

Abstract
Several classes of ribosomally synthesized and post-translationally modified peptides (RiPPs) are comprised of multiple macrocycles. The enzymes that assemble these macrocycles must surmount the challenge of installing a single specific set of linkages out of dozens of distinct possibilities. One class of RiPPs that includes multiple macrocycles are the graspetides, named after the ATP-grasp enzymes that install ester or amide linkages between pairs of nucleophilic and electrophilic side chains. Here, using heterologous expression and NMR spectroscopy, we characterize the connectivity and structure of amycolimiditide, a 29 aa graspetide with a stem-loop structure. The stem includes 4 esters and extends over 20 Å. The loop of amycolimiditide is distinguished by the presence of an aspartimide moiety, installed by a dedicated O-methyltransferase enzyme. We further characterize the biosynthesis of amycolimiditide in vitro, showing that the amycolimiditide ATP-grasp enzyme AmdB operates in a strict vectorial fashion, installing esters starting at the loop and proceeding down the stem. Surprisingly, the O-methyltransferase AmdM that aspartimidylates amycolimiditide prefers a substrate with all four esters installed, despite the fact that the most distal ester is ~30 Å away from the site of aspartimidylation. This study provides insights into the structure and diversity of aspartimidylated graspetides and also provides fresh insights into how RiPP biosynthetic enzymes engage their peptide substrates.
Graphical Abstract
Introduction
The biosynthetic enzymes that assemble ribosomally synthesized and post-translationally peptides (RiPPs) catalyze a unique set of reactions.1–2 One catalytic challenge for these enzymes is that RiPP enzymes must often catalyze the reaction at multiple locations within a single peptide substrate. This challenge is exemplified by the epimerase in polytheonamide B biosynthesis in which a single radical SAM enzyme introduces 18 epimerizations within a 48 aa substrate.3 A subset of RiPPs are defined by multiple macrocyclic linkages including lanthipeptides,4 sactipeptides,5 triceptides,6 darobactins,7 and graspetides.8 The enzymes that make these macrocycles contend with a further catalytic challenge of ensuring that the correct macrocycles are formed. For example, the 24 aa core peptide of the β chain of the lanthipeptide haloduracin9–10 includes 8 Ser/Thr residues, 7 of which are dehydrated. Four cysteine residues within the core peptide react to generate thioether linkages.10 Despite this potential combinatorial diversity, only a single connectivity is observed due to a combination of geometric constraints in the substrate and specificity in the maturation enzymes. The problem of forming only the correct crosslinks in a multi-macrocyclic RiPP can be solved in part by introducing the crosslinks in a specific order. This ordering is observed in the maturation of the haloduracin β chain, in which the four (methyl)lanthionine linkages are introduced in a specific order11 with concomitant changes in enzyme conformation.12
The macrocycles in graspetides are formed via side chain-side chain linkages between amino acids with nucleophilic side chains (Ser, Thr, Lys) and Asp/Glu (Fig. 1A). This family of RiPPs is named after the ATP-grasp enzymes that install the linkages and the most well-studied graspetides are the microviridins.13–15 The 14 aa core peptide of graspetide microviridin J16 includes 4 nucleophilic Ser/Thr/Lys resides and 3 Asp/Glu residues, leading to 24 possible connectivities assuming all three Asp/Glu residues react. Other graspetides have been discovered subsequent to the microviridins, including plesiocin17 and thuringinin,18 which contain only ester linkages and form stem-loop structures (Fig. 1B). The order of ester formation in these peptides has been studied by isolating partially esterified intermediates and analyzing these species using tandem mass spectrometry.8, 18 The picture that emerges from these studies is that the introduction of esters into these core peptides follows a strict order with the first ester to form establishing the loop of the hairpin with subsequent esters forming the hairpin stem (Fig. 1B).
Figure 1: Enzymology and biosynthesis of amycolimiditide.
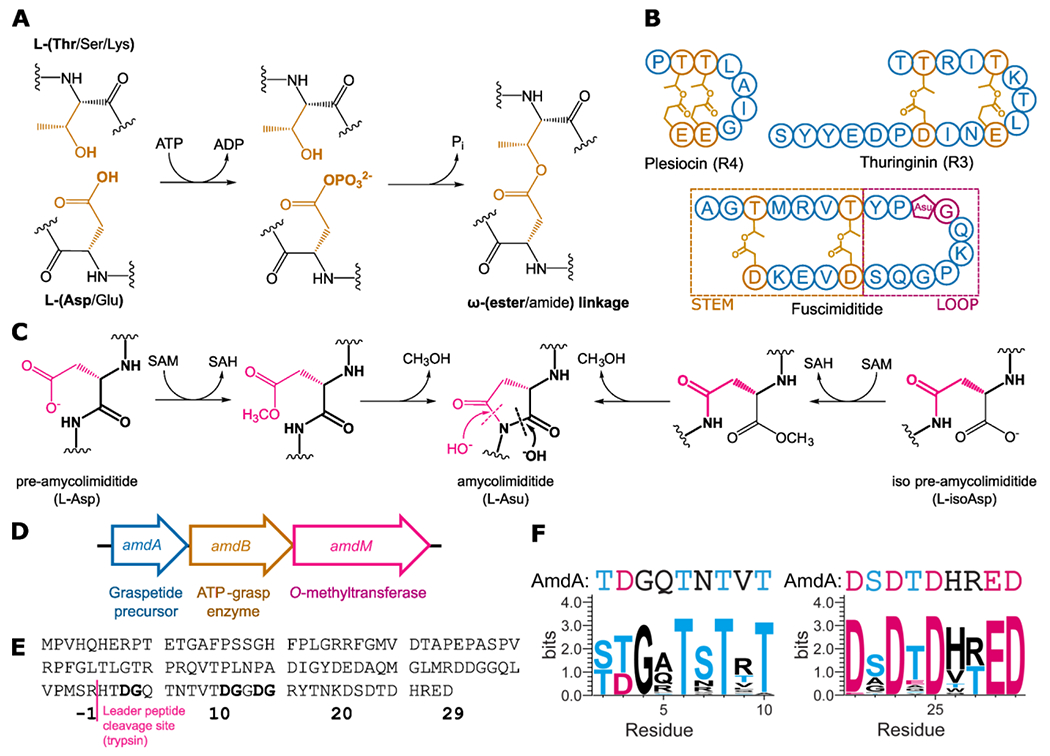
(A) Mechanism of side chain-side chain ester formation catalyzed by an ATP-grasp enzyme. (B) Other ester-linked stem-loop graspetides. For plesiocin and thuringinin one of the multiple, repeated core peptides, repeat 4 (R4) for plesiocin and repeat 3 (R3) for thuringinin, is shown. The Asu in fuscimiditide is aspartimide. (C) O-methyltransferase catalyzed aspartimidylation of pre- and iso pre-amycolimiditide. The O-methyltransferase can recognize and methylate either Asp or isoAsp residues, driving both isomers to aspartimide. (D) The amycolimiditide BGC. (E) Sequence of amycolimiditide precursor AmdA with putative leader and core segments shown. Potential sites for aspartimidylation (DG) are bolded.(F) Sequence logos for the nucleophile-enriched region (left) and the acidic region (right), derived from amycolimiditide and 29 close homologs (Table S1, Fig. S1). The corresponding part of the AmdA sequence is shown above the sequence logos.
Recently our group reported a new example of a graspetide, fuscimiditide, that is comprised of a stem-loop structure with two ester crosslinks in the stem (Fig. 1B).19 Fuscimiditide differs from previously characterized stem-loop graspetides in two ways. First, the loop region of fuscimiditide is longer (10 aa) than the loops of plesiocin (4 aa) and thuringinin (3 aa). Second, the loop of fuscimiditide harbors an additional post-translational modification, the aspartimide, installed by a dedicated O-methyltransferase related to protein isoaspartyl methyltransferases (PIMTs. Fig. 1C). Similar PIMT homologs are present in lasso peptide20 and lanthipeptide21 biosynthetic gene clusters (BGCs). These PIMTs are distinguished from canonical PIMTs by their substrate specificity; RiPP associated PIMTs methylate specific L-Asp side chains while canonical PIMTs methylate L-isoAsp and D-Asp.22 RiPP associated PIMTs also harbor an additional C-terminal domain of >100 aa relative to canonical PIMTs. In our previous work on the PIMT homologs that aspartimidylate lasso peptides,20 we showed that this C-terminal extension to PIMT is important for RiPP substrate recognition.
Our bioinformatic analysis suggested BGCs encoding for many other putative aspartimidylated graspetides throughout diverse actinobacteria.19 In this work we heterologously expressed a novel 29 aa graspetide and determined its structure using 2D NMR experiments. This peptide, named amycolimiditide after its producing organism Amycolatopsis cihanbeyliensis,23 has a loop of 10 aa and an extended stem containing four esters in a twisted ladder conformation that stretches ~24 Å. Like fuscimiditide, amycolimiditide harbors a single aspartimide moiety within its loop. We also reconstitute the biosynthesis of amycolimiditide in vitro giving insights into the order, kinetics, and substrate specificity of ester and aspartimide formation.
Results and Discussion
Discovery and sequence features of amycolimiditide
We identified a BGC with similarities to the fuscimiditide BGC in the genome of Amycolatopsis cihanbeyliensis DSM 45679 with a RiPP precursor gene and genes for an ATP-grasp enzyme and an O-methyltransferase (Fig. 1D). The O-methyltransferase in this BGC is 394 aa, much longer than canonical PIMTs like the E. coli PIMT (208 aa). The graspetide precursor in this BGC, AmdA, has a putative core sequence longer than that of fuscimiditide. The core sequence is enriched in residues capable of forming side chain-side chain esters or amides; 16 of the 28 C-terminal residues of AmdA are capable of participating in ATP-grasp catalyzed crosslinking and there are multiple potential sites for aspartimidylation. To predict the possible locations of the PTMs in this peptide, which we name amycolimiditide, we sought to identify the conserved residues in the C-terminal region of AmdA. We performed protein-protein BLAST (BlastP) on AmdA and curated a list of 29 homologs produced by various actinobacteria (Table S1). All homologs are encoded in the BGCs that follow the “A-B-M” sequence in which the genes are placed, which follows the consensus of the aspartimidylated graspetides (Fig. 1D).19 In the AmdA sequence (Fig. 1E), we identified the nucleophile-enriched and acidic regions and generated their sequence logos from the sequences of AmdA and the homologs. The sequence logos show four Thr residues conserved in the nucleophile-enriched region and four Asp and one Glu residues conserved in the acidic region of AmdA (Fig. 1F). This suggests a maximum of four ω-ester linkages installed by the cognate ATP-grasp enzyme.8 An alignment of AmdA and its 29 homologs suggests one specific DG dipeptide as the possible site of aspartimidylation (Fig. S1). While AmdA contains two additional DG sites, these are not as well conserved in homologous precursors (Fig. S1).
Enzymatic maturation of amycolimiditide in E. coli and in vitro
To assess the extent of PTMs performed by each modifying enzyme, we compared the results of heterologous expressions of amdA, amdAB, and amdABM in E. coli. To do so, we constructed plasmids based on pQE-80 with the gene(s) under the IPTG-inducible T5 promoter. Additionally, a His6-tag was fused upstream of amdA, a SUMO-tag was fused upstream of amdB, and the start codon of amdM was modified to ATG (Fig. S2, see also supplementary text for earlier iterations of this expression system). SUMOylated AmdB is more stable in vitro than AmdB and leads to a greater extent of AmdA esterification than does untagged AmdB (supplementary text, Fig. S3). We used SUMO-AmdB for all subsequent work in E. coli and in vitro, and for brevity’s sake will refer to this enzyme as AmdB. When expressed alone, the purified AmdA protein has an observed average mass of 13,511.6 Da, corresponding to AmdA without the N-terminal methionine (calculated average mass = 13,511.6 Da; Fig. 2A). When coexpressed with AmdB, AmdA has a mass of 13,439.6 Da, corresponding to AmdA with 4-fold dehydration (calculated average mass = 13,439.5 Da; Fig. 2A, Fig. S4). Similarly, we modified AmdA in vitro with AmdB (Fig. S5A) and ATP, resulting in the accumulation of AmdA with four dehydrations (Fig. S6). This indicates that AmdB installs four side chain-side chain linkages on AmdA in total, consistent with our prediction from identifying four conserved Thr residues in the nucleophile-enriched region of the amycolimiditide sequence (Fig. 1F). We name the AmdB-modified AmdA protein, mAmdAB, adapting the nomenclature suggested by Roh et al. where the “m” signifies that the precursor is modified while the superscript indicates which enzyme(s) have modified the precursor.18 Whereas the ATP-grasp enzyme for fuscimiditide has optimal activity at 37 °C,19 AmdB performs poorly at this temperature, instead working best at room temperature (Fig. S7). To locate the putative core peptide region that contains all four ω-ester linkages, we digested mAmdAB with trypsin and observed a 3,134.24 Da fragment corresponding to a 29-aa long C-terminal fragment with 4-fold dehydration and 3 missed trypsin cleavages (calculated monoisotopic mass = 3,134.28 Da; Fig. S8A). We named this fragment pre-amycolimiditide, using the nomenclature adopted for cellulonodin-2, lihuanodin, and fuscimiditide that are not modified by their cognate methyltransferase.19–20 Then, since pre-amycolimiditide contains Lys-20, we tested for the presence of an ω-amide linkage with a hydrolysis test. After incubation at pH 11.0 for 30 hours, pre-amycolimiditide mostly regained 72 Da in mass, indicating that all four crosslinks installed by AmdB are ω-esters (Fig. S9).
Figure 2: Posttranslational modifications to the amycolimiditide precursor.
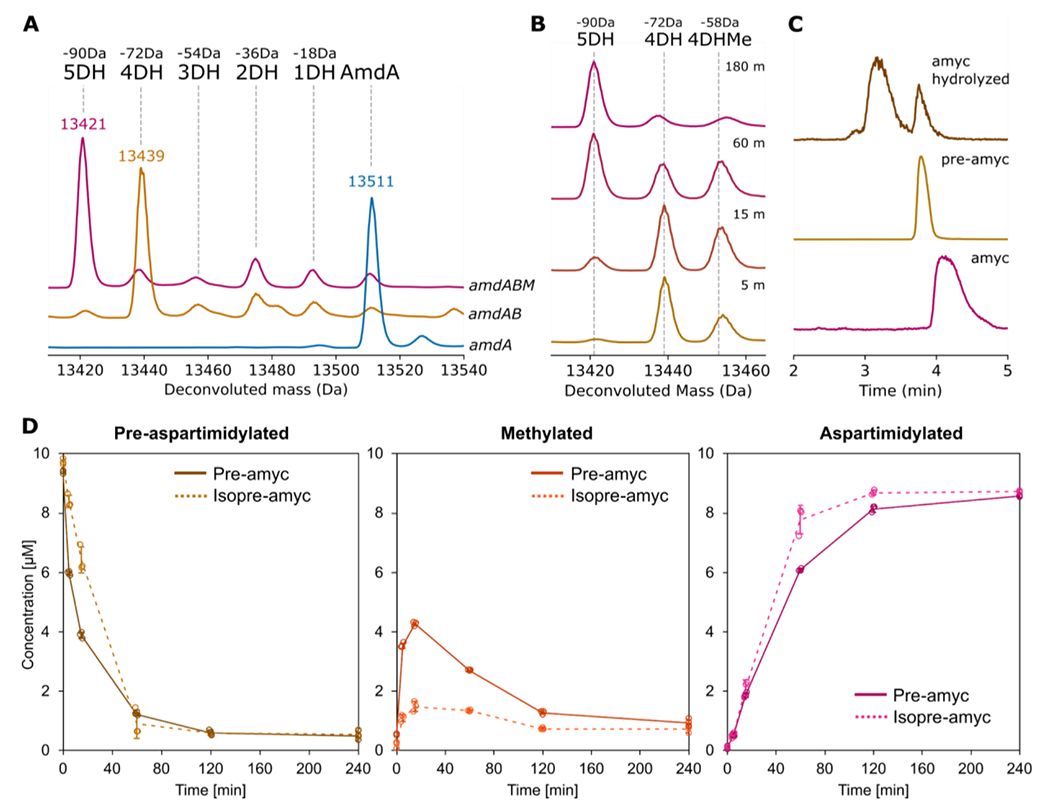
A: Co-expression of amycolimiditide precursor AmdA with ATP-grasp enzyme AmdB leads to 4 dehydrations (4DH) of AmdA while coexpression of AmdA with AmdB and PIMT homolog AmdM leads to 5 dehydrations (5DH) of AmdA. In all cases, AmdB is present as a SUMO fusion protein. Dotted guide lines correspond to the calculated average mass of a given species. B: Time course of aspartimidylation of mAmdAB by AmdM in vitro. Methylated mAmdAB (4DHMe) is present at early times but is converted to the aspartimide within 3 h. C: EIC chromatograms of amycolimiditide (top), its hydrolysis products (middle) and pre-amycolimiditide (bottom) show that amycolimiditide hydrolyzes into two distinct products. D: Time courses of methylation and aspartimidylation of pre-amycolimiditide (containing Asp) and iso pre-amycolimiditide (containing isoAsp). Methylation of pre-amycolimiditide proceeds faster than methylation of iso pre-amycolimiditide, but aspartimide formation is faster for iso pre-amycolimiditide than for pre-amycolimiditide. Experiments were run in triplicate with all data points shown. Error bars represent the standard deviation; some error bars are so small that they are not visible.
When AmdA was coexpressed with AmdB and AmdM, the purified protein, termed mAmdABM, has a mass of 13,421.1 Da, corresponding to AmdA with 5-fold dehydration (calculated average mass = 13,421.5 Da; Fig. 2A). To observe the progression of aspartimidylation, we modified mAmdAB in vitro with His6-AmdM (Fig. S5B) and S-adenosyl methionine (SAM) for different times. Consistent with the aspartimidylation pathway (Fig. 1C), the 13,453.2 Da peak corresponding to the methylated mAmdAB (calculated average mass = 13,453.5 Da) emerged and then disappeared over the first hour of the reaction, while the 13,421 Da peak corresponding to a putatively aspartimidylated mAmdAB (i.e. mAmdABM) accumulated over time (Fig. 2B). No further dehydration was observed during the reaction, indicating that AmdM methylates only one out of three DG sites in the amycolimiditide core peptide. The apparent specificity in the location of the aspartimidylation site for AmdM contrasts with canonical PIMTs, which accept many substrates in which the isoAsp is embedded in different sequences.24 We also monitored the in vitro aspartimidylation of pre-amycolimiditide over time and observed a similar modification rate as the in vitro aspartimidylation of mAmdAB (Fig. S10). This contrasts with the OlvSA-catalyzed aspartimidylation for the lanthipeptide OlvA(BCSA), which becomes aspartimidylated faster with its leader peptide intact.21 In the absence of SAM, AmdM activity was not observed (Fig. S11), demonstrating that the graspetide, lanthipeptide, and lasso peptide methyltransferases are all SAM-dependent methyltransferases.19–21 We monitored the in vitro aspartimidylation of mAmdAB at room temperature, 28 °C, and 37 °C over time and observed similar rates (Fig. S12). Despite being produced by the same organism, AmdM is more thermotolerant than AmdB (Fig. S7, S12).
As mentioned earlier, the methyltransferase-catalyzed aspartimidylation occurs in two steps: Asp methylation and backbone cyclization (Fig. 1C). While the methyltransferase clearly catalyzes the transfer of the methyl group from SAM to the Asp residue of the cognate RiPP substrate, we asked whether the backbone cyclization step also requires the methyltransferase. Methylated pre-amycolimiditide was purified after in vitro aspartimidylation for 1 hr (Fig. S13) and then incubated at 28 °C in 1X PBS, pH 7.4 for 30 minutes with or without AmdM. At both conditions, amycolimiditide was produced at a similar rate (Fig. S14), showing that AmdM does not accelerate the backbone cyclization step appreciably.
As L-aspartimide is known to hydrolyze to L-Asp and L-isoAsp in basic conditions,25 we sought to obtain the amycolimiditide core peptide with isoAsp by hydrolysis. First, we trypsinized mAmdABM and HPLC-purified the 29-aa long C-terminal fragment containing five dehydrations (Fig. S8B and S15A). As mentioned above, we name this peptide amycolimiditide while acknowledging that the leader peptide may be cleaved elsewhere in the native producer.19 After incubation at pH 8 for 48 hours, amycolimiditide was hydrolyzed once to two isobaric species in approximately 7:3 ratio, as observed in MS (Fig. 2C, S16). As the minor species matches the retention time of pre-amycolimiditide, we suspected that the major species contains an isoaspartate moiety. We purified the major species by HPLC (Fig. S17) and allowed it to react with PIMT from H. sapiens, a well-studied enzyme known to catalyze the aspartimidylation of L-isoAsp in peptides and proteins.26 The PIMT successfully aspartimidylated the major species (Fig. S18), confirming it as an isoAsp-containing core peptide, which we name “iso pre-amycolimiditide”. The ~7:3 ratio in the regioselectivity of hydrolysis matches closely to the ratio of the hydrolysates of the lanthipeptide OlvA(BCSA), whereas 95% of fuscimiditide hydrolyzes to iso pre-fuscimiditide and lihuanodin exclusively hydrolyzes to pre-lihuanodin.19–21 This suggests that the regioselectivity of hydrolysis is a characteristic unique to the peptide and not necessarily conserved across different peptides, even from the same class of RiPPs. The difference in the regioselectivity of hydrolysis between fuscimiditide and amycolimiditide may be explained by the presence of two proline residues within the loop of fuscimiditide that limit the conformations achievable by the fuscimiditide loop. The loop of amycolimiditde lacks proline and is expected to be more mobile than the fuscimiditide loop.
Like PIMT, some RiPP-associated O-methyltransferases have been shown to modify substrates containing L-isoAsp.19, 21 Thus, we compared the rates of in vitro aspartimidylation of pre-amycolimiditide and iso pre-amycolimiditide. Initially, though pre-amycolimiditide was methylated slightly faster, iso pre-amycolimiditide was aspartimidylated faster overall, suggesting a more efficient backbone cyclization for iso pre-amycolimiditide (Fig. 2D). On the other hand, the lanthipeptide methyltransferase OlvSA exhibits a much stronger preference for the substrate with isoAsp.21 Like fuscimiditide, the aspartimide of amycolimiditide hydrolyzes rapidly at basic pH and more slowly starting at neutral pH. In cellulo, we expect the aspartimidylated peptide to be the major product as long as SAM is available. If amycolimiditide is secreted into an environment that has a neutral pH or higher, the final disposition of this natural product will be its hydrolyzed forms, pre-amycolimiditide and iso pre-amycolimiditide.
NMR structure of amycolimiditide
After the preliminary analysis of amycolimiditide maturation, we used NMR spectroscopy to determine the residues forming the four ω-ester linkages and the aspartimide. TOCSY, NOESY, 1H-13C HSQC, and 1H-13C HMBC spectra of amycolimiditide were acquired in water at 20 °C (Fig. S19–23). Several proton chemical shifts are diagnostic of esterification or aspartimidylation.13, 27 First, we noticed that five pairs of β-protons of Asp produced widely different chemical shifts compared to the β-protons of other Asp residues. The chemical shifts of the two β-protons of D14, D21, D23, D25, and D29 differ from each other by 0.4–0.6 ppm, whereas those of D3 and D11 differ by less than 0.2 ppm (Fig. S24). Similarly, the chemical shifts of γ-protons of E28, adjacent to the side chain carbonyl carbon, differ only by 0.03 ppm (Fig. S24). On the other hand, the β-protons of T2, T6, T8, and T10 have chemical shifts ranging from 5.14-5.28 ppm, significantly downshifted in comparison to the average value of 4.17 ppm for Thr β-protons.28 Most NMR experiments done on graspetides in past studies also exhibit these chemical shift deviations for Asp, Thr, Ser β-protons, and Glu γ-protons (Table S7). Hence, T2, T6, T8, T10, D14, D21, D23, D25, and D29 are the best candidates for esterification in amycolimiditide.
Then, we examined the HMBC spectrum in search of more direct evidence for the ω-ester linkages and the aspartimide. First, we noticed that the chemical shifts of γ-carbons of D21, D23, D25, and D29 in amycolimiditide are noticeably upshifted to 168.1-169.9 ppm (Table S6). This phenomenon is observed in the ester carbons of fuscimiditide (169.0-169.5 ppm), marinomonasin (171.3 ppm), and microviridin 1777 (169.4-171.0 ppm), suggesting that D21, D23, D25, and D29 are esterified.19, 29–30 Furthermore, the HMBC correlations between the Thr β-proton and the Asp carbonyl carbon of T2 and D29, T6 and D25, T8 and D23, and T10 and D21 were observed, indicating these as pairs for the ω-ester linkages (Fig. 3A). This leaves D3, D11, and D14 as possible aspartimidylation sites, but several observations in the TOCSY and NOESY spectra point to D14 as the aspartimide site. Besides the widely different chemical shifts of D14 β-protons, G4 and G12 amide proton peaks are present, whereas the G15 amide proton peak is missing (Table S6). In the HMBC spectrum, the correlations between the D14 side chain carbonyl carbon and the G15 α-protons provide direct evidence for assigning D14 as the aspartimide site (Fig. 3B). To further verify D14 as the aspartimide, amdAB and amdABM expressions of the AmdA D14N variant resulted in four dehydrations, consistent with our attempt to eliminate the aspartimidylation site (Fig. S25).
Figure 3: NMR structure of amycolimiditide.
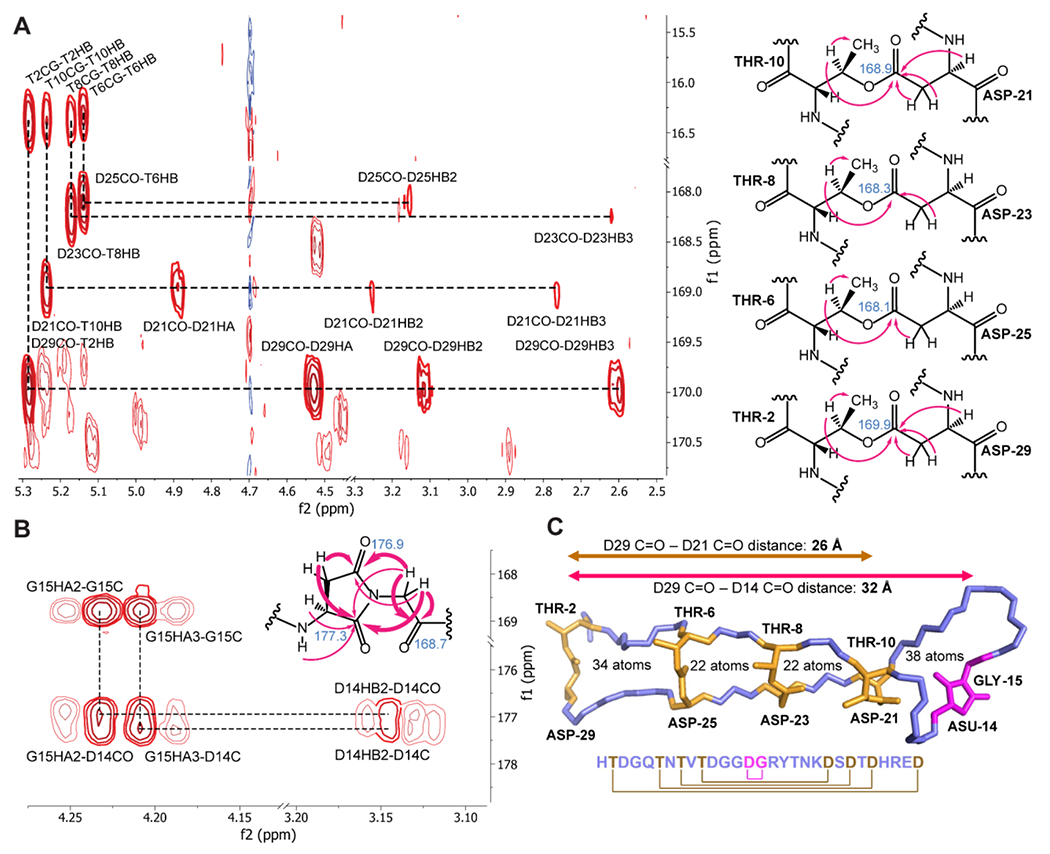
A: Left, portion of the 1H, 13C HMBC spectrum showing key correlations establishing the four ester linkages. Right, structure of the ester linkages with correlations present in the spectrum marked by arrows. The chemical shift of the carbonyl carbon in the ester is noted. B: Portion of the HMBC spectrum with correlations establishing the aspartimide moiety. The key correlations in bold are represented by bold arrows in the structure; normal arrows are additional HMBC correlations. C: Top structural model of amycolimiditide showing four ester linkages (gold) and the aspartimide moiety (magenta). Other side chains are omitted for clarity. The cartoon below the structure shows the connectivity in the peptide. The backbone of the ester-bonded ladder region of amycolimiditide is extended like beta-strands giving amycolimiditide a length in excess of 32 Å.
CYANA 2.131 and Avogadro32 were used to calculate the 20 lowest energy conformers of amycolimiditide, using the NOE correlations as distance constraints (Fig. S26, PDB: 8DYM). The model shows a stem-loop hairpin structure containing four fused macrocycles comprised of 38, 22, 22, and 34 backbone atoms moving from the loop macrocycle to the stem (Fig. 3C). The extended β-strand-like structure of the amycolimiditide stem is consistent with the vicinal amide-alpha proton 3JHNα coupling constants in this region, matching values expected for β-strands (Table S8).33 Additionally, the circular dichroism (CD) spectrum of amycolimiditide resembles the characteristic CD spectra of β-strands, further supporting the NMR structural model (Fig. S27). We note that the ~32 Å long, rigid amycolimiditide structure (Fig. 3C, Fig. S26) differs substantially from other graspetide structures characterized in the literature so far. While graspetides such as plesiocin and chryseoviridin feature multi-loop structures containing one or two ω-ester linkages per loop,17–18, 34 amycolimiditide forms a large single stem-loop containing four ω-ester linkages. This reinforces the ATP-grasp enzyme as an impressive family of enzymes that craft peptides into diverse shapes with ω-ester/amide crosslinks.
Order of ester linkage formation in amycolimiditide
We investigated the order in which AmdB forms the ω-ester linkages on AmdA by examining the linkages present in 1-, 2-, and 3-fold dehydrated pre-amycolimiditide intermediates. In the in vitro time course of AmdB modification of AmdA, 2 hours of reaction yielded the widest distribution of 1, 2, and 3-fold dehydrated intermediates (Fig. S6). To collect a large amount of all three intermediates, we scaled up this 2 h reaction with 200 nmol of AmdA (i.e. a 20 mL reaction), trypsinized the reaction mixture, and HPLC-purified the mixture of AmdA core peptide fragments with different levels of dehydration (Fig. S28).
While methanolysis is a popular method in graspetide studies to label Asp/Glu residues forming ω-ester crosslinks in a NaOMe/MeOH solvent,8 we found that amycolimiditide cannot be methanolyzed because it is insoluble in methanol. Instead, we devised an alternative reaction using hydrazine in an aqueous buffer, acidified to neutral pH to suppress ester hydrolysis which occurs at pH 11 (Fig. S9). In this reaction, we expect the Thr-Asp ω-esters to transform into Thr and aspartyl hydrazide (Ahz) residues, with the latter having a 14 Da increase over Asp residues (Fig. 4A). When the mixture of AmdA core peptides in different dehydration states were hydrazinolyzed, between 1-4 hydrazide adducts were observed in different species (Fig. 4B), consistent with the proposed hydrazinolysis reaction scheme (Fig. 4A). Then, we performed MS/MS experiments on the 1-3-fold hydrazinolyzed peptides, individually selecting each species as the parent ion for fragmentation. The +14 Da adducts were located at D21 on the 1-fold hydrazinolyzed peptide, D21 and D23 on the 2-fold hydrazinolyzed peptide, and D21, D23, and D25 on the 3-fold hydrazinolyzed peptide (Fig. 4C, Fig. S29). No hydrazide adduct was found on the free Asp and Glu residues. The results indicate that AmdB forms the loop first, installing ω-ester linkages down the stem from the T10-D21 linkage towards the T2-D29 linkage. Based on this order, we named the T10-D21 ester as I-ester, the T8-D23 ester as II-ester, the T6-D25 ester as III-ester, and the T2-D29 ester as IV-ester.
Figure 4: Hydrazinolysis maps the order of ester crosslink formation in pre-amycolimiditide.
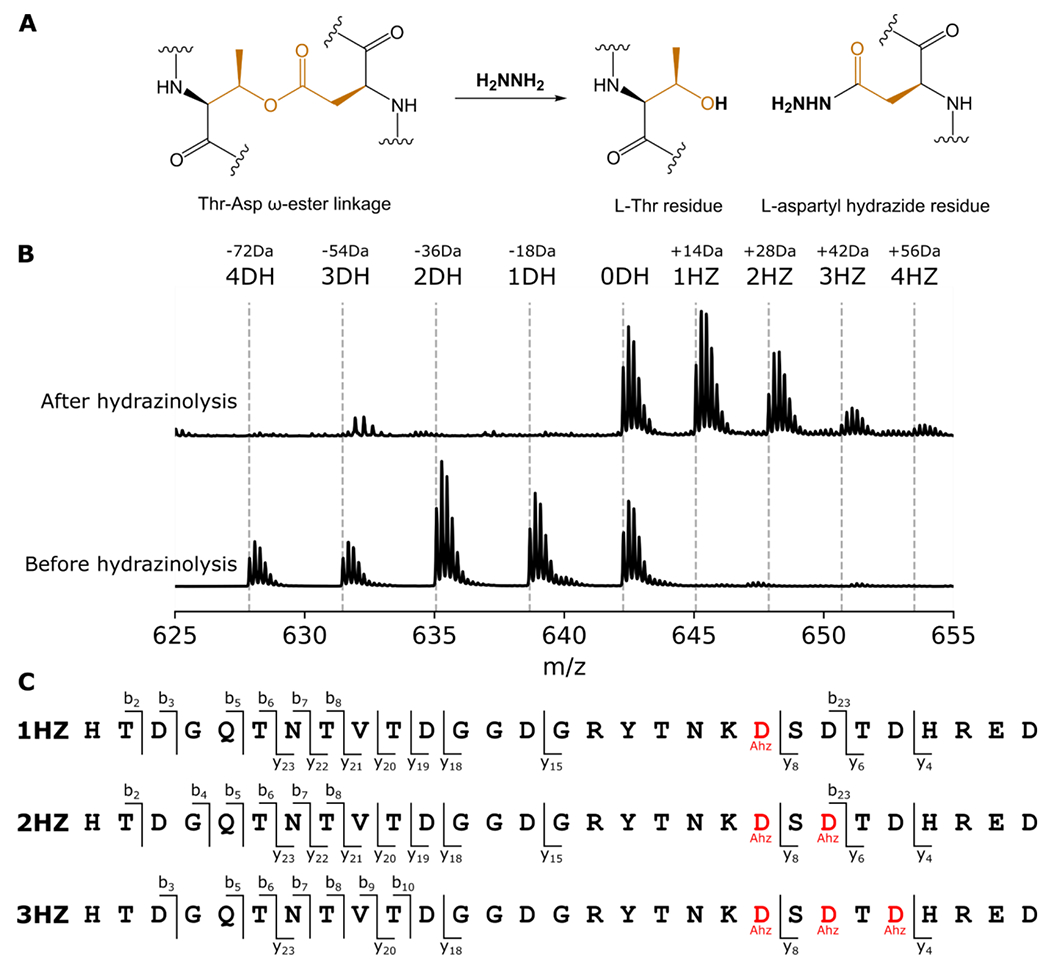
A: Mechanism of ester scission by hydrazine. The L-Thr residue is restored while the Asp residue is marked with a hydrazide. B: A mixture of intermediates of pre-amycolimiditide was generated in vitro and reacted with hydrazine. The mass spectrum shows a distribution of species in different dehydration states (bottom) and quantitative conversion to the hydrazide form (top). Note that the unmodified species (0DH) does not react with hydrazine, showing that the chemistry is specific toward ester linkages. C: Tandem mass spectrometry analysis of the hydrazine-labeled peptide mixture from part B. Hydrazide-labeled Asp residues are denoted as Ahz. In the singly labeled species (1HZ) only Asp21 is labeled with hydrazine whereas the doubly labeled species (2HZ) has Asp21 and Asp 23 labeled. The triply labeled 3HZ species is labeled at Asp21, Asp23, and Asp25, showing a specific order of ester formation. Mass spectra underlying this data are present in Fig. S29.
While AmdB installs the ester crosslinks unidirectionally, an open question is whether the earlier crosslinks must be present for the following ester linkages to be installed. As discussed above, the lanthipeptide maturase HalM2 installs four macrocyclic linkages on its substrate, with the enzyme adopting a different conformation for each intermediate state of the RiPP.12 Similarly, we hypothesized that if AmdB were to adopt a specific conformation when installing each ω-ester linkage of amycolimiditide, then the enzyme must strictly install the I-ester crosslink first and then progress unidirectionally towards the IV-ester crosslink. To answer this question, we generated AmdA variants with each ester crosslink blocked from forming. These variants contain Thr-to-Val and Asp-to-Asn substitutions at the location of each ester linkage: T10V D21N (I-null), T8V D23N (II-null), T6V D25N (III-null), and T2V D29N (IV-null). When co-expressed with AmdB, each variant was not modified, triply dehydrated, doubly dehydrated, and triply dehydrated, respectively (Fig. 5A). Similar co-expression experiments were conducted with AmdA variants that only have a single T-to-V or D-to-N substitution, but some variants designed to block the same ester linkage have different numbers of ester linkages installed by AmdB (Fig. S30). For example, AmdA T10V was mostly dehydrated once, whereas AmdA D21N was mostly dehydrated twice. This may be due to several Thr, Ser, Asp, and Glu residues placed near the four native ester linkages, possibly allowing for incorrect linkages. While others have widely used single amino acid substitutions to study other graspetides,15, 35 the double substitution method provides clearer evidence about the ester crosslinks in amycolimiditide by reducing the possibility of non-native linkages forming. To determine which ester crosslinks are present on the doubly substituted variants, the modified core peptide of each variant was HPLC-purified (Fig. S31) and subjected to hydrazinolysis followed by MS/MS analysis (Fig. S32–34). The I-null variant is incapable of forming any esters while the II-null variant is still able to form the I-, III-, and IV-esters. The I- and II-esters are present in the III-null variant, and I-, II-, and III-esters are present in the IV-null variant (Fig. 5A). These results show that the I-ester must be formed first before any subsequent ester installation. In general, the preceding ester must be present for any further esters to form with the exception of the II-ester, which is dispensable for III-ester formation.
Figure 5: Ester and aspartimide formation in amycolimiditide variants.
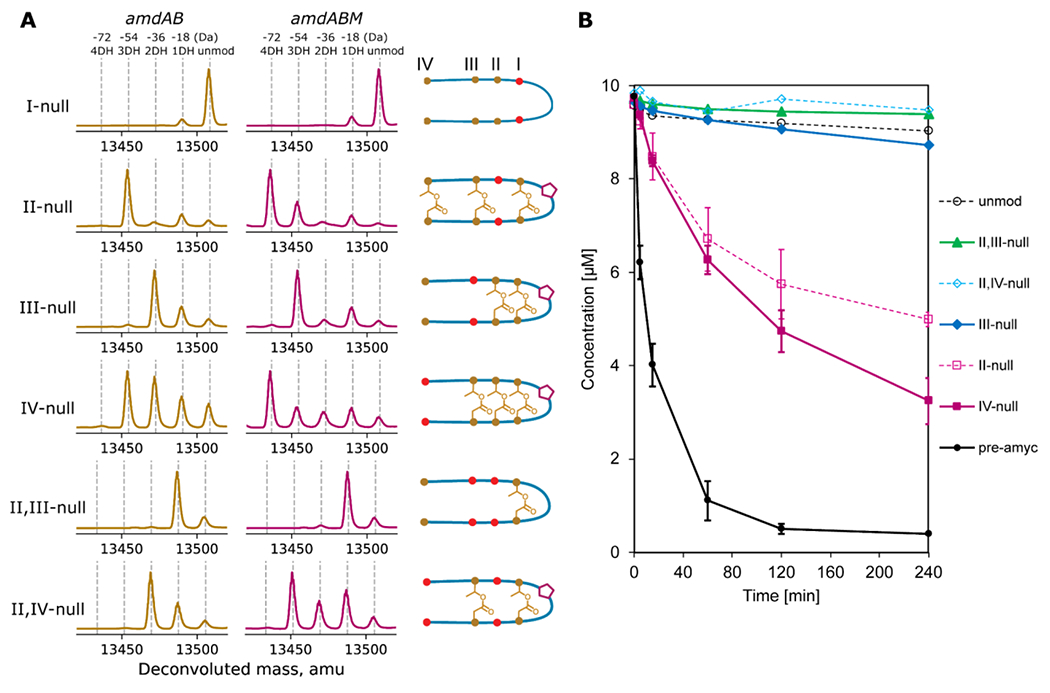
A: Variants of amycolimiditide with different esters disrupted were coexpressed with either AmdB or AmdB and AmdM. The notation I-null indicates that both residues involved in forming the I-ester (T10 and D21) have been substituted with isosteric but non-reactive residues (T10V and D21N). The positions of these substitutions are denoted by red dots in the cartoon, which represent the most probable structure of the variants. The deconvoluted mass spectra in the amdAB column show the number of dehydrations upon coexpression with AmdB while the cartoons for each variant show the position of the esters as determined by hydrazinolysis (Fig. S31–35). The mass spectra in the amdABM column show the number of dehydrations for each of the variants upon coexpression with both AmdB and AmdM. The cartoons also show whether the variants are aspartimidylated by AmdM (pentagon) B: Time course of in vitro aspartimidylation of pre-amycolimiditide (pre-amyc), unmodified core peptide (unmod), and variants. Disruption of any esters, even a distal one like in the IV-null case, has a large negative effect on the rate of aspartimidylation. The experiments on pre-amyc, II-null, and IV-null were repeated in triplicate and error bars reflect the standard deviation.
AmdM activity on pre-amycolimiditide variants
Next, we turned our attention to AmdM activity on pre-amycolimiditide variants with missing ω-ester linkages, determining their importance for aspartimidylation. We generated two additional variants: AmdA II,III-null variant that contains the I-ester linkage and AmdA II,IV-null variant that contains the I- and III-ester linkages (Fig. 5A, S31, S35–36). First, we co-expressed AmdA variants with AmdB and AmdM and examined the number of dehydrations. Only the variants with two or more ester linkages can be aspartimidylated in vivo (Fig. 5A). Surprisingly, the presence of I-ester alone is insufficient for aspartimidylation, despite forming the loop containing the aspartimidylation site, D14. This suggests that the other ester linkages more distal from the AmdM active site also contribute towards efficient aspartimidylation of pre-amycolimiditide. On the other hand, in the in vitro aspartimidylation experiment, only the core peptides with at least three ester crosslinks were aspartimidylated (Fig. 5B). We suspect that the variants with two ester linkages were not aspartimidylated in vitro, possibly due to a lower enzyme-to-substrate ratio in the reaction than in the heterologous expression experiments. Also, the heterologous expression experiments were carried out for 24 h while we stopped the in vitro experiment after 4 h. We repeated the in vitro aspartimidylation reactions at higher enzyme concentrations and observed turnover of substrates with two esters as well, more consistent with the heterologous expression experiments (Fig. S37). These experiments show that the number of ω-ester linkages drastically affects the methylation rate. The initial rates of methylation for the II-null variant, the IV-null variant, and pre-amycolimiditide are 4.4 ± 2.5 µM h−1, 4.3 ± 1.6 µM h−1, and 42.6 ± 4.3 µM h−1, respectively, indicating that the removal of even just one ester linkage leads to about 10-fold slower reaction. Combined with the expression results, this time course study shows that all ester linkages serve as critical scaffolds for substrate recognition by the graspetide methyltransferase, despite being far from its active site.
Aspartimidylation of loop-modified core peptide variants
We further explored the effect of other modifications to the loop of pre-amycolimiditide on their methylation and dehydration. First, we examined the impact of substituting the residue Gly-15, immediately C-terminal to the aspartimidylation site, with a different small residue. In the cases of the aspartimidylated lasso peptides cellulonodin-2 and lihuanodin, the peptides are still aspartimidylated with different residues C-terminal to the aspartimidylation site.20 For amycolimiditide, we tested Ala, Ser, and Thr substitutions of Gly-15 and compared the number of dehydrations after amdAB and amdABM expressions. Only very small amounts of each of these variants were aspartimidylated by AmdM demonstrating that Asp-14 must be followed by Gly-15 for an efficient aspartimidylation by AmdM (Fig. S38).
Then, we modified the size of the loop, inserting a Gly residue before or after D14 (which we named as the +G14, +G16, and +G14+G16 variants). Co-expression with AmdB and AmdM resulted in four dehydrations on the +G14 and +G14+G16 variants and five dehydrations on the +G16 variant (Fig. S39). We surmise that the position of D14 is important for aspartimidylation and that the +G14 variant and +G14+G16 variant cannot be aspartimidylated because the Gly insertion before D14 offsets the aspartimidylation site away from the AmdM active site. These results show that AmdM can still recognize a longer 11 aa loop, as long as D14 remains as the 14th residue of the peptide. This experiment also provides some insight into the substrate specificity of AmdB, namely that this enzyme can remarkably tolerate and efficiently esterify substrates with loop sizes of 10 (the native substrate), 11, or 12 aa.
Finally, we modified the placement of D14-G15–the aspartimidylation site–in the loop. We tested three variants: G12D D14G, G13D D14G, and D14R G15D R16G variants corresponding to the hexapeptide sequences spanned by 11th–16th residues, D[DG]GGR, DG[DG]GR, and DGGR[DG], respectively, as compared to the wild-type sequence DGG[DG]R. Co-expression with AmdB and AmdM resulted in four dehydrations for all variants, showing that AmdM cannot methylate them (Fig. S40). This further demonstrates that the position of the aspartimidylation site is immutable.
Substitution of ω-ester linkages with disulfide bonds
We questioned whether a different crosslink could replace the ω-ester crosslinks and complement their roles in subsequent esterification and aspartimidylation. We decided to use disulfide bonds to replace ester linkages since they are the most common crosslinks in proteins and only require simple mutagenesis to generate them. Additionally, both Thr-Asp ω-ester linkages and Cys-Cys disulfide linkages are comprised of four atoms, only yielding a small structural perturbation. We examined the following variants: T10C D21C (I-disulfide), T8C D23C (II-disulfide), T6C D25C (III-disulfide), and T2C D29C (IV-disulfide).
First, we co-expressed the variants with AmdB. The results resembled those exhibited by the Thr and Asp-substituted null variants (Fig. S41), consistent with a lack of disulfide bond formation in the reducing cytoplasm of E. coli. Next, we attempted the reaction in vitro using purified II-disulfide and III-disulfide variants with dithiothreitol (DTT) added to disrupt disulfide bonds. We digested the reaction products with trypsin and observed that the masses of the core peptides were increased by 2 amu when DTT was added to the reaction as expected (Fig. S42). This shows that we could successfully control the presence of the disulfide bonds with DTT. Strikingly, the ω-ester linkages were formed only when DTT was added to the reaction (Fig. 6A). The disulfide linkages fail to complement the missing ω-ester linkages and interfere with the formation of other ester linkages. This finding suggests that the core peptide region of AmdA does not pre-fold into a well-defined loop before reaching the binding site or the active site of AmdB.
Figure 6: Ester and aspartimide formation in disulfide-substituted amycolimiditide variants.
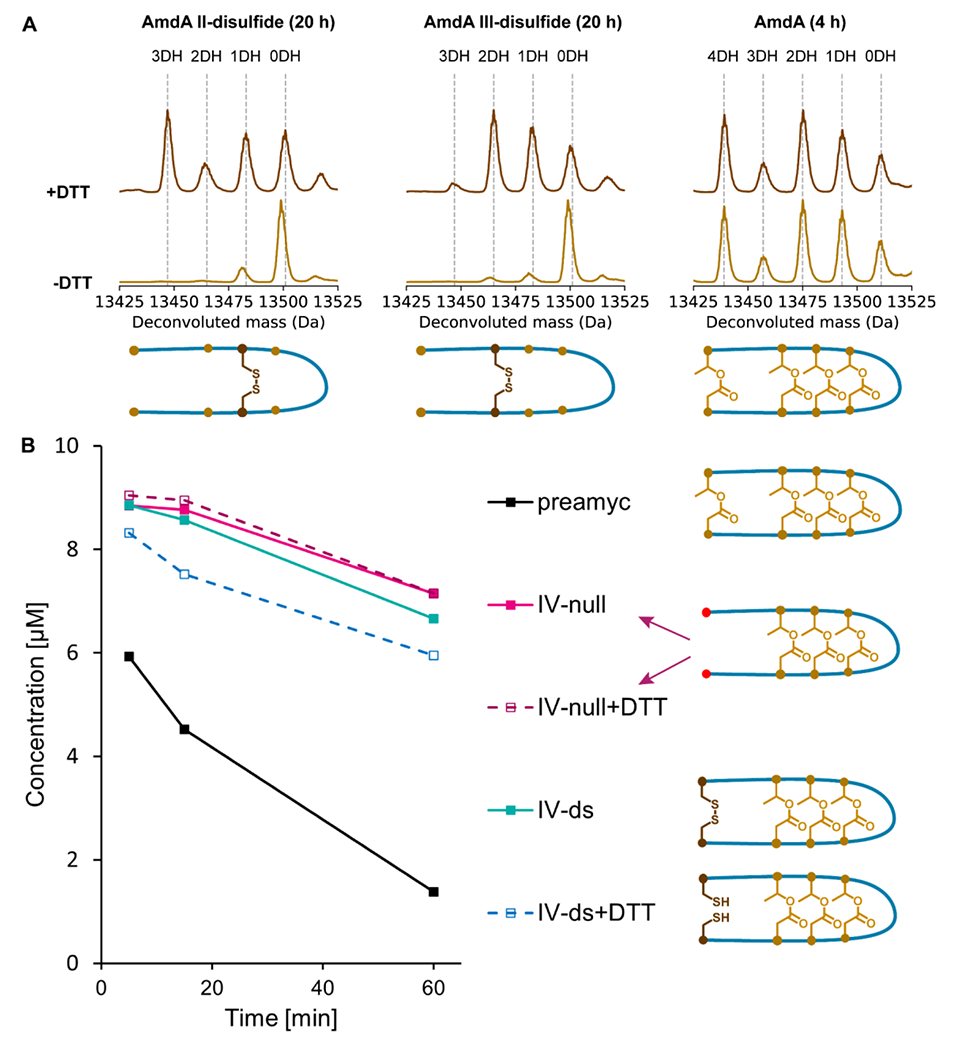
A: Esterification of disulfide-substituted AmdA variants in vitro by AmdB for 20 h at RT. The presence of a disulfide bond that constrains the peptide (-DTT spectra) inhibits esterification whereas reduced, unconstrained substrates are esterified efficiently. A reaction with wild-type AmdA (4 h at RT) is shown as a control. B: Time course of in vitro as partimidylation of pre-amycolimiditide variants with and without disulfide bonds by AmdM. All reactions were initiated with 10 μM peptide, but a mass spectrum was not acquired at the 0 min timepoint. The presence of disulfide bonds has minimal effect on the rate of methylation/aspartimidylation.
We then examined whether a disulfide linkage could complement a missing ω-ester linkage for aspartimidylation. Since we already quantified the effect of the loss of the IV-ester linkage on the methylation rate (Fig. 5B), we decided to analyze the methylation rates of the IV-disulfide variant. The methylation rates of the IV-disulfide and the IV-null variants were similar, indicating that the IV-disulfide linkage cannot complement the missing IV-ester linkage for aspartimidylation (Fig. 6B). Overall, the experiments on the disulfide variants demonstrate that both AmdB and AmdM recognize the substrates crosslinked with ω-ester linkages precisely, not just any type of covalent linkage.
Conclusion
In this paper we have described the biosynthesis and structure of amycolimiditide, a new example of an aspartimidylated graspetide. This peptide is named after its putative producing organism, A. cihanbeyliensis, but close homologs to amycolimiditide are present in additional actinobacteria, primarily in the Pseudonocardiaceae and Micromonosporaceae families (Table S1). Though A. cihanbeyliensis is terrestrial bacterium, it is noteworthy that graspetides similar to amycolimiditide are encoded in the genomes of several Salinospora strains, obligate marine bacteria. The structure of amycolimiditide differs from that of any previously described graspetides; its hairpin structure is crosslinked with four ω-esters forming a >2 nm-long rigid stem attached to a 10 aa loop (Fig. 1B, Fig. 3C). This 10 aa loop is the site of a single aspartimide moiety. Amycolimiditide joins the graspetide fuscimiditide and the lasso peptides cellulonodin-2 and lihuanodin as examples of RiPPs that include stable aspartimide moieties that are intentionally installed via enzymatic activity.19–20 The lanthipeptide OlvA(BCSA) also exists in an aspartimidylated form, but appears to hydrolyze more readily than the graspetide and lasso peptide examples.21 Mapping the linkages in amycolimiditide (four esters and one aspartimide) is challenging due to the high density of different residues in the core peptide that can participate in these linkages (Fig. 1F). Determination of these linkages was facilitated in large part by a high quality HMBC spectrum (Fig. S23). Mass spectrometry is often used to determine the linkages in graspetides,8 but for a structure like amycolimiditide with five different PTMs, the NMR experiment provides more direct evidence.
Studying the biosynthesis of amycolimiditide has offered fresh insights into the catalytic mechanism of ATP-grasp enzymes. Like other graspetides,8, 18 amycolimiditide is synthesized starting at its loop and proceeding down the stem (Fig. 7A). We developed a hydrazinolysis protocol to label the Asp residues present in the ω-esters, which helped to elucidate the order of ester formation (Fig. 4). The hydrazinolysis proceeds in aqueous conditions, and thus is a suitable alternative to the more commonly used methanolysis procedure17 when the substrate peptide has poor solubility in methanol like amycolimiditide. Consistent with the strict ordering of ester formation in amycolimiditide, disruption of the ester by point aa substitutions generally prevents the installation of subsequent esters (Fig. 5A), also observed in microviridins.15 The exception to this rule is that the II-ester can be disrupted while still allowing the formation of the III- and IV-esters (Fig. 5A). More intriguing is our result that prestabilization of the amycolimiditide precursor with disulfide bond is deleterious for ester formation (Fig. 6A). These results suggest one possible model in which the core portion of the amycolimiditide precursor substrate must retain a flexible, random coil structure (Fig. S27) in order to be efficiently recognized and modified by the ATP-grasp enzyme (Fig. 7A). The strict order of ester introduction combined with the inability of “preconstraining” the substrate illustrate that there are likely structural prerequisites in both the substrate and the enzyme before catalysis can occur to form each ester. This model echoes the biosynthesis of the haloduracin β chain discussed in the introduction.
Figure 7: Summary of amycolimiditide biosynthesis. A: Model of amycolimiditide biosynthesis.
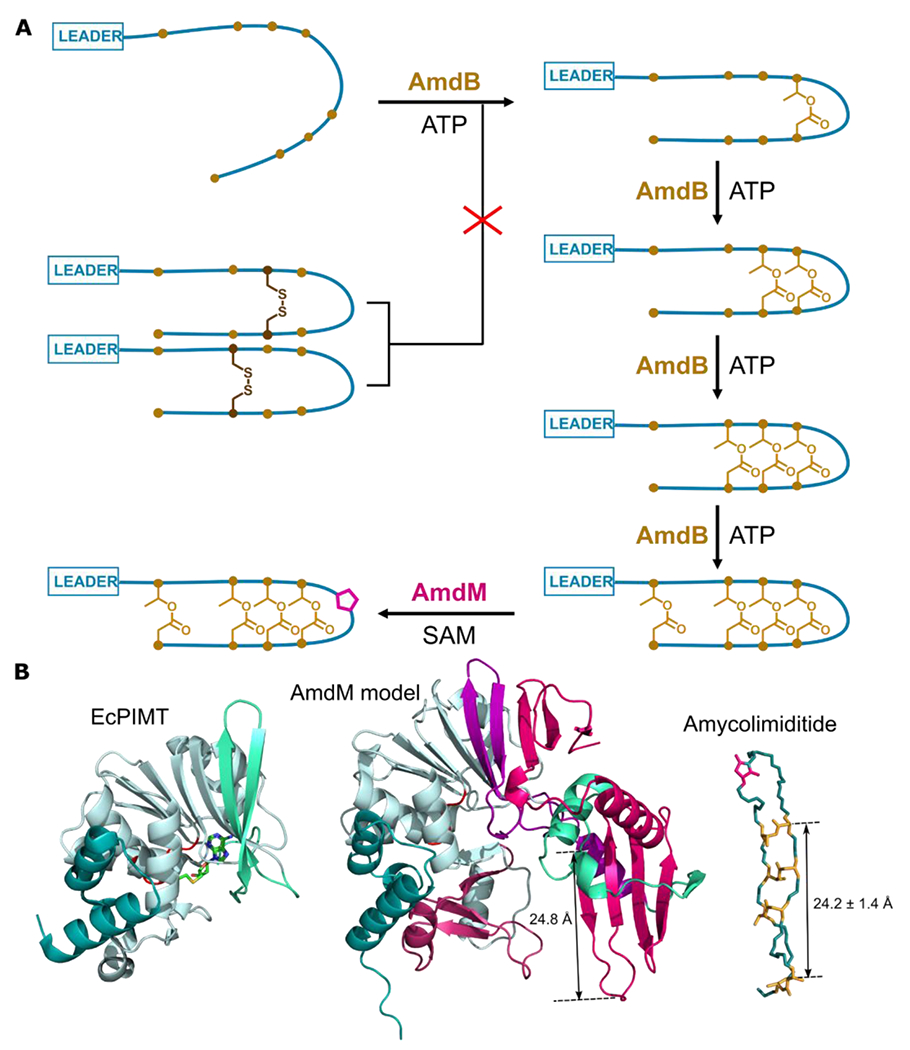
The esters in the amycolimiditide form prior to aspartimidylation. Preconstraining the substrate with disulfide bonds prevents ester formation, suggesting that the substrate must be flexible in order to be recognized by the ATP-grasp enzyme AmdB. The esters are installed in an ordered fashion, starting from the loop and proceeding down the stem. The four-fold esterified product is most efficiently recognized and methylated by the O-methyltransferase AmdM to give the final aspartimidylated graspetide. B: Comparison of E. coli PIMT (EcPIMT, PDB code 3LBF, left) to AlphaFold model of AmdM (middle). Homologous sequence segments are colored the same; see also Fig. S43 for an alignment of these two proteins. A molecule of S-adenosyl homocysteine (SAH) is bound in the methylation active site of EcPIMT. The model of AmdM includes a large C-terminal extension relative to EcPIMT (magenta) that generates a potential substrate binding cleft within the enzyme. The length of a β-strand that may act as a substrate recognition site matches closely the length of amycolimiditide shown on the right.
Amycolimiditide joins a short list of RiPP natural products harboring stable aspartimide moieties,19–20 and our results here provide some new insights into the cascade of methylation and aspartimidylation. By examining pre-amycolimiditide variants with differing numbers of ester linkages, we show that the fully esterified pre-amycolimiditide is by far the best substrate for aspartimidylation (Fig. 5B). This means that the IV-ester at the end of the stem, which is ~30 Å away from the aspartimidylation site in the loop, still influences methyl transfer by AmdM. An emerging picture for O-methyltransferases that aspartimidylate RiPPs is that they require highly specific, constrained structures. The methyltransferases that install aspartimides on lasso peptides only function on lassoed substrates, not on linear or even macrocyclic substrates. In the case of pre-amycolimiditide, the entire stem of the peptide with all four ester linkages is required for the most efficient methyl transfer. Moreover, replacing the ester constraints with another isosteric constraint, the disulfide bond, leads to a loss in catalytic efficiency in the methyltransferase reaction, underscoring the remarkable specificity of the AmdM enzyme. This specificity suggests that perhaps AmdM recognizes the esters via hydrogen bonding which is not possible with the disulfide linkages. Another possibility is that the esters establish a unique extended stem structure with β-strand character, and the enzyme specifically recognizes those β-strands. Perhaps the disulfide bonded substrates have a 3D structure that differs from the ester-linked structure. To attempt to understand the nature of the substrate specificity of AmdM, we generated an Alphafold36 model of AmdM using ColabFold.37 Comparing the AmdM model to E. coli PIMT (Fig. 7B), the conserved methyltransferase fold is clearly present within the N-terminus of AmdM, but the C-terminal portion of AmdM folds into an additional domain. There is a large cleft between these two domains, making it tempting to speculate that the cleft serves to dock pre-amycolimiditide. In particular, there is a four-strand antiparallel β-sheet in the AmdM model that is roughly the same length as the amycolimiditide stem. Engagement between the full stem of pre-amycolimiditide and this β-sheet may explain why fully esterified pre-amycolimiditide is the preferred substrate for AmdM (Fig. 5B, 6B). Another example of the specificity of the AmdM methyltransferase is that it will only methylate one specific DG dipeptide, and only if the Asp is located in one specific position within the loop, 4 aa away from the Thr in the I-ester (Fig. S40). AmdM does however exhibit some promiscuity with regards to loop size; AmdM recognizes and modifies a substrate with an 11 aa loop instead of the native 10 aa loop (Fig. S39). There is still much sequence space to be explored to define the substrate scope of both AmdB and AmdM, but the amycolimiditide loop appears to be readily engineerable.
Supplementary Material
Acknowledgements
We thank I. Pelczer (Princeton University NMR Facility) for help with acquiring NMR spectra and V.G. Vandavasi (Princeton University Biophysics Core Facility) for help with acquiring CD spectra. We thank John Dueber (UC Berkeley) for sharing the pYTK96 plasmid. This work was supported by the National Institutes of Health Grant GM107036 and a grant from Princeton University School of Engineering and Applied Sciences (Focused Research Team on Precision Antibiotics and Innovation Funds). L.C. was supported by an NSF Graduate Research Fellowship Program under Grant DGE-1656466.
Footnotes
Supporting information: Detailed explanations of materials and methodology used, refactoring the biosynthetic gene cluster of amycolimiditide for heterologous expression, and 43 supplementary figures and 16 supplementary tables, mostly showing HPLC, LC-MS(/MS), and NMR data (PDF).
References
- 1.Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian K-D; Fischbach MA; Garavelli JS; Goransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Muller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Sussmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA, Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montalban-Lopez M; Scott TA; Ramesh S; Rahman IR; van Heel AJ; Viel JH; Bandarian V; Dittmann E; Genilloud O; Goto Y; Burgos MJG; Hill C; Kim S; Koehnke J; Latham JA; Link AJ; Martinez B; Nair SK; Nicolet Y; Rebuffat S; Sahl HG; Sareen D; Schmidt EW; Schmitt L; Severinov K; Sussmuth RD; Truman AW; Wang H; Weng JK; van Wezel GP; Zhang Q; Zhong J; Piel J; Mitchell DA; Kuipers OP; van der Donk WA, New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep 2021, 38, 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman MF; Gurgui C; Helf MJ; Morinaka BI; Uria AR; Oldham NJ; Sahl HG; Matsunaga S; Piel J, Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides. Science 2012, 338, 387–390. [DOI] [PubMed] [Google Scholar]
- 4.Sahl HG; Bierbaum G, Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol 1998, 52, 41–79. [DOI] [PubMed] [Google Scholar]
- 5.Kawulka K; Sprules T; McKay RT; Mercier P; Diaper CM; Zuber P; Vederas JC, Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to alpha-carbons of phenylalanine and threonine. J. Am. Chem. Soc 2003, 125, 4726–4727. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen TQN; Tooh YW; Sugiyama R; Nguyen TPD; Purushothaman M; Leow L; Hanif K; Yong RHS; Agatha I; Winnerdy FR; Gugger M; Phan AT; Morinaka BI, Post-translational formation of strained cyclophanes in bacteria. Nature Chemistry 2020, 12, 1042-+. [DOI] [PubMed] [Google Scholar]
- 7.Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma XY; Guo JJ; Makriyannis A; Linares-Otoya L; Boehringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schaberle TF; D’Onofrio A; Lewis K, A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H; Choi M; Park JU; Roh H; Kim S, Genome Mining Reveals High Topological Diversity of omega-Ester-Containing Peptides and Divergent Evolution of ATP-Grasp Macrocyclases. J. Am. Chem. Soc 2020, 142, 3013–3023. [DOI] [PubMed] [Google Scholar]
- 9.Lawton EM; Cotter PD; Hill C; Ross RP, Identification of a novel two-peptide lantibiotic, Haloduracin, produced by the alkaliphile Bacillus halodurans C-125. FEMS Microbiol. Lett 2007, 267, 64–71. [DOI] [PubMed] [Google Scholar]
- 10.McClerren AL; Cooper LE; Quan C; Thomas PM; Kelleher NL; van der Donk WA, Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 17243–17248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uggowitzer KA; Habibi Y; Wei WL; Moitessier N; Thibodeaux CJ, Mutations in Dynamic Structural Elements Alter the Kinetics and Fidelity of the Multifunctional Class II Lanthipeptide Synthetase, HalM2. Biochemistry 2021, 60, 412–430. [DOI] [PubMed] [Google Scholar]
- 12.Habibi Y; Weerasinghe NW; Uggowitzer KA; Thibodeaux CJ, Partially Modified Peptide Intermediates in Lanthipeptide Biosynthesis Alter the Structure and Dynamics of a Lanthipeptide Synthetase. J. Am. Chem. Soc 2022, 144, 10230–10240. [DOI] [PubMed] [Google Scholar]
- 13.Ishitsuka MO; Kusumi T; Kakisawa H; Kaya K; Watanabe MM, Microviridin: A Novel Tricyclic Depsipeptide from the Toxic Cyanobacterium Microcystis viridis. J. Am. Chem. Soc 1990, 112, 8180–8182. [Google Scholar]
- 14.Ziemert N; Ishida K; Liaimer A; Hertweck C; Dittmann E, Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angew. Chem.-Int. Edit 2008, 47, 7756–7759. [DOI] [PubMed] [Google Scholar]
- 15.Philmus B; Guerrette JP; Hemscheidt TK, Substrate Specificity and Scope of MvdD, a GRASP-like Ligase from the Microviridin Biosynthetic Gene Cluster. ACS Chemical Biology 2009, 4, 429–434. [DOI] [PubMed] [Google Scholar]
- 16.Rohrlack T; Christoffersen K; Hansen PE; Zhang W; Czarnecki O; Henning M; Fastner J; Erhard M; Neilan BA; Kaebernick M, Isolation, characterization, and quantitative analysis of microviridin J, a new Microcystis metabolite toxic to Daphnia. J. Chem. Ecol 2003, 29, 1757–1770. [DOI] [PubMed] [Google Scholar]
- 17.Lee H; Park Y; Kim S, Enzymatic Cross-Linking of Side Chains Generates a Modified Peptide with Four Hairpin-like Bicyclic Repeats. Biochemistry 2017, 56, 4927–4930. [DOI] [PubMed] [Google Scholar]
- 18.Roh H; Han Y; Lee H; Kim S, A Topologically Distinct Modified Peptide with Multiple Bicyclic Core Motifs Expands the Diversity of Microviridin-Like Peptides. Chembiochem 2019, 20, 1051–1059. [DOI] [PubMed] [Google Scholar]
- 19.Elashal HE; Koos JD; Cheung-Lee WL; Choi B; Cao L; Richardson MA; White HL; Link AJ, Biosynthesis and characterization of fuscimiditide, an aspartimidylated graspetide. Nature Chemistry 2022, accepted, 10.1038/s41557-022-01022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao L; Beiser M; Koos JD; Orlova M; Elashal HE; Schroder HV; Link AJ, Cellulonodin-2 and Lihuanodin: Lasso Peptides with an Aspartimide Post-Translational Modification. J. Am. Chem. Soc 2021, 143, 11690–11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acedo JZ; Bothwell IR; An LN; Trouth A; Frazier C; van der Donk WA, O-Methyltransferase-Mediated Incorporation of a beta-Amino Acid in Lanthipeptides. J. Am. Chem. Soc 2019, 141, 16790–16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffith SC; Sawaya MR; Boutz DR; Thapar N; Katz JE; Clarke S; Yeates TO, Crystal structure of a protein repair methyltransferase from Pyrococcus furiosus with its L-isoaspartyl peptide substrate. J. Mol. Biol 2001, 313, 1103–1116. [DOI] [PubMed] [Google Scholar]
- 23.Tatar D; Sazak A; Guven K; Cetin D; Sahin N, Amycolatopsis cihanbeyliensis sp nov., a halotolerant actinomycete isolated from a salt mine. International Journal of Systematic and Evolutionary Microbiology 2013, 63, 3739–3743. [DOI] [PubMed] [Google Scholar]
- 24.Johnson BA; Murray ED; Clarke S; Glass DB; Aswad DW, Protein Carboxyl Methyltransferase Facilitates Conversion of Atypical L-Isoaspartyl Peptides to Normal L-Aspartyl Peptides. J. Biol. Chem 1987, 262, 5622–5629. [PubMed] [Google Scholar]
- 25.Geiger T; Clarke S, Deamidation, Isomerization, and Racemization at Asparaginyl and Aspartyl Residues in Peptides. J. Biol. Chem 1987, 262, 785–794. [PubMed] [Google Scholar]
- 26.Maclaren DC; Clarke S, Expression and Purification of a Human Recombinant Methyltransferase That Repairs Damaged Proteins. Protein Expr. Purif 1995, 6, 99–108. [DOI] [PubMed] [Google Scholar]
- 27.Grassi L; Regl C; Wildner S; Gadermaier G; Huber CG; Cabrele C; Schubert M, Complete NMR Assignment of Succinimide and Its Detection and Quantification in Peptides and Intact Proteins. Anal. Chem 2017, 89, 11962–11970. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Wenger RK; Yao HY; Markley JL, BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaweewan I; Nakagawa H; Kodani S, Heterologous expression of a cryptic gene cluster from Marinomonas fungiae affords a novel tricyclic peptide marinomonasin. Appl. Microbiol. Biotechnol 2021, 105, 7241–7250. [DOI] [PubMed] [Google Scholar]
- 30.Sieber S; Grendelmeier SM; Harris LA; Mitchell DA; Gademann K, Microviridin 1777: A Toxic Chymotrypsin Inhibitor Discovered by a Metabologenomic Approach. J. Nat. Prod 2020, 83, 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guntert P, Automated NMR structure calculation with CYANA. Methods Mol. Biol 2004, 278, 353–378. [DOI] [PubMed] [Google Scholar]
- 32.Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pardi A; Billeter M; Wuthrich K, Calibration of the angular dependence of the amide proton-C alpha proton coupling constants, 3JHN alpha, in a globular protein. Use of 3JHN alpha for identification of helical secondary structure. J. Mol. Biol 1984, 180, 741–751. [DOI] [PubMed] [Google Scholar]
- 34.Zhao GX; Kosek D; Liu HB; Ohlemacher SI; Blackburne B; Nikolskaya A; Makarova KS; Sun JD; Barry CE; Koonin EV; Dyda F; Bewley CA, Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic omega-Ester Macrocycles in a New Multicore RiPP Natural Product. J. Am. Chem. Soc 2021, 143, 8056–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramesh S; Guo X; DiCaprio AJ; De Lio AM; Harris LA; Kille BL; Pogorelov TV; Mitchell DA, Bioinformatics-Guided Expansion and Discovery of Graspetides. ACS Chemical Biology 2021, 16, 2787–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Zidek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirdita M; Schutze K; Moriwaki Y; Heo L; Ovchinnikov S; Steinegger M, ColabFold: making protein folding accessible to all. Nature Methods 2022, 19, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.