

Abstract
Akuammine (1) and pseudo-akuammigine (2) are indole alkaloids found in the seeds of the akuamma tree (Picralima nitida). Both alkaloids are weak agonists of the mu opioid receptor (μOR); however, they produce minimal effects in animal models of antinociception. To probe the interactions of 1 and 2 at the opioid receptors, we have prepared a collection of 22 semi-synthetic derivatives. Evaluation of this collection at the μOR and kappa opioid receptor (κOR) revealed structural-activity relationship trends and derivatives with improved potency at the μOR. Most notably, the introduction of a phenethyl moiety to the N1 of 2 produces a 70-fold increase in potency and a 7-fold increase in selectivity for the μOR. The in vitro potency of this compound resulted in increased efficacy in the tail-flick and hot-plate assays of antinociception. The improved potency of these derivatives highlights the promise of exploring natural product scaffolds to probe the opioid receptors.
Keywords: Opioid receptor, pain, natural products
Graphical Abstract
Introduction
Amid the opioid crisis, opioid-related deaths have been on the rise with over 90 Americans dying from overdoses each day.1 Whereas in previous decades opioid deaths have been dominated by analgesics like morphine and its semi-synthetic derivatives, more recently more potent synthetic opioids like fentanyl have been the larger source of fatalities.2 Regardless of the drug class, clinically used and illicit opioids produce their powerful analgesic effects through the potent activation of the mu opioid receptor (μOR).3, 4 This same activation of the μOR also produces life-threatening respiratory depression responsible for opioid-related deaths and along with other adverse effects including constipation, tolerance, and dependence that limit the clinical utility of opioid analgesics.5 Thus, while opioid analgesics remain an essential component of modern medicine, there is a pressing need to develop safer opioid analgesics.
Several promising strategies have emerged to develop safer opioid analgesics including the development of biased agonists,6 mixed pharmacology,7 partial agonism,8–10 peripheral restriction,11 and investigation of delta and kappa opioid receptor (δOR and κOR) agonists.12 The exploration of each of these strategies has been greatly aided by the discovery and design of novel opioid ligands, particularly those that lack the common pharmacophore shared by morphinan and fentanyl-derived analgesics.13 For example, the salvinorin A derivative herkinorin was among the first non-nitrogenous μOR agonists and provided the first evidence for signaling bias with respect to β-Arrestin-2 recruitment at the μOR.14 More recently, of the structure-activity relationships (SAR) studies on the mitragynine alkaloids have revealed how even small changes to the scaffold allow for the fine-tuning of opioid receptor signaling.8, 15–17 In addition to providing new chemical tools to probe the opioid receptor and its function, the development of these novel classes of opioids offers new opportunities to develop potential analgesics.
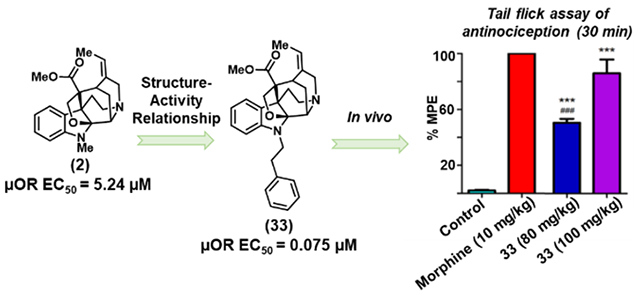
In light of the promising advantages of investigating structurally distinct μOR agonists, we began re-investigating the alkaloids of the akuamma plant (Picralima nitida),18 which has been used in West Africa as a traditional treatment for opioid withdrawal, fever, and pain.19 We recently developed an extraction protocol that allows for the isolation of six monoterpene indole alkaloids known as the akuamma alkaloids directly from commercial P. nitida seeds (Figure 1).20 As a class, these alkaloids are structurally distinct from traditional opioid ligands represented by morphine and fentanyl. Akuammine (1), pseudo-akuammigine (2), and akuammidine (3) act as weakly potent μOR agonists with potencies ranging from 2.6 – 5.2 μM. Due to their agonist activity at the μOR, 1-3 were evaluated in thermal nociception assays in mice. However, contrary to previous studies and the traditional use of P. nitida as an analgesic, 1-3 produced minimal changes to pain-like behavior, which is consistent with their low potency at the μOR.20, 21 Given the low potency of these alkaloids at the μOR, we reasoned that a derivative with increased potency at the μOR may produce more effective antinociceptive effects. To test this hypothesis, here we report the first SAR study of the akuamma alkaloids 1 and 2. These efforts were made possible by the considerable quantities of 1 that can be isolated from our extraction protocol that, in turn, allowed us to employ strategic, chemoselective, late-stage functionalization of the indole nucleus, to introduce modifications at the C10, C11, and N1 positions of 1 and 2. Ultimately, these SAR studies resulted in the discovery of a μOR agonist with 27-fold greater affinity (Ki = 12 nM) and 70-fold greater potency (EC50 = 75 nM) than the parent compound 2. This increase in affinity and agonistic potency highlights the utility of semi-synthesis to discover new ligand classes that can serve as both suitable chemical probes to further understand opioid receptor signaling and promising drug leads to develop much-needed safer opioid analgesics.
Figure 1.
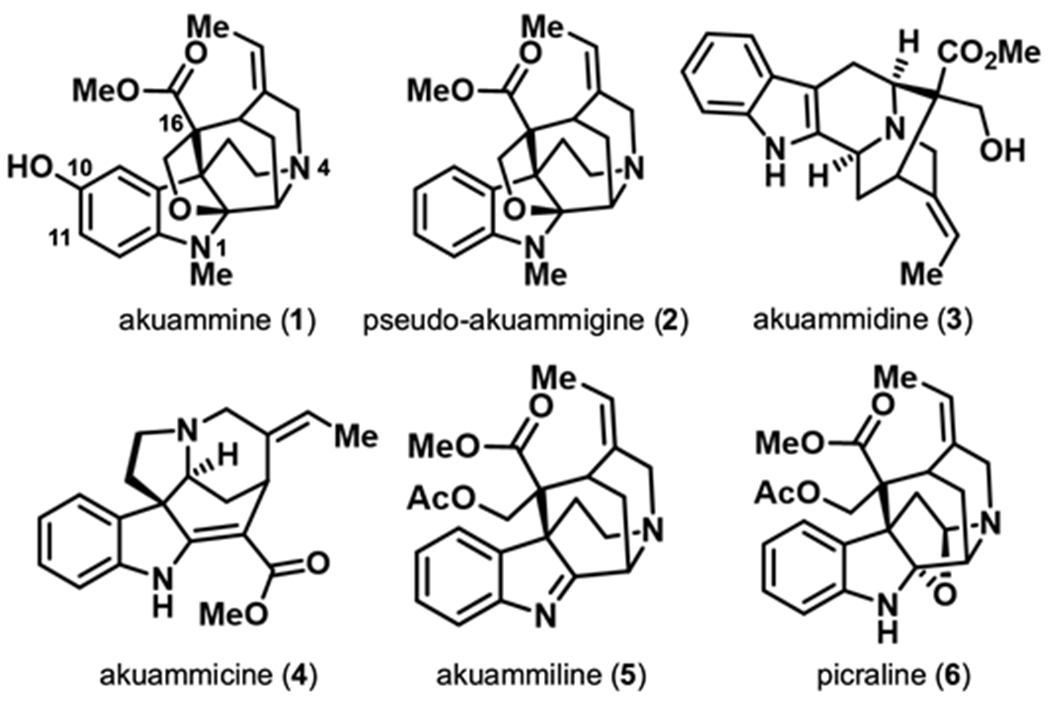
The six major alkaloids (1-6) isolated from akuamma seeds.
Results
The structural complexity of 1 and 2 presents several challenges for investigating their activity at the opioid receptors. Although functional groups like the C10 phenol serve as apparent synthetic handles to alter the scaffold, the complexity of the molecules could also confound the selectivity and reactivity of certain chemical transformations. Therefore, we employed a series of highly chemoselective transformations to diversify three positions on the scaffold that may make important ligand-receptor interactions: the aromatic ring, methyl ester, and indole nitrogen. By evaluating the resulting semi-synthetic derivatives, we gained initial insight into the SAR of 1 and 2.
Aromatic modifications (C10 and C11)
The C10 phenol of 1 represents a logical starting point for our SAR studies; however, we found that the aliphatic N4 tertiary amine is generally more nucleophilic than the corresponding phenoxide anion. Our initial attempts to functionalize the phenol via alkylation with iodomethane resulted in the methylation of the aliphatic nitrogen to produce the corresponding quaternary ammonium ion. However, methylation of 1 with trimethylsilyl diazomethane exclusively produced the methyl ether 7 (Scheme 1A).22 Similarly, the acetylation of 1 with acetic anhydride, but not acetyl chloride, favored O-acylation to produced ester 8.
Scheme 1.
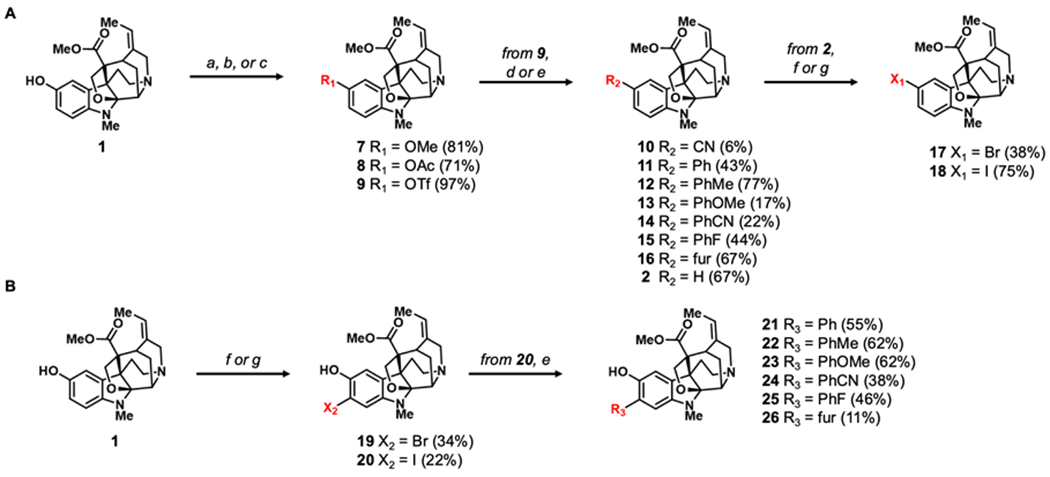
Semi-synthesis of (A) C10 analogues of 2 and (B) C11 analogues of 1. Reagents and conditions: (a) (CH3)2SiCHN2, THF, MeOH, rt, 48 h (b) Ac2O, DMAP, TEA, DCM, rt, 4 h (c) Tf2NPh, DMAP, DCM, rt, 1 h (d) Zn(CN)2, Pd(dppf)Cl2, TEA, DMF, 120 °C, 24 h (e) boronic acid, K2CO3, Pd(PPh3)4, PhMe, MeOH, 80 °C, 4 – 30 h. (f) NBS, TFA, DCM, 0°C to rt, 5 h (g) NIS, TFA, DCM, 0°C to rt, 5 h. Numbers in parentheses represent isolated percent yields. All Suzuki coupling analogues (11 – 15 and 21 – 26) consist of para-substituted aryl rings except for 3-furanyl analogues 16 and 26.
After successful substitution of the phenol, the corresponding aryl triflate was prepared by reacting the phenol with N-phenyl-bis-(trifluoromethane sulfonimide) and 4-dimethylaminopyridine.23 The resulting aryl triflate, 9,24 was leveraged as a pseudohalide intermediate in palladium-catalyzed cross-coupling reactions to compounds 10-16 (Scheme 1A).17 Nitrile analog 10 was synthesized from 9 in a palladium-catalyzed cyanation with Zn(CN)2 as the nitrile source.25 Suzuki-Miyaura cross-couplings were used to append aromatic and heterocyclic rings (11-16).17 In addition to introducing substitutions at C10, reduction of 9 with formic acid as the hydride source provides 2 (Scheme 1A).26 Given the relatively low isolation yields of 2 from P. nitida, this two-step process provides a convenient way to access 2 for additional SAR studies from the more abundant 1.
To further investigate the impact of substitutions on the aromatic ring, halides were introduced at C10 of 2 and C11 of 1 employing an acid-mediated halogenation with N-bromosuccinimide and N-iodosuccinimide (17-20, Scheme 1A–B). Intriguingly, the use of trifluoroacetic acid was necessary for the reaction to progress to completion. In addition to accelerating the reaction, the acidic conditions likely also serve as an in situ protection of the alkyl amines. The aryl iodide 20 was also shown to be a competent coupling partner for Suzuki-Miyaura reactions allowing for the synthesis of a series of C11-substituted derivatives of 1 (21-26, Scheme 1B)
Modifications to the esters
The C16 methyl ester of 1 and 2 were targeted as we anticipated they could be converted into useful points of diversification through reduction to the primary alcohol (Scheme 2, 27-28) or hydrolysis to the carboxylic acid. However, the ester proved remarkably resistant to chemical modifications, presumably due to its attachment to a sterically encumbered quaternary carbon. For example, no reaction is observed when subjecting 1 to lithium aluminum hydride in refluxing THF. Surprisingly, the methyl ester of 2 is readily reduced to the primary alcohol 28 with lithium aluminum hydride at room temperature. At the moment, it is unclear how the phenol of 1 has such a profound impact on the reactivity of the remote ester. Further highlighting the chemical stability of the methyl ester, treatment of 5 with KOH effectively hydrolyzes the acetyl ester to the primary alcohol 29, while leaving the adjacent methyl ester intact.
Scheme 2.
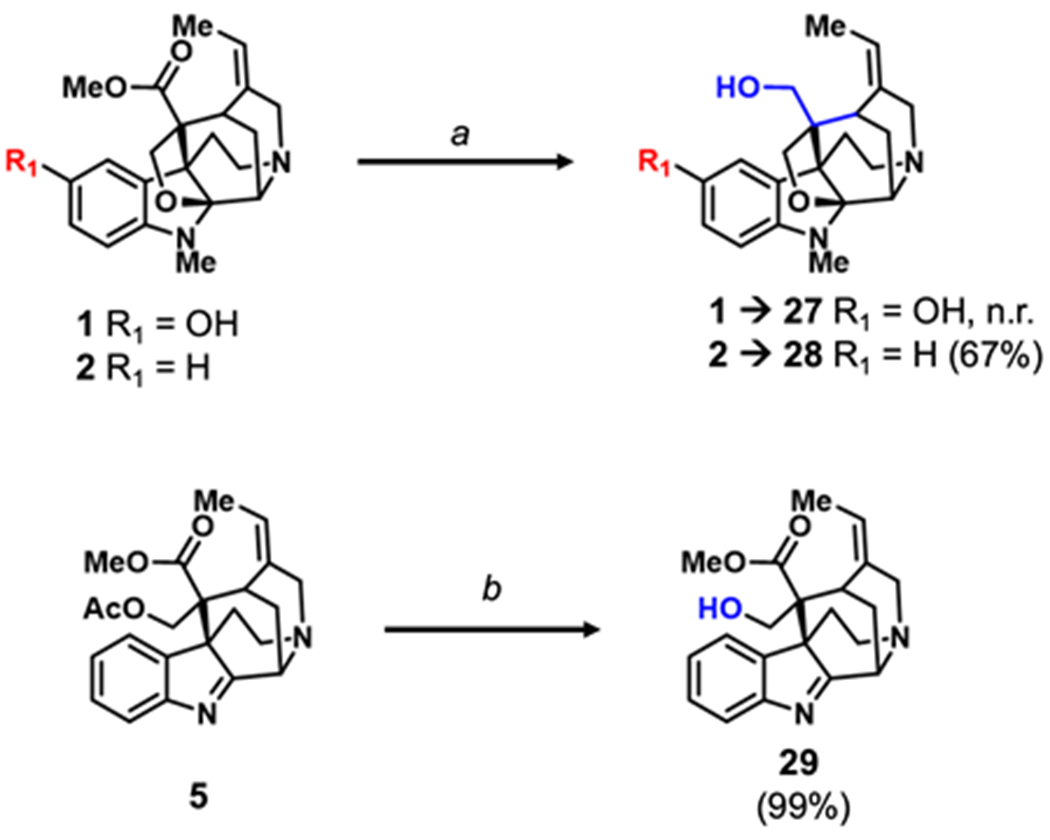
Semi-synthesis of C16 akuamma alkaloid analogues. Reagents and conditions: (a) LAH, TFA, 0 °C to rt, 4 h (b) KOH, MeOH, rt, 1 h. Number in parentheses represent isolated yields.
Indole nitrogen modifications
After modifying the C10 and C11 positions of 1 and 2, we shifted our efforts to substituting the N1 position of the indole ring (Scheme 3). Direct substitution of 1 and 2 are not feasible as the N1 position is substituted with methyl group which, despite multiple attempts, proved difficult to remove.27–30 An alternative to directly substituting 1 or 2 is to alkylate the structurally similar alkaloid 5, which conveniently lacks an N1 methyl group. Notably, the deacetylated analog of 5 (29) exists in equilibrium with the hemiaminal ether 30, which is favored under acidic conditions.31 Recently Zhang et. al. exploited this equilibrium in the total synthesis of 2 utilizing a trioxane, TFA, and triethylsilane in a modified Eschweiler-Clarke reaction to methylate 29.31, 32 Inspired by this finding, we adopted a similar strategy by replacing trioxane with various dimethyl acetals to produce derivatives of 2 bearing alkyl substituents to indolic nitrogen (31-33, Scheme 3).33, 34 The requisite dimethyl acetals were generated from reacting aldehydes with methanol in the presence of catalytic aqueous HCl.35
Scheme 3.
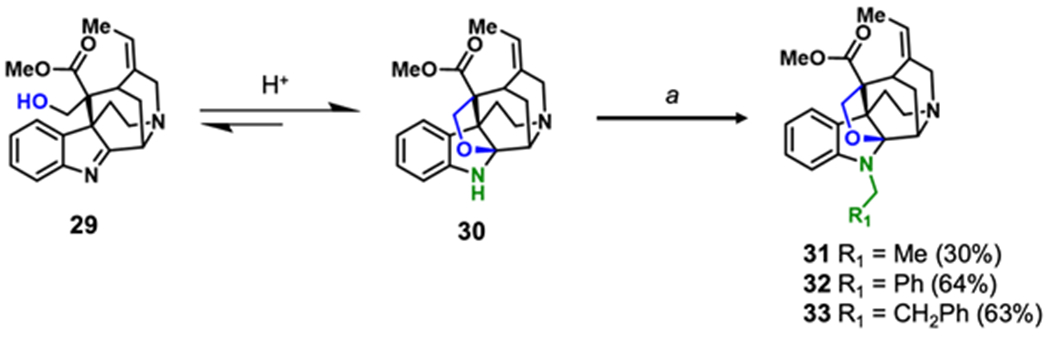
Semi-synthesis of N1 pseudoakuammigine analogues. Reagents and conditions: (a) dimethyl acetal, TFA, TES, DCM, rt, 18 h. Number in parentheses represent isolated yields.
In vitro pharmacology
Having generated a series of 22 novel “akuammalogs”, we looked to study what effects these modifications to the aromatic core of 1 and 2 would have on their activity at the opioid receptors. Since our previous study had determined that 1 and 2 have some affinities for the μOR and κOR, but very limited affinity for the δOR,20 we first examined the derivatives in competitive radioligand binding assays at the μOR and κOR. To streamline our process and more efficiently identify SAR trends, we began by assessing all compounds at 1 μM and 10 μM for their ability to displace [3H]-DAMGO or [3H]-U69,593 from at the μOR and κOR, respectively (Table 1). Compounds that displaced >50% of radioligand at 1 μM in these initial screening assays were then further evaluated in full concentration-response experiments to determine the binding affinity and cAMP inhibition functional activity assays to determine opioid receptor activation (Figure 2).
Table 1.
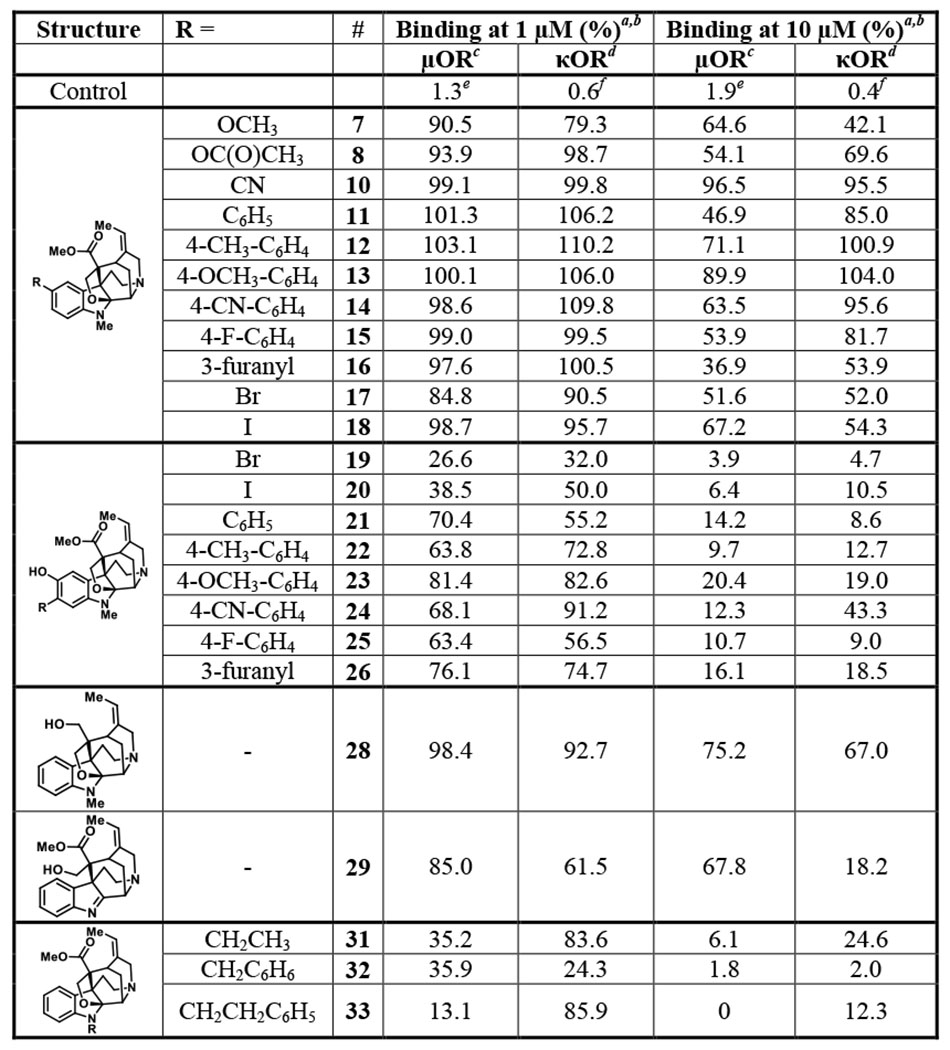
Radioligand displacement of the akuammalogs at the μOR and κOR
|
The akuammalogs were screened at 1 μM and 10 μM in a competitive radioligand binding assay at the μOR and κOR. Data is reported as the percentage of radioligand remaining upon akuammalog binding.
Values reported as the mean of at least 3 independent experiments.
Radioligand [3H]DAMGO was used for the μOR.
Radioligand [3H]U69,593 was used for the κOR.
Control compound DAMGO.
Control compound U50,488.
Figure 2.
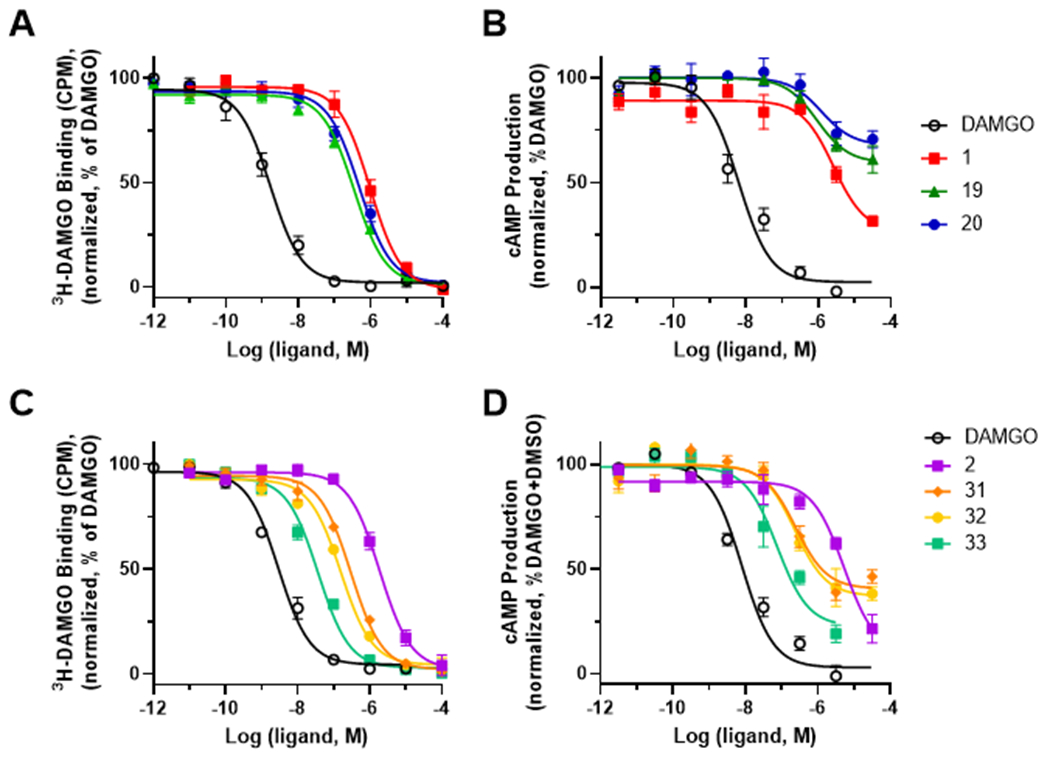
Pharmacological characterization of the akuammalogs at the μOR and κOR. Compounds 19-20 and 31-33 were screened in a full concentration-response radioligand binding assay at the μOR using [3H]DAMGO and akuammine (1) and pseudoakuammigine (2) as controls, respectively (A, C). Inhibition of forskolin-induced cAMP in a GloSensor assay in transfected HEK-293 cells at the μOR (B, D). All curves are representative of the averaged values from a minimum of three independent assays.
When the binding affinity was assessed, compounds bearing modifications at the C10 position (7, 8, and 10-18) displaced <50% of the radioligands and possess considerably less affinity than 1 or 2 (Table 1). These results indicate that C10 substitutions of the akuamma alkaloids are not well tolerated and suggest that the C10 phenol of 1 may make important ligand-receptor interactions with the opioid receptor. This notion was supported by the binding affinity for the C11 substituted derivatives of 1 that retain the C10 phenol. Although the Suzuki-Miyaura coupling products 21-26 do not displace >50% of the radioligand at either the μOR or κOR at 1 μM, they do induce more displacement than the C10 modified analogs (Table 1). Moreover, at 10 μM, 21-26 displace nearly 100% of [3H]DAMGO from the μOR, whereas 10-18 still induce little displacement at this higher concentration (Table 1). This higher concentration also revealed that most of the C11 Suzuki-Miyaura analogs do not retain a preference for the μOR that we observed in 1 and 2 and show similar affinities for the κOR.
In contrast to the other C11-substituted derivatives, halogenation of 1 at C11 (19-20) results in slightly improved affinity for the opioid receptors. Both 19 and 20 induced >50% displacement at 1 μM and possess an increased affinity to the μOR and κOR compared to 1 (Table 1). Completion of a full concentration response radioligand binding assay at the μOR confirmed the increase in binding affinity of 19 (Ki= 0.12 μM) and 20 (Ki= 0.22 μM) as compared to the parent compound 1 (Ki= 0.33 μM) (Figure 2A and Table 2). This improved affinity for the receptor is consistent with the phenol acting as a hydrogen bond donor with the electronegative halogens lowering the pKa of the phenol and increasing its hydrogen bond donating ability. Although 19 and 20 retain a preference for binding to the μOR, their selectivity over the κOR is diminished relative to 1 (Table 2 and Figure S1). We next used the GloSensor cAMP assay to measure G-protein activation 36; 19 and 20 possess increased potency (EC50 = 0.93 μM and 1.2 μM, respectively) at the μOR relative to 1 (Figure 2B and Table 2). In addition to increasing the potency of 1, these modifications also decrease the efficacy relative to the parent compound with compounds 19 and 20 producing partial agonism at the μOR with Emax values of 40% and 32%, respectively (Figure 2B and Table 2).
Table 2.
In vitro pharmacology of akuammalogs using radioligand binding and cAMP inhibition
| μOR Affinity a | κOR Affinity a | κOR/μOR selectivity | cAMP inhibition at μOR b | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Compound | pKi ± SEM c | (Ki, μM) | pKi ± SEM c | (Ki, μM) | pEC50 ± SEM c | EC50 (μM) | Emax % ± SEM c | |
| DAMGO | 9.1 ± 0.1 | 0.0009 | - | - | - | 8.2 ± 0.1 | 0.007 | 96 ± 4 |
| U-50 | - | - | 9.5 ± 0.1 | 0.0005 | - | - | - | - |
| 1 | 6.5 ± 0.1 | 0.30 | 5.8 ± 0.1 | 1.67 | 5.6 | 5.6 ± 0.2 | 2.60 | 62 ± 6 |
| 2 | 6.2 ± 0.1 | 0.33 | 5.6 ± 0.1 | 2.25 | 6.8 | 5.3 ± 0.1 | 5.24 | 82 ± 7 |
| 19 | 6.9 ± 0.1 | 0.12 | 6.6 ± 0.1 | 0.33 | 2.8 | 6.0 ± 0.2 | 0.93 | 40 ± 5 |
| 20 | 6.8 ± 0.1 | 0.22 | 6.2 ± 0.1 | 0.58 | 2.6 | 5.9 ± 0.4 | 1.22 | 32 ± 6 |
| 31 | 7.0 ± 0.1 | 0.10 | 5.9 ± 0.1 | 1.17 | 12 | 6.6 ± 0.2 | 0.24 | 59 ± 4 |
| 32 | 7.3 ± 0.1 | 0.054 | 7.3 ± 0.1 | 0.049 | 0.91 | 6.6 ± 0.2 | 0.23 | 63 ± 4 |
| 33 | 7.9 ± 0.1 | 0.012 | 6.2 ± 0.1 | 0.58 | 48 | 7.1 ± 0.2 | 0.075 | 76 ± 5 |
Affinity at the μOR and κOR was determined by radioligand displacement using [3H]DAMGO and [3H]U69,593 respectively;
cAMP inhibition at the μOR was determined through GloSensor assay;
Mean ± standard error on the mean.
Having assessed the effects of C10 and C11 modifications of the aromatic ring, we moved to investigate the modifications to the indolic nitrogen of 2. Upon the initial binding screen at 1 μM and 10 μM, we saw a significant increase in radioligand displacement at both the μOR and κOR for compounds 31 – 33 (Table 1). Extending the length of the alkyl group from methyl in 2 to an ethyl group in 31 increased binding affinity at the μOR (Ki = 0.10 μM) while retaining selectivity over the κOR (Ki = 1.2 μM) (Figure 2C and Table 2). Appending a phenyl ring to the methyl group of 2 (32) dramatically increased opioid receptor affinity; however, it also resulted in a loss of μOR selectivity with a μOR Ki = 0.054 μM and a κOR Ki = 0.049 μM. Unexpectedly, extending the linker by a single methylene unit as in 33 produced a 27-fold increase in μOR binding affinity relative to 2. This modification also produces a decreased affinity for the κOR relative to 32, resulting in 33 possessing 48-fold selectivity for the μOR (Ki = 0.012 μM) over the κOR (Ki = 0.58 μM). Using the GloSensor cAMP inhibition assay to measure μOR activation, derivatives 31-33 activated the μOR with potencies following a similar trend to that observed in the radioligand binding assays. Most notably, compound 33, bearing the phenethyl substitution on the N1 position was again identified as being considerably more potent than the parent natural product 2 with similar levels of efficacy (EC50 = 0.075 μM, Emax = 76%) (Figure 2D and Table 2).
We sought to further interrogate the selectivity of 33 for the opioid receptors in functional activity assays. Using the GloSensor cAMP inhibition assay to measure δOR and κOR activation, 33 displayed weak agonistic activities at both the δOR (EC50 = 0.61 μM; Emax =74%) and κOR (EC50 = 0.74 μM; Emax = 55%) (Figure 3A–B). The 10-fold difference in potency between 33 and 2 at the κOR is consistent with its reduced binding affinity for the κOR (Table 2 and Figure S1). Although the binding affinity of the akuammalogs at the δOR was not assessed due to the low binding affinity of the parent natural products, the partial agonist activity of 33 at the δOR indicates it does bind to the receptor. Thus, while the conversion of 2 to 33 maintains μOR/κOR selectivity, this modification increases the potency of at the δOR, thereby reducing μOR/δOR selectivity. Nevertheless, 33 does possess moderate selectivity and is 8- and 10-fold more potent at the μOR compared to the δOR and κOR, respectivley.20
Figure 3.

Opioid receptor signaling properties of compound 33. The ability of 33 to inhibit cAMP at δOR (A) and κOR (B) with DPDPE and U50,488 as controls, respectively; Recruitment of β-arrestin-2 in a PathHunter assay at μOR of 33 with DAMGO as a control (C); Recruitment Gα-subtype screening of 2, 33, and DAMGO in hμOR. TRUPATH heatmaps demonstrate 33 and μOR agonists activate the Gi/o-class of transducers with varying levels of potency (D) and efficacy (E). Heatmap colors refer to mean log(EC50) and normalized efficacy values. All curves are representative of the averaged values from a minimum of three independent assays.
Given the potency and selectivity of 33 at the μOR in the GloSensor assay, the signaling properties of 33 at the μOR were examined further by measuring β-arrestin (βArr2) recruitment. It is now well-appreciated that the recruitment of β-arrestins to GPCR is capable of initiating G-protein-independent signaling cascades.37 In the case of the μOR, βArr2-recruitment has been associated in a number of studies with the adverse effects of opioid analgesics including respiratory depression and tolerance.38, 39 So-called G-protein biased agonists that preferentially activate the G-protein pathway have been proposed as a strategy to develop safer opioid analgesics.39–41 We had previously observed that activation of the μOR by the akuamma alkaloids 1-3 does not result in recruitment of βArr2 to the receptor.20 Due to the low potency of 1-3 at the μOR, it is also possible these ligands are capable of inducing βArr2 recruitment but at concentrations higher than were tested. However, 33, which is considerably more potent in the GloSensor assay, yet still does not produce any significant recruitment of βArr2 to the μOR (<10% at 10 μM; Figure 3C), indicating 33 is a G-protein biased agonist. The limited recruitment complicates calculating a reliable bias factor for the compound.
To evaluate the G-protein signaling properties of 33, we employed the recently developed TRUPATH assay (Figure 3D–E).8, 42 The GloSensor assay measures the inhibition of cAMP production by adenylate cyclase as a downstream measure of activation of the Gi/o-signaling cascade induced by agonists activating a GPCR. By relying on the measurement of downstream events, these assays can overestimate the efficacy of ligands at the opioid receptors.43 Conversely, the TRUPATH assay system employs bioluminescence resonance energy transfer (BRET) biosensors to measure the dissociation of the G-protein heterotrimer that initiates the signaling cascade. Importantly, the TRUPATH system can interrogate activation of specific Gαi/o transducers, which in turn, could reveal signaling bias for different ligands.
The novel derivative 33, the natural product 2, and the positive control DAMGO were evaluated using the BRET-based TRUPATH assay to determine their abilities to activate the μOR coupled to the Gαi/o subtypes Gαi-1, Gαi-2, Gαi-3, GαoA, GαoB, and Gαz. Using this assay, both 2 and 33 appear as balanced agonists, producing similar potencies and efficacies in all Gαi/o subtypes. Consistent with the GloSensor assay, natural product 2 is a low potency, partial agonist producing 40-86% of the maximal effect produced by DAMGO (Figure 3D–E). Similarly, compound 33 possessed increased potency compared to 2 with all Gαi/o subtypes, although produced considerably higher levels of efficacy.
In vivo activity
Given the increased potency of 33 relative to the natural products 1 and 2, we elected to evaluate its antinociceptive effects in mice. In the first series of experiments, mice received different doses of 33, and were subjected to the tail-flick 44, 45 and hot-plate46 assays (10-100 mg/kg, s.c.). For the positive control, we used another naturally occurring compound morphine (10 mg/kg) in these assays. Compound 33 at high doses (80 and100 mg/kg) exhibited antinociceptive effect in both warm-water tail-flick assay and hot-plate assay. At 100 mg/kg, 33’s antinociception was comparable to that of the morphine (10 mg/kg) in the tail-flick assay with respect to peak effect and the duration of action, whereas the peak effect time was slightly delayed (Figure 4A). In the subsequent dose response assay, ED50 was estimated to be 77.6 mg/kg (Figure S2). Similar antinociception effect was observed in the hot-plate assay, although the effects at 80 or 100 mg/kg were not as robust as in the tail-flick assay in comparison to the positive control (Figure 4B), with an estimated ED50 of 77.1 mg/kg (Figure S2). The small differences in efficacy are likely due to differences in stimulus intensity and nociceptive pathways. The tail-flick assay is largely a spinal reflex assay whereas the hot-plate assay involves more processing from the supraspinal regions. These results from tail-flick and hot-plate assays clearly demonstrate that the increased potency of 33 relative to the naturally occurring akuamma alkaloids is translated into improved in vivo efficacy. To confirm the observed antinociceptive effects of 33 were mediated by the opioid receptors, mice were treated with the opioid receptor antagonist naloxone (10 mg/kg, i.p.) 15 minutes before treatment with 33 (100 mg/kg, s.c.). The effects of 33 were completely blocked by naloxone in the tail-flick assay (Figure 4C) and in the hot-plate assay (Figure 4D), indicating that the opioid receptors mediate the antinociception effect of 33.
Figure 4.

Antinociceptive effects of Compound 33. (A) Tail-flick assay. (B) Hot-plate assay, and antinociceptive effect of Compound 33 after the pretreatment with naloxone in (C) the Tail-flick assay and the (D) Hot-plate assay. (A-B). Separate groups of 3 mice received different doses of compound 33 (10, 80, 100mg/kg, subcutaneously), morphine (10mg/kg, subcutaneously), or vehicle. Time course of the antinociception response after dosing was measured. Data are expressed in mean ± SEM, *p<0.05, **p<0.01, ***p<0.001 vs the control group. (C-D). Separate groups of 3 mice received naloxone (10 mg/kg, i.p.) or saline, followed by an injection of compound 33 (100 mg/kg, s.c.) or saline 15 min later. *p<0.05, **p<0.01, ***p<0.001 vs naloxone group.
To further investigate the behavioral effects of 33 in mice, we assessed its effects in the accelerating rotarod test. At the high doses (80, 100 mg/kg, s.c.) 33 does appear to impair motor coordination (Figure 5). Although this impaired locomotor activity is significant, other μOR agonists, including morphine, also produce motor incoordination.47 As such, this effect is likely produced through μOR activation and further supports that compound 33 is active in vivo.
Figure 5.
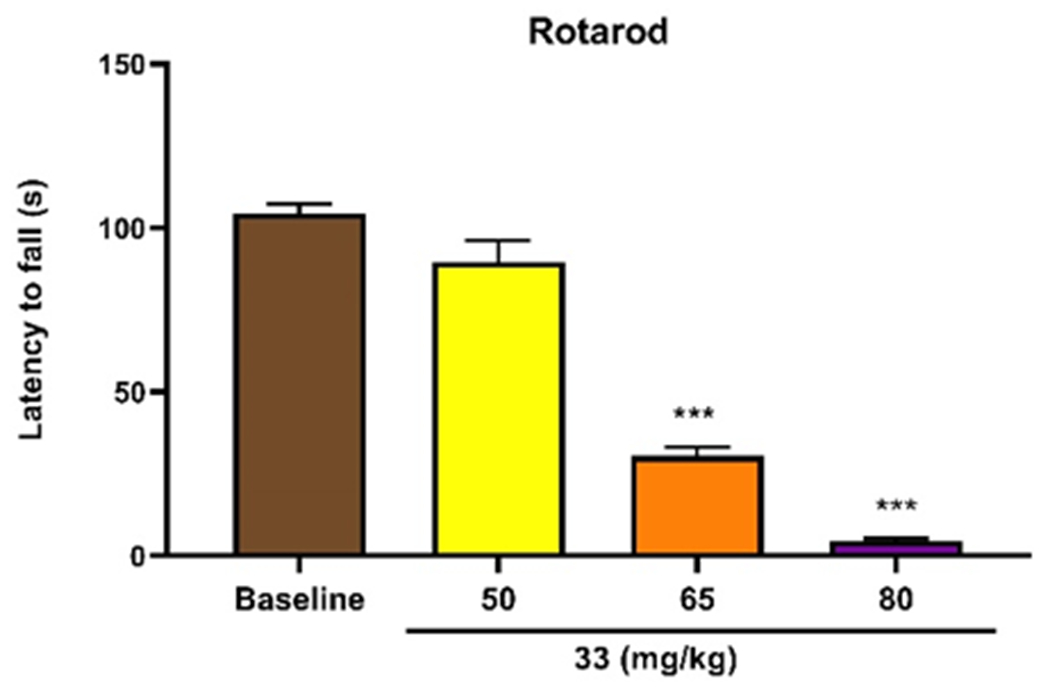
Effect of compound 33 on locomotor activity. Separate groups of 3 mice received different doses of compound 33 (50, 65, 80mg/kg subcutaneously). The mice were placed on an accelerating rotarod (4-40rpm over 300s) and the latency to fall was recorded. Both 65mg/kg and 80mg/kg of compound 33 significantly impaired motor activity. Data are expressed in mean ± SEM, ***p<0.001 vs the control group.
Discussion and Conclusion
Natural products have been used for millennia for the treatment of pain.48 Within the last century, the isolation of bioactive components from these ethnomedicines has provided numerous chemical probes to elucidate many signaling mechanisms in the central nervous system.49 For example, the investigation of the pharmacology of morphine, isolated from Papaver somniferum, resulted in the identification of the opioid receptors.3 In more recent instances, by exploring the SAR of other naturally occurring opioid ligands like salvinorin A and mitragynine, medicinal chemists have unearthed novel compounds with unique pharmacology that have revealed further insight into the molecular mechanisms of analgesia and opioid-induced side effects. 14–16, 50
The results from this SAR study add to this tradition by providing new chemical scaffolds that can engage the μOR binding pocket in unique ways. To rationalize the SAR trends we observed, we used induced-fit docking (Maestro)51 to identify potential binding modes and interactions made between two of the more potent derivatives (19 and 33) and the μOR (Figure 6). When docked into the nanobody-stabilized structure of the active-state μOR, both 19 and 33 adopted similar top-scoring docking poses that share considerable overlap with the co-crystallized morphinan ligand BU72. Notably, the alkyl tertiary amines of 19 and 33 form salt-bridge with Asp147, which have been previously observed to anchor other cationic nitrogens within the opioid binding pocket.52, 53 Similarly, the aromatic rings of 19 and 33 occupy similar space to the phenolic ring of BU72.54 In the case of 19, the phenol moiety engages His297 via a network of water-mediated hydrogen bonds (Figure 6A). This hydrogen-bond interaction between a phenol oxygen and this network of water appears to be a highly conserved interaction and has been observed in both the morphinans BU72 and β-FNA and a tyrosine residue of DAMGO bound to the μOR.55–57 This proposed binding mode of 19 is consistent with the SAR we observed that indicated replacing or substituting the C10 phenol of 1 led to significantly reduced affinity for the μOR. Furthermore, while the binding pocket adjacent to C11 can accommodate the bromine of 19, the steric bulk from the aryl rings of 21-26 prevents the ligands from binding tightly to the μOR.
Figure 6.
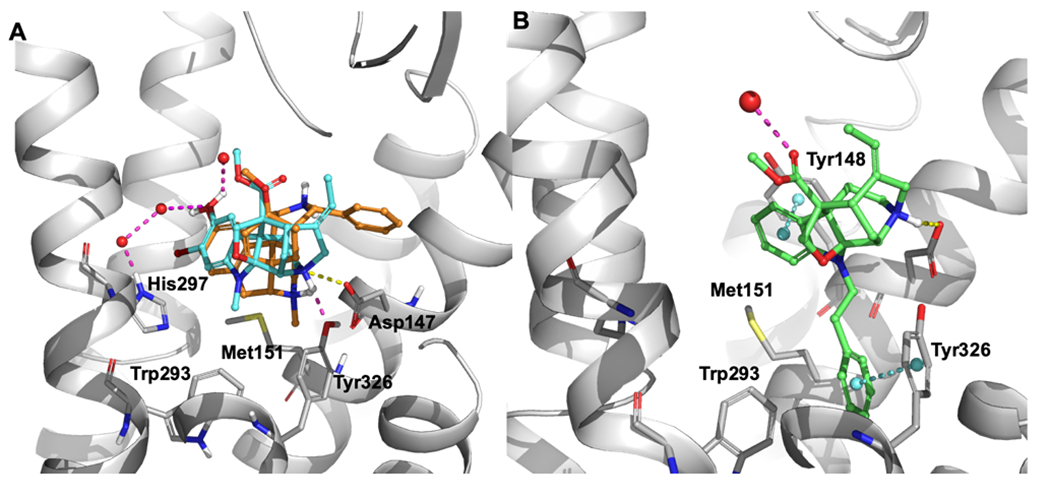
Binding modes and interactions of 19 (cyan) and 33 (green) as compared to BU72 (orange) (A – B, respectively). Crystal structure of μOR co-crystallized with agonist BU-72 (PDB: 5C1M) was used as a starting point for all molecular docking simulation studies. Ligand receptor interactions are depicted as dashed lines: hydrogen bonds (magenta), salt-bridge (yellow), pi-pi stacking (blue). For clarity, ligand-receptor interactions are omitted for BU-72.
In our SAR studies, we observed the installation of a phenethyl moiety to the N1 position of 2 dramatically increased its affinity and potency at the μOR. This improved activity likely indicates that the additional phenethyl moiety in 33 extends into a subpocket of the binding site, allowing it to make additional ligand-receptor interactions. This notion is supported by the docking pose of 33 that suggests the phenethyl group extends into a cavity toward the bottom of the binding site. Interestingly, in the co-crystal structures, this subpocket is occupied by the N-methyl group of BU72 and the cyclopropylmethyl group of β-FNA.58 As exemplified by β-FNA, in the classic “message-address” concept of opioid ligands, longer alkyl at this position generally possesses antagonist activity. However, the relatively high efficacy of 33 (Emax = 76%) suggests that extending further into this pocket or making additional ligand interactions allows for receptor activation. Notably, the binding pose does indicate a possible pi-pi stacking interactions with Tyr326, which has been previously implicated in μOR activation.59, 60 By offering a new scaffold to probe this subpocket by engaging these and other potential ligand-receptor interactions, the akuamma alkaloids may reveal new ways to modulate μOR activation and signaling properties.
In summary, we have conducted the first SAR of the akuamma alkaloids by leveraging highly chemoselective, late-stage functionalization of the indole nucleus of 1 and 2. Through these studies we identified that replacement of the N1-methyl group of 2 with larger alkyl groups engages an allosteric pocket of the μOR, leading to increased affinity. Most notably, we discovered lead compound 33 that possess considerably improved potency and selectivity compared to the parent natural product 2. Compound 33 also exhibited antinociceptive effect in tail-flick and hot-plate assays in mice. Taken together, these in vitro and in vivo results provide further support that the continued investigation of opioid natural products can lead to new classes of potent opioid ligands. Moreover, these initial discoveries provide a foundation for future studies to probe how the akuamma alkaloids interact with the μOR binding pocket.
Experimental
Chemistry
General Experimental Procedures.
All solvents and reagents were purchased from commercial sources and used directly without further purification. Akuamma seed powder was purchased from Relax Remedy and alkaloids were isolated and purified as previously described.20 Bruker 400 MHz, Bruker 400 MHz HD, and Bruker 600 MHz spectrometers were used to record 1H and 13C NMR spectra and they were referenced to the residual solvent peaks (CHCl3: 1H δ=7.26, 13C δ=77.16 ppm). High-resolution mass spectra were obtained on a Shimadzu LCMS-IT-TOF and observed values are within 5.0 ppm of calculated exact masses of the indicated ions. High-performance liquid chromatography (HPLC) was conducted on an Agilent 1260 Infinity II fitted with a DAD detector and a Phenomenox Luna Omega PS-C18 column (100 x 4.6 mm). A gradient of acetonitrile/water (20-45%) each containing 0.1% formic acid with a flow rate of 1 mL/min was used. The purity of all compounds was found to be >95% as determined by HPLC.
O-Methylakuammine (7).
In an oven-dried round bottom flask under nitrogen, 1 (150.5 mg, 0.393 mmol) was dissolved in 10 mL of a 4:1 mixture of THF/MeOH. A solution of TMS-diazomethane in hexanes (2 M, 1.9 mL, 3.8 mmol) was added dropwise and the reaction stirred for 21 hours at room temperature. After 21 hours an additional 5 equivalents of TMS-diazomethane were added. After 48 hours, the reaction was quenched with 1M acetic acid and basified with sodium bicarbonate. The reaction solvent was removed on the rotary evaporator and the aqueous layer was extracted with ethyl acetate (3x50 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography eluting with 2-4% methanol/dichloromethane to afford 126 mg of 7 (81% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.74 (d, J = 2.6 Hz, 1H), 6.66 (dd, J = 8.4, 2.6 Hz, 1H), 6.54 (d, J = 8.4 Hz, 1H), 5.41 (q, J = 6.6 Hz, 1H), 4.17 (s, 1H), 3.96 (s, 1H), 3.94 (d, J = 7.72 Hz, 1H), 3.81 (s, 3H), 3.73 (s, 3H), 3.66 (dd, J = 13.9, 4.3 Hz, 1H), 3.61 (d, J = 7.5 Hz, 1H), 3.48 (s, 1H), 3.36 – 3.25 (m, 1H), 2.85 (d, J = 16.6 Hz, 1H), 2.78 (s, 3H), 2.58 (dd, J = 13.8, 5.9 Hz, 1H), 2.32 (d, J = 13.8 Hz, 1H), 2.05 (d, J = 14.1 Hz, 1H), 1.52 (d, J = 7.0 Hz, 3H), 1.47 (d, J = 15.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 172.17, 154.03, 145.93, 140.59, 118.69, 111.02, 110.98, 110.02, 104.25, 74.17, 58.46, 56.00, 54.65, 53.84, 52.85, 52.09, 50.58, 41.06, 31.10, 29.84, 29.50, 28.04, 13.07. HRMS calculated for C23H29N2O4: [M + H]+: 397.2124 (found); 397.2127 (calcd).
O-Acetylakuammine (8).
In an oven-dried round bottom flask under nitrogen, 1 (50.0 mg, 0.131 mmol) was dissolved in 5 mL dichloromethane. DMAP (16.0 mg, 0.131 mmol), TEA (55 μL, 0.39 mmol), and acetic anhydride (25 μL, 0.26 mmol) were added and the reaction stirred for 4 hours at room temperature. The reaction was diluted with water and basified with sodium bicarbonate. The aqueous layer was extracted with dichloromethane (3x10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography with 1-6% methanol/dichloromethane to afford 39 mg of 8 (71% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 6.82 (d, J = 2.3 Hz, 1H), 6.63 (d, J = 8.5 Hz, 1H), 5.60 (q, J = 7.2 Hz, 1H), 4.54 – 4.49 (s, 1H), 4.16 (d, J = 16.4 Hz, 1H), 3.98 (d, J = 7.9 Hz, 1H), 3.91 – 3.83 (m, 1H), 3.82 (s, 3H), 3.66 (d, J = 7.9 Hz, 1H), 3.60 (s, 1H), 3.38 (ddd, J = 16.1, 14.4, 5.8 Hz, 1H), 3.13 (d, J = 16.4 Hz, 1H), 2.85 (d, J = 5.8 Hz, 1H), 2.83 (s, 3H), 2.47 (ddd, J = 14.5, 4.3, 2.0 Hz, 1H), 2.25 (s, 3H), 2.10 (ddd, J = 14.6, 3.7, 1.9 Hz, 1H), 1.72 (dd, J = 15.9, 4.6 Hz, 1H), 1.57 (dd, J = 7.1, 2.4 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 171.17, 170.02, 148.86, 144.58, 138.60, 134.99, 123.08, 121.06, 116.73, 110.83, 102.59, 74.40, 57.98, 53.94, 53.38, 53.07, 52.50, 50.04, 39.98, 29.58, 28.65, 26.95, 21.24, 13.20. HRMS calculated for C24H29N2O5: [M + H]+: 425.2071 (found); 425.2076 (calcd).
O-Trifluoromethansulfonylakuammine (9).
In an oven dried round bottom flask under nitrogen 1 (100 mg, 0.2618 mmol) was dissolved in 10 mL anhydrous dichloromethane at room temperature. To the solution was added DMAP (96 mg, 0.7854 mmol) and N-phenyl-bis(trifluoromethanesulfonimide) (187 mg, 0.524 mmol). The reaction was stirred at room temperature for 1 hour. The reaction was concentrated via rotary evaporation. The crude mixture was diluted into 30 mL water and extracted with ethyl acetate (3x30 mL). The combine organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified via column chromatography (0-4% MeOH/DCM) to yield 130 mg of the desired product (97% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.03 (d, J = 7.2 Hz, 2H), 6.61 (d, J = 8.5 Hz, 1H), 5.46 (q, J = 7.2 Hz, 1H), 4.25 (s, 1H), 4.01 – 3.93 (m, 2H), 3.82 (s, 3H), 3.66 – 3.52 (m, 3H), 3.34 (td, J = 14.6, 5.7 Hz, 1H), 2.89 (d, J = 16.8 Hz, 1H), 2.85 (s, 3H), 2.67 (dd, J = 13.5, 5.7 Hz, 1H), 2.37 – 2.29 (m, 1H), 2.14 – 2.04 (m, 1H), 1.54 (dd, J = 7.1, 2.4 Hz, 3H), 1.48 (dd, J = 15.3, 4.4 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 170.80, 150.77, 143.42, 139.03, 124.79, 121.45, 117.07, 116.85, 111.66, 111.31, 102.19, 74.66, 57.81, 53.99, 53.47, 53.27, 52.75, 50.21, 39.55, 29.57, 27.96, 26.60, 13.29. HRMS calculated for C23H26N3O3: [M + H]+: 392.1970 (found); 392.1974 (calcd).
Pseudoakuammigine (2).
In an oven-dried round bottom flask under nitrogen, 9 (200 mg, 0.3891 mmol) was dissolved in 3 mL of dry DMF. To the solution at room temperature was added TEA (1.08 mL, 7.7821 mmol), formic acid (0.74 mL, 1.945 mmol), and dpppPd(II)Cl2 (46 mg, 0.07782 mmol). Following the additions, the temperature of the reaction was increased to 80 °C and allowed to stir under N2 for 4 hours. Upon completion, the reaction was cooled to room temperature and diluted with 300 mL H2O. The solution was extracted with 50 mL EtOAc 3X. The combined organic layers were washed with 30 mL H2O 2X and 30 mL bring 1X. The combined organic layers were dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel chromatography eluting with 0–20 % methanol/dichloromethane to afford 97 mg of 7 (69% yield) as a white solid whose 1H and 13C NMR spectra were identical to 2 isolated from P. nitida.20
10-Cyanopseudoakuammigine (10).
In an oven-dried round bottom flask under nitrogen, 9 (200.0 mg, 0.3891 mmol) was dissolved in 10 mL anhydrous DMF at room temperature. To the solution was added anhydrous TEA (5.3 mL, 40 mmol) followed by Pd(dppf)Cl2 (22.9 mg, 0.0389 mmol) and zinc cyanide (23.0 mg, 0.194 mmol). The reaction was stirred at 120 °C for 24 hours. The reaction was diluted with 400 mL water and extracted with ethyl acetate (3x75 mL). The combined organic layers were washed with fresh water (150 mL) three times and 200 mL of brine once. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by recrystallization in ether and ethanol to afford 8.2 mg of the desired product (6 % yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.44 (dd, J = 8.2, 1.7 Hz, 1H), 7.35 (d, J = 1.7 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 5.42 (q, J = 6.9 Hz, 1H), 4.18 (s, 1H), 3.96 (d, J = 7.6 Hz, 1H), 3.91 (d, J = 17.0 Hz, 1H), 3.84 (s, 3H), 3.55 (d, J = 7.6 Hz, 1H), 3.52 (s, 1H), 3.46 – 3.27 (m, 2H), 2.89 (s, 3H), 2.82 (d, J = 17.4 Hz, 1H), 2.61 (dd, J = 12.7, 4.6 Hz, 1H), 2.28 (d, J = 12.6 Hz, 1H), 2.08 (ddd, J = 13.9, 3.7, 2.0 Hz, 1H), 1.53 (dd, J = 7.1, 2.6 Hz, 3H), 1.39 (dd, J = 15.0, 4.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 171.77, 155.55, 141.89, 140.30, 133.29, 126.50, 120.54, 118.92, 109.83, 103.74, 102.05, 74.83, 58.52, 54.60, 54.07, 52.69, 52.38, 51.18, 41.09, 31.30, 28.81, 28.03, 13.03. HRMS calculated for C23H26N3O3: [M + H]+: 392.1970 (found); 392.1974 (calcd).
General Procedure A:
Suzuki-Miyaura Coupling. In an oven-dried round bottom flask under nitrogen, 9 or 20 (1.0 equiv) was dissolved in 3 mL anhydrous toluene and 2 mL anhydrous methanol at room temperature. To the solution was added K2CO3 (2.0 equiv) followed by boronic acid (1.1 equiv) and Pd(PPh3)4 (5 mol%). The reaction was stirred at 80 °C for until consumption of the starting material as indicated by TLC. The reaction was cooled to room temperature concentrated in vacuo. The residue was purified by column chromatography (0 - 4% MeOH/DCM) afford the desired product as a white solid.
10-Phenylpseduoakuammigine (11).
Prepared from 9 (45.0 mg, 0.0875 mmol) according to General Procedure A to yield 16.6 mg of 11 (43% yield). 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 7.1 Hz, 2H), 7.39 (t, J = 7.7 Hz, 3H), 7.35 (s, 1H), 7.28 (d, J = 7.2 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 5.55 (t, J = 7.5 Hz, 1H), 4.41 (s, 1H), 4.09 (d, J = 16.3 Hz, 1H), 3.99 (d, J = 7.8 Hz, 1H), 3.83 (s, 3H), 3.75 (dd, J = 13.8, 5.1 Hz, 1H), 3.68 (d, J = 7.7 Hz, 1H), 3.57 (s, 1H), 3.49 – 3.34 (m, 1H), 3.03 (d, J = 16.5 Hz, 1H), 2.89 (s, 3H), 2.76 (dd, J = 13.5, 5.8 Hz, 1H), 2.43 (d, J = 13.7 Hz, 1H), 2.11 (d, J = 14.2 Hz, 1H), 1.68 (dd, J = 15.6, 4.6 Hz, 1H), 1.57 (d, J = 4.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.75, 150.94, 141.64, 139.24, 138.08, 133.80, 128.86, 126.92, 126.75, 126.55, 121.52, 121.15, 110.58, 103.25, 74.52, 58.20, 54.27, 53.57, 52.95, 52.32, 50.44, 40.50, 29.96, 29.37, 27.48, 13.19. HRMS calculated for C28H31N2O3: [M + H]+: 443.2330 (found); 443.2335 (calcd).
10-(p-Tolyl)-pseduoakuammigine (12).
Prepared from 9 (50.0 mg, 0.0972 mmol) according to General Procedure A to yield 34 mg of 12 (77% yield). 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.1 Hz, 3H), 7.32 (d, J = 1.8 Hz, 1H), 7.21 (d, J = 7.9 Hz, 2H), 6.73 (d, J = 8.1 Hz, 1H), 5.58 (q, J = 7.2 Hz, 1H), 4.46 (s, 1H), 4.13 (d, J = 16.4 Hz, 1H), 4.00 (d, J = 7.8 Hz, 1H), 3.83 (s, 3H), 3.69 (d, J = 7.8 Hz, 1H), 3.60 (s, 1H), 3.44 (td, J = 15.0, 5.8 Hz, 2H), 3.10 (d, J = 16.4 Hz, 1H), 2.88 (s, 3H), 2.81 (dd, J = 13.2, 5.7 Hz, 1H), 2.48 (d, J = 4.0 Hz, 1H), 2.37 (s, 3H), 2.12 (ddd, J = 14.5, 3.8, 1.9 Hz, 1H), 1.72 (dd, J = 15.7, 4.6 Hz, 1H), 1.58 (dd, J = 7.1, 2.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.58, 150.54, 138.88, 138.72, 136.74, 136.34, 134.00, 129.60, 126.82, 126.63, 122.01, 121.35, 110.74, 102.92, 74.49, 58.07, 54.26, 53.43, 53.23, 52.37, 50.43, 40.27, 29.84, 29.42, 27.26, 21.15, 13.23. HRMS calculated for C29H33N2O3: [M + H]+: 457.2479 (found); 257.2491 (calcd).
10-(p-Methoxyphenyl)-pseduoakuammigine (13).
Prepared from 9 (45.0 mg, 0.0875 mmol) according to General Procedure A to yield 7.0 mg of 13 (17% yield). 1H NMR (600 MHz, CDCl3) δ 7.44 – 7.36 (m, 2H), 7.34 (dt, J = 8.1, 2.0 Hz, 1H), 7.29 (t, J = 2.0 Hz, 1H), 6.97 – 6.91 (m, 2H), 6.71 (dd, J = 8.1, 2.7 Hz, 1H), 5.53 (q, J = 7.5 Hz, 1H), 4.38 (s, 1H), 4.08 (d, J = 16.7 Hz, 1H), 3.99 (dd, J = 7.8, 2.7 Hz, 1H), 3.83 (s, 6H), 3.75 (d, J = 13.9 Hz, 1H), 3.67 (d, J = 2.7 Hz, 1H), 3.57 (s, 1H), 3.46 – 3.37 (m, 1H), 3.02 (d, J = 16.5 Hz, 1H), 2.87 (s, 3H), 2.78 – 2.72 (m, 1H), 2.42 (d, J = 14.3 Hz, 1H), 2.14 – 2.08 (m, 1H), 1.66 (dt, J = 15.7, 3.6 Hz, 1H), 1.57 (d, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 171.80, 158.66, 150.47, 139.24, 138.38, 134.33, 133.50, 127.74, 126.41, 121.16, 120.95, 114.31, 110.55, 103.29, 74.47, 58.21, 55.49, 54.36, 53.57, 53.05, 52.30, 50.52, 40.52, 30.05, 29.36, 27.51, 13.18. HRMS calculated for C29H33N2O4: [M + H]+: 473.2439 (found); 473.2440 (calcd).
10-(p-Cyanophenyl)-pseduoakuammigine (14).
Prepared from 9 (50.0 mg, 0.0972 mmol) according to General Procedure A to yield 9.8 mg of 14 (22% yield).1H NMR (400 MHz, CDCl3) δ 7.67 – 7.55 (m, 4H), 7.42 – 7.35 (m, 2H), 6.73 (d, J = 8.1 Hz, 1H), 5.50 – 5.42 (q, 1H), 4.23 (s, 1H), 3.98 (d, J = 7.6 Hz, 1H), 3.93 (s, 1H), 3.83 (s, 3H), 3.63 (d, J = 7.6 Hz, 1H), 3.61 – 3.55 (m, 1H), 3.54 (s, 1H), 3.44 – 3.31 (m, 1H), 2.89 (s, 3H), 2.63 (dd, J = 13.4, 5.6 Hz, 1H), 2.33 (d, J = 13.8 Hz, 1H), 2.14 – 2.06 (m, 2H), 1.54 (dd, J = 7.0, 2.4 Hz, 3H), 1.51 (d, J = 4.2 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 170.99, 151.51, 145.65, 138.54, 132.79, 132.24, 127.94, 127.33, 127.16, 125.18, 121.62, 119.19, 111.49, 110.24, 101.89, 74.59, 57.77, 53.81, 53.31, 52.74, 49.98, 39.62, 29.58, 27.92, 26.58, 13.36. HRMS calculated for C29H30N3O3: [M + H]+: 468.2280 (found); 468.2287 (calcd).
10-(p-Fluorophenyl)-pseduoakuammigine (15).
Prepared from 9 (36.0 mg, 0.0760 mmol) according to General Procedure A to yield 14.1 mg of 15 (44% yield). 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.38 (m, 2H), 7.34 (dd, J = 8.1, 1.9 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H), 7.07 (t, J = 8.7 Hz, 2H), 6.73 (d, J = 8.1 Hz, 1H), 5.57 (q, J = 7.1 Hz, 1H), 4.45 (s, 1H), 4.12 (d, J = 16.5 Hz, 1H), 4.00 (d, J = 7.8 Hz, 1H), 3.83 (s, 3H), 3.78 (dd, J = 13.6, 4.6 Hz, 1H), 3.68 (d, J = 7.8 Hz, 1H), 3.62 – 3.57 (bs, 1H), 3.43 (td, J = 15.0, 5.8 Hz, 1H), 3.07 (d, J = 16.4 Hz, 1H), 2.88 (s, 3H), 2.79 (dd, J = 13.2, 5.6 Hz, 1H), 2.45 (d, J = 14.8 Hz, 1H), 2.11 (d, J = 12.6 Hz, 1H), 1.70 (dd, J = 15.6, 4.5 Hz, 1H), 1.58 (dd, J = 7.1, 2.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.84, 163.52, 161.08, 151.01, 139.35, 137.94, 133.18, 128.45, 128.37, 127.10, 121.98, 121.59, 115.97, 115.76, 110.92, 103.21, 74.71, 58.34, 54.42, 53.73, 53.23, 52.59, 50.57, 40.56, 35.00, 31.92, 30.04, 29.84, 29.61, 27.53, 22.99, 14.45, 13.41. HRMS calculated for C28H30N2O3F: [M + H]+: 461.2237 (found); 461.2240 (calcd).
10-(3’-Furanyl)-pseduoakuammigine (16).
Prepared from 9 (50.0 mg, 0.0972 mmol) according to General Procedure A to yield 28.3 mg of 16 (67% yield).1H NMR (400 MHz, CDCl3) δ 7.59 (t, J = 1.2 Hz, 1H), 7.42 (t, J = 1.7 Hz, 1H), 7.28 (d, J = 1.8 Hz, 1H), 7.22 (d, J = 1.8 Hz, 1H), 6.65 (d, J = 8.1 Hz, 1H), 6.60 (t, J = 1.3 Hz, 1H), 5.47 (q, J = 7.2 Hz, 1H), 4.28 (s, 1H), 4.02 (s, 1H), 3.97 (d, J = 7.7 Hz, 1H), 3.84 (s, 3H), 3.72 – 3.61 (m, 2H), 3.53 (d, J = 3.3 Hz, 1H), 3.43 – 3.30 (m, 1H), 2.92 (d, J = 16.7 Hz, 2H), 2.85 (s, 3H), 2.65 (dd, J = 13.4, 5.8 Hz, 1H), 2.42 – 2.32 (m, 1H), 2.09 (ddd, J = 14.2, 3.7, 1.9 Hz, 1H), 1.59 (d, J = 4.4 Hz, 1H), 1.55 (dd, J = 7.1, 2.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 172.05, 150.90, 143.53, 139.64, 137.48, 126.95, 125.43, 124.59, 120.51, 119.82, 110.36, 109.03, 103.62, 74.47, 58.36, 54.37, 52.70, 52.18, 50.50, 40.81, 30.62, 29.84, 29.25, 27.79, 13.13. HRMS calculated for C26H29N2O4: [M + H]+: 433.2118 (found); 433.2127 (calcd).
General Procedure B:
In an oven-dried round bottom flask under nitrogen, 1 or 2 (1.0 equiv) was dissolved in 1:1 anhydrous dichloromethane and trifluoroacetic acid at 0 °C. Over 1 hour, a cold solution of NBS or NIS (1.1 equiv) dissolved in a 1mL 1:1 DCM/TFA was added to the solution. After the addition of NBS or NIS, the reaction was allowed to stir at 0°C for 5 hours. The reaction was cooled poured into ice water and basified with NaHCO3. The solution was washed with sodium thiosulfate and extracted DCM (3×10 mL). The organic layers were combined, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was then purified by column chromatography (0.5–10% MeOH/DCM) to yield the desired product.
10-Bromopseudoakuammigine (17).
Prepared from 2 (70 mg, 0.1913 mmol) and NBS according to general procedure B to yield 32 mg of 17 (38% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.22 (dd, J = 8.3, 2.1 Hz, 1H), 7.17 (d, J = 2.0 Hz, 1H), 6.51 (d, J = 8.3 Hz, 1H), 5.43 (q, J = 7.2 Hz, 1H), 4.21 (s, 1H), 3.95 (d, J = 7.7 Hz, 1H), 3.93 (s, 1H), 3.83 (s, 3H), 3.60 (d, J = 7.6 Hz, 1H), 3.55 (dd, J = 13.8, 4.5 Hz, 1H), 3.50 (d, J = 3.5 Hz, 1H), 3.30 (ddd, J = 15.3, 14.0, 5.8 Hz, 1H), 2.86 (d, J = 16.7 Hz, 1H), 2.81 (s, 3H), 2.61 (dd, J = 13.4, 5.7 Hz, 1H), 2.31 (d, J = 14.1 Hz, 1H), 2.07 (ddd, J = 14.1, 3.7, 1.9 Hz, 1H), 1.53 (dd, J = 7.1, 2.5 Hz, 3H), 1.47 (dd, J = 15.1, 4.3 Hz, 1H).13C NMR (101 MHz, CDCl3) δ 171.90, 150.97, 141.32, 139.45, 130.33, 126.01, 119.16, 111.84, 111.53, 103.77, 74.51, 58.50, 54.57, 54.07, 52.82, 52.24, 50.81, 41.02, 31.02, 29.15, 27.93, 13.07. HRMS calculated for C22H26N2O3Br: [M + H]+: 445.1125 (found); 445.1127 (calcd).
10-Iodopseudoakuammigine (18).
Prepared from 2 (150 mg, 0.4098 mmol) and NIS according to general procedure B to afford 150.3 mg of 18 (75% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.44 (dd, J = 8.2, 1.8 Hz, 1H), 7.34 (d, J = 1.8 Hz, 1H), 6.45 (d, J = 8.2 Hz, 1H), 5.52 (q, J = 7.4 Hz, 1H), 4.37 (s, 1H), 4.05 (d, J = 16.7 Hz, 1H), 3.97 (d, J = 7.8 Hz, 1H), 3.84 (s, 3H), 3.73 – 3.64 (m, 1H), 3.62 (d, J = 7.7 Hz, 1H), 3.55 (s, 1H), 3.34 (td, J = 14.9, 5.8 Hz, 1H), 2.98 (d, J = 16.5 Hz, 1H), 2.81 (s, 3H), 2.72 (dd, J = 13.2, 5.7 Hz, 1H), 2.38 (d, J = 14.2 Hz, 1H), 2.08 (ddd, J = 14.4, 3.7, 1.9 Hz, 1H), 1.59 (d, J = 3.9 Hz, 1H), 1.55 (dd, J = 7.1, 2.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.48, 151.27, 141.11, 136.75, 131.57, 121.25, 112.68, 102.88, 81.89, 74.53, 58.20, 54.22, 53.72, 52.87, 52.39, 50.44, 40.51, 29.21, 27.41, 22.82, 14.25, 13.16. HRMS calculated for C22H26N2O3I: [M + H]+: 493.0987 (found); 493.0988 (calcd).
11-Bromoakuammine (19).
Prepared from 1 (30.0 mg, 0.0785 mmol) and NBS according to general procedure B to afford 11.9 mg of 19 (34% yield) as a white solid.1H NMR (600 MHz, CDCl3) δ 6.84 (d, J = 2.3 Hz, 1H), 6.65 (d, J = 2.4 Hz, 1H), 5.59 (q, J = 7.2 Hz, 1H), 4.46 (s, 1H), 4.14 (d, J = 16.5 Hz, 1H), 3.98 (d, J = 8.0 Hz, 1H), 3.88 (d, J = 7.8 Hz, 1H), 3.83 (s, 3H), 3.73 (d, J = 7.9 Hz, 1H), 3.56 (s, 1H), 3.35 (td, J = 15.1, 5.8 Hz, 1H), 3.15 – 3.08 (m, 1H), 3.08 (s, 3H), 2.83 – 2.78 (m, 1H), 2.45 (d, J = 14.9 Hz, 1H), 2.11 – 2.06 (m, 1H), 1.56 (dd, J = 7.2, 2.6 Hz, 4H).13C NMR (151 MHz, CDCl3) δ 171.12, 151.65, 142.01, 141.44, 125.28, 119.80, 110.71, 110.12, 105.40, 73.72, 57.96, 53.85, 53.07, 52.58, 49.79, 40.00, 32.62, 29.85, 28.72, 26.99, 14.27, 13.28. HRMS calculated for C22H26N2O4Br: [M + H]+: 461.1078 (found); 461.1076 (calcd).
11-Iodoakuammine (20).
Prepared from 2 (86.0 mg, 0.225 mmol) and NIS according to general procedure B afford 20.1 mg of 19 (22% yield) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.12 (d, J = 2.5 Hz, 1H), 6.66 (d, J = 2.5 Hz, 1H), 5.59 (q, J = 7.3 Hz, 1H), 4.49 (s, 1H), 4.16 (d, J = 16.6 Hz, 1H), 3.96 (d, J = 8.0 Hz, 1H), 3.90 (s, 1H), 3.82 (s, 3H), 3.72 (d, J = 7.9 Hz, 1H), 3.56 (s, 1H), 3.35 (td, J = 15.3, 5.7 Hz, 1H), 3.14 (d, J = 15.3 Hz, 1H), 3.06 (s, 3H), 2.83 (d, J = 12.5 Hz, 1H), 2.46 (d, J = 14.8 Hz, 1H), 2.08 (d, J = 15.2 Hz, 1H), 1.58 (s, 1H), 1.55 (dd, J = 7.1, 2.5 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 171.02, 151.35, 144.90, 141.35, 128.74, 126.48, 114.39, 111.35, 103.10, 75.09, 73.69, 57.81, 53.84, 53.42, 52.63, 52.36, 49.80, 39.91, 33.48, 29.85, 26.86, 13.30. HRMS calculated for C22H26N2O4I: [M + H]+: 509.0938 (found); 509.0937 (calcd).
11-Phenylakuammine (21).
Prepared from 20 (30.0 mg, 0.0590 mmol) according to General Procedure A to yield 14.9 mg of 21 (55% yield). 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 4.4 Hz, 4H), 7.30 (m, 1H), 6.64 (d, J = 2.6 Hz, 1H), 6.50 (d, J = 2.6 Hz, 1H), 5.45 (q, J = 7.3 Hz, 1H), 4.13 (s, 1H), 4.00 (s, 1H), 3.94 (d, J = 7.7 Hz, 1H), 3.85 (d J = 4.3 Hz, 1H), 3.82 (s, 3H), 3.75 (d, J = 7.6 Hz, 1H), 3.48 (d, J = 3.6 Hz, 1H), 3.40 – 3.28 (m, 1H), 2.92 (d, J = 16.7 Hz, 1H), 2.68 (dd, J = 13.2, 5.6 Hz, 1H), 2.33 (s, 1H), 2.28 (s, 3H), 2.08 – 1.99 (m, 1H), 1.63 – 1.54 (m, 1H), 1.54 – 1.47 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 170.78, 150.80, 141.19, 139.61, 139.13, 130.60, 129.18, 128.33, 127.62, 125.45, 117.36, 109.99, 109.51, 102.45, 73.55, 57.55, 53.61, 53.45, 52.75, 52.55, 49.43, 39.21, 33.07, 27.40, 26.35, 13.36. HRMS calculated for C28H31N2O4: [M + H]+: 459.2273 (found); 459.2284 (calcd).
11-(p-Tolyl)-akuammine (22).
Prepared from 20 (30.0 mg, 0.0590 mmol) according to General Procedure A to yield 17.3 mg of 22 (62% yield). 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 7.5 Hz, 2H), 7.16 (d, J = 7.9 Hz, 2H), 6.64 (d, J = 2.6 Hz, 1H), 6.55 (d, J = 2.6 Hz, 1H), 5.58 (q, J = 7.4 Hz, 1H), 4.38 (s, 1H), 4.16 (d, J = 16.5 Hz, 1H), 4.09 – 3.98 (m, 1H), 3.96 (d, J = 7.8 Hz, 1H), 3.85 (s, 3H), 3.78 (d, J = 7.8 Hz, 1H), 3.54 (s, 1H), 3.46 – 3.32 (m, 1H), 3.13 (d, J = 16.4 Hz, 1H), 2.42 (d, J = 15.2 Hz, 1H), 2.37 (s, 3H), 2.29 (s, 3H), 2.04 (dd, J = 14.7, 3.1 Hz, 1H), 1.72 (dd, J = 15.6, 4.5 Hz, 1H), 1.57 (dd, J = 7.1, 2.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 170.98, 150.81, 141.32, 139.81, 137.35, 136.33, 129.09, 124.74, 117.33, 109.37, 102.75, 73.54, 57.67, 53.73, 53.43, 52.70, 52.64, 49.57, 39.42, 33.01, 27.80, 26.55, 21.38, 13.36. HRMS calculated for C29H33N2O4: [M + H]+: 473.2438 (found); 473.2440 (calcd).
11-(p-Methoxyphenyl)-akuammine (23).
Prepared from 20 (30.0 mg, 0.0590 mmol) according to General Procedure A to yield 10.6 mg of 23 (62% yield). 1H NMR (600 MHz, CDCl3) δ 7.27 (s, 2H), 6.89 (d, J = 8.2 Hz, 2H), 6.61 (s, 1H), 6.50 (s, 1H), 5.47 (q, J = 7.4 Hz, 1H), 4.18 (s, 1H), 4.01 (d, J = 16.4 Hz, 1H), 3.94 (d, J = 7.5 Hz, 1H), 3.83 (s, 3H), 3.83 (s, 3H), 3.76 (d, J = 2.5 Hz, 1H), 3.75 (d, J = 1.4 Hz, 1H), 3.50 – 3.48 (m, 1H), 3.35 (s, 1H), 2.94 (d, J = 16.8 Hz, 1H), 2.70 (bs, 1H), 2.34 (s, 1H), 2.31 (s, 3H), 2.03 (d, J = 13.2 Hz, 1H), 1.57 (s, 1H), 1.54 (dd, J = 7.1, 2.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 171.93, 158.81, 150.45, 142.23, 141.42, 132.37, 130.21, 127.87, 120.03, 116.58, 114.28, 113.49, 111.73, 109.49, 73.46, 58.34, 55.41, 54.32, 53.27, 52.88, 52.26, 50.22, 40.61, 32.83, 31.74, 27.68, 13.15. HRMS calculated for C29H33N2O5: [M + H]+: 489.2389 (found); 489.2389 (calcd).
11-(p-Cyanophenyl)-akuammine (24).
Prepared from 20 (39.5 mg, 0.0778 mmol) according to General Procedure A to yield 14.4 mg of 24 (38% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.4 Hz, 2H),7.47 (d, J = 7.7 Hz, 2H), 6.72 (d, J = 2.5 Hz, 1H), 6.52 (d, J = 2.6 Hz, 1H), 5.56 (q, J = 7.4 Hz, 1H), 4.29 (s, 1H), 4.09 (d, J = 16.4 Hz, 1H), 3.96 (d, J = 7.8 Hz, 1H), 3.92 (s, 1H), 3.85 (s, 3H), 3.75 (d, J = 7.7 Hz, 1H), 3.54 (s, 1H), 3.40 (ddd, J = 21.7, 13.8, 6.3 Hz, 1H), 3.06 (d, J = 17.2 Hz, 1H), 2.82 (dd, J = 13.2, 5.5 Hz, 1H), 2.38 (d, J = 14.4 Hz, 1H), 2.28 (s, 3H), 2.05 (dt, J = 14.1, 3.3 Hz, 1H), 1.69 – 1.60 (m, 1H), 1.56 (dd, J = 7.2, 2.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.34, 151.48, 144.77, 141.66, 141.19, 132.01, 126.20, 122.67, 118.91, 116.38, 111.01, 103.58, 73.52, 57.98, 53.91, 53.16, 53.00, 52.57, 49.86, 39.94, 33.58, 29.08, 27.07, 13.26. HRMS calculated for C29H30N3O4: [M + H]+: 484.2235 (found); 484.2236 (calcd).
11-(p-Fluorophenyl)-akuammine (25).
Prepared from 20 (44 mg, 0.0868 mmol) according to General Procedure A to yield 18.8 mg of 25 (46% yield). 1H NMR (600 MHz, CDCl3) δ 7.32 (s, 2H), 7.08 – 7.02 (m, 2H), 6.64 (d, J = 2.8 Hz, 1H), 6.48 (d, J = 2.9 Hz, 1H), 5.46 (q, J = 7.8 Hz, 1H), 4.15 (s, 1H), 3.98 (d, J = 19.2 Hz, 1H), 3.94 (d, J = 7.6 Hz, 1H), 3.83 (s, 3H), 3.83 – 3.77 (m, 1H), 3.74 (d, J = 7.6 Hz, 1H), 3.48 (s, 1H), 3.38 – 3.30 (m, 1H), 2.91 (d, J = 16.9 Hz, 1H), 2.67 (d, J = 13.9 Hz, 1H), 2.32 (s, 1H), 2.29 (s, 3H), 2.04 (dd, J = 14.7, 4.4 Hz, 1H), 1.57 (d, J = 14.0 Hz, 1H), 1.53 (d, J = 4.6 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 171.99, 162.94, 161.30, 150.23, 142.52, 141.80, 136.03, 131.00, 126.84, 119.61, 116.39, 115.03, 110.01, 104.63, 73.50, 58.43, 54.39, 53.38, 52.85, 52.25, 50.31, 40.73, 32.96, 30.73, 27.79, 13.13. HRMS calculated for C28H30N2O4F: [M + H]+: 477.2190 (found); 477.2190 (calcd).
11-(3’-Furanyl)-akuammine (26).
Prepared from 20 (80.0 mg, 0.158 mmol) according to General Procedure A to yield 8.0 mg of 26 (11% yield). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 3.2 Hz, 2H), 6.58 (dd, J = 44.2, 2.5 Hz, 2H), 6.47 (s, 1H), 5.56 (q, J = 7.5 Hz, 1H), 4.37 (s, 1H), 4.12 (d, J = 16.5 Hz, 1H), 3.96 (d, J = 7.9 Hz, 1H), 3.83 (s, 3H), 3.76 (d, J = 7.7 Hz, 1H), 3.54 (s, 1H), 3.40 (dd, J = 15.2, 5.7 Hz, 1H), 3.09 (d, J = 16.4 Hz, 1H), 2.81 (dd, J = 13.2, 5.5 Hz, 1H), 2.53 (s, 3H), 2.42 (d, J = 14.7 Hz, 1H), 2.06 (d, J = 14.4 Hz, 1H), 1.64 (dd, J = 15.9, 4.6 Hz, 1H), 1.56 (d, J = 6.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 171.32, 150.42, 142.66, 142.28, 140.19, 123.69, 119.14, 117.04, 112.47, 110.03, 73.51, 57.95, 53.92, 53.11, 52.82, 52.48, 49.83, 39.93, 32.39, 28.81, 27.02, 13.23. HRMS calculated for C26H29N2O5: [M + H]+: 449.2070 (found); 449.2076 (calcd).
16-Hydroxymethyl pseudoakuammigine (28).
In an oven dried round bottom flask under nitrogen 5 mL of anhydrous THF was added. At 0C, LAH (11 mg, 2 equiv) was added to the reaction flask. A solution of 2 (50 mg, 1 equiv) in 2 mL of anhydrous THF was added dropwise to the reaction at 0 °C. The reaction was stirred for 3 hours at room temperature. LiAlH4 (11 mg, 2 equiv) was added at 0 °C. After 1 hour the reaction was halted over ice by the dropwise addition of 300 uL H2O, 300 uL 15% NaOH, and 1 mL H2O. 3 mL of H2O was added to the reaction and the solution was filtered over celite. The filtrate was extracted with 30 mL EtOAc three times. The organic layers were combined, dried over sodium sulfate, filtered, and concentrated in vacuo. The crude product was purified via column chromatography (0-5% MeOH/DCM) to yield 13 mg (67% yield) of the desired product. 1H NMR (400 MHz, CDCl3) δ 7.13 (td, J = 7.7, 1.3 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), 6.75 (td, J = 7.5, 1.2 Hz, 1H), 6.66 (d, J = 7.8 Hz, 1H), 5.42 (q, J = 7.1 Hz, 1H), 4.39 (d, J = 10.3 Hz, 1H), 4.19 (s, 1H), 4.13 (d, J = 10.3 Hz, 1H), 3.97 (d, J = 16.9 Hz, 1H), 3.91 (d, J = 7.3 Hz, 1H), 3.60 (dd, J = 13.5, 4.3 Hz, 1H), 3.53 (d, J = 7.4 Hz, 1H), 3.46 (s, 1H), 2.85 (s, 3H), 2.82 – 2.76 (m, 2H), 2.56 (dd, J = 13.5, 5.6 Hz, 1H), 2.28 – 2.18 (m, 1H), 2.04 (ddd, J = 14.1, 3.8, 2.0 Hz, 1H), 1.81 (dd, J = 7.1, 2.4 Hz, 3H), 1.22 (dd, J = 14.2, 4.3 Hz, 1H).13C NMR (151 MHz, CDCl3) δ 152.03, 144.09, 139.40, 127.48, 123.49, 119.77, 117.54, 110.22, 103.48, 61.82, 56.42, 55.07, 54.74, 53.18, 50.85, 36.30, 30.57, 29.86, 29.20, 28.17, 13.43.HRMS calculated for C21H27N2O2: [M + H]+: 339.2069 (found); 339.2073 (calcd).
Desacetylakuammiline (29).
In a round bottom flask, 5 (115 mg, 0.2919 mmol) was solubilized in 10 mL of MeOH at room temperature. To the solution was added 8.2 mL of a 10% w/v KOH solution (50 eqiv). After 2 hours, the MeOH was removed by rotary evaporation. The resultant solution was diluted with 10 mL of H2O and extracted with 20 mL DCM 3X. The organic layers were combined, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was then purified by column chromatography (0-5% MeOH/DCM) to yield 103 mg the desired product (99% yield) whose spectra were consistent with the literature.61
General Procedure C: Reductive Alkylation.
In an oven-dried round bottom flask under nitrogen, 29 (1.0 equiv) was dissolved in 5 mL anhydrous dichloromethane. The solution was treated with trifluoroacetic acid (9.0 equiv), triethylsilane (30 equiv), and appropriate dimethyl acetal (10 equiv). The reaction was stirred at room temperature for 18 hours. The solution was neutralized with saturated sodium bicarbonate and extracted DCM (3x20 mL). The organic layers were combined, dried over sodium sulfate, and concentrated in vacuo. The crude product was purified via column chromatography (2% MeOH/DCM) to yield the desired product.
N-1-Ethyl psuedoakuammigine (31).
Prepared from 29 (40 mg, 0.1135 mmol) according to General Procedure C and acetaldehyde dimethyl acetal to yield 13 mg of 31 (30%). 1H NMR (400 MHz, CDCl3) δ 7.17 (t, J = 7.8 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.78 (t, J = 7.4 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 5.68 (q, J = 7.2 Hz, 1H), 4.70 (s, 1H), 4.28 (d, J = 15.9 Hz, 1H), 4.00 (d, J = 7.7 Hz, 1H), 3.85 (s, 3H), 3.71 (d, J = 7.9 Hz, 1H), 3.64 (s, 1H), 3.53 – 3.39 (m, 2H), 3.28 (s, 1H), 3.24 (t, J = 7.5 Hz, 1H), 2.95 (dd, J = 12.7, 5.4 Hz, 1H), 2.59 (d, J = 14.9 Hz, 1H), 2.13 (d, J = 14.8 Hz, 1H), 1.83 – 1.74 (m, 1H), 1.61 (d, J = 7.3 Hz, 4H), 1.31 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 170.82, 149.41, 136.93, 128.68, 128.42, 125.18, 122.50, 120.54, 110.21, 101.48, 74.50, 57.16, 53.63, 52.45, 49.78, 39.25, 38.55, 29.70, 27.42, 26.30, 22.69, 14.71, 13.23. HRMS calculated for C23H29N2O3: [M + H]+: 381.2172 (found); 381.2178 (calcd).
N-1-Benzyl psuedoakuammigine (32).
Prepared from 29 (40 mg, 0.1135 mmol) according to General Procedure C and benzaldehyde dimethyl acetal to yield 32 mg of 32 (64%). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.1 Hz, 2H), 7.34 (dd, J = 8.4, 6.8 Hz, 2H), 7.30 – 7.21 (m, 1H), 7.10 (dd, J = 7.5, 1.3 Hz, 1H), 7.00 (td, J = 7.7, 1.3 Hz, 1H), 6.75 (td, J = 7.5, 1.0 Hz, 1H), 6.49 (d, J = 7.8 Hz, 1H), 5.43 (q, J = 7.1 Hz, 1H), 4.68 (d, J = 16.1 Hz, 1H), 4.23 (dt, J = 3.5, 1.7 Hz, 1H), 4.07 (d, J = 16.2 Hz, 1H), 3.98 (d, J = 7.5 Hz, 1H), 3.94 (d, J = 17.7 Hz, 1H), 3.88 (dd, J = 13.9, 4.6 Hz, 1H), 3.83 (s, 3H), 3.67 (d, J = 7.5 Hz, 1H), 3.50 (d, J = 3.7 Hz, 1H), 3.46 – 3.32 (m, 1H), 2.86 (d, J = 16.8 Hz, 1H), 2.66 (dd, J = 13.6, 5.8 Hz, 1H), 2.37 (ddd, J = 14.1, 4.3, 2.0 Hz, 1H), 2.08 (ddd, J = 14.0, 3.7, 1.9 Hz, 1H), 1.58 (d, J = 4.2 Hz, 1H), 1.54 (dd, J = 7.0, 2.4 Hz, 3H) 13C NMR (101 MHz, CDCl3) δ 172.24, 152.02, 142.17, 139.42, 128.75, 127.52, 127.41, 127.09, 122.53, 120.63, 118.56, 111.37, 104.50, 74.56, 58.71, 54.74, 53.91, 53.60, 52.08, 51.03, 49.62, 41.04, 31.47, 28.28, 13.09. HRMS calculated for C28H31N2O3: [M + H]+: 443.2332 (found); 443.2335 (calcd).
N-1-Phenethyl psuedoakuammigine (33).
Prepared from 29 (30 mg, 0.0851 mmol) according to General Procedure C and phenylacetaldehyde dimethyl acetal to yield 25 mg mg of 33 (63%). 1H NMR (400 MHz, CDCl3) δ 7.35 – 7.27 (m, 4H), 7.25 – 7.17 (m, 1H), 7.16 – 7.06 (m, 2H), 6.75 (t, J = 7.5 Hz, 1H), 6.68 (d, J = 7.8 Hz, 1H), 5.50 (q, J = 7.4 Hz, 1H), 4.42 (s, 1H), 4.07 (d, J = 16.4 Hz, 1H), 3.96 (d, J = 7.7 Hz, 1H), 3.83 (s, 3H), 3.72 – 3.55 (m, 3H), 3.53 (d, J = 3.4 Hz, 1H), 3.43 – 3.29 (m, 2H), 3.09 – 2.93 (m, 3H), 2.69 (dd, J = 13.4, 5.7 Hz, 1H), 2.11 (d, J = 14.2 Hz, 1H), 1.55 (d, J = 7.2 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ172.27, 150.75, 141.84, 139.86, 128.83, 128.70, 127.55, 126.40, 122.70, 120.45, 119.79, 118.72, 109.38, 103.93, 74.57, 58.39, 54.67, 53.79, 53.53, 52.06, 50.98, 46.06, 41.00, 36.28, 31.20, 28.13, 13.08. HRMS calculated for C29H33N2O3: [M + H]+: 457.2486 (found); 457.2491 (calcd).
In Vitro Pharmacology
Drugs.
Forskolin and DMSO were purchased from Sigma-Aldrich (St. Louis, MO, United States).(2S)-2-[[2-[[(2R)-2-[[(2S)-2-Amino-3-(4-hydroxyphenyl)propanoyl]amino]propanoyl]amino]acetyl]-methylamino]-N-(2hydroxyethyl)-3-phenylpropanamide (DAMGO), and 2-(3,4-dichlorophenyl)-Nmethyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl] acetamide (U50,488) were purchased from Tocris Bioscience (Bio-Techne Corporation, Minneapolis, MN, United States). [3H]DAMGO (53.7 Ci/mmol, lot#2376538; 51.7 Ci/mmol, lot#2815607), [3H]U69,593 (60 Ci/mmol, lot#2367921 and lot#2644168; 49.2 Ci/mmol, lot#2791786), were purchased from PerkinElmer (Waltham, MA, United States).
Competitive radioligand binding.
Competitive radioligand binding experiments with full dose-response curves were completed as previously described20, 62 using [3H]-DAMGO and [3H]-U69,593 at μOR and κOR, respectively, with one exception. Instead of incubating the reaction mix for 90 minutes, an incubation time of 180 minutes was used to capture slower binding kinetics of some of the analogs. The same competitive radioligand binding method was used for the binding screens except instead of full dose-response curves, single concentrations of 10 μM and 1 μM were used. In these experiments, the single concentration points were run in duplicate and a concentration-dose response curve of a positive control was included in each assay for data normalization purposes (DAMGO for μOR, U50,488 for κOR). The analogues were solubilized in DMSO to make 10 mM stock solutions. From the stock solutions, a 25X dilution was made using assay buffer, then serial dilutions were performed to obtain working concentrations. Binding screen composites are made up of three individual replicates.
GloSensor cAMP inhibition assay.
GloSensor assays were completed as previously described using HEK cells transiently expressing pGloSensor22F, and either HA-mouse μOR or FLAGmouse κOR.20, 63 For assays shown in Figure 2E, DMSO was added to the buffer for positive controls to control for any solvent-related effects. The analogues were solubilized in DMSO to make 10 mM stock solutions. From the stock solutions, a 25X dilution was made using OptiMEM, then serial dilutions were performed to obtain working concentrations
PathHunter β-Arrestin-2 assay.
β-Arrestin-2 recruitment assays were completed as previously described using PathHunter cell lines stably expressing μOR and β-arrestin-2.20, 63 The analogues were solubilized in DMSO to make 10 mM stock solutions. From the stock solutions, a 20X dilution was made using OptiMEM, then serial dilutions were performed to obtain working concentrations.
TRUPATH assay.
The TRUPATH assay was performed as previously described, using DAMGO as a positive control.42 Cells were plated either in 6-well dishes at a density of 700,000–800,000 cells per well, or 10 cm dishes at 7–8 million cells per dish. Cells were transfected 2–4 h later, using a 1:1:1:1 DNA ratio of receptor:Gα-RLuc8:Gβ:Gγ-GFP2 (100 ng per construct for 6-well dishes, 750 ng per construct for 10 cm dishes), except for the Gγ-GFP2 screen, where an ethanol coprecipitated mixture of Gβ1–4 was used at twice its normal ratio (1:1:2:1). Transit 2020 (Mirus Biosciences) was used to complex the DNA at a ratio of 3 μl Transit per μg DNA, in OptiMEM (Gibco-ThermoFisher) at a concentration of 10 ng DNA per μl OptiMEM. The next day, cells were harvested from the plate using Versene (0.1 M PBS + 0.5 mM EDTA, pH 7.4) and plated in polyD-lysine-coated white, clear-bottom 96-well assay plates (Greiner Bio-One) at a density of 30,000–50,000 cells per well. One day after plating in 96-well assay plates, white backings (PerkinElmer) were applied to the plate bottoms, and growth medium was carefully aspirated and replaced immediately with 60 μl of assay buffer (1× Hank’s balanced salt solution (HBSS) + 20 mM HEPES, pH 7.4), followed by a 10 μl addition of freshly prepared 50 μM coelenterazine 400a (Nanolight Technologies). After a 5 min equilibration period, cells were treated with 30 μl of drug for an additional 5 min. Plates were then read in an LB940 Mithras plate reader (Berthold Technologies) with 395 nm (RLuc8-coelenterazine 400a) and 510 nm (GFP2) emission filters, at integration times of 1 s per well. Plates were read serially six times, and measurements from the sixth read were used in all analyses. BRET2 ratios were computed as the ratio of the GFP2 emission to RLuc8 emission.
Statistics.
Cellular pharmacological data was analyzed using GraphPad 9 (GraphPad Prism software, La Jolla, CA, United States) and is shown as mean ± SEM. For binding, GloSensor and PathHunter assays, composite figures consist of a curve averaged from a minimum of three independent assays that were each normalized to a positive control.
Animal Studies.
Animals.
C57BL/6 mice (20-25 g, mixed sexes, Jackson laboratory) were used in the study. The mice were housed in a 14/10-hour light/dark cycle (5:00 AM on/7:00 PM off) with access to food and water ad libitum. All animal experiments were performed in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals after getting approval from the University of Illinois Institutional Animal Care and Use Committee.
Drugs.
Morphine sulfate injection was purchased from Hospira (Lake Forest, IL). Naloxone was purchased from Sigma-Aldrich (St. Louis, MO). Compound 33 was formulated using 5.4:6.5:0.1 ratio of DMSO, PBS, and 20% aqueous tartaric acid. All injection volumes were calculated according to mouse body weight and did not exceed 200 μL.
Tail-Flick Assay.
To determine basal nociception and antinociception, tail-flick test was performed.45, 64 Distal one-third of the mouse tail was immersed in a water bath (VWR Model 1130S) that was maintained at 52°C. The latency of the mouse to a rapid tail-flick response was recorded. Antinociceptive effects of morphine (10 mg/kg, s.c) and different doses of 33 (10-100 mg/kg, s.c) were evaluated at 15, 30, 60, 120, 180 and 240 min after drug administration. Analogue 33 was administered subcutaneously with the doses 10, 50, 65, 80, and 100 mg/kg (n = 3 for each dose). The antinociceptive effect was expressed as percentage of maximal possible effect (MPE) and the cut-off time was set at 12 seconds to prevent damage to the tail.
Hot-Plate Assay.
Hot-plate test was performed to test basal nociception and antinociception.44 Mice were placed in a glass cylinder on a heated plate (Ugo Basil Hot/Cold Plate 35100) maintained at 55 ± 0.1°C. The latency to hind-paw licking, flinching, withdrawal or jumping was recorded as a response. Antinociceptive effects of morphine (10 mg/kg s.c) and different doses of 33 (mg/kg s.c) were evaluated at 15, 30, 60, 120, 180 and 240 min after drug administration. Analogue 33 was administered subcutaneously with the doses 10, 50, 65, 80, and 100 mg/kg (n = 3 for each dose). The antinociceptive effect was expressed as percentage of maximal possible effect (MPE). Cut-off time was set at 45 seconds to prevent damage to the paws.
Naloxone Inhibition of Antinociceptive Effects.
Mice were pretreated with naloxone (10 mg/kg, i.p.), followed by an injection of 33 (100 mg/kg, s.c.) 15 min later. The tail-flick assay and the hot-plate assay were performed 30, 60, 120, 180 and 240 min after treatment with 33 as described above. Separate groups of control mice received naloxone (10 mg/kg, i.p.) only or 33 (100 mg/kg, s.c.) only.
Rotarod Assay.
The locomotor activity of the mice after the treatment with 33 was tested using the rotarod test.46 On Day 1, mice were placed on the rotarod (Model series 8, IITC Life Science, Woodland Hills, CA) and trained on a fixed speed (4 rpm) for 60 s. The training session from Day 1 was repeated on the second day and the mice that failed to stay on the rotarod for 60 s were excluded from further studies. On test day (Day 3), the mice were placed on an accelerating rotarod (4 - 40 rpm over 300 seconds) and the latencies to fall were recorded 15 min after the administration of 33 (50, 65, 80 mg/kg s.c).
Statistics.
All data are presented as Mean ± S.E.M. MPE% = 100% *(postdrug latency - predrug latency) / (cutoff - predrug latency). Comparisons between groups were analyzed using a one-way analysis of variance followed by post hoc analyses using Dunnett’s t test (multiple groups). For comparison between groups for antinociceptive effect at different time points a two-way repeated measure analysis of variance followed by post hoc analyses using Dunnett’s t test (multiple groups) was used. Statistical significance was established at 95% confidence limit.
Molecular Docking.
The crystal structure of the μOR bound to the agonist BU72 (PDB: 5C1M) was imported and prepared using the Protein Preparation module in Schrödinger Maestro (version 12.9.137) to remove waters outside the binding site, optimize hydrogen bonds, and minimize heavy atoms to RMSD 0.30 Å. Compounds 19 and 33 were imported as SMILES and lowest energy conformations generated using the LigPrep module and the OPLS4 force field. Possible protonation states were determined by Epik at pH = 7.4 ± 2.0. The Induced Fit Docking module 51 was used to generate possible posses with the extended sampling protocol to generate up to 80 initial docking poses. The centroid of BU72 was used as the box center. No hydrogen bonding constraints were applied. Conformational sampling of the ligands was enabled to allow conformations within 2.5 kcal/mol of the LigPrep-generated conformations. Glide docking was performed with a van der Waals scaling of 0.5 to generate initial docking poses. For each docking pose, residue side-chains and backbones within 5.0 Å of the ligand were refined with Prime to generate induced-fit receptors. Ligands were redocked into the top 20 induced-fit structures within 30 kcal/mol of the best structure. The docking pose for each ligand were rank-ordered by docking score and top-scoring pose exported to PyMOL for visualization and figure generation.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Clinical and Translational Sciences KL2 training program (KL2TR002002) and NIH Grant R35GM147005. M.R.H was supported by the NIH T32 Training Grant T32AT007533. This research was supported by funds awarded to AG by the American Foundation of Pharmaceutical Education in the form of a pre-doctoral fellowship; to RvR by the National Institute on Alcohol Abuse and Alcoholism (AA025368, and AA026949) and the National Institute on Drug Abuse (DA045897) of the National Institutes of Health
ABBREVIATIONS
- BRET
bioluminescence resonance energy transfer
- NIS
N-iodosuccinimide
- βArr2
β-arrestin
- β-FNA
β-funaltrexamine
- δOR
delta opioid receptor
- κOR
kappa opioid receptor
- μOR
mu opioid receptor
Footnotes
Supporting Information
The following files are available free of charge.
Additional supplementary figures (Figures S1–3) and table (Table S1).
HPLC Chromatograph of compound 33.
1H NMR and 13C NMR spectra for compounds 7-33.
Molecular Formula Strings and associated activity for compounds 1, 2, 5-8, 10-29, and 31-33.
PDB files of docked binding poses for compounds 19 and 33.
REFERNCES
- 1.Volkow ND; Collins FS The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med 2017, 377, 391–394. [DOI] [PubMed] [Google Scholar]
- 2.Vardanyan RS; Hruby VJ Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future medicinal chemistry 2014, 6, 385–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasternak GW; Pan YX Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 2013, 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Varga BR; Streicher JM; Majumdar S Strategies towards safer opioid analgesics-A review of old and upcoming targets. Br. J. Pharmacol 2021, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paul AK; Smith CM; Rahmatullah M; Nissapatorn V; Wilairatana P; Spetea M; Gueven N; Dietis N Opioid Analgesia and Opioid-Induced Adverse Effects: A Review. Pharmaceuticals 2021, 14, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madariaga-Mazón A; Marmolejo-Valencia AF; Li Y; Toll L; Houghten RA; Martinez-Mayorga K Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov. Today 2017, 22, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Günther T; Dasgupta P; Mann A; Miess E; Kliewer A; Fritzwanker S; Steinborn R; Schulz S Targeting multiple opioid receptors - improved analgesics with reduced side effects? Br. J. Pharmacol 2018, 175, 2857–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakraborty S; DiBerto JF; Faouzi A; Bernhard SM; Gutridge AM; Ramsey S; Zhou Y; Provasi D; Nuthikattu N; Jilakara R; Nelson MNF; Asher WB; Eans SO; Wilson LL; Chintala SM; Filizola M; van Rijn RM; Margolis EB; Roth BL; McLaughlin JP; Che T; Sames D; Javitch JA; Majumdar S A Novel Mitragynine Analog with Low-Efficacy Mu Opioid Receptor Agonism Displays Antinociception with Attenuated Adverse Effects. J. Med. Chem 2021, 64, 13873–13892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virk MS; Arttamangkul S; Birdsong WT; Williams JT Buprenorphine is a weak partial agonist that inhibits opioid receptor desensitization. J. Neurosci 2009, 29, 7341–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillis A; Gondin AB; Kliewer A; Sanchez J; Lim HD; Alamein C; Manandhar P; Santiago M; Fritzwanker S; Schmiedel F; Katte TA; Reekie T; Grimsey NL; Kassiou M; Kellam B; Krasel C; Halls ML; Connor M; Lane JR; Schulz S; Christie MJ; Canals M Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal 2020, 13, eaaz3140. doi: 10.1126/scisignal.aaz3140. [DOI] [PubMed] [Google Scholar]
- 11.Martínez V; Abalo R Peripherally acting opioid analgesics and peripherally-induced analgesia. Behav. Pharmacol 2020, 31, . [DOI] [PubMed] [Google Scholar]
- 12.Vanderah TW Delta and kappa opioid receptors as suitable drug targets for pain. Clin. J. Pain 2010, 26 Suppl 10, 10. [DOI] [PubMed] [Google Scholar]
- 13.Smith MT; Kong D; Kuo A; Imam MZ; Williams CM Analgesic Opioid Ligand Discovery Based on Nonmorphinan Scaffolds Derived from Natural Sources. J. Med. Chem 2022, 65, 1612–1661. [DOI] [PubMed] [Google Scholar]
- 14.Tidgewell K; Harding WW; Lozama A; Cobb H; Shah K; Kannan P; Dersch CM; Parrish D; Deschamps JR; Rothman RB; Prisinzano TE Synthesis of Salvinorin A Analogues as Opioid Receptor Probes. J. Nat. Prod 2006, 69, 914–918. [DOI] [PubMed] [Google Scholar]
- 15.Bhowmik S; Galeta J; Havel V; Nelson M; Faouzi A; Bechand B; Ansonoff M; Fiala T; Hunkele A; Kruegel AC; Pintar JE; Majumdar S; Javitch JA; Sames D Site selective C–H functionalization of Mitragyna alkaloids reveals a molecular switch for tuning opioid receptor signaling efficacy. Nature Communications 2021, 12, 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakraborty S; Majumdar S Natural Products for the Treatment of Pain: Chemistry and Pharmacology of Salvinorin A, Mitragynine, and Collybolide. Biochemistry 2021, 60, 1381–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Váradi A; Marrone GF; Palmer TC; Narayan A; Szabó MR; Le Rouzic V; Grinnell SG; Subrath JJ; Warner E; Kalra S; Hunkele A; Pagirsky J; Eans SO; Medina JM; Xu J; Pan Y; Borics A; Pasternak GW; McLaughlin JP; Majumdar S Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem 2016, 59, 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menzies JR; Paterson SJ; Duwiejua M; Corbett AD Opioid activity of alkaloids extracted from Picralima nitida (fam. Apocynaceae). Eur. J. Pharmacol 1998, 350, 101–108. [DOI] [PubMed] [Google Scholar]
- 19.Toce MS; Chai PR; Burns MM; Boyer EW Pharmacologic Treatment of Opioid Use Disorder: a Review of Pharmacotherapy, Adjuncts, and Toxicity. Journal of medical toxicology : official journal of the American College of Medical Toxicology 2018, 14, 306–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Creed SM; Gutridge AM; Argade MD; Hennessy MR; Friesen JB; Pauli GF; van Rijn RM; Riley AP Isolation and Pharmacological Characterization of Six Opioidergic Picralima nitida Alkaloids. J. Nat. Prod 2021, 84, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duwiejua M; Woode E; Obiri DD Pseudo-akuammigine, an alkaloid from Picralima nitida seeds, has anti-inflammatory and analgesic actions in rats. J. Ethnopharmacol 2002, 81, 73–79. [DOI] [PubMed] [Google Scholar]
- 22.van ‘t Erve TJ; Rautiainen RH; Robertson LW; Luthe G Trimethylsilyldiazomethane: a safe non-explosive, cost effective and less-toxic reagent for phenol derivatization in GC applications. Environ. Int 2010, 36, 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gill D; Hester AJ; Lloyd-Jones G On the preparation of ortho-trifluoromethyl phenyl triflate. Organic & biomolecular chemistry; Org Biomol Chem 2004, 2, 2547–2548. [DOI] [PubMed] [Google Scholar]
- 24.Montesinos-Magraner M; Vila C; Rendón-Patiño A; Blay G; Fernández I; Muñoz MC; Pedro JR Organocatalytic Enantioselective Friedel–Crafts Aminoalkylation of Indoles in the Carbocyclic Ring. ACS Catal. 2016, 6, 2689–2693. [Google Scholar]
- 25.Zhu S; Yu Y; Li S; Wang L; Zhou Q Enantioselective Hydrogenation of α-Substituted Acrylic Acids Catalyzed by Iridium Complexes with Chiral Spiro Aminophosphine Ligands. Angew. Chem. Int. Ed 2012, 51, 8872–8875. [DOI] [PubMed] [Google Scholar]
- 26.Guo S; Liu Y; Zhao L; Zhang X; Fan X Rhodium-Catalyzed Selective Oxidative (Spiro)annulation of 2-Arylindoles by Using Benzoquinone as a C2 or C1 Synthon. Org. Lett 2019, 21, 6437–6441. [DOI] [PubMed] [Google Scholar]
- 27.McCamley K; Ripper JA; Singer RD; Scammells PJ Efficient N-Demethylation of Opiate Alkaloids Using a Modified Nonclassical Polonovski Reaction. J. Org. Chem 2003, 68, 9847–9850. [DOI] [PubMed] [Google Scholar]
- 28.Glotz G; Kappe CO; Cantillo D Electrochemical N-Demethylation of 14-Hydroxy Morphinans: Sustainable Access to Opioid Antagonists. Org. Lett 2020, 22, 6891–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kok GB; Pye CC; Singer RD; Scammells PJ Two-Step Iron(0)-Mediated N-Demethylation of N-Methyl Alkaloids. J. Org. Chem 2010, 75, 4806–4811. [DOI] [PubMed] [Google Scholar]
- 30.Abdel-Monem M; Portoghese PS N-Demethylation of morphine and structurally related compounds with chloroformate esters. J. Med. Chem 1972, 15, 208–210. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X; Kakde B; Guo R; Yadav S; Gu Y; Li A Total Syntheses of Echitamine, Akuammiline, Rhazicine , and Pseudoakuammigine. Angewandte Chemie International Edition 2019, 58, 6053–6058. [DOI] [PubMed] [Google Scholar]
- 32.Popp T; Bracher F N-Methylation of Aromatic Amines and N-Heterocycles under Acidic Conditions with the TTT (1,3,5-Trioxane–Triethylsilane– Trifluoroacetic Acid) System. Synthesis 2015, 47, 3333–3338. [Google Scholar]
- 33.Righi M; Bedini A; Piersanti G; Romagnoli F; Spadoni G Direct, One-Pot Reductive Alkylation of Anilines with Functionalized Acetals Mediated by Triethylsilane and TFA. Straightforward Route for Unsymmetrically Substituted Ethylenediamine. J. Org. Chem 2011, 76, 704–707. [DOI] [PubMed] [Google Scholar]
- 34.Park ES; Lee JH; Kim SJ; Yoon CM One-Pot Reductive Amination of Acetals with Aromatic Amines Using Decaborane (B10H14) in Methanol. null 2003, 33, 3387–3396. [Google Scholar]
- 35.Dong J; Yu L; Xie J A Simple and Versatile Method for the Formation of Acetals/Ketals Using Trace Conventional Acids. ACS Omega 2018, 3, 4974–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang FI; Ding G; Ng GS; Dixon SJ; Chidiac P Luciferase-based GloSensor™ cAMP assay: Temperature optimization and application to cell-based kinetic studies. Methods 2022, 203, 249–258. [DOI] [PubMed] [Google Scholar]
- 37.Mores KL; Cassell RJ; van Rijn RM Arrestin recruitment and signaling by G protein-coupled receptor heteromers. Neuropharmacology 2019, 152, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raehal KM; Bohn LM The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 2011, 60, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowley RS; Riley AP; Alder AF; Anderson RJ 3rd; Luo D; Kaska S; Maynez P; Kivell BM; Prisinzano TE Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Design, Synthesis, and Evaluation of Analogues with Improved Potency and G-protein Activation Bias at the μ-Opioid Receptor. ACS Chem. Neurosci 2020, 11, 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowley RS; Riley AP; Sherwood AM; Groer CE; Shivaperumal N; Biscaia M; Paton K; Schneider S; Provasi D; Kivell BM; Filizola M; Prisinzano TE Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting μ Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem 2016, 59, 11027–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen RHJ; DiBerto JF; English JG; Glaudin AM; Krumm BE; Slocum ST; Che T; Gavin AC; McCorvy JD; Roth BL; Strachan RT TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol 2020, 16, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakraborty S; DiBerto JF; Faouzi A; Bernhard SM; Gutridge AM; Ramsey S; Zhou Y; Provasi D; Nuthikattu N; Jilakara R; Nelson MNF; Asher WB; Eans SO; Wilson LL; Chintala SM; Filizola M; van Rijn RM; Margolis EB; Roth BL; McLaughlin JP; Che T; Sames D; Javitch JA; Majumdar S A Novel Mitragynine Analog with Low-Efficacy Mu Opioid Receptor Agonism Displays Antinociception with Attenuated Adverse Effects. J. Med. Chem 2021, 64, 13873–13892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Y; Chen Y; Tian X; Yang C; Lu J; Xiao C; DeSimone J; Wilkie DJ; Molokie RE; Wang ZJ CaMKIIα underlies spontaneous and evoked pain behaviors in Berkeley sickle cell transgenic mice. Pain 2016, 157, 2798–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang L; Shukla PK; Wang LX; Wang ZJ Reversal of Morphine Antinociceptive Tolerance and Dependence by the Acute Supraspinal Inhibition of Ca2+/Calmodulin-Dependent Protein Kinase II. J. Pharmacol. Exp. Ther 2006, 317, 901. [DOI] [PubMed] [Google Scholar]
- 46.He Y; Wilkie DJ; Nazari J; Wang R; Messing RO; DeSimone J; Molokie RE; Wang ZJ PKCδ-targeted intervention relieves chronic pain in a murine sickle cell disease model. J. Clin. Invest 2016, 126, 3053–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone LS; German JP; Kitto KF; Fairbanks CA; Wilcox GL Morphine and Clonidine Combination Therapy Improves Therapeutic Window in Mice: Synergy in Antinociceptive but Not in Sedative or Cardiovascular Effects. PLOS ONE 2014, 9, e109903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou T; Xu F; Peng X; Zhou H; Zhang X; Qiu M; Wang J; Liu Y; Liang X Label-free cell phenotypic study of opioid receptors and discovery of novel mu opioid ligands from natural products. J. Ethnopharmacol 2021, 270, 113872. [DOI] [PubMed] [Google Scholar]
- 49.Prisinzano TE Natural Products as Tools for Neuroscience: Discovery and Development of Novel Agents to Treat Drug Abuse. J. Nat. Prod 2009, 72, 581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kruegel AC; Grundmann O The medicinal chemistry and neuropharmacology of kratom: A preliminary discussion of a promising medicinal plant and analysis of its potential for abuse. Neuropharmacology 2018, 134, 108–120. [DOI] [PubMed] [Google Scholar]
- 51.Sherman W; Day T; Jacobson MP; Friesner RA; Farid R Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 52.Ellis CR; Racz R; Kruhlak NL; Kim MT; Zakharov AV; Southall N; Hawkins EG; Burkhart K; Strauss DG; Stavitskaya L Evaluating kratom alkaloids using PHASE. PLOS ONE 2020, 15, e0229646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vo QN; Mahinthichaichan P; Shen J; Ellis CR How μ-opioid receptor recognizes fentanyl. Nature Communications 2021, 12, 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manglik A; Kruse AC; Kobilka TS; Thian FS; Mathiesen JM; Sunahara RK; Pardo L; Weis WI; Kobilka BK; Granier S Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cong X; Campomanes P; Kless A; Schapitz I; Wagener M; Koch T; Carloni P Structural Determinants for the Binding of Morphinan Agonists to the μ-Opioid Receptor. PLOS ONE 2015, 10, e0135998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaserer T; Lantero A; Schmidhammer H; Spetea M; Schuster D μ Opioid receptor: novel antagonists and structural modeling. Scientific Reports 2016, 6, 21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lipiński PFJ; Jarończyk M; Dobrowolski JC; Sadlej J Molecular dynamics of fentanyl bound to μ-opioid receptor. Journal of Molecular Modeling 2019, 25, 144. [DOI] [PubMed] [Google Scholar]
- 60.Tian X; Zhang J; Wang S; Gao H; Sun Y; Liu X; Fu W; Tan B; Su R Tyrosine 7.43 is important for mu-opioid receptor downstream signaling pathways activated by fentanyl. Frontiers in Pharmacology 2022, 13, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benayad S; Ahamada K; Lewin G; Evanno L; Poupon E Preakuammicine: A Long-Awaited Missing Link in the Biosynthesis of Monoterpene Indole Alkaloids. Eur. J. Org. Chem 2016, 2016, 1494–1499. [Google Scholar]
- 62.Cassell RJ; Mores KL; Zerfas BL; Mahmoud AH; Lill MA; Trader DJ; van Rijn RM Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsychopharmacol 2019, 29, 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiang T; Sansuk K; van Rijn RM β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol 2016, 173, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He Y; Wang ZJ Nociceptor Beta II, Delta, and Epsilon Isoforms of PKC Differentially Mediate Paclitaxel-Induced Spontaneous and Evoked Pain. J. Neurosci 2015, 35, 4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.