shared by multiple cell types. It reduces high dimensionality into a compact set of actionable features with definitive biological meanings. This distinguishes SCMER from PCA, t-SNE, UMAP, etc., which result in axes (meta-genes) with complex meanings. SCMER is efficiently implemented in Python using PyTorch30, multithreading and GPU acceleration supported, with a user- friendly single-command interface.
3. Results
3.1. The SCMER Approach
In a nutshell, SCMER (Fig. 1a, Methods) examines a data matrix (n cells x D features) and calculates a pairwise cell similarity matrix representing the manifold in . It defines a weight vector w and let . It then calculates another pairwise cell similarity matrix from and quantifies the level of agreement between and using Kullback-Leibler (KL) divergence. Finally, it uses elastic net to find a sparse and robust solution of w that minimizes the KL-divergence using the Orthant-Wise Limited Memory Quasi-Newton (OWL-QN) algorithm31. Features with nonzero weights in are deemed chosen. can also be calculated from a different modality instead of , which enables a “supervised” and multi-omics mode of SCMER.
Fig. 1: The SCMER approach.

(a) Workflow of SCMER. SCMER selects the features that preserve the manifold from a single-cell omics dataset . Features can be selected from either or another co-assayed omics . Vector indicates the selection. is the dataset after feature selection. and are cell-cell similarity matrices for and , respectively.
(b) Applications of SCMER. SCMER selects features that preserve the manifold and retain inter- and intra-cluster diversity, and thus can be applied to discover rich molecular pathways, integrate modalities, and design customized DNA/RNA/antibody panels of restricted sizes.
(c) Capabilities of SCMER compared with mainstream label/cluster-based differential expression (DE) analysis methods and correlation-based methods. The hypothetical branching trajectories contain common progenitors, precursors for A and B, and mature A and B.
A manifold encodes both clusters and continuums of cells. While clusters usually reflect distinct cell types, continuums reflect similar cell types and trajectories of transitioning/differentiating cell states32. SCMER selects optimal features that preserve the manifold and retain inter- and intra-cluster diversity (Fig. 1b). It can be applied to discover rich molecular pathways, identify prognostic genes, and design customized DNA/RNA/antibody panels of restricted sizes for clinical applications.
To elucidate the cell populations and features that SCMER identifies, we simulated a dataset containing a branching trajectory of 4,000 single cells from five major cell types, namely progenitor, precursors of A and B, and mature A and B (Fig. 1c). A total of 180 features are simulated from four categories (Supplementary Note 2), those (I) specific to one cell type/cluster, (II) shared by more than one cell type15, (III) gradually changing over cell states, and (IV) transiently activated (also known as checkpoints33). In addition to major cell type labeling, the cells transitioning from precursor to mature are identified as “RCP-A” and “RCP-B”, which overexpress type-IV features. In as few as 45 selected features, SCMER recalled all types of features. In contrast, the top 45 features determined by a DE analysis are all from type I, while a pseudo-time-based correlation analysis missed type-IV features. As a result, SCMER significantly increased the precision and recall of detecting RCPs, while being comparable to other methods on major cell types (Table 1).
Table 1:
Precision and recall of detecting RCPs on simulated data.
| RCPs | Major cell types | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell types | RCP-A | RCP-B | Progenitor | Precursor-A | Precursor B | Mature-A | Mature-B | |||||||
| Abundance | 2.55% | 2.68% | 21.23% | 22.43% | 19.83% | 16.73% | 15.30% | |||||||
| Precision/recall | Pre. | Rec. | Pre. | Rec. | Pre. | Rec. | Pre. | Rec. | Pre. | Rec. | Pre. | Rec. | Pre. | Rec. |
| SCMER | 0.82 | 0.68 | 0.87 | 0.67 | 0.97 | 0.96 | 0.95 | 0.96 | 0.94 | 0.94 | 0.95 | 0.96 | 0.94 | 0.93 |
| DE analysis | 0.61 | 0.34 | 0.73 | 0.40 | 0.94 | 0.95 | 0.94 | 0.93 | 0.95 | 0.94 | 0.94 | 0.95 | 0.95 | 0.96 |
| Correlation | 0.48 | 0.36 | 0.43 | 0.28 | 0.91 | 0.96 | 0.76 | 0.67 | 0.76 | 0.67 | 0.88 | 0.95 | 0.88 | 0.92 |
Listed are precision (pre.) and recall (rec.) using a K-NN classifier for one cell type at a time using feature selected by SCMER, DE analysis using known cell types, and correlation using pseudo-time (Supplementary Note 2). Higher is better for both metrics. Also shown are the abundances of the cell types.
To comprehensively assess SCMER, we ran it on eight datasets34–41 (Supplementary Table 1) that involve a variety of biological and technological challenges, such as unresolved borderline cells that blur clustering, continuously changing cell states, multicellular and transient cellular programs. For comparison, we used supervised DE analysis and widely-used unsupervised feature selection methods, including highly expressed genes (HXG), highly variable genes (HVG), SCMarker19, Monocle 217, RankCorr12, GiniClust223, EDGE24, and CellSIUS25. SCMER robustly demonstrated the best performance in all the experiments.
3.2. Characterizing cell type and intratumoral heterogeneity
Single-cell datasets derived from cancer samples are often highly complex, containing heterogeneous cell types and states in not only tumor cells but also stromal and immune cells. Supervised analysis of cancer data is challenging as cancer cells are highly plastic42 and can express novel unknown features, which can heavily confound clustering and trajectory-based analysis. We applied SCMER on a scRNA-seq melanoma dataset containing 4,645 cells from 19 human melanoma samples34. Most cells were annotated as malignant cells, B cells, T cells, macrophages, natural killer (NK) cells, endothelial cells, or cancer-associated fibroblasts (CAFs) by the authors based on clustering and DE analysis. However, there were unresolved borderline cells presenting between labeled clusters, which resemble multiple cell types and could be either doublets or RCPs (Fig. 2a). By selecting only 75 genes, SCMER clearly preserved the manifold: the resulting UMAP embedding is very similar to the original and the relations among cell types including the unresolved cells are preserved (Fig. 2b and Supplementary Figures 1–2).
Fig. 2: Results of the data of melanoma patients.

(a) UMAP embedding of the dataset without feature selection. Each dot represents a cell and the cell types are color-coded (Macro: macrophages, Endo: endothelial cells, CAF: cancer-associated fibroblasts, Unres: unresolved cells; labels and dots are colored synchronously by cell types).
(b) UMAP of the dataset using SCMER selected genes.
(c) Recall of gene sets for SCMER, SCMarker, Monocle 2, RankCorr, highly expressed genes (HXG), highly variable genes (HVG), principal component analysis (PCA), and differentially expressed genes (DEG, supervised). X-axis is the number of selected genes and Y-axis is the number of covered gene sets. A gene set is considered recalled when at least one gene in the set is selected. “Random” shows the expected number of gene sets for randomly selected markers. The area corresponds to 1.645 x standard deviation on each side. Results above the area has p < 0.05 based on one-sided z-test.
(d-f) RNA expression levels of genes showing intra-cluster gradients. Cells are in the same locations as in
(a) and overlaid with RNA expression levels (color bar).
(g-i) Overall Kaplan-Meier survival curve for selected markers in TCGA SKCM. High and low include patients in above and under 33% percentile, respectively. Each group includes n = 151 patients.
To understand the biological meanings of the selected genes, we compared them with the 11 gene sets described in the original publication that represent important cell types and pathways in the study. The selected genes compactly covered all the 11 gene sets (Supplementary Table 2). Interestingly, genes belonging to the known drug resistance AXL program and MITF program were also selected by SCMER. These genes do not preferentially express in a specific cluster (e.g., PMEL, TOB1, etc. in Fig. 2d and Supplementary Figure 1). Some genes such as PMEL, PDCD1, and OAS2 appeared predictive of survival outcome in TCGA SKCM patients43 (Fig. 2g–i). The genes selected by SCMER which are not reported by the original publication (i.e., the 11 gene sets), are found to be enriched in EMT, inflammatory abnormality of the skin, T cell exhaustion, and other immune pathways (Supplementary Note 3, Supplementary Data 1–2).
To comprehensively assess the performance of SCMER, we varied the number of selected features and recorded the number of recalled gene sets. SCMER consistently recalled more gene sets than other methods for any given number of features (Fig. 2c). SCMER also showed high performance in recalling genes regardless of which sets they belong (Supplementary Figures 3–5) and end-to-end clustering (Supplementary Note 4, Supplementary Table 3).
We also applied SCMER to a large-scale pan-cancer single-cell transcriptomic study consisting of 198 cell lines and patient samples from 22 cancer types35. SCMER showed high sensitivity in characterizing intra-cluster heterogeneity, identifying recurrent heterogeneous programs shared by most cell lines and by patient tumor samples (Supplementary Result 1, Supplementary Figure 6, Supplementary Data 3).
3.3. Defining cell subtypes and states of immunocytes
We further examined SCMER in a complex setting involving many cell subtypes, subtle intra-cluster structure, and shared pathways. The dataset contains 39,563 gastrointestinal immune cells collected from inflamed tissues from ten Crohn’s disease patients36. As a cancer risk factor, chronic inflammation involves extensive interaction among various immune cell types such as helper T cells (TH) and innate lymphoid cells (ILCs), which are regulated by both shared and cell-type specific TFs and cytokines and are difficult to delineate in high dimensional embeddings. The dataset appeared to include 27 cell types and subtypes/states in the original report. Four major cell types, T cells, B cells, phagocytes, and stromal cells each appeared as a cloud in the original embedding (Fig. 3a) but can be further dissected into subtypes (RCPs). For example, T cells were dissected into eight subtypes/states through further clustering.
Fig. 3: Results of the ileum lamina propria immunocytes data.

(a) UMAP embedding of the original dataset. Each dot represents a cell and the cell types are color coded (T (gd): gamma-delta T cell, Tregs: regulatory T cell, Endo: endothelial cell, TRM: tissue-resident memory T cell, DC: dendritic cell, ILC: innate lymphoid cell).
(b) UMAP embedding of the same dataset based on genes selected by SCMER.
(c-f) Examples of RNA expression levels of the genes selected by SCMER that (c) distinguish major cell types and (d) subtypes, (e) are transcription factors regulating different cell types, and (f) show gradual changes among cell states. Cells are in the same locations as in (a) and overlaid with RNA expression levels (color bar).
(g) The RNA expression level of DUSP1. See Supplementary Figure 7 for DUSP2 and DUSP4.
(h) Distributions of the RNA expression levels in major cell types of the genes above.
(i) Recalls of the gene sets selected by SCMER, SCMarker, Monocle 2, HXG, HVG, PCA, and DEG, similar to Fig. 2c.
Circumventing clustering, SCMER selected 250 features from 3,573 highly variable genes (Supplementary Table 4) with the manifold well preserved. The separability among cell types was comparable with the original embedding, and the manifold of subtypes in each major cell type was maintained (Fig. 3b).
SCMER identified features delineating both clusters and sub-clusters. For example, the well-known lineage features such as CD79A (B cells) and CD7 (T cells) and immune subtype markers such as FCER2 (naïve B cells) and ANKRD28 (TRM) were identified (Fig. 3c–d, Supplementary Figures 7a and 8). Less reported features such as SEPP1 for M2 macrophages were also among the list (Supplementary Figure 7b). The selected features also included genes that encode lysozyme (LYZ), complements (C1QA, C1QB, and C1QC), granulysin (GLNY), and granzymes (GZMA, GZMB, GZMK, and GZMH) (Fig. 3c, Supplementary Figure 7c).
NK and ILC1 cells were mixed together in one cluster and can hardly be further dissected based on unsupervised clustering and DE analysis. However, based on the genes selected by SCMER such as GNLY, CCL4, etc., which displayed dichotomizing levels within the cluster, we were able to further separate NK and ILC1 cells and estimate their abundances (Supplementary Figure 9).
SCMER also found TFs that regulate a wide range of cellular activities, including JUN and FOS (Fig. 3d), which are important for immune cell interactions. These features changed gradually among all the cell types, rather than expressing specifically in certain clusters. Other features such as CD69 (known T cell activation feature) and ODF2L (novel T cell subtype feature) also showed gradual change among subtypes instead of on-and-off patterns (Fig. 3f). Notably, among our selected features that were not reported in the original publication, DUSP1, DUSP2, and DUSP4 (Fig. 3g, Supplementary Figure 7d) were potential key regulators of both innate and adaptive immune responses that are highly relevant to Crohn’s disease (Supplementary Note 5).
SCMER again compared favorably to the other methods that selected various numbers of features (Fig. 3i, Supplementary Note 6). It was evident that the other methods tended to ignore features associated with intra-cluster heterogeneity and multicellular programs. The genes selected by SCMER which do not show in the original publication were also highly enriched in multiple immune pathways44 (humoral immune response, leukocyte migration, complement activation, etc.; Supplementary Data 4). Overall, SCMER sensitively preserved different types and levels of heterogeneity in the original data. Besides continuums of cell subtypes, SCMER also achieved top performance on continuous hematopoietic trajectories (Supplementary Result 2, Supplementary Figures 10–11, Supplementary Tables 5–6, Supplementary Data 5–6).
3.4. Identifying molecular drivers in a cancer treatment
More and more studies using single-cell technologies to investigate heterogeneity of cells in response to a genetic or chemical perturbation45. In these experiments, cell state may transition under complex kinetics.
To investigate the utility of SCMER in studying cellular responses, we applied it on single-cell data derived from dexamethasone (DEX) treated A549 lung adenocarcinoma cell line38. As reported in the original publication, the 1,429 cells sampled at 0, 1, and 3 hours after the DEX treatment formed a continuum in the transcriptomic space (Fig. 4a), indicating heterogeneous responses of the cell population. After running SCMER on the sci-RNA-seq data, 80 genes were selected, with the manifold and treatment states largely preserved (Fig. 4b).
Fig. 4: Results of the A549 lung cancer cell line data.
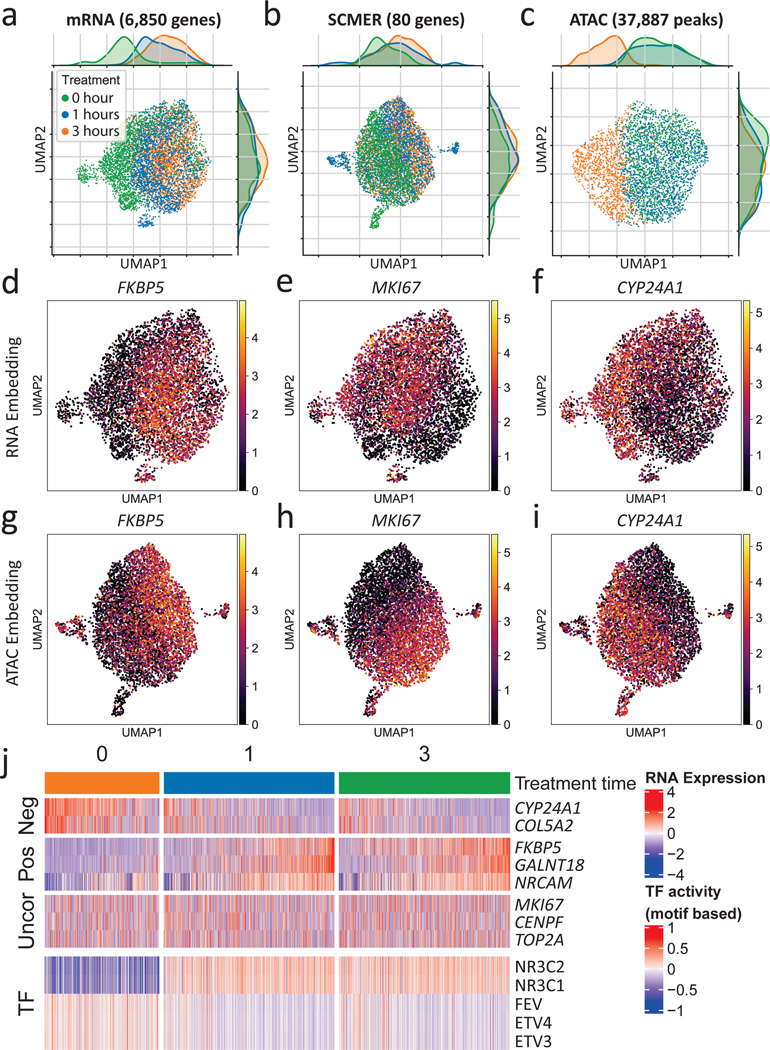
(a-c) UMAP embedding of (a) the original sci-RNA-seq dataset, (b) the sci-RNA-seq dataset on SCMER selected markers, and (c) the sci-ATAC-seq peak dataset. Each dot represents a cell. Treatment time points are color-coded.
(d-i) RNA expression levels of selected genes show in (d-f) RNA space and (g-i) ATAC space. ATAC space only includes co-assayed cells.
(j) Heatmap of RNA expression levels of selected genes and motif-based activity of highly variable transcription factors (TFs). (Uncor: uncorrelated, Pos: positively correlated, Neg: negatively correlated, with regard to NR3C1 and NR3C2.) ETV3 and ETV4 are in the ETS transcription factor family.
We inferred TF activities based on motif enrichment46 in the chromatin accessibility (sci-ATAC-seq) data co-assayed on the same set of cells38 (Methods, Fig. 4c). Among the top 50 highly variable TFs (Supplementary Figure 12), NR3C1, the primary target of DEX38, had the most prominently increasing activity level over treatment time. Other TFs such as FEV47 and the ETS family48, also targets of DEX, had decreasing activity levels.
We then correlated the expression levels of the genes selected by SCMER with the activity levels of the top TFs. We found that FKBP5, GALNT18, NRCAM, etc. were positively correlated with NR3C1, while CYP24A1, COL5A2, etc. were negatively correlated (Supplementary Table 7, Supplementary Figure 13). In particular, FKBP5, a factor in the negative feedback loop of glucocorticoid receptor response and regulator of immune processes49,50, had the highest positive correlation (r = 0.355) in the whole transcriptome; while CYP24A1, which regulates multiple metabolism processes51, was the most negative (r = —0.365). Cells of high FKBP5 expression levels came mostly from 1 and 3 hours (Fig. 4d), with matched polarized distributions in the RNA and the ATAC embeddings (Fig. 4g). Similar patterns were observed between cells of high and those of low CYP24A1 expression levels (Fig. 4f,i). Compared with other feature selection52 and DE analysis methods9, SCMER performed one of the best in recalling DEX target genes (Supplementary Note 7 and Supplementary Figure 14).
Interestingly, SCMER also selected a group of genes uncorrelated with prominent TF activities (Fig. 4j, Supplementary Figure 13). Among them were MKI67 (e.g., r = —0.005 with NR3C1) (Fig. 4e,h), which encodes proliferation marker protein Ki-67, and other cell-cycle genes such as CENPF, TOP2A, RYBP, MLH3, etc. Pathway analysis confirmed that these genes are highly enriched in cell proliferation pathways (Supplementary Data 7), indicating that an appreciable fraction of cells continued proliferating despite the treatment. It is not surprising that the levels of these genes were uncorrelated with chromatin state changes, as it has been shown that cell cycling status has little direct effect on chromatin accessibility53. Also among uncorrelated ones were several cancer cell stemness marker genes44 such as ACTG1, TSC22D1, and FN1, which may indicate that a fraction of cancer cells maintained their stemness during the course of the treatment. These genes would have been missed by a DE analysis supervised by the treatment time.
Taken together, our results demonstrated the superior power of SCMER in discovering features associated with heterogeneous cellular state change in the context of perturbation experiments. It explores alternative explanations and reports the most salient features representing different facets of cells.
3.5. Mapping features across modalities
One challenge in applying scRNA-seq for cell-typing is that expression levels of mRNAs can differ substantially from those of homologous proteins, due to post-transcriptional modifications54. Although performing multi-omics assays may be the ultimate solution, they are currently associated with higher cost and lower throughput. Thus, rather than simply selecting the homologous mRNAs, it is beneficial to identify the set of genes whose expression levels maximally represent cellular diversity at the protein level. This capability can be important for designing targeted, cost-effective assays for preclinical and clinical applications. SCMER is ideally suited for such a purpose, as it allows selecting features in one modality while preserving manifold in another modality.
We ran SCMER on a CITE-seq dataset containing 14,468 bone marrow mononuclear cells (BMNC)39. The protein manifold based on 25 markers was utilized to “supervise” the selection of mRNAs (Methods). CITE-seq, which co-assays mRNA and protein markers from the same set of cells, is ideal for obtaining the optimal mapping between mRNAs and proteins (Fig. 5a,b, Supplementary Figure 15).
Fig. 5: Results of the CITE-seq bone marrow mononuclear cells data.

(a-c) UMAP embedding of the original dataset using (a) protein, (b) all genes, and (c) SCMER selected genes. CD4-like T cells (CD4 TMEM and CD4 Naïve T), CD8-like T cells [Effector CD8 T, CD4 TMEM, CD8 Naïve T, gamma-delta T (gdT) cells, and Mucosal-associated invariant T (MAIT) cells] are framed by dotted circles, respectively, for better visual identification. Three circles are present in the result of RNA because of two separate clusters for CD8-like T cells. Also highlighted are progenitor cells [hematopoietic stem cells (HSCs), lymphoid-primed multipotent progenitors (LMPPs), granulocyte-monocyte progenitor cells (GMPs), and Progenitor (Prog) of B cells, megakaryocytes (Mks), red blood cells (RBCs), and dendritic cell (DCs)]. Fully annotated cell types are shown in Supplementary Figure 10.
(d-f) Levels of (d) proteins, (e) genes encoding the proteins, and (f) genes selected by SCMER. Cells are in the same locations as in (a) and overlaid with RNA expression level (color bar).
As shown, the mRNA expression levels of genes that encode the protein markers, such as CD4 (CD4, a Th cell marker) and NCAM1 (CD56, an NK cell marker), offered low power in delineating the corresponding cell types (Fig. 5d,e). Some markers, e.g., CD45RA (B cells and naïve T cells) and CD45RO (memory T cells) are isoforms of the same gene, PTPRC. Consequently, T cell subtypes were less distinguishable in the RNA space than in the protein space (Fig. 5b). The differences among CD8 T cell subtypes were even bigger than the differences between CD4 and CD8 T cells.
SCMER selected a set of genes that best preserved the diversity at the protein-level, notably the continuum among naïve CD8 T cells, memory CD8 T cells, and effector CD8 T cells (Fig. 5c) (SCMER adjusted Rand index (ARI) 0.544; RNA ARI 0.438; Supplementary Table 8). It identified genes that are non-homologous to the protein markers but better represent the protein level difference, for example, GPR183, KLRF1, CD79B, and S100A4 for CD4, CD56, CD45RA, and CD45RO, respectively (Fig. 5d,f). On the other hand, the SCMER result appeared to better delineate progenitor cells (ARI = 0.489) than the protein markers (ARI = 0.303), which demonstrates a strength of integrating complimentary modalities.
Similar conclusions were drawn when applying SCMER on another smaller PBMC CITE-seq dataset40 with 10 protein markers (Supplementary Result 3, Supplementary Figures 16–19, Supplementary Tables 9–11).
Importantly, the genes selected by SCMER from one donor (14,468 cells) appeared to preserve the cell diversity in another donor (16,204 cells) (Supplementary Figure 15), which validated the applicability of SCMER in designing targeted panels for populational level testing.
4. Discussion
For datasets with multiple samples, SCMER stratifies the samples to find consensus features that prioritize biological but not technical variances (Methods). SCMER can also run in various supervised modes. For example, it can select features from a shortlist (Supplementary Result 2) and find the best “partner” features for preselected features (Supplementary Result 3). The framework appears effective on cell line and patient data generated by various technologies, including scRNA-seq and mass cytometry41 (Supplementary Result 4, Supplementary Figure 20), and can potentially be extended to other modality combinations such as scRNA with scATAC, or mRNA with miRNA.
There are some possible limitations in this study. The evaluations were partly based on the gene sets provided in the publications, which may have some biases. In manifold transferring, SCMER does not provide an explicit mapping from one modality to the other, and thus requires additional analysis to clarify the interaction of features in the two modalities.
SCMER is efficiently implemented. On a dataset with 10,000 cells and 2,000 candidate features, it typically converges in 20 to 40 iterations, which takes 5 to 10 minutes using a 3.20GHz 6-core Intel Core i7–8700 CPU. The time consumption is halved with a middle-end NVidia GTX 960M GPU.
Because SCMER detects informative features that represent wider and more complex biological processes, we expect it to be of interest in projects producing large numbers of unsorted cells, such as the Human Cell Atlas55, the Human BioMolecular Atlas Program (HuBMAP)56, the Precancer Atlas57 and the Human Tumor Atlas Network58. It will be beneficial in various scenarios including biomarker discovery and clinical assay designing. As a feature selection method tailored for biomedical data with complex manifolds, it can potentially be applied to non- single-cell data, for example, bulk RNA expression29, copy number aberration, and genetic and drug screening data in large cohort studies such as TCGA and GTEx59.
5. Methods
5.1. Cell-cell Similarity
SCMER is inspired by three methods: Stochastic Neighbor-Preserving Feature Selection (SNFS)60, t-distributed stochastic neighbor embedding (t-SNE)27 and Uniform Manifold Approximation and Projection (UMAP)28.
t-SNE is one of the most widely used method for data embedding. For a dataset with n cells and D features, the similarity of a cell i to another cell j is defined as
which comprises a cell-cell similarity matrix . σ is a scaling factor. It creates an d-dimensional embedding . It calculates another cell-cell similarity matrix for , whose entries are
The cost function is defined as the Kullback-Leibler (KL) divergence of P and Q, formally
SNFS uses t-SNE formulation directly. Because emerging evidences show that UMAP is more sensitivity to both global relationship between cell groups and local relationship within cell groups29, we borrowed a part of the UMAP formulation, i.e.,
where and . The scaling factor is chosen such that , which may be viewed as constructing a soft nearest neighbor graph. We default it to 100 in our experiments. Similar to UMAP, setting it in the range 10 to 1,000 gives very similar results28.
5.2. Marker Selection by Elastic Net
Different from t-SNE and UMAP, instead of allowing to be an arbitrary matrix, we require each column of to be directly taken from a column of , i.e., to select a feature. To formally model this procedure. We use a vector (initialized as in optimization) to indicate the selection of the features, where 0 means unselected, and set
which set all unselected features to zero in . In terms of calculating the distances, zeroing out the columns is effectively discarding them. Thus, the calculation of using is unchanged. Ideally, to select d features, we optimize
where is the -pseudo-norm, i.e., the number of nonzero entries. However, this question is known to be NP-hard, whose determination requires checking all the possibilities. Thus, we fall back to -norm, the convex approximation of -pseudo-norm, as in
where -norm and is the strength of the regularization. We denote the loss function as L. Because the number of chosen features decreases when gets larger, for a given d, we use a binary search to find a . We used Orthant-wise limited memory quasi-Newton algorithm (OWL-QN, detailed below) to optimize . Due to limitations of precision, the specific d may not always be achievable. In that case, we allow for a few more features to be selected, and discard those that are assigned with the lowest weights (Supplementary Note 8). In the result, the features who have nonzero weights in are considered selected. The specific weight is not used in downstream analysis.
The cost, , is a robust indicator of whether the manifold is successfully retained. A typical range of C is 2.0 – 4.0 when the manifold is reasonably retained. More features (i.e., smaller -regularization) may be needed if the C is greater than 4.0.
Our model also allows an additional -regularization (ridge) to form an elastic net model. It may improve the robustness of the panel by slightly increase the redundancy, so that noise or drop-out in one feature has less effects (Supplementary Note 9 and Supplementary Figure 21).
5.3. Batch Effect Correction by Stratification
Batch effect is a common problem in experiments including multiple samples. For SCMER, the samples are considered a stratum. In specific, a set of and can be constructed for each sample, denoted as and , while is shared by all samples. A cost can thus be calculated for each sample, and collectively form a new objective . Thus, SCMER will ignore features that identify different samples and focuses on features that retain cell-cell similarities in all/most samples.
5.4. Supervised Multi-omics Mode
To transfer the manifold in one matrix () to another (), either between different modalities or subsets of features of the same modality, we simply modify the definition of to . With all other procedures unchanged, the algorithm is now searching for features in that gives a manifold similar to that of . This is also applicable to select features from a shortlist of the original ones.
5.5. Using Preselected features
In the case that a researcher wants to specify a few features that are known to be useful, we slightly modify the regularization to , where is a diagonal matrix. If a feature is considered important a priori, the corresponding entry in is set to 0 to avoid -regularization. In this “softly-supervised” way, SCMER is more likely to select these features, but may still discard some of them if they are contradicting with the manifold. Thus, in addition, we provide a “hard-supervised” way where a set of features are guaranteed to be kept. Other features are selected to supplement them.
5.6. Orthant-Wise Limited Memory Quasi-Newton Algorithm
Limited-memory BFGS (L-BFGS) is an widely-used optimization algorithm in the quasi-Newton methods family61. It approximates the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm with 0(mD) memory, where m can be chosen based on computing resources.
Although L-BFGS usually converge very fast (<20 iterations) for most -regularized regression problems, it will diverge for -regularization, whose partial derivative is undefined at :
It should be noted that setting the undefined point to 0 (or any other value) at does not solve the problem as the discontinuity will also break L-BFGS. SNFS restricts to avoid the discontinuity, but we find it having problems enforcing the sparsity. Instead, a modified version of L-BFGS called orthant-wise limited memory quasi-Newton (OWL-QN) algorithm31 is more suitable for this problem. A modified version of L-BFGS called orthant-wise limited memory quasi-Newton (OWL-QN) algorithm31 solves this problem by introducing pseudo-gradients and restrict the optimization to an orthant without discontinuities in the gradient.
In brief, we first derive the pseudo-gradient, where the pseudo-partial derivative at a discontinuity of the loss function is defined as
where is the pseudo partial derivative and is the short hand of , i.e, the left limit of the partial derivative. Similarly, is the right limit.
Note that the gradient of is continuous, i.e., , and discontinuities of L are all at . Thus, the pseudo-gradient can be simplified to
Then, we confine the search area in each quasi-Newton optimization step so that it does not cross any discontinuity. Specifically, for our problem where all discontinuities are at 0, when updating to . we reset the value of to 0 if . It constrains the optimization to be in the same “orthant” in each iteration.
L-BFGS optimizer is provided in PyTorch30, in which SCMER is implemented. Based on it, we implemented a special case of OWL-QN algorithm for optimization of the model. Two modifications we made are as follows.
5.7. Data Preprocessing
For the melanoma data34, which is TPM based, after removing ERCC spike-ins, we processed the data using the standard workflow of SCANPY62, including quality control (filtering out genes that are detected in less than 3 cells), normalization (10,000 reads per cell), log transformation, highly variable genes detection (with a loose threshold to filter out noisy genes; not to be confused with the DXG we compared with), and scaling.
For the Ileum Lamina Propria Immunocytes data36, bone marrow data37, and A549 data38, which are UMI based, we used the standard workflow of SCANPY, including quality control (filtering out genes that are detected in less than 3 cells), normalization (10,000 reads per cell), log transformation, highly variable genes detection, and scaling. We used the stratified approach to suppress batch effect on the Ileum Lamina Propria Immunocytes data.
For protein data in CITE-Seq39,40, we followed the preprocessing of protein data described in the original publication. For mRNA data in CITE-seq, we follow the standard workflow of SCANPY, as described above, except that we did not filter highly variable genes. We preprocessed protein data as mRNA data, without filtering highly variable genes.
5.8. Inference of TF Activities
Because TFs tend to bind at sites with cognate motifs, accessibility at peaks with the motifs reflects their activity. To estimate transcription factor activity from sci-ATAC-seq data, we use chromVAR46 package with the default setting. It quantifies accessibility variation across single cells by aggregating accessible regions containing a specific TF motif. The observed accessibility of all peaks containing a TF motif is compared with a background set of peaks normalized for known technical confounders.
5.9. Comparison with Other Methods
To identify the highly expressed genes (HXG), we used the standard SCANPY62 workflow. HXG is defined by the total reads of a gene across all cells. To identify the highly variable genes, we followed the standard scoring method in SCANPY62.
SCMarker19 provides a gene list without ranks. It has two parameters, n and k, which affect the number of resulting features. Based on our observation, n has a minor effect on the result. Thus, we fixed n = 50 and tested k from 10 to 1,200 to create feature gene lists of various sizes.
We ran Monocle 217 in unsupervised and supervised manners. For the supervised run, the labels were used directly. The trajectory was inferred using clusters/labels and pseudo-time is calculated. Genes were ranked by the degree they are explained by functions (which were fitted with cubic splines) of pseudo-time. For the unsupervised run, we clustered the cells and visually confirmed the clusters are concordance with the labels.
We ran RankCorr12 in both supervised and unsupervised manner. For the supervised run, we used the label from the data directly. For the unsupervised run, we used the Leiden algorithm63 for clustering which is the recommended method in SCANPY. Default parameters were used, and the clusters are visually checked that they are reasonable.
For random results, we randomly selected gene sets of given sizes. Reported are mean performance and the critical level of statistically significantly better (or worse) than random as defined by single-sample one-sided z-test at 5% significance level.
Data Availability
All original datasets are accessible through the original publications34–41, including the melanoma data (GSE72056), pan-cancer cell line data (https://singlecell.broadinstitute.org/singlecell/study/SCP542 ), immune cell subtypes data (https://singlecell.broadinstitute.org/singlecell/study/SCP359 ), hematopoiesis data (GSE116256), A549 data (GSE128639), CITE-seq data (GSE128639 and GSE100866), and CyTOF data (https://cytobank.org/nolanlab/reports/Levine2015.html). Source Data for Figures 1–5 are available with this manuscript.
Supplementary Material
Acknowledgements
The authors would like to thank Hussein Abbas, Yuanxin Wang, Linghua Wang for their comments. The authors acknowledge the support of the High Performance Computing for research facility at the University of Texas MD Anderson Cancer Center for providing computational resources that have contributed to the research results reported in this paper.
This project has been made possible in part by the Human Cell Atlas Seed Network Grant (CZF2019–002432 and CZF2019–02425) to KC from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation, grant RP180248 to KC and grant RP200520 to WP from Cancer Prevention & Research Institute of Texas, grant U01CA247760 to KC, grant U24CA211006 to LD, and the Cancer Center Support Grant P30 CA016672 to PP from the National Cancer Institute.
Footnotes
Code Availability
The open source implementation of SCMER available at https://github.com/KChen-lab/SCMER under the MIT License. Scripts for reproducing all the results are deposited in Code Ocean64.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable in this study.
Consent for publication
Not applicable in this study.
References
- 1.Merrell AJ & Stanger BZ Adult cell plasticity in vivo: de-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol 17, 413–425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Setty M. et al. Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol 37, 451–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z. et al. Sarcomatoid Renal Cell Carcinoma Has a Distinct Molecular Pathogenesis, Driver Mutation Profile, and Transcriptional Landscape. Clin. Cancer Res 23, 6686–6696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conant JL, Peng Z, Evans MF, Naud S. & Cooper K. Sarcomatoid renal cell carcinoma is an example of epithelial-mesenchymal transition. J. Clin. Pathol 64, 1088–1092 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Lytle NK et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 177, 572–586.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanada Y. et al. Histopathologic Evaluation of Stepwise Progression of Pancreatic Carcinoma with Immunohistochemical Analysis of Gastric Epithelial Transcription Factor SOX2: Comparison of Expression Patterns between Invasive Components and Cancerous or Nonneoplastic Intraductal Components. Pancreas 32, 164–170 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Herreros-Villanueva M. et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2, e61-e61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luecken MD & Theis FJ Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soneson C. & Robinson MD Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Finak G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez R, Regier J, Cole MB, Jordan MI & Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vargo AHS & Gilbert AC A rank-based marker selection method for high throughput scRNA-seq data. BMC Bioinformatics 21, 477 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delaney C. et al. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol. Syst. Biol 15, e9005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 25, 1491–1498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jerby-Arnon L. & Regev A. Mapping multicellular programs from single-cell profiles. bioRxiv 2020.08.11.245472 (2020) doi: 10.1101/2020.08.11.245472. [DOI] [Google Scholar]
- 16.Saelens W, Cannoodt R, Todorov H. & Saeys Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Trapnell C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghazanfar S. et al. Investigating higher-order interactions in single-cell data with scHOT. Nat. Methods 17, 799–806 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Liang S, Kumar T, Navin N. & Chen K. SCMarker: Ab initio marker selection for single cell transcriptome profiling. PLOS Comput. Biol 15, e1007445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Travaglini KJ et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv 742320 (2020) doi: 10.1101/742320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Z, Dai Z. & Locasale JW Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun 10, 3763 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu B. et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun 11, 3155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsoucas D. & Yuan G-C GiniClust2: a cluster-aware, weighted ensemble clustering method for cell-type detection. Genome Biol. 19, 58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun X, Liu Y. & An L. Ensemble dimensionality reduction and feature gene extraction for single-cell RNA-seq data. Nat. Commun 11, 5853 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wegmann R. et al. CellSIUS provides sensitive and specific detection of rare cell populations from complex single-cell RNA-seq data. Genome Biol. 20, 142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angermueller C. et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maaten L. van der & Hinton G. Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 28.McInnes L, Healy J. & Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv180203426 Cs Stat (2020). [Google Scholar]
- 29.Dorrity MW, Saunders LM, Queitsch C, Fields S. & Trapnell C. Dimensionality reduction by UMAP to visualize physical and genetic interactions. Nat. Commun 11, 1537 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paszke A. et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. Adv. Neural Inf. Process. Syst 32, 8026–8037 (2019). [Google Scholar]
- 31.Andrew G. & Gao J. Scalable training of L1-regularized log-linear models. in Proceedings of the 24th international conference on Machine learning 33–40 (Association for Computing Machinery, 2007). doi: 10.1145/1273496.1273501. [DOI] [Google Scholar]
- 32.Karamitros D. et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat. Immunol 19, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McFaline-Figueroa JL et al. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet 51, 1389–1398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tirosh I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinker GS et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet 52, 1208–1218 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin JC et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493–1508.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Galen P. et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176, 1265–1281.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stuart T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoeckius M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine JH et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marjanovic ND et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci 2, e67 (2016). [Google Scholar]
- 44.Chen J, Bardes EE, Aronow BJ & Jegga AG ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixit A. et al. Perturb-seq: Dissecting molecular circuits with scalable single cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pa N, Lk W, Ms S. & Tm O. Follow-up study of a randomized controlled trial of postnatal dexamethasone therapy in very low birth weight infants: effects on pulmonary outcomes at age 8 to 11 years. J. Pediatr 150, 345–350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srivastava S. et al. ETS Proteins Bind with Glucocorticoid Receptors: Relevance for Treatment of Ewing Sarcoma. Cell Rep. 29, 104–117.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zannas AS, Wiechmann T, Gassen NC & Binder EB Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology 41, 261–274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Leary JC, Zhang B, Koren J, Blair L. & Dickey CA The role of FKBP5 in mood disorders: Action of FKBP5 on steroid hormone receptors leads to questions about its evolutionary importance. CNS Neurol. Disord. Drug Targets 12, 1157–1162 (2013). [PMC free article] [PubMed] [Google Scholar]
- 51.Tieu EW, Tang EKY & Tuckey RC Kinetic analysis of human CYP24A1 metabolism of vitamin D via the C24-oxidation pathway. FEBS J. 281, 3280–3296 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Andrews TS & Hemberg M. M3Drop: dropout-based feature selection for scRNASeq. Bioinformatics 35, 2865–2867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma Y, McKay DJ & Buttitta L. Changes in chromatin accessibility ensure robust cell cycle exit in terminally differentiated cells. PLOS Biol. 17, e3000378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogel C. & Marcotte EM Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Regev A. et al. The Human Cell Atlas. eLife 6, 1–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snyder MP et al. The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spira A. et al. Precancer Atlas to Drive Precision Prevention Trials. Cancer Res. 77, 1510–1541 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rozenblatt-Rosen O. et al. The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lonsdale J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei X. & Yu PS Unsupervised Feature Selection by Preserving Stochastic Neighbors. in Artificia Intelligence and Statistics 995–1003 (PMLR, 2016). [Google Scholar]
- 61.Liu DC & Nocedal J. On the limited memory BFGS method for large scale optimization. Math. Program. 45, 503–528 (1989). [Google Scholar]
- 62.Wolf FA, Angerer P. & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Traag VA, Waltman L. & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang S. et al. SCMER: Single-Cell Manifold Preserving Feature Selection [Source Code]. 10.24433/CO.6781338.v1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original datasets are accessible through the original publications34–41, including the melanoma data (GSE72056), pan-cancer cell line data (https://singlecell.broadinstitute.org/singlecell/study/SCP542 ), immune cell subtypes data (https://singlecell.broadinstitute.org/singlecell/study/SCP359 ), hematopoiesis data (GSE116256), A549 data (GSE128639), CITE-seq data (GSE128639 and GSE100866), and CyTOF data (https://cytobank.org/nolanlab/reports/Levine2015.html). Source Data for Figures 1–5 are available with this manuscript.