

Abstract
Rationale
Inflammation drives pulmonary arterial hypertension (PAH). Gut dysbiosis causes immune dysregulation and systemic inflammation by altering circulating microbial metabolites; however, little is known about gut dysbiosis and microbial metabolites in PAH.
Objectives
To characterize the gut microbiome and microbial metabolites in patients with PAH.
Methods
We performed 16S ribosomal RNA gene and shotgun metagenomics sequencing on stool from patients with PAH, family control subjects, and healthy control subjects. We measured markers of inflammation, gut permeability, and microbial metabolites in plasma from patients with PAH, family control subjects, and healthy control subjects.
Measurements and Main Results
The gut microbiome was less diverse in patients with PAH. Shannon diversity index correlated with measures of pulmonary vascular disease but not with right ventricular function. Patients with PAH had a distinct gut microbial signature at the phylogenetic level, with fewer copies of gut microbial genes that produce antiinflammatory short-chain fatty acids (SCFAs) and secondary bile acids and lower relative abundances of species encoding these genes. Consistent with the gut microbial changes, patients with PAH had relatively lower plasma concentrations of SCFAs and secondary bile acids. Patients with PAH also had enrichment of species with the microbial genes that encoded the proinflammatory microbial metabolite trimethylamine. The changes in the gut microbiome and circulating microbial metabolites between patients with PAH and family control subjects were not as substantial as the differences between patients with PAH and healthy control subjects.
Conclusions
Patients with PAH have proinflammatory gut dysbiosis, in which lower circulating SCFAs and secondary bile acids may facilitate pulmonary vascular disease. These findings support investigating modulation of the gut microbiome as a potential treatment for PAH.
Keywords: pulmonary arterial hypertension, microbiome, dysbiosis, metabolites
At a Glance Commentary
Scientific Knowledge on the Subject
There is gut dysbiosis in pulmonary arterial hypertension.
What This Study Adds to the Field
This study provides a mechanistic link between gut dysbiosis, systemic inflammation, and pulmonary arterial hypertension (PAH). Using complementary 16S rRNA and shotgun metagenomic sequencing of stool samples, this study shows that PAH patients have reduced relative abundances of gut bacteria that contain encoding genes for the production of anti-inflammatory metabolites (short-chain fatty acids and secondary bile acids) and increased relative abundances of gut bacteria that contain encoding genes for the production of trimethylamine, a proinflammatory metabolite. Consistent with these gut microbial changes, using targeted metabolomics, this study reveals that PAH patients also have reduced plasma levels of anti-inflammatory short-chain fatty acids and secondary bile acids, which may promote pulmonary vascular disease.
Perivascular inflammation plays a critical role in driving pulmonary vascular remodeling that leads to pulmonary arterial hypertension (PAH) (1). However, the mechanisms that initiate and perpetuate immune dysregulation and perivascular inflammation in PAH remain unclear.
One potential driver of systemic inflammation and a trigger of PAH is an altered gut microbiome, known as gut dysbiosis (2). Failure of the gut microbiota to produce antiinflammatory metabolites, such as short-chain fatty acids (SCFAs), or excessive production of potential toxins, such as hydrogen sulfide, can contribute to a weakened gut barrier and increased circulating concentrations of microbial ligands for microbe-associated molecular patterns (3). In addition, altered composition of products of gut microbiota metabolism, such as secondary bile acids and trimethylamine, can drive systemic immune dysregulation (2, 3). Experimental models of PAH exhibit an altered intestinal microbial community structure (4), while antibiotic treatment and transfer of fecal matter in rats prevents pulmonary vascular remodeling (5, 6). However, little is known about the composition or metabolic output of gut microbiota in patients with PAH (7).
We sought to characterize the gut microbiome, measure systemic concentrations of cytokines and biomarkers of gut barrier function, and perform targeted metabolomics focusing on circulating microbial metabolites associated with immune regulation in deeply phenotyped patients with PAH, their family members, and healthy control subjects. Some of the results of these studies have been previously reported in the form of an abstract (8).
Methods
Study Participants and Procedures
We studied patients with PAH treated at the University of Minnesota Pulmonary Hypertension program between February 2019 and March 2021. PAH was defined as a mean pulmonary arterial pressure (mPAP) >20 mm Hg with a pulmonary arterial wedge pressure ⩽15 mm Hg and a pulmonary vascular resistance (PVR) ≥3 Wood units. Subjects with pulmonary hypertension due to left heart disease, chronic lung disease, chronic thromboembolic disease, and other miscellaneous causes were excluded, as previously described (9). All patients gave informed consent to participate, and the study protocol was approved by the University of Minnesota Institutional Review Board.
During this study period, we included stool samples from 72 patients with PAH, 15 individuals residing with patients with PAH in the same household (family control subjects), and 39 healthy control stool donors from the University of Minnesota Microbiota Therapeutics Program (10). For plasma analysis, we included up to 83 patients with PAH, 7 family control subjects, and 24 age- and sex-matched healthy volunteers with no significant past medical problems who donated blood samples.
We collected clinical metadata, performed 16S ribosomal RNA (rRNA) gene sequencing and shotgun metagenomic sequencing on stool samples, and measured plasma markers of gut permeability, inflammation, and microbial metabolites in patients with PAH, family control subjects, and healthy control subjects.
See the online supplement for detailed methods.
Statistical Analysis
We report categorical variables as frequency and percentage and continuous variables as mean and SD if normally distributed and as median and interquartile range if not. To compare categorical variables, we used chi-square or Fisher exact tests. We compared continuous variables with equal variance using an unpaired t test (for two groups) or one-way ANOVA with Tukey post hoc analysis to correct for multiple comparisons (for more than two groups). We used the Wilcoxon-Mann-Whitney test (for two groups) or Kruskal-Wallis test with Dunn’s multiple-comparisons test (for more than two groups) for continuous data that were not normally distributed. To compare community composition between samples (β diversity), analysis of similarity (11) was used with Bray-Curtis dissimilarity matrices. We compared the relative abundance of taxa that were differentially abundant among the study groups on the basis of linear discriminant analysis of effect size (LEfSe) (12). To assess the independent association of PAH with gut dysbiosis, we performed multivariable regression to adjust for age, sex, body mass index (BMI), recent antibiotic use, type of diet, and smoking status (model 1) and Charlson comorbidity index (CCI; model 2). We determined the association between gut microbiome diversity indices with measures of pulmonary vascular disease and right ventricular (RV) function using simple linear regression analysis. We performed sparse partial least square discriminant analysis of relative abundance data at the genus level of 16S rRNA data using MetaboAnalyst software (13). Hierarchical cluster analysis was conducted to compare the targeted metabolomics, gut microbial gene copies, and relative abundances of species that encode microbial genes among the study groups using MetaboAnalyst software. We used Prism 9.2.0 (GraphPad) and Stata software version 15 (StataCorp LP). A two-sided P value of <0.05 was considered to indicate statistical significance.
Results
Study Subjects
Table 1 lists the demographics and clinical metadata. Table E1 in the online supplement lists the relationship between family control subjects and the cohousing patient with PAH. Compared with healthy control subjects, patients with PAH were older, were more often female, had higher BMI and CCI, and had more significant smoking histories and antibiotic use. Compared with family control subjects, patients with PAH had higher CCI, but there was no significant difference in other demographics. PAH etiologies included the following: 27% idiopathic PAH (IPAH), 48% connective tissue disease–associated PAH (CTD-PAH), 8% congenital heart disease, 7% portopulmonary hypertension, 6% drug-induced PAH, 3% HIV infection, and one patient with pulmonary venoocclusive disease. The majority were prevalent patients on PAH-specific therapies. Patients had moderate PAH, with a mPAP of 38 ± 10 mm Hg, a PVR of 5.3 ± 2.8 Wood units, a mean pulmonary arterial compliance of 2.5 ± 1.2 ml/mm Hg, and a mean cardiac output of 5.5 ± 1.5 L/min. RV function was reduced, with a mean RV fractional area change of 32 ± 12%. The mean REVEAL (Registry to Evaluate Early and Long-Term PAH Disease Management) score was 6.6 ± 3.6, with 53%, 29%, and 18% of patients in the low-, intermediate-, and high-risk categories, respectively.
Table 1.
Baseline Demographics and Clinical Characteristics of the Study Participants
| Characteristic | Healthy Control Subjects (n = 39) | Family Control Subjects (n = 15) | Patients with PAH (n = 73) | P Value* |
|---|---|---|---|---|
| Age, y | 33 ± 9†‡ | 67 ± 13 | 61 ± 14 | <0.0001 |
| Female, n (%) | 20 (51) | 4 (27) | 59 (81) | <0.0001 |
| Body mass index, kg/m2 | 23 ± 3†‡ | 29 ± 5 | 29 ± 8 | <0.0001 |
| Smoking status, n (%) | 0.003 | |||
| Current | 0 (0) | 1 (7) | 1 (1) | — |
| Past smoker | 5 (13) | 3 (20) | 30 (41) | — |
| Never-smoker | 34 (87) | 11 (73) | 42 (58) | — |
| Diet, n (%) | 0.255 | |||
| Omnivore | 34 (87) | 15 (100) | 70 (96) | — |
| Pescatarian | 0 (0) | 0 (0) | 1 (2.6) | — |
| Vegetarian | 0 (0) | 0 (0) | 2 (5.1) | — |
| Vegan | 0 (0) | 0 (0) | 1 (2.6) | — |
| Other | 3 (4.1) | 0 (0) | 1 (2.6) | — |
| Frequent infections§, n (%) | 3 (8) | 2 (13) | 6 (8) | 0.805 |
| Antibiotic use in past 6 mo, n (%) | 2 (5) | 2 (13) | 30 (41) | 0.0001 |
| History of food allergy, n (%) | 5 (15) | 1 (7) | 14 (19) | 0.595 |
| Charlson comorbidity index, median (IQR) | 0 (0 to 0)†‡ | 0 (0 to 1)ǁ | 2 (1 to 3) | 0.0001 |
| Etiology of PAH, n (%) | ||||
| Idiopathic | — | — | 20 (27) | — |
| Connective tissue disease | — | — | 35 (48) | — |
| Congenital heart disease | — | — | 6 (8) | — |
| Portopulmonary hypertension | — | — | 5 (7) | — |
| Drug induced | — | — | 4 (6) | — |
| HIV | — | — | 2 (3) | — |
| Pulmonary venoocclusive disease | — | — | 1 (1) | — |
| Epidemiology, n (%) | ||||
| Prevalent | — | — | 71 (97) | — |
| Incident | — | — | 2 (3) | — |
| NYHA functional class, n (%) | ||||
| I | — | — | 6 (8) | — |
| II | — | — | 36 (49) | — |
| III | — | — | 29 (40) | — |
| IV | — | — | 2 (3) | — |
| 6-min-walk distance, m (n = 67) | — | — | 353 ± 121 | — |
| NT-pro-BNP, pg/dl, median (IQR) (n = 73) | — | — | 321 (115 to 747) | — |
| Right ventricular function by echocardiography | ||||
| Right ventricular fractional area change, % (n = 68) | — | — | 32 ± 12 | — |
| Right ventricular end diastolic area, mm2 (n = 68) | — | — | 27.5 ± 10.2 | — |
| Right ventricular end systolic area, mm2 (n = 68) | — | — | 19.1 ± 9.3 | — |
| Right ventricular global longitudinal strain, % median (IQR) (n = 62) | — | — | −21.5 (−23.6 to −18.5) | — |
| Right ventricular free wall strain, % median (IQR) (n = 62) | — | — | −25.9 (−28.2 to −21.2) | — |
| Hemodynamics | ||||
| Mean right atrial pressure, mm Hg (n = 73) | — | — | 6.0 ± 4 | — |
| Pulmonary arterial systolic pressure, mm Hg (n = 73) | — | — | 61 ± 18 | — |
| Pulmonary arterial diastolic pressure, mm Hg (n = 73) | — | — | 25 ± 7 | — |
| Mean pulmonary arterial pressure, mm Hg (n = 73) | — | — | 38 ± 10 | — |
| Pulmonary arterial wedge pressure, mm Hg (n = 73) | — | — | 11 ± 5 | — |
| Cardiac output, L/min (n = 72) | — | — | 5.1 ± 1.5 | — |
| Pulmonary vascular resistance, Wood units (n = 72) | — | — | 5.3 ± 2.8 | — |
| Pulmonary arterial compliance, ml/mm Hg (n = 72) | — | — | 2.5 ± 1.2 | — |
| REVEAL 2.0 score | — | — | 6.6 ± 3.6 | — |
| REVEAL 2.0 score category, n (%) | ||||
| High | — | — | 21 (29) | — |
| Intermediate | — | — | 13 (18) | — |
| Low | — | — | 39 (53) | — |
Definition of abbreviations: IQR = interquartile range; NT-proBNP = N-terminal pro–brain natriuretic peptide; NYHA = New York Heart Association; PAH = pulmonary arterial hypertension; REVEAL = Registry to Evaluate Early and Long-Term PAH Disease Management.
All continuous variables are presented as mean ± SD unless otherwise noted.
P value from the ANOVA/Kruskal-Wallis test comparing all three groups for continuous variables and chi-square test or Fisher exact test comparing all three groups for categorical variables.
P < 0.05 for healthy control subjects versus patients with PAH.
P < 0.05 for healthy control subjects versus family control subjects.
Sinusitis, pharyngitis, urinary tract infection, and others, requiring three or more courses of antibiotics per year. Antibiotic use and allergy status were reported by patients and did not require a confirmed clinical diagnosis of food allergy.
P < 0.05 for family control subjects versus patients with PAH.
Gut Microbiome Diversity Is Reduced and Distinct in Patients with PAH
Compared with healthy control subjects, patients with PAH had significantly reduced bacterial diversity, as measured by the Shannon diversity index (Figure 1A; P < 0.0001), but no difference in community richness, reflected by the Chao1 index (Figure 1B). On the basis of analysis of similarity using the Bray-Curtis dissimilarity index, the gut microbiome in patients with PAH was distinct from that in healthy control subjects (Figure 1C; P < 0.001; R = 0.246). Compared with family control subjects, patients with PAH also had significantly lower Shannon and Chao1 diversity (Figures 1A and 1B; P = 0.009 and P < 0.001, respectively). However microbial communities from patients with PAH overlapped with those from family control subjects (Figure 1C; P = 0.214; R = 0.063).
Figure 1.
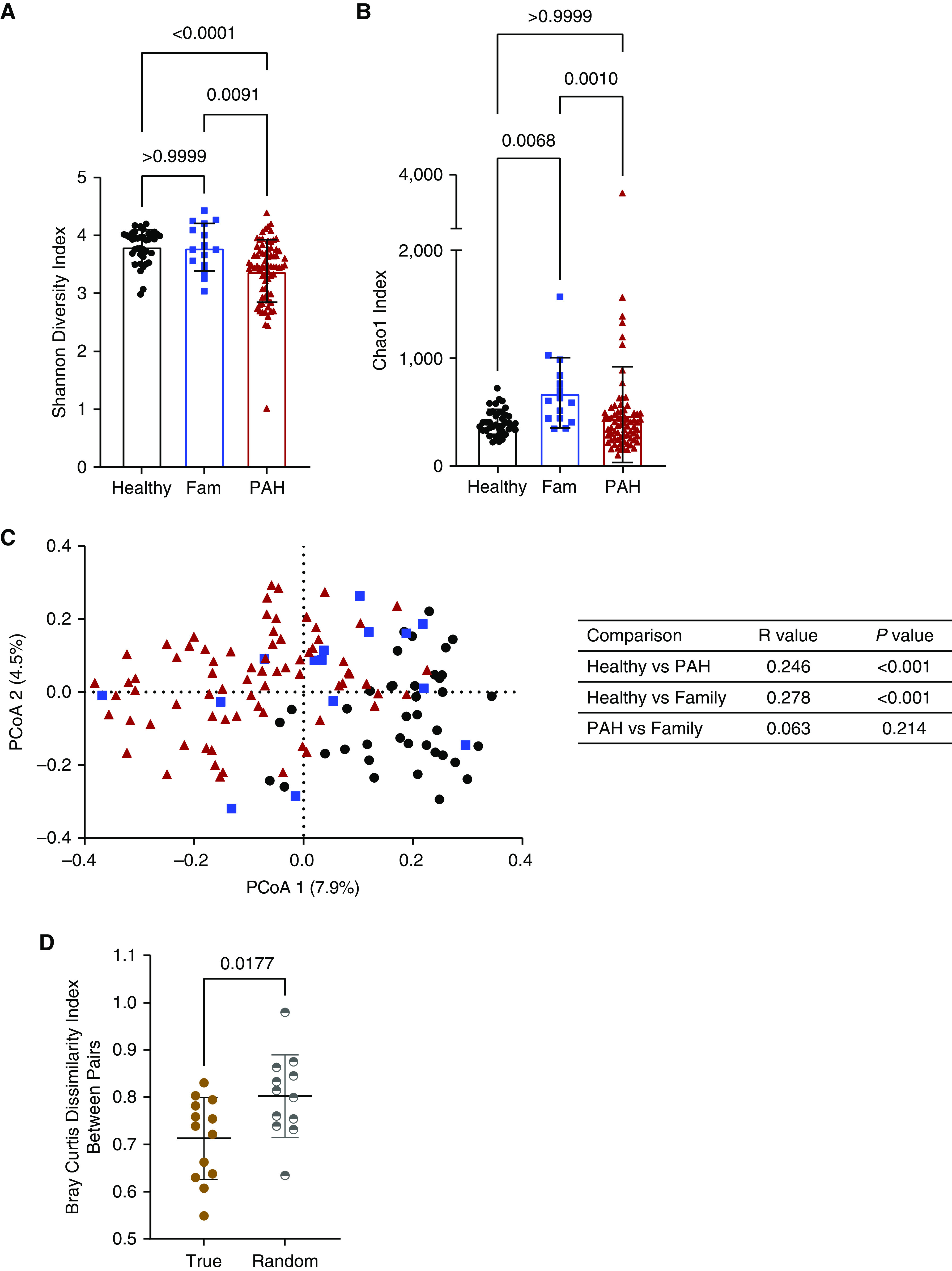
Gut microbiome diversity is reduced and distinct in patients with pulmonary arterial hypertension (PAH) compared with control subjects. (A and B) Shannon diversity (A) and Chao1 (B) indices in healthy and family (Fam) control subjects and patients with PAH. (C) Bray-Curtis pairwise dissimilarity indices are shown in a PCoA plot for healthy control subjects, Fam control subjects, and patients with PAH. Analysis was performed of similarity statistics of Bray-Curtis pairwise dissimilarity indices, comparing all groups (table shows R and P values). (D) Bray-Curtis dissimilarity between PAH–Fam pairs that cohoused (true) was significantly less than the difference between randomly generated PAH–Fam pairs (random). Circles denote healthy control subjects, squares denote Fam control subjects, and triangles denote patients with PAH. PCoA = principal coordinate analysis.
One possible explanation for the greater compositional similarity between gut microbiomes of patients with PAH and family control subjects was sharing of gut microbes in their local environment (14). To test this hypothesis, we calculated the difference in Bray-Curtis dissimilarity between PAH–family pairs that cohoused (true) and randomly generated PAH–family pairs (random). The difference between true pairs was significantly less than the difference between random pairs (Figure 1D; P = 0.017), meaning that the similarity between PAH–family pairs is likely due to cohabitation.
We obtained similar results when we compared the gut microbiomes in patients with CTD-PAH and those with IPAH relative to healthy and family control subjects (see Figures E1A–E1C). Importantly, there were no differences in Shannon diversity, Chao1, and Bray-Curtis dissimilarity indices between patients with IPAH and those with CTD-PAH (see Figures E1A–E1C). These results suggest that patients with PAH have reduced gut microbial diversity and composition compared with healthy control subjects, with some distinct differences from family control subjects, especially those who are not cohabitating.
Patients with PAH Have a Distinct Gut Microbiome Signature at the Phylogenetic Level
LEfSe analysis identified multiple taxonomic differences between patients with PAH and healthy control subjects. Patients with PAH had greater relative abundances of members of the phyla Bacteroidetes and Proteobacteria (see Figures E2A and E3A) and lower relative abundances of members of the phyla Firmicutes and Actinobacteria (see Figures E2A and E3A). The same pattern of greater relative abundance of Bacteroidetes was seen in the gut microbiomes of patients with PAH compared with family members (see Figures E2B and E3B).
To evaluate whether the enrichment of Bacteroidetes in patients with PAH is driven by an uneven expansion of its members, we performed a limited oligotype analysis on the basis of SNPs (15). Despite an increased relative abundance of the family Bacteroidaceae and genus Bacteroides in patients with PAH (see Figures E3A and E3B), the number of distinct oligotypes within these taxa was significantly reduced (see Figures E4A and E4B; P < 0.01 and P < 0.001). The number of distinct oligotypes was also significantly reduced among the Firmicutes families Ruminococcaceae and Lachnospiraceae (see Figures E4C and E4D; P < 0.001 and P < 0.001), mirroring the reduced relative abundances of these families in patients with PAH (see Figure E3A).
LEfSe analysis of shotgun metagenomic data identified multiple taxonomic differences at the species level among patients with PAH and healthy and family control subjects. The gut microbiomes of patients with PAH had greater relative abundances of several species that are generally proinflammatory (Bacteroides thetaiotaomicron, Parabacteroides distasonis, and B. vulgatus) (16, 17) and lower relative abundances of species that are considered antiinflammatory (Faecalibacterium prausnitzii, Eubacterium rectale, Ruminococcus bicirculans, Roseburia sp., and Bifidobacterium adolescentis) (18–22) compared with healthy control subjects (Figure 2A). Similarly, compared with family control subjects, the gut microbiome of patients with PAH had lower relative abundances of several antiinflammatory species (E. rectale, Butyrivibrio sp., B. angulatum, Lachnospira pectinoschiza, and R. torques) (18, 19, 21) (Figure 2B).
Figure 2.

Patients with pulmonary arterial hypertension (PAH) have a distinct gut microbiome signature at the phylogenetic level compared with healthy and family control subjects. (A and B) Linear discriminant analysis of effect size of shotgun metagenomic data between (A) healthy control subjects and patients with PAH and (B) family control subjects and patients with PAH. Gray bars denote healthy control subjects, blue bars denote family control subjects, and red bars denote patients with PAH. CAG = coabundance gene.
Multivariate Analysis for Microbiota Signatures Identified in PAH
To evaluate the independent strength of the associations between PAH patients and the microbiome signatures of control subjects identified above, we performed multivariate linear regression analysis. Patients with PAH had a lower Shannon diversity index compared with healthy and family control subjects, independent of age, sex, BMI, smoking history, antibiotic use, and CCI (Table 2). However, differences between groups for the Chao1 index were no longer significant after these adjustments (Table 2).
Table 2.
Multivariable Analysis of Gut Microbial Diversity Indices in Patients with Pulmonary Arterial Hypertension
| Characteristic | β Coefficient (95% CI) | P Value |
|---|---|---|
| Shannon diversity index | ||
| Model 1 (PAH: referent group) | ||
| Family control subjects | 0.33 (0.05 to 0.61) | 0.021 |
| Healthy control subjects | 0.51 (0.25 to 0.78) | <0.001 |
| Model 2 (PAH: referent group) | ||
| Family control subjects | 0.32 (0.04 to 0.60) | 0.025 |
| Healthy control subjects | 0.58 (0.31 to 0.86) | <0.001 |
| Model 3 (PAH: referent group) | ||
| Family control subjects | 0.32 (0.04 to 0.61) | 0.027 |
| Healthy control subjects | 0.59 (0.30 to 0.87) | <0.001 |
| Model 4 (PAH: referent group) | ||
| Family control subjects | 0.35 (0.06 to 0.64) | 0.020 |
| Healthy control subjects | 0.61 (0.31 to 0.90) | <0.001 |
| Model 5 (PAH: referent group) | ||
| Family control subjects | 0.36 (0.07 to 0.65) | 0.015 |
| Healthy control subjects | 0.39 (0.18 to 0.62) | <0.001 |
| Chao1 index | ||
| Model 1 (PAH: referent group) | ||
| Family control subjects | 179.3 (−42.4 to 401.0) | 0.112 |
| Healthy control subjects | −85.7 (−296.5 to 125.2) | 0.423 |
| Model 2 (PAH: referent group) | ||
| Family control subjects | 176.7 (−46.05 to 399.4) | 0.119 |
| Healthy control subjects | −71.1 (−290.9 to 148.8) | 0.524 |
| Model 3 (PAH: referent group) | ||
| Family control subjects | 189.9 (−37.9 to 417.9) | 0.102 |
| Healthy control subjects | −51.6 (−281.9 to 178.7) | 0.658 |
| Model 4 (PAH: referent group) | ||
| Family control subjects | 180.9 (−53.1 to 415.0) | 0.128 |
| Healthy control subjects | −72.9 (−307.6 to 161.7) | 0.539 |
| Model 5 (PAH: referent group) | ||
| Family control subjects | 184.8 (−41.5 to 411.2) | 0.109 |
| Healthy control subjects | −89.0 (−259.9 to 81.9) | 0.305 |
Definition of abbreviations: CI = confidence interval; PAH = pulmonary arterial hypertension.
Model 1 was adjusted for age and sex. Model 2 was adjusted for model 1 plus body mass index. Model 3 was adjusted for model 2 plus smoking status. Model 4 was adjusted for model 3 plus antibiotic use. Model 5 was adjusted for Charlson comorbidity index.
Likewise, the relative abundances of most taxa that were differentially abundant in patients with PAH compared with healthy control subjects remained significant after adjusting for these covariates (Table 3). The differences in relative abundances of taxa that were differentially abundant between patients with PAH and family control subjects were no longer significant after adjusting for these covariates except for Butyrivibrio sp. coabundance gene 318 and B. angulatum (Table 4).
Table 3.
Comparison of the Relative Abundance of Bacterial Species Differentially Abundant in Patients with Pulmonary Arterial Hypertension Compared with Healthy Control Subjects on Linear Discriminant Analysis of Effect Size
| Model 1* |
Model 2† |
|||||
|---|---|---|---|---|---|---|
| Species Name | PAH Relative Abundance (SD) |
Healthy Relative Abundance (SD) |
β Coefficient (95% CI) | P Value | β Coefficient (95% CI) | P Value |
| Bacterial species enriched in patients with PAH vs. healthy control subjects | ||||||
| B. vulgatus | 10.8 (11.9) | 2.9 (3.7) | 7.8 (1.3 to 14.4) | 0.019 | 9.8 (1.3 to 14.1) | <0.001 |
| P. distasonis | 5.3 (6.9) | 0.9 (1.1) | 3.9 (0.3 to 7.5) | 0.035 | 4.4 (1.7 to 7.2) | 0.002 |
| B. thetaiotaomicron | 4.6 (8.1) | 0.6 (0.4) | 2.7 (−1.4 to 6.8) | 0.199 | 3.3 (0.2 to 6.5) | 0.037 |
| E. coli | 2.3 (6.1) | 0.1 (0.5) | 0.1 (−2.9 to 3.3) | 0.923 | 1.3 (−1.1 to 3.7) | 0.299 |
| K. pneumoniae | 0.7 (2.8) | 0 (0) | 1.3 (−0.6 to 3.1) | 0.178 | 0.8 (−0.6 to 2.1) | 0.274 |
| R. gnavus | 0.6 (1.4) | 0.4 (1.7) | −0.1 (−1.0 to 0.8) | 0.806 | −0.1 (−0.7 to 0.6) | 0.848 |
| P. goldsteinii | 0.3 (1.3) | 0 (0) | 0.4 (−0.2 to 1.1) | 0.203 | 0.03 (−0.5 to 0.5) | 0.888 |
| Bacterial species with reduced abundance in patients with PAH vs. healthy control subjects | ||||||
| F. prausnitzii | 1.8 (2.3) | 7.3 (5.6) | −3.8 (−6.6 to −1.5) | 0.002 | −4.9 (−6.7 to −3.1) | <0.001 |
| E. rectale | 0.6 (1.2) | 6.6 (8.7) | −6.2 (−9.4 to −3.0) | <0.001 | −5.9 (−8.4 to −3.4) | <0.001 |
| R. bicirculans | 0.1 (0.5) | 3.4 (7.7) | −3.1 (−5.8 to −0.4) | 0.023 | −3.2 (−5.2 to −1.1) | 0.002 |
| R. faecis | 0.5 (1.4) | 3.2 (4.9) | −2.4 (−4.4 to −0.5) | 0.015 | −2.6 (−4.0 to −1.1) | 0.001 |
| Prevotella sp. CAG 873 | 0 (0) | 1.7 (9.8) | −4.8 (−8.0 to −1.6) | 0.004 | −1.7 (−4.3 to 0.8) | 0.190 |
| R. inulinivorans | 0.3 (0.6) | 0.7 (0.8) | −0.4 (−0.9 to −0.1) | 0.067 | −0.4 (−0.7 to −0.1) | 0.034 |
| C. comes | 0.1 (0.1) | 0.6 (1.1) | −0.5 (−0.9 to −0.1) | 0.014 | −0.6 (−0.9 to −0.3) | <0.001 |
| B. coprocola | 0 (0) | 0.3 (1.2) | −0.3 (−0.7 to 0.2) | 0.235 | −0.3 (−0.6 to 0.0) | 0.050 |
| B. obeum | 0.2 (0.2) | 0.6 (0.9) | −0.5 (−0.8 to −0.1) | 0.005 | −0.5 (−0.7 to −0.2) | <0.001 |
| Clostridium sp. CAG 167 | 0.1 (0.3) | 0.4 (0.8) | −0.2 (−0.5 to 0.1) | 0.275 | −0.3 (−0.6 to −0.1) | 0.009 |
| Firmicutes sp. CAG 110 | 0.0 (0.1) | 0.3 (1.3) | −0.4 (−0.9 to 0.1) | 0.114 | −0.3 (−0.7 to 0.1) | 0.073 |
| D. succinatiphilus | 0 (0) | 0.1 (0.5) | −0.1 (−0.3 to 0.1) | 0.379 | −0.1 (−0.2 to 0.1) | 0.177 |
| D. formicigenerans | 0.1 (0.1) | 0.4 (0.4) | −0.3 (−0.5 to −0.1) | <0.001 | −0.3 (−0.4 to −0.2) | <0.001 |
| Oscillibacter sp. 57 20 | 0.2 (0.6) | 0.4 (0.5) | −0.3 (−0.7 to 0.1) | 0.136 | −0.1 (−0.4 to 0.1) | 0.316 |
| R. lactaris | 0.1 (0.3) | 0.4 (0.9) | −0.1 (−0.5 to 0.2) | 0.412 | −0.3 (−0.5 to 0.0) | 0.060 |
Definition of abbreviations: B. coprocola = Bacteroides coprocola; B. obeum = Blautia obeum; B. thetaiotaomicron = Bacteroides thetaiotaomicron; B. vulgatus = Bacteroides vulgatus; CAG = coabundance gene; C. comes = Coprococcus comes; CI = confidence interval; D. formicigenerans = Dorea formicigenerans; D. succinatiphilus = Dialister succinatiphilus; E. coli = Escherichia coli; E. rectale = Eubacterium rectale; F. prausnitzii = Faecalibacterium prausnitzii; K. pneumoniae = Klebsiella pneumoniae; PAH = pulmonary arterial hypertension; P. distasonis = Parabacteroides distasonis; P. goldsteinii = Parabactereroides goldsteinii; R. bicirculans = Ruminococcus bicirculans; R. faeci = Roseburia faecis; R. gnavus = Ruminococcus gnavus; R. inulinivorans = Roseburia inulinivorans; R. lactaris = Ruminococcus lactaris.
Adjusted for age, sex, body mass index, smoking status, and recent antibiotic use.
Adjusted for the Charlson comorbidity index.
Table 4.
Comparison of the Relative Abundance of Bacterial Species Differentially Abundant in Patients with Pulmonary Arterial Hypertension Compared with Family Control Subjects on Linear Discriminant Analysis of Effect Size
| Model 1* |
Model 2† |
|||||
|---|---|---|---|---|---|---|
| Species Name | PAH Relative Abundance (SD) |
Family Relative Abundance (SD) |
β Coefficient (95% CI) | P Value | β Coefficient (95% CI) | P Value |
| Bacterial species with reduced relative abundance in patients with PAH vs. family control subjects | ||||||
| B. intestinihominis | 1.3 (2.9) | 1.7 (2.2) | −0.6 (−2.2 to 0.9) | 0.415 | −0.8 (−2.3 to 0.8) | 0.325 |
| E. rectale | 0.6 (1.2) | 6.6 (8.7) | −2.7 (−5.8 to −0.4) | 0.084 | −0.9 (−3.9 to 2.0) | 0.539 |
| E. hallii | 0.2 (0.7) | 0.2 (0.2) | 0.1 (−0.3 to 0.5) | 0.614 | 0.1 (−0.4 to 0.5) | 0.810 |
| Butyrivibrio sp. CAG 318 | 0 (0) | 0.2 (0.8) | −0.3 (−0.5 to −0.1) | 0.004 | 1.6 × 10−16 (−0.1 to 0.1) | 1.000 |
| Prevotella sp. CAG 5226 | 0 (0) | 0.2 (0.6) | −0.2 (−0.7 to 0.3) | 0.516 | 1.7 × 10−16 (−0.5 to 0.5) | 1.000 |
| B. angulatum | 0 (0) | 0.1 (0.4) | −0.1 (−0.2 to −0.1) | 0.004 | 7.2 × 10−16 (−0.1 to 0.1) | 1.000 |
| Oscillibacter sp. 57 20 | 0.2 (0.6) | 0.4 (0.5) | −0.4 (−0.7 to 0.1) | 0.057 | −0.2 (−0.5 to 0.2) | 0.294 |
| L. pectinoschiza | 0.2 (0.5) | 0.2 (0.4) | −0.1 (−0.6 to 0.4) | 0.743 | −0.1 (−0.6 to 0.4) | 0.817 |
| R. torques | 0.3 (0.5) | 0.5 (0.9) | −0.2 (−0.6 to 0.2) | 0.356 | −0.1 (−0.5 to 0.3) | 0.737 |
| Bacterial species with increased relative abundance in patients with PAH vs. family control subjects | ||||||
| S. salivarius | 0.3 (0.6) | 0.2 (0.5) | 0.1 (−0.2 to 0.4) | 0.584 | 0.1 (−0.3 to 0.4) | 0.817 |
Definition of abbreviations: B. angulatum = Bifidobacterium angulatum; B. intestinihominis = Barnesiella intestinihominis; CAG = coabundance gene; CI = confidence interval; E. hallii = Eubacterium hallii; E. rectale = Eubacterium rectale; L. pectinoschiza = Lachnospira pectinoschiza; R. torques = Ruminococcus torques; S. salivarius = Streptococcus salivarius.
Adjusted for age, sex, body mass index, smoking status, recent antibiotic use, and type of diet.
Adjusted for the Charlson comorbidity index.
Gut Dysbiosis Is Associated with the Severity of Pulmonary Vascular Disease but Not with RV Function
Next, to determine whether there is an association between the loss of microbial diversity in the gut and severity of PAH, we performed linear regression analysis of hemodynamic measures of pulmonary vascular disease with the Shannon diversity index. The Shannon diversity index had a significantly negative linear correlation with mPAP (Figure 3A; P = 0.015) and PVR (Figure 3B; P < 0.0001) and a significantly positive linear correlation with pulmonary arterial compliance (Figure 3C; P = 0.035). These results indicate that reduced gut microbial diversity is associated with increasing severity of pulmonary vascular disease. Linear regression analysis revealed no correlation between Shannon diversity index and echocardiographic (RV fractional area change) and hemodynamic (mean right atrial pressure and cardiac output) measures of RV function (Figures 3D–3F). There was also no relationship between Shannon diversity index and serum N-terminal pro–brain natriuretic peptide, RV global longitudinal strain, or RV free wall strain (see Figures E5A–E5C). On sparse partial least squares discriminant analysis, the genus-level data closely clustered together within each REVEAL risk group (see Figure E6), indicating differences in the gut microbial signature at the genus level among patients with PAH on the basis of disease severity.
Figure 3.

Shannon diversity index correlates with the severity of pulmonary vascular disease but not with right ventricular function in patients with pulmonary arterial hypertension. (A–C) There are significant negative linear correlations between Shannon diversity index and (A) mPAP, (B) PVR, and (C) PAC. (D–F) There are no significant linear correlations between Shannon diversity index and (D) RVFAC, (E) CO, and (F) RA pressure. CO = cardiac output; mPAP = mean pulmonary arterial pressure; PAC = pulmonary arterial compliance; PVR = pulmonary vascular resistance; R2 = coefficient of determination; RA = right atrial; RVFAC = right ventricular fractional area change; WU = Wood units.
Patients with PAH Have Increased Circulating Markers of Gut Permeability and Inflammation
To assess whether patients with PAH have increased gut permeability and systemic inflammation, we measured plasma claudin-3 and IL-6 concentrations, respectively. Claudins are transmembrane proteins that participate in the formation of tight junctions, and increased circulating concentrations of claudin-3 indicate increased gut permeability (23). Patients with PAH had higher plasma claudin-3 concentrations compared with family control subjects (see Figure E7A; P < 0.002), but there was no significant difference compared with healthy control subjects (see Figure E7A). Patients with PAH had significantly higher plasma IL-6 concentrations compared with healthy control subjects (see Figure E7B; P < 0.0002) but not compared with family control subjects. Plasma soluble CD14 (cluster of differentiation 14) (sCD14) and endotoxin concentrations did not differ significantly between patients with PAH and healthy control subjects (see Figures E7C and E7D, respectively).
Patients with PAH Have Altered Circulating Gut Microbial Metabolites
To determine whether patients with PAH have altered circulating concentrations of microbial metabolites known to affect inflammation, we next measured plasma concentrations of trimethylamine N-oxide (TMAO), SCFAs, and bile acids. Hierarchical cluster analysis showed a clear separation of patients with PAH from healthy and family control subjects, with reductions in circulating SCFAs and bile acids (Figure 4A). When we compared the individual microbial metabolite plasma concentrations among the study groups, patients with PAH had significantly lower concentrations of glycocholic (P = 0.0153), valeric (P = 0.0261), taurodeoxycholic (P = 0.0016), and glycolithocholic (P = 0.0089) acids compared with healthy control subjects and significantly lower concentrations of butyric (P < 0.0001) and lithocholic (P = 0.0026) acids compared with family control subjects (Figure 4A). There was no significant difference in plasma concentrations of the proinflammatory metabolite TMAO between patients with PAH and healthy control subjects (Figure 4B); however, when patients with PAH were stratified on the basis of REVEAL risk score, high-risk patients had significantly higher TMAO concentrations compared with healthy control subjects (P = 0.0017; see Figure E8).
Figure 4.

Targeted metabolomic analysis of patients with pulmonary arterial hypertension (PAH) and family (Fam) and healthy control subjects. (A) Hierarchical cluster analysis of targeted metabolomics of circulating concentrations of short-chain fatty acids and bile acids in healthy control subjects, Fam control subjects, and patients with PAH shows a clear separation of patients with PAH from healthy and Fam control subjects. The heatmap displays the group averages of the microbial metabolites for each group. The P values were generated by comparing the individual microbial metabolite plasma concentrations among the study groups using one-way ANOVA with a Tukey post hoc analysis to correct for multiple comparisons or a Kruskal-Wallis test with Dunn’s multiple-comparisons test. (B) There was no significant difference in plasma trimethylamine N-oxide (TMAO) concentrations between healthy control subjects and patients with PAH. Data are not available for TMAO concentrations in Fam controls. *P < 0.05, **P ⩽ 0.01, and ****P ⩽ 0.0001. ns = not significant (P ≥ 0.05).
Patients with PAH Have Fewer Copies of Some Gut Microbial Genes Encoding the Synthesis of Antiinflammatory Metabolites
Metagenomic analysis of the relative number of gene copies per million revealed significantly fewer copies of gut microbial genes involved in the synthesis of SCFAs (amidase for propionic and valeric acids [P < 0.001], propionate coenzyme A transferase for acetic acid [P < 0.0001], and phosphate butyryl transferase for butyric acid [P < 0.0005]) and deconjugation of bile acids (choloylglycine hydrolase; P = 0.0009) in patients with PAH compared with healthy control subjects (Figure 5). We did not see higher numbers of gene copies that encode production of trimethylamine in patients with PAH compared with healthy control subjects (Figure 5). No significant differences were seen in the relative number of gene copies for any of the metabolites for patients with PAH versus family control subjects (Figure 5). Hierarchical cluster analysis of microbial gene copies showed a clear separation of patients with PAH from healthy control subjects, with family control subjects in the middle (Figure 5).
Figure 5.
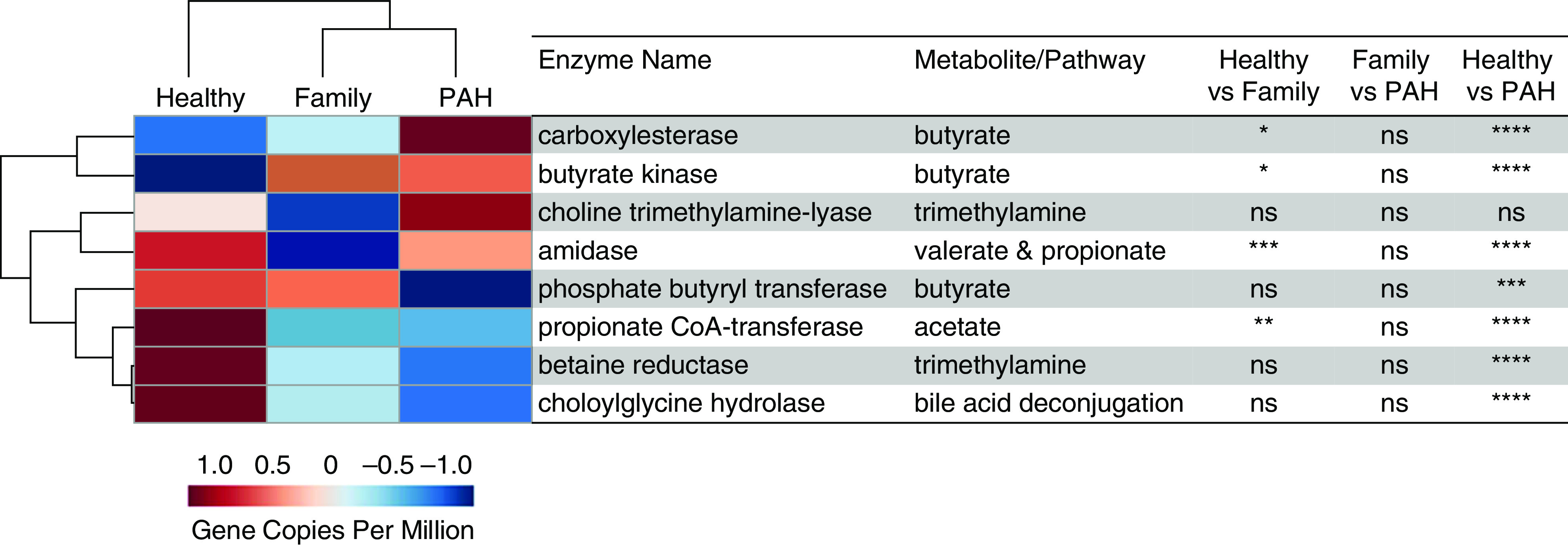
Hierarchical cluster analysis of encoding genes for enzymes that produce short-chain fatty acids, bile acid deconjugation, and trimethylamine from shotgun metagenomic data of the gut microbiomes showed clear separation of patients with PAH from healthy control subjects, with family control subjects in the middle. The heatmap displays the group averages of relative gene copies per million for each group. The P values were generated by comparing the relative number of gene copies per million among the study groups using one-way ANOVA with a Tukey post hoc analysis to correct for multiple comparisons or a Kruskal-Wallis test with Dunn’s multiple-comparisons test. *P < 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001. CoA = coenzyme A; ns = not significant (P ≥ 0.05); PAH = pulmonary arterial hypertension.
Relative Abundances of Species with Encoding Genes for Circulating Microbial Metabolites Significantly Differ among Groups
We examined the significant differences in the relative abundances of species with the encoding genes associated with microbial metabolites among all three experimental groups. Compared with healthy control subjects, patients with PAH had lower relative abundances of several taxa with encoding genes that produce SCFAs (24–28) (E. ramulus, Firmicutes sp. coabundance gene 110, Coprococcus comes, Dorea longicatena, B. adolescentis, Gemmiger formicilis, Fusicatenibacter saccharivorans, E. hallii, Anaerostipes hadrus, Gordonibacter pamelaeae, R. torques, C. catus, C. eutactus, and Blautia obeum; Figures 6 and 7). Compared with healthy control subjects, patients with PAH had a higher relative abundance of several taxa that harbor genes that encode trimethylamine production (29, 30) (Clostridium bolteae, Escherichia coli, and Klebsiella pneumoniae; Figures 8A and 8B) and a lower relative abundance of several taxa that possess the gene that encodes deconjugation of bile acids (31) (Collinsella aerofaciens, C. eutactus, A. hadrus, E. ramulus, B. obeum, E. hallii, R. bicirculans, R. torques, E. eligens, F. saccharivorans, R. faecis, D. longicatena, C. catus, and R. hominis; Figure 8C). Likewise, compared with family control subjects, patients with PAH had a higher relative abundance of C. bolteae (Figure 8A), which encodes TMAO (29), and a lower relative abundance of C. aerofaciens (Figure 8C), which possesses the gene that deconjugates bile acids (31).
Figure 6.

(A–C) Hierarchical cluster analysis of the relative abundance of species with the encoding genes for the enzyme involved in the synthesis of butyrate, including (A) carboxylesterase, (B) butyrate kinase, and (C) phosphate butyryl transferase. The heatmap displays the group averages of relative abundances of species for each group. The P values were generated by comparing the relative abundances of the species among the groups using one-way ANOVA with a Tukey post hoc analysis to correct for multiple comparisons or a Kruskal-Wallis test with Dunn’s multiple comparisons test. *P < 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001. CAG = coabundance gene; Fam = family; ns = not significant (P ≥ 0.05); PAH = pulmonary arterial hypertension.
Figure 7.

(A and B) Hierarchical cluster analysis of the relative abundance of species with the encoding genes for (A) amidase (an enzyme involved in the production of valerate and propionic acid) and (B) propionate CoA transferase (an enzyme involved in acetate production). The heatmap displays the group averages of relative abundances of species for each group. The P values were generated by comparing the relative abundances of the species among the groups using one-way ANOVA with a Tukey post hoc analysis to correct for multiple comparisons or a Kruskal-Wallis test with Dunn’s multiple-comparisons test. *P < 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001. CoA = coenzyme A; Fam = family; ns = not significant (P ≥ 0.05); PAH = pulmonary arterial hypertension.
Figure 8.

(A–C) Hierarchical cluster analysis of the relative abundance of species with the encoding genes for (A) betaine reductase and (B) choline trimethylamine-lyase (both enzymes involved in the synthesis of trimethylamine) and (C) choloylglycine hydrolase (an enzyme involved in bile acid conjugation). The heatmap provides the group averages of relative abundances of species for each group. The P values were generated by comparing the relative abundances of the species between the groups using one-way ANOVA with a Tukey post hoc analysis to correct for multiple comparisons or a Kruskal-Wallis test with Dunn’s multiple-comparisons test. *P < 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001. Fam = family; ns = not significant (P ≥ 0.05); PAH = pulmonary arterial hypertension.
Discussion
Kim and colleagues performed shotgun metagenomics sequencing on fecal samples from 18 patients with PAH and 13 healthy control subjects and showed a lower abundance of bacteria previously reported in the literature to be butyrate and acetate producers and an enrichment of bacteria previously shown in the literature to be TMAO producers (7). Our study adds several novel findings to that study by including a substantially larger cohort of well-characterized patients with PAH. We show that patients with PAH have a distinct gut microbiome, compared not only with healthy control subjects but also with family members, who are a better comparator group, as they are more likely to share some gut microbiota components (32). We included both 16S rRNA and shotgun metagenomics sequencing, which complement each other, given the extra sequencing coverage with 16S rRNA analysis and the direct evaluation of the functional potential of the gut microbiome with shotgun metagenomics. We demonstrate that reduced gut diversity correlates with the severity of pulmonary vascular disease, which further strengthens the association between PAH and gut dysbiosis. Using shotgun functional metagenomics, we show that patients with PAH have a proinflammatory gut microbiome with a lower relative abundance of taxa with gut microbial genes that produce antiinflammatory microbial metabolites and an enrichment of taxa that harbor microbial genes that produce proinflammatory microbial metabolites. Finally, consistent with the proinflammatory gut microbiome, we show that patients with PAH have altered circulating microbial-derived metabolites that favor inflammation. Together these observations provide potential mechanisms by which gut dysbiosis can contribute to the pathogenesis of PAH.
We show that patients with PAH have a greater relative abundance of the phylum Bacteroidetes and its members, including the family Bacteroidaceae and genus Bacteroides, that have been associated with gut inflammation and decreased gut barrier function in genetically susceptible hosts (16, 17). TMAO is a proinflammatory metabolite and causes pulmonary vascular smooth muscle proliferation and remodeling in preclinical models of PAH through macrophage activation (33). Huang and colleagues recently reported higher concentrations of TMAO in intermediate and high-risk PAH patients (33). Although we did not find a difference in plasma TMAO concentrations between all patients with PAH and healthy control subjects, we noted higher plasma concentrations of TMAO in high-risk patients with PAH. In addition, several species that contain genes for trimethylamine production have increased relative abundances in patients with PAH compared with healthy control subjects.
On hierarchical cluster analysis, patients with PAH are distinct from healthy and family control subjects, with relatively lower circulating concentrations of some SCFAs, which have antiinflammatory effects. There are several findings in the literature suggesting potential mechanistic roles for SCFAs in modulating PAH disease. Propionic acid increases regulatory T-cell populations (34), which are protective against the development of PAH (35). Valeric and butyric acids are potent histone deacetylase inhibitors (36, 37). In PAH, expression of histone deacetylase 6 is increased in pulmonary artery smooth muscle cells, and it increases survival and proliferation of these cells (38). In addition, butyrate treatment prevents as well as attenuates the hypoxia-induced increase in RV systolic pressure, RV hypertrophy, and pulmonary vascular remodeling in a Sprague-Dawley rat model of hypoxic pulmonary hypertension (39). Mechanistically, butyrate reduces lung inflammation and modulates cytokine concentrations in the hypoxic rat lung (39). Moreover, the butyrate derivative 4-phenyl butyric acid also prevents as well as reverses pulmonary vascular remodeling in a monocrotaline model of PAH through attenuation of endoplasmic reticulum stress in pulmonary vascular smooth cells (40).
Thus, it is possible that reduced SCFAs promote an environment favorable for pulmonary artery smooth muscle cell proliferation and fibrosis, thereby initiating or worsening PAH disease. Although there is no significant difference in circulating propionic acid concentrations, valeric and butyric acid concentrations are significantly reduced in patients with PAH. Supporting this finding, patients with PAH have fewer copies of the genes encoding the enzyme amidase that produces valerate and propionate (27), together with lower relative abundances of several species that harbor these genes. In addition, metagenomic analysis found that the gene encoding one (phosphate butyryl transferase) out of three enzymes for butyrate production (24–26) is reduced in patients with PAH. We found that the relative abundances for roughly half of the species that have these genes are reduced in patients with PAH. Although the antiinflammatory SCFA acetate (41) is not significantly lower in patients with PAH, the number of gene copies that encode for the enzyme propionate coenzyme A transferase, which produces acetate (28), is significantly reduced in patients with PAH, and all species that encoded this enzyme also have reduced relative abundances. The potential disconnect seen among the numbers of gene copies, relative abundances, and blood concentrations can be due to changes in the host environment and energy use of SCFAs by gut microbiota (42).
Bile acid homeostasis is maintained as a delicate balance between the host and intestinal microbiota metabolism, with the host producing primary bile acids and the microbiome using these to produce secondary bile acids. Secondary bile acids are antiinflammatory, especially lithocholic and taurolithocholic acids, which are potent agonists of TGR5 (Takeda G protein–coupled receptor 5), which can inhibit LPS-induced proinflammatory cytokine production and induce vasodilatory nitric oxide synthesis in endothelial cells (43–45). Likewise, glycodeoxycholic acid reduces vascular responsiveness to norepinephrine in the mesenteric vascular bed in a hypertensive rat model (46). Taurocholic and taurodeoxycholic acids also cause arterial dilation (47). On hierarchical cluster analysis, patients with PAH are distinct, with relatively lower plasma concentrations of bile acids. In addition, patients with PAH have fewer copies of the gene that encodes cholylglycine hydrolase that deconjugates bile acids (31) and lower relative abundances of several species with the gene encoding this enzyme. Multiple prior preclinical studies suggest a potential mechanistic role for bile acids in modulating PAH disease. The receptor for bile acids (farnesoid X receptor) is expressed in pulmonary arterial endothelial cells (45). Ligand activation of this receptor causes downregulation of endothelin-1 and upregulation of endothelial nitric oxide synthase in pulmonary artery endothelial cells (45, 48). In the monocrotaline rat model of PAH, obeticholic acid (a farnesoid X receptor agonist) improves pulmonary function, normalizes treadmill endurance, normalizes lung IL-6 mRNA expression, and reduces RV hypertrophy and pulmonary vascular remodeling (49). These data together suggest that the reduction in microbial-derived bile acids with antiinflammatory and vasodilatory activity can contribute to pathogenesis of PAH.
Although there is preclinical evidence linking SCFAs and secondary bile acids to the pathogenesis of PAH, this has not been studied in patients with PAH. We now show that the circulating concentrations of SCFAs and secondary bile acids are indeed decreased in patients with PAH compared with healthy and family control subjects. More important, with our functional metagenomics data, we link an altered gut microbiome as a potential mechanism for the lower circulating concentrations of SCFAs and secondary bile acids in patients with PAH. In support of this, we demonstrate that the gut microbiome of patients with PAH has fewer copies of microbial genes that produce SCFAs and secondary bile acids and lower relative abundances of species encoding these genes compared with healthy and family control subjects.
Although patients with PAH have a less diverse gut microbiome compared with both healthy and family control subjects, the differences at the phylogenetic level between patients with PAH and family control subjects are not as substantial as the differences between patients with PAH and healthy control subjects. This is likely due to cohabitation, which is associated with a greater resemblance of the gut microbiome (32). In support of this, we saw high gut microbial compositional similarity between patients and cohabitants compared with random family control subjects who are not cohabitating with the patients. Also, family control subjects are less healthy, as indicated by a significantly higher CCI compared with healthy control subjects. There are two possible explanations for the lack of a PAH phenotype in family control subjects despite sharing a large part of the gut microbiome with patients with PAH. Although there is similarity in the gut microbiome between patients with PAH and family control subjects, nevertheless, compared with family control subjects, patients with PAH remain distinct, with an increased relative abundance of the phylum Bacteroidetes and several members of this phylum, for which enrichment of these is associated with gut inflammation and a reduced relative abundance of several antiinflammatory taxa (16, 17). On targeted metabolomic analysis, patients with PAH have significantly lower circulating concentrations of antiinflammatory metabolites (butyrate and lithocholic acids) compared with family control subjects. These differences between patients with PAH and family control subjects may be critical to driving PAH pathogenesis. Alternatively, it is possible that gut dysbiosis acts as a second trigger by inducing inflammation in individuals with a genetic predisposition (i.e., BMPR2 [bone morphogenetic protein receptor type 2] mutant carriers). Consistent with this, chronic LPS administration causes pulmonary hypertension in Bmpr2+/− mice but not in wild-type littermates (50).
It is conceivable that chronic RV failure from PAH can lead to increased intestinal congestion, reduced bowel perfusion, increased intestinal permeability, and gut dysbiosis. However, there is no relationship between Shannon diversity index and various measures of RV function. Conversely, the Shannon diversity index correlates only with the measures of pulmonary vascular disease. These data indicate that gut dysbiosis in PAH is less likely to be due to RV failure. Although our data do not prove a direct cause-and-effect relationship between PAH and gut dysbiosis, preclinical data suggest a mechanistic role for gut dysbiosis in PAH. Gut microbiota modification with antibiotic treatment significantly suppresses pulmonary vascular remodeling and RV hypertrophy in the Sugen-hypoxia model in rats, suggesting a causative role for gut dysbiosis in PAH (5). Moreover, several lines of circumstantial evidence suggest a potential mechanistic link between PAH and gut dysbiosis. First, multiple risk factors for PAH, including HIV infection, Schistosoma spp. infection, and methamphetamine use, have been known to cause gut dysbiosis (51). Second, gut dysbiosis leads to increased gut permeability, allowing translocation of gut bacteria and/or bacterial products, such as endotoxin. Although we did not see differences in plasma claudin-3, endotoxin, and sCD14 concentrations between patients with PAH and healthy control subjects, Ranchoux and colleagues reported higher serum endotoxin and sCD14 concentrations in patients with IPAH and heritable PAH compared with healthy control subjects (52). Patients with PAH in our cohort, however, had higher gut permeability compared with family control subjects. Thus, on the basis of all these observations, it is possible that gut dysbiosis either initiates PAH or facilitates progression of already established PAH by calibrating immune responses.
Our findings have clinical implications. Both our work and the work by Kim and colleagues (7) suggest that patients with PAH have a distinct gut microbiome that favors systemic inflammation. Preclinical models suggest a cause-and-effect relationship between gut dysbiosis and PAH (5, 53). Moreover, alteration of gut microbiome with intermittent fasting improves RV function in the monocrotaline rat model of PAH (54). Thus, modulation of the gut microbiome may not only reduce pulmonary vascular remodeling but also improve RV function, possibly dependent on the timing of intervention. Collectively, these data support further investigations to test whether microbiota transplantation therapy can be used to treat PAH.
There are several limitations to our analyses. Healthy control subjects for microbiome analysis are not age and sex matched to patients with PAH; when we adjusted for these differences in the multivariate modeling, patients with PAH still had a significantly reduced Shannon diversity index, and the relative abundance of majority of the taxa that were differentially abundant in patients with PAH compared with healthy control subjects remained significant. We studied predominantly prevalent patients who were on PAH-specific therapies. It is possible that changes in the gut microbiome may be secondary to these therapies. Future studies should focus on evaluating the gut microbiome in patients with incident, treatment-naive PAH. We studied patients with different PAH etiologies. However, on subgroup analysis, we found no difference in gut microbiome diversity indices between patients with IPAH and those with CTD-PAH, the two most common etiologies of PAH. In addition, we performed only targeted metabolomics. Untargeted metabolomics and proteomics may provide a deeper understanding of the changes in all the circulating microbial products. Although this is the largest study of gut microbiome in PAH this far, it is still relatively small. Therefore, our study is not powered sufficiently to test the correlation among circulating microbial metabolites and disease severity. Finally, we studied fecal microbiota, but the gut microbiome exists within a complex ecological space, and it is possible that small bowel and mucosa-adherent microbiota may be more relevant to PAH pathogenesis.
Conclusions
In this study, we show that PAH is characterized by gut dysbiosis, with enrichment of taxa associated with inflammatory states and lower abundances of beneficial taxa, and that the severity of pulmonary vascular disease is associated with a less diverse gut microbiome. We also demonstrate that patients with PAH have altered circulating microbial metabolites with biological effects that could explain the pathophysiology of PAH. Despite recent advances in determining PAH pathogenesis, diagnosis, and treatment (55–58), our findings provide a compelling rationale to investigate strategies such as microbiota transplantation therapy to target the gut microbiome to treat PAH (59).
Acknowledgments
Acknowledgment
The authors thank Matthew Hamilton, Ph.D., for his help in review and critiques of the microbiome analysis.
Footnotes
Supported by a NIH grant T32 HL144472 (D.M.M.); NIH grants F32 HL154533 and T32 HL144472, a University of Minnesota Clinical Translational Sciences Award (NIH UL1 TR002494), and a University of Minnesota Medical School Academic Investment Educational Program Grant (S.Z.P); NIH grants K08 HL140100, R01 HL158795 and R01 HL162927 and American Lung Association Innovative Award IA-816386 (K.W.P.); Achieving Cures Together (a nonprofit) (A.K.); the Cardiovascular Medical Research and Education Fund, the University of Minnesota Futures Grant, and the Vikki Auzzene Philanthropy Grant (T.T.).
Author Contributions: Conceptualization: D.M.M., E.K.W., A.K., C.S., W.G., and T.T. Resources: T.T. and A.K. Methodology: D.M.M., J.T., S.Z.P., K.W.P., S.L., M.B., L.T., F.K., E.K.W., A.J.K., A.K., and T.T. Investigation: all authors. Formal analysis: D.M.M., S.Z.P., K.W.P., M.B., F.K., and T.T. Writing (original draft): D.M.M. Writing (review and editing): D.M.M., S.Z.P., K.W.P., M.B., C.S., W.G., A.K., E.K.W., and T.T.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202203-0490OC on November 7, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res . 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thenappan T, Khoruts A, Chen Y, Weir EK. Can intestinal microbiota and circulating microbial products contribute to pulmonary arterial hypertension? Am J Physiol Heart Circ Physiol . 2019;317:H1093–H1101. doi: 10.1152/ajpheart.00416.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med . 2016;375:2369–2379. doi: 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- 4. Callejo M, Mondejar-Parreño G, Barreira B, Izquierdo-Garcia JL, Morales-Cano D, Esquivel-Ruiz S, et al. Pulmonary arterial hypertension affects the rat gut microbiome. Sci Rep . 2018;8:9681. doi: 10.1038/s41598-018-27682-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sanada TJ, Hosomi K, Shoji H, Park J, Naito A, Ikubo Y, et al. Gut microbiota modification suppresses the development of pulmonary arterial hypertension in an SU5416/hypoxia rat model. Pulm Circ . 2020;10:2045894020929147. doi: 10.1177/2045894020929147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma RK, Oliveira AC, Yang T, Karas MM, Li J, Lobaton GO, et al. Gut pathology and its rescue by ACE2 (angiotensin-converting enzyme 2) in hypoxia-induced pulmonary hypertension. Hypertension . 2020;76:206–216. doi: 10.1161/HYPERTENSIONAHA.120.14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim S, Rigatto K, Gazzana MB, Knorst MM, Richards EM, Pepine CJ, et al. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension . 2020;75:1063–1071. doi: 10.1161/HYPERTENSIONAHA.119.14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moutsoglou D, Weir KK, Khoruts A, Tatah J, Prisco SZ, Prins KW. Gut dysbiosis and altered circulating microbial metabolites in patients with pulmonary arterial hypertension [abstract] Circulation . 2021;144:A10736. [Google Scholar]
- 9. Prins KW, Archer SL, Pritzker M, Rose L, Weir EK, Sharma A, et al. Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J Heart Lung Transplant . 2018;37:376–384. doi: 10.1016/j.healun.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamilton MJ, Weingarden AR, Sadowsky MJ, Khoruts A. Standardized frozen preparation for transplantation of fecal microbiota for recurrent Clostridium difficile infection. Am J Gastroenterol . 2012;107:761–767. doi: 10.1038/ajg.2011.482. [DOI] [PubMed] [Google Scholar]
- 11. Clarke KR. Non-parametric multivariate analyses of changes in community structure. Austral Ecol . 1993;18:117–143. [Google Scholar]
- 12. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol . 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chong J, Yamamoto M, Xia J. MetaboAnalystR 2.0: from raw spectra to biological insights. Metabolites . 2019;9:57. doi: 10.3390/metabo9030057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song SJ, Lauber C, Costello EK, Lozupone CA, Humphrey G, Berg-Lyons D, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife . 2013;2:e00458. doi: 10.7554/eLife.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eren AM, Maignien L, Sul WJ, Murphy LG, Grim SL, Morrison HG, et al. Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol Evol . 2013;4:1111–1119. doi: 10.1111/2041-210X.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe . 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity . 2014;41:311–324. doi: 10.1016/j.immuni.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature . 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 19. Islam SMS, Ryu HM, Sayeed HM, Byun HO, Jung JY, Kim HA, et al. Eubacterium rectale attenuates HSV-1 induced systemic inflammation in mice by inhibiting CD83. Front Immunol . 2021;12:712312. doi: 10.3389/fimmu.2021.712312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut . 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 21. Ouwehand AC, Bergsma N, Parhiala R, Lahtinen S, Gueimonde M, Finne-Soveri H, et al. Bifidobacterium microbiota and parameters of immune function in elderly subjects. FEMS Immunol Med Microbiol . 2008;53:18–25. doi: 10.1111/j.1574-695X.2008.00392.x. [DOI] [PubMed] [Google Scholar]
- 22. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A . 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sikora M, Chrabąszcz M, Waśkiel-Burnat A, Rakowska A, Olszewska M, Rudnicka L. Claudin-3—a new intestinal integrity marker in patients with psoriasis: association with disease severity. J Eur Acad Dermatol Venereol . 2019;33:1907–1912. doi: 10.1111/jdv.15700. [DOI] [PubMed] [Google Scholar]
- 24. Louis P, Duncan SH, McCrae SI, Millar J, Jackson MS, Flint HJ. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J Bacteriol . 2004;186:2099–2106. doi: 10.1128/JB.186.7.2099-2106.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nardi M, Fiez-Vandal C, Tailliez P, Monnet V. The EstA esterase is responsible for the main capacity of Lactococcus lactis to synthesize short chain fatty acid esters in vitro. J Appl Microbiol . 2002;93:994–1002. doi: 10.1046/j.1365-2672.2002.01793.x. [DOI] [PubMed] [Google Scholar]
- 26. Walter KA, Nair RV, Cary JW, Bennett GN, Papoutsakis ET. Sequence and arrangement of two genes of the butyrate-synthesis pathway of Clostridium acetobutylicum ATCC 824. Gene . 1993;134:107–111. doi: 10.1016/0378-1119(93)90182-3. [DOI] [PubMed] [Google Scholar]
- 27. Brammar WJ, Clarke PH. Induction and repression of Pseudomonas aeruginosa amidase. J Gen Microbiol . 1964;37:307–319. doi: 10.1099/00221287-37-3-307. [DOI] [PubMed] [Google Scholar]
- 28. Lindenkamp N, Schürmann M, Steinbüchel A. A propionate CoA-transferase of Ralstonia eutropha H16 with broad substrate specificity catalyzing the CoA thioester formation of various carboxylic acids. Appl Microbiol Biotechnol . 2013;97:7699–7709. doi: 10.1007/s00253-012-4624-9. [DOI] [PubMed] [Google Scholar]
- 29. Naumann E, Hippe H, Gottschalk G. Betaine: new oxidant in the Stickland reaction and methanogenesis from betaine and l-alanine by a Clostridium sporogenes–Methanosarcina barkeri coculture. Appl Environ Microbiol . 1983;45:474–483. doi: 10.1128/aem.45.2.474-483.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A . 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Batta AK, Salen G, Shefer S. Substrate specificity of cholylglycine hydrolase for the hydrolysis of bile acid conjugates. J Biol Chem . 1984;259:15035–15039. [PubMed] [Google Scholar]
- 32. Finnicum CT, Beck JJ, Dolan CV, Davis C, Willemsen G, Ehli EA, et al. Cohabitation is associated with a greater resemblance in gut microbiota which can impact cardiometabolic and inflammatory risk. BMC Microbiol . 2019;19:230. doi: 10.1186/s12866-019-1602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang Y, Lin F, Tang R, Bao C, Zhou Q, Ye K, et al. Gut microbial metabolite trimethylamine N-oxide aggravates pulmonary hypertension. Am J Respir Cell Mol Biol . 2022;66:452–460. doi: 10.1165/rcmb.2021-0414OC. [DOI] [PubMed] [Google Scholar]
- 34. Duscha A, Gisevius B, Hirschberg S, Yissachar N, Stangl GI, Eilers E, et al. Propionic acid shapes the multiple sclerosis disease course by an immunomodulatory mechanism. Cell . 2020;180:1067–1080.e16. doi: 10.1016/j.cell.2020.02.035. [DOI] [PubMed] [Google Scholar]
- 35. Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res . 2011;109:867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuille S, Reichardt N, Panda S, Dunbar H, Mulder IE. Human gut bacteria as potent class I histone deacetylase inhibitors in vitro through production of butyric acid and valeric acid. PLoS ONE . 2018;13:e0201073. doi: 10.1371/journal.pone.0201073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan X, Feng L, Huang X, Yang Y, Yang C, Gao Y. Histone deacetylase inhibitors promote eNOS expression in vascular smooth muscle cells and suppress hypoxia-induced cell growth. J Cell Mol Med . 2017;21:2022–2035. doi: 10.1111/jcmm.13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boucherat O, Chabot S, Paulin R, Trinh I, Bourgeois A, Potus F, et al. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci Rep . 2017;7:4546. doi: 10.1038/s41598-017-04874-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karoor V, Strassheim D, Sullivan T, Verin A, Umapathy NS, Dempsey EC, et al. The short-chain fatty acid butyrate attenuates pulmonary vascular remodeling and inflammation in hypoxia-induced pulmonary hypertension. Int J Mol Sci . 2021;22:9916. doi: 10.3390/ijms22189916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu Y, Adi D, Long M, Wang J, Liu F, Gai MT, et al. 4-Phenylbutyric acid induces protection against pulmonary arterial hypertension in rats. PLoS ONE . 2016;11:e0157538. doi: 10.1371/journal.pone.0157538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu M, Jiang Z, Wang C, Li N, Bo L, Zha Y, et al. Acetate attenuates inflammasome activation through GPR43-mediated Ca2+-dependent NLRP3 ubiquitination. Exp Mol Med . 2019;51:1–13. doi: 10.1038/s12276-019-0276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, Everard A. Microbial regulation of organismal energy homeostasis. Nat Metab . 2019;1:34–46. doi: 10.1038/s42255-018-0017-4. [DOI] [PubMed] [Google Scholar]
- 43. Wammers M, Schupp AK, Bode JG, Ehlting C, Wolf S, Deenen R, et al. Reprogramming of pro-inflammatory human macrophages to an anti-inflammatory phenotype by bile acids. Sci Rep . 2018;8:255. doi: 10.1038/s41598-017-18305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kida T, Tsubosaka Y, Hori M, Ozaki H, Murata T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb Vasc Biol . 2013;33:1663–1669. doi: 10.1161/ATVBAHA.113.301565. [DOI] [PubMed] [Google Scholar]
- 45. Li J, Wilson A, Kuruba R, Zhang Q, Gao X, He F, et al. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc Res . 2008;77:169–177. doi: 10.1093/cvr/cvm016. [DOI] [PubMed] [Google Scholar]
- 46. Tominaga T, Suzuki H, Ogata Y, Imafuku T, Saruta T. Bile acids are able to reduce blood pressure by attenuating the vascular reactivity in spontaneously hypertensive rats. Life Sci . 1988;42:1861–1868. doi: 10.1016/0024-3205(88)90025-2. [DOI] [PubMed] [Google Scholar]
- 47. Pak JM, Adeagbo AS, Triggle CR, Shaffer EA, Lee SS. Mechanism of bile salt vasoactivity: dependence on calcium channels in vascular smooth muscle. Br J Pharmacol . 1994;112:1209–1215. doi: 10.1111/j.1476-5381.1994.tb13212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. He F, Li J, Mu Y, Kuruba R, Ma Z, Wilson A, et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ Res . 2006;98:192–199. doi: 10.1161/01.RES.0000200400.55539.85. [DOI] [PubMed] [Google Scholar]
- 49. Comeglio P, Filippi S, Sarchielli E, Morelli A, Cellai I, Corno C, et al. Therapeutic effects of the selective farnesoid X receptor agonist obeticholic acid in a monocrotaline-induced pulmonary hypertension rat model. J Endocrinol Invest . 2019;42:951–965. doi: 10.1007/s40618-019-1009-2. [DOI] [PubMed] [Google Scholar]
- 50. Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production: a gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med . 2015;192:859–872. doi: 10.1164/rccm.201408-1509OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oliveira SD. Insights on the gut-mesentery-lung axis in pulmonary arterial hypertension: a poorly investigated crossroad. Arterioscler Thromb Vasc Biol . 2022;42:516–526. doi: 10.1161/ATVBAHA.121.316236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ranchoux B, Bigorgne A, Hautefort A, Girerd B, Sitbon O, Montani D, et al. Gut-lung connection in pulmonary arterial hypertension. Am J Respir Cell Mol Biol . 2017;56:402–405. doi: 10.1165/rcmb.2015-0404LE. [DOI] [PubMed] [Google Scholar]
- 53. Oliveira AC, Yang T, Li J, Sharma RK, Karas MK, Bryant AJ, et al. Fecal matter transplant from Ace2 overexpressing mice counteracts chronic hypoxia-induced pulmonary hypertension. Pulm Circ . 2022;12:e12015. doi: 10.1002/pul2.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prisco SZ, Eklund M, Moutsoglou DM, Prisco AR, Khoruts A, Weir EK, et al. Intermittent fasting enhances right ventricular function in preclinical pulmonary arterial hypertension. J Am Heart Assoc . 2021;10:e022722. doi: 10.1161/JAHA.121.022722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maron BA, Abman SH, Elliott CG, Frantz RP, Hopper RK, Horn EM, et al. Pulmonary arterial hypertension: diagnosis, treatment, and novel advances. Am J Respir Crit Care Med . 2021;203:1472–1487. doi: 10.1164/rccm.202012-4317SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boucly A, Savale L, Jaïs X, Bauer F, Bergot E, Bertoletti L, et al. Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med . 2021;204:842–854. doi: 10.1164/rccm.202009-3698OC. [DOI] [PubMed] [Google Scholar]
- 57. Hong J, Arneson D, Umar S, Ruffenach G, Cunningham CM, Ahn IS, et al. Single-cell study of two rat models of pulmonary arterial hypertension reveals connections to human pathobiology and drug repositioning. Am J Respir Crit Care Med . 2021;203:1006–1022. doi: 10.1164/rccm.202006-2169OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, et al. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am J Respir Crit Care Med . 2018;198:1423–1434. doi: 10.1164/rccm.201710-2079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moutsoglou DM. 2021 American Thoracic Society BEAR Cage winning proposal: microbiome transplant in pulmonary arterial hypertension. Am J Respir Crit Care Med . 2022;205:13–16. doi: 10.1164/rccm.202108-1833ED. [DOI] [PMC free article] [PubMed] [Google Scholar]