

ABSTRACT
RNA-binding proteins are essential regulators of RNA processing and function. Translational repression assays can be used to study how they interact with specific RNA sequences by insertion of such a consensus sequence into the 5’ untranslated region of a reporter mRNA and measuring reporter protein translation. The straightforward set-up of these translational repression assays avoids the need for the isolation of the protein or the RNA providing speed, robustness and a low-cost method. Here, we report the optimization of the assay to function with linear RNA sequences instead of the previously reported hairpin type sequences to allow the study of a wider variety of RNA-binding proteins. Multiplication of a consensus sequence strongly improves the signal allowing analysis by both fluorescence intensity measurements and flow cytometry.
KEYWORDS: Translation, RNA-binding proteins, Protein-RNA interactions, Binding studies, Translational repression assay
Introduction
RNA-binding proteins (RBPs) are essential in the processing and function of all RNA types and therefore play a prominent role in physiology and disease [1–3]. Studying how RBPs interact with specific RNA sequences is relevant to learn more about their function and selectivity and to identify novel therapeutic targets [4,5]. Experiments using purified proteins and RNA can lead to novel insights but can be affected by the lack of the components of the intracellular environment. Furthermore, obtaining both protein and RNA in good purity and sufficient quantities can be challenging and laborious. These challenges can be overcome by performing protein-RNA interaction assays in bacterial cells and various assays have been reported to do so including the bacterial three-hybrid assay, antitermination-based assays, and assays based on translational repression [6–10]. Although the bacterial three-hybrid assay has been shown to be effective at coupling a protein-RNA interaction to a read-out, it requires three components to be constructed and has been reported to only be effective for high-affinity interactions [7]. The antitermination-based assays are hard to adapt to new protein-RNA interactions due to conformational restrictions and also require very high affinities to produce a read-out [11,12]. The systems based on translational repression require only two designed components and have been demonstrated to work in bacterial, yeast, and mammalian cells [13–17]. In contrast to the other two systems, the RBP is conformationally free to interact with the target RNA rather than being part of a larger complex making the system more flexible and straightforward to design. Various read-outs have been tested in such repression assays including survival-based selectors, or read-outs based on colorimetric (i.e.: LacZ), luminescent (i.e.: luciferase), or fluorescent reporter proteins (i.e.: GFP). A major advantage of the translational repression assay is that the repression levels appear to correlate with binding affinity allowing the study of protein-RNA interactions without purification of the individual components [10,17].
The translational repression system relies on an mRNA encoding for a reporter gene which is modified with an RBP binding site (see Figure 1A) by inserting it in the 5’ UTR upstream of the Shine-Dalgarno (SD) sequence in prokaryotic systems or the Kozak sequence in eukaryotic systems [10,14,15,18]. Alternatively, insertion of the RBP recognized sequence after the start codon and in frame with the reporter protein has been reported in both prokaryotic and eukaryotic systems [16,17]. Binding of an RBP to the RBP binding site leads to a blockade of reporter protein translation which can be used as a measure for the protein-RNA interaction (see Figure 1B). By fusing the RBP to a fluorescent protein compatible with the reporter protein, the RBP expression levels can be monitored simultaneously. In previously described versions of the system, the RBP binding site has always been a stable hairpin RNA to facilitate a high affinity interaction (e.g. MS2 with the MS2 hairpin) [14]. However, in our studies, we are interested in alternative splicing factors that interact with unstructured linear RNA sequences. Until now, only the core splicing component U1A has been tested in this assay, which has a very high affinity for the U1 snRNA hairpin. We, therefore, sought to adapt the system, so it accepts such interactions and allow straightforward modification and interaction analysis.
Figure 1.
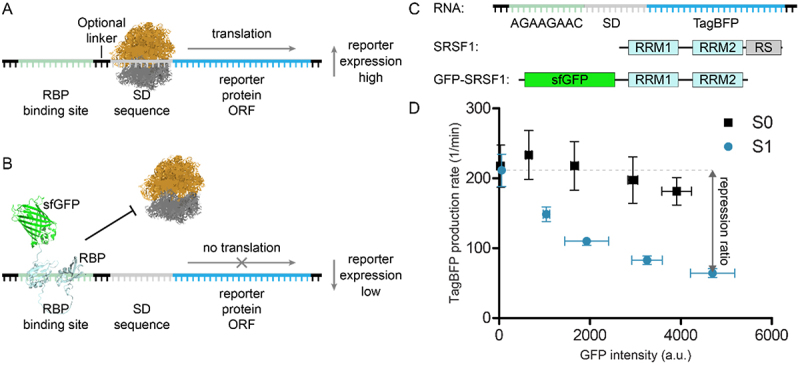
Translational repression assay. A) in the absence of the RBP, translation of the reporter protein can take place. B) in the presences of the RBP-sfGFP fusion construct, translation is blocked. C) Schematic representation of the modified TagBFP mRNA reporter used as starting point for assay development, schematic representation of the SRSF1 domain structure, and the used SRSF1 protein construct GFP-SRSF1. D) Repression of TagBFP observed upon increasing expression of GFP-SRSF1 using a reporter with consensus sequence for SRSF1 (S1) or without (S0). The repression ratio is calculated by dividing the highest with the lowest TagBFP production rates.
Here, we report a bacterial variation of the translational repression system that allows the study of the interaction between an RBP and a linear RNA sequence. The RNA sequence is inserted in front of the SD sequence of a blue fluorescent protein (TagBFP) reporter so that RBP binding interferes with TagBFP translation. We found that TagBFP repression can be increased by multiplication of the RNA sequence and varying the distance between the insert and the SD sequence. We demonstrate the use of the assay for two different splicing factors (SRSF1 and hnRNP A2/B1) and used both a plate reader-based read-out as well as flow cytometry. The assay is performed in E. coli to avoid a background signal arising from endogenously expressed proteins since the splicing factors of interest are not expressed in this organism. The correlation between binding affinity and translational repression was further studied using fluorescence polarization assays. Our findings demonstrate that the assay can report on binding affinity, but for a strong signal-to-noise ratio it is important to make sure no secondary structures are formed in the 5’-UTR of the reporter in close proximity to the SD sequence.
Materials and methods
Protein expression and purification
MBP-tagged hnRNP A2/B1 (aa1–251) was expressed and purified at the Protein Chemistry Facility (PCF) at the MPI Dortmund. HnRNP A2/B1 (1–251) was sub-cloned into the pOPIN-His-MBP multihost expression vector. The MBP fusion was chosen to increase expression yield and solubility [19]. MBP-hnRNP A2/B1 was expressed in E. coli BL21 CodonPlus (DE3) RIPL. Bacteria with the respective plasmid were cultured in Terrific Broth with 0.01% lactose, 2 mM MgSO4, 100 µg/ml ampicillin and 50 µg/ml chloramphenicol. Protein expression was auto-induced, with incubation of the starter-culture (starting OD of ~0.05) at 37°C for 4 h, followed by an overnight incubation (20-24 h) at 25°C. Bacteria were harvested by centrifugation and lysed (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, 1 mM TCEP, pH 8) using a Celldisrupter TS 0.75 (Constant Systems) at 1350 bar. Protein purification was performed on a HisTrap FF crude 5 ml Ni-based column using an ÄKTA Xpress System (Cytiva, former GE Healthcare). For that a wash buffer (50 mM HEPES, 300 mM NaCl, 30 mM imidazole, 1 mM TCEP at pH 8) and elution buffer (50 mM HEPES, 300 mM NaCl, 500 mM imidazole, 1 mM TCEP at pH 8) were used. The protein was then further purified using size exclusion chromatography (HiLoad 26/60 Superdex 75 prep grade column) in storage buffer (50 mM HEPES, 100 mM NaCl, 1 mM TCEP at pH 8.0) at 4°C. Fractions were collected and concentrated with a 50 kDa molecular mass cut-off Amicon spin filter.
SRSF1 RRM1 + 2 (aa1–195) was sub-cloned into the pOPIN-His multihost expression vector by SLIC, expressed in E. coli BL21 (DE3) and purified using a protocol adapted from Cléry et al. [19,20] Bacteria with the respective plasmid were cultured in LB medium with 100 µg/ml ampicillin. Protein expression was induced at OD600 = 0.6 with 1 mM isopropyl β-D-thiogalactoside (IPTG) overnight at 18°C. Cells were harvested by centrifugation and lysed (50 mM Na2HPO4, 300 mM KCl, 50 mM L-Arg, 50 mM L-Glu, 1.5 mM MgCl2, 1 mM PMSF at pH 8) using a microfluidizer. Protein purification was performed on a HisTrap HP 5 ml Ni-based column (Cytiva, former GE Healthcare) using an ÄKTA Explorer System (Cytiva, former GE Healthcare). The protein was dialysed overnight at 4°C into wash buffer (50 mM Na2HPO4, 300 mM KCl, 50 mM L-Arg, 50 mM L-Glu, 1.5 mM MgCl2, 40 mM Imidazole at pH 8), and a second Ni-based column purification was performed. The protein was again dialysed at 4°C into wash buffer and treated with His-tagged 3C protease. After overnight cleavage, the protein was loaded onto a Ni-based column for reversed purification. The protein was finally dialysed at 4°C in the storage buffer (20 mM NaH2PO4, 150 mM KCl, 50 mM L-Arg, 50 mM L-Glu,m 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM TCEP at pH 7) and concentrated with a 3 kDa molecular mass cut-off Amicon spin filter.
Translational repression assay procedure
A 1 mL preculture of E. coli Top10F‘ cotransformed with assay plasmids was prepared in LB with corresponding antibiotics (kanamycin 50 µg/mL, chloramphenicol 34 µg/mL) in a 96-deep well plate. The plate was sealed with a semipermeable seal and the cultures were grown overnight at 37°C in a shaking incubator (160 rpm). Next day the cultures were diluted 1:19 in M9 minimal medium with respective antibiotics in a total volume of 190 µL in a black, 96-well plate with clear bottom. Optical density at 600 nm (OD600) was monitored until a value of ~0.2 was reached. The assay was induced with IPTG to a final concentration of 1 mM and arabinose to a final concentration of 0, 0.125, 0.25, 0.5 or 1%. Fluorescence of TagBFP (402 nm/457 nm) and sfGFP (485 nm/510 nm) in addition to OD600 were measured every 20 minutes over a time course of ~7 h at 30°C with a TECAN Spark plate reader. Data evaluation was performed according to the report by Katz et al.[21]
Flow cytometry analysis
A 1 mL preculture of E. coli Top10F‘ cotransformed with assay plasmids was prepared in LB with corresponding antibiotics (kanamycin 50 µg/mL, chloramphenicol 34 µg/mL) in a 96-deep well plate. The plate was sealed with a semipermeable seal and the cultures were grown overnight at 37°C in a shaking incubator (160 rpm). Next day the cultures were diluted 1:19 in LB medium with respective antibiotics in a total volume of 1 mL in a 96-deep-well plate. OD600 was monitored until a value of ~0.2 was reached. The assay was induced with 1 mM IPTG and 0.125% arabinose for 4 h and 30 minutes at 30°C. The plate was centrifuged at 4500 g for 5 minutes in a tabletop centrifuge. Medium was removed and cells were washed twice with 500 µL PBS. Finally, cells were centrifuged and resuspended in 800 µL PBS and placed on ice until analysis.
Samples were analysed on a SH800SFP Cell Sorter (Sony Biotechnology, Weybridge, U. K.) using a 70 μm microfluidic chip (Sony Biotechnology), and TagBFP and sfGFP fluorescence intensities recorded and compensated using the respective single-colour controls (see supplemental figure S32). Each measurement was replicated twice.
Fluorescence Polarization Assay
The RNA oligonucleotides were purchased from Integrated DNA Technologies or SigmaAldrich. Oligonucleotides were dissolved in nuclease-free water at 100 µM according to the manufacturers’ instructions and kept at −20°C until use. The assay was performed in protein storage buffer with 0.01% Triton, in black, 384 well-plates (Corning), with a total volume of 20 μL per well. The analysed, 6-FAM labelled RNAs were tested at a final concentration of 1 nM, and the appropriate unlabelled protein was titrated as two-fold dilution series. The plates were analysed after 20 minutes incubation at room temperature using a plate reader (Tecan Spark) with (ex/em) 490/520 nm for 6-FAM. The assay was performed in three replicates for SRSF1 and two replicates for hnRNP A2/B1.
Reverse Transcription quantitative Polymerase Chain Reaction (RT-qPCR)
A 2-mL preculture of E. coli Top10F’ cotransformed with assay plasmids in LB medium with corresponding antibiotics (kanamycin 50 µg/mL, chloramphenicol 34 µg/mL) was prepared and grown overnight at 37°C in a shaking incubator (160 rpm). The next day the cultures were diluted 1:19 in M9 medium with respective antibiotics in a total volume of 1.9 mL. OD600 was monitored until a value of ~0.2 was reached. The assay plasmids were induced with 1 mM IPTG and 0, 0.125, 0.25, 0.5 or 1% arabinose for 4 h and 30 minutes at 30°C. 500 µL of cell suspension was treated with 1 mL of RNAprotect Bacterial Reagent (Qiagen), incubated for 5 minutes and then centrifuged for 10 minutes at 5000 × g. The supernatant was decanted and the cells were either stored overnight at 20°C or total RNA was isolated immediately according to the manufacturers’ protocol (RNAprotect Bacterial Reagent Handbook protocol 1, 7 and appendix B (Qiagen)). Total RNA was reverse-transcribed using the High Capacity cDNA Reverse-Transcription Kit (Applied Biosystems). Quantitative PCR was performed using the PowerUp™ SYBR™ Green Master Mix (Applied Biosystems). Fold expression of genes was calculated with the ΔΔCt-method and normalized to the gapA gene [22]. The assay was performed in two duplicates on two independent days (quadruplicates). Primer sequences, amplicon sizes and efficiencies are reported in supplemental table 5.
Results
Development of the Assay for SRSF1.
To test the use of linear sequences in the translational repression system we initially used SRSF1 as our RBP of choice. SRSF1 is a splicing factor that interacts with exonic splicing enhancer (ESE) sequences to influence splicing events [23,24]. Two RNA-recognition motif (RRM) domains drive its RNA binding and are followed by a C-terminal unstructured arginine-serine rich (RS) domain (see Figure 1C) [20,23]. To prepare an SRSF1 construct for use in the translation repression assay, we fused sfGFP to the SRSF1 N-terminus (see Figure 1C) and removed the RS domain as this was previously shown not to participate in RNA binding (GFP-SRSF1) [25]. As the RBP binding site in the TagBFP reporter, we chose a consensus sequence (AGAAGAAC) previously identified to be recognized by SRSF1 via SELEX experiments [26]. In contrast to the method described by Katz et al., we introduced the RBP binding site in front of the SD sequence (construct S1, see Table 1) rather than after the start codon (see Figure 1C). By doing so, we avoided the need for a design that stays in frame with the TagBFP ORF. Both sfGFP and TagBFP intensity were measured under increasing concentrations of the inducing agent (i.e. arabinose) for GFP-SRSF1 while the RNA was constantly induced with IPTG (1 mM) (see Figure 1D). To measure repression, we used the method described by Katz et al. which determines the fluorescent protein production rate rather than steady state levels to avoid saturation of the system [17,21]. TagBFP production rate is represented by the ratio of TagBFP levels divided by the integral of cell density in a time interval within the linear growth phase. sfGFP expression levels were normalized to cell density and averaged by the number of time points within the chosen time interval. Next, the TagBFP production rates are plotted against the sfGFP expression levels. To facilitate straightforward comparison of different RNA-RBP pairs, we calculated a repression ratio by dividing the basal TagBFP production rate (non-induced sfGFP-RBP) by the TagBFP production rate at the highest expression level (at 1% arabinose) of the sfGFP-RBP fusion (see Figure 1D). Such repression ratios have been used previously and allow straightforward comparison of different RNA/RBP pairs [14,18,27].
Table 1.
Different reporter constructs used for translational repression assays with SRSF1.
| Con-struct | RBP binding sequence | Linker | Repression ratio | Basal TagBFP production rate (1/min) |
|---|---|---|---|---|
| S1 | AGAAGAAC | - | 3.3 ± 0.3 | 211.5 ± 23.1 |
| S2 | AGAAGAACAGAAGAAC | - | 9.0 ± 2.2 | 260.4 ± 57.3 |
| S3 | AGAAGAACAGAAGAACAGAAGAAC | - | 15.1 ± 2.2 | 280.1 ± 15.2 |
| S4 | AGAAGAAC | AUA | 4.8 ± 1.0 | 194.5 ± 33.1 |
| S5 | AGAAGAACAGAAGAAC | AUA | 12.5 ± 1.0 | 314.4 ± 14.8 |
| S6 | AGAAGAACAGAAGAACAGAAGAAC | AUA | 17.0 ± 0.4 | 301.5 ± 12.3 |
| S7 | AGAAGUACAGAAGAACAGAAGAAC | AUA | 14.2 ± 2.1 | 321.0 ± 20.2 |
| S8 | AGAAGAACAGAAGUACAGAAGAAC | AUA | 16.1 ± 4.0 | 305.3 ± 20.9 |
| S9 | AGAAGAACAGAAGAACAGAAGUAC | AUA | 11.8 ± 1.3 | 316.7 ± 18.8 |
| S10 | AGAAGUACAGAAGUACAGAAGUAC | AUA | 3.2 ± 0.4 | 219.4 ± 6.0 |
| S0 | - | - | 1.2 ± 0.0 | 217.5 ± 30.2 |
| S6–4 | AGAAGAACAGAAGAACAGAAGAAC | (AU)2 | 14.5 ± 1.6 | 283.3 ± 22.5 |
| S6–5 | AGAAGAACAGAAGAACAGAAGAAC | (AU)2A | 13.5 ± 1.1 | 276.2 ± 20.8 |
| S6–6 | AGAAGAACAGAAGAACAGAAGAAC | (AU)3 | 11.9 ± 2.3 | 294.1 ± 17.7 |
| S6–7 | AGAAGAACAGAAGAACAGAAGAAC | (AU)3A | 11.1 ± 2.7 | 265.0 ± 18.5 |
| S6–8 | AGAAGAACAGAAGAACAGAAGAAC | (AU)4 | 9.2 ± 1.6 | 256.5 ± 16.1 |
| S6–9 | AGAAGAACAGAAGAACAGAAGAAC | (AU)4A | 8.4 ± 3.1 | 201.5 ± 13.1 |
| S6–10 | AGAAGAACAGAAGAACAGAAGAAC | (AU)5 | 6.5 ± 0.8 | 155.4 ± 9.9 |
In the first reporter construct S1 (see Figure 1C/D and Table 1), we introduced the SRFS1 RNA consensus sequence directly upstream of the SD sequence and we measured a repression ratio of 3.3 ± 0.3. When the S1 construct was compared to the S0 construct that has no insert (repression ratio 1.2 ± 0.0, see Table 1), a significant difference was observed. As a control, we fused sfGFP to the RRM domains 3 and 4 of the RNA-binding protein PTBP1 (GFP-PTBP1) which recognizes polypyrimidine sequences rather than the purine rich S1 sequence. Expression of this protein led to a 1.5 ± 0.2 fold repression (see supplemental Table 1 and supplemental Figure 1), which likely represents increased competition for protein synthesis resources and similar results were observed in the presence of sfGFP alone (supplemental Table 1 and supplemental Figure 2). Although repression was stronger in the presence of the SRSF1 construct, a bigger dynamic range would allow a clearer discrimination. The Saito group demonstrated that duplication of a hairpin-shaped RBP binding site improved translational repression of a reporter gene [28]. Furthermore, a recent report describing oligonucleotides with potent SRSF1 binding indicated that repeating a consensus sequence two or more times significantly improved binding affinity [29]. To explore whether this phenomenon also improved translational repression in our system, we multiplied the RNA sequence two or three times and observed a significant increase in repression ratio of 9.0 (construct S2) and 15.1 (construct S3) as shown in Table 1. The increase amounted to more than the sum of the repression ratio observed for a single insert possibly indicating an avidity effect.
Figure 2.
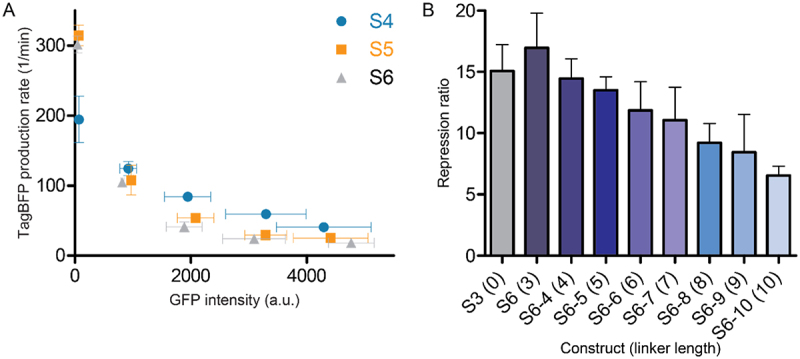
A) Repression curves of constructs S4-S6 in the presence of increasing concentrations of GFP-SRSF1. Repression ratios of each experiment are reported in table 1. B) Effect of increasing linker length for construct S6 (S6, S6-4 –S6-10) between the RBP binding site and the SD sequence on the repression ratio.
Previous descriptions of translational repression systems with similar designs reported that the distance between the RBP binding site and the SD sequence can influence the repression [17]. We therefore introduced a short three nucleotide linker to see whether the repression effect improves. Addition of the AUA linker indeed improved the repression for all three different constructs (S4-S6) albeit to a small degree (see Figure 2A, Table 1 and supplemental Figure 3). To gain further insight into the effect of the linker, we increased the length to 10 nucleotides with single nucleotide steps. The results clearly showed that increasing this distance reduces the repressive effect as would be expected (see Figure 2B, Table 1, and supplemental Figure 4). Since the S6 sequence displayed the highest repression ratio we tested it in the presence of an SRSF1 mutant (GFP-SRSF1mut) designed to have a strongly reduced affinity for RNA by introducing previously reported mutations in both RRMs, i.e.: F56D and F58D in RRM1, and Q135A and K138A in RRM2 [20,30]. For this combination, we observed a repression ratio of 1.6 ± 0.2 (see supplemental Table 1 and supplemental Figure 3) which is similar to the repression ratio observed for GFP-PTBP1 with this reporter (1.7 ± 0.1, see supplemental Table 1) indicating that the repression is driven by RBP binding.
Figure 3.
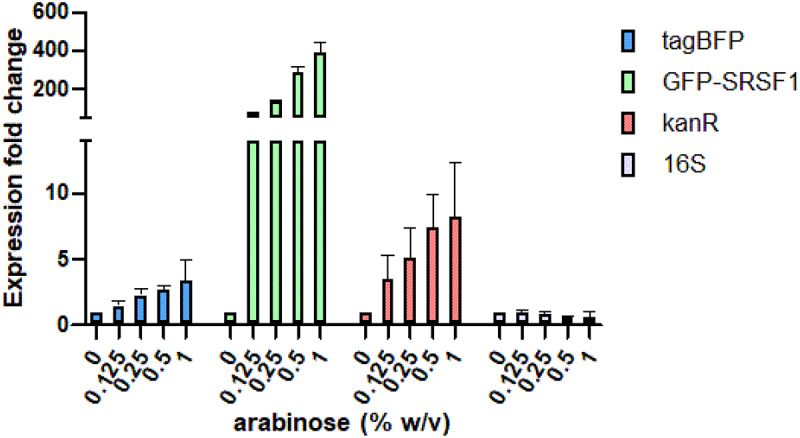
RT-qPCR analysis of gene expression in cells harbouring both the S6 and GFP-SRSF1 expressing plasmids. Expression levels were analysed at varying arabinose concentrations but at identical IPTG concentrations.
Figure 4.
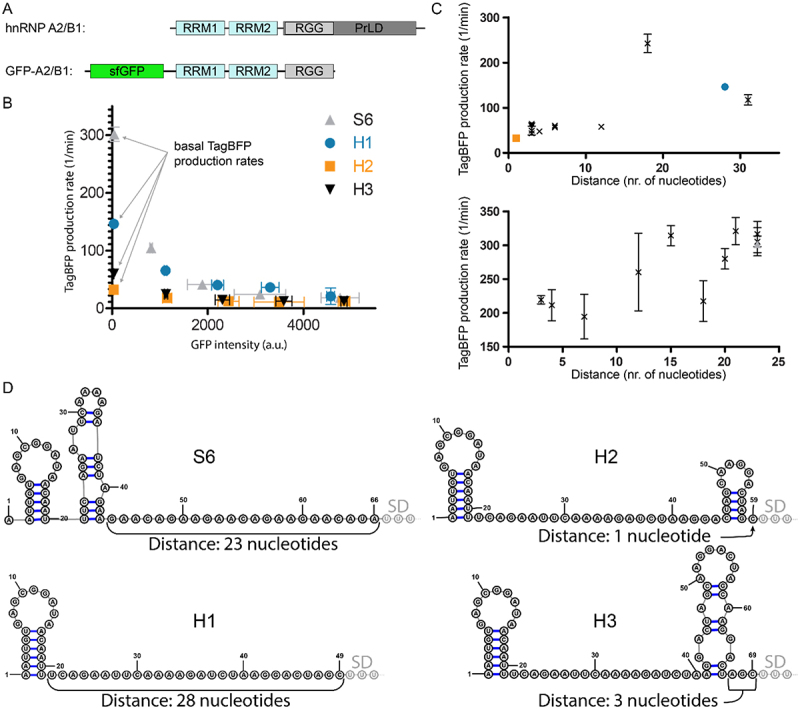
A) Schematic representation of hnRNP A2/B1 and the GFP-A2/B1 protein construct. B) Translational repression assay results for RNA reporters H1-H3 in the presence of GFP-A2/B1 in comparison with RNA reporter S6 in the presence of GFP-SRSF1 C) Correlation between TagBFP production rate and the distance between a secondary structure and the SD sequence for GFP-A2/B1 (top) and GFP-SRSF1 (bottom). D) Predicted secondary structures of the inserted RBP binding sequences for 5’-UTRs of the reporters S6 and H1-H3 using RNAfold. The first three nucleotides of the SD sequence are indicated as well as the distance between the SD and the closest predicted secondary structure.
Next, to gain insight into how multiplication of the binding sequences affected the interaction, we introduced a point mutation in the S6 sequence. We prepared variants where the mutation occurred at either the first, second or third repeat (S7-S9, see Table 1 and supplemental Figure 5) while the other two repeats maintained the original sequence. These constructs indicated that if the mutation was in the copies more distant from the SD sequence that the effect was minimal. However, when the mutation was right next to the SD sequence the repression ratio dropped to a value similar to that of S5. When the mutation was inserted in all repeats (construct S10) the repression was significantly decreased and similar to S1. However, the S10 sequence did have some propensity to form a hairpin like structure which could influence this repression value (see supplemental figure S13). All RNA inserts were also tested for translational repression in the presence of GFP-PTBP1 as described above and no significant repression was observed (see supplemental Table 1 and supplemental Figures 1–5).
Figure 5.
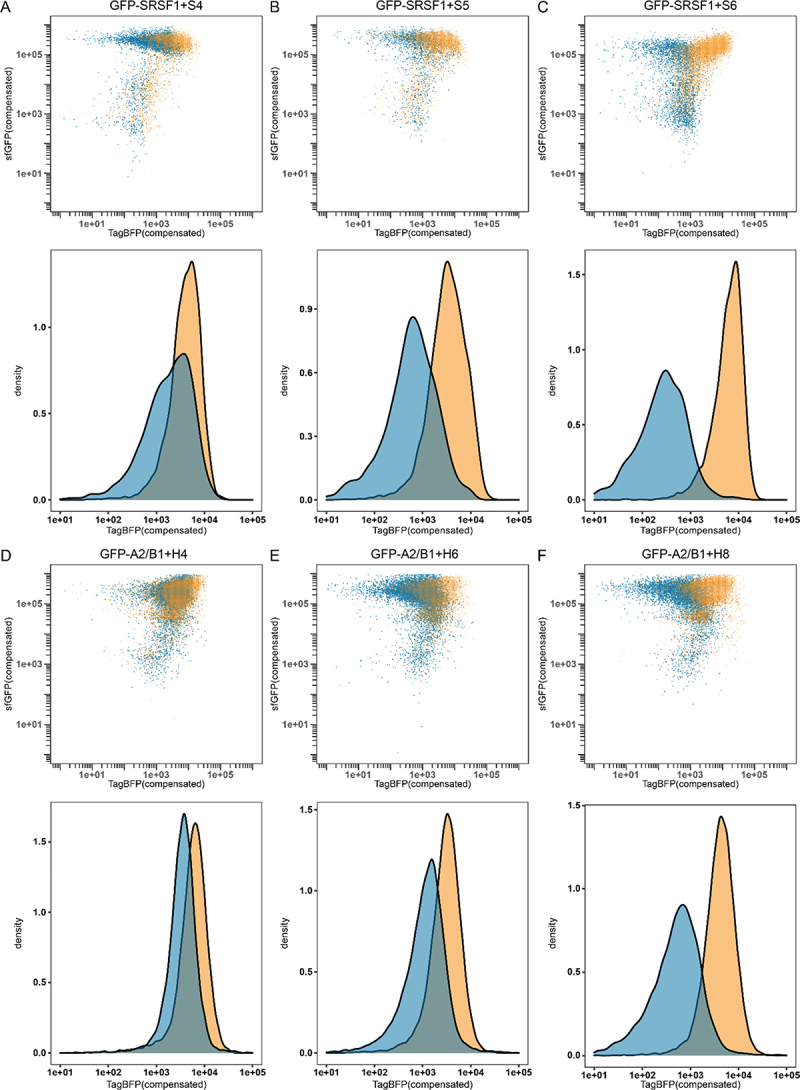
Flow cytometry results for GFP-SRSF1 (blue) or GFP-PTBP1 (orange) in combination with reporter S4 (A), reporter S5 (B) or reporter S6 (C). As well as GFP-A2/B1 (blue) or GFP-PTBP1 (orange) in combination with reporter H4 (D), reporter H6 (E), or reporter H8 (F). Density graphs were produced by analysing all events >1e05 in the sfGFP channel.
To verify that the reduction in TagBFP expression was caused by a reduction in translation rather than reduced mRNA levels, we performed RT-qPCR experiments using cells with the S6 construct. The cells were induced with increasing concentrations of arabinose to generate increasing levels of GFP-SRSF1 while maintaining the same concentration of inducing agent for the S6 reporter. The results as shown in Figure 3 indicate that the levels of TagBFP mRNA actually increase rather than decrease in the presence of increasing concentrations of GFP-SRSF1. The increase was also observed for the Kanamycin resistance gene (kanR) encoded on the same plasmid as TagBFP, but since this gene is expressed under a constitutive promoter, we hypothesize that the increase could be caused by an increase of plasmid stability or replication. The E. coli derived 16S gene remained stable, further confirming this hypothesis.
Development of the Assay for hnRNP A2/B1.
To explore the general applicability of using linear RNA sequences as RBP recognition sites in the translational repression assay, we also explored the splicing factor hnRNP A2/B1. Similar to the SRSF1 construct, we prepared an N-terminally sfGFP fused construct containing the residues 1–251 (GFP-A2/B1) which are similar to a truncation that was previously described to have high binding affinity for various consensus sequences as determined by isothermal titration calorimetry (see Figure 4A) [31]. We chose to use the RNA sequence AAGGACUAGC which was previously reported to have an affinity of 26.5 nM [31]. The sequence was introduced in the same position in the TagBFP reporter mRNA as single, double and triple repeats as we did for the SRSF1 sequence and repression ratios were measured (H1-H3, see Table 2 and supplemental figure S6). Constructs H1-H3 were also tested in the presence of GFP-PTBP1 and GFP alone which resulted in low repression values similar as for the SRSF1 experiments (see supplemental Table 1 and supplemental figures S6 and S7). Surprisingly, multiplication of the recognition sequence did not improve the repression ratio as was observed for the SRSF1 system. Adding the AUA linker (H4-H6, see Table 2 and supplemental figure S8) as done for the S-constructs did not improve the repression ratio and still no trend was observable. It is possible that direct connection of the consensus sequence does not provide enough spacing for two sfGFP-A2/B1 molecules to bind so we inserted a GGG spacer in between two replicated sequences (H7). Construct H7 indeed demonstrated an improved repression ratio over construct H2 and adding the AUA linker (H8) improved it even further (see Table 2 and supplemental figure S9). However, a similar improvement was not observed for the introduction of a spacer in the triplicate sequence (compare H6 and H9 in Table 2 and supplemental figures S8 and S9). Introducing a mutation (sequence H10 and H11, see Table 2 and supplemental figure S10) that was previously reported to reduce the affinity approximately 8-fold, only weakly reduced the repression ratio (compared to H8). The minimal effect is likely due to the mutation only occurring in one of the sequences, maintaining a high affinity sequence as well. Again, as a control we measured the repression of the unmodified TagBFP reporter plasmid in the presence of GFP-A2/B1 (entry H0, see Table 2) and measured a repression ratio of 1.3 ± 0.2 indicating that the reporters do respond to hnRNP A2/B1 binding. Furthermore, all constructs were evaluated in the presence of the non-binding GFP-PTBP1 resulting in repression ratios varying from 1.1 to 1.8 further indicating the reporters do respond to hnRNP A2/B1 binding (see supplemental figures S6 and 8–10).
Table 2.
Different reporter constructs used for translational repression assays with hnRNP A2/B1.
| Construct | RBP binding sequence | Linker | Repression ratio | Basal TagBFP production rate (1/min) |
|---|---|---|---|---|
| H1 | AAGGACUAGC | - | 5.3 ± 0.4 | 146.4 ± 7.3 |
| H2 | AAGGACUAGCAAGGACUAGC | - | 2.7 ± 0.4 | 32.3 ± 2.9 |
| H3 | AAGGACUAGCAAGGACUAGCAAGGACUAGC | - | 5.0 ± 1.1 | 60.0 ± 3.3 |
| H4 | AAGGACUAGC | AUA | 3.7 ± 0.2 | 117.4 ± 11.3 |
| H5 | AAGGACUAGCAAGGACUAGC | AUA | 2.6 ± 0.6 | 47.7 ± 5.0 |
| H6 | AAGGACUAGCAAGGACUAGCAAGGACUAGC | AUA | 4.6 ± 0.7 | 56.7 ± 5.4 |
| H7 | AAGGACUAGCGGGAAGGACUAGC | - | 4.6 ± 0.2 | 43.3 ± 2.2 |
| H8 | AAGGACUAGCGGGAAGGACUAGC | AUA | 6.4 ± 0.1 | 60.2 ± 3.6 |
| H9 | AAGGACUAGCGGGAAGGACUAGCGGGAAGGACUAGC | AUA | 5.0 ± 1.7 | 49.5 ± 9.7 |
| H10 | AAGCACUAGCGGGAAGGACUAGC | AUA | 4.6 ± 0.3 | 58.1 ± 6.4 |
| H11 | AAGGACUAGCGGGAAGCACUAGC | AUA | 6.0 ± 0.5 | 61.4 ± 4.6 |
| H0 | - | - | 1.3 ± 0.2 | 242.5 ± 20.5 |
Analysis of the Effect of Secondary Structures in the 5’-UTR of the Reporter mRNA.
We observed that for several of the H-reporters, the basal TagBFP production rate was significantly lower than for most of the S-reporters (see Tables 1 and 2, and Figure 4B). Both the S0 and H0 experiments showed that the basal TagBFP production rate was above 200 1/min, and while most S-reporters were above this level, most H-reporters were significantly below it. The lowered basal TagBFP production rate could potentially be caused by secondary structure formation, and we therefore used the RNAfold algorithm to predicted whether any secondary structures formed in the 5’-UTR of the reporter RNA up until the SD sequence [32]. In all 5’-UTRs, there was at least one hairpin but several contained more and in different locations along the sequence (see Figure 4D and supplemental figures S11–18). When the distance between any formed secondary structure and the SD sequence was plotted against the basal TagBFP production rate, a trend became evident that showed that a longer spacing is favoured (see Figure 4C). Although the differences were not so big for the S-reporters, the H-reporter series shows there is a maximum around 18 nucleotides. These results indicate that a reporter should be designed so that it provides a high basal production and therefore sufficient dynamic range to accurately analyse changes in RNA-binding after mutation.
Correlation of Translational Repression with Binding Affinity.
To investigate whether increased repression correlated with binding affinity, we performed fluorescence polarization (FP) experiments with fluorescein amine (6-FAM) labelled RNA sequences and recombinantly expressed SRSF1 and hnRNP A2/B1 proteins spanning the same residues as used in the translational repression assay but without the N-terminal GFP fusion. Instead, hnRNP A2/B1 was modified with an MBP-tag to improve yields during expression and purification while SRSF1 was untagged (see supplemental table 4). For SRSF1, we tested the RNA sequences equivalent to S1, S2 and S3 and for hnRNP A2/B1 the sequences used for H1, H2 and H3 (see Table 3, supplemental Table 3 and supplemental figures S21 and 22). For SRSF1, we were surprised to observe that there was no binding between the protein and the single repeat sequence. The lack of affinity could be caused by the RNA sequence being too short to accommodate both RRM domains. Therefore, the sequence was extended on both the 5’ and 3’ end with the three flanking nucleotides that were used in the S1 construct (FAM-S1ext, see supplemental Table 3). Good affinity was observed for this RNA as well as the FAM-S2 and FAM-S3 sequences, and the results follow the logic of the repression experiments where an increased affinity was observed if the consensus sequence is repeated. The hill-slopes of the measured curves indicate that FAM-S1ext can accommodate a single protein while the FAM-S2 and FAM-S3 sequence accommodate multiple copies of SRSF1. The FP experiments for hnRNP A2/B1 also correlate with the repression assay where H1 and H3 show good binding affinity but a reduction for H2 is observed. The reduced affinity for H2 is somewhat unexpected but is likely caused by secondary structure formation and we therefore also predicted the structures of these isolated RNA sequences using RNAfold (see supplemental figures S19 and 20). Indeed, both FAM-H1 and FAM-H3 possess the AAGGACU sequence in non-base paired form which is recognized by the two RRMs according to the reported crystal structure [31]. In contrast, the binding sequence is embedded into the stem of FAM-H2 probably leading to a balance between the energy required to disrupt this and the binding energy for the linear sequence explaining its reduced affinity. When binding affinity as measured by FP was plotted against the repression ratio as measured by the translational repression assay a good correlation was observed for both proteins (see supplemental figure S23).
Table 3.
Affinities of SRSF1 and hnRNP A2/B1 for representative RNA sequences measured by fluorescence polarization.
| RNA | Protein | KD (nM) | Hill slope |
|---|---|---|---|
| FAM-S1 | SRSF1 | >25000 | - |
| FAM-S1ext | SRSF1 | 92.3 ± 44.1 | 1.33 ± 0.32 |
| FAM-S2 | SRSF1 | 56.1 ± 17.5 | 0.62 ± 0.30 |
| FAM-S3 | SRSF1 | 42.5 ± 33.9 | 0.53 ± 0.11 |
| FAM-H1 | hnRNP A2/B1 | 51.7 ± 9.0 | 0.75 ± 0.09 |
| FAM-H2 | hnRNP A2/B1 | 134.8 ± 9.0 | 0.66 ± 0.05 |
| FAM-H3 | hnRNP A2/B1 | 24.0 ± 8.1 | 0.52 ± 0.04 |
Analysis of Translational Repression by Flow Cytometry.
To evaluate whether the repression of TagBFP translation could also be observed as an endpoint assay, we used flow cytometry to analyse different populations. Flow cytometry was previously used to analyse translational repression assays but only by evaluating the fluorescent reporter output [14]. Here, we combined both the sfGFP signal with the TagBFP signal to provide a clear analysis of the fluorescent properties within the bacterial cell population. We used the constructs S4, S5 and S6 in combination with GFP-SRSF1 and compared it to a combination of the same reporters with GFP-PTBP1 to see whether the increase in repression ratio was also reflected in the flow cytometry data. Indeed, with increasing repression ratio we observe a larger difference between the repressed and non-repressed populations (see Figure 5a–c). In the case of S6, we observed that the populations were almost entirely separated from one another allowing for very clear analysis of the different populations. We also analysed constructs S0-S3 and S7-S10 and observed population changes correlating with the measured repression ratios (see supplemental figures S24–27).
We then analysed several constructs in combination with either GFP-A2/B1 or GFP-PTBP1 and found that similar shifts were observable. Construct H4 showed very little difference between the populations (see Figure 5D) while the constructs with the higher repression ratios (H6 and H8) showed increasingly larger differences correlating with the repression ratios (see Figure 5e and f). The results are more obvious when histograms are made of all events of a sufficiently high GFP level (>1 × 105) to reflect good expression levels of the RBPs (see Figure 5 and supplemental figures S29–30). These histograms also indicate that a relatively high repression ratio is required for good discrimination between the populations in flow cytometry, further emphasizing that replication of binding sites while avoiding secondary structure formation is essential for a clear read-out. The remaining H-constructs were analysed as well and also demonstrated shifts that correlated with the repression ratios (supplemental figures S28–31).
Discussion
Here, we demonstrate that translational repression-based protein-RNA interaction analysis is also feasible using non-structured RNA sequences and can be optimized to analyse protein-RNA pairs with low affinity. The possibility to do so opens this assay up to a wide variety of RNA-binding proteins beyond those that have very high affinity for their target sequences (i.e.: MS2, U1A). Repetition of the RNA sequence led to an increase in signal possibly due to an avidity affect leading to increased affinity. The findings resemble the observation that multiplication of SRSF1 binding sites in model RNAs increases splicing efficiency and that enrichment of SRSF1 binding sequences is observed near splice sites [33–35]. Furthermore, repetitive binding sequences were found in long non-coding RNAs and demonstrated to be bound by SRSF1 [36,37]. When the RNA insert does form a secondary structure in close proximity to the SD sequence accurate determination of repression values tend to be obstructed due to a reduced basal translation rate. However, using secondary structure prediction algorithms it is possible to design suitable reporters before cloning and testing in the assay. Alternatively, it is possible that the consensus sequence used for hnRNP A2/B1 is recognized by an endogenous RNA-binding protein. The fact that single repeats still show relatively high basal TagBFP expression levels while multiplication lowers it could be an indicator for this hypothesis. It is perhaps wise to explore a variety of inserts when multiple consensus sequences are known for the RBP of interest.
We demonstrate that repression values determined using time-course assays correlate with end-point analysis using flow cytometry. Although the FP experiments seem to correlate with repression, the basal expression level of the reporter plays a key role in the signal-to-noise ratio. It is therefore important to evaluate the effectiveness of the assay for new protein-RNA pairs by monitoring this parameter. Furthermore, since the consensus sequences have to be repeated to get a good signal-to-noise ratio, it is important to introduce RNA mutations in all repeats if these are to be studied. Simple manipulation of the assay plasmids by straightforward cloning techniques allows the comparative study of variations in RNA sequence or mutations in the RBP. The various constructs described here as well as the design rules provided for the assay allow it to be adapted to other RBPs that bind linear sequences to study the effect of mutation on both the RNA as well as the protein side. The assay is fast and low in cost and avoids the need for the isolation and purification of protein and RNA. It also has the potential to be used in fluorescence assisted cell sorting experiments to select for optimized RNA sequences for a given RBP or vice versa. Furthermore, the effect of reduction in basal reporter protein production rate could potentially be used to investigate the secondary structure of the RNA insert.
Supplementary Material
Acknowledgments
We kindly acknowledge the Protein Chemistry Facility of the Max Planck Institute and in particular Jan-Erik Hoffmann for assistance with protein expression and purification. We also thank Pim Huis in ‘t Veld for providing the sfGFP plasmid and Oleg Sitsel for the TagBFP plasmid.
Funding Statement
This work was funded via the Chemical Genomics Centre of the Max Planck Society which is supported by AstraZeneca, Merck KGaA, Pfizer Inc, and the Max Planck Society. Gulshan Amrahova was funded by a scholarship from the German Academic Exchange Service (DAAD).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2192553
Data availability statement
Raw data were generated at the Max Planck Institute of Molecular Physiology. Derived data supporting the findings of this study are available from the corresponding author P.H. on request.
References
- [1].Gebauer F, Schwarzl T, Valcárcel J, et al. RNA-binding proteins in human genetic disease. Nat Rev Genet. 2021;22(3):185–198. [DOI] [PubMed] [Google Scholar]
- [2].Hentze MW, Castello A, Schwarzl T, et al. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19(5):327–341. [DOI] [PubMed] [Google Scholar]
- [3].Pereira B, Billaud M, Almeida R.. Trends in Cancer. Trends Cancer. 2017;3(7):506–528. [DOI] [PubMed] [Google Scholar]
- [4].Marchese D, de Groot NS, Lorenzo Gotor N, et al. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2016;7(6):793–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mohibi S, Chen X, Zhang J. Cancer the‘rbp’eutics–rna-binding proteins as therapeutic targets for cancer. Pharmacology & Therapeutics. 2019;203:107390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Berry KE, Hochschild A. A bacterial three-hybrid assay detects Escherichia coli Hfq–sRNA interactions in vivo. Nucleic Acids Res. 2018;46(2):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stockert OM, Gravel CM, Berry KE. A bacterial three-hybrid assay for forward and reverse genetic analysis of RNA–protein interactions. Nat Protoc. 2022;17(4):941–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wilhelm JE, Vale RD. Genes to Cells. Genes Cells. 1996;1(3):317–323. [DOI] [PubMed] [Google Scholar]
- [9].Harada K, Martin SS, Frankel AD. Selection of RNA-binding peptides in vivo. Nature. 1996;380(6570):175–179. [DOI] [PubMed] [Google Scholar]
- [10].Jain C, Belasco JG. A Structural Model for the HIV-1 Rev–RRE complex deduced from altered-specificity rev variants isolated by a rapid genetic strategy. Cell. 1996;87(1):115–125. [DOI] [PubMed] [Google Scholar]
- [11].Horiya S, Inaba M, Koh C-S, et al. Replacement of the λ boxB RNA-N peptide with heterologous RNA-peptide interactions relaxes the strict spatial requirements for the formation of a transcription anti-termination complex. Mol Microbiol. 2009;74(1):85–97. [DOI] [PubMed] [Google Scholar]
- [12].Peled-Zehavi H, Horiya S, Das C, et al. Selection of RRE RNA binding peptides using a kanamycin antitermination assay. RNA. 2003;9(2):252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peabody DS. Translational repression by bacteriophage MS2 coat protein expressed from a plasmid. A system for genetic analysis of a protein-RNA interaction. J Biol Chem. 1990;265(10):5684–5689. [PubMed] [Google Scholar]
- [14].Paraskeva E, Atzberger A, Hentze MW. A translational repression assay procedure (TRAP) for RNA–protein interactions in vivo. Proceedings of the National Academy of Sciences. 1998;95:951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goldfless SJ, Belmont BJ, de Paz AM, et al. Direct and specific chemical control of eukaryotic translation with a synthetic RNA–protein interaction. Nucleic Acids Res. 2012;40(9):e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Saito H, Kobayashi T, Hara T, et al. Synthetic translational regulation by an L7Ae–kink-turn RNP switch. Nat Chem Biol. 2010;6(1):71–78. [DOI] [PubMed] [Google Scholar]
- [17].Katz N, Cohen R, Solomon O, et al. An in vivo binding assay for rna-binding proteins based on repression of a reporter gen. ACS Synth Biol. 2018;7(12):2765–2774. [DOI] [PubMed] [Google Scholar]
- [18].Nie M, Htun H. Different modes and potencies of translational repression by sequence-specific RNA–protein interaction at the 5′-UTR. Nucleic Acids Res. 2006;34(19):5528–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Berrow NS, Alderton D, Sainsbury S, et al. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007;35(6):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clery A, Sinha R, Anczukow O, et al. Isolated pseudo–RNA-recognition motifs of SR proteins can regulate splicing using a noncanonical mode of RNA recognition. Proceedings of the National Academy of Sciences. 2013:110:E2802–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Katz N, Cohen R, Atar R, et al. Synthetic 5′ UTRs Can Either Up- or Downregulate Expression upon RNA-Binding Protein Binding. J Visualized Exp. 2019;2019(1):1–9. [DOI] [PubMed] [Google Scholar]
- [22].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [23].Cho S, Hoang A, Chakrabarti S, et al. Nucleic Acids Research. Nucleic Acids Res. 2011;39(21):9413–9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Das S, Krainer AR. Molecular Cancer Research. Mol Cancer Res. 2014;12(9):1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cho S, Hoang A, Sinha R, et al. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tacke R, Manley JL. The EMBO Journal. Embo J. 1995;14(14):3540–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bouvet P, Jain C, Belasco JG, et al. RNA recognition by the joint action of two nucleolin RNA-binding domains: genetic analysis and structural modeling. Embo J. 1997;16(17):5235–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ono H, Kawasaki S, Saito H. Orthogonal Protein-Responsive mRNA Switches for Mammalian Synthetic Biology. ACS Synth Biol. 2020;9(1):169–174. [DOI] [PubMed] [Google Scholar]
- [29].Denichenko P, Mogilevsky M, Cléry A, et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat Commun. 2019;10(1):1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Caceres JF, Krainer AR. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. Embo J. 1993;12(12):4715–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu B, Su S, Patil DP, et al. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9(1):420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gruber AR, Lorenz R, Bernhart SH, et al. Nucleic Acids Research. Nucleic Acids Res. 2008;36(Web Server):W70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jobbins AM, Reichenbach LF, Lucas CM, et al. The mechanisms of a mammalian splicing enhancer. Nucleic Acids Res. 2018;46(5):2145–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang J, Smith PJ, Krainer AR, et al. Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res. 2005;33(16):5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sanford JR, Wang X, Mort M, et al. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19(3):381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Khan M, Hou S, Azam S, et al. Nucleic Acids Research. Nucleic Acids Res. 2021;49(11):6420–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ninomiya K, Adachi S, Natsume T, et al. Ln RNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. Embo J. 2020;39(3):e102729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were generated at the Max Planck Institute of Molecular Physiology. Derived data supporting the findings of this study are available from the corresponding author P.H. on request.