

Abstract
Porous silicon (pSi) nanoparticles are loaded with Immunoglobulin A-2 (IgA2) antibodies, and the assembly is coated with pH-responsive polymers on the basis of the Eudragit family of enteric polymers (L100, S100, and L30-D55). The temporal release of the protein from the nanocomposite formulations is quantified following an in vitro protocol simulating oral delivery: incubation in simulated gastric fluid (SGF; at pH 1.2) for 2 h, followed by a fasting state simulated intestinal fluid (FasSIF; at pH 6.8) or phosphate buffer solution (PBS; at pH 7.4). The nanocomposite formulations display a negligible release in SGF, while more than 50% of the loaded IgA2 is released in solutions at a pH of 6.8 (FasSIF) or 7.4 (PBS). Between 21 and 44% of the released IgA2 retains its functional activity. A capsule-based system is also evaluated, where the IgA2-loaded particles are packed into a gelatin capsule and the capsule is coated with either EudragitL100 or EudragitS100 polymer for a targeted release in the small intestine or the colon, respectively. The capsule-based formulations outperform polymer-coated nanoparticles in vitro, preserving 45–54% of the activity of the released protein.
Keywords: biologic antibacterial therapeutics, Eudragit polymer, pH-responsive drug delivery, oral drug delivery
Graphical Abstract
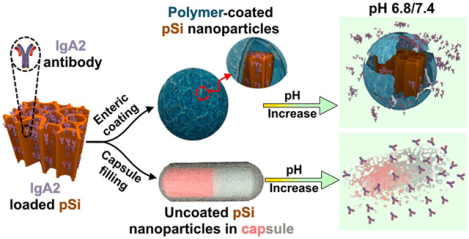
INTRODUCTION
Biologic-based therapeutics (i.e., proteins, peptides, nucleic acids) have become increasingly popular as treatment options due to their narrow target spectrum and their biocompatibility.1 However, biologics pose serious formulation challenges and most of the current biologic drugs are used as parenteral formulations because of poor bioavailability.2 Oral administration is a preferred, common therapeutic administration route, but the delivery of proteins through the digestive tract faces numerous challenges, such as degradation in the acidic and proteolytic conditions of the stomach and low site specificity.3–5 These obstacles result in high dosage requirements that substantially increase the cost and the potential for toxicity and have led the pharmaceutical industry to invest in the development of delivery systems. Carrier systems using polymers,6,7 liposomes,8–10 silica,11–15 and other nano-16,17 and microcarriers18–20 have been developed for oral protein delivery, but the complexity of the human gut and stringent requirements for clinical translation have limited their deployment.21,22 Additionally, the delivery systems developed to date suffer from low loading capacity (<10 wt %), limited control of release profiles, and denaturation or deactivation of the loaded proteins.
In recent years, porous silicon (pSi) has been studied as a delivery system for a variety of drug payloads.23–28 In particular, previous work has demonstrated the ability of pSi particles to load and release active proteins.29–39 Of relevance to oral delivery, pSi has been shown to protect the sensitive Cry5B anthelmintic protein from the highly acidic and proteolytic conditions in simulated gastric fluid (SGF),40 although the release kinetics were too slow to be effective in a hamster model of hookworm infection. To increase the residence time in a gastrointestinal (GI) system, Salonen, Santos, and co-workers developed a polymer/biopolymer modified pSi particle system as a mucoadhesive drug carrier for the oral delivery of insulin for the treatment of type I diabetes.41–43 One advantage of such hybrid mesoporous particle/polymer systems is that the mesoporous host material can protect the protein payload from the harsh or otherwise incompatible conditions typically needed to process the polymeric component of the drug delivery system.37 Despite these advances, incompatibilities between the protein and the delivery system, uncontrolled release kinetics, and a lack of site-specific release remain key challenges for the oral delivery of biologics.
The most common approach to achieve the site-specific oral delivery of drugs has been to use pH-responsive enteric polymers such as the commercially available Eudragit or Aqoat classes. These materials exploit the sharp pH variations in the gastrointestinal (GI) tract, where the stomach is highly acidic (pH 1–2), the small intestine is mildly acidic (pH 5.5–6.8), and the colon is neutral to basic (pH >7).43 Of relevance to the present study, the well-established Eudragit family of enteric polymers displays a range of favorable pH-responsive dissolution characteristics.44,45 These materials are composed of copolymers of methacrylic acid and methyl methacrylate, which undergo dissolution upon ionization of the carboxylic acid groups on the methacrylic acid chains. Thus, varying the ratio of methacrylic acid and methyl methacrylate enables a precise tuning of the dissolution pH. For the present work, we selected two organic-soluble members of this family and one that is available as an aqueous dispersion. The two organic-soluble enteric polymers were as follows: (1) Eudragit L100, which has a 1:1 methacrylic acid to methyl methacrylate ratio and dissolves at pH 5 for release in the small intestine; and (2) Eudragit S100, which has a 1:2 methacrylic acid to methyl methacrylate ratio and dissolves at pH 7 for release in the colon.46,47 The aqueous dispersed polymer was Eudragit L30 D-55, an anionic copolymer with a 1:1 ratio of methacrylic acid and ethyl acrylate. Although these polymers are ideal carriers for small molecule delivery, their macromolecular nature and the need to formulate them in organic solvents causes denaturation of biologic-based therapeutics. On the basis of the results with polycaprolactone and poly(lactide-co-glycolide) and related polymer formulations that used porous Si nanoparticles to protect the protein payloads,36–39 we reasoned that a similar composite structure using an enteric polymer might be able to protect a protein payload of relevance to oral delivery.
IgA2 was selected as the protein payload for its central role in gut microbiota homeostasis and defense against enteropathogens and their toxins at the mucosal surface.48 IgA2 monoclonal antibodies (mAbs) act through steric hindrance, agglutination, and immune exclusion, forming an immunological barrier by binding to pathogens or toxins and impeding their passage across the intestinal epithelial lining into the body.49 On the basis of these features, IgA is a potential therapeutic for the treatment of gut infections from pathogenic bacteria and their toxins, which cause acute diarrhea and the most infantile deaths in developing countries.50 However, IgA cannot survive the harsh conditions of the stomach; a delivery system that can protect it in this environment and deliver it to the lower GI tract would be a significant milestone in the development and commercialization of this class of therapeutics for bacterial or protozoal infections.
For the nanocarrier system developed here, porous Si nanoparticles acted as a carrier to hold and protect the protein payload and the Eudragit enteric coatings enabled pH-controlled site-specific release. We explored two methods employing Eudragit coatings to protect these protein-loaded nanoparticles: The first involved directly coating individual IgA2-loaded nanoparticles with the Eudragit polymer (Scheme 1a), and the second involved loading powders of the IgA2-loaded nanoparticles into gelatin capsules and then coating the capsules with Eudragit (Scheme 1b). The rationale for the capsule approach is that it physically separates the enteric polymer from the IgA2-loaded pSiNPs, avoiding direct exposure of the IgA2-loaded pSiNPs to the organic solvent used in the Eudragit coating step. As a control, we also explored the possibility of delivering the “free” IgA2 in a capsule format, by loading IgA2 directly into a capsule without the agency of porous Si nanoparticles. A potential disadvantage of the capsule approach is that it might be expected to release the nanoparticles (or the free drug) in a localized bolus, whereas the individually coated nanoparticles of Scheme 1a might be expected to be better dispersed throughout the lower GI tract. The persistent diarrhea that typically accompanies an enteric infection results in fast gastric clearance, and therefore, a capsule filled with “free” IgA2 or with a nanoparticle formulation of IgA2 might be cleared from the lumen too quickly to effectively agglutinate the enteric bacteria and toxins that cause the infection. It is known that certain nano- or microparticles can transit more slowly than freely dissolved molecules due to an enhanced retention of the particles by the infected and inflamed tissues in the gastrointestinal tract.51,52 Therefore, while an enteric polymer on its own may protect a protein payload from gastric degradation, a motivation for combining this with a nanoparticle carrier is to prolong the transit time through the gut. Taken together, these concepts might contribute to a more effective oral IgA2 therapy for enteric infections.
Scheme 1. (a) Schematic Illustrating the Preparation and the Proposed Fate of Polymer-Coated Porous Silicon (pSi) Nanoparticles for Oral Delivery of IgA2a and (b) Schematic Illustrating the Preparation of IgA2-Loaded pSi Nanoparticles for Capsule-Based Deliveryb.
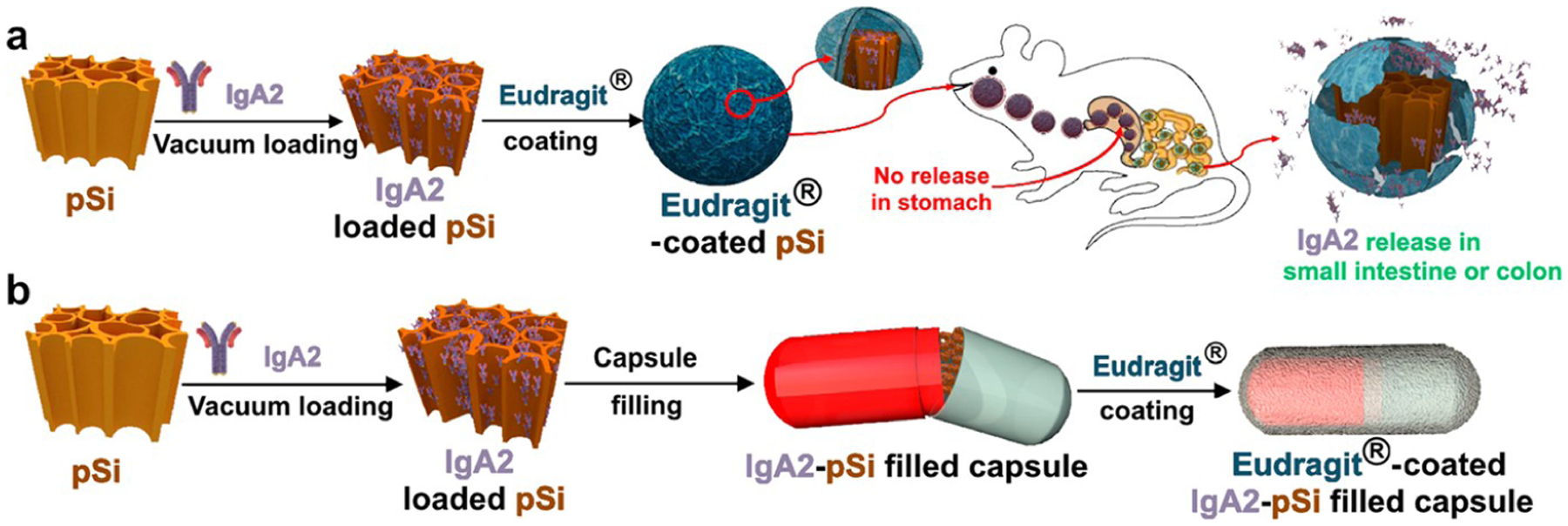
a First, the nanoparticles are loaded with the protein therapeutic (IgA2) through a vacuum infiltration process, and then, they are coated with a member of the Eudragit family of pH-responsive enteric polymers. The Eudragit L100 and Eudragit L30 D-55 polymers selected for this study dissolve at ~pH 6 (targeting release in the small intestine), and Eudragit S100 dissolves at pH 7 (for release in the colon). The role of the polymers is to protect the loaded antibody from exposure to the acidic conditions commonly encountered in the stomach. bRather than coating the individual nanoparticles with an enteric polymer as in (a), the uncoated, IgA2-loaded pSi nanoparticles are loaded into a commercial biodegradable capsule, which is then coated with either EudragitL100 or Eudragit S100 pH-responsive enteric polymers.
2. MATERIALS AND METHODS
2.1. Materials.
Highly boron-doped p-type silicon wafers, with 1.2 mΩ·cm resistivity and single-side polished on the (100) face, were obtained from Sil’Tronix Silicon Technologies. Concentrated hydrofluoric acid (48% aqueous, ACS grade) was obtained from Fisher Scientific. Absolute ethanol, methanol, poly(vinyl alcohol) (PVA), pepsin (from porcine gastric mucosa), sodium hydroxide, sodium chloride, maleic acid, and potassium hydroxide were obtained from Sigma-Aldrich. The SGF medium was prepared by mixing 7 mL of hydrochloric acid, 2 g of sodium chloride, and 3.2 g of pepsin (Sigma-Aldrich) in 1 L of DI water. Fasting state simulated intestinal fluid (FasSIF) powder was purchased from Biorelevant Media, and the medium was prepared according to the instructions provided by the manufacturer and using the composition calculator on the Biorelevant Media Web site (https://biorelevant.com/fassif-v2/how-to-make/). Briefly, 1.392 g of sodium hydroxide pellets (NaOH), 2.220 g of maleic acid (Sigma-Aldrich), and 4.010 g of sodium chloride (NaCl) were added to 1 L of ultrapure water and mixed thoroughly. Once solubilized, 1.790 g of FaSSIF-V2 powder was added to this solution and stirred until dissolved. Finally, pancreatin (final concentration 10 mg/mL) was added to obtain the solution referred to as FasSIF in this work. Phosphate buffered saline (PBS) was purchased from Gibco. Eudragit S100 was purchased from Evonik Industries, while Eudragit L100–55 (referred to as Eudragit L100 throughout this manuscript) and Eudragit L30-D55 were generously provided as test samples by Evonik Industries. Deionized (18 MΩ) water was used for the preparation of all the buffers and dissolution mediums. Monomeric IgA2 of the human monoclonal antibody F425A1g8 was prepared and purified as described previously.53
2.2. Fabrication of Porous Silicon Nanoparticles.
Porous silicon nanoparticles with an approximate diameter of 550 nm were fabricated using the perforation etching method described previously.54 Briefly, a single crystal silicon wafer (100) with a resistivity of 1.2 mΩ·cm and a thickness of 525 ± 25 μm was anodically etched using an aqueous HF-based electrolyte (CAUTION: HF is highly toxic and corrosive, and proper care should be exerted to avoid contact with skin, eyes, or lungs). The silicon wafer was packed in a custom-designed Teflon etch cell with an exposed area of 8.2 cm2 and the ability to hold 24 mL of etching solution. A sacrificial etch layer was prepared first in 3:1 (v/v) 48% aqueous HF/absolute ethanol electrolyte by application of a current density of 10 mA·cm−2 (Keithley 2651a Sourcemeter) for 30 s, which was removed by immersion in a 2 M aqueous potassium hydroxide solution (KOH). The perforation etch was carried out by applying a current density of 50 mA·cm−2 for 20 s followed by 200 mA·cm−2 for 0.5 s in an electrolyte solution consisting of 1:1 (v/v) 48% aqueous HF/absolute ethanol. In a typical etch, this process was repeated 50 times to generate 50 layers of alternating porosities. The high current density pulse introduced a high porosity “perforation” layer that acted as fracture points during ultrasonication. The fabricated pSi layer was detached from the underlying bulk Si by electropolishing the sample in a 1:10 (v/v) 48% aqueous HF/absolute ethanol solution at 4 mA· cm−2 for 250 s. The free-standing pSi layer was washed three times with ethanol and collected in a glass vial with ethanol. For particle preparation, four free-standing pSi layers were collected in a glass vial with 10 mL of ethanol and ultrasonicated for 20 h. The brown suspension collected after ultrasonication was centrifuged at 11 000 rpm for 10 min, and the bottom pellet was collected and the supernatant was discarded. The pellet was resuspended in ethanol and centrifuged three additional times to remove the smaller size fraction of particles. Next, the particles were centrifuged at 2000 rpm for 3 min and the supernatant was collected to eliminate the large particles. The particles in the supernatant were of the required size (~550 nm) on the basis of dynamic light scattering (DLS) measurement. The particles were stored in ethanol until further use.
2.3. IgA2 Loading onto pSi Nanoparticles.
The IgA2 antibodies were loaded onto the prepared pSi nanoparticles using a vacuum loading method. In this process, 1 mg of pSi nanoparticles was washed three times with water to remove the ethanol used during ultrasonication of the pSi films. The washed particles were incubated overnight with 100 μL of IgA2 solution (7 mg·mL−1) in PBS at 4 °C. The mixture of IgA2 and pSi nanoparticles then was vacuum-dried to evaporate the buffer solution. The vacuum-dried IgA2-pSi formulation was removed from the tube with a spatula.
2.4. Eudragit Enteric Coating on IgA2-Loaded pSi Nanoparticles.
The IgA2-loaded pSi nanoparticles were coated with three different polymers from the Eudragit family: L100–55, S100, or L30 D-55. The Eudragit L100–55 polymer (referred to in this work as Eudragit L100) was selected for payload release in the small intestine (pH > 5.5), while Eudragit S100 was intended for payload release in the colon (pH > 7). The Eudragit L100 and Eudragit S100 polymers are soluble in organic solvents such as methanol. To coat the Eudragit polymer onto IgA2-loaded pSi nanoparticles, a 2.5% (w/v) solution of the polymer was prepared in methanol and 100 μL of the polymer solution was added to 1 mg of IgA2-loaded pSi nanoparticles. The particles were quickly mixed and added to 5 mL of an acidic (pH 3) aqueous solution containing 1% by mass PVA with vigorous stirring for 20 min. The sample was then centrifuged at 3000 rpm for 3 min and washed with simulated gastric fluid (SGF). The Eudragit S100 polymer was coated onto IgA2-loaded pSi nanoparticles using a similar protocol as that with the Eudragit L100 polymer. A 2.5% (w/v) methanol solution (100 μL) of Eudragit S100 was used with 1 mg of pSi, followed by the addition of 1 w/v % PVA (at pH 3) and the centrifugation steps described above. In the case of Eudragit L30 D-55 (referred to in this work as Eudragit L30)-coated IgA2-loaded pSi nanoparticles, 200 μL of aqueous Eudragit L30 was added to 1 mg of IgA2-loaded pSi nanoparticles and mixed quickly, This mixture was then rapidly added to 5 mL of an acidic (pH 3) aqueous solution of PVA (1 wt %) with vigorous stirring for 20 min. Similar to in the other two formulations, the Eudragit L30-coated pSi nanoparticles were washed and collected through centrifugation in SGF. The control formulations with Eudragit pure polymers (i.e., L100, S100, L30) were prepared using the same protocols, where vacuum-dried pure IgA2 was used in place of the IgA2-loaded pSi nanoparticles. The amount of protein remaining in the wash solutions was measured using micro bicinchoninic acid (BCA) protein assay according to the procedure provided with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The amount of loaded protein was calculated by subtracting the unloaded protein from total protein in the loading solution (700 μg, unless indicated otherwise). The amount of protein loaded per milligram of particles was used to calculate the loading capacity for all the formulations.
2.5. Capsule Formulations of IgA2-Loaded pSi.
The capsule formulations were prepared by manually filling size 4 gelatin capsules with 1.7 mg of IgA2-loaded pSi nanoparticles (i.e., 1 mg of pSi + 0.7 mg of IgA2). The filled capsules were then coated with Eudragit S100 or Eudragit L100 enteric polymers. A dip coating method was employed, where the capsule was dipped into a 10% (w/v) solution of polymer in methanol and dried under a N2 flow. To obtain sufficiently thick coatings, the procedure was repeated three times for each capsule. Uncoated capsules charged with IgA2-loaded pSi particles served as the control. In addition, 210 μg of pure vacuum-dried IgA2 (without pSi) was also filled into gelatin capsules, followed by coating with the enteric polymer as a second set of controls.
2.6. In Vitro IgA2 Release from Eudragit Polymers-Coated pSi Formulations.
The in vitro IgA2 release experiments were carried out by incubating the test formulations in 5 mL of the relevant media maintained at 37 °C in a shaker–incubator. The formulations were first incubated in simulated gastric fluid (SGF) at pH 1.2–1.8 for 2 h, and then, they were isolated and incubated for 3 h in either the fasting state simulated intestinal fluid (FasSIF, pH 6.8) or in the phosphate buffer solution (PBS, pH 7.4) depending on the experiment. A release time point was collected every hour up to 5 h by centrifugation of the formulations at 3000 rpm for 5 min and collection of the release medium. Fresh release medium was added to the formulations at each time point. Concentrations of the released IgA2 in the release media were determined using the BCA protein assay. Control BCA assay measurements from empty pSi particles incubated in the three separate media were subtracted from the test data to correct for protein concentrations.
2.8. Activity Testing for the in Vitro Released IgA2.
Aliquots of IgA2 released from the different formulations at specified time points were analyzed by enzyme-linked immunosorbent assay (ELISA) in order to quantify the amount of bioactive (i.e., not denatured) IgA2. All the samples were analyzed by ELISA to determine the bioactivity of IgA2 released from the relevant formulations, and the bioactivity of IgA2 in some samples (Table 1) was confirmed with an HIV neutralization-based functional assay previously described.51
Table 1.
Mass Loading, Release, and Activity of IgA2 Antibody in Polymer-Coated Porous Si Particle Formulations
| polymer coating | dissolution mediuma | intended release region | IgA2 loadingb (%) | release 3h @ pH 6.8/7.4c(%) | preserved activityd (%) |
|---|---|---|---|---|---|
| Eudragit S100 | SGF→PBS | colon (pH >7) | 29 ± 1 | 70 ± 7 | 44 ± 10 |
| Eudragit L100 | SGF→PBS | small intestine (pH > 5.5) | 29 ± 1 | 61 ± 2 | 21 ± 6 |
| SGF→FasSIF | 48 ± 2 | 32 ± 13 | |||
| Eudragit L30 D-55 | SGF→PBS | small intestine (pH > 5.5) | 31 ± 2 | 56 ± 2 | 20 ± 6 |
| SGF→FasSIF | 41 ± 3 | 24 ± 11 |
SGF is simulated gastric fluid, pH = 1.2; PBS is phosphate buffered saline solution, pH 7.4; FasSIF is fasting state simulated intestinal fluid, pH = 6.8.
Mass loading of IgA2 protein in the porous Si carrier, prior to polymer coating; defined as (mass of IgA2)/(mass of IgA2 + pSi carrier), given as a percentage. Errors are 3 SD.
Percent by mass of IgA2 released from the particle formulation into the indicated release medium, after 3 h of exposure. Measured by BCA assay. Errors given are 3 SD.
Activity of IgA2 measured by both ELISA and HIV neutralization assay as percent of total IgA2 released, after 3 h of exposure to the pH 6.8/7.4 solution, corresponding to the 5 h time point in Figure 2. Errors given are 3 SD.
3. RESULTS AND DISCUSSION
3.1. Synthesis and Characterization of IgA2-Loaded Porous Si Particle Formulations.
The present study explored two strategies for preparing materials suitable for the delivery of a protein therapeutic to the lower GI tract, as outlined in Scheme 1; both involved loading of the protein into porous silicon (pSi) nanoparticles as a means of preserving the activity of the biologic. The pSi nanoparticles were prepared from single crystal silicon wafers using a perforation etch process previously described,52 which yielded nanoparticles approximately 550 nm in diameter. The etch parameters were adjusted to achieve mean pore diameters of 58 ± 14 nm. The average particle size and the notional pore diameter were measured by dynamic light scattering (DLS, Figure S1) and by scanning electron microscopy (SEM, Figure 1), respectively. The particles were then loaded with the IgA2 antibody (MW 162 kDa, Stokes radius 6.2–8.7 nm55) by vacuum infiltration. This process involved soaking of the pSi nanoparticles in a cold aqueous solution of IgA overnight and then evaporation of the solvent at room temperature under a vacuum. To ensure the rapid release of the protein payload, as-prepared pSi nanoparticles were used. The as-prepared pSi nanoparticles possess a Si–H and Si–Si surface chemistry that is readily oxidized and hydrolyzed in water,56 and they rapidly dissolve under physiological conditions. In order to configure the particles to survive the stomach conditions and release payload in conditions simulating the intestinal lumen,40 the nanoparticles were then protected with enteric polymer coatings, either by coating individual nanoparticles (Scheme 1a) or by placing a bolus of uncoated nanoparticles in capsules that were then coated with an enteric polymer (Scheme 1b). An illustration of the type of nanoparticles prepared and the in vitro carrier dissolution/drug release protocol is provided in Scheme 2.
Figure 1.

Scanning electron microscope (SEM) images of (a) as-etched pSi nanoparticles with an average pore diameter of 60 nm and an average diameter of 550 nm (DLS number density histogram shown in the inset), (b) empty pSi nanoparticles after coating with Eudragit L100, and (c) empty pSi nanoparticles after coating with Eudragit S100. The scale bars in all images are 1 μm.
Scheme 2. Description of the IgA2-Loaded Nanoparticles and the Release Protocol Used in This Studya.
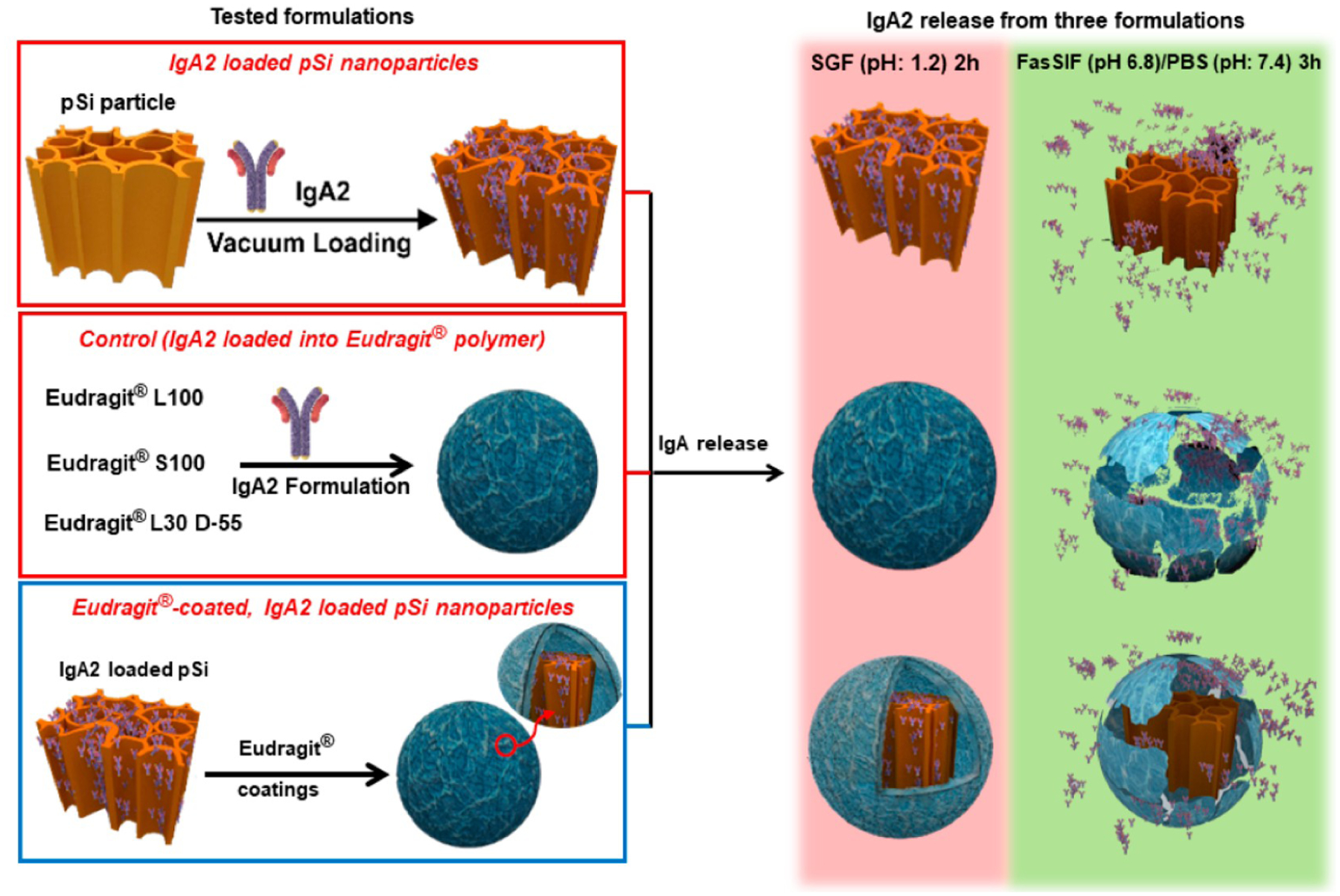
a The porous Si nanoparticles are loaded with the antibody via a vacuum infiltration procedure (left panel, top), which yields a mass loading of ~29%. The control involves a pure Eudragit enteric polymer bead containing IgA2 (left panel, middle). To prepare nanoparticles individually coated with the enteric polymer (left panel, bottom), the IgA2-loaded pSi nanoparticles are dispersed in a methanol solution of the Eudragit polymer and quickly added acidic aqueous solution of PVA (1 wt %) with rapid stirring to induce deposition of the coating. The drug release protocol is carried out over a total period of 5 h; for the first 2 h, the samples are incubated in SGF at pH 1.2 (right panel, red backdrop) followed by either FasSIF (pH 6.8) for the formulations using Eudragit L100 and Eudragit L30 D-55 or PBS (pH 7.4) for the formulations using Eudragit S100 (right panel, green backdrop).
For the first delivery strategy involving coating of individual nanoparticles with Eudragit (Scheme 1a), the IgA2-loaded pSi nanoparticles were coated with either Eudragit L100 or Eudragit S100, which are polymers that have been engineered for specific delivery to the small intestine or the colon, respectively. The coating was achieved by the rapid addition of acidified water to a dispersion of the nanoparticles in a methanol solution of the polymer. The deposition solution contained 1% by mass poly(vinyl alcohol) (PVA) stabilizer. The SEM images (Figure 1) revealed what appeared to be a relatively conformal coating of polymer on the pSi nanoparticles. The DLS data showed an increase in the average particle size, to approximately 1100 nm for Eudragit L100 and 1300 nm for Eudragit S100. Multiple images of multiple samples indicated that a subset (<5%) of the pSi nanoparticles were incompletely coated with polymer.
We next measured the dissolution characteristics of the nanoparticle preparations. For these experiments, we followed an in vitro protocol designed to mimic oral delivery conditions: Samples were measured over a period of 5 h, where, for the first 2 h, they were exposed to simulated gastric fluid (SGF) of a low pH (1.2) and containing the digestive enzyme pepsin at a physiologically relevant concentration and, then, for the following 3 h, they were incubated in either fasting state simulated intestinal fluid (FasSIF, pH 6.8) or in phosphate buffer solution (PBS, pH 7.4), depending on the experiment. The uncoated pSi nanoparticles (loaded with IgA2) displayed a slow decrease in diameter during the 2 h at pH 1.2, and this was followed by a more pronounced decrease in diameter (from approximately 550 to 100 nm) when the particles were transitioned to pH 7.4 (Figure S1a, Supporting Information). This is consistent with the known behavior of porous Si; the material is relatively stable in acidic solutions and it dissolves more rapidly in neutral or alkaline aqueous media.57
The polymer coatings slowed the aqueous dissolution of the nanoparticles in acidic media while maintaining the rapid dissolution behavior in neutral solutions. Thus, the average particle size of the Eudragit L100-coated pSi nanoparticles (approximately 950 nm) remained stable for 2 h in SGF at pH 1.2, and it decreased markedly (to 300 nm) after the medium was replaced with FasSIF at pH 6.8 (Figure S1b, Supporting Information). Similarly, the average particle size of the Eudragit S100-coated pSi nanoparticles was maintained (between 1440 and 1720 nm) for 2 h in SGF, and the size decreased (to ~450 nm) when held for 3 h in PBS at pH 7.4 (Figure S1c, Supporting Information).
3.2. In Vitro IgA2 Loading and Release of Eudragit-Coated pSi Particles under Simulated Oral Delivery Conditions.
In vitro IgA2 loading and release studies were then carried out to assess the potential performance of the Eudragit–pSi composite system. Loading of IgA2 into the pSi particles was carried out under vacuum-drying conditions. On average, the mass loading of IgA2 in the pSi particles (determined by assaying the wash solutions postcoating) was 29 wt % (i.e., 0.4 mg Iof gA2 loaded into 1 mg of pSi particles; the final mass of IgA2 + pSi particles = 1.4 mg) for both of the Eudragit-coated pSi formulations studied. In vitro IgA2 release from the Eudragit-pSi constructs was carried out under the simulated gastrointestinal protocol described above, and the total protein released was quantified using the BCA protein assay (Figure 2). As anticipated, the Eudragit L100–pSi–IgA2 formulation showed minimal release in the acidic SGF (pH 1.2) phase of the protocol; less than 2% of the total loaded IgA2 was released during the 2 h incubation period, attributed to residual IgA2 present on the surface of the particles (Figure 2a). When the solution pH was increased by the introduction of FasSIF (pH 6.8) buffer, approximately 50% of the loaded IgA2 was released in the ensuing 3 h. The release behavior was consistent with the performance of the Eudragit L100 coating, which is engineered to dissolve in the small intestine at pH values >5.5. In another set of experiments, the IgA2 release behavior from this same formulation was measured using PBS (pH 7.4) in place of FasSIF. The total released IgA2 was somewhat larger in this case, approaching 61% (Figure S2, Supporting Information). Similarly, the Eudragit S100-coated formulation showed no significant release of IgA2 in SGF, while nearly 70% of the IgA2 payload was released during the 3 h incubation in PBS (Eudragit S100–pSi–IgA2 formulation, Figure 2c). The Eudragit S100 polymer dissolves preferentially at pH > 7, as it is designed for site-specific release in the colon. The release of silicon from the pSi–polymer formulations tracked IgA2 release, whereas minimal silicon was released into the solution during the low pH phase of the test; once the pH was raised to the point where polymer dissolution occurred, the silicon pSi nanomaterial began to dissolve (Figure 2a,c).
Figure 2.
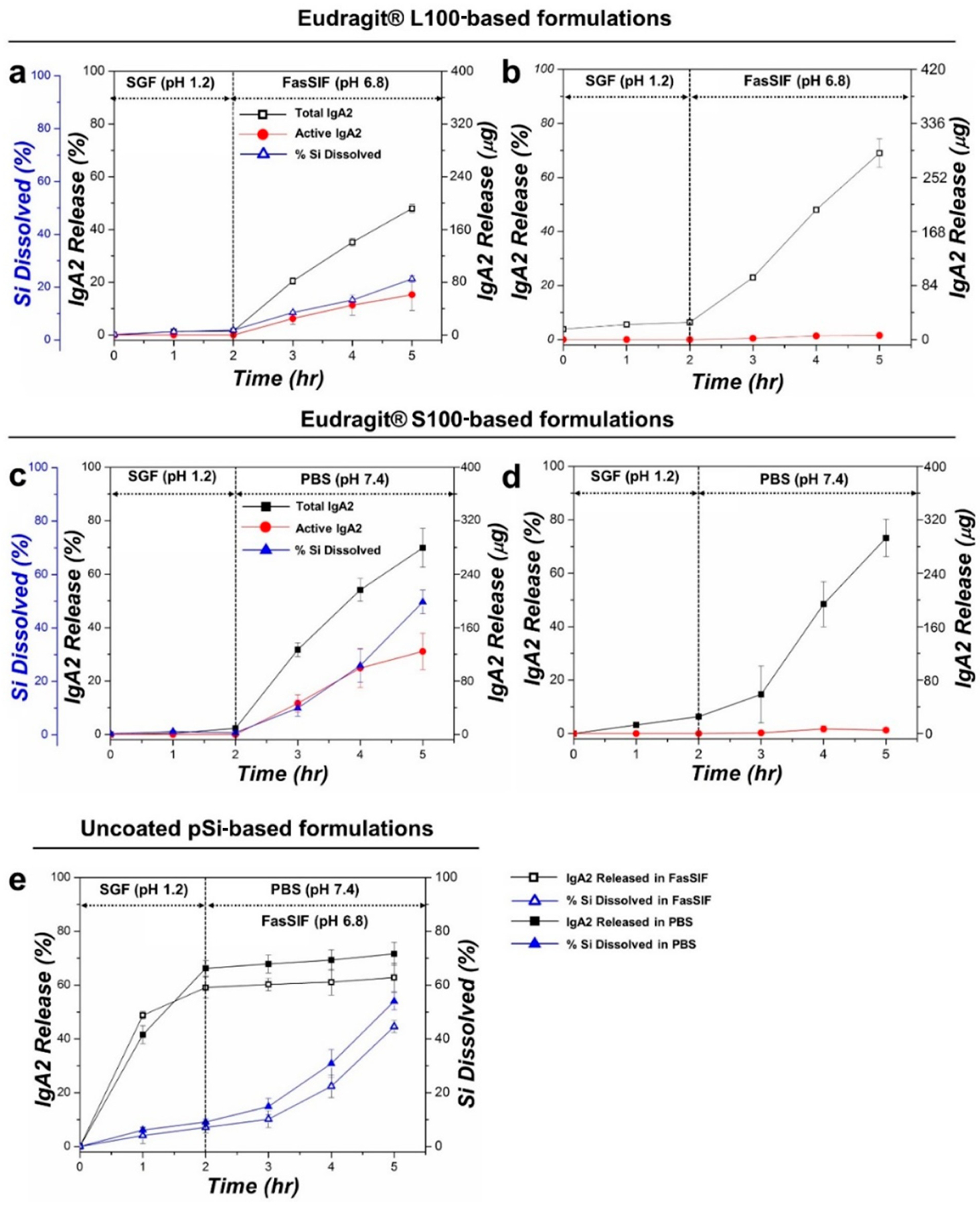
Release of IgA2 from enteric polymer-coated pSi particle formulations and from controls, comparing total protein (Total IgA2) with protein activity (Active IgA2) as a function of time: (a) Eudragit L100-coated pSi particles, (b) Eudragit L100 polymer control (IgA2 loaded into polymer only), (c) Eudragit S100-coated pSi particles, (d) Eudragit S100 polymer control, and (e) uncoated pSi particle control. For the compositions that used the pSi carrier, the percent of dissolved Si measured in the elution solution is also given. These temporal release profiles were obtained following an elution protocol mimicking oral delivery to either the small intestine or to the colon, involving a 2 h incubation in simulated gastric fluid (SGF, pH 1.2) followed by either fasting-state simulated intestinal fluid (FasSIF, pH 6.8) or phosphate buffered saline (PBS, pH 7.4) for 3 h. The FasSIF condition was designed to simulate delivery to the small intestine, whereas the PBS condition simulated delivery to the colon. The temperature throughout the experiments was maintained at 37 °C. The total protein was measured by BCA assay, the total active IgA2 antibody was measured by ELISA (for Eudragit–pSi formulations, activity was further confirmed with a stringent HIV neutralization assay), and the % Si present in eluents was measured by molybdenum blue assay.
Control experiments involving IgA2 loaded into the pure polymers, without the pSi vehicle, were performed. The polymer-only formulations of Eudragit L100 and Eudragit S100 displayed similar elution characteristics under the physiological release conditions (Figure 2b,d). Both formulations displayed minimal (6–8%) release in SGF in the 2 h period of the elution protocol, and approximately 70% of the total loaded IgA2 was released during the 3 h incubation period in FasSIF (for Eudragit L100) or in PBS (for Eudragit S100). The IgA2 release profiles for the two control polymer formulations and for the formulations in which IgA2 was contained in the pSi carriers embedded in the polymers were qualitatively similar, suggesting that the rate of IgA2 release was primarily dictated by the state of the enteric polymer. In the case of Eudragit L100, the release of IgA2 from the pSi-containing formulation was somewhat slower than what was observed from the pure polymer (Figure 2a,b), indicating that the nanomaterial exerted some control over IgA2 elution.
In a second set of control experiments, we measured the release of IgA2 from uncoated pSi particles. In the absence of the polymer coatings, the particles displayed a rapid, burst-like release of the protein payload into the SGF medium (Figure 2e), releasing 50–60% of the payload within 2 h of incubation in SGF. Little dissolution of silicon was observed in this pH range, which is consistent with the known stability of pSi and its surface oxide in acidic media. These results suggest that a majority of the protein payload was held within the pSi host via relatively weak electrostatic or van der Waals forces, such that the protein rapidly leached from the pSi matrix in the low pH aqueous medium in the absence of a polymer coat. The pSi carrier dissolved more rapidly upon transition to the higher pH FasSIF or PBS media, and the release of IgA2 continued, although it slowed substantially, suggesting that the release of protein in the higher pH range was tied more to the breakdown of the pSi matrix than was observed at a low pH.
In summary, the in vitro release data for both enteric polymer types confirmed that they effectively coated the IgA2-loaded particles and prevented premature payload release in the low pH, proteolytic environment of SGF, which in these in vitro experiments approximated the conditions of the stomach, whereas dissolution of the pSi carrier and release of IgA2 was triggered in the higher pH conditions simulating either the small intestine or the colon, for which the respective enteric polymers were designed.
3.3. Activity of IgA2 Released from the Formulations.
Whereas the above data demonstrated that the coated nanoparticles were effective at preventing premature release of the IgA2 protein therapeutic into the degradative conditions of the stomach, the more important question is whether the protein payload, once released in the target compartment, retains its function. The conditions used to process the Eudragit class of polymers typically involve organic solvents that can denature proteins, and the polymers themselves and their hydrolysis chemistries can also induce protein denaturation.58 Indeed, this was the main motivation for using the pSi particles as a carrier, as it has previously been shown that the preloading of proteins into pSi particles can preserve their activity through the processing steps needed to incorporate them into a biodegradable polymer.36–39
The first issue evaluated here was what effect the vacuum loading procedure, which was used to load IgA2 into the pSi carriers (Scheme 2, top panel), might have on the activity of the protein. We assessed the procedure by vacuum drying 30 μL aliquots of IgA2 solution (7 mg/mL), dissolved in the various release media (PBS, FasSIF, or SGF), and then dispersing the dried protein back into the relevant media. SGF was used as a negative control, as the low pH and presence of pepsin in that medium was expected to lead to a complete degradation of the protein. Vacuum-dried IgA2 redispersed quickly in all three media, and the protein concentration was confirmed by BCA assay. The activity of the IgA2 redispersed in PBS, FasSIF, or SGF was quantified using a functional ELISA involving the F425A1g8 (IgA2)-specific gp120-CD4 complex antigen immobilized on ELISA plates (Figure S3, Supporting Information).51 As expected, the data showed a complete loss of IgA2 activity in the SGF control. However, a significant loss of activity for IgA2 was also observed in the PBS and FasSIF buffers; only 60 and 50% IgA2 activity was retained upon redissolution in PBS and FasSIF, respectively. These activity numbers likely represent an upper bound of the activity that might be expected from the loading method used in this work.
We next assessed the activity of IgA2 released into eluent solutions from the fully formulated Eudragit-coated IgA2-loaded pSi particles (Scheme 2, bottom panel) using a functional ELISA and confirmed it using an HIV neutralization assay, a very stringent assay used to determine the bioactivity of IgA.53 As a control, IgA2 released from the polymer-only Eudragit formulations (without pSi, Scheme 2, middle panel) showed no detectable activity under any of the release conditions tested (Figure 2b,d), indicating that direct formulation of the protein into the Eudragit polymer from methanol solutions is not a viable approach. A second control, using the pSi carrier but no polymer coating (Scheme 2, top panel), was also tested. The uncoated pSi particles released essentially all of the IgA2 payload in the low pH (SGF) phase of the elution (Figure 2e). No activity was observed from IgA2 that was released in this phase, as can be expected due to the presence of the proteolytic enzyme pepsin in the SGF medium. By contrast, the release of IgA2 from pSi particles into the pH 1.2 buffer simulating gastric conditions was substantially hindered by the Eudragit polymer coatings, as discussed above. Upon transition to the higher pH FasSIF buffer simulating intestinal conditions, IgA2 released from the Eudragit L100–pSi formulation retained 32% of its activity (61 μg of active IgA2 out of 192 μg of total protein released, Figure 2a). Similarly, IgA2 from the Eudragit S100–pSi formulation retained around 44% of its activity (124 μg of active IgA2 out of 279 μg of total protein released, Figure 2c) when released into PBS buffer simulating conditions in the colon. For completeness, similar experiments using the Eudragit L100–pSi formulation but releasing the IgA2 payload into PBS (pH 7.4) media were also performed using the same protocol and are presented in Figure S2 (Supporting Information). Here, 20% activity was retained in the IgA2 released.
The bioactivity data thus demonstrates that the polymer-coated pSi delivery system was capable of pH-responsive release of protein and that a substantially greater percentage of the protein remained active relative to the polymer-only delivery system but some protein activity was lost upon coating of the nanoparticles with enteric polymer. The retention of 20–44% activity with this system was markedly greater than those of previously reported delivery systems including polymer, liposome, silica, and other nanocarrier-based formulations (summarized in Table S1, Supporting Information). Despite its improved performance relative to these other benchmark systems, the Eudragit–pSi formulations still showed a substantial loss in activity of the IgA2 protein, as (at most) only 44% activity was retained. The reason for the loss in activity of the released IgA2 is attributed to the loading and polymer processing conditions, as discussed above.
3.4. Changes in Morphology of Polymer–Particle Composites under Release Conditions.
The triggered release of IgA2 upon dissolution of the polymer coating was confirmed by DLS measurements and SEM observations. The DLS measurements (Figure S1, Supporting Information) show that the average nanoparticle size increased upon coating with Eudragit L100 and Eudragit S100 (to 1000 and 1650 nm, respectively). The particle size did not change during the 2 h incubation of these coated nanoparticles in SGF. After replacing the medium to FasSIF or PBS, the average particle size decreased substantially, indicating dissolution of the polymer shell and of the particle within. The trend in size reduction was similar for both Eudragit L100–pSi and Eudragit S100–pSi formulations. However, the overall size reduction was greater for Eudragit S100–pSi, where the average particle size reduced to ~350 nm after a total of 5 h of incubation in the release medium (2 h in SGF and then 3 h in PBS). This result is consistent with the Si dissolution trend of Figure 2a,c, and it is attributed to a more rapid rate of dissolution of the pSi core at the higher pH of PBS (7.4) relative to FasSIF (6.8); the increase in the rate of dissolution of pSi with increasing pH is well documented.59–61
The SEM micrographs obtained at different times during incubation in release media support the DLS data and confirm the fate of the enteric polymer coatings (Figure 3). The as-etched (uncoated) pSi particles showed no evidence of changes in particle size or pore morphology when incubated for 2 h in the highly acidic (pH 1.2) simulated gastric fluid, SGF (Figure 3a). By contrast, the incubation of uncoated pSi particles in PBS buffer (pH 7.4) showed evidence of dissolution of the pSi matrix that increased with increasing time of exposure to the buffer (Figure 3b,c). The polymer-coated pSi particles performed as expected in the buffers for which the enteric polymers were designed. Thus, the Eudragit L100–pSi formulation showed no obvious signs of dissolution after 2 h of incubation in SGF (Figure 3d), but it dissolved rapidly upon switching to the less acidic (pH 6.8) FasSIF buffer (Figure 3e,f). The SEM images make it clearly evident that the underlying pSi particles are unveiled as the Eudragit L100 coating dissolves. The data also confirm, as suggested by the DLS measurements and by the protein release and dissolved Si data of Figure 2, that the pSi carrier dissolved more slowly than the Eudragit L100 polymer at pH 6.8.
Figure 3.

Representative ex situ SEM images monitoring morphological changes in polymer-coated or uncoated pSi particles at various stages of exposure to eluent solutions of different pH values. In this experiment, particles were sequentially exposed to SGF for 2 h followed by SIF or PBS for 3 h and a 5 μL sample aliquot was taken at specified time point for SEM imaging. Uncoated (as-etched) pSi particles after (a) 2 h incubation in SGF (pH 1.2), (b) 1.5 h incubation in PBS (pH 7.4), or (c) 3 h incubation in PBS (pH 7.4). The uncoated particles are stable at pH 1.2 but show signs of slow dissolution at pH 7.4. Eudragit L100-coated pSi after (d) 2 h in SGF (pH 1.2), (e) 1.5 h in FasSIF (pH 6.8), or (f) 3 h incubation in FasSIF (pH 6.8). The Eudragit L100 coating is stable at pH 1.2 but readily dissolves at pH 6.8 to expose the underlying pSi particles. Eudragit S100-coated pSi particles after (g) 2 h in SGF (pH 1.2), (h) 1.5 h in PBS (pH 7.4), or (i) 3 h in PBS (pH 7.4). The Eudragit S100 coating is stable at pH 1.2 but dissolves at pH 7.4 to expose the underlying pSi particles. Scale bars for all images are 200 nm.
In case of the Eudragit S100–pSi formulation, 2 h of incubation in SGF (Figure 3g) resulted in no apparent dissolution of the polymer coating, whereas extensive dissolution of the Eudragit polymer and the pSi particles occurred upon incubation in PBS at pH 7.4 (Figure 3h,i). In this case, vestiges of the enteric polymer coat could be seen in the SEM images of the formulation after 1.5 h of exposure to the pH 7.4 buffer (Figure 3h), whereas after 3 h of exposure the coating was substantially less evident (Figure 3i).
In order to assess if using a water-processable enteric polymer might provide better performance, Eudragit L30 D-55 was coated onto pSi particles and in vitro release was assessed. Unlike the other two Eudragit polymers used in the present study, which were processed in methanol, Eudragit L30 D-55 is soluble in water, and so, the coating process was performed in water. The data for a Eudragit L30 D-55–pSi formulation and control (Eudragit L30 D-55 without pSi) are presented in Figure S4 (Supporting Information). The formulation showed an in vitro release trend similar to the other two composite formulations, and the released IgA2 displayed only 20% activity (measured by ELISA; HIV neutralization assay was used to confirm the activity for the Eudragit L30 D-55–pSi formulation). The low activity for this formulation is attributed to the increased interaction of IgA2 with polymer chains in the aqueous medium. The in vitro release and activity data for the three enteric polymer-coated pSi formulations studied in this work (see Scheme 1a) are summarized in Table 1.
3.5. In Vitro IgA2 Release and Activity Using Eudragit-Coated Capsules Filled with pSi Particles.
Because the process of coating the enteric polymers on the pSi particle-based carriers reduced the overall activity of the IgA2 payload somewhat, we explored the possibility of avoiding the polymer coating on each individual pSi particle altogether by placing a bolus of IgA2-loaded, uncoated pSi particles into standard size 4 gelatin capsules, which were then coated with the same Eudragit enteric polymers (Scheme 1b). The size 4 gelatin capsules used in this study were loaded with 1.7 mg of drug-loaded pSi particles, corresponding to 700 μg of IgA2 (based on 41 wt % loading on unwashed particles) in a single capsule. The size 4 capsules had a volume sufficient to hold ~85 mg of particles, but they were under-filled for experimental convenience. The capsule was then coated with either Eudragit L100 or Eudragit S100. The Eudragit polymer concentration (5, 10, and 15 wt/v %). The coating process (i.e., number of dipping cycles used to coat the capsules with the Eudragit polymer) was optimized using capsules filled with the indicator dye Rhodamine 6G (R6G; 5 mg per capsule), which provided a convenient means to monitor leakage. The conditions were selected on the basis of the ability to obtain a conformal coating that (1) protected the capsule from premature disintegration in the low pH SGF conditions and (2) dissolved quickly at intestinal pH (i.e., pH 6.8 in FasSIF or pH 7.4 in PBS). R6G release was monitored visually under simulated physiological conditions (Figure S5, Supporting Information). The optimal concentration was determined to be 10% (mass/volume) for Eudragit L100 and 15% (mass/volume) for Eudragit S100. The gelatin capsules were filled with IgA2-loaded pSi particles, sealed, and then coated with the Eudragit polymer of interest.
The in vitro release studies were performed following the same protocol as was used for the polymer-coated pSi particles. The capsules showed no IgA2 release into SGF during the initial 2 h of incubation for either the Eudragit L100 or the Eudragit S100 coatings. Since the target release site for Eudragit L100-coated capsules is the small intestine, for the Eudragit L100 experiments, the release medium was switched to FasSIF (pH = 6.8). In this medium, 60% (i.e., 420 ± 14 μg) of the total loaded IgA2 was released within 3 h (Figure 4a). The release from the same system was also measured in PBS (pH 7.4) after 2 h of initial incubation in SGF, and 70% (i.e., 490 ± 89 μg) of the total loaded IgA2 eluted during the 3 h incubation (Figure S6, Supporting Information). The difference between the quantities of IgA2 released with these two buffers can be attributed to the increased rate of dissolution of pSi at the higher pH of PBS relative to FasSIF. The gelatin capsule disintegrated quickly in either of these buffers. Of the IgA released into solution, 53% (i.e., 223 ± 20 μg) retained functional activity in FasSIF buffer and 54% (i.e., 265 ± 77 μg) retained the activity in PBS buffer (Figure 4a and Figure S6, respectively).
Figure 4.
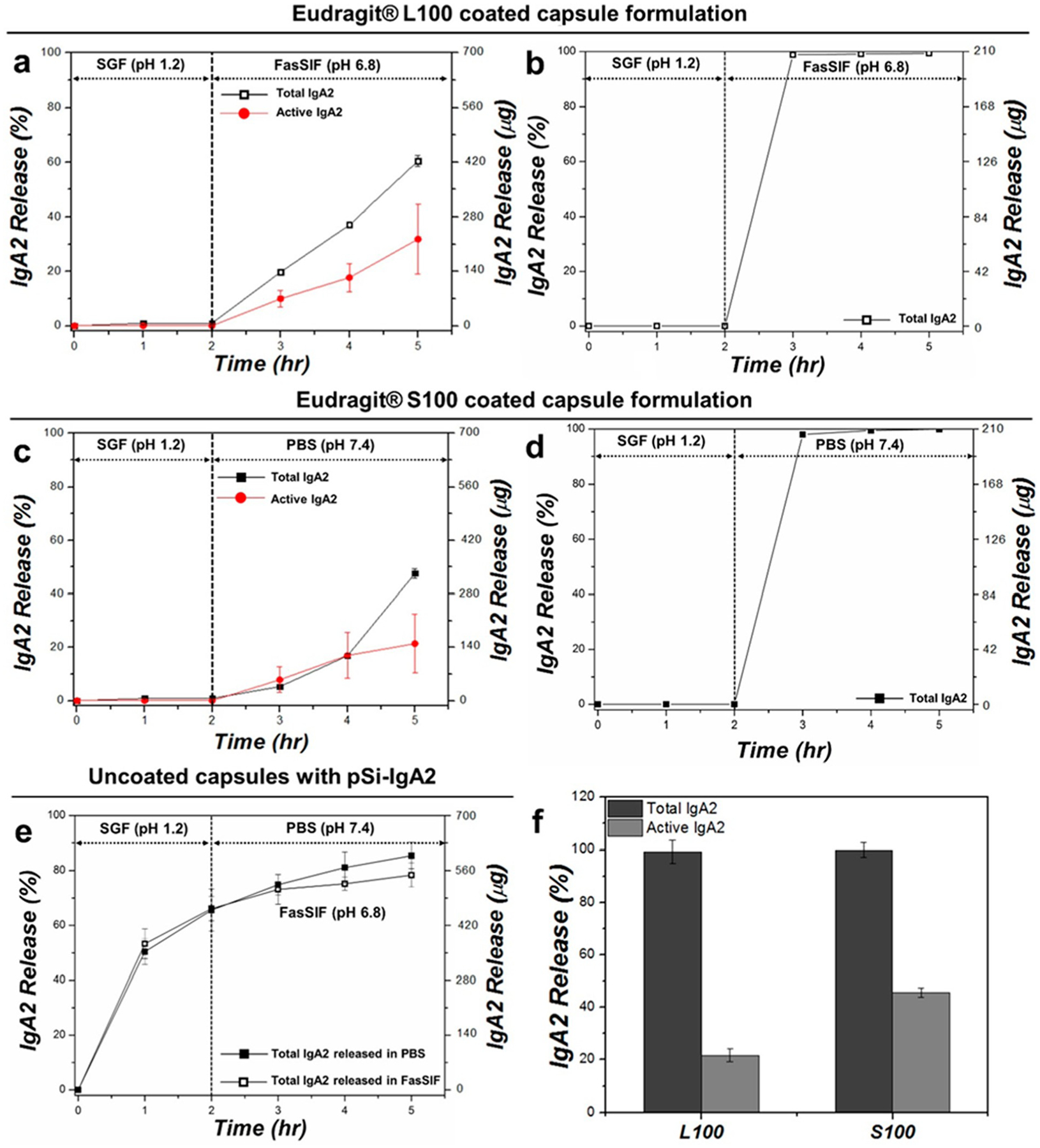
Release of IgA2 from capsule-packed formulations and from controls, comparing total protein (Total IgA2) with protein activity (Active IgA2) as a function of time: (a) total and active IgA2 released from Eudragit L100-coated capsule packed with IgA2-loaded pSi particles, (b) total IgA2 released from Eudragit L100-coated capsule packed with vacuum-dried IgA2 without pSi particles, (c) total and active IgA2 released from Eudragit S100-coated capsule packed with IgA2-loaded pSi particles, (d) total IgA2 released from Eudragit S100-coated capsule packed with vacuum-dried IgA2 without pSi particles, (e) total IgA2 released from uncoated capsule packed with IgA2-loaded pSi particles in SGF for 2 h followed by release in either FasSIF (pH 6.8; open black squares) or PBS (pH 7.4; closed black squares), and (f) total (BCA assay) and active (ELISA assay) IgA2 released at 3 h time point from (b) and (d) (i.e., IgA2-packed Eudragit polymer-coated capsules without pSi). Since negligible quantities of IgA2 were released in SGF during the first 2 h of incubation and approximately 99% of the IgA2 packed in the capsule was released during the following 3 h time point, activity was determined at this point only.
Since the target release site for Eudragit S100-coated capsules is the colon, the Eudragit S100 experiments involved 2 h in SGF followed by 3 h in PBS (pH = 7.4). The in vitro temporal release data for the Eudragit S100-coated capsules are plotted in Figure 4c. Similar to the Eudragit L100-coated capsule system, the Eudragit S100 system did not show any release of IgA2 in SGF over 2 h of incubation, but upon changing the release medium to PBS, approximately 47% (i.e., 333 ± 12 μg) of the total loaded IgA2 was released within 3 h. The activity analysis using functional ELISA showed that 45% (i.e., 150 ± 79 μg) of the released IgA2 retained activity. The IgA2-loaded pSi particles packed in an uncoated gelatin capsule disintegrated in SGF during the first 2 h of incubation and released 60% of the total loaded IgA2 into the SGF buffer; after 3 h in FasSIF or PBS, a total of 85% of the loaded IgA2 had been released (Figure 4e).
As the controls, vacuum-dried IgA2 (not loaded into pSi particles) was packed into capsules that were then coated with Eudragit polymers and tested for in vitro release behavior (Figure 4b,d). The Eudragit L100- and Eudragit S100-coated capsules showed similar dissolution trends, with no noticeable capsule degradation or rupture during the 2 h of incubation time in SGF and complete disintegration of the capsule within 1 h of switching the release medium to FasSIF or PBS. At this point, the free IgA2 released from the Eudragit L100-coated capsules retained 25% activity, while the Eudragit S100-coated capsules yielded 45% activity for the released IgA2 (Figure 4f). The lower activity in IgA2 released from the Eudragit L100-coated capsules is attributed to the interaction between the released protein and the locally high concentration of Eudragit L100 and gelatin components dissolved in the release medium. By contrast, at the same 3 h time point (1 h postexposure to FasSIF or PBS), the activity of IgA2 released from Eudragit-coated pSiNPs (Figure 2) was 30 ± 11 or 37% ± 10 for Eudragit L100 (FasSIF eluent) or Eudragit S100 (PBS eluent) coatings, respectively; the activity of IgA2 released from Eudragit-coated capsules containing IgA2-loaded pSiNPs (Figure 4) was 50 ± 16 or 147% ± 62 for Eudragit L100 (FasSIF eluent) or Eudragit S100 (PBS eluent) coatings, respectively. Thus, in terms of retention of IgA2 activity, the Eudragit S100-coated capsules containing IgA2-loaded pSiNPs showed the best performance and the Eudragit-coated particles performed comparable to the capsules containing free IgA2, within the error limits of the measurements. The superior performance observed for capsules packed with IgA2-loaded pSi particles is attributed to the ability of the pSi carrier to shield the protein from interacting with the relatively high local concentration of polymer dissolved in the elution medium during the initial release time points. These data are consistent with the performance throughout the 5 h elution experiments and indicate that uncoated IgA2-loaded nanoparticles loaded into a capsule offers more controlled release with a higher protein bioactivity overall. Because nanoparticles can passively target sites of inflammation in the GI tract whereas orally administered molecular therapeutics tend to clear quickly through the gut,51 a nanoparticle formulation such as the present one may show a greater retention and thus longer duration of action for its therapeutic payload.
CONCLUSIONS
This work is the first example of a Eudragit-enabled porous silicon (pSi) oral delivery system. The unique feature of the approach is that the pSi nanoparticles provided protection and site-specific delivery for the IgA2 antibody therapeutic payload, with a high mass loading of 29%, defined as (mass of drug)/(mass of pSi particle + drug) and substantial retention of biologic activity. Two approaches were used to protect the pSi particles and their drug payload against degrading gastric conditions were explored using Eudragit enteric polymers: coating of individual pSi particles and coating of a standard gelatin capsule containing a bolus of uncoated pSi particles. Both approaches provided protection of the protein payload under acidic gastric conditions and both released >50% of the protein payload within 3 h of entering the target neutral pH range for which the Eudragit polymer was engineered. The approach involving placing uncoated protein-loaded pSi nanoparticles into coated capsules was the most effective at delivering the highest quantity of protein with the highest retention of activity; only 21–44% of activity was retained in IgA2 released from the individually coated pSi nanoparticles, whereas 45–54% of activity was retained in IgA2 released from pSi nanoparticles delivered by capsule. The capsule formulation also delivered a larger amount of IgA2 on a mass percentage basis. Overall, the small intestine- and colontargeted systems were able to provide selective pH-responsive releases and retain substantial activity of the released proteins, demonstrating promise for oral delivery of biologics targeted to the lower GI tract.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded in part by the National Institutes of Health (Grant R01 AI132413-01), by the National Science Foundation under grant CBET-1603177, by the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement HR0011-13-2-0017, and by the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC), supported by the National Science Foundation (Grant DMR-2011924). The authors would like to acknowledge support to M.K. from the Defense Advanced Research Project Agency (DARPA-BAA-13-03). T.K. would like to thank the National Health and Medical Research Council of Australia for an Early Career Fellowship (GNT1143296) and the University of New South Wales-Sydney for a Scientia grant. J.W. would like to thank the National Institutes of Health for support from training grant T32 CA153915-06. This work was performed in part in the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148). The content of the information within this document does not necessarily reflect the position or the policy of the Government.
The authors declare the following competing financial interest(s): M.J.S. is a scientific founder and a member of the board of directors, and he holds an equity interest of Spinnaker Biosciences and Impilo Therapeutics. He also has a financial interest (as a consultant, shareholder, scientific advisor, and/or board member) with Bejing ITEC Technologies, Cend Therapeutics, Illumina, Matrix Technologies, NanoVision Bio, Pacific Integrated Energy, TruTag Technologies, and Well-Healthcare Technologies. M.J.S. is a Guest Professor at Zhejiang University, China. Although one or more of the grants that supported this research has been identified for conflict of interest management based on the overall scope of the project and its potential benefit to these companies, the research findings included in this particular publication may not necessarily relate to their interests. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomaterials.0c01313.
Figures of time-resolved DLS measurements, in vitro temporal release data, in vitro evaluation of the effect of the vacuum drying procedure, in vitro temporal release curves, optimization coating of Eudragit enteric polymer on gelatin capsules photos, and total IgA2 and active IgA2 released from the capsule over a period of 5 h and table of oral protein delivery systems reported in the literature (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsbiomaterials.0c01313
Contributor Information
Tushar Kumeria, Department of Chemistry and Biochemistry, University of California, San Diego, California 92093, United States; School of Materials Science and Engineering, University of New South Wales-Sydney, Sydney, NSW 2052, Australia.
Joanna Wang, Materials Science and Engineering Program, University of California, San Diego, California 92093, United States.
Byungji Kim, Materials Science and Engineering Program, University of California, San Diego, California 92093, United States.
Ji-Ho Park, Department of Bio and Brain Engineering, KAIST, Daejeon 34141, Korea.
Jonathan M. Zuidema, Department of Chemistry and Biochemistry, University of California, San Diego, California 92093, United States
Mark Klempner, MassBiologics of the University of Massachusetts Medical School, Boston, Massachusetts 02126, United States.
Lisa Cavacini, MassBiologics of the University of Massachusetts Medical School, Boston, Massachusetts 02126, United States.
Yang Wang, MassBiologics of the University of Massachusetts Medical School, Boston, Massachusetts 02126, United States.
Michael J. Sailor, Department of Chemistry and Biochemistry, University of California, San Diego, California 92093, United States
REFERENCES
- (1).Leader B; Baca QJ; Golan DE Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug Discovery 2008, 7, 21–39. [DOI] [PubMed] [Google Scholar]
- (2).Yun Y; Cho YW; Park K Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv. Drug Delivery Rev 2013, 65, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Park K; Kwon IC; Park K Oral protein delivery: Current status and future prospect. React. Funct. Polym 2011, 71, 280–287. [Google Scholar]
- (4).Morishita M; Peppas NA Is the oral route possible for peptide and protein drug delivery? Drug Discovery Today 2006, 11, 905–910. [DOI] [PubMed] [Google Scholar]
- (5).Brown TD; Whitehead KA; Mitragotri S Materials for oral delivery of proteins and peptides. Nat. Rev. Mater 2020, 5, 127–148. [Google Scholar]
- (6).Lee KY; Yuk SH Polymeric protein delivery systems. Prog. Polym. Sci 2007, 32, 669–697. [Google Scholar]
- (7).Tabata Y; Miyao M; Ozeki M; Ikada Y Controlled release of vascular endothelial growth factor by use of collagen hydrogels. J. Biomater. Sci., Polym. Ed 2000, 11, 915–930. [DOI] [PubMed] [Google Scholar]
- (8).Margulis BA; Sandler S; Eizirik DL; Welsh N; Welsh M Liposomal Delivery of Purified Heat Shock Protein hsp70 Into Rat Pancreatic Islets as Protection Against Interleukin 1β–Induced Impaired β-Cell Function. Diabetes 1991, 40, 1418–1422. [DOI] [PubMed] [Google Scholar]
- (9).Iwanaga K; Ono S; Narioka K; Kakemi M; Morimoto K; Yamashita S; Namba Y; Oku N Application of surface-coated liposomes for oral delivery of peptide: Effects of coating the liposome’s surface on the GI transit of insulin. J. Pharm. Sci 1999, 88, 248–252. [DOI] [PubMed] [Google Scholar]
- (10).Kaneda Y Virosomes: evolution of the liposome as a targeted drug delivery system. Adv. Drug Delivery Rev 2000, 43, 197–205. [DOI] [PubMed] [Google Scholar]
- (11).Zhao Y; Trewyn BG; Slowing II; Lin VS-Y Mesoporous silica nanoparticle-based double drug delivery system for glucose-responsive controlled release of insulin and cyclic AMP. J. Am. Chem. Soc 2009, 131, 8398–8400. [DOI] [PubMed] [Google Scholar]
- (12).Yang Y; Niu Y; Zhang J; Meka AK; Zhang H; Xu C; Lin CXC; Yu M; Yu C Biphasic Synthesis of Large-Pore and Well-Dispersed Benzene Bridged Mesoporous Organosilica Nanoparticles for Intracellular Protein Delivery. Small 2015, 11, 2743–2749. [DOI] [PubMed] [Google Scholar]
- (13).Abeer MM; Rewatkar P; Qu Z; Talekar M; Kleitz F; Schmid R; Lindén M; Kumeria T; Popat A Silica nanoparticles: A promising platform for enhanced oral delivery of macromolecules. J. Controlled Release 2020, 326, 544–555. [DOI] [PubMed] [Google Scholar]
- (14).Abeer MM; Meka AK; Pujara N; Kumeria T; Strounina E; Nunes R; Costa A; Sarmento B; Hasnain SZ; Ross BP; et al. Rationally designed dendritic silica nanoparticles for oral delivery of exenatide. Pharmaceutics 2019, 11, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lamson NG; Berger A; Fein KC; Whitehead KA Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nat. Biomed. Eng 2020, 4, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ellis-Behnke RG; Liang Y-X; You S-W; Tay DK; Zhang S; So K-F; Schneider GE Nano neuro knitting: peptide nanofiber scaffold for brain repair and axon regeneration with functional return of vision. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 5054–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kim J-Y; Choi WI; Kim YH; Tae G Brain-targeted delivery of protein using chitosan-and RVG peptide-conjugated, pluronic-based nano-carrier. Biomaterials 2013, 34, 1170–1178. [DOI] [PubMed] [Google Scholar]
- (18).Sinha VR; Trehan A Biodegradable microspheres for protein delivery. J. Controlled Release 2003, 90, 261–280. [DOI] [PubMed] [Google Scholar]
- (19).Koppolu B. p.; Smith SG; Ravindranathan S; Jayanthi S; Suresh Kumar TK; Zaharoff DA Controlling chitosan-based encapsulation for protein and vaccine delivery. Biomaterials 2014, 35, 4382–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Santhosh KT; Alizadeh A; Karimi-Abdolrezaee S Design and optimization of PLGA microparticles for controlled and local delivery of Neuregulin-1 in traumatic spinal cord injury. J. Controlled Release 2017, 261, 147–162. [DOI] [PubMed] [Google Scholar]
- (21).Hamman JH; Enslin GM; Kotzé AF Oral Delivery of Peptide Drugs. BioDrugs 2005, 19, 165–177. [DOI] [PubMed] [Google Scholar]
- (22).Cao S.-j.; Xu S; Wang H.-m.; Ling Y; Dong J; Xia R.-d.; Sun X.-h. Nanoparticles: Oral Delivery for Protein and Peptide Drugs. AAPS PharmSciTech 2019, 20, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Salonen J Drug Delivery With Porous Silicon. In Handbook of Porous Silicon; Canham LT, Ed.; Springer: Switzerland, 2014; p 909. [Google Scholar]
- (24).Tzur-Balter A; Shtenberg G; Segal E Porous silicon for cancer therapy: from fundamental research to the clinic. Rev. Chem. Eng 2015, 31, 193–207. [Google Scholar]
- (25).Canham LT Porous silicon for medical use: from conception to clinical use. In Porous Silicon for Biomedical Applications; Santos HA, Ed.; 2014; Vol. 68, pp 3–20. [Google Scholar]
- (26).Bonanno LM; Segal E Nanostructured porous silicon-polymer-based hybrids: from biosensing to drug delivery. Nanomedicine 2011, 6, 1755–1770. [DOI] [PubMed] [Google Scholar]
- (27).McInnes SJP; Voelcker NH Silicon-polymer hybrid materials for drug delivery. Future Med. Chem 2009, 1, 1051–1074. [DOI] [PubMed] [Google Scholar]
- (28).Kumeria T; McInnes SJP; Maher S; Santos A Porous silicon for drug delivery applications and theranostics: recent advances, critical review and perspectives. Expert Opin. Drug Delivery 2017, 14, 1407–1422. [DOI] [PubMed] [Google Scholar]
- (29).Andrew JS; Anglin EJ; Wu EC; Chen MY; Cheng L; Freeman WR; Sailor MJ Sustained Release of a Monoclonal Antibody from Electrochemically Prepared Mesoporous Silicon Oxide. Adv. Funct. Mater 2010, 20, 4168–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tanaka T; Mangala LS; Vivas-Mejia PE; Nieves-Alicea R; Mann AP; Mora E; Han HD; Shahzad MMK; Liu XW; Bhavane R; Gu JH; Fakhoury JR; Chiappini C; Lu CH; Matsuo K; Godin B; Stone RL; Nick AM; Lopez-Berestein G; Sood AK; Ferrari M Sustained Small Interfering RNA Delivery by Mesoporous Silicon Particles. Cancer Res. 2010, 70, 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Anglin EJ; Cheng L; Freeman WR; Sailor MJ Porous silicon in drug delivery devices and materials. Adv. Drug Delivery Rev 2008, 60, 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Foraker AB; Walczak RJ; Cohen MH; Boiarski TA; Grove CF; Swaan PW Microfabricated porous silicon particles enhance paracellular delivery of insulin across intestinal Caco-2 cell monolayers. Pharm. Res 2003, 20, 110–116. [DOI] [PubMed] [Google Scholar]
- (33).Kovalainen M; Mönkäre J; Kaasalainen M; Riikonen J; Lehto V-P; Salonen J; Herzig K-H; Järvinen K Development of porous silicon nanocarriers for parenteral peptide delivery. Mol. Pharmaceutics 2013, 10, 353–359. [DOI] [PubMed] [Google Scholar]
- (34).Kovalainen M; Mönkäre J; Mäkilä E; Salonen J; Lehto V-P; Herzig K-H; Järvinen K Mesoporous silicon (PSi) for sustained peptide delivery: effect of PSi microparticle surface chemistry on peptide YY3–36 release. Pharm. Res 2012, 29, 837–846. [DOI] [PubMed] [Google Scholar]
- (35).Pastor E; Matveeva E; Valle-Gallego A; Goycoolea FM; Garcia-Fuentes M Protein delivery based on uncoated and chitosancoated mesoporous silicon microparticles. Colloids Surf., B 2011, 88, 601–609. [DOI] [PubMed] [Google Scholar]
- (36).Zuidema JM; Kumeria T; Kim D; Kang J; Wang J; Hollett G; Zhang X; Roberts DS; Chan N; Dowling C; Blanco-Suarez E; Allen NJ; Tuszynski MH; Sailor MJ Oriented Nanofibrous Polymer Scaffolds Containing Protein-Loaded Porous Silicon Generated by Spray Nebulization. Adv. Mater 2018, 30, 1706785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zuidema JM; Dumont CM; Wang J; Batchelor WM; Lu Y-S; Kang J; Bertucci A; Ziebarth NM; Shea LD; Sailor MJ Porous Silicon Nanoparticles Embedded in Poly(lactic-co-glycolic acid) Nanofiber Scaffolds Deliver Neurotrophic Payloads to Enhance Neuronal Growth. Adv. Funct. Mater 2020, 30, 2002560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kang RH; Jang J-E; Huh E; Kang SJ; Ahn D-R; Kang JS; Sailor MJ; Yeo SG; Oh MS; Kim D; Kim HY A brain tumor-homing tetra-peptide delivers a nano-therapeutic for more effective treatment of a mouse model of glioblastoma. Nanoscale Horiz. 2020, 5, 1213–1225. [DOI] [PubMed] [Google Scholar]
- (39).Rosenberg M; Zilony N; Shefi O; Segal E Designing Porous Silicon Films as Carriers of Nerve Growth Factor. J. Visualized Exp 2019, 143, e58982. [DOI] [PubMed] [Google Scholar]
- (40).Wu C-C; Hu Y; Miller M; Aroian RV; Sailor MJ Protection and delivery of anthelmintic protein Cry5B to nematodes using mesoporous silicon particles. ACS Nano 2015, 9, 6158–6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Shrestha N; Araújo F; Shahbazi MA; Mäkilä E; Gomes MJ; Herranz-Blanco B; Lindgren R; Granroth S; Kukk E; Salonen J; et al. Thiolation and Cell-Penetrating Peptide Surface Functionalization of Porous Silicon Nanoparticles for Oral Delivery of Insulin. Adv. Funct. Mater 2016, 26, 3405–3416. [Google Scholar]
- (42).Shrestha N; Shahbazi M-A; Araújo F; Zhang H; Mäkilä EM; Kauppila J; Sarmento B; Salonen JJ; Hirvonen JT; Santos HA Chitosan-modified porous silicon microparticles for enhanced permeability of insulin across intestinal cell monolayers. Biomaterials 2014, 35, 7172–7179. [DOI] [PubMed] [Google Scholar]
- (43).Shrestha N; Shahbazi M-A; Araújo F; Mäkilä E; Raula J; Kauppinen EI; Salonen J; Sarmento B; Hirvonen J; Santos HA Multistage pH-responsive mucoadhesive nanocarriers prepared by aerosol flow reactor technology: A controlled dual protein-drug delivery system. Biomaterials 2015, 68, 9–20. [DOI] [PubMed] [Google Scholar]
- (44).Sato Y; Kawashima Y; Takeuchi H; Yamamoto H In vitro evaluation of floating and drug releasing behaviors of hollow microspheres (microballoons) prepared by the emulsion solvent diffusion method. Eur. J. Pharm. Biopharm 2004, 57, 235–243. [DOI] [PubMed] [Google Scholar]
- (45).Sonaje K; Chen Y-J; Chen H-L; Wey S-P; Juang J-H; Nguyen H-N; Hsu C-W; Lin K-J; Sung H-W Enteric-coated capsules filled with freeze-dried chitosan/poly (γ-glutamic acid) nanoparticles for oral insulin delivery. Biomaterials 2010, 31, 3384–3394. [DOI] [PubMed] [Google Scholar]
- (46).Kadian SS; Harikumar S Eudragit and its pharmaceutical significance; 2016.
- (47).Nikam VK; Kotade KB; Gaware VM; Dhamak R; Somwanshi SB; Khadse AN Eudragit a versatile polymer: a review. Pharmacologyonline 2011, 1, 152–164. [Google Scholar]
- (48).Snoeck V; Peters IR; Cox E The IgA system: a comparison of structure and function in different species. Vet. Res 2006, 37, 455–467. [DOI] [PubMed] [Google Scholar]
- (49).Woof J; Russell M Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590. [DOI] [PubMed] [Google Scholar]
- (50).Qadri F; Svennerholm A-M; Faruque A; Sack RB Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin. Microbiol. Rev 2005, 18, 465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nehoff H; Parayath NN; Domanovitch L; Taurin S; Greish K Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int. J. Nanomed 2014, 9, 2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Collnot E-M; Ali H; Lehr C-M Nano- and microparticulate drug carriers for targeting of the inflamed intestinal mucosa. J. Controlled Release 2012, 161, 235–246. [DOI] [PubMed] [Google Scholar]
- (53).Yu X; Duval M; Lewis C; Gawron MA; Wang R; Posner MR; Cavacini LA Impact of IgA constant domain on HIV-1 neutralizing function of monoclonal antibody F425A1g8. J. Immunol 2013, 190, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Qin Z; Joo J; Gu L; Sailor MJ Size control of porous silicon nanoparticles by electrochemical perforation etching. Part. Part. Sys. Char 2014, 31, 252–256. [Google Scholar]
- (55).Björk I; Lindh E Gross conformation of human secretory immunoglobulin A and its component parts. Eur. J. Biochem 1974, 45, 135–145. [DOI] [PubMed] [Google Scholar]
- (56).Sailor MJ Porous Silicon in Practice: Preparation, Characterization, and Applications; Wiley-VCH: Weinheim, Germany, 2012; p 249. [Google Scholar]
- (57).Anderson SHC; Elliott H; Wallis DJ; Canham LT; Powell JJ Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Phys. Status Solidi A-Appl. Res 2003, 197, 331–335. [Google Scholar]
- (58).Tran V-T; Karam J-P; Garric X; Coudane J; Benoît J-P; Montero-Menei CN; Venier-Julienne M-C Protein-loaded PLGA–PEG–PLGA microspheres: A tool for cell therapy. Eur. J. Pharm. Sci 2012, 45, 128–137. [DOI] [PubMed] [Google Scholar]
- (59).Chen X; Wo F; Jin Y; Tan J; Lai Y; Wu J Drug-Porous Silicon Dual Luminescent System for Monitoring and Inhibition of Wound Infection. ACS Nano 2017, 11, 7938–7949. [DOI] [PubMed] [Google Scholar]
- (60).Bimbo LM; Mäkilä E; Laaksonen T; Lehto V-P; Salonen J; Hirvonen J; Santos HA Drug permeation across intestinal epithelial cells using porous silicon nanoparticles. Biomaterials 2011, 32, 2625–2633. [DOI] [PubMed] [Google Scholar]
- (61).Park J-H; Gu L; Von Maltzahn G; Ruoslahti E; Bhatia SN; Sailor MJ Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat. Mater 2009, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.