

Abstract
Neutrophil extracellular traps (NETs) occur when chromatin is decondensed and extruded from the cell, generating a web-like structure. NETs have been implicated in the pathogenesis of several sterile disease states and thus are a potential therapeutic target. Various pathways have been shown to induce NETs, including autophagy, with several key enzymes being activated like peptidyl arginine deiminase 4 (PAD4), an enzyme responsible for citrullination of histones, allowing for DNA unwinding and subsequent release from the cell. Pre-clinical studies have already demonstrated that chloroquine (CQ) and hydroxychloroquine (HCQ) are able to reduce NETs and slow disease progression. The exact mechanism as to how these drugs reduce NETs has yet to be elucidated. CQ and HCQ decrease NET formation from various NET activators, independent of their autophagy inhibitory function. CQ and HCQ were found to inhibit PAD4 exclusively, in a dose-dependent manner, confirmed with reduced CitH3+ NETs after CQ or HCQ treatment. Circulating CitH3 levels were reduced in pancreatic cancer patients after HCQ treatment. In silico screening of PAD4 protein structure identified a likely binding site interaction at Arg639 for CQ and Trp347, Ser468, and Glu580 for HCQ. SPR analysis confirmed the binding of HCQ and CQ with PAD4 with KD values of 54.1 µM (CQ) and 88.1 µM (HCQ). This data provide evidence of direct PAD4 inhibition as a mechanism for CQ/HCQ inhibition of NETs. We propose that these drugs likely reduce NET formation through multiple mechanisms; the previously established TLR9 and autophagy inhibitory mechanism and the novel PAD4 inhibitory mechanism.
Keywords: neutrophil extracellular traps, pancreatic adenocarcinoma, hydroxychloroquine, citrullinated histone H3
Proposed mechanisms of neutrophil extracellular trap (NET) inhibition by CQ and HCQ. Chloroquine (CQ) and hydroxychloroquine (HCQ) inhibit NET formation through direct inhibition of PAD4, which is responsible for unwinding and release of DNA from neutrophils. When considered with regard to the established function of CQ/HCQ in autophagy inhibition and of the role of autophagy in NETs, we suggest two independent mechanisms for NET inhibition by these drugs, through blockade of autophagy or inhibition of PAD4.
Graphical Abstract
Graphical Abstract.

Introduction
Neutrophil extracellular traps (NETs) are a function of neutrophils involving the extrusion of decondensed chromatin with associated proteins to form a lattice-like structure. Originally, NETs were described as a means to capture and kill bacteria, preventing dissemination in the setting of infection and sepsis [1]. During the process of NET formation, inflammatory stimuli result in neutrophil activation, leading to the release of several key enzymes, including neutrophil elastase and myeloperoxidase, which result in cytosolic and nuclear membrane degradation. Another key enzyme is peptide arginine deiminase 4 (PAD4), which is responsible for converting arginine to citrulline on histones, allowing for chromatin to unwind [2]. Once the nuclear membrane is dissolved and the chromatin decondensed, the neutrophil bursts, forming the lattice structure with chromatin and associated proteins. Since their discovery, NETs have been further implicated in several different sterile inflammatory conditions including autoimmunity, sepsis, atherosclerosis, and cancer [3–6]. Due to their expanding role in the pathogenesis of several different diseases, NETs have become promising new therapeutic targets.
Chloroquine (CQ) was discovered in the 1950s and was originally used as malarial prophylaxis and treatment. Hydroxychloroquine (HCQ) was formulated soon after as a safer alternative with less adverse side effects for patients [7–9]. Since their use as anti-malarial drugs, chloroquine and hydroxychloroquine have been discovered to have therapeutic potential in numerous diseases, including lupus, rheumatoid arthritis, pancreatitis, and cancer [10–12]. These beneficial effects are thought to be driven largely by the ability of the drugs to inhibit autophagy, a metabolic recycling mechanism that allows for cell survival during conditions of cellular stress [13–16]. Importantly, chloroquine and hydroxychloroquine reduce NET markers in several diseases including pancreatitis and cancer [11, 17]. The exact mechanism(s) as to how chloroquine and hydroxychloroquine mediate their NET inhibitory function is unknown. We set out to evaluate the effectiveness of these drugs inhibiting NETs in both murine models and patients and generate a better mechanistic understanding of how CQ and HCQ block NET formation.
Methods and materials
Chemical reagents and enzymatic assays
Chloroquine, Hydroxychloroquine, and Bafilomycin A1 were obtained from Sigma Aldrich. LPS was obtained from Novus Biologicals and reconstituted at 1 mg/ml. Platelet-activating factor (PAF) was obtained from Fisher. PAD4, PAD2, and myeloperoxidase (MPO) inhibition kits were purchased from Cayman Chemical. Neutrophil Elastase (NE) inhibition kit was purchased from Sigma Aldrich. All assays were performed according to the manufacturer’s protocol.
Ex vivo NET assay
Murine neutrophils were extracted from healthy C57BL/6J mouse bone marrow and isolated using density gradient centrifugation as previously reported [17]. Briefly, bone marrow was collected and cells were washed with RPMI-10 with 10% FBS and 1% Penicillin/Streptomycin. The cells were collected after density centrifugation using histiopaque 1077 and histiopaque 1119 (Sigma #10771, #11191). Cells were washed again and then plated into a 96-well plate at 1.5 × 104 cells per well and incubated for 30 min at 37°C to allow for attachment. NETs were induced either by PAF (50 µM, Fisher) or LPS (10 ug/ml) and incubated for 2 h to allow for NET formation. NETs were quantified by measuring cell-free DNA in the supernatant using the PicoGreen QuanTi kit according to the manufacturer’s protocol.
Immunofluorescence (IF) staining
Murine neutrophils were extracted and plated in a 96-well plate at 1.5 × 104 cells per well. The neutrophils were incubated at 37°C for 30 min to allow for attachment to the well. Control media, CQ (1 mM), HCQ (1 mM), or BlfA (100 nM) were added to their respective wells followed by the NET activators, control media, PAF, or LPS. After the neutrophils were incubated for 2 h at 37°C, the cells were fixed with a 4% paraformaldehyde (PFA) solution. Immunoflourescence (IF) staining was conducted for anti-citrullinated histone 3 (Abcam, #5103) with an anti-rabbit secondary conjugated to FITC (Abcam, #7086) and DNA was stained using Hoechst. Images were collected using a Zeiss microscope and quantified using ImageJ.
Modeling of PAD4/CQ/HCQ interaction and surface plasmon resonance analysis
Molecular modeling studies to investigate the binding interaction of the compounds with PAD4 were initiated using the MOE2020.09 (ChemComp) modeling software. The protein crystal structure with a bound ligand was downloaded from the Protein Data Bank (1WDA.pdb), and prepared for docking by adding hydrogens and partial charges at pH 7.4. An induced-fit docking method was used for each compound. CQ and HCQ structures were obtained from PubChem and used in the docking studies [18].
CQ and HCQ were reconstituted in distilled water at 1 mg/ml concentration. Human recombinant PAD4 enzyme was obtained from Cayman Chemical. SPR analysis for CQ, HCQ and PAD4 was performed on a Biacore T200 instrument. PAD4 was directly immobilized on the CM5 chip using an amine coupling kit (GE Healthcare Life Sciences). Before immobilization, the CM5 sensor surface was activated using a mixture of 400 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 100 mM N-hydroxysuccinimide (NHS). PAD4 diluted into 60 μg/ml using the immobilization buffer was then injected into FC4 at a flow rate of 10 μl/min. The amount of PAD4 immobilized was about 15 000 RU. The chip was deactivated by 1 M Ethanolamine hydrochloride-NaOH (GE Healthcare Life Sciences) at a flow rate of 10 μl/min for 420 s. The reference surface FC3 channel underwent similar procedures but without injecting PAD4 ligand. HCQ and CQ were serially diluted with the running buffer to give a concentration of 100, 50, 25, 12.5, 6.25, 3.125, 1.563, 0.781, 0.391, and 0 nM, respectively. HCQ and CQ were then injected into FC3 and FC4, respectively, at a flow rate of 30 μl/min, with a contact time of 60 s and a dissociation time of 90 s. Data analysis was performed on the Biacore T200 computer and with the Biacore T200 evaluation software 3.0. Reports were generated and the binding affinity and KD was determined.
Human serum analysis of CitH3
Patients with pancreatic adenocarcinoma treated in a randomized phase II clinical trial at the Hillman Cancer Center at the University of Pittsburgh Medical Center (Pittsburgh, PA) registered with clinicaltrials.gov (NCT01978184) were assessed for circulating markers of NET formation. Patients were randomly assigned to either chemotherapy or chemotherapy plus HCQ treatment groups. Gemcitabine with nab-paclitaxel was given to all patients with or without HCQ, 600 mg twice daily, followed by surgical resection. Human serum was collected through centrifugation of human blood obtained pre and post treatment. The serum was aliquoted and frozen at −80°C until needed for analysis. Serum was thawed overnight at 4°C, and CitH3 concentration was analysed using Cayman Chemical Citrullinated Histone 3 ELISA (Clone 11D3) kit. Serum was diluted 1:2 and the ELISA was performed according to the manufacture protocol. Absorbance was read on a Molecular Devices plate reader and CitH3 concentration was calculated.
Statistical analysis
Data are expressed mean ± SD of at least two independent experiments performed in duplicate. The box and whisker plot for the median change in CitH3 reports the minimum and maximum range for the whiskers. Statistical analysis was performed using student t-test, or one-way ANOVA with a Tukey post-hoc test; P values <0.05 were considered statistically significant.
Results
Chloroquine and hydroxychloroquine inhibit ex vivo NET formation from different activators
We tested the ability of CQ and HCQ to inhibit NETs stimulated by activators that function through different NET induction pathways [19, 20]. CQ and HCQ reduced NET formation by both PAF and LPS measured as reduced cell-free DNA in the supernatant compared to PAF or LPS alone (Fig. 1a–c). This led us to hypothesize that these drugs are inhibiting NETs by working through a central pathway involved with NET formation.
Figure 1.

CQ and HCQ inhibit NETs induced with various NET activators. (a) Murine neutrophils were isolated from bone marrow using density gradient centrifugation, pre-treated with vehicle, CQ (1 mM), or HCQ (1 mM) then stimulated to form NETs with either PAF (50 µM) or LPS (10 µg/ml) for 2 h. DNA was stained with Hoechst (red) to visualize extracellular DNA and images were obtained to visualize NET structures at 20× using a Zeiss microscope. Both CQ and HCQ led to a subjective decrease in visualized extracellular DNA, consistent with reduced NET formation. (b and c) NET formation was quantified by measuring cf-DNA released into the supernatant with QuantiT Pico Green kit per the manufacturer’s protocol. CQ and HCQ led to a significant reduction in cf-DNA in both PAF and LPS-induced NET assays.*P value < 0.05.
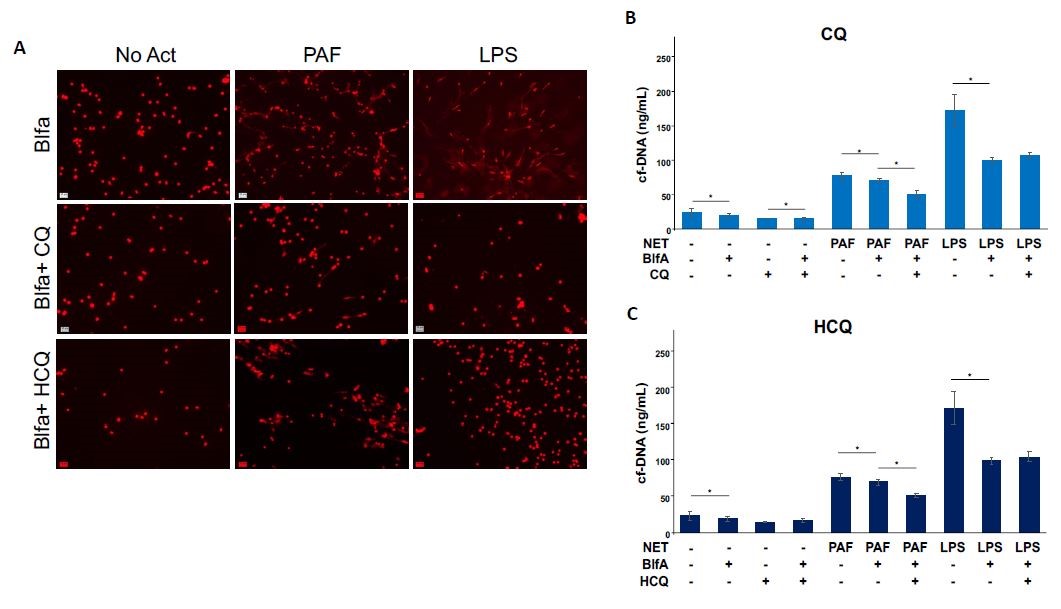
CQ and HCQ may inhibit NETs independent of their autophagy inhibitory mechanism
Activation of autophagy is just one of the many processes that have been associated with NET formation [6, 21]. Since CQ and HCQ are known autophagy inhibitors, we set out to determine if the NET inhibitory effect was dependent on this function. To do so, we pre-treated isolated murine neutrophils with BlfA, an autophagy inhibitor, followed by CQ or HCQ. When autophagy is inhibited with BlfA treatment, we see a reduction in NET formation measured through cfDNA release. When autophagy inhibition (BlfA treatment) is combined with CQ and HCQ treatment, we see even a greater reduction in NET formation for PAF-induced NETs, but not in LPS-induced NETs (Supplemental Fig. 1a–c). This led us to hypothesize that CQ and HCQ are working through PAD4 inhibition, not just autophagy, to inhibit NET formation in PAF-induced NETs and perhaps working through autophagy alone in LPS-induced NETs.
CQ and HCQ inhibit PAD4 in a dose-dependent manner
We hypothesized that CQ and HCQ are working on a pathway central to NET formation independent of autophagy. We tested several different key enzymes involved in NET formation including MPO, NE, and PAD4. PAD4 was the only enzyme that showed a dose-dependent response with CQ and HCQ treatment (Fig. 2a). Unlike other nonspecific PAD inhibitors that are currently commercially available [22, 23], PAD2 was not inhibited by these drugs, suggesting CQ/HCQ function through a PAD4-specific mechanism (Fig. 2b). We sought to validate this finding of specific PAD4 inhibition by CQ/HCQ through IF staining of the NETs for citrullinated histone 3 (citH3), a product of PAD4 activity that results from PAD4 mediated exchange of arginine for citrulline on histones during NET formation. CQ or HCQ treatment led to a reduction of citH3 NETs compared to untreated controls (Fig. 2c and d), validating PAD4-specific inhibition by CQ/HCQ treatment.
Figure 2.

CQ and HCQ inhibits PAD4 in a dose-dependent manner. (a) PAD4 inhibition was measured through Cayman Chemical PAD4 Inhibitor Screening Assay Kit (Ammonia) per manufacturer’s instructions after treatment with positive control, Cl-Amidine, CQ, or HCQ at the concentrations indicated, demonstrating dose-dependent inhibition of PAD4 for both compounds. (b) Measurement of PAD2 inhibition through Cayman Chemical PAD2 Inhibitor Screening Assay Kit after treatment with Cl-Amidine, CQ, and HCQ showing lack of PAD2 inhibition with CQ and HCQ. (c) NETs were stained with Hoechst for DNA (Blue) and CitH3 (Red, anti-citrullinated histone 3 with an anti-rabbit secondary conjugated to FITC) after stimulation with PAF with and without treatment of hydroxychloroquine or chloroquine and a representative image is shown. CitH3 positive NETs are marked with white arrows and CitH3 negative NETs with green arrows. These findings demonstrated a reduction in CitH3 in HCQ/CQ-treated neutrophils. (d) Quantification of 9 wells from each treatment measuring CitH3 positive NETs.
HCQ reduces circulating NET markers in patients with pancreatic adenocarcinoma
We set out to determine the effectiveness of HCQ in reducing circulating NET markers in patients with pancreatic cancer, specifically the marker of PAD4-dependent NETs, citH3. Patients with pancreatic adenocarcinoma in a randomized phase II clinical trial were assigned to either chemotherapy only or chemotherapy with HCQ [24]. Patients were given two months of gemcitabine plus nab-paclitaxel with or without HCQ (600 mg twice daily) followed by surgical resection. Serum collected pre and post treatment was analysed for CitH3 levels. Patients in the chemotherapy alone group saw no difference in CitH3 after treatment. Chemotherapy combined with HCQ treatment resulted in a significant reduction in circulating CitH3 levels (Fig. 3a and b). Although a majority of patients had a decrease in CitH3 with HCQ treatment, eight patients (30%) treated with HCQ had an increase in CitH3. These patients had significantly lower pre-treatment CitH3 levels when compared to those that had a decrease with HCQ treatment (1207 vs. 3198 pg/ml, P < 0.05). There were no statistically significant differences in clinical outcomes related to the level of CitH3 reduction in HCQ-treated patients, although the study was not powered to detect these differences. These correlative findings validate the NET inhibitory function of HCQ on NET formation and provide evidence for inhibition of PAD4, as evident by the reduction in CitH3 levels in patients with pancreatic cancer treated with HCQ.
Figure 3.

HCQ treatment reduces circulating CitH3 levels in PDAC patients. (a) Patients with pancreatic adenocarcinoma were enrolled in a randomized clinical trial of neoadjuvant gemcitabine/nab-paclitaxel plus HCQ versus gemcitabine/nab-paclitaxel alone. CitH3 concentration was measured in patient plasma using ELISA and pre treatment (pre-neoadjuvant) versus post treatment (post-neoadjuvant) concentration was compared in the chemotherapy only (n = 26) and the chemotherapy plus HCQtreatment groups (n = 27). There was a significant reduction in CitH3 in patients treated with chemotherapy plus HCQ. (b) Median change of CitH3 levels in the chemotherapy versus chemotherapy + HCQ treatment group demonstrating reduction in CitH3 in the chemotherapy +_HCQ group (P value = 0.01). Whisker bars represent the range of minimum and maximum within each group.
CQ and HCQ bind to PAD4
To determine how CQ and HCQ inhibited PAD4, we performed computational modeling of their interactions and found that both drugs had potential binding sites with PAD4. iTASSER interaction analysis identified a hydrogen bond interaction between CQ and Arg639, while HCQ predicted to interact with Trp347, Ser468, and Glu580 residues of PAD4 via a hydrogen bond (Fig. 4a and b). SPR analysis confirmed that chloroquine and hydroxychloroquine binding to PAD4 with a KD of 54.1 µM (CQ) and 88.1 µM (HCQ) (Fig. 4c and e). These data support CQ and HCQ inhibiting the NET formation process in part by binding to PAD4 and inhibiting the enzyme activity. When considering our data in light of existing literature, we propose a multi-mechanism model to explain how CQ and HCQ reduce NET formation, through TLR9, autophagy, and our newly discovered PAD4 inhibition pathway.
Figure 4.

Binding of CQ and HCQ to the PAD 4 enzyme. (a and b) Predicted binding sites of CQ (a) and HCQ (b) through computational docking studies (1 WDA.pdb). CQ is predicted to bind to amino acid residue Arg639 and HCQ to Trp347, Ser468, and Glu580 in PAD4. (c–e) SPR analysis determining binding potential of CQ (c) and HCQ (d) to PAD4 with KD values at 54.1 µM, and 88.1 µM respectively (e).
Discussion
We have shown that CQ and HCQ directly inhibit PAD4 to block NET formation. When combined with known literature regarding the function of CQ and HCQ in NET pathways, particularly autophagy and TLR9 inhibition, these data suggest a multi-mechanistic explanation for NET inhibition by CQ and HCQ. CQ and HCQ inhibited ex vivo NET formation induced by differing stimuli and for the first time, we have shown that HCQ blocks NET formation in patient samples, as demonstrated through reduction of circulating CitH3 levels. Direct binding of CQ and HCQ to PAD4 was predicted through iTASSAR modeling and confirmed by SPR analysis. This evidence warrants consideration for the inclusion of CQ/HCQ to therapeutic regimens of diseases where NETs are pathologic, such as in cancer and auto-immune disease. Importantly, these drugs are orally available with an established safety profile given years of clinical use, and therefore they have a distinct advantage over current PAD4 inhibitors currently commercially available. However, the multiple biological mechanisms that CQ/HCQ can functionally affect, in addition to NET/PAD4 inhibition, is a clear limitation of their translation to clinical settings where isolated NET inhibition is desired.
Neutrophils can be activated to induce NET formation by a variety of substances. The exact signalling cascade resulting in NETs is still not fully known but appears to be activator specific [25]. For example, PAF has been shown to activate PKC and PHOX resulting in a signaling cascade to induce NET formation [20]. LPS, on the other hand, activates TLR4 leading to the activation of the JNK sensing mechanism promoting NET formation [19]. All stimuli that induce NET formation result in the activation of several key enzymes including NE, MPO, PAD4, autophagy mediators, reactive oxygen species (ROS), and several others [6, 26]. These enzymes and mediators are required for membrane degradation, decondensation of chromatin, and finally, formation of the NET structure after the cytosolic membrane bursts [25, 27]. We confirmed that both PAF and LPS lead to the activation autophagy, as inhibiting autophagy with BlfA resulted in a significant reduction in NETs produced by both stimuli. The current data suggest that PAF may also induce PAD4 expression to a greater degree than LPS based on the disparate response to CQ/HCQ after BlfA treatment, but this requires more focused studies measuring PAD4 activation and CitH3 production from both of these NET inducers.
CQ and HCQ are drugs that have been used for over 50 years with a multitude of potential therapeutic uses. Originally, CQ, and then HCQ due to its reduced toxicity, were used as a malarial treatment and prophylaxis as they were shown to prevent proper food vacuole processing for the parasite [7]. Over the years since its discovery, several other mechanisms have been elucidated and HCQ has been used extensively in the treatment of other diseases. One of the most commonly known mechanisms of these drugs is their ability to inhibit autophagy through prevention of autophagic vessel processing [28]. Because of their ability to affect many different cellular processes, CQ and HCQ have been popular choices for off label prescribing for the treatment of various diseases. HCQ is currently being used to treat systemic erythematous lupus and rheumatoid arthritis with promising results in reducing disease severity [8]. Preclinical models have shown promising results in CQ/HCQ treatment for sepsis, ischemia injury prevention, pancreatitis, and cancer [10, 11, 29]. Several clinical trials are currently testing CQ or HCQ with chemotherapy to treat various cancer types, including breast cancer and glioblastoma [30–34]. These studies were designed mainly to test HCQ as an autophagy inhibitor when combined with chemotherapy (including the referenced pancreatic cancer trials in the current manuscript). The impact of HCQ appears to be context dependent, with trials demonstrating mixed results across different cancer types. Further study to better understand these disparate results is required. Zhang et al. showed that HCQ treatment reduced NETs in a hepatic ischemia model by demonstrating that HCQ bound to TLR9, blocking its signaling cascade and preventing the activation of several key mediators during the NET process. Here, we have further contributed to this understanding further by providing evidence that HCQ/CQ prevents NETs through a novel mechanism of PAD4 inhibition with preliminary data to suggest that this occurs independent of autophagy. Separating out these different mechanisms experimentally is challenging. Further studies should be done to fully understand the multiple mechanisms that are potentially involved in HCQ NET inhibition. Additional testing evaluating autophagy inhibition, PAD4 inhibition, and TLR9 signaling would be interesting to validate PAD4 inhibition as an independent mechanism. We are also the first to demonstrate this PAD4 inhibitory effect of HCQ in patients by showing a reduction in circulating CitH3, a product of PAD4, after HCQ treatment in pancreatic cancer patients. Although a majority of patients had a reduction in CitH3, 30% of patients had an increase in CitH3 with HCQ treatment. Interestingly, this cohort of patients had significantly lower baseline levels of CitH3, suggesting that HCQ mediates its greatest NET inhibitory effect in patients who have larger induction of NETs at baseline. The clinical trials of HCQ in pancreatic cancer referenced in the current study were both positive trials. The initial phase I/II trial demonstrated the safety and efficacy of HCQ with gemcitabine given in the pre-operative setting [35]. Their median PFS was 11 months and OS was 31 months, with a 5-year OS of 31%, which is encouraging compared with historical controls [36]. A follow-up randomized trial of gemcitabine/nab-paclitaxel with or without HCQ demonstrated significant improvement in histopathologic tumour response and reduction in the pancreatic cancer biomarker Ca 19-9 [24]. In the current analysis, we are unable correlate CitH3 levels with clinical outcomes including survival, however, these studies were not powered to detect these correlations. In fact, at the time these trials were conducted, the effects of HCQ on NETs and PAD4 were not yet recognized. While these trials were ongoing, we recognized a critical role for NETs in the pathogenesis of pancreatic cancer and pancreatitis and identified that CQ/HCQ inhibits NETs in these preclinical models [10, 11, 17, 37]. This promoted our interest in returning to the clinical trial samples for the current analysis. These promising data support future studies designed specifically to analyse the impact of NET inhibition on clinical outcomes including histopathologic response and survival.
The limitation of this work includes the challenges associated with isolating a single pathway involved in the NET formation and focusing on its role independently, particularly given the significant overlap in signaling cascades. Furthermore, the field currently has an incomplete understanding of NET induction pathways, which limits the possible conclusions from the current experiments. For example, there are some stimuli that are not dependent on ROS generation, once thought to be a very important mediator of NET formation [38]. Although we demonstrate novel PAD4 inhibition with HCQ, we recognize that the current data supports but does not definitively prove that PAD4 inhibition is completely independent of other HCQ mechanisms. Proving this experimentally is challenging and beyond the scope of the current manuscript. While we do suggest preliminarily that the PAD4 inhibition may occur independent of autophagy, our main conclusion to be gained from these studies is that CQ/HCQ inhibits PAD4 as one mechanism to block NET formation as demonstrated through SPR analysis and in vitro enzymatic assays. This work has stemmed further study into the structure of CQ/HCQ, which can serve as a starting point to generate novel compounds with stronger affinity to PAD4 to develop new treatments for diseases where NET formation has been implicated in the pathogenesis.
Through this and previous works, we hypothesize a multi-mechanistic model for how CQ and HCQ block NET formation and, despite the limitations outlined above, this work provides a better understanding of how CQ and HCQ function to inhibit NETs. NETs are involved in the pathophysiology of several different inflammatory processes and therefore represent an excellent new therapeutic target for numerous diseases. There are several different NET inhibitors being used in pre-clinical models that still need to be fully investigated before they are ready for use in patients. CQ and HCQ are excellent candidates to explore NET inhibition strategies as they are already FDA approved and are orally available. Because CQ/HCQ inhibition NETs at several different points in the NET formation pathway, they are ideal for diseases in which the principle pathway for the upregulation of NETs is unknown.
Conclusion
CQ and HCQ inhibit PAD4 through direct binding and inhibited NET formation both ex vivo and in patient samples. This study not only provides a better understanding of how these drugs function to inhibit NETs but also gives insight as to what pathways are driving the upregulation of NETs in different diseases.
Supplementary data
Supplementary data is available at Clinical and Experimental Immunology online.
Supplemental Figure 1. CQ and HCQ inhibit NETs independent of their autophagy inhibitory mechanism. (a) Murine neutrophils isolated from bone marrow, pre-treated with BlfA (100 nM) and either CQ (1 mM) or HCQ (1 mM) then stimulated with either PAF (50 µM) or LPS (10 µg/ml) for 2 h. Images (20×) of neutrophils after stimulation, 4% PFA fixed, DNA stained with Hoechst and imaged with a Zeiss microscope. DNA is visualized in red. (b and c) Quantification of NET formation measured through cell-free DNA released in the supernatant with QuantiT Pico Green kit. *P value < 0.05.
{kind=link}
Abbreviations
- ANOVA
analysis of variance
- BlfA
bafilomycin A; CitH3: citrullinated histone H3
- CQ
chloroquine
- ELISA
enzyme linked immunosorbent assay
- FBS
fetal bovine serum
- HCQ
hydroxychloroquine
- IF
immunoflourescence
- KD
equilibrium dissociation constant
- LPS
lipopolysaccharide
- MPO
myeloperoxidase
- NE
neutrophil elastase
- NETs
neutrophil extracellular traps; PAD4: peptidyl arginine deiminase 4
- PAF
platelet-activating factor
- RPMI
Roswell Park Memorial Institute
- SPR
surface plasmon resonance; TLR9: toll-like receptor 9
Contributor Information
Abby D Ivey, Cancer Cell Biology, West Virginia University, Morgantown, WV, USA.
B Matthew Fagan, Department of Surgery, West Virginia University, Morgantown, WV, USA.
Pranav Murthy, Department of Surgery, University of Pittsburgh, Pittsburgh, PA, USA.
Michael T Lotze, Department of Surgery, University of Pittsburgh, Pittsburgh, PA, USA.
Herbert J Zeh, III, Department of Surgery, UT Southwestern, Dallas, TX, USA.
Lori A Hazlehurst, Cancer Cell Biology, West Virginia University, Morgantown, WV, USA; Pharmaceutical Sciences, West Virginia University, Morgantown, WV, USA.
Werner J Geldenhuys, Pharmaceutical Sciences, West Virginia University, Morgantown, WV, USA; Neuroscience, West Virginia University, Morgantown, WV, USA.
Brian A Boone, Cancer Cell Biology, West Virginia University, Morgantown, WV, USA; Department of Surgery, West Virginia University, Morgantown, WV, USA; Microbiology, Immunology and Cell Biology, West Virginia University, Morgantown, WV, USA.
Animal research
The animal research adheres to the ARRIVE guidelines.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Conflict of interests
The authors have no competing interests to declare.
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 5U54GM104942-04 (BAB) and the National Cancer Institute under award number 1R01 CA181450 (MTL and HJZ) and P20GM109098 (WJG). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
All authors reviewed, provided critical feedback and approved the final version of the manuscript. Conceptualization: ADI, MTL, HJZ, LAH, WJG, and BAB. Methodology: ADI, MTL, HJZ, LAH, WJG, and BAB. Analysis and interpretation of data: ADI, BMF, PM, and BAB. Performed investigations: ADI, BMF, and PM. Writing/editing manuscript: ADI, BMF, PM, MTL, HJZ, LAH, WJG, and BAB. Project administration: MTL, HJZ, and BAB. Funding acquisition: ADI, MTL, HJZ, and BAB.
Clinical Trial Registration
This manuscript contains correlative data taking from a clinical trial registered at clinicaltrials.gov (NCT01978184).
References
- 1. Volker Brinkmann UR, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, et al. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–5. [DOI] [PubMed] [Google Scholar]
- 2. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L,. et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 2012, 3, 307. doi: 10.3389/fimmu.2012.00307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV.. The emerging role of neutrophil extracellular traps (NETs) in tumor progression and metastasis. Front Immunol 2020, 11, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight 2020, 5, e134796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis 2018, 9, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park SY, Sanjeeb S, Young-Jin Y, Jun-Kyu K, Shin-Yeong K, Hyun Jung K, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med 2017, 196, 577–89. [DOI] [PubMed] [Google Scholar]
- 7. Askarian F, Firoozi Z, Ebadollahi-Natanzi A, Bahrami S, Rahimi HR.. A review on the pharmacokinetic properties and toxicity considerations for chloroquine and hydroxychloroquine to potentially treat coronavirus patients. Toxicol Res 2021, 38, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plantone D, Koudriavtseva T.. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig 2018, 38, 653–71. [DOI] [PubMed] [Google Scholar]
- 9. Jorge A, Ung C, Young LH, Melles RB, Choi HK.. Hydroxychloroquine retinopathy - implications of research advances for rheumatology care. Nat Rev Rheumatol 2018, 14, 693–703. [DOI] [PubMed] [Google Scholar]
- 10. Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. doi: 10.1186/s12885-018-4584-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI, et al. Enhanced neutrophil extracellular trap formation in acute pancreatitis contributes to disease severity and is reduced by chloroquine. Front Immunol 2019, 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M.. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun 2004, 323, 264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ireland JM, Unanue ER.. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med 2011, 208, 2625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rand JH, Wu XX, Quinn AS, Ashton AW, Chen PP, Hathcock JJ, et al. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: evidence for a novel effect for an old antimalarial drug. Blood 2010, 115, 2292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Restrepo JF, Del Rincon I, Molina E, Battafarano DF, Escalante A.. Use of hydroxychloroquine is associated with improved lipid profile in rheumatoid arthritis patients. J Clin Rheumatol 2017, 23, 144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Floris A, Piga M, Mangoni AA, Bortoluzzi A, Erre GL, Cauli A.. Protective effects of hydroxychloroquine against accelerated atherosclerosis in systemic lupus erythematosus. Mediators Inflamm 2018, 2018, 3424136. doi: 10.1155/2018/3424136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi A D, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther 2015, 22, 326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M.. Structural basis for Ca(2+)-induced activation of human PAD4. Nat Struct Mol Biol 2004, 11, 777–83. [DOI] [PubMed] [Google Scholar]
- 19. Khan MA, Farahvash A, Douda DN, Licht JC, Grasemann H, Sweezey N, et al. JNK activation turns on LPS- and Gram-negative bacteria-induced NADPH oxidase-dependent suicidal NETosis. Sci Rep 2017, 7, 3409. doi: 10.1038/s41598-017-03257-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 2011, 7, 75–7. [DOI] [PubMed] [Google Scholar]
- 21. Liang X, Liu L, Wang Y, Guo H, Fan H, Zhang C, et al. Autophagy-driven NETosis is a double-edged sword – review. Biomed Pharmacother 2020, 126, 110065. doi: 10.1016/j.biopha.2020.110065 [DOI] [PubMed] [Google Scholar]
- 22. Kawalkowska J, Quirke AM, Ghari F, Davis S, Subramanian V, Thompson PR, et al. Abrogation of collagen-induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell-mediated immune responses. Sci Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol 2011, 186, 4396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zeh HJ, Bahary N, Boone BA, Singhi AD, Miller-Ocuin JL, Normolle DP, et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin Cancer Res 2020, 26, 3126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brinkmann V. Neutrophil extracellular traps in the second decade. J Innate Immun 2018, 10, 414–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2011, 21, 290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaplan MJ, Radic M.. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 2012, 189, 2689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu R, Ji Z, Xu C, Zhu J.. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Medicine 2018, 97, e12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang S, Zhang Q, Wang F, Guo X, Liu T, Zhao Y, et al. Hydroxychloroquine inhibiting neutrophil extracellular trap formation alleviates hepatic ischemia/reperfusion injury by blocking TLR9 in mice. Clin Immunol 2020, 216, 108461. doi: 10.1016/j.clim.2020.108461 [DOI] [PubMed] [Google Scholar]
- 30. Anand K, Niravath P, Patel T, Ensor J, Rodriguez A, Boone T, et al. A phase II study of the efficacy and safety of chloroquine in combination with taxanes in the treatment of patients with advanced or metastatic anthracycline-refractory breast cancer. Clin Breast Cancer 2021, 21, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arnaout A, Robertson SJ, Pond GR, Lee H, Jeong A, Ianni L, et al. A randomized, double-blind, window of opportunity trial evaluating the effects of chloroquine in breast cancer patients. Breast Cancer Res Treat 2019, 178, 327–35. [DOI] [PubMed] [Google Scholar]
- 32. Compter I, Eekers DBP, Hoeben A, Rouschop KMA, Reymen B, Ackermans L, et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy 2021, 17, 2604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sotelo J, Briceno E, Lopez-Gonzalez MA.. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2006, 144, 337–43. [DOI] [PubMed] [Google Scholar]
- 35. Boone BA, Bahary N, Zureikat AH, Moser JA, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol 2015, 22, 4402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. AlMasri S, Zenati MS, Desilva A, Nassour I, Boone BA, Singhi AD, et al. Encouraging long-term survival following autophagy inhibition using neoadjuvant hydroxychloroquine and gemcitabine for high-risk patients with resectable pancreatic carcinoma. Cancer Med 2021, 10, 7233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, et al. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology 2019, 8, e1605822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Douda DN, Khan MA, Grasemann H, Palaniyar N.. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A 2015, 112, 2817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.