

Summary
While inbred mice have informed most of what we know about the immune system in the modern era, they have clear limitations with respect to their ability to be informative regarding genetic heterogeneity or microbial influences. They have also not been very predictive as models of human disease or vaccination results. While there are concerted attempts to compensate for these flaws, the rapid rise of human studies, driven by both technical and conceptual advances, promises to fill in these gaps, as well providing direct information about human diseases and vaccination responses. Work on human immunity has already provided important additional perspectives on basic immunology such as the importance of clonal deletion to self tolerance, and while many challenges remain, it seems inevitable that “the human model” will continue to inform us about the immune system and even allow the discovery of new mechanisms.
Introduction
While the “Delbrück School” of bacteriophage(Judson, 1996) and the structure of DNA were key to the rapid advancement of molecular biology, inbred mice have long been the Rosetta Stone for immunologists in the modern era, allowing decades of immunological phenomena to be deciphered and reduced to mechanistic insights, or at least workable hypotheses. Powerful methodologies hae ben developed and used to understand antibody and T cell receptor diversity, major histocompatibility complex (MHC) restriction, the differences between innate and adaptive detection of self and non-self antigens and ensuing effector systems. Methods like monoclonal antibodies, the discovery of various inflammatory pathways and checkpoint inhibition developed in mice have been widely adopted as therapeutics for cancer, infectious disease and autoimmunity, as well as being a mainstay of ongoing basic research in many areas of biology. But immunology is one of the least static of any area of modern biology and so one must always ask what’s next? The purpose of this Perspectivew is to suggest that human immunology is the answer, not as a replacement to mice, but as an increasingly rich field in itself, taking advantage of many new technologies and events(like the ongoing pandemic) to expand our immunological universe into places that mice cannot go, such as the interplay of the immune system with thousands of human diseases, genetic and environmental diversity, and the effects of the many medical treatments on peoples’ immune systems(Pulendran and Davis, 2020). There is also the distinct possibility that entirely new immunological mechanisms will be discovered in human beings, given the many unique genes that are known and the fact that human infants have to survive many more years than mice in sufficient numbers to allow the species to survive. Within this context, we will discuss the various ways immunologists have tried to compensate for some of the deficiencies in the inbred mouse model, humanized mice, collaborative cross mice, pet shop mice etc. and how these have definite advantages and yet significant difficulties in terms of wide adoption and utility. Which is not to say that human immunology exists in isolation, since in most cases one must continually go to mice to rigorously test hypotheses. An exception to this are human genetic lesions that effect the immune system, where there is a clear cause and effect, although phenotypes can vary widely. Human genetics has even led the way in some cases, such as the cloning the autoimmune regulator (AIRE) gene in patients with a rare autoimmune disease (Nagamine et al., 1997) (Aaltonen et al., 1997) leading to an explosion of work on the equivalent gene in mice and the elucidation of its key role in immune tolerance (Mathis and Benoist, 2007). Similarly the identification of another key gene in tolerance, FoxP3 was discovered in humans (Bennett et al., 2001) just slightly before the mouse equivalent was identified (Brunkow et al., 2001), and an even more prodigious amount of work defining role of this gene (Rudensky, 2011). In any case, the technology to analyze human immune responses is advancing rapidly and has tremendous potential since there is so much unexplored territory. But also important is the way human immunology provides a different and consequently broader perspective on basic immune mechanisms. Longer term, the many differences seen between mice and human immune responses and the structural differences in skin and spleens highlighted here, suggest that new mechanisms may emerge from human work, especially as it relates to the lengths infectious pathogens will go to evade the human immune system.
Human immunology provides a valuable alternative perspective
The first time one visits a country and culture outside one’s own, you immediately become more aware of the many normally “hidden assumptions” about your own country and culture, and thus develop a more informed perspective. In a similar way, because studies of inbred mice have been so successful and dominated the field for over sixty years, we have not had much empirical input from other species to shed light on what might be important hidden assumptions in contemporary immunology. Here, human studies are an ideal complement because they have the genetic diversity that inbred mice lack, and they have the continuous exposure to infectious diseases that are scrupulously avoided in modern mouse facilities. A very vivid example of this to one of us (MMD) is that when Yu Wong in my group asked what the pre-immune frequency of pathogen specific CD8+ T cells might be in blood bank donors, following the ground-breaking work on mice from Jenkins and colleagues who used a sensitive tetramer staining enrichment method (Day et al., 2003) to quantitate the pre-immuneT cell receptor (TCR) repertoire. As a control, we also analyzed self-specific T cells and to our surprise found that the frequencies were very similar (Yu et al., 2015) (Su et al., 2013). We also looked at the Y chromosome encoded SMCY antigen-specific T cells in males and females and found a 2 to 50–65% reduction in the former vs the latter, and almost no difference between male and female mice (using the famous H-Y antigen). These results were confirmed a short time later with results in mice by Moon and colleagues (Legoux et al., 2015), who identified Cre-specific CD4+ T cells and showed that, if Cre was expressed ubiquitously there was only a 50% reduction in frequency, but in cases of organ specific expression, there was no detectable deletion. These results have been shocking because of classic experiments with super antigens and especially TCR transgenics in later 1980’s that have shown that self-specific T cells are massively deleted in the thymus of mice, and thus are a major component of what was called “central tolerance” (Herman et al., 1991) (Von Boehmer, 1990). While there was also persistent evidence of clonal anergy (Goodnow et al., 1988; Goodnow et al., 1989; Miller, 1993; Miller and Morahan, 1992; Pike et al., 1982) being a factor, negative selection has been widely accepted as the major mechanism of self-tolerance for 25+ years and seems to confirm Burnet’s earlier speculation that to avoid autoimmunity, self-reactive immune cells are eliminated during fetal development(Burnet, 1959).
But clearly, while negative thymic selection is a real phenomenon, as confirmed by the papers cited, the magnitude of its effects has been greatly exaggerated by implanting specific TCRs into the mice, which cause them to be expressed earlier in thymic development. For the superantigen results, this was probably due to the abundance of these particular self-antigens and the triggering of many TCRs at once.
It is important to note, though, that these experiments were the only way this issue could be addressed at the time. Tetramers, and the sensitivity needed, were many years in the future. But most importantly, these results force a re-evaluation of how important negatively selection is to prevent autoimmunity. Clearly, it’s not as important as it was thought to be, since the evidence is that even healthy people have a significant fraction of self-specific T cells, and not just of a low affinity since many can bind peptide-MHC tetramers perfectly well. What this re-evaluation has led to is the fundamental question of what shapes the TCR repertoire-if efficient negative selection is not important, what takes precedence? Following Dobzhansky’s dictum that “Nothing in biology makes sense except in the light of evolution” (Dobzhansky, 1973) leads to the realization that autoimmunity could not be a driver of TCR repertoire, since it has such a modest effect on selection in children and young adults (<1%). In contrast, infectious diseases, before widespread vaccination, antibiotics and sanitation, have killed almost half of children under 5 and many older children and young adults as well(Casanova and Abel, 2005; Su et al., 2013). This was the case for many generations, and probably started soon after the invention of agriculture, when people started living in fixed, dense communities and trading between these communities. And this degree of child mortality was even worse in the plague years. If infectious diseases are the primary driver of immune repertoire, it makes sense to have peripheral T cells expresssing as many specificities as they can and find other ways to hold self-specific T cells in check, unless they are needed, as Yu et al provided evidence for (Yu et al., 2015). Indeed a number of classic papers in murine immunology show that this is the case-that self-specific T cells are quiescent, unless triggered by an infection that includes their cognate antigen(Ohashi et al., 1991) (Röcken et al., 1992).
There are other examples, but the lesson here is that even when the basic immune mechanisms are the same between mice and humans, the fact that we often must approach human immunology in ways that are different than mice, can add an important perspective. In some cases, such as the one cited, this can completely change our understanding of the phenomenon.
Mouse Models of Disease
Since the development of inbred mouse strains by Snell in the late 1940’s (Gorer et al., 1948), mouse models of cancer have been a major activity by immunologists, soon joined by those interested in autoimmunity and infectious diseases. By the 1980’s the advent of recombinant DNA methodology and transgenics and then genetic albation etc. made mouse models of disease the premier route to understanding the role of the immune system in virtually every disease where this was thought to be relevant. And it became a given, that if you were doing serious science, and especially medically relevant “translation” of that science, you had to be working on a mouse model. There also arose the prejudice that good science was “hypothesis driven and mechanistic” and that “descriptive” became an epithet to describe work that was primitive and hopelessly old fashioned. This implicitly discouraged work on human beings, which, since there were few methods that could be applied ethically to those studies, most work could only be descriptive and correlative. But this ignores the fact that all science starts with description, and close and thorough observation is key to discovering new phenomena and formulating useful hypotheses. A mechanistic understanding is, of course, the ultimate goal, but it can take many years of observation and experiments to get there. Even then, an actual mechanism can be elusive. Even in physics one often settles for an accurate formula. The failure of many of these mouse translational models was noted many years ago in autoimmunity (von Herrath and Nepom, 2005) cancer immunology (Steinman and Mellman, 2004), and stem cell biology (Payne and Crooks, 2007). But also, aside from these critiques, in the academic world, one rarely hears about the many failures of mouse models to “translate” into effective clinical treatments. This is because this work is almost entirely done by biotech or pharmaceutical companies, and failures are rarely published, since no one wants to spend the time on that, nor do journals or reviewers welcome them. This results in a classic “publication bias” where only the good news gets out there. But those of us who advise companies widely (MMD) know that mouse models are viewed with great skepticism by the people who actually need to find useful drugs. Earlier on, these models were almost the only source of information, but recent advances in technology have created other options. I asked Frank Nestle, Global Head of Research and Chief Scientific Officer at Sanofi to comment on how mouse models are viewed in Biotech and Pharma: “The translational drug discovery paradigm is changing from a mouse first to a human first approach. We are in the age of human immunology, where we are tapping into unprecedented orthogonal data sets (Genetics, single cell transcriptomics, proteomics, metabolomics, real world clinical data et al) to drive our target discovery and translational research engine. While some mechanism based mouse models retain relevance, disease mimicking mouse models are becoming more and more the tools of the last decade.”
Nevertheless, even though mouse models of disease have often been disappointing as a seamless path to human treatments, as noted earlier, they have been a gold mine of information about how the immune system works, and will continue to be of great value, especially as more details of human diseases are uncovered, and we can determine exactly where the systems converge. Progress on finding these convergences informatically has been reported by Shen-Orr and colleagues (Normand et al., 2018). Even better would be a broad program of comparing human immune deficiencies and their detail phenotypes with mice with the same genetic lesions, while a number of discordances have been noted, there has been no systematic program to date.
Similarities and Differences
Humans and mice share many, although not all of the genes and cell types we know as important in human function, the same types of V, D, and J gene segments in T cell receptors and immunoglobulin genes, as well as the same rearrangement apparatus that goes back to early vertebrates. Also, many of the same or similar cytokine receptors and innate apparatus toll-like reeptors (TLR’s) etc., and MHC molecules also, basically all the nuts and bolts of adaptive and innate immunology that we know about at this time, but with some exceptions. One such example is well known, which is the expression of Class II human leukocyte antigens (HLA) molecules on activated human T cells, which is not seen in mice. This suggests that these human T cells can serve as “professional antigen-presenting cells” and act on CD4+ T cells to promote specific responses independently of B cells or dendritic cells. Whether this has any major consequences remains to be seen. Benoist and colleagues at the IMMGEN consortium have done a deep dive into the thousands of genes that they and others have identified as being expressed in immune cells, beyond those having generic functions, and find that the vast majority of homologs are expressed in the same cell subsets (Shay, et al., PNAS 2013). But a considerable number are expressed differently, at least 169 genes, using very stringent criteria. This is not a trivial number, and may contribute to the many differences seen phenotypically between the species. Also it appears that there are differences in the magnitude of expression that could make a substantial difference, as indicated by the activation of CD4+ T cells between the two species (Shay, et al., PNAS 2013). There may also be major differences in posttranslational modifications, but that is much more difficult to assess comprehensively. In addition, many immunological cues in the form of cytokines and chemokines are provided by non immunological tissues, and so this is another potential source of divergence to be investigated, which may be particularly relevant to differences in tissue or organ architecture, as discussed below.
To this point, the structure of tissues and organs relevant to the immune system between humans and mice suggests functional differences. One of these is skin, a major starting point and target of many pathogens. As shown in Figure 1(Zomer and Trentin, 2018), human skin is much thicker (~4x) and has 5–10 cellular layers, versus 2–3 for mice(Zomer and Trentin, 2018). The epidermal layers of skin in both species contain dendritic, antigen presenting Langerhans, together with CD4+ and CD8+ T cells, but only mice have the mysterious dendritic epidermal T cells (DETCs) with their uniform γδ T cell receptors(Allison and Havran, 1991; Johnson et al., 2020) apparently performing a specific sensory function, but also lacking in αβ T cells. The dermis layer in both species contains a broader mix of immune cells, αβ and γδ T cells, innate lymphocytic cells (ILCs), dendritic cells, and macrophages. The thickness of human skin also allows a direct connection to the lymphatic system(Pasparakis et al., 2014). Another striking difference in structure has emerged from the analysis of human spleens compared to mice, which show some striking anatomical differences. Figure 2. from Eisenbarth and Lewis (Lewis et al., 2019), with data also drawn from (Steiniger, 2015) shows how anatomically different human spleens are from mice. While both have red pulp and white pulp regions, the lymphocyte rich areas (white) where most of the adaptive immune response occurs, have a very different distribution of T cells which arediffuse in human germinal centers (GCs), versus discrete clusters in mice, and the absence of a clear bridging outer layer in human spleens. Whether these striking differences have any impact on immune responses is not clear as yet.
Figure 1. A comparison of mouse and human skin (Zomer and Trentin, 2018).
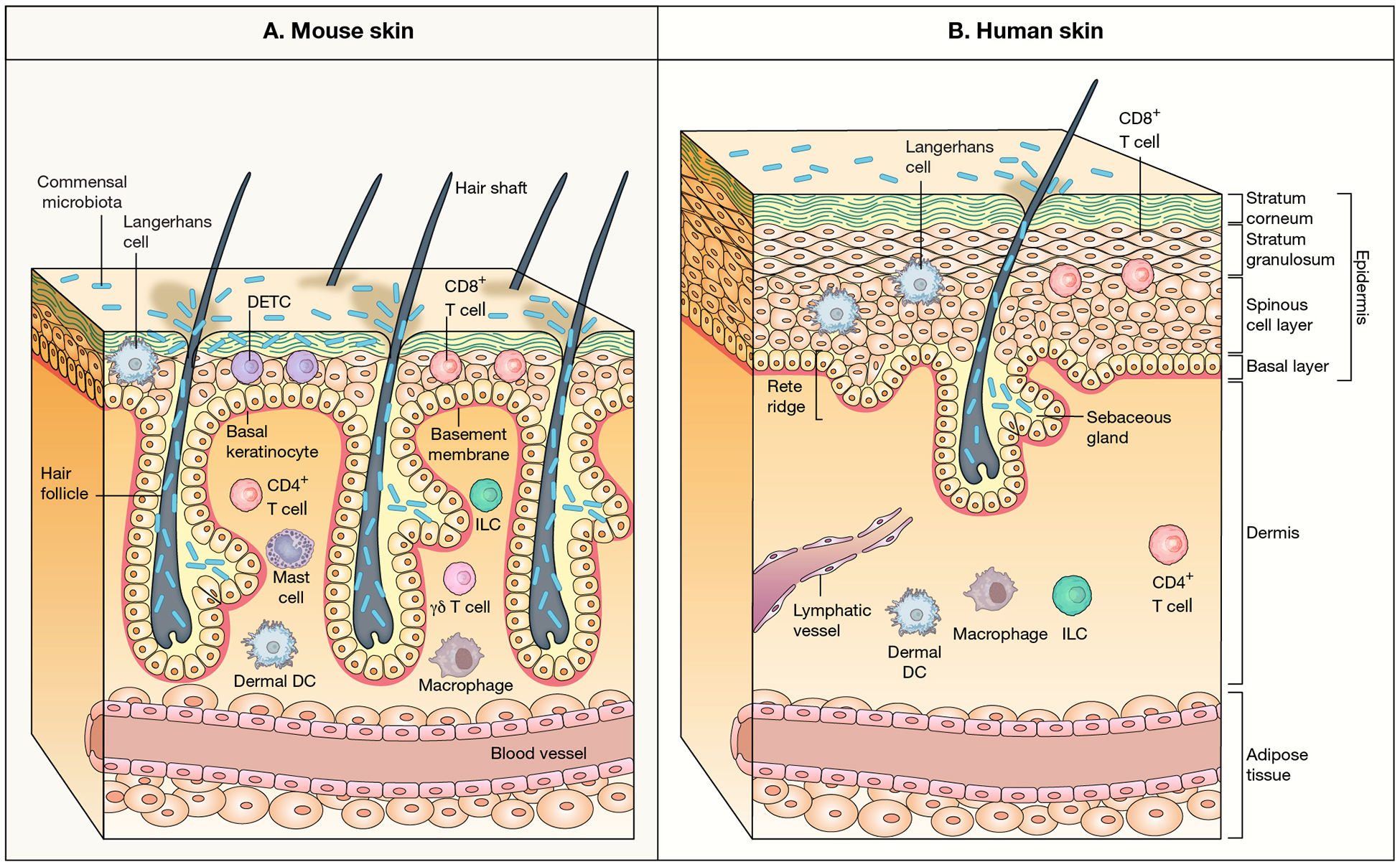
Human skin is much thicker and is more complex than mouse skin. In their immune composition they differ in their epidermal layer, where mice have the unique DETC cells with their uniform γδ T cell T cell receptors, and share with humans Langerhans and CD8 T cells(Pasparakis et al., 2014). Adapted with permissionfrom Nature (Pasparakis el al., 2014) copyright 2014.
Figure 2. Mouse and human splenic immune cellular architecture at steady state.
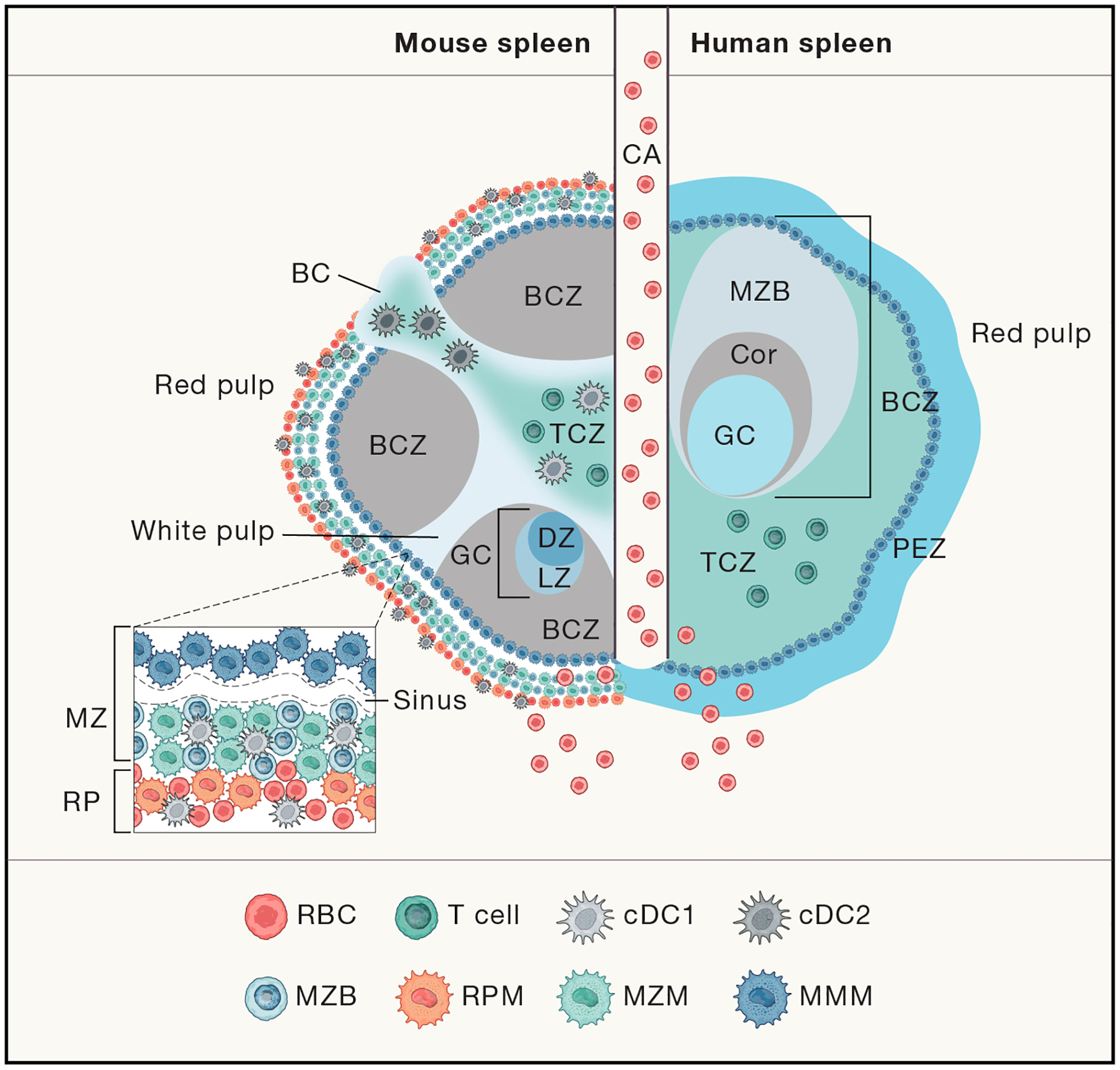
There are structural differences between the murine (left) and human (right) splenic immune system, most notably, the organizationof T cell zone (TC, turquoise; also known as PALS) and B cell zone (BCZ) follicles (gray and shades of blue, shown with light zone, LZ, and dark zone, DZ, organization in mouse spleen) within the WP and the border between the WP and RP, the MZ (marginal zone) in mouse or perifollicular zone (PFZ) in human (dark blue outer ring). Because applications of advanced imaging techniques to the human spleen have been limited, the extent to which the mouse MZ and human PFZ are analogous remains unknown. For example, the precise layering and composition of macrophage subsets in the MZ is known for mice (see bottom left box) CD169+ MMMs (dark blue) form a concentric ring around the WP with MZMs (light blue) and MB cells(darker blue)-but not for humans. In humans, MZB cells surround activated B cells, containing a GC (light blue in the human spleen on the right) and Corona (gray,”Cor”). The homeostatic location of dendritic cell (DC) subsets in mice is shown (with cDC2s in the bridging channel, BC, and cDC1s in the TCZ, MZ and RP, red pulp). Release of blood into the MZ of the WP from a central arteriole (CA) is shown. From, Science Immunology (Lewis et al., 2019). Adapted with permission from AAAS.
DeBoer and colleagues (van Hoeven et al., 2017) have also noted a striking difference in thymic output and T cell half-life between mice and humans, with the mice having a much shorter half-life than humans. Which makes sense in terms of the huge difference in life spans between the species. And if other immunological properties are speeded up this way in mice, it means that human responses might play out over a much longer time scale. Other anomalies have emerged from the study of human immune deficiencies, in cases where they can be directly compared to the equivalent mouse model. In the case of CD28, a key co-stimulatory molecule, mice lacking this gene have very severe immune dysfunction, with susceptibility to multiple pathogens, whereas humans with this deficiency are largely healthy, apparently only susceptible to skin papilloma viruses (Béziat et al., 2021). Another discordant gene is ISG15, an interferon inducible, ubiquitin-like intracellular protein, which is necessary for protection against viruses in mice, but human beings deficient in this gene seem to have reasonably effective anti viral responses, but deficient microbial defenses (Bogunovic et al., 2012) (Zhang et al., 2015).
These comparisons of the same genetic lesions in both species give us some of the clearest evidence we have of important functional differences between the species, although with the important caveat that humans lead much more complex lives in terms of environmental exposure than inbred mice. But in many cases pursuing information about a gene from humans to mice has been very fruitful, and shed new light and context as to its function. This is particularly true in the case of GCN2, whose expression unexpectedly has arisen in an analysis of a human cohort exposed to a yellow fever vaccine and correlates with an enhanced CD8+ T cell response (Querec et al., 2009). Analyzing the effects of deleting the murine homolog, Pulendran and colleagues have found that this gene is involved in the amino acid starvation response and the suppression of intestinal inflammation, which leads to an enhanced immune response (Ravindran et al., 2016) and insights into the control of autophagy and reactive oxygen species, (ROS). Thus, the mouse system can provide much greater depth to a human result when the systems are in alignment. Currently there are 485 known immunodeficiency genes that have been identified in human beings (Tangye et al., 2022). In addition, it has become clear that a number of older individuals make autoantibodies against particular cytokines, which attenuate pathogen responses that depend on those cytokines, with sometimes fatal results, as shown in recent work on COVID-19 patients (Bastard et al., 2020) (Chang et al., 2021).
The effects of environment and genetic diversity.
To reduce genetic variation, immunologistshave embraced inbred mice, originally developed by Snell, et al (Gorer et al., 1948) in the 1950’s. More recently, since mouse disease outbreaks can wreak havoc on immunological experiments, increasingly cleaner and more stringent mouse husbandry methods have been introduced, up to and including ultraclean methodologies that involve rederiving all mouse lines by cesarian delivery.
Both of these efforts make human comparisons problematic, especially for genetics, where most of the many gene manipulated mice are within the same strain, C57 BL/6. A modern fix to this is the collaborative cross system (Welsh et al., 2012) which consists of 5 inbred strains of mice, and three from wild mice, that have been crossed and then backcrossed extensively to fix each of the scrambled genomes into a separate, homozygous strains (Iraqi et al., 2008) (Shorter et al., 2019). Over 160 separate strains have been produced, with most of these having their genomes sequenced (Shorter et al., 2019), these have been very valuable in showing how different gene polymorphisms in mice can alter infectious disease responses (Noll et al., 2020) (Martin et al., 2020). This system has also been used to show how some of these strains mimic human drug responses (Zeiss et al., 2019). These are very valuable studies that show how the standard inbred strains give us only a very limited view of genetic influences on important phenomenon, but they involve analyzing sometimes over a hundred different mice (Martin et al., 2020) and so it’s a major commitment with no guarantee of relevance to human immunology. Research on these mice also cannot benefit from the many examples of genetic ablation models and other manipulations without a great deal of effort.
The lack of constant microbial exposure is also a problem, as clearly demonstrated by Masopust and colleagues, who very cleverly introduced mice obtained from pet shops to cohabitate with C57BL/6 mice and has shownthat those that survived, had acquired tissue resident T cells (Beura et al., 2016). But this would be difficult to do in modern mouse houses, as it would require a separate containment facility, so as not to endanger many of the users, animals, and studies.
Finally In 1988, McCune, Weissman and colleagues (McCune et al., 1988) introduced severe combined immunodeficiency-Human (SCID-Hu) mice, a novel effort to reconstitute immunodeficient mice with a human immune system in order to create ‘humanized’ mice that could faithfully model human responses. Despite many improvements over the years, (McCune, 1996) (Yong et al., 2018) (Rongvaux et al., 2013) there are still issues with getting good B cell responses, although many aspects seem to model human immune activities adequately. Again, this approach is not able to employ the power of genetic modifications that have made inbred mouse strains so powerful.
Do humans have novel immune mechanisms?
There is a reasonable hope that human beings might have unique immune mechanisms, because they are much less fecund and take much longer to reach sexual maturity. Thus, it’s fine if the average mouse lives six months or so, but it’s a major problem for survival of the species if enough humans don’t make it to 25–30 years. This means that if the risk of a fatal infectious disease is relatively constant, then humans are ~50x more vulnerable over the average lifetime. Thus, we may find additional ‘immunological defense’ mechanisms in human beings if we look hard enough. How might we find these, if they exist? One way might be the repurposing of genes that are shared between mice and humans, as discussed above. And thus those 169 genes with different expression patterns between mice and humans might mediate novel functions (Shay, et al., PNAS 2013) cited earlier. But another way to look for human or primate-specific mechanisms would be to identify human-specific genes versus mice. Here we have used the Ensemble Program to search for likely genes that are unique to humans vs mice (Table 1).
Table 1.
Shows genes in human immune cells that are not found in inbred mice, and thus may mediate novel mechanisms. But note many are shared with rats, and thus are likely not related to longevity, but nonetheless may mediate important functions in human beings.
| Gene | Immune population | The human protein atlas tissue specificity | Group | Top blastp against mouse | ||||
|---|---|---|---|---|---|---|---|---|
| GLYATL1B | B naive | breast, lymphoid tissue | glycine-N-acyltransferase-like 1B | 0.412 | primates | – | – | – |
| ZNF860 | B naive + memory | lymphoid tissue | zinc fingers C2H2-type | 0.438 | primates | – | – | – |
| PLEKHG7 | B naive + memory | cervix, epididymis, fallopian tube | Pleckstrin homology domain containing/Dbl family Rho GEFs | 0.258 | mammals | rats | sea urchins | – |
| PLAAT2 | plasmablasts | intestine | phospholipase A and acyltransferase family | primates | – | – | – | |
| ZNF683 | T CD8 TE | testis | zinc fingers C2H2-type | 0.56 | mammals | rats | – | – |
| TRABD2A | T naive | intestine, lymphoid tissue, ovary | TraB domain containing 2A | 0.648 | broad | fish | – | – |
| FAAH2 | Tfh + Th | multiple tissues including lymphoid | fatty acid amide hydrolase 2 | 0.304 | broad | fish | Drosph | xeno |
| GNLY | T, NK | bone marrow, lymphoid tissue | granulysin, cytotoxic T cells, NK cells | 0.292 | broad | – | – | – |
| KIR2DP1 | NK | lymphoid tissue | killer cell immunoglobulin like receptors | 0.401 | mammals | – | – | – |
| KLRF1 | NK | bone marrow, lymphoid tissue | killer cell lectin like receptors/C-type lectin domain containing | 0.303 | mammals | – | – | – |
| LGALS9B | NK | esophagus, intestine, stomach 1 | galectins | 0.685 | – | – | – | – |
| LGALS9C | NK | esophagus, intestine, stomach 1 | galectins | 0.688 | – | – | – | – |
| CD1B | mDCs | lymphoid tissue | CD molecules/C1-set domain containing | 0.489 | mammals | – | – | – |
| CD1E | mDCs | lymphoid tissue | CD molecules/C1-set domain containing | 0.521 | mammals | bats | chinchilla | – |
| CLIC2 | mDCs | multiple tissues including lymphoid | chloride intracellular channels | 0.662 | broad | rats | – | – |
| KCNK17 | pDCs | lung, thyroid gland | potassium two pore domain channel subfamily K | 0.42 | broad | Drosophila | – | – |
| NLRP7 | pDCs | testis | NLR family/pyrin domain containing | 0.408 | mammals | – | – | – |
| CTSV | pDCs | lymphoid tissue | cathepsins | 0.792 | mammals | mole rat | – | – |
| PROC | pDCs | liver | receptor ligands/Gla domain containing | 0.695 | mammals | rats | – | – |
| SLC9C2 | pDCs | brain, choroid plexus, testis | solute carriers | 0.292 | mammals | rats | – | – |
| ADGRE3 | granulocytes LD | bone marrow, lymphoid tissue | adhesion G protein-coupled receptors, subfamily E | 0.459 | mammals | – | – | – |
| NT5DC4 | monocytes I | testis | 50-nucleotidase domain containing 4 | 0.654 | broad | – | – | – |
| CASP5 | monocytes NC | brain, intestine, lymphoid tissue | caspases|caspase recruitment domain containing | 0.636 | broad | – | – | – |
| LILRA1 | monocytes NC + I | lymphoid tissue | CD molecules/activating leukocyte immunoglobulin like receptors | 0.507 | primates | – | – | – |
| LILRB2 | monocytes NC + I | bone marrow, lung, lymphoid tissue | CD molecules/inhibitory leukocyte immunoglobulin-like receptors/Ig-like cell adhesion molecule family | 0.452 | mammals | rats | – | – |
| LILRA2 | myeloid | bone marrow, lymphoid tissue | CD molecules/activating leukocyte immunoglobulin like receptors | 0.513 | primates | – | – | – |
| SIGLEC14 | myeloid | bone marrow, lymphoid tissue | V-set domain containing/sialic acid binding Ig-like lectins | 0.498 | mammals | |||
| – | – | lung, lymphoid tissue | – | – | – | – | – | – |
| OR14L1P | basophils LD | brain | olfactory receptors, family 14 | 0.53 | mammals | reptiles | – | – |
| OR6K3 | basophils LD | bone marrow | olfactory receptors, family 6 | 0.784 | mammals | rats | – | – |
| OXER1 | basophils LD | liver | leukotriene receptors | 0.418 | mammals | – | – | – |
| TRIM49D1 | basophils LD | undetected | ring finger proteins/tripartite motif containing | 0.364 | primates | – | – | – |
| TRIM49D2 | basophils LD | undetected | ring finger proteins/tripartite motif containing | 0.364 | primates | – | – | – |
| TRIM51 | basophils LD | undetected | ring finger proteins/tripartite motif containing | 0.367 | – | – | – | – |
| TRIM64 | basophils LD | placenta | ring finger proteins/tripartite motif containing | 0.348 | – | – | – | – |
| TRIM64B | basophils LD | placenta | ring finger proteins/tripartite motif containing | 0.348 | – | – | – | – |
| FCAR | neutrophils LD | bone marrow, lung, lymphoid tissue | CD molecules/immunoglobulin-like domain containing/Fc receptors | 0.327 | – | – | – | – |
| H2AC19 | neutrophils LD | multiple tissues including lymphoid | H2A histones | 1 | – | – | – | – |
| CSNK1A1L | neutrophils LD | testis | casein kinase 1 family | 0.91 | broad | Drosophila | – | – |
| LINC02218 | neutrophils LD | undetected | long intergenic non-protein coding RNAs | 0.441 | – | – | – | – |
| PEAK3 | neutrophils LD | bone marrow, lymphoid tissue | PEAK family member 3 | 0.268 | broad | fish | – | – |
| PI3 | neutrophils LD | esophagus, lymphoid tissue, vagina | WAP four-disulfide core domain containing | 0.941 | broad | chkns | – | – |
| S100A12 | neutrophils LD | bone marrow | S100 calcium binding proteins/EF-hand domain containing | 0.388 | mammals | rats | – | – |
| SIRPB2 | neutrophils LD | lymphoid tissue | V-set domain containing/signal regulatory proteins | 0.348 | mammals | rats | – | – |
| SIRPD | neutrophils LD | testis | V-set domain containing/signal regulatory proteins | 0.48 | mammals | rats | – | – |
| ST20 | neutrophils LD | multiple tissues including lymphoid | suppressor of tumorigenicity 20 | 0.724 | ? | – | – | – |
| TMEM272 | neutrophils LD | brain | transmembrane protein 272 | 0.368 | mammals | – | – | – |
The program compares sequence homologies to determine whether a human gene is sufficiently different from mice to be considered unique. The list is not long, and while it may just reflect divergent evolution and minor tweaking of functions that are well known from the many intensive studies of mouse immunology, but given the many apparent differences seen for years between mouse models and clinical outcomes, these genes and those identified by Shay and colleagues (Shay, et al., PNAS 2013) may be well worth following up for clues regarding novel immunological mechanisms.
Although the above examples show that even with respect to the same molecules and organs, humans and mice exhibit clear differences, another way to ask the question is to identify genes that are unique in humans, since these may lead to even more profound differences and mechanisms. Thus, we have taken advantage of a rich data set of RNA seq data for 29 different human immune cell subsets that have been isolated and analyzed for gene expression (Monaco et al., 2019).
The table listing all human genes based on Genome Reference Consortium human build (GRCh38.p13) was downloaded from Ensembl BioMart (Kinsella et al., 2011). Genes whose “gene type” is “protein coding” were selected from the table. Next, the table listing mouse genes based on GRCm39 and their human homologs were downloaded from Ensembl BioMart. Human genes from the former table that do not have any mouse homologs in the latter were identified as candidate unique human protein-coding genes. In addition, the NHBL website (link) collates a very extensive collection of species data on all known genes and with this resource we have been able to eliminate many genes which did have identified homologs in mice, and other species. We also used the UniProt blastp program using the protein sequence to search the mouse genome to make sure there was not any mouse sequence homologies of significance(Bateman et al., 2021). The resulting summary of this work is presented in Table 1, together with some of the information about cell type distribution, and other species that had this particular gene. Many of these genes have rat homologs, suggesting that their lack in mice was not due to very different functionality. But a few were exclusively in primates, which would fit with novel immune functions related to increased longevity and low fecundity. It is also noteworthy the large number of unique genes in human dendritic cells, neutrophils and basophils, which may correlate with the difficulty encountered in modeling allergy and other human pathologies involving those cells in mice.
Also, this list should not be considered the last word, we in fact missed intercellular adhesion molecule-3 (ICAM3) and TLR10 in this survey, which came up anecdotally, so there are likely others.
The entire workflow of identifying the human genes, matching with immune population genes, and running blastp can be found in GitHub repositories: https://github.com/KwatMDPhD/immune_populations.pro and https://github.com/KwatMDPhD/human_genes.pro.
It was also beneficial to vet candidate genes on the very helpful National Heart Lung and Blood Institute website which compares particular genes across many species.
In the table, it is interesting to note the relatively large number of human genes in dendritic cells, neutrophils and basophils. This may be an explanation for why many of the anomalies between mice and humans occur in the innate immune mechanisms that these cells mediate. Specifically dendritic cells play a pivotal role in gathering and presenting antigens to T cells and have long been thought to be somewhat different between humans and mice, but exactly how and what the consequences might be, is unclear. For example, in humans, TLR7 and 9 are mostly expressed in plasmacytoid DCs and thus agonists for these TLRs can only activate these cells, whereas in mice TLRs7 and 9 are more broadly expressed on other subsets of DCs TLR7 and 9 are mostly expressed in pDCs and thus agonists for these TLRs can only activate pDCs. In contrast, in mice TLRs7 and 9 are more broadly expressed on other subsets of DCs too which can be activated by such agonists(Pulendran and Davis, 2020). The power of recent advances in single cell transcriptional analysis has great potential in illuminating human-mouse differences, but the limitations are also evident in a recent analysis of dendritic cells in the two species by Rudensky and colleagues (Brown et al., 2019). They note clear differences in subsets, but because a functional readout is not readily available, whether these differences are consequential is not clear; which is a general problem for all of these genes that don’t have a mouse homolog, pointing to the need for a complete human system that can be analyzed. Here humanized mice or immune organoids are possible answers.
The number of unique neutrophil genes is also interesting, and may underlie some of observed discordances between mice and humans, especially given the very different composition of these cells in circulating white blood cells, 60–70% in humans vs only ~30% in inbred mice (Nauseef, 2014) (Junhee et al., 2013; Nauseef, 2019), but again, until we can probe more deeply into a human assay system, it remains a mystery how significant these are.
Lastly, the number of basophil genes is interesting and potentially explains the difficulties in modeling human allergic reactions in mice.
Our main hope in presenting these results is not to resolve questions about their importance, but to tantalize readers with the possibility that some of these genes might mediate novel immune mechanisms. We also caution about thinking about individual genes in isolation, as they always work in pathways where they can be influenced by polymorphisms in other genes or by different environmental factors, such as pathogens or microbiome components.
Another way we are likely to discover additional mechanisms in immunology, is by taking advantage of the vast number of human pathogens and learning what their strategies are for avoiding or distracting the immune system. These mechanisms may not be unique to humans, but the sheer number of human pathogens compared to the few commonly used in mouse models suggest that there is a wealth of insight to be gained, and enabled with recently developed technologies, to expect important discoveries. Indeed, Rolf Zinkernagel (Zinkernagel, 1996), entitled a famous review “Immunology taught by viruses” to make this point years ago, but only now do we have the ability to analyze human responses to pathogens in depth. Some pathogen strategies are well known, with HIV for example, uses an error prone polymerase to produce an extreme degree of genomic diversity, producing thousands of quasi-species in each infected individual, eventually wearing down the immune systems’ ability to keep up.(Bbosa et al., 2019)
T. brucei, a parasite which causes sleeping sickness, is well known for employing thousands of possible surface proteins, where immune pressure against one type, leads to another one becoming dominant(Cross, 1990). But these strategies are very simple compared to the complexities of other pathogens. Cytomegalovirus (CMV) is remarkable in both its lifelong staying power, and its ability to enhance immune responses against other viruses, in both mice and humans (Furman et al., 2015). Additionally, it has a number of mechanisms to subvert the immune system, as reviewed by F. Goodrun,W. Britt and ES Mocarski “Cytomegalovirus” chapter 12 Fields Virology 2021 (Goodrun et al., 2021).
But recent work has shown even greater subtilty, where work by Picker and colleagues has shown that vaccine vector that they made was remarkably effective against both SIV and TB in monkeys (Hansen et al., 2010) (Hansen et al., 2016) (Malouli et al., 2021) (Verweij et al., 2021) and this was accompanied by a novel HLA-E restricted T cell response, and class II MHC restricted CD8+ T cells. Just last year it was reported that the class II restricted CD8+ T cells were not important for these remarkable vaccine responses, indicating that the HLA-E ones were. Also telling was the finding that no less than seven CMV genes seemed to be engaged in diverting T cell responses away from HLA-E (Yang et al., 2021). Thus, while T cell responses against CMV antigens are quite active, accounting for an estimated 10–20% of all CD8+ T cells in carriers, there are no known cases of the virus being cleared from anyone. Another interesting fact is that in 16 monozygotic twins discordant for cytomegalovirus (CMV) seropositivity, there have been massive changes in immune system variables in the positives vs the negatives (Brodin et al., 2015). The implications of all this are not very clear, other than that the virus has things under control, and we have a lot to learn about what it is up to. And probably from many of our other pathogens as well.
The way forward.
Almost 14 years ago, one of us (MMD) wrote in this journal a plea for the immunology community to not just pay lip service to human immunity, but to put serious effort into welcoming it to the fold as a distinct, but vitally important complement to the juggernaut that is mouse immunology (Davis, 2008). There is still a long way to go to catch up to that standard, but human immunology is definitely getting there. And it isn’t just conceptual or abstract anymore, because in the last 2+ years we have all been affected by this horrendous pandemic and it has had such a wide range of health effects, from negligible to painful death. It has shown that serious, life-threatening infections are not just something that happens to unfortunate countries far away from most of us, but close and very real. And as immunologists, this has to hit close to home, and make clear the urgency of why understanding the human side of immunology, with its complexity and wide variations, is critical. And that even as this pandemic fades, as it might, another is brewing, somewhere.
Clearly, inbred mice and the amazing technology built up over the past 60+ years are such a powerful and useful system that it will continue to be a mainstay of the field for the foreseeable future. Even when investigating new human phenomenon, being able to go to mice to investigate mechanisms and/or confirm hypotheses has been invaluable to my group and many others. But it is also important not to ignore the many variances, genetic, anatomical and in gene expression. It unlikely that all of these represent minor tweaks in the human immune system, and so we could be missing important aspects of human immunity. While humanized mice might represent part of the answer in exploring these anomalies, there will always be questions about how faithfully these hybrid animals can represent human immunity, especially given the constraints of mouse husbandry. Work on monkeys is one solution, which has been used to great effect in many studies, but they are extremely expensive and in short supply. Thus, the advent of human immune organoids using discarded tonsils or spleens may be the key to being to be able to do mechanistic and hypothesis-driven experiments that will be needed (Wagar et al., 2021). The advent of gene modifications using CRIPR-Cas9 is also introducing powerful ways to screen for important genes such as the genome-wide screen of CD8 T cells by Marson and colleagues or early work on immune and intestinal organoids (Shifrut et al., 2018) (Fujii et al., 2015) (Wu et al., 2018).
In conclusion, while human immunology is still very much a work in the progress, there is so much potential and uncharted territory to explore, and with such a strong foundation from mouse immunology and continued interactivity between the two systems, that one can’t help but to be optimistic about what we will learn in the coming decades.
ACKNOWLEDGEMENTS
We thank J.L. Casanova, Bali Pulendran, Paul Kubes, Stephanie Eisenbarth, Karan Kathuria, Juliana Idoyaga, Guangbo Chen, Chen Wang, Xin Chen, Ed Mocarski, John Boothroyd. We thank the Howard Hughes Medical Institute, the Open Philantropy Foundation, and NIAID AIO57229 for support.
REFERENCES
- Finnish-German APECED Consortium, Björses P, Perheentupa J, Horelli-Kuitunen N, Palotie A, Peltonen L, Lee Su Y, Francis F, Hennig S, Thiel C, et al. (1997). An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet 17, 399–403. 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- Allison JP, and Havran WL (1991). The immunobiology of T cells with invariant γδ antigen receptors. Annual review of immunology. [DOI] [PubMed] [Google Scholar]
- Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V, et al. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585. 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, et al. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic acids research 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bbosa N, Kaleebu P, and Ssemwanga D (2019). HIV subtype diversity worldwide. Current opinion in HIV and AIDS 14, 153–160. 10.1097/COH.0000000000000534. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genetics 27, 20–21. 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516. 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Rapaport F, Hu J, Titeux M, Bonnet des Claustres M, Bourgey M, Griffin H, Bandet É, Ma CS, Sherkat R, et al. (2021). Humans with inherited T cell CD28 deficiency are susceptible to skin papillomaviruses but are otherwise healthy. Cell 184, 3812–3828.e3830. 10.1016/j.cell.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. (2012). Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688. 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJL, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, et al. (2015). Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47. 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature Genetics 27, 68–73. 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Burnet FM (1959). The Clonal Selection Theory of Acquired Immunity (Vanderbilt University Press; ). [Google Scholar]
- Casanova JL, and Abel L (2005). Inborn errors of immunity to infection: The rule rather than the exception. J. Exp. Med 202, 197–201. 10.1084/jem.20050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, Barman L, Bennett K, Chakraborty S, Chang I, et al. (2021). New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nature communications 12, 5417. 10.1038/s41467-021-25509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross GAM (1990). Glycolipid anchoring of plasma membrane proteins. Annual Review of Cell Biology 6, 1–39. 10.1146/annurev.cb.06.110190.000245. [DOI] [PubMed] [Google Scholar]
- Davis MM (2008). A prescription for human immunology. Immunity 29, 835–838. 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CL, Seth NP, Lucas M, Appel H, Gauthier L, Lauer GM, Robbins GK, Szczepiorkowski ZM, Casson DR, Chung RT, et al. (2003). Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J. Clin. Invest 112, 831–842. 10.1172/JCI200318509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T (1973). Nothing in Biology Makes Sense except in the Light of Evolution. American Biology Teacher 35, 125–129. 10.2307/4444260. [DOI] [Google Scholar]
- Fujii M, Matano M, Nanki K, and Sato T (2015). Efficient genetic engineering of human intestinal organoids using electroporation. Nature protocols 10, 1474–1485. 10.1038/nprot.2015.088. [DOI] [PubMed] [Google Scholar]
- Furman D, Jojic V, Sharma S, Shen-Orr SS, Angel CJL, Onengut-Gumuscu S, Kidd BA, Maecker HT, Concannon P, Dekker CL, et al. (2015). Cytomegalovirus infection enhances the immune response to influenza. Sci. Transl. Med 7, 281ra43. 10.1126/scitranslmed.aaa2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. (1988). Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682. 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Jorgensen H, Brink RA, and Basten A (1989). Induction of self-tolerance in mature peripheral B lymphocytes. Nature 342, 385–391. 10.1038/342385a0. [DOI] [PubMed] [Google Scholar]
- Goodrun F, Britt W, and Mocarski ES (2021). Cytomagalovirus. In Fields Virology. [Google Scholar]
- Gorer PA, Lyman S, and Snell GD (1948). Studies on the genetic and antigenic basis of tumour transplantation Linkage between a histocompatibility gene and ‘fused’ in mice. Proceedings of the Royal Society Biological Sciences, 499–505. [Google Scholar]
- Hansen SG, Powers CJ, Richards R, Ventura AB, Ford JC, Siess D, Axthelm MK, Nelson JA, Jarvis MA, Picker LJ, and Früh K (2010). Evasion of CD8+ T Cells Is Critical for Superinfection by Cytomegalovirus. Science 328, 102–106. 10.1126/science.1185350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SG, Wu HL, Burwitz BJ, Hughes CM, Hammond KB, Ventura AB, Reed JS, Gilbride RM, Ainslie E, Morrow DW, et al. (2016). Broadly targeted CD8+ T cell responses restricted by major histocompatibility complex E. Science 351, 714–720. 10.1126/science.aac9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman A, Kappler JW, Marrack P, and Pullen AM (1991). Superantigens: Mechanism of T-cell stimulation and role in immune responses. Annual review of immunology. [DOI] [PubMed] [Google Scholar]
- Iraqi FA, Churchill G, and Mott R (2008). The Collaborative Cross, developing a resource for mammalian systems genetics: A status report of the Wellcome Trust cohort. Mammalian Genome 19, 379–381. 10.1007/s00335-008-9113-1. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Witherden DA, and Havran WL (2020). The Role of Tissue-resident T Cells in Stress Surveillance and Tissue Maintenance. Cells 9. 10.3390/cells9030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson HF (1996). The Eighth Day of Creation; Makers of the revolution in biology (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Junhee S, Warren HS, Alex GC, Michael NM, Henry VB, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America 110, 3507–3512. 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. (2011). Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, bar030. 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legoux FP, Lim JB, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, Sparwasser T, Way SS, and Moon JJ (2015). CD4+ T Cell Tolerance to Tissue-Restricted Self Antigens Is Mediated by Antigen-Specific Regulatory T Cells Rather Than Deletion. Immunity 43, 896–908. 10.1016/j.immuni.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SM, Williams A, and Eisenbarth SC (2019). Structure and function of the immune system in the spleen. Science immunology 4, eaau6085. 10.1126/sciimmunol.aau6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malouli D, Hansen SG, Hancock MH, Hughes CM, Ford JC, Gilbride RM, Ventura AB, Morrow D, Randall KT, Taher H, et al. (2021). Cytomegaloviral determinants of CD8+T cell programming and RhCMV/SIV vaccine efficacy. Science immunology 6, eabg5413. 10.1126/SCIIMMUNOL.ABG5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Sompallae R, Winborn CS, Harty JT, and Badovinac VP (2020). Diverse CD8 T Cell Responses to Viral Infection Revealed by the Collaborative Cross. Cell reports 31, 107508. 10.1016/j.celrep.2020.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D, and Benoist C (2007). A decade of AIRE. Nat. Rev. Immunol 7, 645–650. 10.1038/nri2136. [DOI] [PubMed] [Google Scholar]
- McCune JM (1996). Development and applications of the SCID-hu mouse model. Seminars in immunology 8, 187–196. 10.1006/smim.1996.0024. [DOI] [PubMed] [Google Scholar]
- McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, and Weissman IL (1988). The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 241, 1632–1639. 10.1126/science.2971269. [DOI] [PubMed] [Google Scholar]
- Miller JFAP (1993). Self-nonself discrimination and tolerance in T and B lymphocytes. Immunologic Research 12, 115–130. 10.1007/BF02918299. [DOI] [PubMed] [Google Scholar]
- Miller JFAP, and Morahan G (1992). Peripheral T cell tolerance. Annual review of immunology. [DOI] [PubMed] [Google Scholar]
- Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carré C, Burdin N, Visan L, Ceccarelli M, Poidinger M, et al. (2019). RNA-Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types. Cell reports 26, 1627–1640.e1627. 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJE, Lalioti MD, Mullis PE, Antonarakis SE, et al. (1997). Positional cloning of the APECED gene. Nature Genetics 17, 393–398. 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- Nauseef WM (2014). Proteases, neutrophils, and periodontitis: The NET effect. J. Clin. Invest 124, 4237–4239. 10.1172/JCI77985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM (2019). The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Current opinion in immunology 60, 130–140. 10.1016/j.coi.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll KE, Whitmore AC, West A, McCarthy MK, Morrison CR, Plante KS, Hampton BK, Kollmus H, Pilzner C, Leist SR, et al. (2020). Complex Genetic Architecture Underlies Regulation of Influenza-A-Virus-Specific Antibody Responses in the Collaborative Cross. Cell reports 31, 107587. 10.1016/j.celrep.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, and Hengartner H (1991). Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell 65, 305–317. 10.1016/0092-8674(91)90164-T. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Haase I, and Nestle FO (2014). Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol 14, 289–301. 10.1038/nri3646. [DOI] [PubMed] [Google Scholar]
- Payne KJ, and Crooks GM (2007). Immune-Cell Lineage Commitment: Translation from Mice to Humans. Immunity 26, 674–677. 10.1016/j.immuni.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Pike BL, Boyd AW, and Nossal GJV (1982). Clonal anergy: the universally anergic B lymphocyte. Proceedings of the National Academy of Sciences of the United States of America 79, 2013–2017. 10.1073/pnas.79.6.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B, and Davis MM (2020). The science and medicine of human immunology. Science 369, eaay4014. 10.1126/science.aay4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, et al. (2009). Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nature immunology 10, 116–125. 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, Machiah DK, Lawson B, Hakimpour P, Wang YC, et al. (2016). The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature 531, 523–527. 10.1038/nature17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röcken M, Urban JF, and Shevach EM (1992). Infection breaks T-cell tolerance. Nature 359, 79–82. 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, and Manz MG (2013). Human hemato-lymphoid system mice: Current use and future potential for medicine. Annual review of immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky AY (2011). Regulatory T cells and Foxp3. Immunological reviews 241, 260–268. 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, and Marson A (2018). Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971.e1915. 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter JR, Najarian ML, Bell TA, Blanchard M, Ferris MT, Hock P, Kashfeen A, Kirchoff KE, Linnertz CL, Sebastian Sigmon J, et al. (2019). Whole genome sequencing and progress toward full inbreeding of the mouse collaborative cross population. G3: Genes, Genomes, Genetics 9, 1303–1311. 10.1534/g3.119.400039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger BS (2015). Human spleen microanatomy: Why mice do not suffice. Immunology 145, 334–346. 10.1111/imm.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, and Mellman I (2004). Immunotherapy: Bewitched, bothered, and bewildered no more. Science 305, 197–200. 10.1126/science.1099688. [DOI] [PubMed] [Google Scholar]
- Su L, Kidd B, Han A, Kotzin J, and Davis M (2013). Virus-Specific CD4+ Memory-Phenotype T Cells Are Abundant in Unexposed Adults. Immunity 38, 373–383. 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, et al. (2022). Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology. 10.1007/s10875-022-01289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoeven V, Drylewicz J, Westera L, den Braber I, Mugwagwa T, Tesselaar K, Borghans JA, and de Boer RJ (2017). Dynamics of recent thymic emigrants in young adult mice. Frontiers in immunology 8, 933. 10.3389/fimmu.2017.00933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij MC, Hansen SG, Iyer R, John N, Malouli D, Morrow D, Scholz I, Womack J, Abdulhaqq S, Gilbride RM, et al. (2021). Modulation of MHC-E transport by viral decoy ligands is required for RhCMV/SIV vaccine efficacy. Science 372, eabe9233. 10.1126/science.abe9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Boehmer H (1990). Developmental biology of T cells in T cell-receptor transgenic mice. Annual review of immunology. [DOI] [PubMed] [Google Scholar]
- von Herrath MG, and Nepom GT (2005). Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. The Journal of experimental medicine 202, 1159–1162. 10.1084/jem.20051224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagar LE, Salahudeen A, Constantz CM, Wendel BS, Lyons MM, Mallajosyula V, Jatt LP, Adamska JZ, Blum LK, Gupta N, et al. (2021). Modeling human adaptive immune responses with tonsil organoids. Nature medicine 27, 125–135. 10.1038/s41591-020-01145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh CE, Miller DR, Manly KF, Wang J, McMillan L, Morahan G, Mott R, Iraqi FA, Threadgill DW, and De Villena FPM (2012). Status and access to the Collaborative Cross population. Mammalian Genome 23, 706–712. 10.1007/s00335-012-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CM, Roth TL, Baglaenko Y, Ferri DM, Brauer P, Zuniga-Pflucker JC, Rosbe KW, Wither JE, Marson A, and Allen CDC (2018). Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. Journal of immunological methods 457, 33–40. 10.1016/j.jim.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rei M, Brackenridge S, Brenna E, Sun H, Abdulhaqq S, Liu MKP, Ma W, Kurupati P, Xu X, et al. (2021). HLA-E-restricted, Gag-specific CD8+T cells can suppress HIV-1 infection, offering vaccine opportunities. Science immunology 6, eabg1703. 10.1126/SCIIMMUNOL.ABG1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong KSM, Her Z, and Chen Q (2018). Humanized Mice as Unique Tools for Human-Specific Studies. Archivum Immunologiae et Therapiae Experimentalis 66, 245–266. 10.1007/s00005-018-0506-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Jiang N, Ebert PJR, Kidd BA, Müller S, Lund PJ, Juang J, Adachi K, Tse T, Birnbaum ME, et al. (2015). Clonal Deletion Prunes but Does Not Eliminate Self-Specific αβ CD8+ T Lymphocytes. Immunity 42, 929–941. 10.1016/j.immuni.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiss CJ, Gatti DM, Toro-Salazar O, Davis C, Lutz CM, Spinale F, Stearns T, Furtado MB, and Churchill GA (2019). Doxorubicin-induced cardiotoxicity in Collaborative Cross (CC) mice recapitulates individual cardiotoxicity in humans. G3: Genes, Genomes, Genetics 9, 2637–2646. 10.1534/g3.119.400232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. (2015). Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517, 89–93. 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM (1996). Immunology taught by viruses. Science 271, 348–350. [DOI] [PubMed] [Google Scholar]
- Zomer HD, and Trentin AG (2018). Skin wound healing in humans and mice: Challenges in translational research. Journal of Dermatological Science 90, 3–12. 10.1016/j.jdermsci.2017.12.009. [DOI] [PubMed] [Google Scholar]