

Abstract
Significant progress has been achieved in the treatment of metastatic colorectal cancer (mCRC) patients during the last 20 years. There are currently numerous treatments available for the first-line treatment of mCRC. Sophisticated molecular technologies have been developed to reveal novel prognostic and predictive biomarkers for CRC. The development of next-generation sequencing and whole-exome sequencing, which are strong new tools for the discovery of predictive molecular biomarkers to facilitate the delivery of customized treatment, has resulted in tremendous breakthroughs in DNA sequencing technology in recent years. The appropriate adjuvant treatments for mCRC patients are determined by the tumor stage, presence of high-risk pathologic characteristics, microsatellite instability status, patient age, and performance status. Chemotherapy, targeted therapy, and immunotherapy are the main systemic treatments for patients with mCRC. Despite the fact that these novel treatment choices have increased overall survival for mCRC, survival remains optimal for individuals with non-metastatic disease. The molecular technologies currently being used to support our ability to practice personalized medicine; the practical aspects of applying molecular biomarkers to regular clinical practice; and the evolution of chemotherapy, targeted therapy, and immunotherapy strategies for the treatment of mCRC in the front-line setting are all reviewed here.
Keywords: Systemic treatment, Metastatic colorectal cancer, Personalized medicine, Biomarkers, Chemotherapy, Targeted therapy, Immunotherapy
Core Tip: Advances in the molecular profiling of metastatic colorectal cancer allow treatment to be tailored to the biologic characteristics of the tumor for certain patient subgroups. Although cures are still rare, more people can expect to live longer. Genomic profiling enables therapy selection, allowing more individuals to benefit while exposing fewer to the harm of ineffective medicines. An important component in determining treatment results is the choice of an effective first-line therapy, which should consider both clinical considerations and molecular indicators. The systemic treatments used in the first-line regimen determine the second-line regimen. Third-line therapy, which includes epithelial growth factor receptor inhibitors for patients with rat sarcoma virus wild-type, should consider molecular profiling. Patients with high microsatellite instability illnesses may be candidates for immunotherapy with pembrolizumab or nivolumab plus ipilimumab.
INTRODUCTION
The third most commonly occurring cancer in humans is colorectal cancer (CRC). More than 2 million individuals are diagnosed with this cancer each year worldwide. This year, about one million individuals will die from CRC. The liver is the most common target of CRC hematogenous metastasis, as well as the site most responsible for death from this common malignancy. When patients are diagnosed in the late stage of disease, their prognosis remains poor[1,2]. The majority of individuals who have a CRC diagnosis are over 50, whereas only 12% of all new CRC diagnoses are found in those under 50. Overall, the lifetime risk of developing CRC is about 1 in 23 (4.3%) for men and 1 in 25 (4.0%) for women. According to statistics from the Surveillance, Epidemiology, and End Results Program, at the time of diagnosis, 38% of patients have localized disease, 35% have regional disease, 21% have distant disease, and 6% have no stage[3]. CRC diagnoses have decreased overall since 2000 as a result of increased screening efforts, although it has increased in young people under 50 since the 1990s. Preventive measures, such as routine colonoscopies, remain the most effective way to combat CRC. With a rising interest in non-invasive biomarkers, many additional approaches have been developed. However, nearly 50% of patients are still diagnosed at an advanced stage. Metastatic CRC (mCRC) has a poor prognosis with a 5-year survival rate of 14%, and this number has remained constant over the past 5 years. Until the late twentieth century, mCRC was thought to always be fatal. The discovery that metastatic cancer is mostly localized to the liver in postmortem investigations and radiologic examinations utilizing computer-assisted tomography scanners led early pioneers to resect liver metastasis. To establish liver resections for metastatic carcinoma as an acceptable treatment, pioneers conducted and published data[4]. Only one standard regimen to manage CRC has been shown to be inefficient, resulting in high rates of treatment failure and disease resistance. Recently, the clinical outcomes of patients with mCRC have improved dramatically as a result of the discovery of prognostic and predictive molecular biomarkers and the subsequent individualization of treatment options. High genetic heterogeneity, including but not limited to chromosomal instability (CIN), microsatellite instability (MSI), and the CpG island methylator phenotype (CIMP), has been identified by molecular profiling of CRC. Different CRC subtypes have varying prognoses and therapeutic outcomes. Promising improvements for the use of systemic therapy and precision medicine in mCRC have resulted from recent advancements in our understanding of the molecular signaling networks that control intestinal regeneration and homeostasis[5].
The discovery of the key molecular drivers in CRC pathogenesis, coupled with the ability to screen tissue for measurable mutations crucial to disease progression, led to the development of an innovative treatment model for patients with advanced CRC. Despite substantial breakthroughs in tumor biology understanding, these have not entirely translated into proven novel therapies for all patients, since the chemotherapeutic strategy is still built around combination cytotoxic regimens targeted at proliferative epithelium. Although there have been some achievements with targeted therapy, such as the synergistic effect of protein kinase B-Raf (BRAF) inhibitors and epithelial growth factor receptor (EGFR) inhibitors in BRAF-mutant CRC, certain medications have not been able to offer clinically meaningful improvements for other pathways. As evidenced by the effect of immune checkpoint inhibition in microsatellite unstable CRC, therapeutic exploitation of intercompartmental signaling in the malignant epithelium may represent an important new drug paradigm in CRC. This is because we are becoming more aware of the signaling crosstalk that controls intestinal cell fate in health and disease, as well as the function of the tumor microenvironment (TME)[6,7].
We have split these important signaling pathways into those that control the destiny of cancer cells or intestinal epithelium directly and those that function indirectly by leveraging the TME in this review. Additionally, we explore how signaling affects each cancer cell’s destiny and comment on some possible treatment prospects that result from effective pathway modulation for each.
RISK FACTORS FOR CRC
Behaviors, diets, and lifestyle
Only a small proportion of CRCs are associated with germline mutations or discovered in the presence of a strong family history; the majority of CRCs are random[8,9]. The significance of environmental exposure has been further demonstrated by the variation in CRC risks throughout the world and the discovery that younger generations are at a higher risk of CRC in westernized nations. Numerous studies have been conducted in an effort to identify and quantify the environmental and dietary risk factors for CRC. Global studies have shown a 45-fold variation in the age-standardized incidence of CRC worldwide[10]. Gambia and other non-industrialized nations have the lowest rates of CRC, whereas westernized nations have the highest rates. The prevalence of CRC has been seen to rise over time when a nation industrializes and starts to follow a westernized lifestyle and diets low in fiber[11,12]. A westernized diet, or one that is low in fiber, fruits, and vegetables and heavy in processed meats, sugary drinks, and refined grains, is linked to greater risks of CRC. It has been challenging to pinpoint everything that increases the risk of CRC in a westernized diet. This diet’s many components are probably a factor in the greater prevalence of CRC. Studies have repeatedly shown that diets rich in processed foods and red meat are linked to higher risks of CRC. For every 100 g of red and processed meat consumed, CRC incidence increases by 12%, according to a recent meta-analysis of 111 studies involving 400 individuals[13] (Table 1). Smoking increases the likelihood of both serrated polyps and colorectal adenomas[14]. According to large observational studies, more pack-years result in higher CRC rates[15]. Recent research suggests that smoking is marginally related to MSI-high (MSI-H) tumors, increases the rates of rectal and proximal CRC, and is correlated with BRAF-mutant malignancies. Similar to smoking, drinking alcohol is a recognized risk factor for CRC, and recent pooled studies have demonstrated that even occasional drinking increases the risk of CRC[16].
Table 1.
Modifiable and non-modifiable risk factors and preventive factors for colorectal cancer
| Modifiable | Non-modifiable | |
| Risk factors | Excess body fat | Family history of CRC |
| Sedentary life-style | Advanced polyps | |
| Westernized diet | Polyposis syndrome (FAP, MAP) | |
| Processed meats | Lynch syndrome (HNPCC) | |
| Red meats | Black people | |
| Older age | ||
| Male | ||
| Preventive factors | High fiber diet | Female |
| Whole grain diet | ||
| No alcohol | ||
| No smoking |
CRC: Colorectal cancer; FAP: Familial adenomatous polyposis; HNPCC: Hereditary non-polyposis colorectal cancer; MAP: MUTYH-associated polyposis.
Numerous studies have shown that an increased risk of CRC is related to obesity and decreased physical activity. CRC has repeatedly proven to be correlated with excess body fat, which is most typically quantified using body mass index and waist circumference. Sedentary activity, such as extended sitting or TV viewing, is linked to a higher risk of CRC. In populations over 50, the majority of research verifying obesity and a lack of physical exercise as risk factors for CRC has been demonstrated. Attempts to quantify the impact of obesity and physical activity on rates of early-onset CRC have rekindled attention in light of the rising burden of young-onset CRC in developed nations[17,18]. These and other investigations support the advice from the American Cancer Society and other cancer organizations that maintaining a healthy weight and engaging in more physical exercise are crucial for lowering the risk of CRC.
Genetic factors
A family history of cancer without a specific condition is thought to be the cause of 25% of CRCs, while hereditary cancer syndromes are thought to be the cause of 5% of CRCs. The hereditary component of CRC is predicted to be 35%-40%. Hereditary non-polyposis CRC (Lynch syndrome), familial adenomatous polyposis (FAP), and MUTYH-associated polyposis (MAP) are the most prevalent hereditary cancer syndromes. About 2%-4% of all CRCs are caused by Lynch syndrome, the most prevalent hereditary CRC condition. Patients with Lynch syndrome are susceptible to endometrial, ovarian, stomach, small intestine, hepatobiliary tract, pancreatic, ureter, and renal pelvis malignancies. Lynch syndrome is caused by mutations in the DNA mismatch repair genes hMLH1, hMSH2, hMSH6, and hPMS2[19,20]. The lifetime risk of CRC for these people is between 5% and 85%. FAP makes up roughly 1% of CRC cases and is the second most prevalent hereditary CRC syndrome. A germ line mutation in the adenomatous polyposis coli (APC) gene that causes a shortened APC protein is the secondary cause of FAP. With a 90% inheritance, it is inherited in an autosomal dominant manner. For these people, thousands of polyps form in the gastrointestinal system, the majority of which are in the colon. By the third or fourth decade, CRC will manifest in all FAP-affected individuals without preventative colectomy[21]. The lifetime risk of CRC for these MAP individuals is expected to be 43%-100%. Serrated polyposis syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome are further polyposis syndromes associated with a higher risk of CRC. These are all uncommon disorders that together only contribute to 1% of CRC incidence. Race, age, and sex are other unmodifiable risk factors for CRC in addition to family history. Male patients are generally at a higher risk of developing CRC than female patients; ideas explaining this include the fact that women typically have less visceral fat and benefit from estrogen's general preventive properties against CRC. Men are also less likely to pursue screening, and they may be more exposed to environmental risks, including drinking, smoking, and unhealthy diets[22].
Polyps
Most CRCs develop from a harmless precursor polyp. Therefore, people who have a considerable personal or family history of high-risk polyps are at a higher risk of developing CRC. There are many different forms of polyps, some of which are non-neoplastic, including hyperplastic polyps, mucosal polyps, inflammatory polyps, and harmartomatous polyps. The remaining polyps, including adenomatous and serrated polyps, have cancerous potential. Adenomatous polyps are where the vast majority of sporadic CRCs originate. By the time they are 50, roughly one-third of individuals are predicted to develop polyps. However, the majority of these will not progress to CRC. Villous histology, high-grade dysplasia, and polyps larger than 1 cm all enhance the likelihood that they may develop into CRC. Serrated polyps are believed to be antecedents for up to 10%-15% of sporadic CRCs, albeit less frequent[23,24].
COLORECTAL CARCINOGENESIS
Three molecular pathways have been hypothesized for colorectal carcinogensis, two of which center on the growth of polyps into cancerous tumors. The traditional pathway depicts the long-term evolution of normal cells to adenomas and finally to carcinomas (the adenocarcinoma sequence). This causes 85%-90% of all sporadic CRCs and is mostly related to the growth of tumors that are CIN. It is frequently accompanied by an early APC gene mutation, activation of the growth-promoting oncogenes Kristin rat sarcoma virus (KRAS) or BRAF, and additional mutations that cause cancer to proceed. About 10%-15% of sporadic CRCs are caused by the CIMP/serrated pathway[25,26]. Due to these changes in the methylation of gene promoter regions and general hypomethylation, many genes are silenced. Early BRAFV600E mutations are frequently seen in these tumors, which activate the mitogen-activated protein kinase (MAPK) pathway and cause the growth of hyperplastic polyps[27]. The MSI-H pathway is the third route. More than 95% of the malignancies related to Lynch syndrome are caused by this important pathway. Outside of Lynch syndrome, MSI is a rare condition that is caused by a lack of DNA mismatch repair genes, which eventually results in altered DNA sequences (Figure 1).
Figure 1.
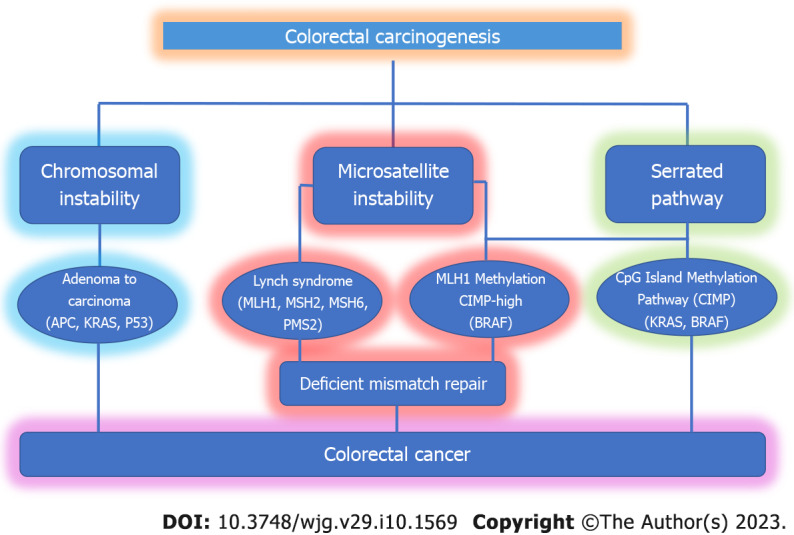
Three molecular pathways can lead to colorectal cancer. For colorectal carcinogenesis, three molecular pathways have been proposed: the chromosomal instable or classic pathway, the microsatellite instability pathway, and the serrated pathway. APC: Adenomatous polyposis coli; BRAF: Protein kinase B-Raf; CIMP: CpG island methylator phenotype; KRAS: Kristin rat sarcoma virus.
KRAS
A proto-oncogene called KRAS, which produces a GTPase protein, is essential for intracellular signal transduction downstream of membrane-bound receptors like EGFR. Uncontrolled cell growth, proliferation, survival, migration, and invasion ensue from a mutation in KRAS because it causes constitutive activation of the MAPK pathway, regardless of independent activation of the upstream EGFR receptor. Activating KRAS mutations have been reported in a variety of cancers and have been found between 30% and 50% of CRC patients[28,29]. KRAS is a crucial biomarker for prognosis and prediction in the management of mCRC. KRAS-mutated tumors in mCRC have a poorer prognosis and are more likely to exhibit aggressive biology, spread metastatically, recur, and result in mortality. KRAS mutation is independently linked to poorer progression-free survival (PFS) and overall survival (OS) in individuals who underwent hepatic resection for mCRC, according to data from those patients. KRAS codon 12 has been linked to lower recurrence-free survival in variations of KRAS mutations throughout all stages of mCRC[30]. Data suggest that curative resection may not be advantageous in the metastatic scenario in KRAS mutant patients who also have other poor clinical prognostic characteristics, such as node-positive disease or extensive metastases, due to the prognostic consequences of KRAS mutations[31]. Additionally, the existence of a KRAS mutation acts as a biomarker for the therapeutic efficacy of some therapies, such as EGFR inhibitor therapy. Anti-EGFR treatment has been shown to be effective in treating KRAS wild-type (WT) cancers in several clinical studies. Due to the independent constitutive activation of KRAS downstream of EGFR, which persistently encourages cell growth and division, anti-EGFR treatment is not helpful in patients with KRAS mutations[32]. Although EGFR inhibition is effective in treating the majority of KRAS WT tumors, some patients continue to have resistance, necessitating more research. Mutations in other RAS family oncogenes, such as neuroblastoma RAS (NRAS) and Harvey RAS, identify tumors that are resistant to anti-EGFR treatment. Other phosphorylation pathway genes that work downstream of EGFR, such as phosphoinositide 3-kinases, phosphatase and tensin homolog, MAPK, and mitogen-activated protein kinase kinase (MEK), have not been demonstrated to be accurate predictors of the EGFR response for mCRC, and research into the causes of anti-EGFR resistance in this patient group is still underway[33].
BRAF
BRAF, a serine/threonine-protein kinase, is an essential component of the MAPK signaling cascade downstream of KRAS. In less than 10% of CRC patients, BRAF mutations have been found. The same MAPK pathway that KRAS uses for BRAF signaling also uses it, and functional mutations in either of these genes have identical effects on phenotypic and treatment implications. As a predictive biomarker for mCRC, the BRAF mutation is linked to worse outcomes, shorter survival, and a greater incidence of peritoneal and distant lymph node involvement. In patients with mCRC undergoing curative-purpose hepatectomy, the BRAF mutation is associated with poorer survival compared to both BRAF WT and KRAS mutated tumors[34,35]. BRAF also functions as a prognostic marker. Vemurafenib, a direct BRAF inhibitor, was first discovered through early attempts at targeted medication treatment for melanoma. Studies have been conducted to determine if BRAF inhibition has comparable effects on CRC. BRAF inhibition and EGFR inhibitors together produced an OS improvement. In patients with BRAF mutant mCRC, encorafenib in conjunction with EGFR inhibition is a potent form of newer generation BRAF inhibition therapy[36].
Human epidermal growth factor 2
A further promising target for mCRC is human epidermal growth factor 2 (HER2), which is essential for intracellular signal transduction. HER2 plays a role in the development of breast cancer, and trastuzumab and other HER2 inhibitors can be used to specifically treat the disease. Because there are so many targeted treatments available, many researchers have concentrated on HER2 mutations in the mCRC population, despite the fact that only a small percentage of patients (10%) overexpress HER2. Trastuzumab with pertuzumab, trastuzumab with lapatinib, and fam-trastuzumab deruxtecannxki are currently available as HER2 inhibitors. Even though it only accounts for a small proportion of all mCRC patients, the HER2 amplified condition serves as a predictive biomarker for HER2 targeted therapy with the potential for a therapeutic response[37-39].
SYSTEMIC TREATMENT FOR MCRC
A surgeon may be able to completely remove a few metastatic foci of mCRC, which are often located in the liver or lung. When the main tumor and all metastases can be completely removed surgically, mCRC is said to be resectable. However, nodal infiltration and covert micrometastatic spread are frequent in these individuals. Less than 20% of individuals with mCRC who undergo resection are permanently cured. Oncologists from surgical and medical branches should work together to develop treatment strategies when mCRC may be resectable. If the main tumor is in the rectum, radiation oncologists should be consulted. The main therapy for mCRC is systemic chemotherapy. The United States Food and Drug Administration (FDA) has authorized many medications for the treatment of mCRC. However, for the majority of patients, mCRC is still incurable. Despite the rarity of a cure for mCRC, recent major clinical trials with patients who could tolerate chemotherapy have demonstrated that patients can live for 2 to 3 years with intense treatment and numerous systemic medicines. Survival is influenced by the molecular subtype, which provides information about the prognosis by describing the natural history of a tumor and the therapies that are and are not likely to be successful. The median OS for the 50% of patients with KRAS/NRAS/BRAF WT mCRC is about 30 mo with survival rates of 80% at 1 year, 40% at 3 years, and 20% at 5 years after the start of first-line chemotherapy (Table 2).
Table 2.
Chemotherapy and targeted therapies for metastatic colorectal cancer patients
|
Ref.
|
Country
|
Drug(s)
|
Number of patients
|
Study phase
|
ORR, %
|
Mean OS in mo
|
Mean PFS in mo
|
Results
|
| Resectable CLM with no extra-hepatic metastasis | ||||||||
| Gruenberger et al[42] | Austria | Capecitabine, oxaliplatin plus bevacizumab | 56 | 2 | 73.2 | - | - | Bevacizumab can be safely administered until 5 wk before liver resection in patients with metastatic CRC without increasing the rate of surgical or wound healing complications or the severity of bleeding |
| Bridgewater et al[43] | United Kingdom | Oxaliplatin, L-folinic acid, fluorouracil or capecitabine, oxaliplatin plus cetuximab | 257 | 3 | - | 81.0/55.4 (Chemo/Chemo+) | 22.2/15.5 (Chemo/Chemo+) | In the perioperative setting, patients with operable diseases are at a disadvantage in terms of OS; hence, cetuximab should not be used in this setting |
| Unresectable CLM with potential for resection and no extra-hepatic metastasis | ||||||||
| Wong et al[44] | United Kingdom | Capecitabine, oxaliplatin plus bevacizumab | 46 | - | 78.0 | - | - | A high response rate for patients with CLMs with poor-risk features not selected for upfront resection and converted 40% of patients to resectability |
| Gruenberger et al[45] | United Kingdom, Austria, France, and Spain | Bevacizumab plus FOLFOX-6 (5-fluorouracil folinic acid oxaliplatin) or FOLFOXIRI (5-fluorouracil folinic acid oxaliplatin irinotecan) | 80 | 2 | 81/62 (BF/BF6) | - | 18.6/11.5 (BF/BF6) | In patients with CLM that were originally unresectable, bevacizumab-FOLFOXIRI was correlated with better response and resection rates as well as a longer PFS than bevacizumab-mFOLFOX-6 |
| Garufi et al[47] | Italy | Cetuximab plus chrono-IFLO (chrono-irinotecan 5-fluorouracil leucovorin oxaliplatin) | 43 | 2 | - | 37 | - | Cetuximab in combination with chrono-IFLO resulted in 60% full resectability of CLM patients |
| Folprecht et al[48] | Germany | Cetuximab plus FOLFOX (5-fluorouracil folinic acid oxaliplatin) or FOLFOXIRI (5-fluorouracil folinic acid oxaliplatin irinotecan) | 56/55 (CFX/CFI) | - | - | 35.7/29.0 (CFX/CFI) | 10.8/10.5 (CFX/CFI) | Both FOLFOX/FOLFIRI with cetuximab appear to be effective conversion therapy regimens in patients with KRAS codon 12/13/61 wild-type tumors. Thus, liver surgery can be deemed curative or as a second line of therapy in people who are not cured |
| Unresectable CLM | ||||||||
| Rivera et al[50] | Spain, Germany, United States, Belgium, Switzerland, and Italy | mFOLFOX6 plus panitumumab or bevacizumab | 170/156 (RAS WT/RAS WT/BRAF WT) | - | RAS WT 65/60, RAS WT/BRAF WT 65/62 (FXP/FXB) | RAS WT 36.9/28.9, RAS WT/BRAF WT 46.3/28.9 (FXP/FXB) | RAS WT 12.8/10.1, RAS WT/BRAF WT 13.1/10.1 (FXP/FXB) | Panitumumab in combination with mFOLFOX6 is a successful first-line therapy for individuals with RAS WT and RAS WT/BRAF WT mCRC |
| Stintzing et al[53] | Germany | FOLFIRI (5-fluorouracil leucovorin irinotecan) plus cetuximab or bevacizumab | 400 | 3 | 65.3/58.7 (FIC/FIB) | 33.1/25.0 (FIC/FIB) | 10.3/10.2 (FIC/FIB) | In the first-line therapy of patients with RAS wild-type metastatic colorectal cancer, FOLFIRI with cetuximab may be preferable than FOLFIRI plus bevacizumab |
| Pfeiffer et al[58] | Denmark | Trifluridine plus tipiracil hydrochloride (TAS-102) or TAS-102 plus bevacizumab | 47/46 (T/TB) | 2 | 51/67 (T/TB) | 6.7/9.4 (T/TB) | 2.6/4.6 (T/TB) | In terms of efficacy and safety, bevacizumab may be an effective companion for TAS-102 in patients with chemorefractory metastatic colorectal cancer |
| Cremolini et al[59] | Italy | Cetuximab plus irinotecan | 13/12 (RWT/RMT) | 2 | - | 12.5/5.2 (RWT/RMT) | 4.0/1.9 (RWT/RMT) | In patients with RAS and BRAF wild-type mCRC who have acquired resistance to first-line irinotecan and cetuximab-based treatment, a re-challenge approach with cetuximab and irinotecan may be effective. The examination of RAS mutational status on cDNA may aid in the selection of potential patients |
| Sartore-Bianchi et al[60] | Italy | Panitumumab (re-challenge) | 25 | - | 30 | 13.7 | 4 | Interventional liquid biopsies can be used efficiently and safely to guide anti-EGFR re-challenge treatment with panitumumab in patients with mCRC |
| Kopetz et al[63] | United States | Irinotecan cetuximab or irinotecan cetuximab plus vemurafenib | 50/50 (IC/ICV) | - | - | 12/12 (IC/ICV) | 2.0/4.2 (IC/ICV) | Vemurafenib in combination with cetuximab and irinotecan is an active combination that increases PFS. This is a well-planned research based on a solid grasp of the mechanisms of adaptive resistance in mCRC |
| Tabernero et al[64] | United States, France, Italy, Spain, United Kingdom, Germany, Australia, Hong Kong, Norway, Switzerland, Japan, South Korea, and the Netherlands | Encorafenib cetuximab binimetinib or encorafenib cetuximab or FOLFIRI cetuximab | 224/220/221 (3D/2D/CD) | 3 | 26.8/19.5/1.8 (3D/2D/CD) | 9.3/9.3/5.9 (3D/2D/CD) | - | When compared to conventional chemotherapy, encorafenib plus cetuximab improved OS, ORR, and progression-free survival in previously treated patients with metastatic disease. Encorafenib with cetuximab is a new standard-of-care regimen for previously treated patients with BRAF V600E mCRC, according to main and revised studies |
| Siena et al[68] | United States, Italy, United Kingdom, Spain, and Japan | Trastuzumab deruxtecan (DS-8201) | 53/7/18 (A/B/C) | 2 | 45.3 (A) | 5.4 (A) | 6.9 (A) | Trastuzumab deruxtecan shown promising and sustained effect in HER2-positive mCRC that was resistant to conventional therapy, as well as a favorable safety profile |
| Mayer et al[71] | Belgium, Italy, Japan, Spain, Australia, and United States | Trifluridine plus tipiracil hydrochloride (TAS-102) or placebo | 800 (2:1) | - | 1.6/0.4 (TAS/placebo) | 7.1/5.3 (TAS/placebo) | 2.0/1.7 (TAS/placebo) | A significant number of Japanese and Western patients with mCRC who had had a lot of prior treatment, including those whose condition was resistant to fluorouracil, were shown to respond clinically to TAS-102 |
2D: Encorafenib plus cetuximab; 3D: Encorafenib plus cetuximab plus binimetinib; A: Cohort A; B: Cohort B; BF: Bevacizumab plus FOLFOXIRI; BF6: Bevacizumab plus FOLFOX-6; BRAF: Protein kinase B-Raf; C: Cohort C; CD: FOLFIRI plus cetuximab; CFI: Cetuximab plus FOLFOXIRI; CFX: Cetuximab plus FOLFOX; CRC: Colorectal cancer; EGFR: Epidermal growth factor receptor; FIB: FOLFIRI plus bevacizumab; FIC: FOLFIRI plus cetuximab; FOLFIRI: Folinic acid, fluorouracil, and irinotecan hydrochloride; FXB: mFOLFOX6 plus bevacizumab; FXP: mFOLFOX6 plus panitumumab; HER2: Human epidermal growth factor receptor 2; IC: Irinotecan plus cetuximab; ICV: Irinotecan plus cetuximab plus vemurafenib; mCRC: Metastatic colorectal cancer; ORR: Objective response rate; OS: Overall survival; PFS: Progression-free survival; RAS: Rat sarcoma virus; RMT: Rat sarcoma virus mutate-type; RWT: Rat sarcoma virus wild-type; T: Trifluridine plus tipiracil hydrochloride; TB: Trifluridine plus tipiracil hydrochloride plus bevacizumab; WT: Wild-type.
SYSTEMIC TREATMENT FOR LIVER METASTASIS CRC
Nearly half of individuals with initial CRC develop liver metastatic diseases from CRC or colorectal liver metastasis (CLM). The resectability of CLM determines how to manage it, and interdisciplinary approaches are frequently used. Conversion treatment (CT), a kind of systemic treatment, is used for liver metastases that are initially incurable. Both the number (4 vs > 4) and size (diameter < 6 cm vs ≥ 6 cm) of CLMs are independent variables linked to successful CT[40,41].
Targeted treatment
FOLFOX (5-fluorouracil [5-FU], leucovorin, and oxaliplatin) and FOLFIRI (5-FU, leucovorin, and irinotecan) are the mainstays of systemic chemotherapy used to treat mCRC. EGFR inhibitors (EGFRis) for RAS WT tumors (cetuximab [Cet] and panitumumab [Pan]) and anti-vascular endothelial growth factor (VEGF) (bevacizumab [Bev]) are the two main groups of medications now added to these chemotherapy regimens.
Resectable CLM with no extrahepatic metastasis
There is debate regarding the benefits of adding a targeted treatment, however, the addition of Bev or Cet to FOLFOX or FOLFIRI is tolerable in resectable liver metastases. However, a single-arm phase 2 study found that the addition of Bev to the capecitabine and oxaliplatin combination (CAPOX) (six cycles with no Bev on the final cycle), before surgery in high-risk CRC, had a remarkable objective response rate (ORR) of 73%[42]. In 2020, Bridgewater et al[43] conducted a multicenter, open-label, randomized, controlled, phase 3 trial to investigate the effects of Cet plus chemotherapy compared with those given chemotherapy alone in 257 adult patients (aged ≥ 18 years) with KRAS WT (codons 12, 13, and 61) resectable or suboptimally resectable. At a median follow-up of 66 mo after the last patient was recruited, this analysis was conducted. In the chemotherapy alone group, the median PFS was 22.2 mo, whereas in the chemotherapy plus Cet group, it was 15.5 mo (P = 0.304). In the chemotherapy alone group, the median OS was 81.0 mo, but in the chemotherapy plus Cet group, it was 55.4 mo (P = 0.036). The status of pathological resection or the preoperative response were secondary outcomes that did not significantly differ between groups. High-risk CRC in this study included those with synchronous liver metastases, metastatic disease discovered within a year of initial resection, primary tumors with positive lymph nodes, CLMs > 1 or > 5 cm, and positive carcinoembryonic antigen (CEA) levels. They concluded that Cet had no effect when used with perioperative treatment. As a result, targeted therapy is deemed ineffective in CRC patients with resectable CLM, whereas Bev may be beneficial in high-risk patients. FOLFOX adjuvant treatment is advised following resection.
Unresectable CLM with potential for resection and no extrahepatic metastasis
In high-risk CRC (> 4 metastases, diameter > 5 cm, poor viable liver function if undertaking upfront resection, or inability to maintain liver vascular supply), neoadjuvant treatment (NAT) with CAPOX plus Bev achieved a 78% ORR[44]. Of the 46 patients included, 40% of the individuals with unresectable disease upon diagnosis got a resection. Four patients responded so well that they were kept under surveillance without having surgery. In comparison to FOLFOX, Bev with FOLFOXIRI (combination of 5-FU/leucovorin (LV), oxaliplatin, and irinotecan) had a greater resection rate (61% vs 49%), R0 resection rate (49% vs 29%), ORR (81% vs 62%), and mean PFS (18.6 m vs 11.5 m)[45]. After a hepatic artery infusion chemotherapy pump has been installed, adding Bev to the chemotherapy has no survival benefit. On the other hand, it worsens liver toxicity (hyperbilirubinemia > 3 mg/dL) and is not advised[46]. In the POCHER study, 60% (25/43) of the 43% of patients with unresectable CLM who received chronomodulated irinotecan (Iri) and 5-FU/LV on days 2-6 every 2 wk as NAT experienced full resections[47]. Twenty-nine participants with more than four lesions and nine patients with more than 5 cm in diameter made up the study population. The 2-year survival rate was 68% for the whole population. In people who have had surgery, it may reach 80%. As a result, research was conducted to evaluate the role of EGFRi in NAT or POT. When Cet was given to FOLFOX or FOLFIRI in patients with unresectable CLM at diagnosis who were eligible (with KRAS WT codons 12, 13, and 61), it improved long-term survival[48].
Unresectable CLM
First-line plus chemotherapy: Bev is advised as first-line treatment for RAS-mutated tumors in mCRC with CLM and without surgical consideration. A meta-analysis of two randomized control trials and three observational studies revealed that Cet had a higher OS, ORR, and complete response rate than Bev in RAS-WT malignancies (with chemotherapy as the backbone). The same trial found no appreciable differences between the two medications in PFS, disease control rate, or partial response rate[49]. In the PEAK study, Pan outperformed Bev in terms of PFS while maintaining the same OS[50]. In the past 5 years, a lot of research has been done on the function of the main tumor side (right vs left). Regardless of the CLM status, two meta-analyses have conclusively demonstrated that left-sided cancers react better to EGFRi than Bev[51,52]. However, there was no statistically significant difference in OS or PFS between Bev and EGFRi in tumors on the right side. In the PEAK study and the FIRE-3 trial, EGFRi with chemotherapy produced deeper responses in RAS-WT tumors than Bev plus chemotherapy. Tolerability is a crucial consideration when choosing a medication. Patients with a history of thromboembolic illness, uncontrolled hypertension, proteinuria, significant bleeding risk, or gastrointestinal perforation are not advised to get anti-VEGF medication. As a result, EGFRi (Cet or Pan) is preferable over Bev plus chemotherapy in left-sided tumors in RAS-WT mCRC with CLM, although either of them can be administered in right-sided tumors[53,54]. There is not any conclusive proof that Pan or Cet is superior to the other. Patients who experience adverse reactions to Cet, a mouse-based monoclonal antibody (mab), frequently prefer Pan since it is a humanized mab.
Second-line plus chemotherapy: The continuation of Bev in the second line after first-line treatment improved PFS and OS without a worsening in side events. There is insufficient information to definitively conclude that changing eligible patients from Bev to Cet or Pan following clinical progression would be beneficial[55,56]. There is no advantage to switching Bev to another VEGF medication. Patients who progress on oxaliplatin-based therapy (FOLIRI-naive) benefit from ziv-aflibercept or ramucirumab when used in conjunction with FOLIRI[57]. Bev may also be used in the third or fourth line of TAS-102 (trifluridine and tipiracil hydrochloride)[58]. Continuing EGFRi has no advantage for OS if Cet or Pan are used as the first-line treatments[59]. Circulating tumor DNA can be used to identify acquired resistance, which may manifest in a small number of patients. It is anticipated that this resistance would fade with time. As a result, switching to Bev is advised[60,61]. After the disease progressed, switching from Cet to Pan or vice versa is not useful. If not used in the first-line setting, EGFRi may be administered either alone (monotherapy) or with Iri, FOLFIRI, or FOLFOX but not with CAPEOX, or to patients who cannot handle chemotherapy[62]. In ongoing studies, they are being used with additional medications such as immune checkpoint inhibitor (ICI), BRAF, and tyrosine kinase inhibitors.
Patients with resistant mCRC who have BRAF mutations, notably BRAF V600E and KRAS WT, may benefit from a combination therapy that combines BRAF inhibitors with Cet or Pan[63]. In the interim analysis of the BEACON trial, triple treatment with the MEK inhibitor (binimetinib) demonstrated a survival benefit over the control group (Cet plus Iri or FOLFIRI) and combination therapy. A more recent investigation, however, found no benefit of triplet treatment over doublet treatment compared to the control group[64]. In the initial studies with Pan, other BRAF inhibitors, dabrafenib and vemurafenib, with/without MEK inhibitors (trametinib), showed favorable outcomes[65]. In approximately 6% of CRCs, HER2 is amplified or overexpressed[66]. Trastuzumab, a HER2 inhibitor, in combination with pertuzumab (an mAb that prevents dimerization of HER2 and HER3) or lapatinib (inhibitor that binds to the cytoplasmic ATP-binding site inhibitor EGFR/HER1 and HER2 receptors), is well tolerated in refractory mCRC patients with HER2 amplification and RAS/BRAF WT[67]. HER2-amplified, a humanized anti-HER2 Ab combined with a topoisomerase I inhibitor, known as trastuzumab deruxtecan (T-dx), demonstrated an excellent ORR (45.3%) after a median follow-up of 27.1 wk in refractory mCRC[68]. Patients who were resistant to HER2 inhibitors also showed activity when given T-dx. Neurotropic tyrosine receptor kinase (NTRK) genes code for the tyrosine kinase receptors that control cell growth, and rearrangements result in unregulated cell proliferation. It was first discovered in CRC and occurs in just 0.3% of solid tumors[69]. There was 7% CRC in the studies that administered NTRK inhibitors to solid tumors, such as entrectanib and larotectinib. Patients with NTRK mutations may choose this as a treatment option[70].
A multinational, randomized, placebo-controlled, phase 3 trial of a multikinase tyrosine kinase inhibitor is called the CORRECT trial. The regorafenib therapy group outlived the placebo group by 1.4 years (mOS for regorafenib is 6.4 years vs 5 years for placebo; P = 0.005). The use of a regulator was associated with higher medication toxicity (93% in the regorafenib vs 61% in the placebo). Hand-foot skin reactions were the most common adverse event (AE) (83%) observed, followed by tiredness (48%) and hypertension (36%). ICIs including ipilimumab/nivolumab, pembrolizumab, and EGFRi are now being explored in conjunction with it. Its usage is frequently contrasted with that of the TAS-102, which had an OS improvement of 1.8 m above placebo. Combinations of ICI and EGFRi are being investigated[71] (Figure 2).
Figure 2.

Targeted therapy for metastatic colorectal cancer. AKT: Ak strain transforming; D: Down regulation pathway; EGFR: Epidermal growth factor receptor; HER2: Human epidermal growth factor receptor 2; IRS1/2: Insulin receptor substrates 1 or 2; MAPK: Mitogen activated protein kinase; mTOR: Mammalian target of rapamycin; MEK: Mitogen-activated protein kinase kinase; NTRK: Neurotrophic tyrosine receptor kinase; PTEN: Phosphatase and tensin homolog; PI3K: Phosphoinositide 3-kinases; RAF: Rapidly accelerated fibrosarcoma; RAS: Rat sarcoma virus; SOS: Son of sevenless.
IMMUNOTHERAPY
The goal of immunotherapy is to use the immune system to fight cancer. For patients with mCRC that is mismatch-repair-deficient (dMMR) or MSI-H (dMMR/MSI-H mCRC), ICIs have emerged as a very effective treatment. ICIs modify the interaction of T cells, antigen-presenting cells, and tumor cells to help unleash suppressed immune responses. The FDA approved pembrolizumab and nivolumab (with or without Ipilimumab) for the treatment of these patients due to their effective, stable, and long-lasting responses. The fundamental difficulty is to offer the advantage of immunotherapy to the great majority of mCRC patients who are mismatch-repair-proficient (pMMR), microsatellite-stable (MSS), or have low MSI (MSI-L), since mCRC is characterized by an inadequate number of mutant tumor antigens[72].
ICIs-based immunotherapy
Use of ICIs in DMMR/MSS mCRC: To preserve DNA integrity, MMR is essential. CRC can be classified as dMMR or pMMR CRC based on the detection of the MMR proteins MLH1, MSH2, MSH6, or PMS2 utilizing immunohistochemical staining. Moreover, insertions and deletions can cause MSI, which can be precisely identified by PCR or next-generation sequencing, resulting in a change in microsatellite length. Major histocompatibility complex (MHC) class I peptide complexes, including mutant peptides that might be identified as neoantigens and subsequently increase immune cell priming and infiltration, are present on the surface of tumor cells in dMMR-MSI-H malignancies. T helper 1 CD4+ T cells, macrophages, and CD8+ tumor-infiltrating lymphocytes (TILs) enter the TME and produce interferon gamma. Programmed cell death ligand-1, cluster of differentiation 80 (CD80), and CD86 of the B7 family are examples of T cell inhibitory ligands that dMMR-MSI-H tumor cells persistently upregulate to support immune escape[73-75]. The percentage of dMMR-MSI-H CRCs, which accounts for about 15% of all CRCs, is correlated with tumor stage. dMMR-MSI-H cancers make up about 5%-20% of stage 2 and 11% of stage 3, but only 5% of stage 4. Additionally, dMMR-MSI-H is a predictive biomarker for individuals at various phases of their condition. Patients with dMMR-MSI-H tumors have a much better prognosis than those with pMMR-MSI-L cancers in stages 2 and 3. Surprisingly, individuals with stage 4 dMMR-MSI-H have a poor prognosis yet respond well to immune checkpoint inhibition[76,77].
Le et al[78] conducted a phase 2 trial in 41 patients with progressive mCRC with or without dMMR to investigate the clinical efficacy of pembrolizumab, an anti- Programmed cell death protein 1 (PD-1) ICI, in 2015. Pembrolizumab was given intravenously every 14 d at a dosage of 10 mg/kg body weight to patients with dMMR CRC, pMMR, and patients with dMMR who were not colorectal. The immune-related ORR and PFS for dMMR CRC were 40% and 78%, respectively, and 0% and 11% for pMMR CRC. In the group with dMMR CRC, the median PFS and OS were not attained, but in the cohort with pMMR CRC, they were 2.2 and 5.0 mo, respectively (hazard ratio [HR] for PFS and death = 0.10 and 0.22). Responses in patients with dMMR non-CRC were comparable to those in individuals with dMMR CRC. High somatic mutation loads were related to longer PFS (P = 0.02), and whole-exome sequencing found that dMMR tumors had an average of 1782 somatic mutations per tumor, compared to 73 somatic mutations in pMMR tumors (P = 0.007). They concluded that MMR status predicted the therapeutic benefit of immune checkpoint inhibition with pembrolizumab (Figure 3).
Figure 3.

Immune check point inhibitors treatment for metastatic colorectal cancer. CTLA4: Cytotoxic T-lymphocyte-associated protein 4; MHC: Major histocompatibility complex; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand; TCR: T cell receptor.
In 2020, the KEYNOTE-164 study analyzed pembrolizumab's effectiveness in 124 patients with dMMR/MSI-H mCRC who had undergone treatment. The ORR was 32% among the 63 patients examined, and the median PFS was 4.1 mo. The overall median survival rate has not yet been reached. The percentages of OS and PFS at 1 year were 41% and 76%, respectively. A single ICI therapy for individuals with dMMR/MSI-H mCRC demonstrated sustained anticancer efficacy[79]. In a phase 3, open-label study, 307 mCRC patients with dMMR/MSI-H who had not previously received treatment were enrolled to assess the effectiveness of PD-1 blockers or chemotherapy as first-line treatments. They were given a 1:1 random assignment to undergo chemotherapy (5-FU-based treatment with or without bevacizumab or cetuximab) every 2 wk, or pembrolizumab at a dosage of 200 mg every 3 wk. After advancement of the condition, patients taking chemotherapy could switch to pembrolizumab therapy. Of the 307 patients enrolled, 153 received single-agent pembrolizumab and 154 received chemotherapy. The median PFS time was 16.5 mo in the pembrolizumab group and 8.2 mo in the chemotherapy group at a median follow-up of 32.4 mo (HR = 0.60; P < 0.001). Significant differences were seen between the pembrolizumab group’s 12- and 24-mo PFS values, which were 55% and 48%, respectively, vs 37% and 19% in the chemotherapy group. These data show that, compared to chemotherapy, pembrolizumab demonstrates more stable anticancer activity and fewer treatment-related AEs[80]. Based on the compelling evidence from this trial, the FDA approved pembrolizumab for the first-line treatment of patients with dMMR/MSI-H or advanced, unresectable, or metastatic CRC in June 2020.
In certain clinical studies, the use of nivolumab, another PD-1 inhibitor that targets dMMR/MSIH CRC, is also being investigated. Nivolumab’s effectiveness was examined in the phase 2 study CheckMate 142 on 74 patients with dMMR/MSI-H mCRC. The ORR was 31%, and 69% of patients had disease control for 12 wk or longer. The median duration of response was not attained. The PFS and OS rates during the past 12 mo were 50% and 73%, respectively. Nivolumab's safety profile in this cohort study was consistent with that previously reported in other solid tumor trials, and there were no additional AEs noted[81]. Nivolumab was approved by the FDA in August 2017 for the treatment of dMMR/MSI-H mCRC in adults and children older than 12-years-old on the basis of these research findings[82]. Nivolumab's administration in conjunction with the cytotoxic T-lymphocyte-associated protein 4 mAb ipilimumab was also investigated in the CheckMate142 trial. Nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg was given once every 3 wk (four doses) to a total of 119 patients with dMMR/MSI-H mCRC who had not responded to conventional therapy. Nivolumab 3 mg/kg was then given once every 2 wk. The ORR was 55% at a median follow-up of 13.4 mo. In 80% of patients, the disease was under control for at least 12 wk; however, the median PFS was not met. The PFS rates for the 9th and 12th mo were 76% and 71%, respectively. The 9- and 12-mo OS rates were 87% and 85%, respectively, but the median OS was not met[83]. The FDA expedited the approval of nivolumab in combination with ipilimumab for the treatment of patients with dMMR/MSI-H mCRC in July 2018 based on the findings of this trial. Furthermore, the most recent information from the 2-year follow-up was used to update the study outcomes. The ORR and disease control rates determined by the study were 69% and 84%, respectively, over a median follow-up of 29 mo. The 2-year PFS and OS rates were 74% and 79%, respectively, but the median PFS and OS was not attained[84].
After first-line chemotherapy failed, Kim et al[85] conducted a prospective, open-label, multicenter phase 2 trial in 2020 to assess the effectiveness and safety of avelumab in 30 mCRC patients who had dMMR)/MSI-H and 3 mCRC patients who had polymerase-epsilon (POLE) mutations. All of the respondents were dMMR/MSI-H, and the ORR was 24.2%. At a median follow-up time of 16.3 mo, the median PFS and OS for all patients was 3.9 and 13.2 mo, respectively. They concluded that in patients with previously treated mCRC carrying dMMR/MSI-H, avelumab demonstrated anticancer efficacy with controllable toxicity. To assess the effectiveness and safety of cetuximab re-challenge treatment combined with avelumab in 71 MSS, 3 MSI-H, and 3 patients with uncertain microsatellite status with mCRC, Martinelli et al[86] conducted a single-arm, multicenter phase 2 study in 2021. The patients were given cetuximab (400 mg/m2, then 250 mg/m2 weekly) and avelumab (10 mg/kg every 2 wk) until the disease progressed or the side effects became intolerable. With a median OS of 11.6 mo and a median PFS of 3.6 mo, the trial accomplished its primary aim; 4% of grade 3 AEs were diarrhea, and 14% of them were skin eruptions. There were 48 people with WT illnesses and 19 people with mutations. Those with RAS/BRAF WT cDNA had a median OS of 17.3 mo, opposed to patients with mutations, who had a median OS of 10.4 mo. In contrast to patients with mutant cDNA, those with RAS/BRAF WT had a median PFS of 4.1 mo opposed to 3.0 mo. They concluded that an active, well-tolerated challenge treatment for RAS WT mCRC is cetuximab plus avelumab.
Use of ICIs in PMMR/MSS mCRC: pMMR/MSS CRC, which makes up about 95% of all mCRCs, is referred to as a "cold tumor." Single-agent ICI had no effect on pMMR/MSS CRCs in contrast to inflammatory tumors of dMMR/MSI-H. The results of recent investigations on the use of combination ICIs have raised the prospect of enhancing immunotherapy efficacy in this population. ICIs in conjunction with systemic chemotherapy were proven to dramatically increase tumor treatment response in refractory mCRC treated with conventional chemotherapy and immunotherapy, especially in pMMR/MSS CRCs. Existing data show that immunogenic chemotherapy might improve the efficacy of ICIs by increasing tumor infiltration of CD4+ and CD8+ T cells and interrupting the function of immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells[87,88]. To investigate ways to make cold tumors heated to boost sensitivity to immunotherapy, a number of ICI-based combination treatment studies tested ICIs in conjunction with chemotherapy, targeted therapy, ICI therapy, and radiation.
Pembrolizumab combined with modified FOLFOX6 was tested in a single-arm, multicenter phase 1b study by Herting et al[89] for the treatment of mCRC. In this study, 87% of the participants had pMMR/MSS mCRC. At a median follow-up of 19.9 mo among the 30 patients, the investigators noted an ORR of 57% and the mean time it took for the responding patients to respond was 37.57 wk. The median PFS time was 8.8 mo, and the median OS was not reached. Recently, two phase 2 trials, AtezoTRIBE and MAYA, evaluating combinations of ICIs with chemotherapy, revived optimism for the use of immunotherapy in pMMR/MSS mCRC patients, indicating a significant breakthrough and a potential basis for future research in this scenario.
AtezoTRIBE trial: Regardless of microsatellite status, 218 patients with unresectable and chemo-naive mCRC were randomized in a 1:2 ratio to receive FOLFOXIRI. The control group received first-line FOLFOXIRI (intravenous 165 mg/m2 irinotecan, 85 mg/m2 oxaliplatin, 200 mg/m2 leucovorin, and 3200 mg/m2 fluorouracil as a 48-h infusion) plus bevacizumab (5 mg/kg intravenously), and the atezolizumab group received the same regimen plus atezolizumab (840 mg intravenously) in the AtezoTRIBE phase 2 multicenter, open-label, comparative study. According to the randomized arm, both treatments were given for up to eight cycles, then 5-FU with bevacizumab, with or without atezolizumab, was given until the condition progressed, there were unacceptable side effects, or the patient withdrew their consent. PFS was the main endpoint, with a one-sided alpha error of 0.10 and an 85% power. A median follow-up of 19.9 mo was being used. In the atezolizumab group, the median PFS was 13.1 mo, compared to 11.5 mo in the control group (HR = 0.69; P = 0 012). Neutropenia (42% of 142 patients in the atezolizumab group vs 36% of 72 individuals in the control group), diarrhea (15% vs 13%), and febrile neutropenia (10% vs 10%) were the most common all-cause grade 3-4 AEs. A total of 39 patients (27%) in the atezolizumab group and 19 patients (26%) in the control group suffered serious AEs. Acute myocardial infarction and bronchopulmonary hemorrhage caused two (1%) treatment-related fatalities in the atezolizumab group; none were recorded in the control group. They concluded that first-line FOLFOXIRI plus bevacizumab with atezolizumab added was safe and enhanced PFS in patients with mCRC who had not previously received treatment[90].
MAYA trial: MAYA is a prospective single-arm phase 2 trial that included patients with chemo-resistant mCRC who had centrally confirmed MSS status, O6-methylguanine-methyltransferase (MGMT) silence determined by promoter methylation of the MGMT gene, and total immunohistochemical loss of MGMT protein. Only in situations where disease control is obtained during phase 1 are participants to receive two cycles of temozolomide (TMZ) (phase 1), followed by the addition of nivolumab and low-dose ipilimumab (phase 2). The 8-mo PFS rate in patients included in phase 2 of the trial served as the study's main objective. The MAYA trial's design is supported by an intriguing biological theory. In short, the MGMT gene plays a role in repairing DNA damage brought on by alkylating drugs like TMZ[91]. Sensitivity to TMZ is increased when MGMT is inactivated by hypermethylation of its promoter. As demonstrated in studies evaluating the efficacy of TMZ alone or in combination with other chemotherapeutic agents, such as capecitabine and irinotecan, retrospective data revealed that sensitivity to TMZ was primarily restricted to pMMR/MSS tumors with complete MGMT protein loss detected with immunohistochemistry[92]. After the first disease response, MGMT re-expression, the selection of sub-clones that express MGMT, or a hypermutated condition resulting from acquired mutations in MMR genes that might make mCRC sensitive to ICIs can all lead to secondary resistance to TMZ[93]. In the MAYA study, 204 of 703 assessed patients (29%) were found to be molecularly suitable. Overall, 142 of 703 (19%) patients were enrolled in phase 1, with just 33 (5% of the initial 703 screened patients) progressing to phase 2. The 8-mo PFS rate was 36% after a median follow-up of 23.1 mo. The median PFS and OS were 7.0 and 18.4 mo, respectively, with a 45% response rate. Skin rash (6%), colitis (3%), and hypophysitis (3%), were all immune-related side effects of grade 3-4 severity. There were no unanticipated AEs or treatment-related fatalities recorded. They concluded that TMZ priming followed by a combination of low-dose ipilimumab and nivolumab might result in long-term therapeutic benefit in MSS and MGMT-silenced mCRC. These findings should be considered with caution due to the lack of a control arm testing the effectiveness of TMZ monotherapy, which prevented the investigators from distinguishing the effect of immunotherapy addition vs TMZ alone. Given that not all patients who are initially susceptible to TMZ develop a hypermutated phenotype, only a subset of individuals may benefit from immunotherapy. Future analyses separating the ORR observed during phase 1 (with TMZ) from the ORR reported during phase 2 (with TMZ plus ipilimumab plus nivolumab), as well as current translational investigations, might reduce this issue.
Despite the need for more mature follow-up data, the good findings from the AtezoTRIBE and MAYA trials bring an end to a long period of stagnation and dismal outcomes in the landscape of immunotherapy in pMMR/MSS mCRC. In the first-line scenario, chemotherapy escalation and TMZ delivery in MGMT-silenced chemo-refractory patients are capable of sensitizing immune-deficient or cold mCRCs to immunotherapy, perhaps rewiring an inflamed/hot TME, then unleashed against the tumor by ICIs. Larger confirmatory and translational trials, however, are required to identify people who benefited the most from these therapies[94].
ADOPTIVE CELL THERAPY
Adoptive cell therapy (ACT), a crucial component of tumor immunotherapy, entails the introduction of immunologically active cells that have been grown and altered in vitro to have direct anticancer action against the cancer-stricken host. Chimeric antigen receptor T (CAR-T) cells, TILs, and T cell receptor-engineered T cells are the three ACT types currently being researched for the treatment of cancer. CAR-T cell therapy entails modifying T cells in vitro in an MHC-independent manner so that they can target tumor antigens and produce an anticancer immune response. With its tremendous effectiveness in treating leukemia, multiple myeloma, some forms of lymphoma, and mCRC, CAR-T cell therapy has a lot of room to grow[95]. Numerous clinical investigations on the safety and efficacy of CAR-T cell treatment are now being conducted in order to determine its therapeutic potential in the field of CRC. One of the first human trials using CAR-T cells to treat metastatic CRC was published by Hege et al[96]. It consisted of two phase 1 experiments with the same CART72 cells (C9701 and C9702). As first-generation CAR-T cells with a CD3-zeta intracellular signaling domain that specifically targeted the tumor-associated glycoprotein-72, CART72 cells were created. The way CART72 was administered in the two studies was different. In trial C9702, patients with CLM received direct hepatic artery infusions, whereas in trial C9701, CART72 was administered intravenously in increasing dosages. Despite a brief blood persistence and modest trafficking to tumor tissue, the data indicated a good safety profile. In addition, rapid clearance following CAR-T cell infusion was linked to CART72 immunogenicity.
Guanylylcyclase2C (GUCY2C) was mentioned by Magee et al[97,98] as a potential CAR-T cell target. In a mouse model lacking autoimmunity, they demonstrated that GUCY2C CAR-T cells may cure parenchymal CRC metastases. Additionally, they showed that GUCY2C targeted CAR-T cell treatment works well against metastatic cancers in mouse models and in human CRC xenograft models. Zhang et al[99] conducted a phase 1 study using CEA-positive CRC patients to create and assess CEA CAR-T cell treatment in 10 resistant and relapsed CRC patients with metastases. CAR-T cells were administered at five increasing dosage levels (1 × 105 to 1 × 108/CAR+/kg cells) to these individuals. The findings demonstrated that there were no significant side effects of CAR-T treatment. Seven of the ten patients—those with progressing illness throughout prior therapies—had stable disease following CAR-T cells therapy. Two patients had tumors removed, and two others had stable illnesses for more than 30 wk. They concluded that CEA CAR-T cell treatment, even at large dosages, was well tolerated in CEA+ CRC patients and that the majority of the treated patients showed some effectiveness.
Several ongoing trials are investigating the use of CAR-T cell in the treatment of CRC[100]. These included the safety, cellular kinetics, and efficacy of CYAD-101, an allogenic CAR-T cell therapy targeting ligands of NKG2D that was administered concurrently with FOLFOX, the efficiency and safety of NKR-2 CAR-T cells, EGFR and EGFR IL 12 CAR-T cell safety and feasibility, the use of anti-carcinoembryonic antigen targeted CAR-T cells, and investigating the efficacy and safety of HER2 chimeric antigen receptor-modified adenovirus-specific cytotoxic T lymphocytes administered in association with intratumoral injection of CAdVEC in patients with unresectable mCRC.
CONCLUSION
With the discovery and comprehension of several molecular and anatomical indicators, the landscape of systemic therapy for mCRC has significantly changed. To find the most effective treatment solution for mCRC patients, a baseline, thorough molecular study is now required. The effect of RAS, BRAF, HER2, POLE, MMR, MSS, and MSI status on therapy choice is summarized in this article. The selection of an efficient first-line therapy, which should consider both clinical factors and molecular signs, is crucial for deciding treatment outcomes. The second-line regimen is chosen based on the systemic therapies utilized in the first. For patients with RAS WT, third-line treatment, which includes EGFR inhibitors, should consider molecular profiling. Immunotherapy with pembrolizumab or nivolumab plus ipilimumab may be an option for patients with high microsatellite instability diseases.
The TME has been identified as a crucial player in CRC tumor growth and metastasis. This process involves all of the components from both bacteria and the host. Although each component has a unique function in CRC growth and metastasis, the majority of them act as a double-edged sword, promoting or inhibiting tumor expansion depending on the setting. TME-modulating treatment methods are showing promise. Many researches have confirmed that altering the TME can result in greater anti-tumor actions. Clinical investigations have indicated that TME remodeling has a high potential for improving medication therapeutic efficacy. Furthermore, because tumors tend to acquire resistance, monotherapy is frequently insufficient. Combining TME remodeling techniques with other potential therapies, such as targeted treatment and immunotherapy, is another component we need to investigate in the near future to reduce treatment resistance.
Footnotes
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: November 29, 2022
First decision: February 8, 2023
Article in press: February 27, 2023
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: Thailand
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Luo Y, China; Wang Z, China S-Editor: Zhang H L-Editor: Filipodia P-Editor: Zhang H
Contributor Information
Wattana Leowattana, Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok 10400, Thailand. wattana.leo@mahidol.ac.th.
Pathomthep Leowattana, Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok 10400, Thailand.
Tawithep Leowattana, Department of Medicine, Faculty of Medicine, Srinakharinwirot University, Bangkok 10110, Thailand.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin . 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Stewart CL, Warner S, Ito K, Raoof M, Wu GX, Kessler J, Kim JY, Fong Y. Cytoreduction for colorectal metastases: liver, lung, peritoneum, lymph nodes, bone, brain. When does it palliate, prolong survival, and potentially cure? Curr Probl Surg . 2018;55:330–379. doi: 10.1067/j.cpsurg.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. American Cancer Society: About Colorectal Cancer (2022). [cited 4 November 2022]. Available from: https://www.cancer.org/cancer/colon-rectal-cancer/about.html/
- 4.Hughes KS, Rosenstein RB, Songhorabodi S, Adson MA, Ilstrup DM, Fortner JG, Maclean BJ, Foster JH, Daly JM, Fitzherbert D. Resection of the liver for colorectal carcinoma metastases. A multi-institutional study of long-term survivors. Dis Colon Rectum . 1988;31:1–4. doi: 10.1007/bf02552560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baidoun F, Elshiwy K, Elkeraie Y, Merjaneh Z, Khoudari G, Sarmini MT, Gad M, Al-Husseini M, Saad A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr Drug Targets . 2021;22:998–1009. doi: 10.2174/1389450121999201117115717. [DOI] [PubMed] [Google Scholar]
- 6.Santos AJM, Lo YH, Mah AT, Kuo CJ. The Intestinal Stem Cell Niche: Homeostasis and Adaptations. Trends Cell Biol . 2018;28:1062–1078. doi: 10.1016/j.tcb.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chacón-Martínez CA, Koester J, Wickström SA. Signaling in the stem cell niche: regulating cell fate, function and plasticity. Development . 2018;145 doi: 10.1242/dev.165399. [DOI] [PubMed] [Google Scholar]
- 8.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology . 2010;138:2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu H, Hemminki K. Genetic epidemiology of colorectal cancer and associated cancers. Mutagenesis . 2020;35:207–219. doi: 10.1093/mutage/gez022. [DOI] [PubMed] [Google Scholar]
- 10.Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol . 2019;16:713–732. doi: 10.1038/s41575-019-0189-8. [DOI] [PubMed] [Google Scholar]
- 11.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut . 2017;66:683–691. doi: 10.1136/gutjnl-2015-310912. [DOI] [PubMed] [Google Scholar]
- 12.GBD 2019 Colorectal Cancer Collaborators. Global, regional, and national burden of colorectal cancer and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol Hepatol . 2022;7:627–647. doi: 10.1016/S2468-1253(22)00044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vieira AR, Abar L, Chan DSM, Vingeliene S, Polemiti E, Stevens C, Greenwood D, Norat T. Foods and beverages and colorectal cancer risk: a systematic review and meta-analysis of cohort studies, an update of the evidence of the WCRF-AICR Continuous Update Project. Ann Oncol . 2017;28:1788–1802. doi: 10.1093/annonc/mdx171. [DOI] [PubMed] [Google Scholar]
- 14.Figueiredo JC, Crockett SD, Snover DC, Morris CB, McKeown-Eyssen G, Sandler RS, Ahnen DJ, Robertson DJ, Burke CA, Bresalier RS, Church JM, Church TR, Baron JA. Smoking-associated risks of conventional adenomas and serrated polyps in the colorectum. Cancer Causes Control . 2015;26:377–386. doi: 10.1007/s10552-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang PS, Chen TY, Giovannucci E. Cigarette smoking and colorectal cancer incidence and mortality: systematic review and meta-analysis. Int J Cancer . 2009;124:2406–2415. doi: 10.1002/ijc.24191. [DOI] [PubMed] [Google Scholar]
- 16.Choi YJ, Myung SK, Lee JH. Light Alcohol Drinking and Risk of Cancer: A Meta-Analysis of Cohort Studies. Cancer Res Treat . 2018;50:474–487. doi: 10.4143/crt.2017.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong Y, Zhou J, Zhu Y, Luo L, He T, Hu H, Liu H, Zhang Y, Luo D, Xu S, Xu L, Liu J, Zhang J, Teng Z. Abdominal obesity and colorectal cancer risk: systematic review and meta-analysis of prospective studies. Biosci Rep . 2017;37 doi: 10.1042/BSR20170945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu PH, Wu K, Ng K, Zauber AG, Nguyen LH, Song M, He X, Fuchs CS, Ogino S, Willett WC, Chan AT, Giovannucci EL, Cao Y. Association of Obesity With Risk of Early-Onset Colorectal Cancer Among Women. JAMA Oncol . 2019;5:37–44. doi: 10.1001/jamaoncol.2018.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graff RE, Möller S, Passarelli MN, Witte JS, Skytthe A, Christensen K, Tan Q, Adami HO, Czene K, Harris JR, Pukkala E, Kaprio J, Giovannucci EL, Mucci LA, Hjelmborg JB. Familial Risk and Heritability of Colorectal Cancer in the Nordic Twin Study of Cancer. Clin Gastroenterol Hepatol . 2017;15:1256–1264. doi: 10.1016/j.cgh.2016.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, Goldberg RM, Paskett E, Shields PG, Freudenheim JL, Stanich PP, Lattimer I, Arnold M, Liyanarachchi S, Kalady M, Heald B, Greenwood C, Paquette I, Prues M, Draper DJ, Lindeman C, Kuebler JP, Reynolds K, Brell JM, Shaper AA, Mahesh S, Buie N, Weeman K, Shine K, Haut M, Edwards J, Bastola S, Wickham K, Khanduja KS, Zacks R, Pritchard CC, Shirts BH, Jacobson A, Allen B, de la Chapelle A, Hampel H Ohio Colorectal Cancer Prevention Initiative Study Group. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol . 2017;3:464–471. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talseth-Palmer BA. The genetic basis of colonic adenomatous polyposis syndromes. Hered Cancer Clin Pract . 2017;15:5. doi: 10.1186/s13053-017-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Augustus GJ, Ellis NA. Colorectal Cancer Disparity in African Americans: Risk Factors and Carcinogenic Mechanisms. Am J Pathol . 2018;188:291–303. doi: 10.1016/j.ajpath.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinhart K, Bannert C, Dunkler D, Salzl P, Trauner M, Renner F, Knoflach P, Ferlitsch A, Weiss W, Ferlitsch M. Prevalence of flat lesions in a large screening population and their role in colonoscopy quality improvement. Endoscopy . 2013;45:350–356. doi: 10.1055/s-0032-1326348. [DOI] [PubMed] [Google Scholar]
- 24.Guo TJ, Chen W, Chen Y, Wu JC, Wang YP, Yang JL. Diagnostic performance of magnifying endoscopy with narrow-band imaging in differentiating neoplastic colorectal polyps from non-neoplastic colorectal polyps: a meta-analysis. J Gastroenterol . 2018;53:701–711. doi: 10.1007/s00535-018-1436-4. [DOI] [PubMed] [Google Scholar]
- 25.Malki A, ElRuz RA, Gupta I, Allouch A, Vranic S, Al Moustafa AE. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int J Mol Sci . 2020;22 doi: 10.3390/ijms22010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukund K, Syulyukina N, Ramamoorthy S, Subramaniam S. Right and left-sided colon cancers - specificity of molecular mechanisms in tumorigenesis and progression. BMC Cancer . 2020;20:317. doi: 10.1186/s12885-020-06784-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol . 2015;6:660–667. doi: 10.3978/j.issn.2078-6891.2015.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer . 2021;20:143. doi: 10.1186/s12943-021-01441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan C, Du X. KRAS mutation testing in metastatic colorectal cancer. World J Gastroenterol . 2012;18:5171–5180. doi: 10.3748/wjg.v18.i37.5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margonis GA, Spolverato G, Kim Y, Karagkounis G, Choti MA, Pawlik TM. Effect of KRAS Mutation on Long-Term Outcomes of Patients Undergoing Hepatic Resection for Colorectal Liver Metastases. Ann Surg Oncol . 2015;22:4158–4165. doi: 10.1245/s10434-015-4587-z. [DOI] [PubMed] [Google Scholar]
- 31.Passot G, Denbo JW, Yamashita S, Kopetz SE, Chun YS, Maru D, Overman MJ, Brudvik KW, Conrad C, Aloia TA, Vauthey JN. Is hepatectomy justified for patients with RAS mutant colorectal liver metastases? Surgery . 2017;161:332–340. doi: 10.1016/j.surg.2016.07.032. [DOI] [PubMed] [Google Scholar]
- 32.Vale CL, Tierney JF, Fisher D, Adams RA, Kaplan R, Maughan TS, Parmar MK, Meade AM. Does anti-EGFR therapy improve outcome in advanced colorectal cancer? Cancer Treat Rev . 2012;38:618–625. doi: 10.1016/j.ctrv.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Shaib W, Mahajan R, El-Rayes B. Markers of resistance to anti-EGFR therapy in colorectal cancer. J Gastrointest Oncol . 2013;4:308–318. doi: 10.3978/j.issn.2078-6891.2013.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schirripa M, Bergamo F, Cremolini C, Casagrande M, Lonardi S, Aprile G, Yang D, Marmorino F, Pasquini G, Sensi E, Lupi C, De Maglio G, Borrelli N, Pizzolitto S, Fasola G, Bertorelle R, Rugge M, Fontanini G, Zagonel V, Loupakis F, Falcone A. BRAF and RAS mutations as prognostic factors in metastatic colorectal cancer patients undergoing liver resection. Br J Cancer . 2015;112:1921–1928. doi: 10.1038/bjc.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tie J, Desai J. Targeting BRAF mutant metastatic colorectal cancer: clinical implications and emerging therapeutic strategies. Target Oncol . 2015;10:179–188. doi: 10.1007/s11523-014-0330-0. [DOI] [PubMed] [Google Scholar]
- 36.Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, Qian ZR, Morikawa T, Shen J, Meyerhardt JA, Fuchs CS, Ogino S. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst . 2013;105:1151–1156. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, Bertotti A, Siravegna G, Torri V, Amatu A, Ghezzi S, Marrapese G, Palmeri L, Valtorta E, Cassingena A, Lauricella C, Vanzulli A, Regge D, Veronese S, Comoglio PM, Bardelli A, Marsoni S, Siena S. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol . 2016;17:738–746. doi: 10.1016/S1470-2045(16)00150-9. [DOI] [PubMed] [Google Scholar]
- 38.Wang G, He Y, Sun Y, Wang W, Qian X, Yu X, Pan Y. Prevalence, prognosis and predictive status of HER2 amplification in anti-EGFR-resistant metastatic colorectal cancer. Clin Transl Oncol . 2020;22:813–822. doi: 10.1007/s12094-019-02213-9. [DOI] [PubMed] [Google Scholar]
- 39.Keam SJ. Trastuzumab Deruxtecan: First Approval. Drugs . 2020;80:501–508. doi: 10.1007/s40265-020-01281-4. [DOI] [PubMed] [Google Scholar]
- 40.van der Pool AE, Damhuis RA, Ijzermans JN, de Wilt JH, Eggermont AM, Kranse R, Verhoef C. Trends in incidence, treatment and survival of patients with stage IV colorectal cancer: a population-based series. Colorectal Dis . 2012;14:56–61. doi: 10.1111/j.1463-1318.2010.02539.x. [DOI] [PubMed] [Google Scholar]
- 41.Ma R, Li T. Conversion therapy combined with individualized surgical treatment strategy improves survival in patients with colorectal cancer liver metastases. Int J Clin Exp Pathol . 2021;14:314–321. [PMC free article] [PubMed] [Google Scholar]
- 42.Gruenberger B, Tamandl D, Schueller J, Scheithauer W, Zielinski C, Herbst F, Gruenberger T. Bevacizumab, capecitabine, and oxaliplatin as neoadjuvant therapy for patients with potentially curable metastatic colorectal cancer. J Clin Oncol . 2008;26:1830–1835. doi: 10.1200/JCO.2007.13.7679. [DOI] [PubMed] [Google Scholar]
- 43.Bridgewater JA, Pugh SA, Maishman T, Eminton Z, Mellor J, Whitehead A, Stanton L, Radford M, Corkhill A, Griffiths GO, Falk S, Valle JW, O'Reilly D, Siriwardena AK, Hornbuckle J, Rees M, Iveson TJ, Hickish T, Garden OJ, Cunningham D, Maughan TS, Primrose JN New EPOC investigators. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis (New EPOC): long-term results of a multicentre, randomised, controlled, phase 3 trial. Lancet Oncol . 2020;21:398–411. doi: 10.1016/S1470-2045(19)30798-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong R, Cunningham D, Barbachano Y, Saffery C, Valle J, Hickish T, Mudan S, Brown G, Khan A, Wotherspoon A, Strimpakos AS, Thomas J, Compton S, Chua YJ, Chau I. A multicentre study of capecitabine, oxaliplatin plus bevacizumab as perioperative treatment of patients with poor-risk colorectal liver-only metastases not selected for upfront resection. Ann Oncol . 2011;22:2042–2048. doi: 10.1093/annonc/mdq714. [DOI] [PubMed] [Google Scholar]
- 45.Gruenberger T, Bridgewater J, Chau I, García Alfonso P, Rivoire M, Mudan S, Lasserre S, Hermann F, Waterkamp D, Adam R. Bevacizumab plus mFOLFOX-6 or FOLFOXIRI in patients with initially unresectable liver metastases from colorectal cancer: the OLIVIA multinational randomised phase II trial. Ann Oncol . 2015;26:702–708. doi: 10.1093/annonc/mdu580. [DOI] [PubMed] [Google Scholar]
- 46.Kemeny NE, Jarnagin WR, Capanu M, Fong Y, Gewirtz AN, Dematteo RP, D'Angelica MI. Randomized phase II trial of adjuvant hepatic arterial infusion and systemic chemotherapy with or without bevacizumab in patients with resected hepatic metastases from colorectal cancer. J Clin Oncol . 2011;29:884–889. doi: 10.1200/JCO.2010.32.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garufi C, Torsello A, Tumolo S, Ettorre GM, Zeuli M, Campanella C, Vennarecci G, Mottolese M, Sperduti I, Cognetti F. Cetuximab plus chronomodulated irinotecan, 5-fluorouracil, leucovorin and oxaliplatin as neoadjuvant chemotherapy in colorectal liver metastases: POCHER trial. Br J Cancer . 2010;103:1542–1547. doi: 10.1038/sj.bjc.6605940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Folprecht G, Gruenberger T, Bechstein W, Raab HR, Weitz J, Lordick F, Hartmann JT, Stoehlmacher-Williams J, Lang H, Trarbach T, Liersch T, Ockert D, Jaeger D, Steger U, Suedhoff T, Rentsch A, Köhne CH. Survival of patients with initially unresectable colorectal liver metastases treated with FOLFOX/cetuximab or FOLFIRI/cetuximab in a multidisciplinary concept (CELIM study) Ann Oncol . 2014;25:1018–1025. doi: 10.1093/annonc/mdu088. [DOI] [PubMed] [Google Scholar]
- 49.Zheng B, Wang X, Wei M, Wang Q, Li J, Bi L, Deng X, Wang Z. First-line cetuximab vs bevacizumab for RAS and BRAF wild-type metastatic colorectal cancer: a systematic review and meta-analysis. BMC Cancer . 2019;19:280. doi: 10.1186/s12885-019-5481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rivera F, Karthaus M, Hecht JR, Sevilla I, Forget F, Fasola G, Canon JL, Guan X, Demonty G, Schwartzberg LS. Final analysis of the randomised PEAK trial: overall survival and tumour responses during first-line treatment with mFOLFOX6 plus either panitumumab or bevacizumab in patients with metastatic colorectal carcinoma. Int J Colorectal Dis . 2017;32:1179–1190. doi: 10.1007/s00384-017-2800-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold D, Lueza B, Douillard JY, Peeters M, Lenz HJ, Venook A, Heinemann V, Van Cutsem E, Pignon JP, Tabernero J, Cervantes A, Ciardiello F. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol . 2017;28:1713–1729. doi: 10.1093/annonc/mdx175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holch JW, Ricard I, Stintzing S, Modest DP, Heinemann V. The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta-analysis of first-line clinical trials. Eur J Cancer . 2017;70:87–98. doi: 10.1016/j.ejca.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 53.Stintzing S, Modest DP, Rossius L, Lerch MM, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, Held S, Giessen-Jung C, Moehler M, Jagenburg A, Kirchner T, Jung A, Heinemann V FIRE-3 investigators. FOLFIRI plus cetuximab vs FOLFIRI plus bevacizumab for metastatic colorectal cancer (FIRE-3): a post-hoc analysis of tumour dynamics in the final RAS wild-type subgroup of this randomised open-label phase 3 trial. Lancet Oncol . 2016;17:1426–1434. doi: 10.1016/S1470-2045(16)30269-8. [DOI] [PubMed] [Google Scholar]
- 54.Piawah S, Venook AP. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer . 2019;125:4139–4147. doi: 10.1002/cncr.32163. [DOI] [PubMed] [Google Scholar]
- 55.Masi G, Salvatore L, Boni L, Loupakis F, Cremolini C, Fornaro L, Schirripa M, Cupini S, Barbara C, Safina V, Granetto C, Fea E, Antonuzzo L, Boni C, Allegrini G, Chiara S, Amoroso D, Bonetti A, Falcone A BEBYP Study Investigators. Continuation or reintroduction of bevacizumab beyond progression to first-line therapy in metastatic colorectal cancer: final results of the randomized BEBYP trial. Ann Oncol . 2015;26:724–730. doi: 10.1093/annonc/mdv012. [DOI] [PubMed] [Google Scholar]
- 56.Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Van Cutsem E, von Moos R, Viéitez JM, Bouché O, Borg C, Steffens CC, Alonso-Orduña V, Schlichting C, Reyes-Rivera I, Bendahmane B, André T, Kubicka S ML18147 Study Investigators. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol . 2013;14:29–37. doi: 10.1016/S1470-2045(12)70477-1. [DOI] [PubMed] [Google Scholar]
- 57.Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, Ciuleanu TE, Portnoy DC, Van Cutsem E, Grothey A, Prausová J, Garcia-Alfonso P, Yamazaki K, Clingan PR, Lonardi S, Kim TW, Simms L, Chang SC, Nasroulah F RAISE Study Investigators. Ramucirumab vs placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol . 2015;16:499–508. doi: 10.1016/S1470-2045(15)70127-0. [DOI] [PubMed] [Google Scholar]
- 58.Pfeiffer P, Yilmaz M, Möller S, Zitnjak D, Krogh M, Petersen LN, Poulsen LØ, Winther SB, Thomsen KG, Qvortrup C. TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: an investigator-initiated, open-label, randomised, phase 2 trial. Lancet Oncol . 2020;21:412–420. doi: 10.1016/S1470-2045(19)30827-7. [DOI] [PubMed] [Google Scholar]
- 59.Cremolini C, Rossini D, Dell'Aquila E, Lonardi S, Conca E, Del Re M, Busico A, Pietrantonio F, Danesi R, Aprile G, Tamburini E, Barone C, Masi G, Pantano F, Pucci F, Corsi DC, Pella N, Bergamo F, Rofi E, Barbara C, Falcone A, Santini D. Rechallenge for Patients With RAS and BRAF Wild-Type Metastatic Colorectal Cancer With Acquired Resistance to First-line Cetuximab and Irinotecan: A Phase 2 Single-Arm Clinical Trial. JAMA Oncol . 2019;5:343–350. doi: 10.1001/jamaoncol.2018.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sartore-Bianchi A, Pietrantonio F, Lonardi S, Mussolin B, Rua F, Crisafulli G, Bartolini A, Fenocchio E, Amatu A, Manca P, Bergamo F, Tosi F, Mauri G, Ambrosini M, Daniel F, Torri V, Vanzulli A, Regge D, Cappello G, Marchiò C, Berrino E, Sapino A, Marsoni S, Siena S, Bardelli A. Circulating tumor DNA to guide rechallenge with panitumumab in metastatic colorectal cancer: the phase 2 CHRONOS trial. Nat Med . 2022;28:1612–1618. doi: 10.1038/s41591-022-01886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parseghian CM, Loree JM, Morris VK, Liu X, Clifton KK, Napolitano S, Henry JT, Pereira AA, Vilar E, Johnson B, Kee B, Raghav K, Dasari A, Wu J, Garg N, Raymond VM, Banks KC, Talasaz AA, Lanman RB, Strickler JH, Hong DS, Corcoran RB, Overman MJ, Kopetz S. Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol . 2019;30:243–249. doi: 10.1093/annonc/mdy509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol . 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 63.Kopetz S, Guthrie KA, Morris VK, Lenz HJ, Magliocco AM, Maru D, Yan Y, Lanman R, Manyam G, Hong DS, Sorokin A, Atreya CE, Diaz LA, Allegra C, Raghav KP, Wang SE, Lieu CH, McDonough SL, Philip PA, Hochster HS. Randomized Trial of Irinotecan and Cetuximab With or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406) J Clin Oncol . 2021;39:285–294. doi: 10.1200/JCO.20.01994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, Desai J, Ciardiello F, Loupakis F, Hong YS, Steeghs N, Guren TK, Arkenau HT, Garcia-Alfonso P, Elez E, Gollerkeri A, Maharry K, Christy-Bittel J, Kopetz S. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J Clin Oncol . 2021;39:273–284. doi: 10.1200/JCO.20.02088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huijberts SC, van Geel RM, Bernards R, Beijnen JH, Steeghs N. Encorafenib, binimetinib and cetuximab combined therapy for patients with BRAFV600E mutant metastatic colorectal cancer. Future Oncol . 2020;16:161–173. doi: 10.2217/fon-2019-0748. [DOI] [PubMed] [Google Scholar]
- 66.Ross JS, Fakih M, Ali SM, Elvin JA, Schrock AB, Suh J, Vergilio JA, Ramkissoon S, Severson E, Daniel S, Fabrizio D, Frampton G, Sun J, Miller VA, Stephens PJ, Gay LM. Targeting HER2 in colorectal cancer: The landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer . 2018;124:1358–1373. doi: 10.1002/cncr.31125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meric-Bernstam F, Hurwitz H, Raghav KPS, McWilliams RR, Fakih M, VanderWalde A, Swanton C, Kurzrock R, Burris H, Sweeney C, Bose R, Spigel DR, Beattie MS, Blotner S, Stone A, Schulze K, Cuchelkar V, Hainsworth J. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol . 2019;20:518–530. doi: 10.1016/S1470-2045(18)30904-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siena S, Di Bartolomeo M, Raghav K, Masuishi T, Loupakis F, Kawakami H, Yamaguchi K, Nishina T, Fakih M, Elez E, Rodriguez J, Ciardiello F, Komatsu Y, Esaki T, Chung K, Wainberg Z, Sartore-Bianchi A, Saxena K, Yamamoto E, Bako E, Okuda Y, Shahidi J, Grothey A, Yoshino T DESTINY-CRC01 investigators. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol . 2021;22:779–789. doi: 10.1016/S1470-2045(21)00086-3. [DOI] [PubMed] [Google Scholar]
- 69.Lange AM, Lo HW. Inhibiting TRK Proteins in Clinical Cancer Therapy. Cancers (Basel) . 2018;10 doi: 10.3390/cancers10040105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, Blakely CM, Seto T, Cho BC, Tosi D, Besse B, Chawla SP, Bazhenova L, Krauss JC, Chae YK, Barve M, Garrido-Laguna I, Liu SV, Conkling P, John T, Fakih M, Sigal D, Loong HH, Buchschacher GL Jr, Garrido P, Nieva J, Steuer C, Overbeck TR, Bowles DW, Fox E, Riehl T, Chow-Maneval E, Simmons B, Cui N, Johnson A, Eng S, Wilson TR, Demetri GD trial investigators. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol . 2020;21:271–282. doi: 10.1016/S1470-2045(19)30691-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero J, Komatsu Y, Sobrero A, Boucher E, Peeters M, Tran B, Lenz HJ, Zaniboni A, Hochster H, Cleary JM, Prenen H, Benedetti F, Mizuguchi H, Makris L, Ito M, Ohtsu A RECOURSE Study Group. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med . 2015;372:1909–1919. doi: 10.1056/NEJMoa1414325. [DOI] [PubMed] [Google Scholar]
- 72.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest . 2015;125:3335–3337. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X, Lu Y. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci . 2021;17:3837–3849. doi: 10.7150/ijbs.64077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol . 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, Vogelstein B, Sears CL, Anders RA, Pardoll DM, Housseau F. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov . 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou C, Jiang T, Xiao Y, Wang Q, Zeng Z, Cai P, Zhao Y, Zhao Z, Wu D, Lin H, Sun C, Zhang R, Xiao W, Gao Y. Good Tumor Response to Chemoradioimmunotherapy in dMMR/MSI-H Advanced Colorectal Cancer: A Case Series. Front Immunol . 2021;12:784336. doi: 10.3389/fimmu.2021.784336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Golas MM, Gunawan B, Cakir M, Cameron S, Enders C, Liersch T, Füzesi L, Sander B. Evolutionary patterns of chromosomal instability and mismatch repair deficiency in proximal and distal colorectal cancer. Colorectal Dis . 2022;24:157–176. doi: 10.1111/codi.15946. [DOI] [PubMed] [Google Scholar]
- 78.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med . 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, Burge M, O'Neil B, Kavan P, Yoshino T, Guimbaud R, Taniguchi H, Elez E, Al-Batran SE, Boland PM, Crocenzi T, Atreya CE, Cui Y, Dai T, Marinello P, Diaz LA Jr, André T. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol . 2020;38:11–19. doi: 10.1200/JCO.19.02107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med . 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 81.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, Goldberg MV, Cao ZA, Ledeine JM, Maglinte GA, Kopetz S, André T. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol . 2017;18:1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.André T, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, Sawyer MB, Hendlisz A, Neyns B, Abdullaev S, Memaj A, Lei M, Dixon M, Kopetz S, Overman MJ. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann Oncol . 2022;33:1052–1060. doi: 10.1016/j.annonc.2022.06.008. [DOI] [PubMed] [Google Scholar]
- 83.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A, Sawyer MB, Hendlisz A, Neyns B, Svrcek M, Moss RA, Ledeine JM, Cao ZA, Kamble S, Kopetz S, André T. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol . 2018;36:773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 84.Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, García-Alfonso P, Neyns B, Luppi G, Cardin DB, Dragovich T, Shah U, Abdullaev S, Gricar J, Ledeine JM, Overman MJ, Lonardi S. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J Clin Oncol . 2022;40:161–170. doi: 10.1200/JCO.21.01015. [DOI] [PubMed] [Google Scholar]
- 85.Kim JH, Kim SY, Baek JY, Cha YJ, Ahn JB, Kim HS, Lee KW, Kim JW, Kim TY, Chang WJ, Park JO, Kim J, Kim JE, Hong YS, Kim YH, Kim TW. A Phase II Study of Avelumab Monotherapy in Patients with Mismatch Repair-Deficient/Microsatellite Instability-High or POLE-Mutated Metastatic or Unresectable Colorectal Cancer. Cancer Res Treat . 2020;52:1135–1144. doi: 10.4143/crt.2020.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martinelli E, Martini G, Famiglietti V, Troiani T, Napolitano S, Pietrantonio F, Ciardiello D, Terminiello M, Borrelli C, Vitiello PP, De Braud F, Morano F, Avallone A, Normanno N, Nappi A, Maiello E, Latiano T, Falcone A, Cremolini C, Rossini D, Santabarbara G, Pinto C, Santini D, Cardone C, Zanaletti N, Di Liello A, Renato D, Esposito L, Marrone F, Ciardiello F. Cetuximab Rechallenge Plus Avelumab in Pretreated Patients With RAS Wild-type Metastatic Colorectal Cancer: The Phase 2 Single-Arm Clinical CAVE Trial. JAMA Oncol . 2021;7:1529–1535. doi: 10.1001/jamaoncol.2021.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lote H, Starling N, Pihlak R, Gerlinger M. Advances in immunotherapy for MMR proficient colorectal cancer. Cancer Treat Rev . 2022;111:102480. doi: 10.1016/j.ctrv.2022.102480. [DOI] [PubMed] [Google Scholar]
- 88.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity . 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herting CJ, Farren MR, Tong Y, Liu Z, O'Neil B, Bekaii-Saab T, Noonan A, McQuinn C, Mace TA, Shaib W, Wu C, El-Rayes BF, Shahda S, Lesinski GB. A multi-center, single-arm, phase Ib study of pembrolizumab (MK-3475) in combination with chemotherapy for patients with advanced colorectal cancer: HCRN GI14-186. Cancer Immunol Immunother . 2021;70:3337–3348. doi: 10.1007/s00262-021-02986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Antoniotti C, Rossini D, Pietrantonio F, Catteau A, Salvatore L, Lonardi S, Boquet I, Tamberi S, Marmorino F, Moretto R, Ambrosini M, Tamburini E, Tortora G, Passardi A, Bergamo F, Kassambara A, Sbarrato T, Morano F, Ritorto G, Borelli B, Boccaccino A, Conca V, Giordano M, Ugolini C, Fieschi J, Papadopulos A, Massoué C, Aprile G, Antonuzzo L, Gelsomino F, Martinelli E, Pella N, Masi G, Fontanini G, Boni L, Galon J, Cremolini C GONO Foundation Investigators. Upfront FOLFOXIRI plus bevacizumab with or without atezolizumab in the treatment of patients with metastatic colorectal cancer (AtezoTRIBE): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol . 2022;23:876–887. doi: 10.1016/S1470-2045(22)00274-1. [DOI] [PubMed] [Google Scholar]
- 91.Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, Murgioni S, Randon G, Palermo F, Antonuzzo L, Pella N, Racca P, Prisciandaro M, Niger M, Corti F, Bergamo F, Zaniboni A, Ratti M, Palazzo M, Cagnazzo C, Calegari MA, Marmorino F, Capone I, Conca E, Busico A, Brich S, Tamborini E, Perrone F, Di Maio M, Milione M, Di Bartolomeo M, de Braud F, Pietrantonio F. Temozolomide Followed by Combination With Low-Dose Ipilimumab and Nivolumab in Patients With Microsatellite-Stable, O(6)-Methylguanine-DNA Methyltransferase-Silenced Metastatic Colorectal Cancer: The MAYA Trial. J Clin Oncol . 2022;40:1562–1573. doi: 10.1200/JCO.21.02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pietrantonio F, Lobefaro R, Antista M, Lonardi S, Raimondi A, Morano F, Mosconi S, Rimassa L, Murgioni S, Sartore-Bianchi A, Tomasello G, Longarini R, Farina G, Petrelli F, Gori S, Randon G, Corallo S, Pagani F, Guarini V, Palermo F, Martinetti A, Macagno M, Barault L, Perrone F, Tamborini E, Milione M, Di Nicolantonio F, Di Maio M, Fucà G, Di Bartolomeo M, de Braud F. Capecitabine and Temozolomide vs FOLFIRI in RAS-Mutated, MGMT-Methylated Metastatic Colorectal Cancer. Clin Cancer Res . 2020;26:1017–1024. doi: 10.1158/1078-0432.CCR-19-3024. [DOI] [PubMed] [Google Scholar]
- 93.Germano G, Lamba S, Rospo G, Barault L, Magrì A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G, Siravegna G, Mussolin B, Frapolli R, Montone M, Morano F, de Braud F, Amirouchene-Angelozzi N, Marsoni S, D'Incalci M, Orlandi A, Giraudo E, Sartore-Bianchi A, Siena S, Pietrantonio F, Di Nicolantonio F, Bardelli A. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature . 2017;552:116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 94.Germani MM, Moretto R. Immune Checkpoint Inhibitors in Mismatch Repair Proficient/Microsatellite Stable Metastatic Colorectal Cancer Patients: Insights from the AtezoTRIBE and MAYA Trials. Cancers (Basel) . 2021;14 doi: 10.3390/cancers14010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Z, Cao YJ. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front Immunol . 2020;11:176. doi: 10.3389/fimmu.2020.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, Sherwin SA. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer . 2017;5:22. doi: 10.1186/s40425-017-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P, Waldman SA, Snook AE. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology . 2016;5:e1227897. doi: 10.1080/2162402X.2016.1227897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Magee MS, Abraham TS, Baybutt TR, Flickinger JC Jr, Ridge NA, Marszalowicz GP, Prajapati P, Hersperger AR, Waldman SA, Snook AE. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol Res . 2018;6:509–516. doi: 10.1158/2326-6066.CIR-16-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, Zhang R, Xiong Z, Wei Z, Shen J, Luo Y, Zhang Q, Liu L, Qin H, Liu W, Wu F, Chen W, Pan F, Zhang X, Bie P, Liang H, Pecher G, Qian C. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther . 2017;25:1248–1258. doi: 10.1016/j.ymthe.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang Q, Xia J, Wang L, Wang X, Ma X, Deng Q, Lu Y, Kumar M, Zhou Z, Li L, Zeng Z, Young KH, Yi Q, Zhang M, Li Y. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol . 2018;11:58. doi: 10.1186/s13045-018-0600-x. [DOI] [PMC free article] [PubMed] [Google Scholar]