

Abstract
ZNF462 haploinsufficiency is linked to Weiss-Kruszka Syndrome, a genetic disorder characterized by neurodevelopmental defects including Autism. Though conserved in vertebrates and essential for embryonic development the molecular functions of ZNF462 remain unclear. We identified its murine homolog ZFP462 in a screen for mediators of epigenetic gene silencing. Here, we show that ZFP462 safeguards neural lineage specification of mouse embryonic stem cells (ESCs) by targeting the H3K9-specific histone methyltransferase complex G9A/GLP to silence mesoendodermal genes. ZFP462 binds to transposable elements (TEs) that are potential enhancers harboring ESC-specific transcription factor (TF) binding sites. Recruiting G9A/GLP, ZFP462 seeds heterochromatin, restricting TF binding. Loss of ZFP462 in ESCs results in increased chromatin accessibility at target sites and ectopic expression of mesoendodermal genes. Taken together, ZFP462 confers lineage- and locus-specificity to the broadly expressed epigenetic regulator G9A/GLP. Our results suggest that aberrant activation of lineage non-specific genes in the neuronal lineage underlies ZNF462-associated neurodevelopmental pathology.
Introduction
A totipotent zygote develops into a multicellular organism via cell specialization 1–3. Cell fate specification is primarily controlled by transcription factors (TFs) that activate lineage-specific genes 4, but the silencing of lineage non-specific genes is also important. Silencing often involves histone methlytransferases (HMTases) that establish repressive chromatin modifications, including Histone H3 Lysine 9 di- and trimethylation (H3K9me2/3) 5. H3K9me2/3 binds Heterochromatin protein 1 isoforms (HP1α, β, γ) 6, and chromatin-bound HP1 can recruit additional heterochromatin modifiers, thus promoting chromatin compaction and transcriptional silencing 7,8. Epigenetic feedback mechanisms promote stable inheritance of heterochromatin, gene silencing, and cell identity 5,9–13.
H3K9me2 is catalysed by an HMTase complex comprising G9A (also known as Euchromatic Histone Methyltransferase 2 – EHMT2) and G9-like protein (GLP, also known as Euchromatic Histone Methyltransferase 1 - EHMT1) 14, which is essential for embryogenesis 15. G9A/GLP-dependent heterochromatin modifications at promoters and enhancers have been proposed to block TF binding, thereby preventing aberrant expression of lineage non-specific genes 16–19. Indeed, G9A/GLP-dependent heterochromatin silences the pluripotency-linked gene Oct3/4 (also known as Pou5f1) during embryonic stem cell (ESC) differentiation, preventing its expression in somatic tissues 20,21. Inactivation of G9A facilitates Oct3/4 induction during iPSC reprogramming of somatic cells 22. G9A/GLP is proposed to regulate neurodevelopment 23 by interacting with the RE1-silencing TF (REST also known as NRSF) to target and repress neuronal genes in non-neuronal cells 24–26. G9A/GLP also has important and distinct functions in silencing both non-neuronal and early neuron progenitor genes in mature neurons 27,28. Notably, de-novo mutations in genes encoding either GLP or proteins that cooperate in facultative heterochromatin formation cause the neurodevelopmental disorder Kleefstra syndrome 29. Although the biochemical functions and developmental importance of G9A/GLP are well-established, the mechanisms underlying its precise spatiotemporal silencing of lineage non-specific genes remain largely unclear 14,30,31. G9A/GLP are broadly expressed and lack sequence-specificity, therefore heterochromatin mediated by G9A/GLP requires yet-unknown cell-type specific interactions with TFs.
Here, we identified Zinc finger protein 462 (ZFP462) in a CRISPR genetic screen for modifiers of heterochromatin-mediated silencing of Oct3/4. ZFP462 is a vertebrate-specific, putative TF of unknown function. Notably, its human ortholog, ZNF462, was recently identified as a high-confidence risk gene for a neurodevelopmental disorder (OMIM#: 618619) 32–34. We demonstrate that ZFP462 is required to silence inappropriate meso-endodermal gene expression in mouse ESCs and neural progenitor cells (NPCs). ZFP462 silences lineage non-specific genes by recruiting G9A/GLP to deposit heterochromatin modifications at enhancers, thereby limiting their accessibility to pluripotency TFs. Thus, ZFP462 precisely targets formation of G9A/GLP-dependent heterochromatin to prevent unscheduled activation of meso-endodermal genes and maintain cell fate identity. Our findings illuminate the developmental regulation of G9A/GLP-dependent gene silencing and suggest that neural phenotypes associated with ZNF462 haploinsufficiency arise from defects in cell fate specification during embryogenesis.
Results
Zfp462 encodes a regulator of heterochromatic gene silencing
To dissect the mechanisms underlying heterochromatin-mediated silencing of a lineage non-specific gene, we focused on Oct3/4 20,22,35. Oct3/4 is highly expressed in ESCs encoding OCT4 TF essential for pluripotency and self-renewal 36, but is inactivated and acquires H3K9me2 upon differentiation. Subsequent HP1 binding and DNA methylation irreversibly silence Oct3/4 21,22,35.
Tethering the chromoshadow domain of HP1α (HP1) to a genetically modified Oct3/4 allele in mouse ESCs is sufficient to recruit H3K9-specific HMTases and recapitulate heterochromatin-mediated gene silencing without affecting the wild-type allele13. To enable efficient, reversible HP1 tethering via the Tet-OFF system we established an ESC clone with Tet operator sites (TetO) and a Puromycin-BFP reporter gene downstream of the Oct4-GFP reporter allele (Fig. 1a) that expresses HP1 fused to a FLAG-Tet repressor domain (TetR-FLAG-HP1) (Extended Data Fig. 1a). This ESC clone also expressed hCas9 to enable CRISPR-based screening in ESCs.
Fig. 1: CRISPR screen identifies heterochromatin regulators required for heritable Oct3/4 gene silencing.
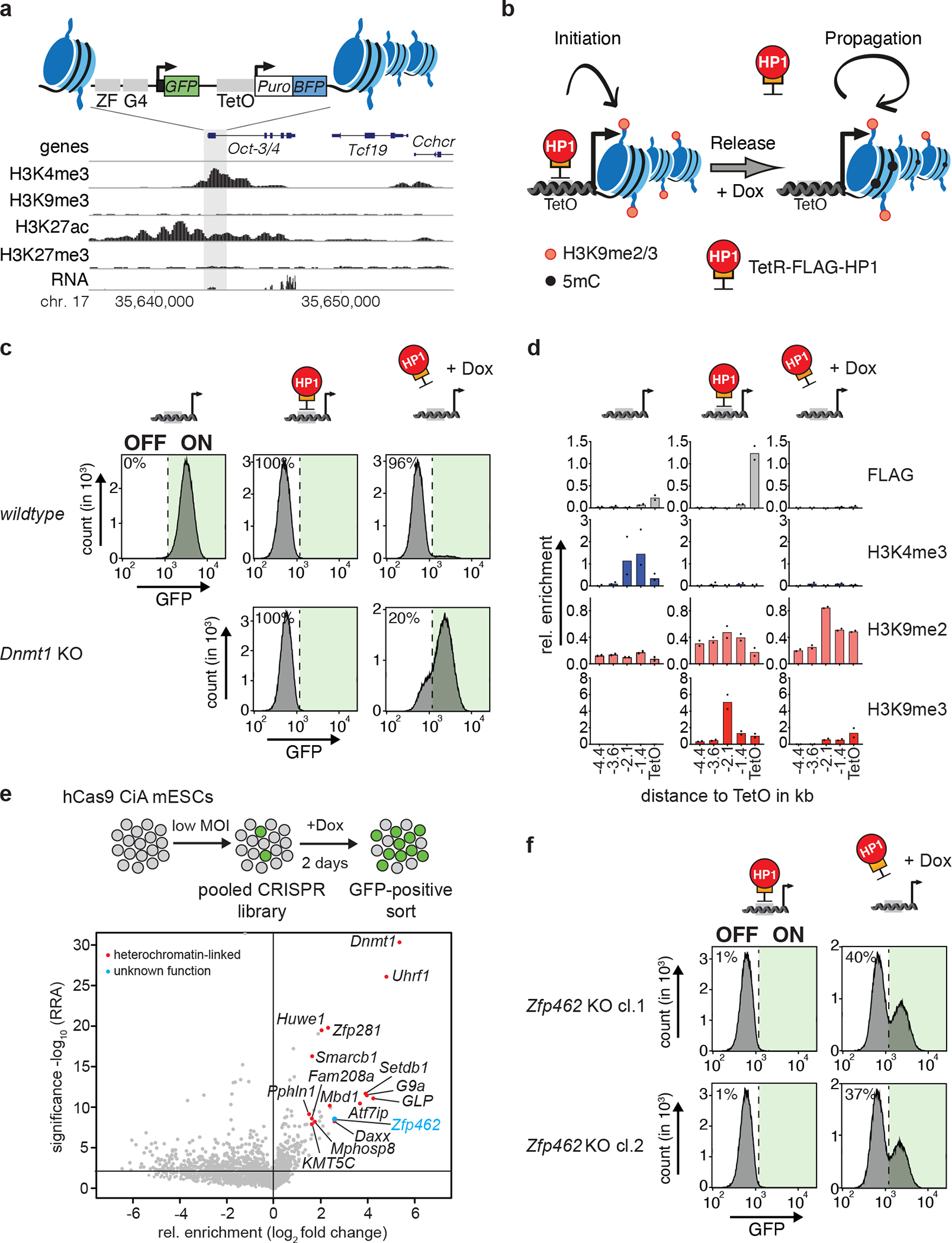
a) Design of the CiA Oct4 dual reporter locus in ESCs. One of the Oct3/4 alleles was modified in ESCs by inserting seven Tet Operator sites (TetO) flanked by a GFP and a BFP reporter gene on either side. GFP expression is under control of the Oct3/4 promoter whereas a PGK promoter drives BFP expression. The genomic screen shot (below) shows histone modifications and RNA expression at the Oct3/4 locus in wild-type ESCs. b) Scheme of the experimental design. TetR facilitates reversible HP1 tethering to TetO binding sites to establish heterochromatin and silence both GFP and BFP reporters. Doxycycline (Dox) addition releases TetR binding to distinguish heritable maintenance of chromatin modifications and gene silencing in the absence of the sequence-specific stimulus. c) Flow cytometry histograms of wild-type and Dnmt1 KO CiA Oct4 dual reporter ESCs show GFP expression before TetR-FLAG-HP1 tethering, in the presence of TetR-FLAG-HP1 and after four days of Dox-dependent release of TetR-FLAG-HP1. Percentages indicate fraction of GFP-negative cells. d) ChIP-qPCR shows relative enrichment of TetR-HP1 (FLAG) and histone modifications surrounding TetO before TetR-FLAG-HP1 tethering, in the presence of TetR-FLAG-HP1 and after four days of Dox-dependent release of TetR-FLAG-HP1. n = 2 independent biological replicates. e) Scheme of CRISPR screen design. MOI refers to multiplicity of infection. Volcano plot shows enrichment (log fold change GFP-pos. sorted vs unsorted cells) and corresponding significance (−log10 MAGeCK significance score) of genes in CRISPR screen (n = mean of three independent experiments). f) Flow cytometry histograms show GFP expression of two independent Zfp462 −/− CiA Oct4 dual reporter cell lines in the presence of TetR-FLAG-HP1 and after four days of Dox-dependent release of TetR-FLAG-HP1. Percentages indicate fraction of GFP-positive cells.
TetR-FLAG-HP1 binds to TetO sites with high affinity, but Doxycycline (Dox) addition abolishes this interaction (Fig. 1b). TetR-FLAG-HP1, but not TetR-FLAG alone, resulted in transcriptional silencing of the Oct4-GFP reporter (Fig. 1c and Extended Data Fig. 1c), concomitant with loss of the active mark H3K4me3, acquisition of the inactive marks H3K9me2 and H3K9me3 (Fig. 1d), and enrichment of endogenous HP1γ (Extended Data Fig. 1d)13. Importantly, transcriptional silencing and inactive histone modifications were maintained even after the addition of Dox (Fig. 1c–d and Extended Data Fig. 1d). In agreement with previous results using small molecule inhibition of DNA methyltransferases 13, we found that genetic Dnmt1 deletion and loss of global DNA methylation (Extended Data Fig. 1e) did not affect the initiation of heterochromatin-dependent reporter gene silencing but compromised its maintenance upon Dox addition (Fig. 1c). We infer that ectopic heterochromatin recapitulates G9A/GLP-induced silencing of the endogenous Oct3/4 gene in somatic cells20, including the stable inheritance of heterochromatin.
To identify genes required for heterochromatin-dependent silencing of Oct3/4, we performed a pooled CRISPR screen with unique molecular identifiers (UMIs), which enables single-cell analysis of mutant phenotypes 37. The UMI CRISPR library contained approx. 27,000 sgRNAs targeting all annotated mouse nuclear protein-coding genes with four sgRNAs per gene. Each sgRNA was paired with thousands of barcodes representing UMIs improving signal-to-noise ratio and hit calling. We transduced with the pooled library into the hCas9-expressing CiA reporter cells, selected with G418 for five days, treated with Dox for two days, and then isolated GFP-positive cells by FACS (Fig. 1e). The unsorted population served as background control. Relative enrichment of sgRNAs was determined by sequencing UMIs in both populations followed by statistical analysis using MAGeCK38. 130 genes were significantly enriched in the GFP-positive cell population (P-value <0.001) (Fig. 1e and Supplementary Table 1), including Dnmt1, Uhrf1, Setdb1, G9a, GLP, Atf7ip, Daxx, Atrx, KMT5C and genes encoding members of the HUSH complex. These factors have all been linked to DNA methylation or H3K9me2/3-dependent gene silencing14,39,40, supporting the known crosstalk between these pathways in heterochromatin 13,39
The uncharacterized Zfp462 gene scored very highly in the screen but has not been associated with heterochromatin regulation. We validated loss of heritable Oct4-GFP silencing in Zfp462 mutant CiA ESCs by independent CRISPR-Cas9 targeting (Fig. 1f and Extended Data Fig. 1f).
ZFP462 represses through interaction with G9A/GLP and HP1γ
To further investigate ZFP462, we engineered ESCs with an Avi-GFP-FLAG tag inserted into the endogenous Zfp462 gene (Avi-Zfp462) (Fig. 2a) and identified ZFP462-interacting proteins in ESCs. Several well-known co-repressors affinity purified with ZFP462 (Fig. 2b and Supplementary Table 2), including HP1γ, G9A, GLP and WIZ, a known G9A/GLP interacting protein 41. Similarly, ZFP462, HP1γ, G9A and WIZ affinity purified with Avi-fused endogenous GLP (Avi-GLP) (Fig. 2c and Supplementary Table 3)41. Thus, ZFP462 interacts with co-repressor proteins, suggesting it is involved in heterochromatin regulation.
Fig. 2: ZFP462 elicits silencing function through interaction with G9A/GLP and HP1γ.
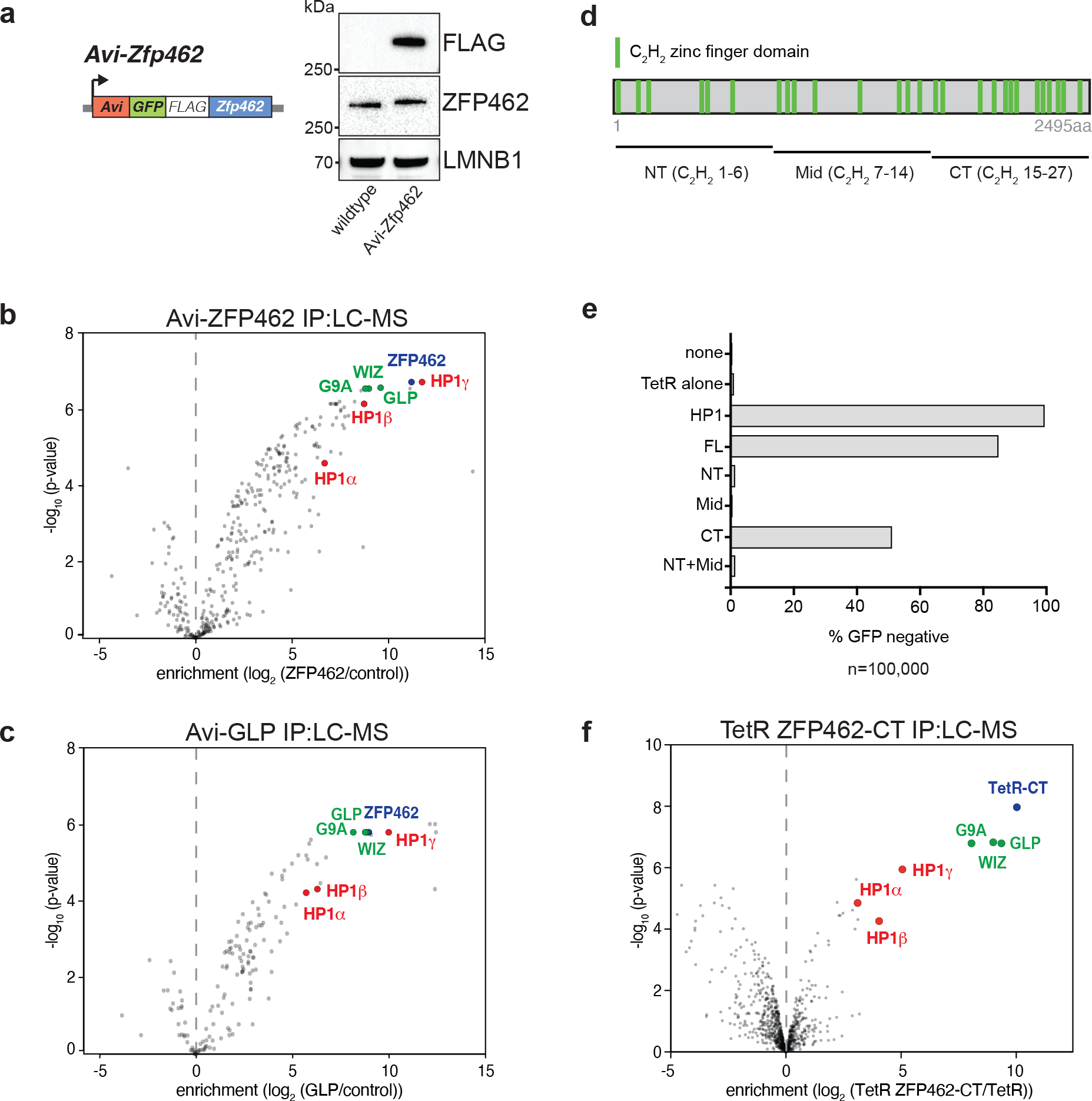
a) Design of Avi-Zfp462 ESCs and western blot validation. Mouse ESCs expressing Biotin ligase (BirA) were used to modify the endogenous Zfp462 gene by inserting the Avi-GFP-3XFLAG tag downstream of the translation start codon. Western blot with FLAG and ZFP462 antibodies confirms ZFP462 tagging. b) and c) LC-MS analysis of Avi-tagged ZFP462 and Avi-tagged GLP ESCs. Volcano plots show enrichment and corresponding significance of co-purified proteins. (n = three replicates). d) Scheme of ZFP462 protein depicts locations of 27 C2H2 zinc finger domains (green bars). Fragments used to generate TetR fusions for tethering in CiA Oct4 dual reporter assay are indicated below. e) Bar plot shows percentage of GFP-negative CiA Oct4 ESCs measured by flow cytometry in response to ectopic TetR fusion protein expression (y-axis). f) Volcano plot of LC-MS analysis compares enrichment and corresponding significance of co-purified proteins between TetR-FLAG-ZFP462-CT and TetR-FLAG (n = three replicates).
ZFP462 is a highly conserved, vertebrate-specific C2H2-type zinc finger protein (Fig. 2d and Extended Data Fig. 2a) C2H2-type zinc fingers are typically associated with DNA binding activity but may also contribute to protein-protein interactions41,42. To determine if ZFP462 recruitment is sufficient to silence the Oct3/4 reporter, we transduced CiA ESCs with TetR-FLAG fusions of full-length ZF462 (FL), or of N-terminal (NT), mid (Mid), C-terminal truncation (NT+Mid) and C-terminal (CT) fragments (Fig. 2d and Extended Data Fig. 2b). TetR-FLAG-ZFP462-FL targeting led to loss of GFP expression in over 80% of CiA reporter cells (Fig. 2e and Extended Data Fig. 2c). TetR-FLAG-ZFP462-CT also resulted in substantial GFP silencing and interacted with G9A, GLP, WIZ and HP1γ by co-immunoprecipitation (Fig. 2f and Supplementary Table 4). In contrast, GFP silencing was not observed in CiA ESCs expressing TetR-FLAG fusions of ZFP462-NT, ZFP462-Mid, or ZFP462-NT+Mid. Thus, ZFP462 associates with G9A/GLP and HP1γ via its C-terminal domain and is sufficient to initiate transcriptional gene silencing.
ZFP462 represses primitive endoderm differentiation
To investigate the role of ZFP462 during mouse embryogenesis, we first examined available scRNA-seq data of three developmental stages representing exit from pluripotency and primary germ-layer specification43. Consistent with its role in regulating heterochromatin in ESCs, Zfp462 was initially expressed in epiblast stem cells (embryonic day (E)4.5). At gastrulation onset, Zfp462 displayed almost exclusive expression in pluripotent and neural cells (E6.5–7, Extended Fig. 3a).
Next, we generated homozygous Zfp462 mutant ESCs using CRISPR-Cas9 (Zfp462 −/−). Additionally, we engineered premature stop codons into one allele of Zfp462 using CRISPR-Cas9 assisted homology-dependent repair (Zfp462 +/Y1195*and Zfp462 +/R1257*). Zfp462 +/R1257* mutant ESCs mimic human ZNF462 haploinsufficiency associated with Weiss-Kruzska Syndrome (ZNF462 +/R1263*)32. We isolated two independent Zfp462 mutant clones for each genotype and confirmed reduction and loss of ZFP462 protein (Fig. 3a) and heterozygous point mutation (Extended Data Fig. 3b). Compared to wild-type ESCs, heterozygous and homozygous Zfp462 mutant ESCs displayed morphological changes with dispersed, refractile cells spreading out of characteristically densely packed ESC colonies (Fig. 3b and Extended Data Fig. 3c). Further, Zfp462 mutant ESC colonies showed reduced expression of the pluripotency marker alkaline phosphatase, suggesting a function for ZFP462 in ESC self-renewal. RNA-seq revealed differential expression of ~1800 genes in homozygous Zfp462 mutant ESCs and ~1400 genes in heterozygous Zfp462 mutant ESCs (cutoff: LFC ≥ 1, padj. ≤ 0.01) (Fig. 3c and Extended Data Fig. 3d, e). Gene Ontology terms of differentially expressed genes were related to developmental processes, consistent with the spontaneous ESC differentiation observed upon Zfp462 deletion (Fig. 3d and Extended Data Fig. 3f). Several key developmental TFs were upregulated in both heterozygous and homozygous Zfp462 mutant ESCs, including Bmp4, Gata6, Gata4, and Sox17, required for cell fate specification towards meso- and endodermal lineages (Fig. 3c, e). Gata6 and Sox17 expression is restricted to primitive endoderm in early embryos, and overexpression of these TFs in ESCs promotes their differentiation into primitive/extraembryonic endoderm-like cells 44–46 with similar phenotypes to heterozygous and homozygous mutant Zfp462 ESCs44. Thus, Zfp462 is required to maintain ESC pluripotency and Zfp462 haploinsufficiency upregulates key developmental genes including TFs that promote meso-endodermal lineage-specification.
Fig. 3: Depletion of Zfp462 leads to aberrant expression of lineage specifying genes.
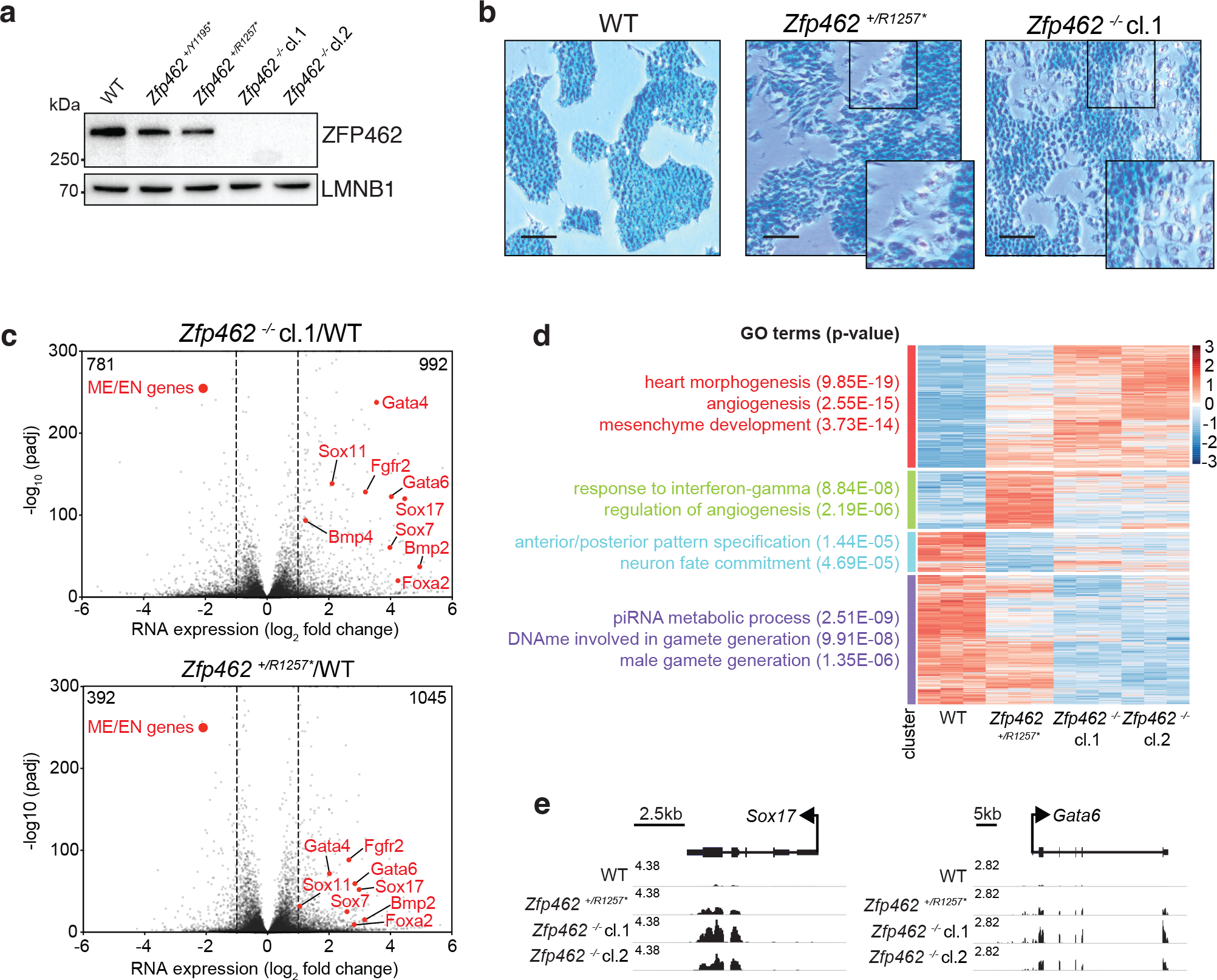
a) Western blot shows ZFP462 protein expression in wild-type (WT), two heterozygous and two homozygous Zfp462 mutant ESC lines. LMNB1 serves as loading control and reference for relative ZFP462 quantification (numbers below). b) Alkaline phosphatase staining of WT, heterozygous and homozygous Zfp462 mutant ESCs. Enlarged region is marked as square in the image. Scale bar = 100μm. c) Volcano plots show gene expression changes in homozygous (top) and heterozygous (bottom) Zfp462 mutant ESCs compared to WT ESCs (n = three replicates). Indicated are the numbers of significantly up- or down regulated genes. (padj. ≤ 0.05; LFC ≥ 0.5). d) Heatmap show cluster analysis of differentially expressed genes (padj. ≤ 0.05; LFC ≥ 1) in WT and Zfp462 mutant ESCs. Top gene ontology (GO) terms and corresponding significance are indicated for each cluster (left). e) Genomic screen shots of Sox17 and Gata6 show mRNA expression levels in WT and Zfp462 mutant ESCs. All RNA-seq profiles are normalized for library size.
ZFP462 represses endodermal genes in neural differentiation
Based on the link between Zfp462/ZNF462 haploinsufficiency and defects in mouse and human neurodevelopment32,33,47, we investigated the role of Zfp462 in neural differentiation. To reduce variability prior to differentiation, we tested if enforcing the naïve ground state of ESCs using inhibitors against MAPK and GSK3 (termed 2i) could restore self-renewal of Zfp462 mutant ESCs. Culturing Zfp462 mutant ESCs under 2i conditions (2i/S/L) suppressed their spontaneous differentiation (Fig. 4a and Extended Data Fig. 4a, b) and ESCs grown in 2i/S/L medium displayed lower ZFP462 expression (Extended Data Fig. 4c, d). We used an established protocol to induce neuronal differentiation starting from naïve ground state ESCs 48. Sequential withdrawal of 2i and LIF initiated ESC differentiation into embryoid bodies (EBs) containing progenitors of ecto-, meso- and endodermal lineages. Subsequent addition of retinoic acid (RA) stimulated ectoderm expansion followed by enrichment of NPCs. ZFP462 expression levels increased during neural differentiation with highest levels in NPCs, consistent with previous reports (Extended Data Fig. 4c, d)47. Importantly, heterozygous and homozygous Zfp462 mutants gave rise to EBs and NPCs with reduced size compared to wild-type ESCs (Fig. 4a and Extended Data Fig. 4b). RT-qPCR analysis revealed that Nanog and Oct4 were downregulated and Pax6 and Ngn2 were upregulated, indicating successful exit from pluripotency and induction of neural differentiation in wild-type and mutant cells (Fig. 4b). However, endodermal markers Gata6 and FoxA2 were strongly induced in Zfp462 −/− ESCs upon 2i withdrawal and remained high even in NPCs (Fig. 4b). Although heterozygous Zfp462 mutant cells showed similar marker gene expression levels to wild-type cells at the onset of differentiation, they failed to downregulate endoderm-specific genes in NPCs similar to KO cells.
Fig. 4: Zfp462 mutant cells show abnormal cell fate specification during neuronal differentiation.
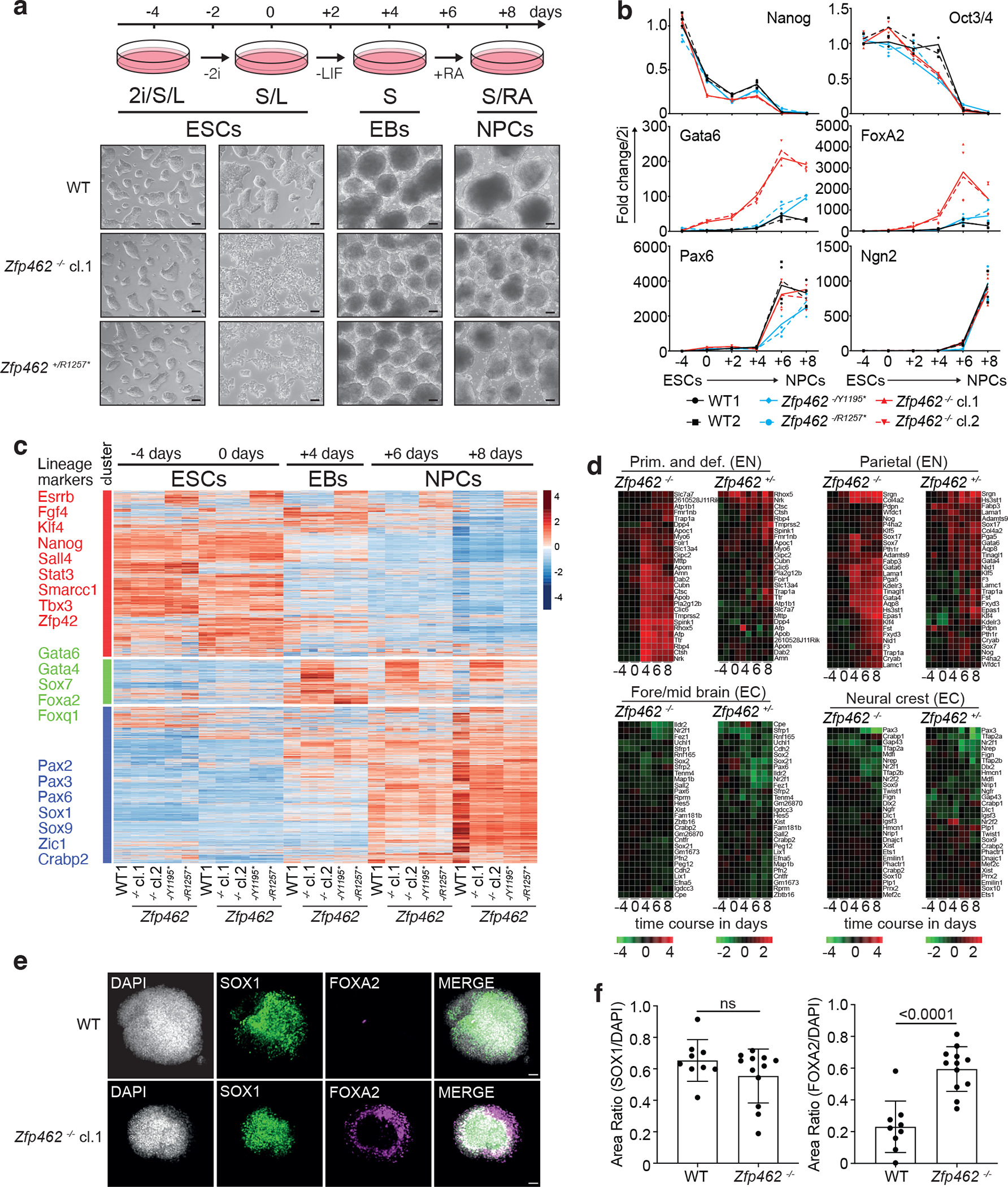
a) Scheme shows design of neural differentiation experiment from ESCs to neural progenitor cells (NPCs) (top). Stepwise withdrawal of 2i inhibitors (2i) and Leukaemia Inhibitory Factor (LIF) leads to formation of cellular aggregates called embryoid bodies (EBs). Subsequent treatment with retinoic acid (RA) induces enrichment of NPCs. Representative bright field images show WT and Zfp462 mutant cells at corresponding stages of neural differentiation. Scale bar = 100μm. b) Line plots shows RT-qPCR analysis of lineage marker expression during neural differentiation (n=two replicates). Expression levels are shown relative to ESCs (2i/S/L). c) Heatmap shows cluster analysis of differentially expressed genes (padj. ≤ 0.05; LFC ≥ 1) in WT and Zfp462 mutant cells during neural differentiation (n = two replicates). Selected lineage marker genes are indicated for each cluster (left). d) Heatmaps show differential expression of selected marker genes specific for endodermal and neural lineages in heterozygous and homozygous Zfp462 mutant cells during neural differentiation. e) Immunohistochemistry analysis shows SOX1 and FOXA2 expression in WT and homozygous Zfp462 mutant at day 8 of neural differentiation. Cell aggregates are counterstained with DAPI. Scale bar = 50μm. f) Bar plots show quantification of SOX1 and FOXA2 immunofluorescence in WT (n = 9 cell aggregates over 2 independent experiments) and Zfp462 −/− cell (n = 12 cell aggregates over 3 independent experiments). Data are presented as mean values +/− SD. P values derived from a two-tailed t-test are indicated.
Next, we profiled global gene expression changes throughout the differentiation time course (Fig. 4c). Cluster analysis of differentially regulated genes yielded three separate groups based on distinct expression kinetics. Genes in cluster 1 comprising ESC-specific markers, were highly expressed in ESCs under 2i/S/L and S/L conditions and were downregulated in EBs and NPCs. Cluster 2 contained ~1300 genes including meso- and endodermal lineage markers. These genes were induced in EBs and gradually decreased in NPCs derived from wild-type ESCs. In contrast, in heterozygous and homozygous Zfp462 mutant cells cluster 2 genes were highly induced in EBs and remained overexpressed in NPCs (Fig. 4c–d and Extended Data Fig. 4e). Addition of RA induced upregulation of cluster 3 genes including key neural TFs. Compared to wild-type cells, upregulation of neural genes was lower in Zfp462 mutant NPCs suggesting a delay in neural differentiation and/or maturation (Fig. 4c–d and Extended Data Fig. 4e). Although deregulation of meso- and endodermal genes was less severe, neural gene expression was similarly impaired in heterozygous and homozygous mutant cells. This is consistent with significant neurodevelopmental defects seen in patients with ZNF462 haploinsufficiency32,33. Finally, immunohistochemistry in NPCs revealed that the deregulation of genes extended to the protein level (Fig. 4e–f). Most wild-type NPCs were SOX1-positive whereas FOXA2 expression was barely detected, reflecting efficient neural differentiation49. In contrast, enrichment and distribution of FOXA2-positive cells was strongly increased in Zfp462 −/− NPCs. Thus, Zfp462 deletion results in misspecification towards meso-endodermal lineages under conditions that normally induce neural cell identity. We infer that ZFP462 is required to silence expression of lineage non-specific genes and promotes specification into neural cell lineages, consistent with developmental delay and neural defects of heterozygous Zfp462 KO mice47.
ZFP462 mediates sequence-specific G9A/GLP recruitment
To investigate how ZFP462 contributes to lineage specification, we used ChIP-seq to profile binding of Avi-tagged ZFP462 genome-wide, which revealed 16,264 significantly enriched sites (Fig. 5a and Extended Data Fig. 5a). Most ZFP462 peaks (86%) were found in intergenic and intronic regions, distal to transcription start sites. Since the related family of Krüppel-associated box (KRAB) domain-containing zinc finger proteins (KZFP) target heterochromatin modifiers to silence transposable elements (TE)50, we analyzed ZFP462 binding at repeat DNA families and uncovered a large overlap (Fig. 5b). Among the various repeat DNA classes, ZFP462 binding was strongly enriched at mouse-specific long terminal repeat (LTR) families of endogenous retroviruses (ERVs) (58.11%). Specifically, ZFP462 frequently occupied RLTR9a, RLTR9d, RLTR9d2, RLTR9a2 and RLTR9e families, suggesting a role in repressing these TE subfamilies via G9A/GLP recruitment (Fig. 5c).
Fig. 5: ZFP462 establishes H3K9me2 containing heterochromatin by recruiting GLP and WIZ proteins.
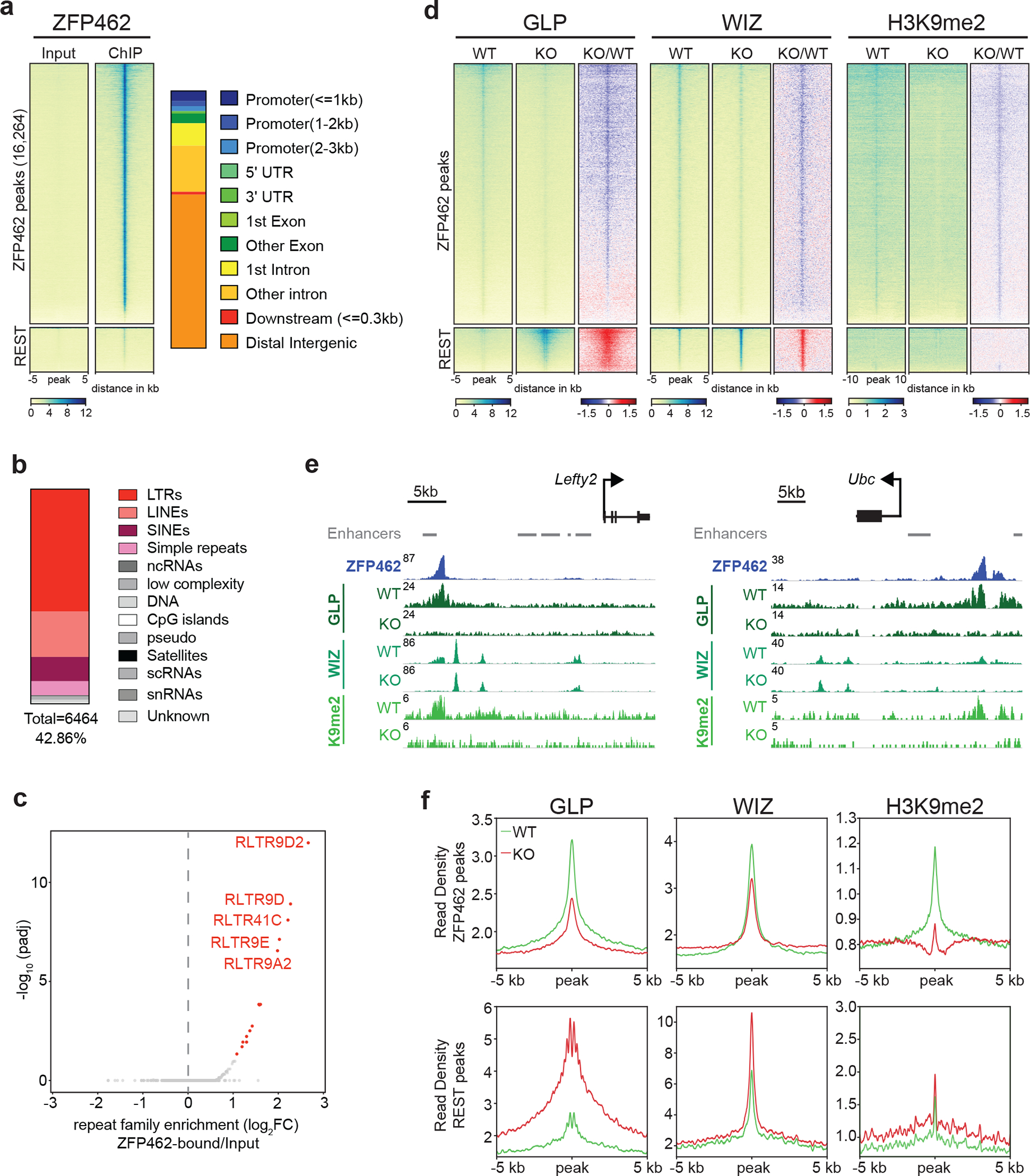
a) Heatmaps of ZFP462 ChIP-seq enrichment at significant ZFP462 peaks (16,264) (top cluster) and REST peaks (bottom cluster) in ESCs. Each row represents a 10 kb window centred on peak midpoint, sorted by ZFP462 ChIP signal. Input signals for the same windows are shown on the left. Below are scale bars. (n = average distribution of two replicates). Bar plot, on the right, shows percentage of genomic features overlapping with ZFP462 peaks. b) Bar plot shows fraction of repetitive DNA types overlapping with ZFP462 peaks. 39.76 % of ZFP462 peaks are associated with repeat elements. c) Volcano plot shows enrichment and corresponding significance of TE families overlapping with ZFP462 peaks. d) Heatmaps of GLP, WIZ and H3K9me2 ChIP-seq enrichment at ZFP462 and REST peaks in WT and Zfp462 KO ESCs. Blue-to-red scaled heatmap represents corresponding ChIP-seq enrichment ratio between Zfp462 KO versus WT (KO/WT). All heat maps are sorted by GLP enrichment signal in WT. For GLP and WIZ, each row represents a 10 kb window centred on ZFP462 peak midpoints. For H3K9me2, each row represents a 20 kb window (n = average distribution of two replicates). e) Screen shots of two selected genomic regions with ZFP462 peaks display GLP, WIZ and H3K9me2 ChIP-seq signals in WT and Zfp462 KO ESCs. Grey bars indicate ENCODE enhancer annotations. ChIP-seq profiles are normalized for library size. f) Metaplots show average GLP, WIZ and H3K9me2 signal at ZFP462 and REST peaks in WT and Zfp462 KO ESCs. For each plot, read density is plotted at 10 kb window centred on peak midpoints.
To determine its role in targeting the G9A/GLP complex to chromatin, we used CRISPR-mediated deletion to remove ZFP462 in Avi-GLP expressing ESCs and profiled GLP, WIZ and H3K9me2 (Extended Data Fig. 5b–d). In wild-type ESCs, ZFP462 targets were co-occupied by GLP and WIZ, similar to REST binding sites, and displayed modest levels of H3K9me2 (Fig. 5d–f). HMTase subunits and histone methylation were markedly reduced at ZFP462 targets in ESCs lacking ZFP462. By comparison, REST TF binding sites displayed increased GLP and WIZ ChIP-seq signals upon Zfp462 deletion. Since global G9A levels remained unchanged in the mutant (Extended Data Fig. 4c), we conclude that the gain at REST binding sites reflects genomic redistribution of the H3K9-specific HMTase in the absence of ZFP462 interaction. Next, we used CRISPR to delete ZFP462 in Avi-HP1γ expressing ESCs and tested its role in HP1γ chromatin targeting (Extended Data Fig. 5c). Compared to known binding sites such as ADNP targets 51, Avi-HP1γ showed only modest enrichment at ZFP462 peaks (Extended Data Fig. 5e). Moreover, the low HP1γ signal remained largely unchanged by Zfp462 deletion arguing against a significant role of ZFP462 in directly recruiting HP1γ to its target loci. Thus, ZFP462 is primarily required for sequence-specific targeting of the G9A/GLP complex to induce heterochromatin modifications at its targets.
G9A/GLP restricts DNA accessibility at ZFP462 peaks
Transcriptional silencing by G9A/GLP is associated with chromatin compaction 5. We investigated whether failure to target G9A/GLP and H3K9me2 leads to increased DNA accessibility in Zfp462 KO ESCs using ATAC-seq. Despite G9A/GLP occupancy and H3K9me2, most ZFP462 peaks were already accessible in wild-type ESCs (Fig. 6a, b and Extended Data Fig. 6a). Nonetheless, DNA accessibility was further increased at target loci upon loss of G9A/GLP and H3K9me2 in Zfp462 KO ESCs. In contrast, REST binding sites remained unchanged (Fig. 6a–c).
Fig. 6: ZFP462 targeted heterochromatin restricts DNA accessibility and TF binding.
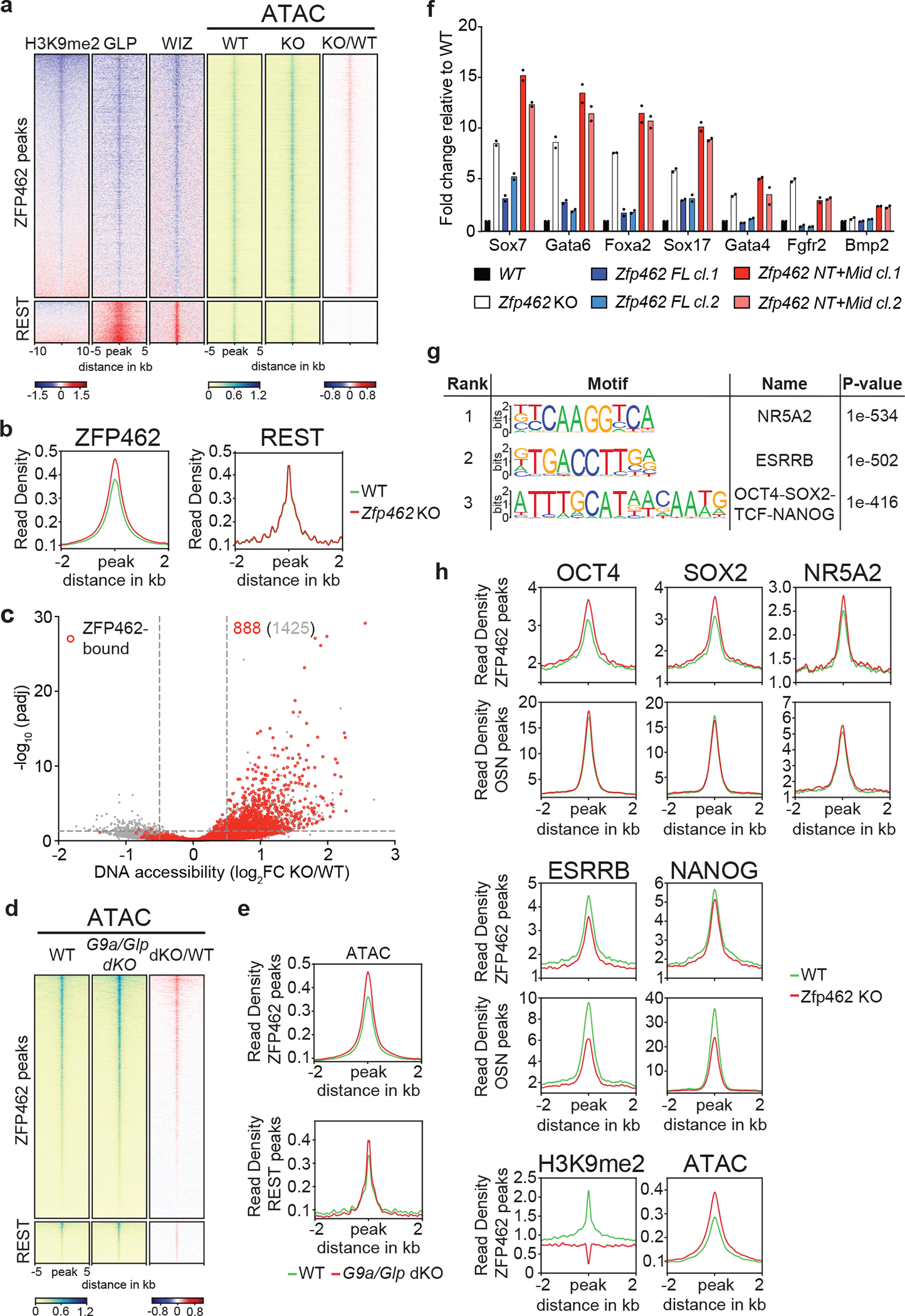
a) Heatmaps of GLP, WIZ and H3K9me2 ChIP-seq enrichment ratios between Zfp462 KO versus WT (KO/WT) ESCs at ZFP462 and REST peaks. On the right, heatmaps of ATAC-seq signal at ZFP462 and REST peaks in WT and Zfp462 KO ESCs. Blue-to-red scaled heatmap represent corresponding enrichment ratios between Zfp462 KO versus WT (KO/WT) ESCs. GLP, WIZ and ATAC-seq heatmaps represent a 10kb window, H3K9me2 heatmap represent a 20 kb window centred on peak midpoints, sorted by H3K9me2 KO/WT enrichment ratio (n = average distribution of two ChIP-seq replicates and average distribution of three ATAC-seq replicates). b) Metaplots show average profiles of ATAC-seq signal in WT (green) and Zfp462 KO (red) ESCs at ZFP462 peaks (left) and REST peaks (right). c) Volcano plot shows DNA accessibility changes between WT and Zfp462 KO ESCs. X-axis represents fold change in accessibility and corresponding significance on Y-axis. Differentially accessible sites bound by ZFP462 are highlighted in red. Indicated in grey is the number of loci with increased DNA accessibility and in red the number of ZFP462-bound loci with increased DNA accessibility with significance < 0.05. d) Heatmaps of ATAC-seq signal at ZFP462 and REST peaks in WT and G9a/Glp dKO ESCs. Blue-to-red scaled heatmap represent corresponding enrichment ratios between G9a/Glp dKO versus WT (KO/WT) ESCs. Each row represents a 10 kb window centred on peak midpoints, sorted by dKO/WT enrichment ratio (n = average distribution of two ATAC-seq replicates). e) Metaplots show average profiles of ATAC-seq signal in WT (green) and G9a/Glp dKO (red) ESCs at ZFP462 peaks (above) and REST peaks (below). f) RT-qPCR expression analysis of meso-endodermal marker genes in WT, Zfp462 KO and Zfp462 KO ESCs expressing ZFP462 FL or ZFP462 NT+Mid. n = 2 independent biological replicates. g) HOMER analysis of known DNA sequence motifs enriched at significant ZFP462 peaks. Top ranked DNA sequence motifs and respective significance values are shown in the table. h) Metaplots show average pluripotency TF ChIP-seq signal in WT and Zfp462 KO ESCs at ZFP462 peaks with LFC ≥ 1 in KO/WT H3K9me2 ChIP signal loss and at shared OCT4-SOX2-NANOG (OSN) peaks that are not bound by ZFP462. (n = average distribution of two replicates).
To examine if G9A/GLP-dependent chromatin modifications directly contribute to restricting DNA accessibility at ZFP462 peaks, we analyzed available ATAC-seq data of G9a/Glp double knockout (G9a/Glp dKO) ESCs 52. As expected, G9A/GLP loss led to increased DNA accessibility at REST targets. DNA accessibility was also increased at ZFP462 peaks arguing that G9A/GLP also acts as transcriptional co-repressor at these targets (Fig. 6d, e).
To determine if G9A/GLP regulation of DNA accessibility is required for ZFP462-dependent silencing of lineage non-specific genes, we compared differential gene expression between G9a/Glp dKO ESCs and Zfp462 KO ESCs which revealed an overall positive, but moderate correlation (Extended Data Fig. 6b). We speculate that the discrepancy is likely because in ESCs G9A/GLP interacts with multiple TFs including REST, enforcing repression of diverse sets of target genes.
To address whether ZFP462-mediated recruitment of the G9A/GLP complex is required for meso-endodermal gene silencing, we used CRISPR-Cas9 to introduce sequences encoding either full-length ZFP462 (ZFP462 FL) or CT-truncated ZFP462 (ZFP462-NT+Mid) at the endogenous locus in Zfp462 KO ESCs, isolating two independent ESC clones for each Zfp462 rescue construct (Extended Data Fig. 6c–e). Unlike ZFP462-NT+Mid, expression of ZFP462 FL rescued ESC morphology (Extended Data Fig. 6f). Furthermore, ATAC-seq analysis showed that ZFP462-FL, but not ZFP462-NT+Mid, mostly restored wild-type levels of DNA accessibility at ZFP462 peaks (Extended Data Fig. 6g). Finally, RT-qPCR showed that ZFP462-FL but not ZFP462-NT+Mid restored silencing in Zfp462 KO ESCs (Fig. 6f). ZFP462-FL expression also restored endodermal marker gene silencing during NPC differentiation, whereas Gata6 and FoxA2 expression remained high upon rescue with ZFP462-NT+Mid (Extended Data Fig. 6h). We infer that ZFP462 recruits G9A/GLP to restrict DNA accessibility and enforce silencing of lineage non-specific genes, potentially by affecting TF occupancy at cis-regulatory sequences.
ZFP462-dependent heterochromatin restricts TF binding
TEs contribute up to 25% of binding sites for pluripotency TFs and play an integral role in the core regulatory network of ESCs53,54. Indeed, the mouse-specific RLTR9 subfamilies contain sequence motifs for pluripotency TFs including OCT4 and SOX2 and promote gene expression in ESCs 55–57. Intriguingly, consensus sequences for several pluripotency TFs were enriched at ZFP462 peaks (Fig. 6g). ChIP-seq profiling of OCT4, SOX2, NANOG, NR5A2 and ESRRB confirmed robust binding at ZFP462 peaks overlapping with H3K9me2 in wild-type ESCs (Extended Data Fig. 7a). Notably, ZFP462 also colocalized with OCT4, SOX2 and NANOG at upstream regulatory sequences of the Oct3/4 gene suggesting that it limits gene expression by targeting G9A/GLP (Extended Data Fig. 7b).
The precise regulation of pluripotency TFs is essential to maintain ESC identity 58; a less than twofold increase in OCT4 expression is sufficient to trigger spontaneous differentiation of ESCs into primitive endoderm and mesoderm 36,59–62. To investigate if heterochromatin mediated by G9A/GLP affects TF interactions, we analyzed TF binding changes at ZFP462 targets (Fig. 6h, Extended Data Fig. 7a). OCT4, SOX2 and NR5A2 displayed increased enrichment at ZFP462 targets in KO ESCs, concomitant with loss of H3K9me2. Notably, binding of these TFs was unaffected at OSN peaks that do not overlap with ZFP462, indicating specificity. Consistent with spontaneous differentiation into primitive endoderm, NANOG and ESRRB were downregulated and had reduced occupancy at OSN peaks (Fig. 6h, Extended Data Fig. 7a). In contrast, NANOG and ESRRB binding remained higher at ZFP462 peaks in KO ESCs, consistent with a relative increase in DNA accessibility. OCT4 binding was also increased at ZFP462 target site upstream of the Oct3/4 gene (Extended Data Fig. 7b). We infer that ZFP462-dependent recruitment of G9A/GLP restricts TF binding at its target sites.
ZFP462 targets heterochromatin to repress enhancers in ESCs
To examine the role of ZFP462 at cis-regulatory sequences we used the ChromHMM annotation of ESC chromatin states 63,64. Nearly half of ZFP462 peaks overlapped with genomic regions categorized as active enhancers (44.6%, Fig. 7a). ZFP462-bound TEs represented 35.37% of ESC enhancers (Extended Data Fig. 7c). In contrast, less than 13% of ZFP462 peaks were associated “repressed chromatin” and “constitutive heterochromatin” categories. ChIP-seq profiling of euchromatic histone modifications, H3K4me1, H3K4me3 and H3K27ac, confirmed that many ZFP462 peaks localized at active enhancers in ESCs (Fig. 7b). Notably, G9A/GLP levels were comparable at all ZFP462 peaks, but H3K9me2 abundance was low at active ESC enhancers and accumulated mostly at ZFP462 peaks devoid of euchromatic chromatin modifications. Loss of G9A/GLP and H3K9me2 in Zfp462 KO ESCs led to increased H3K27ac at active ESC enhancers and at a subset of intergenic ZFP462 peaks marked primarily by H3K4me1, potentially revealing aberrant activation of silent enhancers (Fig. 4b).
Fig. 7: ZFP462 represses meso-endodermal enhancers in ESCs.
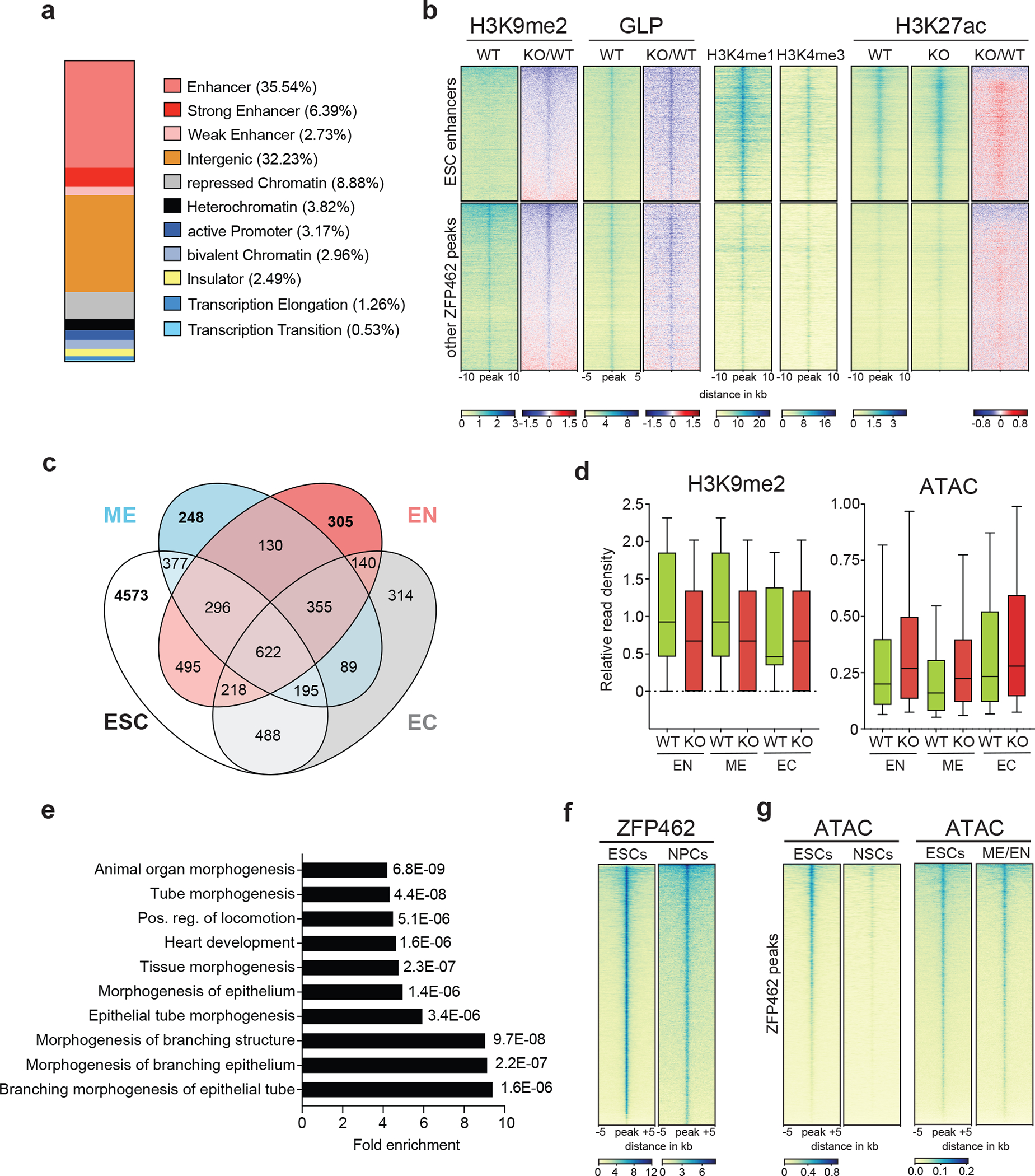
a) Bar plot shows percentage of ZFP462 ChIP-seq peaks overlapping with ChromHMM states in ESCs. b) Heatmaps of GLP, H3K9me2, H3K4me1, H3K4me3 and H3K27ac ChIP-seq signals at ZFP462 peaks separated into two clusters: ESC enhancers and other ZFP462 peaks. Blue-to-red scaled heatmap represent corresponding enrichment ratios between Zfp462 KO versus WT (KO/WT). GLP heatmap represent 10kb and histone modifications heatmaps represent a 20 kb window centred on peak midpoints, sorted by H3K9me2 KO/WT enrichment ratio (n = average distribution of two ChIP-seq replicates). c) Venn diagram shows overlap of ZFP462 peaks with ChromHMM-annotated enhancers in ESC, Endoderm (EN), Mesoderm (ME) and Ectoderm (EC). d) Box plots shows enrichment of H3K9me2 ChIP signal and ATAC-seq signal in WT and Zfp462 KO ESCs at ZFP462 peaks overlapping with Endoderm (EN)-, Mesoderm (ME)- and Ectoderm (EC)-specific enhancers. n = 305 (EN), n = 248 (ME), n = 314 (EC). Shown are median (horizontal line), 25th to 75th percentiles (boxes), and 90% (whiskers). Outliers are excluded. e) Bar plot shows GO term analysis of biological processes of genes with significant differential expression (KO/WT, LFC ≥ 1 and padj. ≤ 0.05) located proximal to ZFP462 peaks annotated as meso-endoderm-specific enhancers77. f) Heatmap of ZFP462 ChIP-seq enrichment at ZFP462 peaks in ESCs and NPCs. ChIP-seq rows represent 10 kb window centred on ZFP462 peak midpoints, sorted by ZFP462 ChIP-seq signal intensity (n = average distribution of two replicates). g) Heatmaps of ATAC-seq signal at ZFP462 peaks in ESCs and NSCs (left). Heatmaps of ATAC-seq signal at ZFP462 peaks in ESCs and meso-endoderm (ME/EN) cells 66. ATAC-seq rows represent 10 kb window centred on ZFP462 peak midpoints, sorted by ESCs ATAC-seq signal intensity.
To determine if ZFP462-dependent heterochromatin is required to repress mesoderm- and endoderm-specific enhancers in ESCs, we identified ZFP462 peaks overlapping with enhancers in endoderm (EN), mesoderm (ME) and ectoderm (EC). We used ChomHMM annotation based on epigenomic profiling of mouse fetal tissues of E14 65 and identified 4,272 ZFP462 peaks (26.3%) overlapping with enhancers that are active in at least one of these three primary germ layers. From this set we selected enhancers specific for one germ layer and inactive in ESCs to analyze H3K9me2 and DNA accessibility changes in wild-type and Zfp462 KO ESCs (Fig 7c). EN- and ME-specific enhancers displayed ZFP462-dependent H3K9me2 enrichment and DNA accessibility in ESCs (Fig. 7d), in contrast with EC-specific enhancers. To determine if loss of ZFP462-dependent heterochromatin led to increased TF binding at EN- and ME-specific enhancers, we first performed a motif analysis to identify the consensus sequences with the highest enrichment at ZFP462 peaks overlapping ME/EN-specific enhancers. We uncovered NR5A2 and ESRRB motifs (Extended Data Fig. 7d), whereas OCT4-SOX2-NANOG motifs were not enriched. Our TF ChIP-seq data showed that NR5A2 occupancy was markedly increased at ME/EN-specific enhancers, but unchanged at EC-specific enhancers, in Zfp462 KO relative to wild-type ESCs (Extended Data Fig. 7e). Consistent with its global downregulation at ZFP462 peaks and OSN targets, ESRRB binding decreased at all germ layer-specific enhancers. We conclude that inactivation of a set of ME/EN-specific enhancers in ESCs relies on ZFP462-dependent heterochromatin to limit DNA accessibility and aberrant TF binding.
Finally, we examined if elevated TF binding and aberrant activation of EN- and ME-specific enhancers at ZFP462 peaks impacts transcriptional regulation. Genes proximal to ZFP462 peaks overlapping with activated EN- and ME-specific enhancers were more frequently upregulated, arguing that enhanced DNA accessibility and TF binding promotes aberrant gene expression (Extended Data Fig. 7f). To test if proximal gene regulation is linked to spontaneous meso-endodermal differentiation, we performed GO term analysis. Notably, the most enriched GO terms were related to developmental processes, in particular cardiovascular system development (Fig. 7e). These results suggest that ZFP462 binds to enhancers and controls their regulatory activity in ESCs, particularly preventing unscheduled activation of EN- and ME-specific enhancers. In the absence of ZFP462, these enhancers have increased DNA accessibility and TF binding, leading to aberrant expression of proximal genes involved in meso-endoderm differentiation.
ZFP462 represses ME/EN enhancers in neuronal cells
Our data argue that ZFP462 is required for normal neuro-differentiation and development by regulating enhancers in neural cell lineages. To determine if ZFP462 occupies enhancers later in development, we differentiated Avi-ZFP462-expressing ESCs and mapped its genome-wide distribution in NPCs. ZFP462 occupancy was largely conserved between ESCs and NPCs (Fig. 7f). To investigate DNA accessibility at ZFP462 targets in neural cells, we performed ATAC-seq in neural stem cells (NSCs) isolated from the subventricular zone of adult mice. ZFP462 was abundant in NSCs (Extended Data Fig. 7g), and we found that ZFP462-bound enhancers were mostly inaccessible in wild-type NSCs by ATAC-seq (Fig. 7g). These data suggest that G9A/GLP-dependent heterochromatin efficiently restricts inappropriate TF binding in NSCs (Fig. 7g).
Finally, we interrogated public ATAC-seq data to compare DNA accessibility in meso-endoderm progenitors that lack ZFP462 expression (Extended Data Fig. 3a) 66. In contrast to NSCs, meso-endoderm progenitors displayed substantial DNA accessibility at ZFP462 targets, indicative of lineage-specific enhancer activity (Fig. 7g). We infer that ZFP462 controls endoderm-specific enhancers by recruiting G9A/GLP and establishing heterochromatin in ESCs and cells of the neural lineage. Our data suggest that ZFP462 promotes robust differentiation along the neural lineage by preventing lineage non-specific gene expression.
Discussion
Our CRISPR genetic screen for regulators of heritable H3K9me2/3-dependent gene silencing in mouse ESCs uncovered Zfp462, an essential gene encoding a C2H2 zinc-finger TF, previously of unknown function. Heterozygous mutations of the human ortholog, ZNF462, cause the neurodevelopmental disorder Weiss-Kruszka syndrome 33, but the underlying molecular mechanism is unknown. We demonstrate that ZFP462 regulates cell identity by preventing aberrant expression of endoderm-specific genes in ESCs and during neural differentiation.
ZFP462 binds to enhancer sequences that contain ESC-specific TF binding motifs and represses these loci by recruiting G9A/GLP, which catalyzes H3K9me2. Establishing heterochromatin at ZFP462 target sites restricts DNA accessibility and thereby controls TF binding and enhancer activity. Failure to target G9A/GLP-dependent heterochromatin in heterozygous and homozygous Zfp462 deletion mutants results in mis-expression of meso-endoderm-specifying TF, which impairs self-renewal and interferes with timely establishment of neural-specific gene expression during NPC differentiation. Neural differentiation is similarly impaired in heterozygous and homozygous Zfp462 mutant cells, consistent with the neurodevelopmental defects observed in patients with ZNF462 haploinsufficiency.
ZFP462 maintains closed chromatin at a subset of ME/EN-specific enhancers, blocking NR5A2 binding which otherwise may promote meso-endoderm cell fates 67–69. In contrast, most ZFP462 targets are co-occupied by pluripotency TFs and show hallmarks of enhancer activity in ESCs despite the presence of G9A/GLP-dependent heterochromatin modifications. The capacity of OCT4 and SOX2 to engage with sequence motifs in the context of heterochromatin is a unique feature of pioneer TFs and a critical first step in iPSC reprogramming 70,71. Nevertheless, OCT4 and SOX2 binding is limited by positioning the binding motif on the nucleosome surface and chromatin accessibility 72,73. For example, to engage with its target sites in closed chromatin, OCT4 requires the chromatin remodeling factor Brg1 to mobilize nucleosomes 74. Here, we show that ZFP462-dependent heterochromatin also contributes to limiting DNA accessibility for OCT4 binding at enhancers preventing aberrant gene expression. Precise levels of OCT4 are critical for ESC self-renewal and govern cell type specification during early embryogenesis 36. Thus, by modulating OCT4 binding at ESC-specific enhancers, ZF462 may contribute to the quantitative readout of OCT4 and define one of the earliest steps of cell fate specification during embryogenesis.
Although ZFP462 is conserved across vertebrates, it predominantly bound to evolutionarily young, species-specific TEs 55. It is possible that ZFP462-bound TEs contain DNA sequence motifs that are much “older” that the rest of the TE sequence. Indeed, mouse-specific RLTR9 subfamilies harbor conserved binding sites for key pluripotency TFs including OCT4 and SOX255,56. Adaptation of pluripotency TF binding sites bestows TEs with enhancer activity specific for germ cells and preimplantation embryos optimizing the likelihood to persist by vertical transmission54,75,76. We surmise that ZFP462/ZNF462 emerged in vertebrates to repress TEs by recognizing a DNA sequence that overlaps with pluripotency TF binding motifs. Therefore, TE-derived enhancers were co-opted to contribute not only to the ESC-gene regulatory network via pluripotency TFs, but also to cell specification via ZFP462.
Based on our findings, we propose that the human ortholog ZNF462 regulates G9A/GLP and targets non-neural genes for repression during neurogenesis. Failure to silence lineage non-specific genes due to a ZNF462 heterozygous LOF mutation might impair neural cell differentiation and underlie neurodevelopmental pathology. However, given the species-specificity of ZFP462-regulated TEs in mouse, we predict that ZNF462 will have different human targets controlling a distinct gene regulatory network compared to mice. Nevertheless, our findings predict that ZNF462-linked pathology involves loss of heterochromatin silencing at enhancers and interference in neural differentiation by lineage non-specific gene expression. Our results argue that Weiss-Kruszka syndrome arises from deregulation of neural lineage-specific gene expression early in embryogenesis, consistent with the broad defects affecting cells of neuronal and neural crest origin.
Methods
The experiments in this manuscript are in complience with relevant guidelines and ethical regulations.
Cell culture
Mouse Embryonic Stem Cells (ESCs) (TC1/RRID:CVCL_M350) were cultured without feeders on gelatin-coated dishes in DMEM supplemented with 15% fetal bovine serum (FBS; F7524/Sigma), 1x non-essential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), 2 mM L-glutamine (Gibco), 0.1 mM 2-mercaptoethanol (Sigma), 50 mg ml−1 penicillin, 80 mg ml−1 streptomycin and homemade LIF, at 37 °C in 5% CO2. Cells were regularly tested for mycoplasma. Neural Stem Cells (NSCs) were isolated from subventricular zone of 7–8 week mouse brain and cultured as previously described 78.
Generation of TetO Oct3/4 reporter ESCs and delivery of TetR-FLAG-fusion constructs
TetO Oct3/4 reporter ESCs were derived from the original Oct4 reporter (CiA:Oct4) ESCs13 by excising the floxed Neomycin gene cassette with Cre-mCherry. After confirming successful excision by genotyping PCR, CRISPR-assisted homologous recombination was employed using electroporation of a sgRNA/Cas9 ribonucleoprotein (RNP) complex and the donor plasmid YR9 (Supplementary Table 5) to introduce a TetO-BFP reporter cassette (7xTetO-PGK-Puro-BFP-pA) downstream of the Oct4-GFP reporter. Finally, a TetO Oct4 reporter ESCs clone expressing GFP and BFP was isolated by FACS analysis and genotyping. constructs encoding TetR-3xFLAG-GOI-P2A-mCherry were transduced into TetO Oct4 reporter ESCs using lenti virus.
CRISPR genetic screen
A TetO Oct4 reporter ESC clone with stable Cas9 expression was generated for CRISPR genetic screening. Next, the Cas9-expressing ESC clonal line was transduced with lenti virus encoding TetR-3XFLAG-HP1-P2A-mCherry and mCherry-positive ESCs were isolated by FACS for clonal expansion. ESC clone #H5 was isolated based on robust initiation and maintenance Oct4 reporter gene silencing. A retroviral sgRNA library targeting nuclear factors was used for CRISPR screening as previously described 37 with minor modifications. After 5 days and completion of G418 selection, cells were cultured in ESC medium containing doxycycline for 2 days. The GFP positive cell populations of doxycycline-treated cells were sorted on a FACSAria II cell sorter (BD Biosciences). Unsorted mutant populations were served as background controls. Following genomic DNA isolation from GFP-positive sorted and unsorted cells, sgRNA cassettes were amplified by PCR and subjected to NGS sequencing on an Illumina HiSeq 2500. Data was analysed as previously described 37. Gene enrichment was determined using MAGeCK 38.
Generation of Zfp462 endogenously tagged ESC line
For endogenous Zfp462 tagging, 5×106 Rosa26:BirA-V5-expressing cells 79 were electroporated with sgRNA/Cas9 ribonucleoprotein complex (sgRNA pre-incubated with Cas9 in cleavage buffer) mixed with 15μg of donor plasmid containing the Tag (Avi-GFP-3XFLAG) sequence flanked by ~500bp of homology arms (Supplementary Table 5) using Mouse Embryonic Stem Cell Nucleofector™ Kit (Lonza). GFP-positive ESCs were isolated by FACS and seeded for clonal expansion after 48 hours. Individual ESC clones were screened by genotyping and western blot.
Editing of endogenous Zfp462 to generate mutant and rescue ESCs
Zfp462−/− ESCs were generated with CRISPR-Cas9 using two different sgRNAs (Supplementary Table 5). The sgRNA sequences were cloned into SpCas9-Thy1.1 plasmid which was electroporated using Mouse Embryonic Stem Cell Nucleofector™ Kit (Lonza). After 36 hours, Thy1.1-positive ESCs were isolated by FACS and seeded for clonal expansion. Individual clones were screened by genotyping and western blot.
Heterozygous mutant ESCs were generated as previously described 80 with few modifications. ESCs were electroporated with sgRNA/CAS9 ribonucleoprotein (RNP) complex and a mix of 1:1 ratio WT/mutant donor single-stranded oligodeoxynucleotides carrying PAM mutation (Supplementary Table 5) using Mouse Embryonic Stem Cell Nucleofector™ Kit (Lonza). 24 hours after transfection, ESCs were seeded for clonal expansion. Individual ESC clones were screened by PCR followed by Restriction Fragment Length Polymorphism (RFLP). In case of Zfp462+/Y1195*, WT allele is resistant to MseI and mutant allele is sensitive to MseI. In case of Zfp462+/R1257*, WT allele is sensitive to TaqI and mutant allele is resistant to TaqI. Final ESC clones were also confirmed by Sanger sequencing and western blot.
Zfp462-FL and Zfp462-NT+Mid rescue ESCs were generated by transfecting sgRNA1/Cas9 and sgRNA6/Cas9 ribonucleoprotein (RNP) complex (1:1 ratio mix) with a repair template carrying Zfp462-FL-GFP-3xFLAG-Zfp462 3’UTR-pA or Zfp462-NT+Mid-GFP-3xFLAG-Zfp462 3’UTR-pA rescue construct into Zfp462 −/− ESCs using Mouse Embryonic Stem Cell Nucleofector™ Kit (Lonza) (Extended Data Fig. 6c, Supplementary Table 5). GFP-positive ESCs were isolated by FACS after 36 hours and seeded for clonal expansion. Individual ESC clones were screened by genotyping PCR and western blot.
Western blot analysis
Cells were grown to confluency on 10 cm plates, collected in PBS, pelleted by 3 min centrifugation at 300g, and cell pellets were then resuspended in 5ml of buffer 1 (10 mM Tris-HCl at pH 7.5, 2 mM MgCl2, 3 mM CaCl2, Complete protease inhibitor (Roche)), incubated for 20 min at 4 °C followed by a centrifugation step. The cell pellet was resuspended in buffer 2 (10 mM Tris-HCl at pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.5% IGEPAL CA-630, 10% glycerol, Roche Complete Protease Inhibitor), incubated for 10 min at 4 °C followed by centrifugation. Isolated nuclei were lysed in buffer 3 (50mM HEPES-KOH, pH7.3, 200mM KCl, 3.2mM MgCl2, 0.25% Triton X-100, 0.25% NP-40, 0.1% Na-deoxycholate and 1mM DTT and Complete protease inhibitor (Roche)), 2ul of Benzonase (E1014, Millipore) was added and incubated for 1 hour at 4 °C. The lysate was cleared by centrifugation at 16,000g for 10 min at 4 °C, and the protein concentration in the nuclear extract was determined using the Broadford protein assay. For western blotting, 30 μg of protein was resolved on NuPAGE-Bis-Tris 4–12% mini protein gels (Invitrogen), which were transferred on to polyvinylidene fluoride (PVDF) membrane, blocked for 30 min in 5% non-fat dry milk in TBS with 0.1% Tween 20 (TBST), and stained with primary antibodies at 4 °C overnight. The primary antibodies used for western blotting were mouse anti-Flag (1:1,000, Sigma clone M2), rabbit anti-ZNF462(1:500, PA5–54585 - Invitrogen), rabbit anti-Lamin B1(1:5,000, ab16048 - abcam), rabbit anti-G9a (1:1,000, CST# 68851 - Cell Signaling) and mouse anti-H3K9me2 (1:1,000, ab1220 - Abcam), Signal was detected with corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies and Clarity Western ECL substrate (170–5061, Bio-Rad).
Differentiation of ESCs to Neural Progenitor Cells (NPCs) and immunostaining
Differentiation was performed as previously described 48 with few modifications. ESCs are cultured on gelatin coated plates. In addition, cells are cultured in ESC medium containing 2i inhibitors (3 μM glycogen synthase kinase (GSK) inhibitor and 10 μM MEK inhibitor) for five passages before starting the differentiation experiment. For immunostaining, four-day retinoic acid treated EBs were fixed with 4% PFA (Thermo Scientific, #28906) for 30 min, washed 3x with PBS and stored at 4 °C until further processing. EBs were incubated in blocking solution, which consisted of PBS (Gibco, #14190094), 4% goat/donkey serum (Bio-Rad Laboratories, #C07SA/Bio-Rad Laboratories, #C06SB) and 0.2% Triton X-100 (Sigma-Aldrich, #T8787) for at least 15 min before application of primary antibody in the same blocking solution. The primary antibody was incubated with the EBs for 2 days at 4C while shaking. Used primary antibodies were SOX1 (R&D Systems, #AF3369, 1:200) and FOXA2 (Cell Signaling Technologies, #8186T, 1:200). Subsequently, EBs were washed twice for 15 min each using a PBS/Tween20 solution (0.1% Tween20, Sigma-Aldrich, #P1379). A secondary antibody was then applied 1:500 in blocking solution for another 2 days at 4C, while shaking. Secondary antibodies used were: Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 (Invitrogen, #A32814) and Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 (Invitrogen, # A32733). DAPI (Sigma-Aldrich, #9542) was added to the secondary antibody staining solution. Afterward, EBs were washed once with PBS/Tween20 solution for 15 min followed by a second wash with the same solution for 24h at 4C while shaking. Finally, EBs were stored at 4C prior to imaging on glass slides, in FocusClear (CellExplorer Labs, #FC-101) clearing solution. Image acquisition was carried out using Spinning Disk Confocal Olympus.
Transcriptome analysis by RT–qPCR, RNA-seq and QuantSeq
For RT-qPCR experiments, total RNA was extracted from cells using the Qiagen RNeasy mini kit with on-column DNase digestion step. Total RNA (500 ng) was reverse transcribed using the SuperScript™ III Reverse Transcriptase (Invitrogen). RT–qPCR was performed on a CFX96 Real-Time PCR System (Bio-Rad) using the Promega GoTaq® qPCR Master Mix (Ref: A6001) with RT-qPCR primers described in Supplementary Table 5. Relative RNA levels were calculated from Ct values according to the ΔCt method and normalized to Tbp mRNA levels. For RNA-seq, total RNA was subjected to polyA enrichment using NEBNext® Poly(A) mRNA Magnetic Isolation Module followed by library construction using the NEBNext® Ultra™ II RNA Library Prep Kit for Illumina. QuantSeq libraries are generated using QuantSeq 3’ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen) according to manufacturer instructions. RNA-seq libraries were sequenced on Illumina NovaSeq machine with 100bp single-end sequencing and QuantSeq libraries are sequenced on NextSeq550 machine with 75bp single-end sequencing.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described with minor modifications to allow quantification using spike-in of human chromatin81. Chromatin from human embryonic stem cells (hESCs) was prepared according to the mouse ESC protocol and 1% (relative to mouse chromatin) was spiked into mouse ESC chromatin. Combined chromatin was used for immunoprecipitation as follows: Chromatin equivalent of 50 μg DNA was incubated overnight in 1x IP buffer (final: 50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, 0.1% SDS) with 5ul antibody (rabbit anti-H3K4me1/ab8895/abcam, rabbit anti-H3K4me3/ab8580/abcam, mouse anti-H3K9me2/ab1220/abcam, rabbit anti-H3K9me3/ab8898/abcam, rabbit anti-H3K27ac/ab4729/abcam, mouse anti-Flag/F1804/Sigma, mouse anti-HP1γ/Millipore/05–690, rabbit anti-WIZ/NBP1–80586/Novus, goat anti-OCT4/AF1759/R&D systems, goat anti-SOX2/AF2018/R&D systems, rabbit anti-NANOG/ab80892/Abcam, rabbit anti-NR5A2/ABE2867/SIGMA and mouse anti-ESRRB/PP-H6705–00/R&D systems) at 4 °C on a rotating wheel. Subsequent immunoprecipitation, washes and DNA isolation were performed as previously described81.
ZFP462 and HP1γ ChIP was performed as previously described 79. Avi-tagged cells cross-linking and chromatin preparation was done as described above. Chromatin equivalent of 100 μg DNA was incubated overnight in 1× IP buffer (final: 50 mM HEPES/KOH pH 7.5, 140mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, 0.1% SDS) with 40ul of pre blocked Streptavidin M-280 Dynabeads (Invitrogen) at 4 °C on a rotating wheel. Beads were subsequently washed 2 times with 2% SDS in 1xTE, 2 times with high salt buffer (final: 50 mM HEPES/KOH pH 7.5, 500 mM NaCl, 1mM EDTA, 1% Triton-X100, 0.1% DOC, 0.1% SDS), 2 times with DOC buffer (10 mM Tris pH 8, 0.25 mM LiCl, 1 mM EDTA, 0.5% NP40, 0.5% DOC) and 1 time with 1xTE. The beads were resuspended in elution buffer, treated with RNase A and Proteinase K and reverse crosslinked overnight at 65 °C. Next day beads were separated with magnet and supernatant was phenol/chloroform extracted and IP DNA is ethanol precipitated.
GLP ChIP was performed as previously described 82. Avitag-GLP cells are cross linked and nuclei preparation was done as described above. Nuclei were washed once in 5 ml NUC buffer (15 mM HEPES pH 7.5, 60 mM KCl, 15 mM NaCl, and 0.32 mM sucrose) and resuspended in 1 ml of NUC buffer supplemented with Complete protease inhibitors, 3.3 μl of 1 M CaCl2, and 50U of Micrococcal Nuclease (LS004798, Worthington). Enzymatic activity was induced for 15 min at 37°C and shaking at 1,000 rpm and stopped by addition of 50 μl of STOP solution (250 mM EDTA and 500 mM EGTA) and 110 μl of 10× ChIP buffer (500 mM HEPES/KOH pH 7.5, 1.4 M NaCl, 10 mM EDTA, 10% Triton X-100, 1% DOC and 1% SDS) with a further incubation for 5 min on ice. Nuclei were gently disrupted by sonication in Diagenode 15-ml Falcon tubes for eight cycles (5 s ON, 5 s OFF) in ice-cold water using a Bioruptor Plus. Crude chromatin lysate was clarified by spinning at 20,000 × g at 4 °C for 1 min to separate insoluble debris. Chromatin equivalent of 50 μg DNA was incubated overnight in 1× IP buffer (final: 50 mM HEPES/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% DOC, 0.1% SDS) with 40ul of pre blocked Streptavidin M-280 Dynabeads (Invitrogen) at 4 °C on a rotating wheel. Beads were subsequently washed 2 times with 2% SDS in 1xTE, 2 times with high salt buffer (final: 50 mM HEPES/KOH pH 7.5, 500 mM NaCl, 1mM EDTA, 1% Triton-X100, 0.1% DOC, 0.1% SDS), 2 times with DOC buffer (10 mM Tris pH 8, 0.25 mM LiCl, 1 mM EDTA, 0.5% NP40, 0.5% DOC) and 1 time with 1xTE. The beads were resuspended in elution buffer, treated with RNase A and Proteinase K and reverse crosslinked overnight at 65 °C. Next day beads were separated with magnet and supernatant was phenol/chloroform extracted and IP DNA is ethanol precipitated.
ChIP–qPCR analysis and ChIP–seq libraries preparation
ChIP DNA was subjected to qPCR analysis on a CFX96 Real-Time PCR System (Bio-Rad) using the Promega GoTaq® qPCR Master Mix (Ref: A6001) with ChIP-qPCR primers described in Supplementary Table 5. Relative ChIP enrichment was calculated by percent input method. For ChIP–seq sample preparation, library construction was performed using the NEBNext Ultra-II kit (New England Biolabs) following manufacturer recommendations. Libraries were sequenced on Illumina NextSeq550 machines, with 75-bp single-end sequencing.
Protein Co-Immunoprecipitation (Co-IP)
Cells were grown to confluency on 15cm plates, collected in PBS, pelleted by 3 min centrifugation at 300g. For each Co-IP, 30 million equivalent cell pellets were then resuspended in 5ml of buffer 1 (10 mM Tris-HCl at pH 7.5, 2 mM MgCl2, 3 mM CaCl2, Complete protease inhibitor (Roche)), incubated for 20 min at 4°C followed by a centrifugation step. The cell pellet was resuspended in buffer 2 (10 mM Tris-HCl at pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.5% IGEPAL CA-630, 10% glycerol, Complete Protease Inhibitor (Roche)), incubated for 10 min at 4°C followed by centrifugation. Isolated nuclei were lysed in buffer 3 (50mM HEPES-KOH, pH7.3, 200mM KCl, 3.2mM MgCl2, 0.25% Triton X-100, 0.25% NP-40, 0.1% Na-deoxycholate and 1mM DTT and Roche Complete protease inhibitor), 4ul of Benzonase (E1014, Millipore) was added and incubated for 1 hour at 4°C. The lysate was cleared by centrifugation at 16,000g for 10 min at 4 °C. The lysate was cleared by centrifugation and incubated for 2 hours at 4°C with Tag specific magnetic beads (Dynabeads™ M-280 Streptavidin/Invitrogen for immunoprecipitation of Avitag-ZFP462 and Avitag-GLP. Anti-FLAG® M2 magnetic beads/Sigma for immunoprecipitation of TetR-3xFLAG-CT-ZFP462). The beads were washed four times for 10 min with buffer 4 (50mM HEPES-KOH, pH7.3, 300mM KCl, 3.2mM MgCl2, 0.25% Triton X-100, 0.25% NP-40, 0.1% Na-deoxycholate and 1mM DTT). Beads are further washed four times with Tris buffer (20mM Tris pH7.5, 137mM Nacl) and used for mass spectrometry analysis.
Mass spectrometry analysis
Mass spectrometry and identification of co-immunoprecipitated proteins was performed as previously described 83.
ATAC-seq
ATAC-seq was performed on WT, Zfp462 knockout, Zfp462-FL and Zfp462-NT+Mid rescue ESCs, and WT mouse NSCs following a previously published protocol 84. The experiment was performed in biological replicates. Libraries were paired-end sequenced using an Illumina NextSeq550/Novaseq sequencer.
DNA methylation analysis by mass spectrometry
1ug of pure genomic DNA was digested using DNA Degradase plus (Zymo Research) for 6 hours at 37 degrees. For determination of the methyl-dC content, samples were analyzed with LC-MS/MS using a triple quadrupole mass spectrometer, employing the selected reaction monitoring (SRM) mode of the instrument. The following transitions were used in the positive ion mode: dC m/z 228.1 → m/z 112.1 and methyl-dC m/z242.1 → m/z 126.1. Data were interpreted using the Trace Finder software suite (Thermo Fisher Scientific) and manually validated. Calibration curves of defined mixtures of dC and methyl-dC were acquired using the same procedure and employed for calculating the molar percentage of methyl-dC.
Phylogenetic analysis
ZFP462 orthologs were collected in a convergent PSI-BLAST v2.10.0 search against the NCBI non-redundant protein database using amino-acids 632–835 of NP_766455.2 as a query. The obtained sequence set was filtered, removing identical and partial sequences, keeping only sequences with identity below 95%. The remaining sequences were aligned using mafft v7.427, phylogeny was reconstructed with IQ-TREE 85 version 1.6.1 using parameters -alrt 1000 -bb 1000, and the phylogenetic tree was visualized using iTOL v6 86.
RNA-seq and QuantSeq data analysis
RNA-seq reads were trimmed using Trim Galore v0.5.0, whereas QuantSeq reads are trimmed using BBDuk v38.06. Trimmed reads mapping to abundant sequences included in the iGenomes Ensembl GRCm38 bundle (mouse rDNA, mouse mitochondrial chromosome, phiX174 genome, adapter) were removed using bowtie2 v2.3.4.1 alignment87. Remaining reads were analyzed using genome and gene annotation for the GRCm38/mm10 assembly obtained from Mus musculus Ensembl release 94. Reads were aligned to the genome using STAR v2.6.0c and reads in genes were counted with featureCounts (subread v1.6.2) with parameter -s 2 for RNAseq and using strand-specific read counting for QuantSeq samples with parameter -s 188. Differential gene expression analysis on raw counts and variance-stabilized transformation of count data for heatmap visualization were performed using DESeq2 v1.18.189. Functional annotation enrichment analysis of differentially expressed genes was conducted using clusterProfiler v3.6.0 in R v3.4.190.
ChIP-seq data analysis
ChIP-seq reads were trimmed using Trim Galore v0.4.4 and thereafter aligned to the mm10 reference genome using BWA MEM v0.7.17. Duplicated reads were marked and excluded with Picard MarkDuplicates v2.23.4 and the obtained bam files were used to generate bigwig files using deepTools bamCoverage v3.5.0 with the parameter -normalizeUsing RPKM. Peaks were called from the sorted BAM using MACS2 v2.1.1 with default settings91. To analyze overlapping and non-overleaping peaks, bedtools intersect command was used. Peaks in blacklist regions were identified using bedtools intersect v2.27.1 and mm10.blacklist.bed.gz v1. Spike-in normalization of ChIP data was performed as previously described92. Repeat analysis was performed using RepEnrich2 following alignment of trimmed reads with bowtie2 v2.2.9. Heatmaps and metaplots are generated using deepTools version 3.1.293.
ATAC-seq data analysis
ATAC-seq reads were processed using nfcore/atacseq v1.0.0 with standard settings. Reads were trimmed using trim-galore v0.5.0, aligned to the mm10 genome using BWA MEM v0.7.17 and duplicated reads were marked with Picard MarkDuplicates v2.19.0. Alignment was filtered using samtools v1.9 thereby removing multimapping and duplicated reads. Peaks were called from the filtered and sorted BAM using MACS2 v2.1.2 with parameters --broad --BAMPE --keep-dup all --nomodel. Consensus peaks were obtained by merging peak calls using bedtools mergeBed v2.27.1. Heatmaps and metaplots are generated using deepTools version 3.1.2. DNA sequence motif analysis in ATAC-seq peaks was performed using HOMER version homer/4.10-foss-2018b94.
Statistics and reproducibility
All experiments were independently repeated at least two times (unless otherwise stated) with similar results, and representative results are shown throughout all figures. No statistical method was used to predetermine sample size. No data were excluded from the analyses. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Fig. 1: Characterization of WT and mutant CiA Oct4 dual reporter cells.
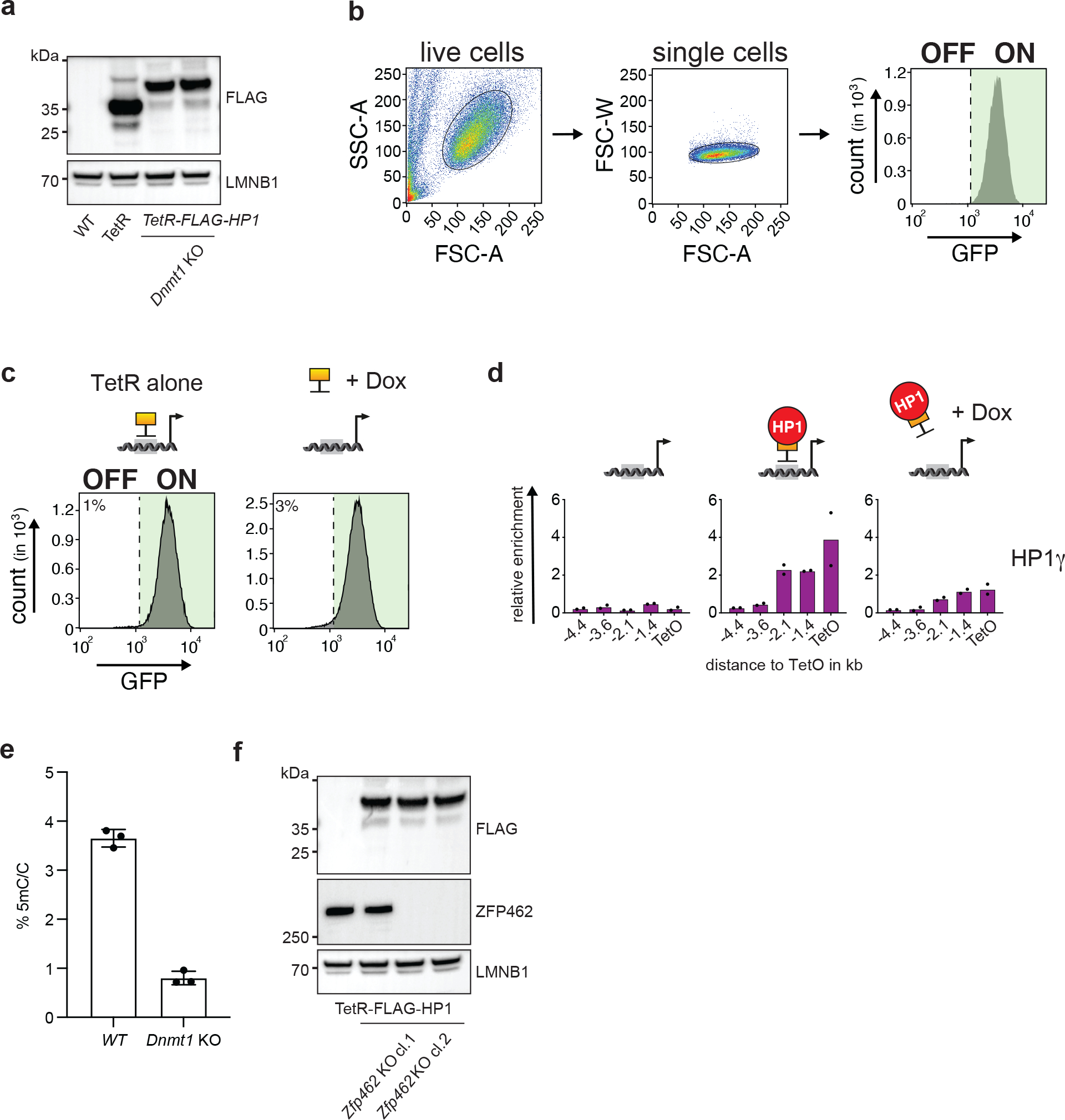
a) Western blot confirms expression of TetR-FLAG and TetR-FLAG-HP1 proteins in WT and Dnmt1 knockout (KO) CiA Oct4 dual reporter ESCs. LMNB1 is used as protein loading control. b) FACS gating strategy used to analyse the GFP fluorescence in cells. c) Flow cytometry histograms show GFP expression in CiA Oct4 dual reporter cells after TetR-FLAG recruitment and after TetR-FLAG release following Dox addition for four days. d) ChIP-qPCR shows relative enrichment of HP1γ upstream of the TetO site before tethering, in the presence of TetR-FLAG-HP1 and after Dox-dependent release of TetR-FLAG-HP1 for four days. n = 2 independent biological replicates. e) Bar plot shows fraction of cytosine methylation (5mC) in WT and Dnmt1 KO CiA Oct4 dual reporter ESCs measured by LC-MS. n = 3 independent biological replicates. Data are presented as mean values +/− SD. f) Western blot shows expression of TetR-FLAG-HP1 and ZFP462 in WT and two independent Zfp462 KO CiA Oct4 dual reporter ESC lines. LMNB1 is used as loading control.
Extended Data Fig. 2: ZFP462 is conserved across vertebrates and acts as transcriptional repressor.
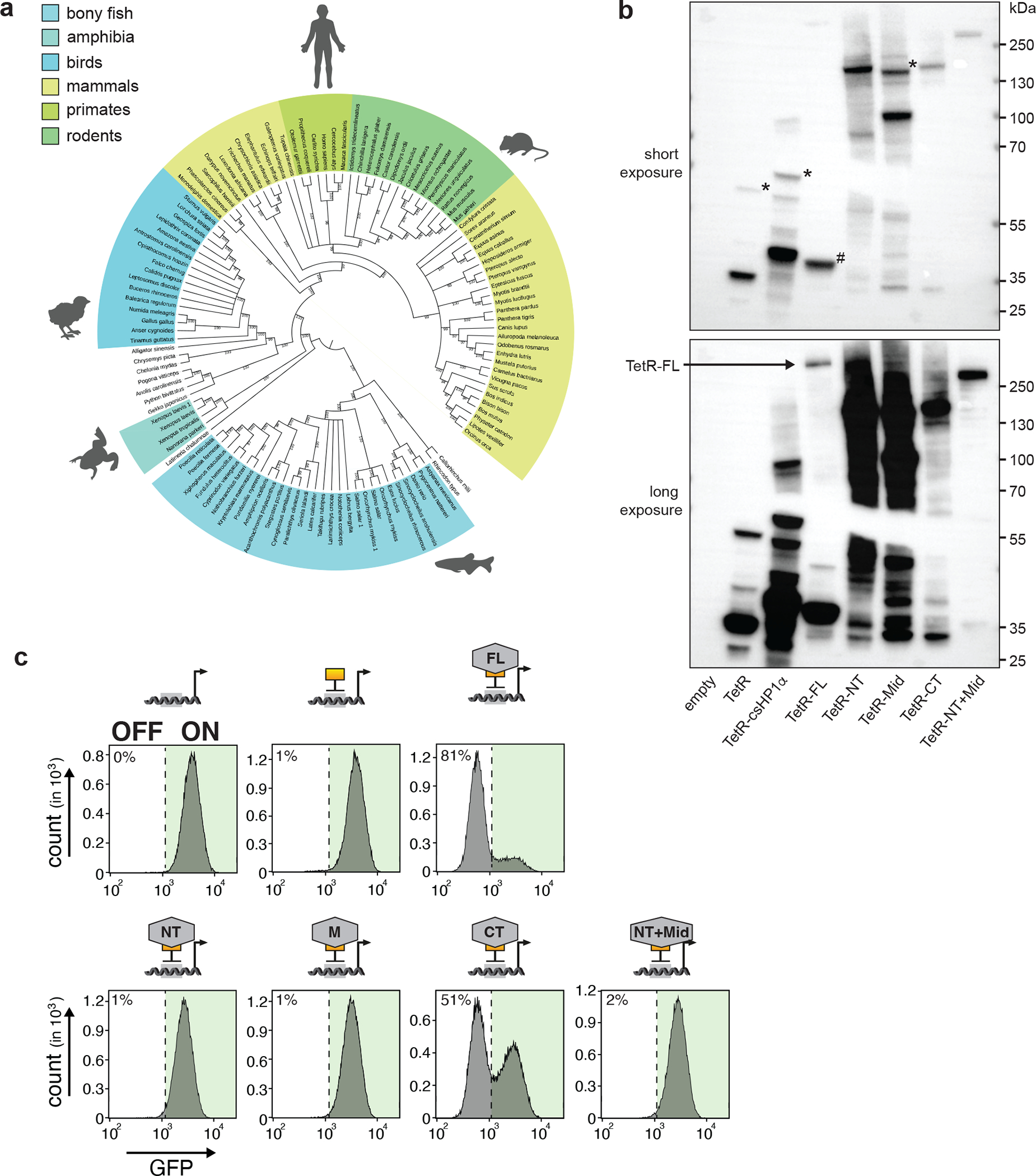
a) Phylogenetic tree of ZFP462 protein orthologues in vertebrate species. Bootstrap values are shown on branches. b) Western blot shows expression ZFP462 fusions with TetR-FLAG in CiA Oct4 dual reporter ESCs. Bottom image shows long exposure. Hashtag indicates cleaved TetR-FLAG protein from TetR-FLAG-ZFP462-FL (full-length). TetR-FLAG fusions initially express mCherry for selection which is cleaved via P2A signal. Asterisk marks P2A uncleaved protein product. c) Representative flow cytometry histograms show GFP expression in CiA Oct4 dual reporter ESCs expressing TetR-FLAG fusions with full-length or truncated ZFP462 proteins. Each histogram is average profile of 100,000 analysed cells.
Extended Data Fig. 3: Zfp462 expression analysis and impact of Zfp462 deletion on ESC morphology and gene expression.
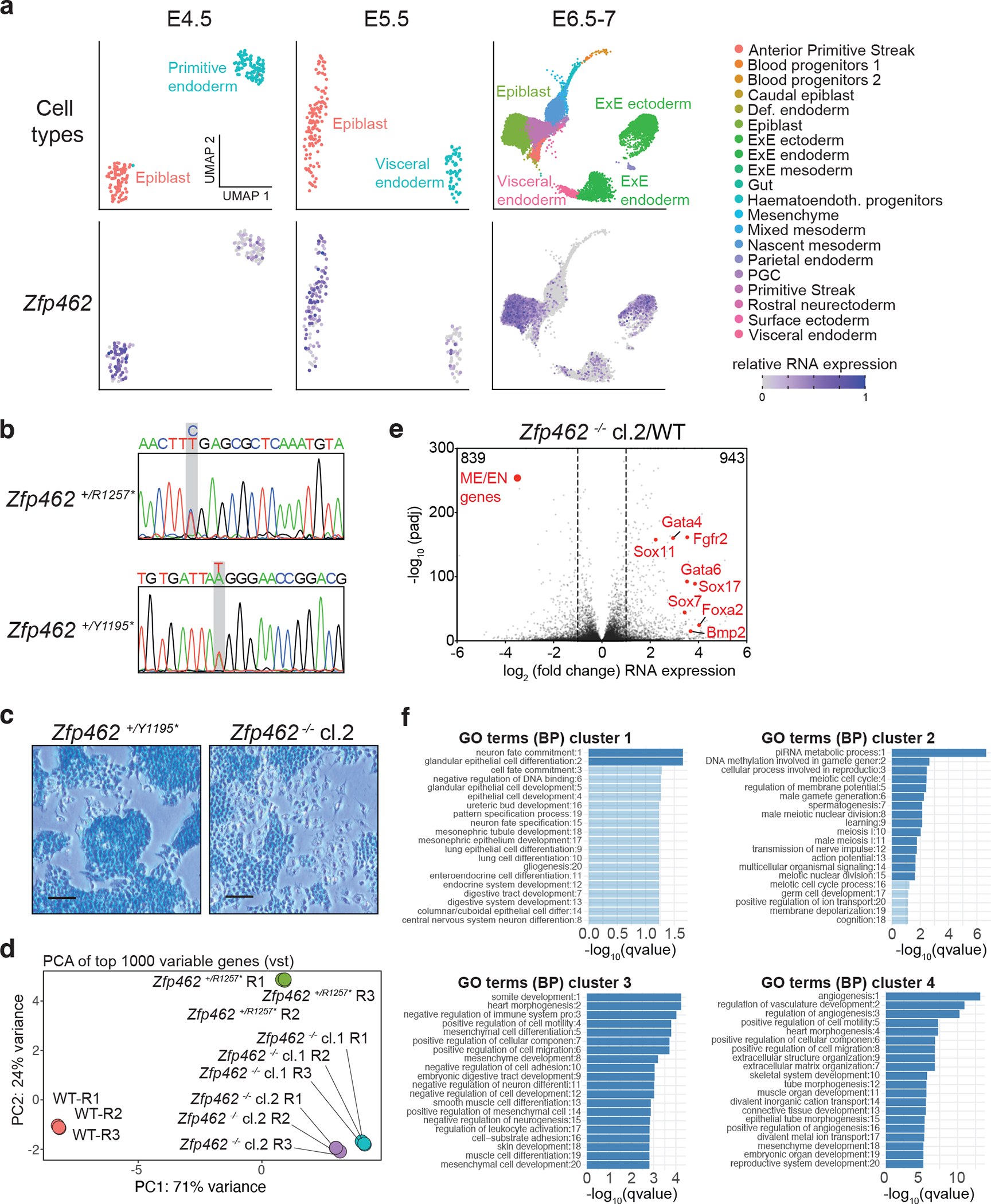
a) UMAP plots visualize lineage assignments of mouse embryonic developmental stages (embryonic day (E) E4.5 cells, E5.5 cells and E6.5–7 cells) as previously described (Wolf Reik et.al) (top). UMAP plots of Zfp462 RNA expression at corresponding developmental stages (bottom). b) Sanger sequence chromatograms of heterozygous Zfp462 mutants. Heterozygous non-sense mutations are highlighted in grey. c) Alkaline phosphatase staining of Zfp462 +/Y1195* and Zfp462 −/− cl.2 ESCs (scale bar = 100 μm). d) Correlation plot shows principal component analysis (PCA) of replicate RNA-seq experiments from WT, two Zfp462 +/− and two Zfp462 −/− ESC lines. e) Volcano plot shows differential gene expression of Zfp462 −/− cl.2 ESCs compared to WT ESCs. (n = three replicates). f) Bar plots show gene ontology (GO) terms enriched in the four clusters of heatmap in Fig. 3e.
Extended Data Fig. 4: Lineage-specifying genes are deregulated during neuronal differentiation.
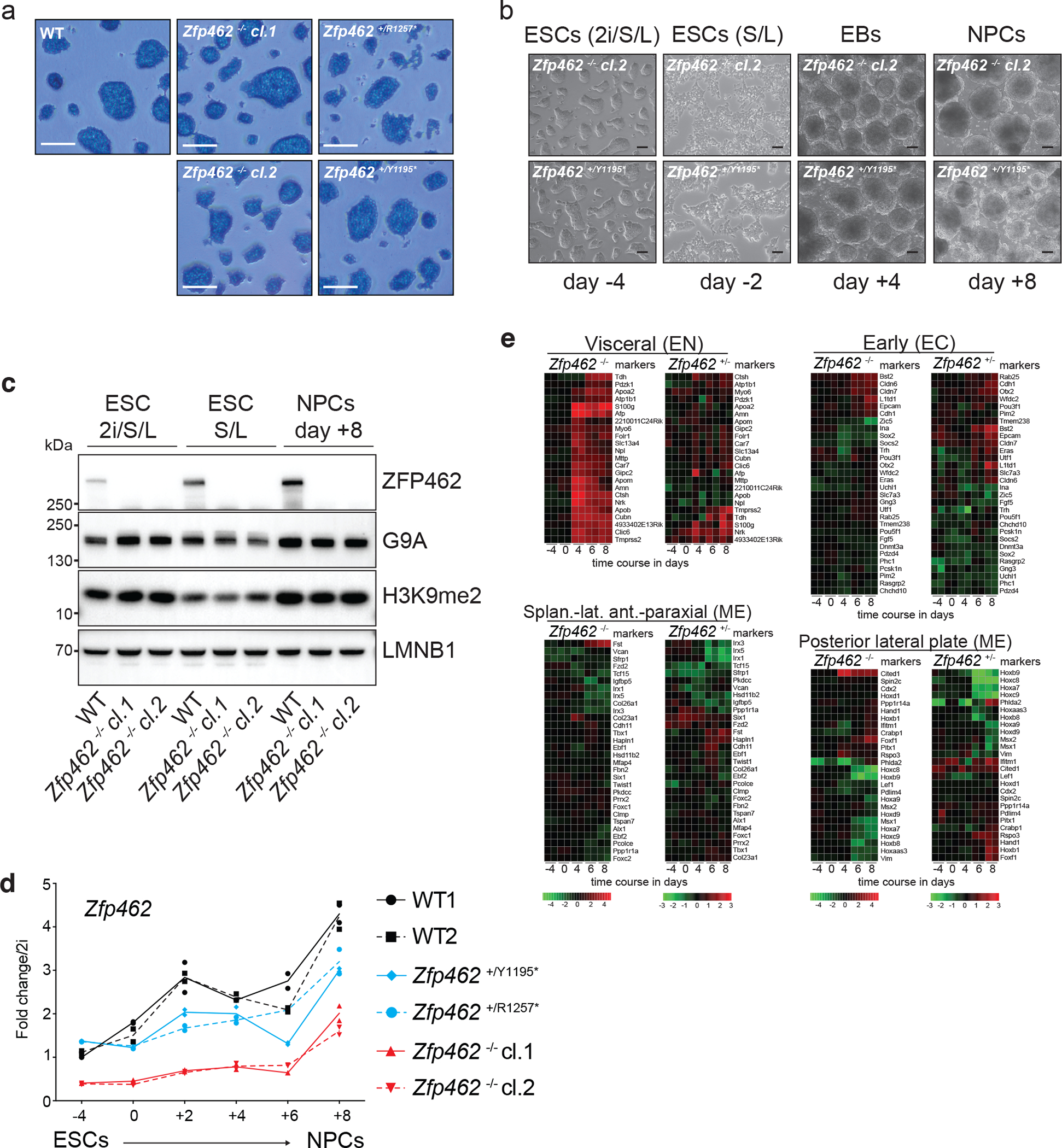
a) Alkaline phosphatase staining of WT and Zfp462 mutant ESCs cultured in 2i/LIF/Serum medium (scale bar = 180 μm). b) Representative bright field images show Zfp462+/Y1195* and Zfp462 −/− cl.2 at corresponding stages of neural differentiation. Scale bar = 100μm. c) Western blot analysis shows levels of ZFP462, G9A and H3K9me2 in WT and Zfp462 −/− ESCs during neural differentiation. LMNB1 is used as loading control. d) Line plot shows RT-qPCR analysis of Zfp462 RNA expression during neural differentiation (n = two replicates). Expression level is shown relative to ESCs (2i/S/L). e) Heatmaps show differential expression of selected marker genes specific for endodermal (EN), mesodermal (ME) and neural lineages (EC) in heterozygous and homozygous Zfp462 mutant cells during neural differentiation.
Extended Data Fig. 5: ChIP-seq profiles of ZFP462, REST, ADNP, HP1γ and H3K9me2 in WT and Zfp462 KO ESCs.
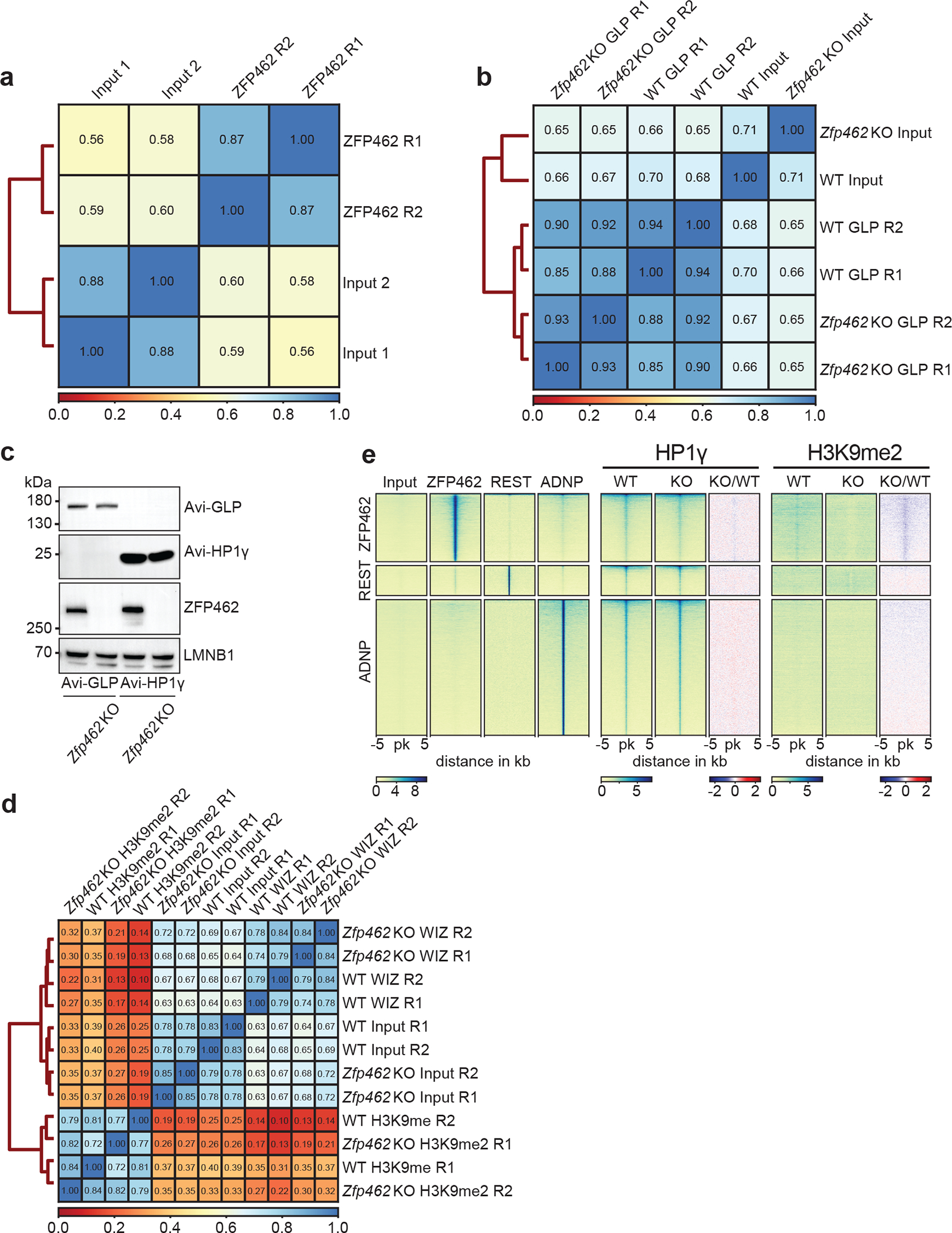
a) Heatmap shows Spearman correlation between input controls and two independent ZFP462 ChIP-seq experiments. b) Heatmap shows Spearman correlation between input controls and two independent ChIP-seq experiments of GLP and HP1γ in WT and Zfp462 KO ESCs. c) Western blot shows ZFP462 expression in WT and Zfp462 KO Avi-FLAG-tagged GLP and HP1γ ESCs. d) Heatmap shows Spearman correlation between input controls and two independent ChIP-seq experiments of WIZ and H3K9me2 in WT and Zfp462 KO ESCs. e) Heatmaps of ZFP462, REST, ADNP, HP1γ and H3K9me2 ChIP-seq signals at ZFP462, REST and ADNP peaks in WT and Zfp462 KO ESCs. Blue-to-red scaled heatmap represents ChIP-seq enrichment ratios between Zfp462 KO versus WT (KO/WT). Each heatmap represents a 10 kb window centred on peak midpoints, sorted by ZFP462, REST and ADNP signal in their respective clusters. Below are scale bars (n = average distribution of two replicates).
Extended Data Fig. 6: Rescue of Zfp462 KO ESCs with ZFP462 full-length and ZFP462 C-terminal truncation.
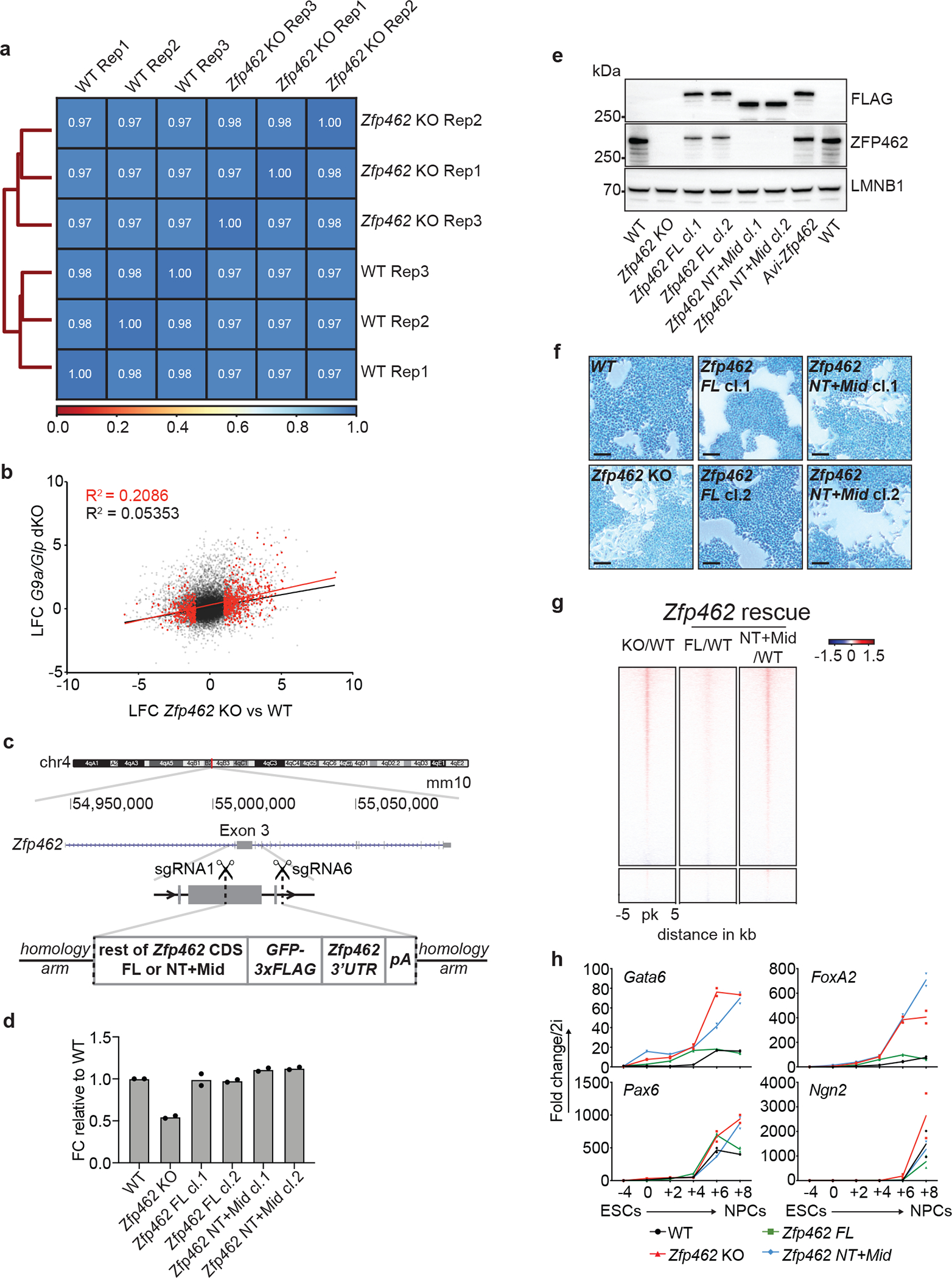
a) Heatmap shows Spearman correlation between three independent ATAC-seq experiments in WT and Zfp462 KO ESCs. b) Scatter plot shows pairwise correlation of gene expression changes between Zfp462 KO vs WT and G9a/Glp dKO vs WT. coefficient of determination (R2): black – all genes, red – differentially regulated genes in Zfp462 KO vs WT (padj. ≤ 0.05, LFC ≥ 1). c) Cartoon depicts CRISPR strategy to knock-in coding sequences for ZFP462 full-length and ZFP462 NT+Mid proteins at the Zfp462 gene locus. Insertions were targeted in-frame with exon 3. d) Bar plot shows RT-qPCR analysis of Zfp462 RNA transcript levels in WT, KO and rescue ESCs. n = 2 independent biological replicates. e) Western blot analysis shows ZFP462 protein levels in WT, Zfp462 KO and rescue ESCs. LMNB1 is used as loading control. f) Alkaline phosphatase staining of WT and Zfp462 KO and rescue ESCs (scale bar = 100μm). g) Heatmaps of ATAC-seq signal ratios at ZFP462 and REST peaks of Zfp462 KO vs WT (KO/WT) and Zfp462 rescue ESCs (ZFP462 FL/WT) or (ZFP462 NT+Mid/WT). Each row represents a 10 kb window centred on peak midpoints, sorted by KO/WT enrichment ratio (n = average distribution of two ATAC-seq replicates). h) Line plots show RT-qPCR analysis of lineage marker expression during neural differentiation (n=two replicates) in WT, Zfp462 KO and rescue ESCs. Expression levels are shown relative to ESCs (2i/S/L).
Extended Data Fig. 7. Correlation between genomic distribution of ZFP462 and pluripotency transcription factors.
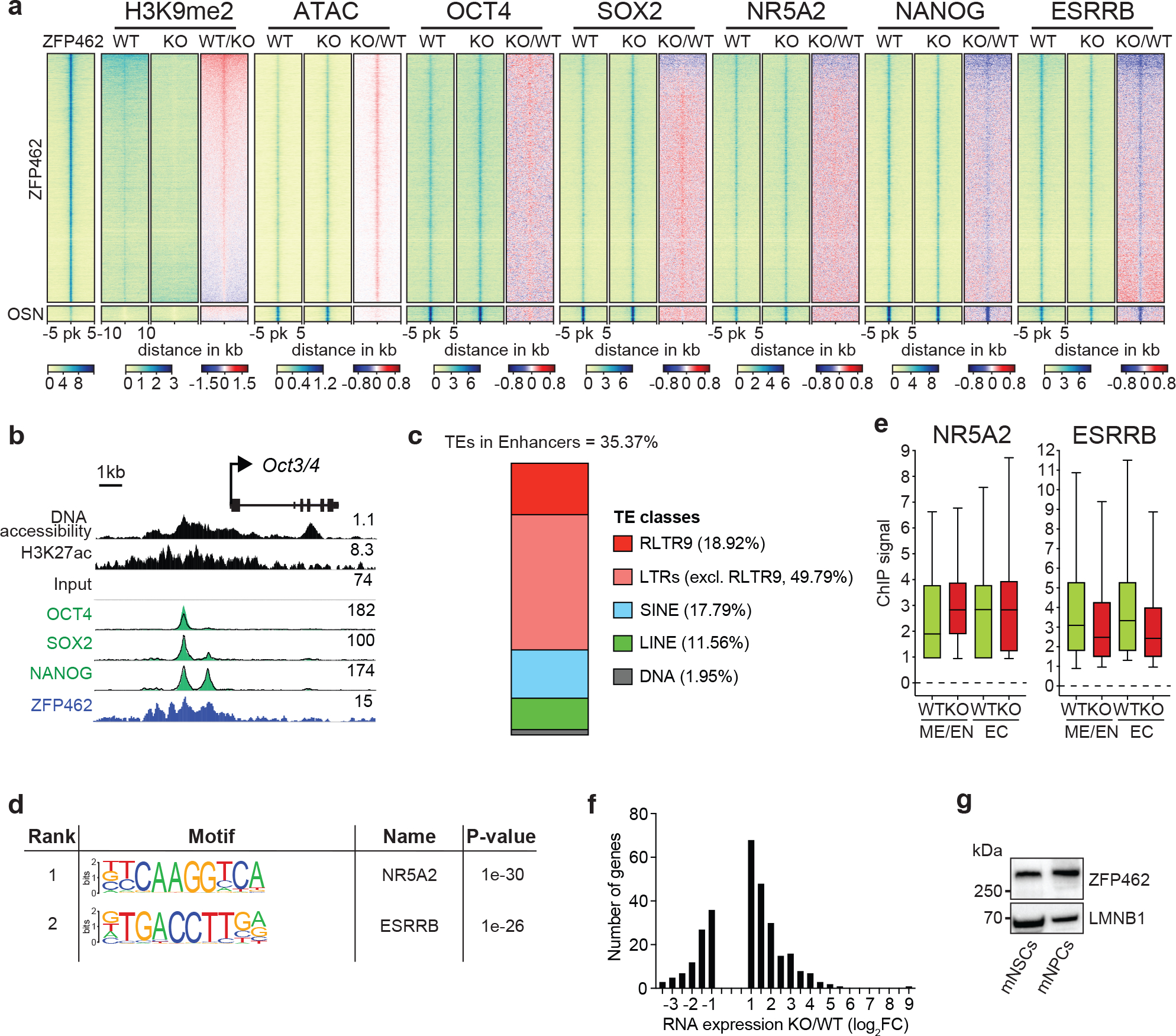
a) Heatmaps show ATAC-seq and ChIP-seq signal of ZFP462, H3K9me2, OCT4, SOX2, NANOG, ESRRB and NR5A2 at peaks of ZFP462, OSN (shared OCT4-SOX2-NANOG peaks not overlapping with ZFP462 peaks) and REST. Red-to-blue scaled heatmaps represent signal ratios between Zfp462 KO versus WT (KO/WT). Each row represents a 10 kb window centred on peak midpoints, sorted by H3K9me2 KO/WT ChIP signal loss. (n = average distribution of two ChIP-seq replicates / ATAC-seq three replicates). b) Genomic screenshot shows DNA accessibility (ATAC-seq) and ChIP-seq signals of H3K27ac and ZFP462 at the Oct3/4 locus in WT ESCs. ChIP-seq signals of OCT4, SOX2 and NANOG in WT (black line) and Zfp462 KO ESCs (green fill) are superimposed. ATAC-seq and ChIP-seq profiles are normalized to library size. c) Bar plot shows percentage of ZFP462-bound TE families overlapping with ChromHMM-annotated enhancers in ESCs. ZFP462-bound TEs contribute a total of 35.37% of ChromHMM-annotated enhancers in ESCs. d) HOMER analysis of known DNA sequence motifs enriched at ZFP462 peaks overlapping ChromHMM-annotated ME/EN-specific enhancers. Top ranked DNA sequence motifs and respective significance values are shown in the table. e) Box plots shows enrichment of NR5A2 and ESRRB ChIP signal in WT and Zfp462 KO ESCs at ZFP462 peaks overlapping with Mesoderm (ME)/Endoderm (EN)- and Ectoderm (EC)-specific enhancers. n = 553 (EN/ME), n = 314 (EC). Shown are median (horizontal line), 25th to 75th percentiles (boxes), and 90% (whiskers). Outliers are excluded. f) Bar plot shows frequency distribution of significantly up- and down-regulated genes located proximal to ZFP462 peaks annotated as ME/EN-specific enhancers (KO/WT, LFC ≥ 1 and padj. ≤ 0.05). g) Western blot shows ZFP462 protein expression in NSCs isolated from mouse brain and NPCs differentiated from WT ESCs. LMNB1 is used as loading control.
Supplementary Material
Acknowledgements
We are grateful to all members of the Bell and Brennecke laboratories for support, feedback and discussions. HP1γ and GLP Avi-tagged cell lines are kind gifts from Marc Bühler and Joerg Betschinger labs at FMI, Switzerland. We thank Noelia Urban for providing mouse NSCs. We thank Christa Buecker, Fabio Mohn and Kristoffer Jensen for sharing unpublished date, experimental advice and helpful discussions. We thank Frederic Berger, Luisa Cochella and Jakob Schnabl for feedback on the manuscript. We thank the Vienna Biocenter Core Facility Next Generation Sequencing. The GMI/IMBA/IMP Scientific Service units, especially the BioOptics facility and the Mass Spectrometry unit provided outstanding support. DNA methylation analysis by LC-MS/MS was performed by the Metabolomics Facility at Vienna BioCenter Core Facilities (VBCF), funded by the City of Vienna through the Vienna Business Agency. We thank Life Science Editors for editorial assistance. We apologize to colleagues whose work could not be cited due to space limitations. Funding: O.B., J.B., U.E. and S.M. were supported by the Austrian Academy of Sciences. O.B. was supported by the New Frontiers Group of the Austrian Academy of Sciences (NFG-05), the Human Frontiers Science Programme Career Development Award (CDA00036/2014-C), the National Institute of Mental Health (R01MH122565) and the Norris Comprehensive Cancer Center of USC and its NCI Award (P30CA014089). R.Y. was supported by EMBO Long-Term Fellowship (ALTF 256–2015). D.S. acknowledges support from the Novartis Research Foundation and the European Research Council under the European Union’s (EU) Horizon 2020 research and innovation program grant agreement (ReadMe-667951). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests Statement
The authors declare that they have no competing interests.
Data Availability
High-throughput sequencing data produced in this study is deposited at the Gene Expression Omnibus (GEO) database under super series accession number GSE175369 (RNA-seq: GSE176321, QuantSeq: GSE176319, ATAC-seq: GSE176322 and ChIP-seq: GSE177058). G9a/GLP DKO ESCs ATAC-seq and RNA-seq data is obtained from GSE13810252. REST and ADNP ChIP-seq data are obtained from GSE2714895 and GSE9794551 respectively. Mesoendoderm cells ATAC-seq data is obtained from GSE11626266. Processed scRNAseq data of mouse embryo stage E4.5 to E7.0 is obtained from ftp://ftp.ebi.ac.uk/pub/databases/scnmt_gastrulation 43. Mass spectrometry data have been deposited in ProteomeXchange with the primary accession code PXD037238. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
References
- 1.Eckersley-Maslin MA, Alda-Catalinas C & Reik W Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 19, 436–450 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Burton A & Torres-Padilla M-E Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell Biol. 15, 723–735 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Gökbuget D & Blelloch R Epigenetic control of transcriptional regulation in pluripotency and early differentiation. Development 146, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwafuchi-Doi M & Zaret KS Cell fate control by pioneer transcription factors. Development 143, 1833–1837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Padeken J, Methot SP & Gasser SM Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. (2022) doi: 10.1038/s41580-022-00483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishibuchi G & Nakayama J Biochemical and structural properties of heterochromatin protein 1: understanding its role in chromatin assembly. J. Biochem. 156, 11–20 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Larson AG & Narlikar GJ The Role of Phase Separation in Heterochromatin Formation, Function, and Regulation. Biochemistry 57, 2540–2548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A & Kono H Heterochromatin protein 1 (HP1): interactions with itself and chromatin components. Biophys. Rev. 12, 387–400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicetto D & Zaret KS Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Curr. Opin. Genet. Dev. 55, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45, 34–42 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Ragunathan K, Jih G & Moazed D Epigenetic inheritance uncoupled from sequence-specific recruitment. Science (80-. ). 348, 1258699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Audergon PNCB et al. Restricted epigenetic inheritance of H3K9 methylation. Science (80-. ). 348, 132 LP – 135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hathaway NA et al. Dynamics and Memory of Heterochromatin in Living Cells. Cell 149, 1447–1460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shinkai Y & Tachibana M H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 25, 781–788 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tachibana M et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lienert F et al. Genomic Prevalence of Heterochromatic H3K9me2 and Transcription Do Not Discriminate Pluripotent from Terminally Differentiated Cells. PLOS Genet. 7, e1002090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen B, Wu H, Shinkai Y, Irizarry RA & Feinberg AP Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 41, 246–250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu N et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 29, 379–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zylicz JJ et al. G9a regulates temporal preimplantation developmental program and lineage segregation in blastocyst. Elife 7, e33361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldman N et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol. 8, 188–194 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Kellner S & Kikyo N Transcriptional regulation of the Oct4 gene, a master gene for pluripotency. Histol. Histopathol. 25, 405–412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epsztejn-Litman S et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 15, 1176–1183 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benevento M, van de Molengraft M, van Westen R, van Bokhoven H & Nadif Kasri N The role of chromatin repressive marks in cognition and disease: A focus on the repressive complex GLP/G9a. Neurobiol. Learn. Mem. 124, 88–96 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Roopra A, Qazi R, Schoenike B, Daley TJ & Morrison JF Localized Domains of G9a-Mediated Histone Methylation Are Required for Silencing of Neuronal Genes. Mol. Cell 14, 727–738 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Mozzetta C et al. The Histone H3 Lysine 9 Methyltransferases G9a and GLP Regulate Polycomb Repressive Complex 2-Mediated Gene Silencing. Mol. Cell 53, 277–289 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Mulligan P et al. CDYL Bridges REST and Histone Methyltransferases for Gene Repression and Suppression of Cellular Transformation. Mol. Cell 32, 718–726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer A et al. Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron 64, 678–691 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deimling SJ, Olsen JB & Tropepe V The expanding role of the Ehmt2/G9a complex in neurodevelopment. Neurogenesis 4, e1316888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleefstra T et al. Loss-of-Function Mutations in Euchromatin Histone Methyl Transferase 1 (EHMT1) Cause the 9q34 Subtelomeric Deletion Syndrome. Am. J. Hum. Genet. 79, 370–377 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramer JM Regulation of cell differentiation and function by the euchromatin histone methyltranserfases G9a and GLP. Biochem. Cell Biol. 94, 26–32 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Scheer S & Zaph C The Lysine Methyltransferase G9a in Immune Cell Differentiation and Function. Frontiers in Immunology vol. 8 429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss K et al. Haploinsufficiency of ZNF462 is associated with craniofacial anomalies, corpus callosum dysgenesis, ptosis, and developmental delay. Eur. J. Hum. Genet. 25, 946–951 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruszka P et al. Phenotype delineation of ZNF462 related syndrome. Am. J. Med. Genet. Part A 179, 2075–2082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González-Tarancón R et al. A novel mutation in the ZNF462 gene c.3306dup; p.(Gln1103Thrfs*10) is associated to Weiss-Kruszka syndrome. A case report. Acta Clin. Belg. 1–4 (2020) doi: 10.1080/17843286.2020.1780391. [DOI] [PubMed] [Google Scholar]
- 35.Athanasiadou R et al. Targeting of De Novo DNA Methylation Throughout the Oct-4 Gene Regulatory Region in Differentiating Embryonic Stem Cells. PLoS One 5, e9937 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niwa H, Miyazaki J & Smith AG Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24, 372–376 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Michlits G et al. CRISPR-UMI: single-cell lineage tracing of pooled CRISPR–Cas9 screens. Nat. Methods 14, 1191–1197 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose NR & Klose RJ Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta - Gene Regul. Mech. 1839, 1362–1372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren W et al. DNMT1 reads heterochromatic H4K20me3 to reinforce LINE-1 DNA methylation. Nat. Commun. 12, 2490 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueda J, Tachibana M, Ikura T & Shinkai Y Zinc Finger Protein Wiz Links G9a/GLP Histone Methyltransferases to the Co-repressor Molecule CtBP*,. J. Biol. Chem. 281, 20120–20128 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Olsen JB et al. G9a and ZNF644 Physically Associate to Suppress Progenitor Gene Expression during Neurogenesis. Stem Cell Reports 7, 454–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Argelaguet R et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald ACH, Biechele S, Rossant J & Stanford WL Sox17-Mediated XEN Cell Conversion Identifies Dynamic Networks Controlling Cell-Fate Decisions in Embryo-Derived Stem Cells. Cell Rep. 9, 780–793 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Wamaitha SE et al. Gata6 potently initiates reprograming of pluripotent and differentiated cells to extraembryonic endoderm stem cells. Genes Dev. 29, 1239–1255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrode N, Saiz N, Di Talia S & Hadjantonakis A-K GATA6 Levels Modulate Primitive Endoderm Cell Fate Choice and Timing in the Mouse Blastocyst. Dev. Cell 29, 454–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang B et al. Zfp462 deficiency causes anxiety-like behaviors with excessive self-grooming in mice. Genes, Brain Behav. 16, 296–307 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Bibel M, Richter J, Lacroix E & Barde Y-A Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat. Protoc. 2, 1034–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Venere M et al. Sox1 marks an activated neural stem/progenitor cell in the hippocampus. Development 139, 3938–3949 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ecco G, Imbeault M & Trono D KRAB zinc finger proteins. Development 144, 2719–2729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ostapcuk V et al. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature 557, 739–743 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Jiang Q et al. G9a Plays Distinct Roles in Maintaining DNA Methylation, Retrotransposon Silencing, and Chromatin Looping. Cell Rep. 33, 108315 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Kunarso G et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Sundaram V & Wysocka J Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philos. Trans. R. Soc. B Biol. Sci. 375, 20190347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sundaram V et al. Functional cis-regulatory modules encoded by mouse-specific endogenous retrovirus. Nat. Commun. 8, 14550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Todd CD, Deniz Ö, Taylor D & Branco MR Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. Elife 8, e44344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bourque G et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 18, 1752–1762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niwa H How is pluripotency determined and maintained? Development 134, 635–646 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Thomson M et al. Pluripotency Factors in Embryonic Stem Cells Regulate Differentiation into Germ Layers. Cell 145, 875–889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Oron E, Nelson B, Razis S & Ivanova N Distinct Lineage Specification Roles for NANOG, OCT4, and SOX2 in Human Embryonic Stem Cells. Cell Stem Cell 10, 440–454 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Le Bin GC et al. Oct4 is required for lineage priming in the developing inner cell mass of the mouse blastocyst. Development 141, 1001–1010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frum T et al. Oct4 Cell-Autonomously Promotes Primitive Endoderm Development in the Mouse Blastocyst. Dev. Cell 25, 610–622 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pintacuda G et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 68, 955–969.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ernst J & Kellis M Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12, 2478–2492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gorkin DU et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cernilogar FM et al. Pre-marked chromatin and transcription factor co-binding shape the pioneering activity of Foxa2. Nucleic Acids Res. 47, 9069–9086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nissim S et al. Iterative use of nuclear receptor Nr5a2 regulates multiple stages of liver and pancreas development. Dev. Biol. 418, 108–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seitz C et al. The orphan nuclear receptor LRH-1/NR5a2 critically regulates T cell functions. Sci. Adv. 5, eaav9732 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olivieri D et al. Cooperation between HDAC3 and DAX1 mediates lineage restriction of embryonic stem cells. EMBO J. 40, e106818 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soufi A et al. Pioneer Transcription Factors Target Partial DNA Motifs on Nucleosomes to Initiate Reprogramming. Cell 161, 555–568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morgunova E & Taipale J Structural insights into the interaction between transcription factors and the nucleosome. Curr. Opin. Struct. Biol. 71, 171–179 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Michael AK et al. Mechanisms of OCT4-SOX2 motif readout on nucleosomes. Science (80-. ). 368, 1460–1465 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Isbel L, Grand RS & Schübeler D Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat. Rev. Genet. (2022) doi: 10.1038/s41576-022-00512-6. [DOI] [PubMed] [Google Scholar]
- 74.King HW & Klose RJ The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. Elife 6, e22631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frank JA & Feschotte C Co-option of endogenous viral sequences for host cell function. Curr. Opin. Virol. 25, 81–89 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bourque G Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr. Opin. Genet. Dev. 19, 607–612 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Carbon S et al. AmiGO: online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blomfield IM et al. Id4 promotes the elimination of the pro-activation factor Ascl1 to maintain quiescence of adult hippocampal stem cells. Elife 8, e48561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baubec T, Ivánek R, Lienert F & Schübeler D Methylation-Dependent and -Independent Genomic Targeting Principles of the MBD Protein Family. Cell 153, 480–492 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Paquet D et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Moussa HF et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 10, 1931 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mayer D et al. Zfp281 orchestrates interconversion of pluripotent states by engaging Ehmt1 and Zic2. EMBO J. 39, e102591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Batki J et al. The nascent RNA binding complex SFiNX licenses piRNA-guided heterochromatin formation. Nat. Struct. Mol. Biol. 26, 720–731 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nguyen L-T, Schmidt HA, von Haeseler A & Minh BQ IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Letunic I & Bork P Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 89.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omi. A J. Integr. Biol. 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fursova NA et al. Synergy between Variant PRC1 Complexes Defines Polycomb-Mediated Gene Repression. Mol. Cell 74, 1020–1036.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heinz S et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arnold P et al. Modeling of epigenome dynamics identifies transcription factors that mediate Polycomb targeting. Genome Res. 23, 60–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data produced in this study is deposited at the Gene Expression Omnibus (GEO) database under super series accession number GSE175369 (RNA-seq: GSE176321, QuantSeq: GSE176319, ATAC-seq: GSE176322 and ChIP-seq: GSE177058). G9a/GLP DKO ESCs ATAC-seq and RNA-seq data is obtained from GSE13810252. REST and ADNP ChIP-seq data are obtained from GSE2714895 and GSE9794551 respectively. Mesoendoderm cells ATAC-seq data is obtained from GSE11626266. Processed scRNAseq data of mouse embryo stage E4.5 to E7.0 is obtained from ftp://ftp.ebi.ac.uk/pub/databases/scnmt_gastrulation 43. Mass spectrometry data have been deposited in ProteomeXchange with the primary accession code PXD037238. All other data supporting the findings of this study are available from the corresponding author on reasonable request.