

Abstract
Background:
Minimal change disease (MCD) is the major cause of childhood idiopathic nephrotic syndrome, which is characterized by massive proteinuria and debilitating edema. Proteinuria in MCD is typically rapidly reversible with corticosteroid therapy, but relapses are common, and children often have many adverse events from the repeated courses of immunosuppressive therapy. The pathobiology of MCD remains poorly understood. Prior clinical observations suggest that abnormal T-cell function may play a central role in MCD pathogenesis. Based on these observations, we hypothesized that T-cell responses to specific exposures or antigens lead to a clonal expansion of T-cell subsets, a restriction in the T-cell repertoire, and elaboration of specific circulating factors that trigger disease onset and relapses.
Methods:
To test these hypotheses, we sequenced T-cell receptors in 14 MCD, four focal segmental glomerulosclerosis (FSGS), and four membranous nephropathy (MN) patients with clinical data and blood samples drawn during active disease and during remission collected by the Nephrotic Syndrome Study Network. We calculated several T-cell receptor diversity metrics to assess possible differences between active disease and remission states in paired samples.
Results:
Median productive clonality did not differ between MCD active disease (0.0083; range: 0.0042, 0.0397) and remission (0.0088; range: 0.0038, 0.0369). We did not identify dominant clonotypes in MCD active disease, and few clonotypes were shared with FSGS and MN patients.
Conclusions:
While these data do not support an obvious role of the adaptive immune system T-cells in MCD pathogenesis, further study is warranted given the limited sample size.
Keywords: minimal change disease, NEPTUNE study, immunosequencing, T-cell receptor, adaptive immunity
Introduction
Minimal change disease (MCD), characterized by heavy proteinuria greater than 3.5 grams per day, is often accompanied by the typical clinical manifestations of nephrotic syndrome including edema, hypoalbuminemia, and hyperlipemia [1, 2]. Despite being a rare kidney disease, among pediatric patients MCD is the most common form of idiopathic nephrotic syndrome [3]. Named for the lack of discernable glomerular pathology visible by light microscopy, MCD is diagnosed by the effacement of podocyte foot processes that is evident under electron microscopy [4, 5]. The incidence of MCD is approximately two to seven new cases per 100,000 children [3]. When untreated, MCD is associated with increased risk for infection [6, 7], thromboembolism [8], and cardiovascular diseases [9, 10], leading to a greater mortality risk. The proteinuria and symptoms associated with MCD are often responsive to steroid therapy; however, relapses are common and long-term use of immunosuppressive therapies is associated with undesirable health consequences [11, 12].
The cause and pathogenesis of MCD are unknown, but several lines of evidence suggest the involvement of the adaptive immune system. In 1974, Shalhoub suggested that circulating factors released by abnormal T-cells may be involved in the disease pathogenesis [13]. Several lines of evidence were offered to support this hypothesis, including the well-recognized observation that MCD is generally responsive to glucocorticoids [14, 15], which have broad immunosuppressive effects that includes T-cells [16]. Early case reports compiled in the mid-to-late 1940s describe remission of proteinuria and the symptoms of nephrotic syndrome in pediatric patients with MCD with measles infection [17-19]. Interestingly, these phenomena were recognized well enough that two studies reported outcomes of several individuals who were intentionally inoculated with measles to treat nephrotic syndrome [18, 20]. Subsequent studies have suggested multiple possible mechanisms for the suppression of T-cell immune responses mediated by the measles virus [21].
The contribution of T cell activation and the adaptive immune response in MCD pathogenesis remains unclear. Based on earlier observations described in the literature, we hypothesized that the onset of MCD is triggered by unknown environmental exposure(s) or antigen(s), which results in T-cell receptor (TCR) activation and clonal expansion of select T-cells, resulting in reduced overall TCR diversity. To test this hypothesis, we compared the TCR repertoires during active disease and remission of 14 MCD, four focal segmental glomerulosclerosis (FSGS), and four membranous nephropathy (MN) patients ascertained by Nephrotic Syndrome Study Network (NEPTUNE) [22]. After comparing TCR sequence diversity metrics, we did not identify intra-individual differences for active disease-remission MCD patient pairs. We also did not identify TCR clonotypes shared among and specific to MCD patients compared with FSGS and MN patients. While this small sample does not support a strong response to a single antigen among MCD patients, further study is needed to identify the factors contributing to the pathogenesis of this rare kidney disease.
Materials and Methods
Biospecimens and clinical data were acquired from the Nephrotic Syndrome Study Network (NEPTUNE), a North American multicenter collaborative consortium consisting of 26 sites conducting clinical and translational research for MCD, FSGS and MN [3, 23, 24]. As previously described [22], NEPTUNE is a longitudinal cohort study of patients with primary nephrotic syndrome based on kidney histopathology. Detailed clinical data (including immunosuppresive therapy status), kidney biopsy tissue, blood, and urine are collected by each NEPTUNE clinical center for each consented patient. Immunosuppressive therapy includes steroids, calcineurin inhibitors, cyclophosphamide, mycophenolate mofetil, and rituximab [25]. For pediatric patients, parents provided written consent and, when appropriate, assent was obtained. This ancillary study was approved by the NEPTUNE Ancillary Study Committee and Steering Committee as well as local Institutional Review Boards.
For the present study, we selected MCD patients who had a blood sample drawn during a time of disease activity defined as nephrotic range proteinuria (urine to creatinine ratio (UPCR) >3.5; “active disease”) and during complete remission (UPCR <0.3; “remission”). A total of 14 MCD patients met the clinical criteria for this study and who had sufficient amounts of DNA (~5 micrograms) extracted from both blood samples for immunosequencing. We also selected other causes of nephrotic syndrome for comparison, which included four FSGS and four MN patients with paired blood samples and UPCRs that fit the active disease and remission definition and eGFRs that were as closely matched as possible to the MCD cohort (i.e., < 15 ml/min). Age matching was not practically possible as FSGS and MN have late ages of onset on average compared with MCD.
Genomic DNA was extracted and purified from whole blood samples using Qiagen’s QIAsymphony (Hilden, Germany) per manufacturer’s protocols. For each genomic DNA sample, the TCR beta chain CDR3 regions were amplified and barcoded in a two-step multiplex PCR using Adaptive Biotechnologies’ immunoSEQ kit per manufacturer’s protocol [26]. Amplicons were sequenced with six replicates using Illumina’s (San Diego, California) NextSeq sequencing platform. Amplification and sequencing were performed by the University of Miami’s Center for Genome Technology. Sequencing data were transferred to Adaptive Biotechnologies (Seattle, Washington) for quality control, alignment, and further processing using their bioinformatics pipeline ANALYZER.
For each DNA sample, the Adaptive Biotechnologies’ ANALYZER outputs TCR amino acid sequences and a set of TCR diversity metrics, including total templates (the sum of the number of biological molecules assayed known as “templates” for all productive and non-productive rearrangements), total productive templates (the sum of template counts regardless of uniqueness for all productive rearrangements that are capable of producing a functional peptide, e.g., the amino acid sequences are in-frame relative to the conserved cysteine and phenylalanine and are sans stop codons), the fraction of productive templates (total number of productive templates divided by the total number of templates), the number of rearrangements (the sum of productive and non-productive unique nucleotide sequences generated through V(D)J recombination and representative of a unique clonal lineage), the number of productive rearrangements (the sum of unique clonal lineages capable of producing a functional peptide), among others. The ANALZYER pipeline also computes the productive Simpson clonality and the Morisita-Horn index. Derived from the Simpson’s diversity metric [27], productive Simpson clonality is a proxy for TCR diversity that describes both clonal frequency “evenness” (low and high evenness indicative of the presence or absence of dominant clonotypes, respectively) and overall TCR sequence “richness” (number of unique clonotypes). The value of productive Simpson clonality ranges from 0 to 1, with a higher value indicating a single dominant clone (representing no diversity such as that typically observed in patients with a T-cell malignancy, for example) and a lower value indicating completely even clonal distribution (reflecting maximum diversity in healthy individuals, for example). As compared to the Shannon clonality, the productive Simpson clonality tends to be more stable and less affected by variations in the sample input material or the T-cell fraction [28]. The Morisita-Horn index [29] is a TCR repertoire similarity metric that also ranges from 0 to 1, with a higher value indicating a higher level of the pairwise TCR similarity. T-cell repertoires were characterized for each patient both during active disease and remission.
Whole-genome sequencing (WGS) data at 30x were available for nine out of the fourteen MCD patients, three out of four FSGS patients, and all MN patients. We used the HLA Genotype Imputation With Attribute Bagging (HIBAG) [30, 31] to infer the 4-digit HLA alleles from WGS to characterize the adaptive immune system’s MHC genomic region in these patients. HIBAG leverages attribute bagging accompanied by the haplotype inference from the unphased single nucleotide polymorphisms (SNPs) and HLA alleles. In the process, the HLA-type posterior probabilities are averaged over the classifiers constructed from bootstrap samples to make the predictions. We relied on the published parameter estimates for population-specific models for African Americans and European Americans [31], respectively.
Statistical analyses were performed using PLINK (1.90) [32], R, and VDJtools [33]. We used unadjusted paired tests and ANOVAs to test for differences in TCR repertoire diversity between active disease and remission among MCD, FSGS, and MN patients, respectively. The VDJtools software was employed to perform the comparative analysis of TCR repertoires. The hierarchical clustering of the pairwise TCR similarity and the clonotype overlap for the patients was constructed and determined using VDJtools software.
Results
We sequenced the TCR beta chain using genomic DNA extracted from blood samples drawn during active disease, defined as nephrotic range proteinuria with urine protein creatinine ratio (UPCR) > 3.5, and complete remission, defined as UPCR < 0.3, for 14 patients with MCD. For comparison, we also sequenced paired samples for patients with FSGS (n=4) and MN (n=4), two causes of nephrotic syndrome with distinct histopathologic patterns from MCD. The median time between active disease and remission patient pair sampling was ~12, 6, and 16 months for MCD, FSGS, and MN patients, respectively. The MCD patient group was majority male and European-descent with median UPCR of 15.14 and 0.07 at active disease and remission, respectively (Table 1). The majority of MCD patients (64%) had received immunosuppressive therapies prior to NEPTUNE enrollment (Table 1). The majority of MCD patients were also receiving immunosuppresive therapy during active disease (71%) and remission (86%). Only one MCD patient was free of any therapy at the time of exam during active disease and remission. Age at baseline visit was statistically different across the three groups, where MCD patients had the lowest median (6.5 years) compared with FSGS and MN patients. The median UPCR for MCD patients during active disease is higher than FSGS and MN patients, while the median UPCR for patients during complete remission is similar across three groups. Among MCD patients with available WGS (n=9), we observed 13, 13, and 10 distinct HLA-A, HLA-B, and HLA-C Class I alleles, respectively, and 13, 10, 9, and 8 distinct HLA-DRB1, HLA-DQA1, HLA-DPA1, and HLA-DPB1 Class II alleles, respectively. No single HLA allele dominated either Class I or Class II distributions among MCD, FSGS, or MN cases with WGS (Table 1).
Table 1. Demographic and clinical characteristics of patients in the present study.
Data shown are from patients with minimal change disease (MCD; n =14), focal segmental glomerulosclerosis (FSGS; n = 4), and membranous nephropathy (MN; n = 4) ascertained by the Nephrotic Syndrome Study Network (NEPTUNE). Paired observations of urine protein to creatinine ratio (UPCR) are available for each patient: one during active disease and the other during remission. Active disease is defined as UPCR > 3.5 and UPCR < 0.3 is considered to be complete remission according to the NEPTUNE definition. Whole-genome sequencing data were available for nine of fourteen MCD patients, three of four FSG patients, and all MN patients included in this study. We inferred major HLA Class I HLA-A, HLA-B, HLA-C) and Class II (HLA-DRB1, HLA-DQA1, HLA-DQB1, and HLA-DPB1) alleles using HIBAG as described in the main text. Values are presented as median [interquartile range] unless otherwise indicated.
| MCD (n = 14) |
FSGS (n =4) |
MN (n = 4) |
|
|---|---|---|---|
| Age at baseline visit (years) | 6.50 [3.25, 10.00] |
9.50 [4.00, 29.75] |
63.50 [58.25, 69.25] |
| Female (%) | 35.7 | 100.0 | 50.0 |
| European-descent (%) | 57.1 | 25.0 | 75.0 |
| UPCR | |||
| During active disease | 15.14 [7.95, 16.78] |
4.43 [3.21, 6.90] |
3.56 [3.10, 4.97] |
| During remission | 0.07 [0.04, 0.15]] |
0.11 [0.09, 0.16] |
0.03 [0.03, 0.04] |
| Immunosuppressive therapy | |||
| Previous to enrollment (%) | 64.29 | 25.0 | 0 |
| During active disease (%) | 71.43 | 50.0 | 25.0 |
| During remission (%) | 85.71 | 75.0 | 50.0 |
| HLA-A (%) | |||
| 01:01 | 10.71 | 25.00 | 25.00 |
| 02:01 | 10.71 | 25.00 | 12.50 |
| Other | 42.86 | 25.00 | 62.50 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-B (%) | |||
| 08:01 | 10.71 | 12.50 | 25.00 |
| 53:01 | 10.71 | 12.50 | 12.50 |
| Other | 42.86 | 50.00 | 62.50 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-C (%) | |||
| 04:01 | 10.71 | 0.00 | 12.50 |
| 07:01 | 10.71 | 12.50 | 25.00 |
| Other | 42.86 | 62.50 | 62.50 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-DRB1 (%) | |||
| 03:01 | 10.71 | 12.50 | 50.00 |
| 07:01 | 10.71 | 25.00 | 0.00 |
| 13:01 | 10.71 | 0.00 | 0.00 |
| Other | 32.14 | 37.50 | 50.00 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-DQA1 (%) | |||
| 02:01 | 17.86 | 12.50 | 0.00 |
| 05:01 | 10.71 | 12.50 | 50.00 |
| Other | 35.71 | 50.00 | 50.00 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-DQB1 (%) | |||
| 02:01 | 17.86 | 12.50 | 50.00 |
| 02:02 | 10.71 | 25.00 | 0.00 |
| 03:01 | 10.71 | 0.00 | 25.00 |
| Other | 25.00 | 37.50 | 25.00 |
| Unknown | 35.71 | 25.00 | 0.00 |
| HLA-DPB1 (%) | |||
| 01:01 | 17.86 | 25.00 | 25.00 |
| 04:01 | 10.71 | 12.50 | 25.00 |
| Other | 35.71 | 37.50 | 50.00 |
| Unknown | 35.71 | 25.00 | 0.00 |
We evaluated the diversity of the TCR repertoire for MCD, FSGS, and MN cases during remission and during active disease using two metrics. First, we focused on basic overall clonotype population diversity metrics, including the number of templates, rearrangements, and productive Simpson’s clonality (Table 2). In paired analyses, none of these metrics were significantly different when active disease is compared with remission status for any of the three patient groups (Table 2). Overall clonotype population diversity metrics were similar across MCD, FSGS, and MN during active disease (ANOVA; p>0.05 for all comparisons). During remission, however, productive Simpson clonality was lower in MCD compared with FSGS and MN (ANOVA, p=0.0217; Supplementary Table 1).
Table 2. TCR repertoire diversity metrics for patients, both during active disease and during remission.
The table presents the observations of major diversity metrics for TCR repertoire computed for the clonotype population based on immunosequencing of minimal change disease (MCD; n=14), focal segmental glomerulosclerosis (FSGS; n=4), and membranous nephropathy (MN; n=4) patients, accompanied by the bioinformatic processing using the Adaptive Biotechnologies’ ANALYZER pipeline. Total templates refer to the sum of templates for all rearrangements, while productive templates only indicate the sum of templates for all productive rearrangements regardless of uniqueness that are capable of producing a functional peptide. Productive rearrangements represent the sum of unique productive rearrangements in the sample. Among these metrics, productive Simpson clonality is the primary measure of TCR diversity, which originated from the Simpson diversity metrics. Note that a lower value of the productive Simpson clonality represents a more diverse TCR repertoire while a higher value suggesting enriched oligoclonal. The values of productive clonality are rounded to four digits. Values are presented as median (range) unless otherwise indicated. P-values for pair-wise t-tests are given for comparisons between active disease and remission within cases of MCD, FSGS, and MN.
| MCD (n = 14) | FSGS (n = 4) | MN (n = 4) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Active Disease | Remission | p- value |
Active Disease | Remission | p- value |
Active Disease | Remission | p-value | |
| Total templates | 68,726 (11,084, 112,639) |
71,143 (21,095, 172,633) |
0.0991 | 112,159 (33,269, 212,885) |
96,621 (37,138, 151,382) |
0.7181 | 68,355 (40,949, 127,034) |
115,236 (81,551, 128,533) |
0.1029 |
| Productive templates | 52,640 (8,769, 90,134) |
57,247 (16,688, 137,486) |
0.0972 | 95,932 (26,684, 175,562) |
78,777 (31,879, 125,632) |
0.7050 | 53,637 (33,207, 108,010) |
94,498 (65,869, 101,891) |
0.1067 |
| Productive Rearrangements | 46,611 (8,186, 82,174) |
51,318 (13,844, 131,427) |
0.0939 | 76,769 (23,845, 159,348) |
61,699 (27,696, 114,720) |
0.7112 | 36,344 (27,984, 79,497) |
60,323 (46,451, 72,178) |
0.1472 |
| Productive Simpson Clonality | 0.0083 (0.0042, 0.0397) |
0.0088 (0.0038, 0.0369) |
0.7673 | 0.0122 (0.0049, 0.0736) |
0.0126 (0.0060, 0.0476) |
0.4736 | 0.0311 (0.0118, 0.1013) |
0.0389 (0.0133, 0.0960) |
0.8772 |
While lower productive Simpson clonality (higher TCR diversity) among MCD patients compared with FSGS and MN patients may be expected given the age differences in the patient population [34, 35], the absence of differences between active disease and remission samples was somewhat unexpected. A closer examination of productive Simpson clonality among MCD patients revealed outliers in both disease and remission samples (Fig. 1). None of the outliers can be explained by obvious patterns related to age or status of immunosuppressive therapy (data not shown). Stratification by immunosuppressive therapy status at the time of sampling did not reveal differences in median productive Simpson clonality (Supplementary Table 2).
Fig. 1. Productive Simpson clonality for minimal change disease patients during active disease and during remission (n = 14).
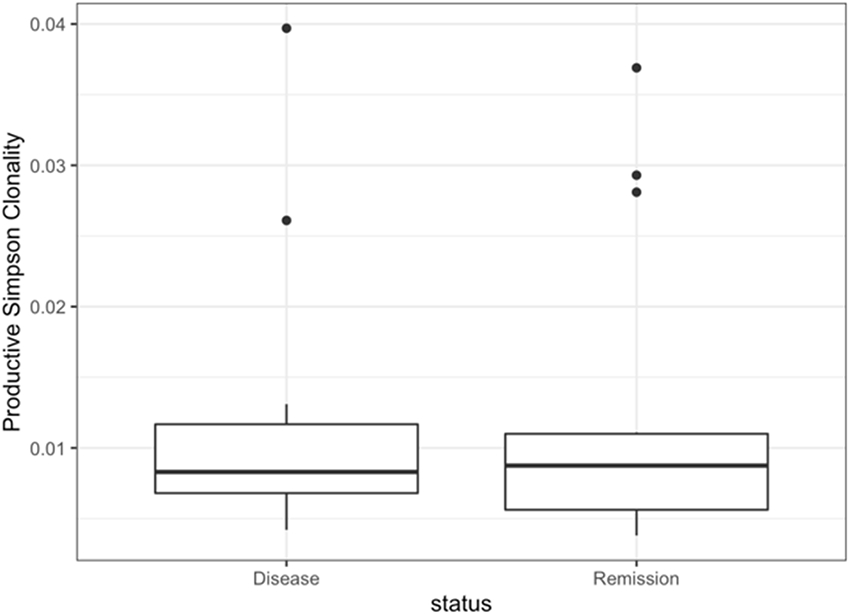
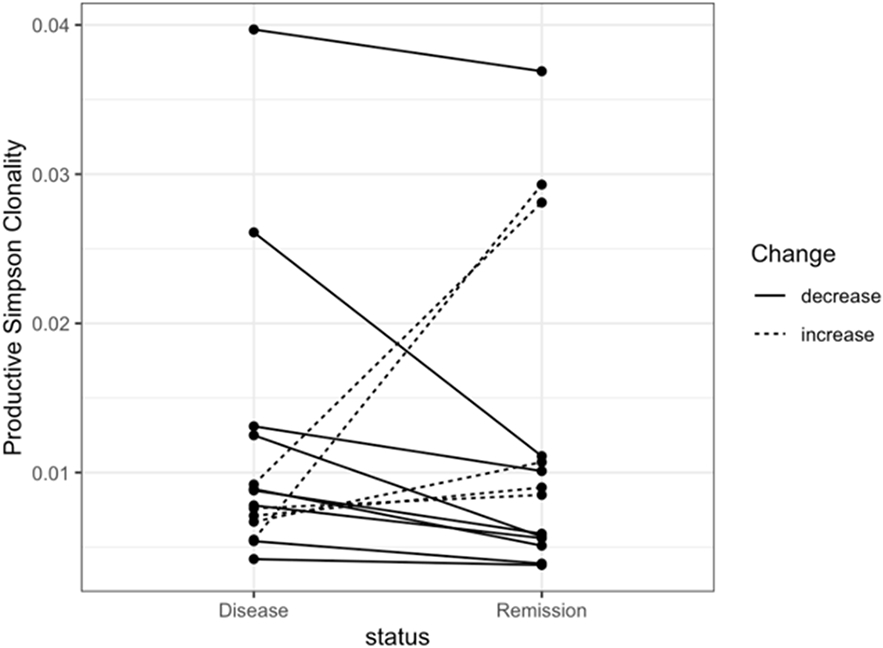
Fig. 1. a The box plot of productive Simpson clonality visualizes the comparison of the mean TCR diversity between the samples drawn from MCD patients during active disease and those during complete remission. Fig. 1. b The line plot of productive Simpson clonality shows changes of the TCR diversity between the sample drawn from MCD patients during active disease and the corresponding sample during complete remission from the same patient. Each dot represents a sample and the line links the samples from the same individual. A dotted line denotes an increase in productive Simpson clonality from active disease to remission for the paired sample, while a solid line denotes a decrease in productive Simpson clonality from active disease to remission for the paired sample.
Although there was no consistent change in the TCR repertoire diversity evenness stratified by disease status, we hypothesized that there may be greater similarity of clonotypes present in the setting of active disease compared with remission. To examine this possibility, we calculated pairwise TCR repertoire similarity using the Morisita-Horn index to describe intra-individual (active disease-remission pairs) and inter-individual (between individual MCD patients) clonotype diversity. We visualized this intra- and inter- individual clonotype similarity by constructing a dendrogram of the Morisita-Horn index for the MCD, FSGS, and MN patients both during active disease and during complete remission. From this visualization (Fig. 2), we observed that the paired samples from the same MCD, FSGS, and MN patients tend to cluster together, suggesting a higher similarity among clonotypes in each individual regardless of disease status as compared to the samples from different patients with the same disease regardless of remission or active disease status. Overall, relatively few clones (38,751) are shared between active disease (774,955) and remission (874,814) MCD patient clonotypes (Table 3) or between MCD-FSGS combined patient groups and MN clonotypes (data not shown).
Fig. 2. Hierarchical clustering of Morisita-Horn index for the MCD, FSGS, and MN patients both during active disease and during remission.

The dendrogram is constructed of the Morisita-Horn index, a metric to quantify the pairwise TCR similarity, for the 14 minimal change disease (MCD) patients, four focal segmental glomerulosclerosis (FSGS) patients, and four membranous nephropathy (MN) patients during active disease and during remission. Morisita-Horn index takes a value between 0 and 1, with the higher value indicating a more similar TCR repertoire.
Table 3. Shared clones comparing the joint samples from minimal change disease patients in active disease and remission.
The table shows the shared clones comparing samples from patients with minimal change disease (MCD; n=14) in active disease to those from MCD patients in complete remission. The clonotype table exported from the Adaptive Biotechnologies’ ANALYZER was employed by VDJtools to perform the analysis of clonotype sharing, providing detailed information regarding normalized count, normalized frequency, nucleotide sequence, amino acids sequence, and frequency at corresponding sample for the top 20 shared clones. Of the 774,955 and 874,814 clonotypes identified in the pooled sample of MCD patients in active disease and remission, respectively, only 38,751 clonotypes overlapped or were shared.
| Count* | Freq** | Representative CD3 Nucleotide Sequence | Representative CD3 Amino Acids Sequence |
Samples in Active Disease (MCD) |
Samples in Remission (MCD) |
|---|---|---|---|---|---|
| 770 | 0.00722454 | TGTGCCAGCTCACCAGAGGGATCGAACACTGAAGCTTTCTTT | CASSPEGSNTEAFF | 0.00149863 | 3.74E-04 |
| 579 | 0.00544107 | TGTGCCAGCACTCGTGCGGGAACAACTAATGAAAAACTGTTTTTT | CASTRAGTTNEKLFF | 6.59E-04 | 4.82E-04 |
| 479 | 0.00449917 | TGTGCCACCAGCAGCGGGAGCACCCAAGAGACCCAGTACTTC | CATSSGSTQETQYF | 0.00116243 | 1.87E-04 |
| 453 | 0.00425098 | TGTGCCAGCAGCCAAGCTCGGGGGATGAGCAGTACTTCG | CASSQARGMSSTS | 9.75E-04 | 1.99E-04 |
| 409 | 0.00384307 | TGTGCCAGCAGTTATCAGGGGATCGCTGAAGCTTTCTTT | CASSYQGIAEAFF | 5.08E-04 | 3.12E-04 |
| 374 | 0.00351007 | TGCGCCACCGGACAGGGCAATCAGCCCCAGCATTTT | CATGQGNQPQHF | 3.33E-04 | 3.97E-04 |
| 330 | 0.00310272 | TGTGCCAGCAGCTCCCCCCGGACAGGATCCCTCTCCTACGAGCAGTACTTC | CASSSPRTGSLSYEQYF | 2.73E-04 | 3.78E-04 |
| 313 | 0.00294084 | TGTGCCAGCAGTTTAGGAAGGGAGACCCAGTACTTC | CASSLGRETQYF | 8.97E-05 | 0.0010353 |
| 301 | 0.00282488 | TGTGCCAGCAGCAACCCACCTCCGGGACATTTACGAGCAGTACTT | CASSNPPPGHLRAVL | 2.24E-04 | 3.82E-04 |
| 283 | 0.00266365 | TGTGCCAGCAGCCAACGAGGACAGGGAAACTATGGCTACACCTTC | CASSQRGQGNYGYTF | 3.03E-04 | 2.52E-04 |
| 277 | 0.00260288 | TGTGCCAGCAGCTTGGGGGACTCCGCCTACGAGCAGTACTTC | CASSLGDSAYEQYF | 5.91E-04 | 1.23E-04 |
| 259 | 0.00243343 | TGCGCCAGCAGCTATCGCGACCAGCCCCAGCATTTT | CASSYRDQPQHF | 3.60E-04 | 1.77E-04 |
| 259 | 0.00243193 | TGTGCCAGCAGCTTTTATGTCGGGATAAGCTCCTACGAGCAGTACTTC | CASSFYVGISSYEQYF | 5.28E-04 | 1.20E-04 |
| 255 | 0.00239921 | TGTGCCAGCAGCTTAAAGTACAGGGGAAATAGTGCTCCACTTTGG | CASSLKYRGNSAPLW | 4.60E-04 | 1.34E-04 |
| 248 | 0.00233427 | TGTGCCAGCAGCTCACAGGGTTTAGACACCATATATTTT | CASSSQGLDTIYF | 4.48E-04 | 1.30E-04 |
| 243 | 0.00228337 | TGTGCCAGCAGTGGGACCGGCCAAGGGGGACTCTATGGCTACACCTTC | CASSGTGQGGLYGYTF | 1.98E-04 | 2.83E-04 |
| 241 | 0.00226165 | TGTGCCAGTAGTATCTGGGAAAGCGTAGGTCTCCGGGGCTACACCTTC | CASSIWESVGLRGYTF | 5.71E-04 | 9.62E-05 |
| 234 | 0.00220174 | TGTGCCAGCAGCAAAGGGGTTGAGCTCTGGAAACACCATATATTT | CASSKGVELWKHHIF | 8.46E-05 | 6.15E-04 |
| 227 | 0.00213146 | TGTGCCAGCAGTCTCGGGGCGGGGCAAGCCAAAAACATTCAGTACTTC | CASSLGAGQAKNIQYF | 4.32E-04 | 1.13E-04 |
| 222 | 0.0020891 | TGTGCCAGCAGTTACGCCGGGGCTTACGAGCAGTACTTC | CASSYAGAYEQYF | 5.01E-04 | 9.34E-05 |
Normalized clonotype count
Normalized clonotype frequency
Discussion
MCD has been described as a disease of T-cell dysfunction [13]. Although the cause of MCD is not yet known, several observations suggest the involvement of the adaptive immune system. Corticosteroids, which at high dose suppress T-cell function, are the first line effective treatments for patients with steroid sensitive nephrotic syndrome [36]. These observations evoked the hypothesis that the presence of a soluble factor suppresses T-cell proliferation, resulting in steroid sensitive nephrotic syndrome or MCD. While this soluble factor has yet to be identified, recent genome-wide association studies have further implicated the adaptive immune system with the identification of common MHC class II variants associated with steroid sensitive nephrotic syndrome [37-42].
Given the evidence for the involvement of T-cells in MCD pathogenesis, we sought to characterize the TCR repertoire during active disease and remission among participants diagnosed with MCD compared to individuals with FSGS, and MN. If MCD disease status were triggered by an environmental or soluble factor, we would expect that TCR sequences would demonstrate 1) greater diversity for MCD patients during remission as compared to the paired sample during active disease; and 2) shared TCR clones among the MCD patients during active disease, leading to a higher level of similarity. However, despite the evidence that T cell dysregulation and/or dysfunction are closely associated with MCD pathogenesis [43-46], we did not detect differences in TCR diversity between MCD patients in complete remission versus active disease. We also observed higher TCR repertoire similarity within the same individual regardless of nephrotic syndrome diagnosis or disease activity.
While the data presented here do not support a clonal T-cell response in active MCD, there are several notable limitations of the study. First, the sample size for each group is small, limiting statistical power. TCR diversity in healthy participants suggests metrics such as Simpson’s clonality can vary widely (for example, 0.002 to 0.44) [28] and is inversely correlated with age. Simpson’s clonality observed in this sample of MCD participants is very low and is within the range reported for healthy children. The per-person change in clonality between active disease and remission is small, although within the range reported for response to influenza vaccination [47]. Larger samples sizes will be necessary to detect evidence of clonal expansion for an MCD pediatric patient population given the low clonality values and small changes between states.
The small sample sizes also precluded adjustment for confounding by age, immunosuppressive treatment status, and variation in time between active disease and remission sampling. Increased age is associated with a contraction in the TCR repertoire diversity as expected [34, 35]. In addition, MCD is a heterogeneous condition, and the MCD patients had different statuses regarding the immunosuppression therapy during active remission and/or remission as well as differences in time between the two blood draws. Prior studies have shown immunosuppression can affect the TCR repertoire. Most of the MCD patients in the present study were on immunosuppressive therapy both during active disease and remission (Supplementary Table 2), making it difficult to characterize the effects, if any, of therapy on the TCR repertoires observed here. Larger sample sizes are required for stratified adjusted analyses that incorporate potentially important variables such as age as well as immunosuppressive therapy.
Similarly, we were unable to stratify TCR diversity metrics by demographics or HLA status. WGS data were only available for a subset of the patients with TCR sequences. When comparing the inferred HLA alleles across MCD, FSGS, and MN, we did not observe specific patterns nor dominant alleles. In general, the confidence for inferring HLA alleles from the WGS data for African Americans in this study was lower across all three nephrotic syndrome groups, presumably due to the increased nucleotide diversity in this population compared with European-descent populations. Also, the samples used to construct the African American model are limited as compared to that for the European Americans: the former was built on the HLARES data from GlaxoSmithKline clinical trials of African ancestry (n = 173) and African Americans of HapMap Phase 2 (n = 60) while the latter was generated from HLARES data from GlaxoSmithKline clinical trials data of European ancestry (n = 2,668) [31], which would further affect the confidence of the inferring process.
Our inability to further examine the MHC region, alone and combined with TCR clones in the present study, may have contributed to the lack of differences observed between paired active disease and remission samples. The initiation and regulation of the adaptive immune responses are related to the binding between TCRs and antigens presented by MHC molecules [48]. The MHC region, which is mapped to the short arm of chromosome 6, harbors the HLA genes in humans. The genes in the MHC regions encode the cell surface proteins, the MHC molecules, which are important for the adaptive immune responses and are involved in multiple human diseases, such as autoimmune diseases [49-51] and infectious disease [52]. Notably, the adaptive immune system is highly variable as a tailored response to the diversity of pathogens or antigens encountered by humans. The ability for the active immune system to diversify to fight different pathogens could be attributed to a diverse TCR repertoire and also related to the germline variations within the MHC.
It is possible that TCR sequence differences between active disease and remission groups and/or across MCD, FSGS, and MN exist but were not detected in this study. Rare clones may have been missed or undercounted, and this along with expected stochastic sampling bias and experimental error may have impacted the observed diversity metrics. We employed a strict exact match comparison of TCR amino acid sequences, which resulted in a small number of identified shared clonotypes. Also, we immunosequenced DNA extracted from whole blood; future studies may benefit from T-cell sorting prior to the immunosequencing to better differentiate the types of T cells that may be involved in MCD pathogenesis.
The heterogeneity of nephrotic syndrome and its low frequency in the general population make the study of MCD and other causes of nephrotic syndrome difficult. Definitive diagnosis of nephrotic syndrome is invasive, compounding the challenges to amassing sample sizes sufficient for study. Here we leveraged NEPTUNE, one of the largest ongoing cohort studies of nephrotic syndrome with biospecimes and genomic data available for study. While NEPTUNE has one of the largest sample sizes from which to draw, study designs such as the one described here that require repeated biospecimen collection during the clinical course of the disease further restrict the already limited sample size. Further, the protocol used for immunosequencing requires micrograms of DNA, a large amount that precluded inclusion of participants who met the study criteria clinically but who did not have sufficient amounts of DNA extracted from both blood draws. Larger samples made possible by consortia such as NEPTUNE as well as advancements in immunosequencing technologies are needed to assess the adaptive immune response in patients with nephrotic syndrome.
Supplementary Material
Acknowledgement
We thank the participants and their families without whom this study would not be possible. The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services; nor does mention by trade names, commercial practices, or organizations imply endorsement by the U.S. Government. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Members of the Nephrotic Syndrome Study Network (NEPTUNE)
NEPTUNE Enrolling Centers
Cleveland Clinic, Cleveland, OH: K Dell*, J Sedor**, M Schachere#, J Negrey#
Children’s Hospital, Los Angeles, CA: K Lemley*, S Tang#
Children’s Mercy Hospital, Kansas City, MO: T Srivastava*, S Morrison#
Cohen Children’s Hospital, New Hyde Park, NY: C Sethna*, M Pfaiff#
Columbia University, New York, NY: P Canetta*, A Pradhan#
Emory University, Atlanta, GA: L Greenbaum*, C Wang**, E Yun#
Harbor-University of California Los Angeles Medical Center: S Adler*, J LaPage#
John H. Stroger Jr. Hospital of Cook County, Chicago, IL: A Athavale*, M Itteera
Johns Hopkins Medicine, Baltimore, MD: M Atkinson*, T Dell#
Mayo Clinic, Rochester, MN: F Fervenza*, M Hogan**, J Lieske*, V Chernitskiy#
Montefiore Medical Center, Bronx, NY: F Kaskel*, M Ross*, P Flynn#
NIDDK Intramural, Bethesda MD: J Kopp*, J Blake#
New York University Medical Center, New York, NY: L Malaga-Dieguez*, O Zhdanova**, F Modersitzki#, S Vento#
Stanford University, Stanford, CA: R Lafayette*, B Yeung#
Temple University, Philadelphia, PA: I Lee*, S Quinn-Boyle#
University Health Network Toronto: H Reich *, M Hladunewich**, P Ling#, M Romano#
University of Miami, Miami, FL: A Fornoni*, C Bidot#
University of Michigan, Ann Arbor, MI: M Kretzler*, D Gipson*, A Williams#, C Klida#
University of North Carolina, Chapel Hill, NC: V Derebail*, K Gibson*, A Froment#, J Ormond-Foster#
University of Pennsylvania, Philadelphia, PA: L Holzman*, K Meyers**, K Kallem#, A Swenson#
University of Texas Southwestern, Dallas, TX: K Sambandam*, Z Wang#, M Rogers#
University of Washington, Seattle, WA: A Jefferson*, S Hingorani**, K Tuttle**§, M Bray #, E Pao#, A Cooper#§
Wake Forest University Baptist Health, Winston-Salem, NC: JJ Lin*, Stefanie Baker#
Data Analysis and Coordinating Center: M Kretzler*, L Barisoni**, G Alter, H Desmond, S Eddy, D Fermin, C Gadegbeku**, B Gillespie**, D Gipson**, L Holzman**, V Kurtz, M Larkina, S Li, S Li, CC Lienczewski, J Liu, T Mainieri, L Mariani**, M Sampson**, J Sedor**, A Smith, A Williams, J Zee.
Digital Pathology Committee: Carmen Avila-Casado (University Health Network, Toronto), Serena Bagnasco (Johns Hopkins University), Joseph Gaut (Washington University in St Louis), Stephen Hewitt (National Cancer Institute), Jeff Hodgin (University of Michigan), Kevin Lemley (Children’s Hospital of Los Angeles), Laura Mariani (University of Michigan), Matthew Palmer (University of Pennsylvania), Avi Rosenberg (Johns Hopkins University), Virginie Royal (University of Montreal), David Thomas (University of Miami), Jarcy Zee (University of Pennsylvania) Co-Chairs: Laura Barisoni (Duke University) and Cynthia Nast (Cedar Sinai).
*Principal Investigator; **Co-investigator; #Study Coordinator
§Providence Medical Research Center, Spokane, WA
Funding Sources
The Nephrotic Syndrome Rare Disease Clinical Research Network III (NEPTUNE) is part of the Rare Diseases Clinical Research Network (RDCRN), funded by the National Institutes of Health (NIH) and led by the National Center for Advancing Translational Sciences (NCATS) through its Office of Rare Diseases Research (ORDR). NEPTUNE (U54DK083912) is funded under a collaboration between NCATS and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Additional funding and/or programmatic support is provided by the University of Michigan, NephCure Kidney International and the Halpin Foundation. All RDCRN consortia are supported by the RDCRN Data Management and Coordinating Center (DMCC) (U2CTR002818). Funding support for the DMCC is provided by NCATS and the National Institute of Neurological Disorders and Stroke (NINDS). Additional funding and/or programmatic support for this project has also been provided by the University of Michigan, the NephCure Kidney International and the Halpin Foundation. This publication was made possible by R01 DK108329, R01 DK097836, and the Clinical and Translational Science Collaborative of Cleveland, UL1TR0002548 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research.
Footnotes
Statement of Ethics
This ancillary study was approved by the NEPTUNE Ancillary Study Committee and Steering Committee as well as local Institutional Review Boards. Written informed consent was obtained for adult participants. For pediatric patients, parents provided written consent and, when appropriate, assent was obtained.
Conflict of Interest Statement
J.R.S. has received research funding for randomized clinical trials in nephrotic syndrome from Goldfinch, Novartis, and Calliditas. He has also consulted for Maze, Goldfinch, Boehringer Ingelheim in the least three years and receives royalties from Sanofi for transgenic mice. The other authors have no conflicts of interest to declare.
Data Availability Statement
The underlying data are available through the NEPTUNE Study by ancillary study application.
References
- 1.Niaudet P, Boyer O (2009) Idiopathic Nephrotic Syndrome in Children: Clinical Aspects. In: Avner E, Harmon W, Niaudet P, Yoshikawa N (eds) Pediatric Nephrology: Sixth Completely Revised, Updated and Enlarged Edition. Springer Berlin Heidelberg, Berlin, Heidelberg, pp 667–702 [Google Scholar]
- 2.Mak SK, Short CD, Mallick NP (1996) Long-term outcome of adult-onset minimal-change nephropathy. Nephrol Dial Transplant 11:2192–2201 [DOI] [PubMed] [Google Scholar]
- 3.Eddy AA, Symons JM (2003) Nephrotic syndrome in childhood. The Lancet 362:629–639 [DOI] [PubMed] [Google Scholar]
- 4.Garin EH, Laflam PF, Muffly K (2006) Proteinuria and fusion of podocyte foot processes in rats after infusion of cytokine from patients with idiopathic minimal lesion nephrotic syndrome. Nephron Exp Nephrol 102:e105–112 [DOI] [PubMed] [Google Scholar]
- 5.Farquhar MG, Vernier RL, Good RA (1957) An electron microscope study of the glomerulus in nephrosis, glomerulonephritis, and lupus erythematosus. J Exp Med 106:649–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIntyre P, Craig JC (1998) Prevention of serious bacterial infection in children with nephrotic syndrome. Journal of paediatrics and child health 34:314–317 [DOI] [PubMed] [Google Scholar]
- 7.Huang JJ, Hsu SC, Chen FF, Sung JM, Tseng CC, Wang MC (2001) Adult-Onset Minimal Change Disease among Taiwanese. American Journal of Nephrology 21:28–34 [DOI] [PubMed] [Google Scholar]
- 8.Hoyer PF, Gonda S, Barthels M, Krohn HP, Brodehl J (1986) Thromboembolic complications in children with nephrotic syndrome. Risk and incidence. Acta paediatrica Scandinavica 75:804–810 [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishnan J, Appel AS, Valeri A, Appel GB (1993) The nephrotic syndrome, lipids, and risk factors for cardiovascular disease. Am J Kidney Dis 22:135–142 [DOI] [PubMed] [Google Scholar]
- 10.Ashoor IF, Mansfield SA, O’Shaughnessy MM, Parekh RS, Zee J, Vasylyeva TL, Kogon AJ, Sethna CB, Glenn DA, Chishti AS, Weaver DJ, Helmuth ME, Fernandez HE, Rheault MN (2019) Prevalence of Cardiovascular Disease Risk Factors in Childhood Glomerular Diseases. J Am Heart Assoc 8:e012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kyrieleis HA, Löwik MM, Pronk I, Cruysberg HR, Kremer JA, Oyen WJ, van den Heuvel BL, Wetzels JF, Levtchenko EN (2009) Long-term outcome of biopsy-proven, frequently relapsing minimal-change nephrotic syndrome in children. Clin J Am Soc Nephrol 4:1593–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fenton A, Smith SW, Hewins P (2018) Adult minimal-change disease: observational data from a UK centre on patient characteristics, therapies, and outcomes. BMC Nephrol 19:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shalhoub R (1974) Pathogenesis of lipoid nephrosis: a disorder of T-cell function. The Lancet 304:556–560 [DOI] [PubMed] [Google Scholar]
- 14.Luetscher JA Jr., Deming QB, Harvey J, Lew W, Poo LJ (1950) Treatment of nephrosis with cortisone. The Journal of Clinical Investigation 29:1576–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn D, Hodson EM, Willis NS, Craig JC (2015) Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database of Systematic Reviews [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fakhouri F, Bocquet N, Taupin P, Presne C, Gagnadoux MF, Landais P, Lesavre P, Chauveau D, Knebelmann B, Broyer M, Grünfeld JP, Niaudet P (2003) Steroid-sensitive nephrotic syndrome: from childhood to adulthood. Am J Kidney Dis 41:550–557 [DOI] [PubMed] [Google Scholar]
- 17.Blumberg RW, Cassady HA (1947) Effect of measles on the nephrotic syndrome. Am J Dis Child 73:151–166 [DOI] [PubMed] [Google Scholar]
- 18.Hutchins G, Janeway CA (1947) Observations on the relationship of measles and remissions in the nephrotic syndrome. Am J Dis Child 73:242. [PubMed] [Google Scholar]
- 19.Rosenblum AH, Lander HB, Fisher RM (1949) Measles in the nephrotic syndrome. The Journal of Pediatrics 35:574–584 [DOI] [PubMed] [Google Scholar]
- 20.Meizlik EH, Carpenter AM (1948) Beneficial effect of measles on nephrosis; report of three cases. Am J Dis Child 76:83–90 [DOI] [PubMed] [Google Scholar]
- 21.Kerdiles YM, Sellin CI, Druelle J, Horvat B (2006) Immunosuppression caused by measles virus: role of viral proteins. Reviews in medical virology 16:49–63 [DOI] [PubMed] [Google Scholar]
- 22.Gadegbeku CA, Gipson DS, Holzman LB, Ojo AO, Song PXK, Barisoni L, Sampson MG, Kopp JB, Lemley KV, Nelson PJ, Lienczewski CC, Adler SG, Appel GB, Cattran DC, Choi MJ, Contreras G, Dell KM, Fervenza FC, Gibson KL, Greenbaum LA, Hernandez JD, Hewitt SM, Hingorani SR, Hladunewich M, Hogan MC, Hogan SL, Kaskel FJ, Lieske JC, Meyers KEC, Nachman PH, Nast CC, Neu AM, Reich HN, Sedor JR, Sethna CB, Trachtman H, Tuttle KR, Zhdanova O, Zilleruelo GE, Kretzler M (2013) Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney International 83:749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orth SR, Ritz E (1998) The nephrotic syndrome. N Engl J Med 338:1202–1211 [DOI] [PubMed] [Google Scholar]
- 24.Hull RP, Goldsmith DJA (2008) Nephrotic syndrome in adults. BMJ 336:1185–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gipson DS, Troost JP, Lafayette RA, Hladunewich MA, Trachtman H, Gadegbeku CA, Sedor JR, Holzman LB, Moxey-Mims MM, Perumal K, Kaskel FJ, Nelson PJ, Tuttle KR, Bagnasco SM, Hogan MC, Dell KM, Appel GB, Lieske JC, Ilori TO, Sethna CB, Fervenza FC, Hogan SL, Nachman PH, Rosenberg AZ, Greenbaum LA, Meyers KEC, Hewitt SM, Choi MJ, Kopp JB, Zhdanova O, Hodgin JB, Johnstone DB, Adler SG, Avila-Casado C, Neu AM, Hingorani SR, Lemley KV, Nast CC, Brady TM, Barisoni-Thomas L, Fornoni A, Jennette JC, Cattran DC, Palmer MB, Gibson KL, Reich HN, Mokrzycki MH, Sambandam KK, Zilleruelo GE, Licht C, Sampson MG, Song P, Mariani LH, Kretzler M (2016) Complete Remission in the Nephrotic Syndrome Study Network. Clinical Journal of the American Society of Nephrology 11:81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung M-W, Parsons JM, Steen MS, LaMadrid-Herrmannsfeldt MA, Williamson DW, Livingston RJ, Wu D, Wood BL, Rieder MJ, Robins H (2013) Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun 4 [DOI] [PubMed] [Google Scholar]
- 27.Simpson EH (1949) Measurement of Diversity. Nature 163:688–688 [Google Scholar]
- 28.Emerson RO, DeWitt WS, Vignali M, Gravley J, Hu JK, Osborne EJ, Desmarais C, Klinger M, Carlson CS, Hansen JA, Rieder M, Robins HS (2017) Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nature Genetics 49:659–665 [DOI] [PubMed] [Google Scholar]
- 29.Horn HS (1966) Measurement of “Overlap” in Comparative Ecological Studies. The American Naturalist 100:419–424 [Google Scholar]
- 30.Zheng X (2018) Imputation-Based HLA Typing with SNPs in GWAS Studies. In: Boegel S (ed) HLA Typing: Methods and Protocols. Springer New York, New York, NY, pp 163–176 [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, Weir BS (2014) HIBAG—HLA genotype imputation with attribute bagging. The Pharmacogenomics Journal 14:192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, Kirgizov KI, Skorobogatova EV, Chudakov DM (2015) VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLOS Computational Biology 11:e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E (2005) The influence of age on T cell generation and TCR diversity. J Immunol 174 [DOI] [PubMed] [Google Scholar]
- 35.Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY (2014) Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A 111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodson EM, Willis NS, Craig JC (2007) Corticosteroid therapy for nephrotic syndrome in children. The Cochrane database of systematic reviews:Cd001533. [DOI] [PubMed] [Google Scholar]
- 37.Sanchez-Rodriguez E, Southard CT, Kiryluk K (2021) GWAS-Based Discoveries in IgA Nephropathy, Membranous Nephropathy, and Steroid-Sensitive Nephrotic Syndrome. Clinical Journal of the American Society of Nephrology 16:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debiec H, Dossier C, Letouzé E, Gillies CE, Vivarelli M, Putler RK, Ars E, Jacqz-Aigrain E, Elie V, Colucci M, Debette S, Amouyel P, Elalaoui SC, Sefiani A, Dubois V, Simon T, Kretzler M, Ballarin J, Emma F, Sampson MG, Deschênes G, Ronco P (2018) Transethnic, Genome-Wide Analysis Reveals Immune-Related Risk Alleles and Phenotypic Correlates in Pediatric Steroid-Sensitive Nephrotic Syndrome. Journal of the American Society of Nephrology 29:2000–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, Voinescu C, Patel N, Pearce K, Hubank M, Stephens HA, Laundy V, Padmanabhan S, Zawadzka A, Hofstra JM, Coenen MJ, den Heijer M, Kiemeney LA, Bacq-Daian D, Stengel B, Powis SH, Brenchley P, Feehally J, Rees AJ, Debiec H, Wetzels JF, Ronco P, Mathieson PW, Kleta R (2011) Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 364:616–626 [DOI] [PubMed] [Google Scholar]
- 40.Jia X, Horinouchi T, Hitomi Y, Shono A, Khor S-S, Omae Y, Kojima K, Kawai Y, Nagasaki M, Kaku Y, Okamoto T, Ohwada Y, Ohta K, Okuda Y, Fujimaru R, Hatae K, Kumagai N, Sawanobori E, Nakazato H, Ohtsuka Y, Nakanishi K, Shima Y, Tanaka R, Ashida A, Kamei K, Ishikura K, Nozu K, Tokunaga K, Iijima K, Japan ftRCoGoCINSi (2018) Strong Association of the HLA-DR/DQ Locus with Childhood Steroid-Sensitive Nephrotic Syndrome in the Japanese Population. Journal of the American Society of Nephrology 29:2189–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dufek S, Cheshire C, Levine AP, Trompeter RS, Issler N, Stubbs M, Mozere M, Gupta S, Klootwijk E, Patel V, Hothi D, Waters A, Webb H, Tullus K, Jenkins L, Godinho L, Levtchenko E, Wetzels J, Knoers N, Teeninga N, Nauta J, Shalaby M, Eldesoky S, Kari JA, Thalgahagoda S, Ranawaka R, Abeyagunawardena A, Adeyemo A, Kristiansen M, Gbadegesin R, Webb NJ, Gale DP, Stanescu HC, Kleta R, Bockenhauer D (2019) Genetic Identification of Two Novel Loci Associated with Steroid-Sensitive Nephrotic Syndrome. Journal of the American Society of Nephrology 30:1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia X, Yamamura T, Gbadegesin R, McNulty MT, Song K, Nagano C, Hitomi Y, Lee D, Aiba Y, Khor S-S, Ueno K, Kawai Y, Nagasaki M, Noiri E, Horinouchi T, Kaito H, Hamada R, Okamoto T, Kamei K, Kaku Y, Fujimaru R, Tanaka R, Shima Y, Araki Y, Nagaoka Y, Okamoto T, Sato Y, Hayashi A, Takahashi T, Aoyagi H, Ueno M, Nakanishi M, Toita N, Uetake K, Kobayashi N, Fujita S, Tsuruga K, Kumagai N, Kudo H, Tanaka E, Omori T, Okada M, Hatai Y, Udagawa T, Motoyoshi Y, Ishikura K, Kamei K, Ogura M, Sato M, Kano Y, Hattori M, Miura K, Harita Y, Kanda S, Sawanobori E, Kobayashi A, Kojika M, Ohwada Y, Yan K, Hataya H, Hamada R, Terano C, Harada R, Hamasaki Y, Hashimoto J, Ito S, Machida H, Inaba A, Matsuyama T, Goto M, Shimizu M, Ohta K, Ikezumi Y, Yamada T, Suzuki T, Tamamura S, Mori Y, Hidaka Y, Matsuoka D, Kinoshita T, Noda S, Kitahara M, Fujita N, Hibino S, Iijima K, Nozu K, Kaito H, Minamikawa S, Yamamura T, Nagano C, Horinouchi T, Nakanishi K, Fujimura J, Sakakibara N, Aoto Y, Ishiko S, Tanaka R, Kanda K, Inaguma Y, Hashimura Y, Ishimori S, Kamiyoshi N, Shibano T, Takeshima Y, Fujimaru R, Ueda H, Ashida A, Matsumura H, Kubota T, Kitaoka T, Okuda Y, Sawai T, Sakai T, Shima Y, Hama T, Fujieda M, Ishihara M, Itoh S, Iwaki T, Shimizu M, Nagatani K, Kagami S, Urushihara M, Kaku Y, Nishimura M, Yoshino M, Hatae K, Hinokiyama M, Kuroki R, Ohtsuka Y, Oka M, Nishimura S, Sato T, Tanaka S, Zaitsu A, Nakazato H, Tamura H, Nakanishi K, Baek J, Kang HG, Ha I-S, Han KH, Yang EM, Cho MH, Ha T-S, Cheong HI, Kang HG, Ha I-S, Kim JH, Park PG, Cho MH, Han KH, Yang EM, Abeyagunawardena A, Lane B, Chryst-Stangl M, Esezobor C, Solarin A, Quiroga A, Moudgil A, Chavers B, Kwon C, Bowers C, Gipson D, Chand D, Weaver DJ, Abraham E, Janjua H, Lin J-J, Greenbaum L, Kallash M, Rheault M, De Jeus Gonzalez N, Brophy P, Gbadegesin R, Nagaraj S, Massengill S, Srivastava T, Hunley T, Cai Y, Omoloja A, Silva C, Adeyemo A, Thalgahagoda S, Kari JA, El Desoky S, Dossier C, Deschênes G, Abdelhadi M, Akil R, Azib S, Basmaci R, Benoist G, Bensaid P, Blanc P, Boyer O, Bucher J, Chace A, Chalvon A, Cheminee M, Chendjou S, Daoud P, Deschênes G, Dossier C, Elias O, Gagliadone C, Gajdos V, Galerne A, Aigrain EJ, Sanchez LJ, Khaled M, Khelfaoui F, Laoudi Y, Larakeb A, Limani T, Mahdi F, Mandelcwaijg A, Muller S, Nacer K, Nathanson S, Pellegrino B, Pharaon I, Roudault V, Rouget S, Saf M, Simon T, Tahiri C, Ulinski T, Zenkhri F, Vivarelli M, Debiec H, Ishikura K, Matsuo M, Nozu K, Ronco P, Cheong HI, Sampson MG, Tokunaga K, Iijima K (2020) Common risk variants in NPHS1 and TNFSF15 are associated with childhood steroid-sensitive nephrotic syndrome. Kidney International 98:1308–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunard R, Kelly CJ (2002) T cells and minimal change disease. J Am Soc Nephrol 13:1409–1411 [DOI] [PubMed] [Google Scholar]
- 44.van den Berg JG, Weening JJ (2004) Role of the immune system in the pathogenesis of idiopathic nephrotic syndrome. Clinical science (London, England : 1979) 107:125–136 [DOI] [PubMed] [Google Scholar]
- 45.Zhang SY, Audard V, Fan Q, Pawlak A, Lang P, Sahali D (2011) Immunopathogenesis of idiopathic nephrotic syndrome. Contributions to nephrology 169:94–106 [DOI] [PubMed] [Google Scholar]
- 46.Elie V, Fakhoury M, Deschênes G, Jacqz-Aigrain E (2012) Physiopathology of idiopathic nephrotic syndrome: lessons from glucocorticoids and epigenetic perspectives. Pediatr Nephrol 27:1249–1256 [DOI] [PubMed] [Google Scholar]
- 47.Herati RS, Muselman A, Vella L, Bengsch B, Parkhouse K, Del Alcazar D, Kotzin J, Doyle SA, Tebas P, Hensley SE, Su LF, Schmader KE, Wherry EJ (2017) Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Science immunology 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janeway CA, Travers P, Walport M, Shlomchik MJ (2001) The major histocompatibility complex and its functions. Immunobiology: the immune system in health and disease. Garland Science, New York [Google Scholar]
- 49.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD (2008) Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet 4:e1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones EY, Fugger L, Strominger JL, Siebold C (2006) MHC class II proteins and disease: a structural perspective. Nat Rev Immunol 6:271–282 [DOI] [PubMed] [Google Scholar]
- 51.Rioux JD, Goyette P, Vyse TJ, Hammarström L, Fernando MM, Green T, De Jager PL, Foisy S, Wang J, de Bakker PI, Leslie S, McVean G, Padyukov L, Alfredsson L, Annese V, Hafler DA, Pan-Hammarström Q, Matell R, Sawcer SJ, Compston AD, Cree BA, Mirel DB, Daly MJ, Behrens TW, Klareskog L, Gregersen PK, Oksenberg JR, Hauser SL (2009) Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc Natl Acad Sci U S A 106:18680–18685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shiina T, Hosomichi K, Inoko H, Kulski JK (2009) The HLA genomic loci map: expression, interaction, diversity and disease. Journal of Human Genetics 54:15–39 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The underlying data are available through the NEPTUNE Study by ancillary study application.