

Abstract
Development of genetic tests for rare genetic diseases has traditionally focused on individual diseases. Similarly, development of new therapies occurred one disease at a time. With 7,000 rare genetic diseases, this approach is not feasible. Diagnosis of genetic disorders has already transcended old paradigms as whole exome and genome sequencing have allowed expedient interrogation of all relevant genes in a single test. The growth of newborn screening has allowed identification of diseases in pre-symptomatic babies. Similarly, the ability to develop therapies is rapidly expanding due to technologies that leverage platform technology that address multiple diseases. However, movement from the basic science laboratory to clinical trials is still hampered by a regulatory system rooted in traditional trial design, requiring a fresh assessment of safe ways to obtain approval for new drugs. Ultimately, the number of nucleic acid-based therapies will challenge the ability of clinics focused on rare diseases to deliver them safely with appropriate evaluation and long term follow up. This manuscript summarizes discussions arising from a recent NIH conference on nucleic acid therapy, with a focus on scaling technologies for diagnosis of rare disorders and provision of therapies across the age and disease spectrum.
Keywords: Newborn screening, Recommended Uniform Screening Panel (RUSP), Whole exome sequencing, Whole genome sequencing, Gene therapy
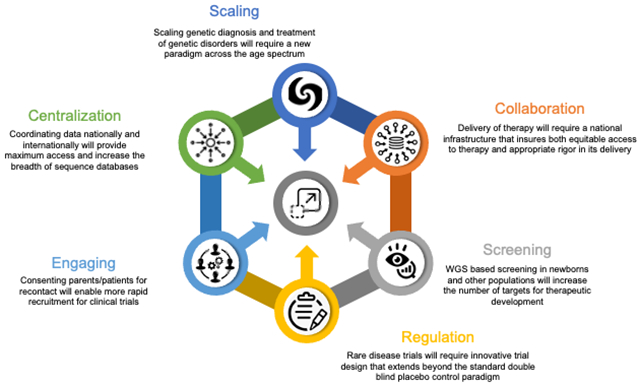
Graphical Abstract.
Scaling technologies for diagnosis and treatment of rare genetic disorders will take a coordinated effort across many disciplines to deliver therapies equitably and efficiently.
1. Introduction
The most common model for approval of new therapies by the U.S. Food and Drug Administration (FDA) is for one drug to be tested for efficacy for one disorder (or a group of disorders with similar pathophysiology) with a decision made based on double-blind placebo control trials (U.S. Department of Health and Human Services, 2019). The process is slow (taking many years), inefficient (with only a tiny fraction of candidate compounds in early studies ever reaching approval), and expensive (costing tens of millions of dollars to bring a drug to market). The economics of this system has been a major stumbling block to developing new drugs for rare disorders. For the last decade of the 20th and first decade of the 21st centuries, investigational new drug applications increased slowly with approval of only a handful of such medications per year (Lapteva et al., 2020). The number of gene therapies lagged even further behind due to a variety of practical, regulatory, and ethical issues, with the death of Jesse Gelsinger in a gene therapy trial for ornithine transcarbamylase deficiency an especially notable and tragic event that stalled progress for many years (Raper et al., 2003). The 2010s saw an upswing in IND applications for gene therapies, reaching 161 and 160 in 2019 and 2020, respectively. However, with >7,000 rare disorders as candidates for gene therapy, even with the recent increase in INDs, the trajectory to development of new treatments still leads to the inevitable conclusion that it will be many decades before therapies are available for the majority of genetic disorders. Platform approaches to drug development seek approval for a base therapeutic molecule with disease-specific modifications that leverage the early approval stages, such as toxicology studies, to reduce the amount of time and effort for approval of new members of the platform. For example, in the context of gene therapy, a common viral vector could be viewed as the platform base with different expression inserts to treat different disorders as the platform members (Luther et al., 2018; Wang et al., 2019). Subsequently, adaptive trial designs that account for the rare nature of most genetic disorders and deviate from the double-blind placebo control paradigm, have been suggested as mechanisms to reduce the number of subjects and time to reach an approval endpoint (Jahanshahi et al., 2021; Tandon & Kakkis, 2021). While such approaches have indeed been recognized as possible by the FDA, in practice, implementation to date has been halting.
Diagnosis of rare diseases has also been a challenge to treatment. Many patients come to diagnosis only after a long odyssey that begins with the development of symptoms and requires multiple referrals and testing before an answer is found. The advent of clinical whole exome or whole genome sequencing (WES and WGS, respectively) can significantly shorten this odyssey, but only if patients are seen in a clinic where they are available (Lavelle et al., 2022; Malinowski et al., 2020; Manickam et al., 2021). In this setting, the opportunity to affect disease progression and improve quality of life is significantly greater when a patient is treated pre-symptomatically rather than after symptoms develop. This phenomenon is perhaps best demonstrated by newborn screening (NBS), which was developed in the 1960’s upon the recognition that early treatment of patients with phenylketonuria (PKU) improved outcomes (Vockley et al., 2014). Moreover, patients with PKU identified and treated at birth due to the diagnosis of a previous affected sibling subsequently had normal intellectual development. Since then, NBS programs in the United States and many other developed nations have expanded to include dozens of disorders leading to improved outcomes in identified babies (McCandless & Wright, 2020; Watson, 2006). NBS has traditionally been performed with tests that measure a functional marker of gene function. However, most recently, disorders such as severe combined immune deficiencies and spinal muscular atrophy have joined the newborn recommended uniform screening panel (RUSP) in the US with primary molecular tests (Butterfield, 2021; Kobrynski, 2021), while second-tier testing of functional test abnormalities through gene sequencing is becoming a standard part of some screening programs (Furnier et al., 2020). Reduction in cost and increase in speed of WGS have brought us to an inflection point in NBS. Since clinically, most genetic disorders are not identified by functional testing but by sequencing, the use of WGS as a primary genetic screening tool has emerged as a possibility (Powell, 2018; Roman et al., 2020; Woerner et al., 2021). While the technology remains in development, the implications of such screening are currently being explored. Timely return of clinically actionable results, and the ethical implications of and strategies for dealing with variants of uncertain significance (VUS) and late-onset disease identified in a newborn loom over the field. Additionally, making WGS data available for clinical diagnostic purposes later in life brings new challenges in data storage, transfer, and access.
This manuscript summarizes discussions arising from a recent NIH conference on nucleic acid therapy, with a focus on scaling technologies and resources to ultimately optimize development and utilization of nucleic acid-based therapies across the age and disease spectrum.
2. Scaling Newborn Screening
Newborn screening is the largest population-based screening program and is offered to all newborns in the United States to aid in early identification of those at risk for a panel of treatable, often genetic diseases. Owing to its success in timely detection and treatment, newborn screening was named one of the ten greatest public health achievements of the first decade of the 21st century by the U.S. Centers for Disease Control and Prevention (Centers for Disease & Prevention, 2011). Since its inception in 1963, newborn screening programs have had tremendous growth not only in the number of diseases being screened for, but in the complexity of the testing being utilized to assess disease risk. While historically, expansion of newborn screening has been limited by assay technology; today, expansion of newborn screening is limited more by the lack of therapeutic availability for many rare diseases (Tarini, 2007).
Addition to newborn screening panels relies heavily on criteria originally outlined in a World Health Organization commissioned report on the principles of preventative screening programs. These criteria are more commonly known as the Wilson and Jungner criteria and include ten key tenants of a disease that makes it suitable for population screening (Figure 1) (Watson et al., 2006). Briefly, these criteria include that the disorder should be an important health problem, there should be some understanding of the natural history of disease, a suitable test, an agreed upon policy on whom to treat, and there should be an accepted and effective treatment. Though these criteria were developed in 1968, they very much remain the basis for addition of diseases to newborn screening panels today. In the US, because newborn screening is mandated at the state level, the continued use of criteria for disease addition remains paramount in ensuring ongoing trust in the screening system (McCandless & Wright, 2020).
Figure 1. Criteria for newborn screening.
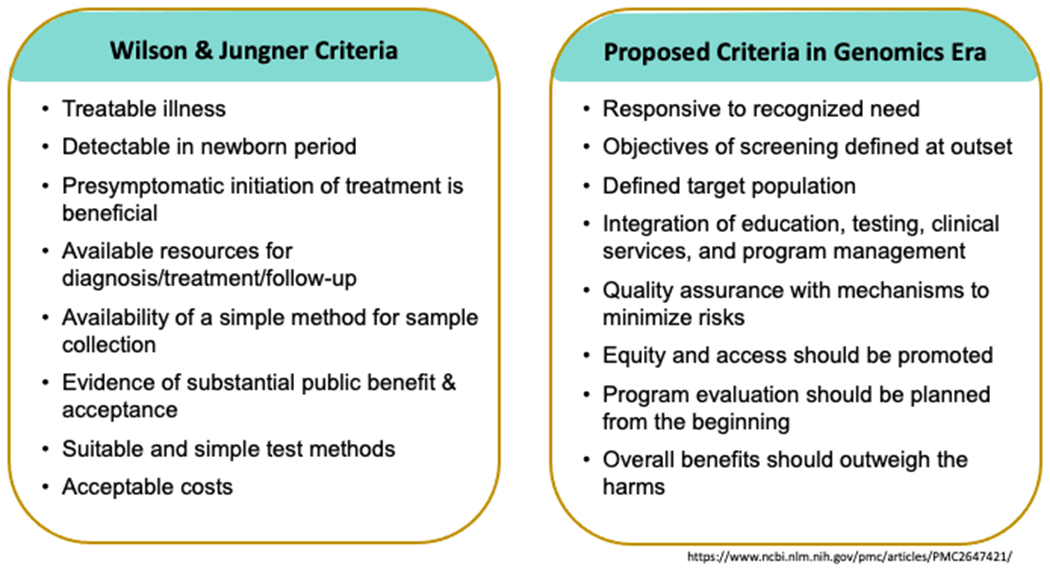
Wilson and Jungner criteria for disease screening as applied to newborn screening (left) can be mapped to clinical needs for implementation of genomic technologies for newborn screening (right).
Over the first three decades of newborn screening, diseases typically were added to newborn screening panels one at a time utilizing a one disease-one assay approach (Woerner et al., 2021). This paradigm was disrupted with the application of tandem mass spectrometry to newborn screening, which allowed for the multiplexing of numerous analytes and the potential for a many disease-one assay approach (Figure 2). The ability to add many diseases more rapidly, while beneficial to screening programs, began to result in disparities across the country whereby vast variations in newborn screening panels arose between states. Efforts to mitigate this led to the creation of a federal RUSP and a structured approach to reviewing potential diseases for inclusion on newborn screening panels. However, because state NBS programs still individually decide whether to add diseases, even if on the RUSP, disparities still remain among programs (Health Resources & Services Administration).
Figure 2. Scaling technologies for newborn screening.
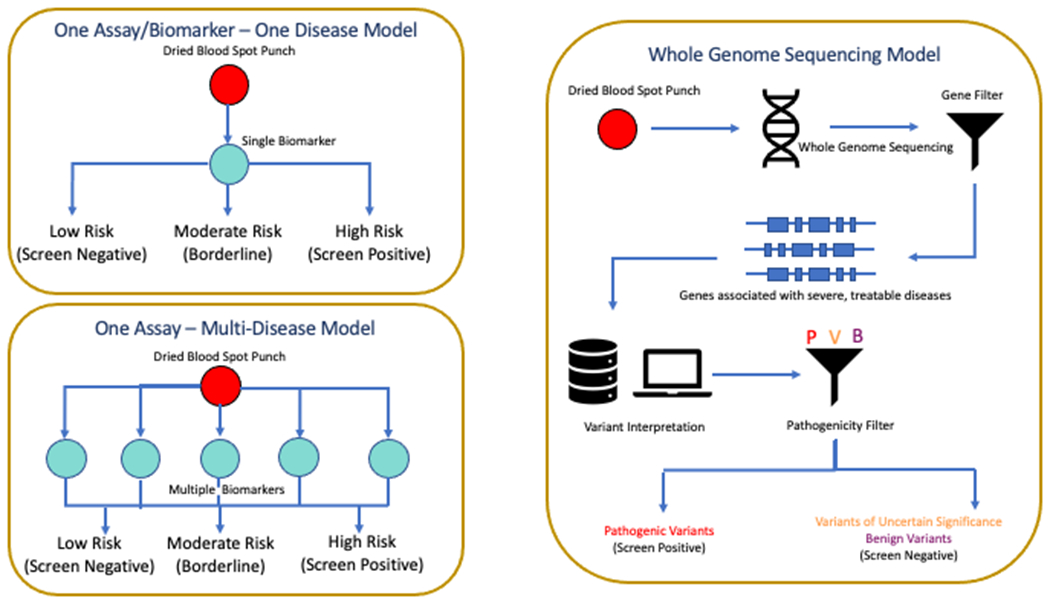
Traditional newborn screens were developed one test at a time and focused on a single disease (top left), with later technologies such as tandem mass spectrometry expanding the approach to evaluate many analytes at a time (bottom left). Both led to identification of a limited scope of diseases and were added to newborn screening panels in a methodical, but often slow, fashion. In contrast, whole genome sequencing in theory can identify any and all diseases in a single test, but at a cost that includes the need for broader, pan-ethnic data bases of genomic sequence to allow definitive classification of variants as pathogenic or benign. The sequence can also be carried forward to allow screening or testing for diseases across a lifetime. In reality, a combination of molecular and functional testing is likely to provide the most robust outcomes in a comprehensive NBS program.
Today, despite growing screening panels and advancements in technological capabilities, newborn screening still relies heavily on functional biochemical biomarkers to assess risk of disease (Longo et al., 2022). However, molecular assays increasingly are being incorporated into newborn screening programs, giving rise to another potential paradigm shift for newborn screening (Bailey et al., 2021). Molecular applications within newborn screening programs have arisen for several reasons and can generally be described in three categories: 1) No functional biomarker exists for the disease. This is the case for newborn screening for severe combined immunodeficiency (SCID) and spinal muscular atrophy (SMA) where RT-PCR is used to look simply for the presence or absence of genetic material; 2) Only a poor functional biomarker exists for the disease. For example, immunoreactive trypsinogen is neither a sensitive nor specific marker for cystic fibrosis, and thus, many NBS programs have implemented a second-tier CFTR gene variant panel into their screening algorithms; 3) Additional information regarding severity or treatment strategies can be provided through the delineation of underlying genotype. Though there has been a slow and steady increase in molecular technologies in newborn screening, it is still relatively limited.
Recent advances in the diagnosis of critically ill newborns in the neonatal intensive care unit (NICU) and the pediatric intensive care unit (PICU) have opened the door for advances in newborn screening by whole genome sequencing (NBS-WGS) (Clark et al., 2019; Farnaes et al., 2018; Smith et al., 2015). As detailed elsewhere, rapid whole genome sequencing has been tremendously effective in diagnosing infants in crisis, leading to an answer is 30-50% of cases (Maron et al., 2021). This clinical utility has been coupled to clear demonstrations of its cost effectiveness, which has led to multiple states reimbursing for the test, and the potential for its use to expand nationwide (Farnaes et al., 2018; Tan et al., 2017). Multiple efforts are now underway to apply the underlying technologies of rapid whole genome sequencing to newborn screening, potentially enabling identification of hundreds of diseases simultaneously (Adhikari et al., 2020). Efforts led by Genomics England, and Rady Children’s Institute for Genomics Medicine (RCIGM), plan to deliver screens for genetically driven diseases where treatments are available. Just as with traditional newborn screening, diseases are being considered for this approach based on the Wilson and Jungner criteria (Owen et al., 2022). With a dramatic decrease in the cost of genomic sequencing over time, the cost effectiveness is increasing and is soon likely to reach a price point where broad implementation will be possible (Berg et al., 2017; Dimmock et al., 2021). Of course, just as with traditional NBS, confirmatory testing of some sort will be needed to confirm screening results.
One example of a project to utilize WGS for NBS is a pilot program called BeginNGS currently being developed at RCIGM (Kingsmore et al., 2022). This project will progress in multiple stages in order to iteratively integrate new knowledge into the program. The project, currently being guided by a group involving multiple stakeholders, ultimately looks to evolve to a consortium supported by academic groups, patient advocacy groups, and pharmaceutical and biotech companies. Stage 1 Prototype / Feasibility has just been completed. A retrospective analysis was performed on 454,707 UK Biobank sequences and 4,376 critically ill children with suspected genetic disorders and their parents. Simulated Newborn Screening by rapid Whole Genome Sequencing (NBS-rWGS) investigating 388 disorders had a specificity of 99.7% after root cause analysis, and a sensitivity of 87% versus the previously performed diagnostic rWGS. In Stage 2 and 3 technical and logistic limitations of sequencing first with hundreds and then thousands of patients will be explored. While limitations still exist, the potential of the technologies involved has grown rapidly. The sensitivity and specificity of NBS rWGS may rival that of current clinical WGS methods that cost thousands of dollars, with limitations largely being knowledge based due to variants of uncertain significance. As more information is accumulated on disease-causing variants, NBS sensitivity and specificity will increase. For example, in one study, analysis of WGS data focused on set of genes that play a role in cancer development with a new analytical platform identified over three times as many “actionable” variants, with an increase of 50% in sensitivity (Chunn et al., 2020). Note that all WGS based diagnostic approaches are limited by our understanding of the genetics of the disease. Indeed, current molecular NBS for spinal muscular atrophy has been successful because of a robust genotype phenotype correlation between the copy numbers of the SMN2 gene and disease severity (De Vivo et al., 2019). In contrast, most current NBS tests have sensitivities that approach 100% under normal conditions, though with variable specificity, and lower sensitivity in premature newborns (Adhikari et al., 2020).
Even with these advances, it is important to understand that use of WGS as a primary screening tool is a shift from traditional newborn screening based on biochemical markers that can, in most cases, identify up to 99% of the individuals with significant risk for a rare disease (Adhikari et al., 2020; Woerner et al., 2021). Although current WGS techniques cannot match this performance, it can interrogate essentially any gene and an associated disorder regardless of the availability of a known biomarker or age of onset. Of course, identification of late-onset disease or a variant of uncertain significance in a healthy newborn have their own ethical ramifications that will be discussed later. In a very real way, traditional biomarker-based and WGS NBS complement each other. Infants with a confirmed diagnosis after traditional newborn screening are usually studied genetically in order to predict severity of disease (if known genotype-phenotype relationships exist) in a specific patient and ultimately to obtain a richer understanding of the genetics of that rare disease in aggregate patients. Such studies further improve the sensitivity of WGS as VUS are eventually recognized as either pathogenic or benign. In contrast, blood spots from patients screened positive by WGS can serve as a valuable resource for developing novel functional NBS assays for follow up and/or specific traditional screening methods when appropriate.
Specificity is another major challenge in newborn screening. Since, by design, <1% of infants will test positive for a traditional biomarker based genetically determined rare disease, even a small number of false positives is a burden for parents and the medical system. To circumvent this problem, tiered screening with reflex second-tier testing prior to reporting have been developed allowing for extremely high specificity. Early experience with WGS-based NBS suggest that this level of specificity might be a challenge. However, as noted above, a recent retrospective study examining 454,707 individuals from a UK Biobank supports that the needed specificity can be achieved. Here, the false positive rate across a set of almost 400 diseases was predicted to be three per thousand, which is comparable to rates expected with traditional newborn screening.
While the current WGS-based NBS trials are still in their infancy, the technique is developing rapidly. As it evolves, approaches to appropriately consent patients for recontact, deposition of data into widely accessible genomic databases to allow refinement of variant status, and data storage for future diagnostic inquiries will be critical to address. Advances in NBS will be accelerated if the existing efforts are able to appropriately aggregate data to leverage new information generated nationally and internationally. Importantly, the initial focus of most WGS NBS efforts has been on severe treatable disorders with neonatal onset. However, beyond the newborn period, the data obviously remain, and thus, are available to screen for diseases or variants with onset later in life. In anticipation of such programs, participants in neonatal screening could be consented for recontact in future.
Providing pharmaceutical companies access to the precise genetic information on genes of interest from millions of people would unquestionably accelerate their ability to identify variations that may need to be corrected. However, such a use of population-based WGS data, especially NBS, also raises the need for considerable caution. WGS that identifies a disease for treatment inevitably can identify untreatable genetic disease. Thus, appropriate personal and legal safeguards must be developed to prevent discrimination on the basis of genetic information.
3. Scaling Therapeutic Development
While advances in the use of WGS for diagnosis and NBS/population screening are exciting, it is an unfortunate reality that most genetic disorders have no available therapy. Today, fewer than 400 diseases meet Wilson and Jungner criteria, which begs the question, “How do we scale treatment availability to serve over 7000 rare diseases?” For WGS to have maximum health impact, it is critical to consider how to expand the number of diseases with effective treatment.
Much of classical drug development has focused on a relatively physiologic agnostic approach with screening of large chemical libraries that affect a specific biomarker in vivo or in vitro and subsequent optimization of effect through multiple rounds of chemical modification. However, understanding of cellular pathophysiology in genetic disorders is typically robust and allows a more directed approach to drug development. Here, gene-targeted therapies appear to be a natural answer; however, development presents multiple challenges (Aiuti et al., 2007). Researchers and companies need to identify the precise genetic defect that is responsible for a disease, develop therapies to overcome that defect, and then prove in pre-clinical experiments and clinical trials that the therapy works. For gene therapy, cellular and animal models of disease provide the opportunity for more a more rapid pre-clinical phase based on vector expression and biomarker studies. In this context, a drug development pipeline is best viewed as a collaborative effort across the public, private, and academic sectors. Most genetic disease experts are faculty at academic institutions, but public funding, which may have served well to help build the knowledge necessary to move therapy for a rare disease to clinical trials, typically is insufficient to pay for those clinical trials. In contrast, private pharmaceutical companies, and even therapy-focused venture capital firms, are more likely to have access to the financial resources to implement a clinical trial, but lack the disease-specific expertise to develop it. Bringing a new gene therapy to approval ultimately represents an intersection of advocacy around specific disorders (often championed by patients and parents), academic scientists and clinicians, and industry. Furthermore, the availability of a treatment is likely to drive awareness of the disease and improvement of techniques to diagnose the disease early enough to successfully intervene for maximum effect. Indeed, such diagnostics may well even drive implementation of newborn screening assuming availability of a suitable test. If families consent at the time of NBS to being re-contacted if new therapies emerge for disorders not originally included in the original screening report, recruitment of patienshort cutts for clinical trials could then be filled in a fraction of the current time (and cost), incentivizing a rapid increase in the number of gene therapies under development.
There are significant hurdles to scaling development and implementation of gene therapies for rare disease. A timeline that can stretch to a decade and costs that drive post-approval pricing to $1-2 million per treatments are anathema to the development of therapies for large numbers of diseases. Thus, novel pathways to development that do not compromise patient safety are crucial to breaking these barriers. One such option is the utilization of platform design for nucleic acid-based therapies and more ready recognition of them by regulators (Figure 3). For example, for gene therapies this would entail the approval of a standard vector based on extensive pre-clinical and toxicity studies with subsequent submission of that vector with disease-specific expression inserts as new drugs with a reduced need for such studies. Similarly, a broader understanding of the toxicities of oligonucleotides in general could eventually shorten the path to approval for patient-specific molecules. Other toxicities such as immunologic response to viral vectors and the expressed proteins will require additional attention but were not addressed by this conference.
Figure 3. Scaling development of gene therapies.
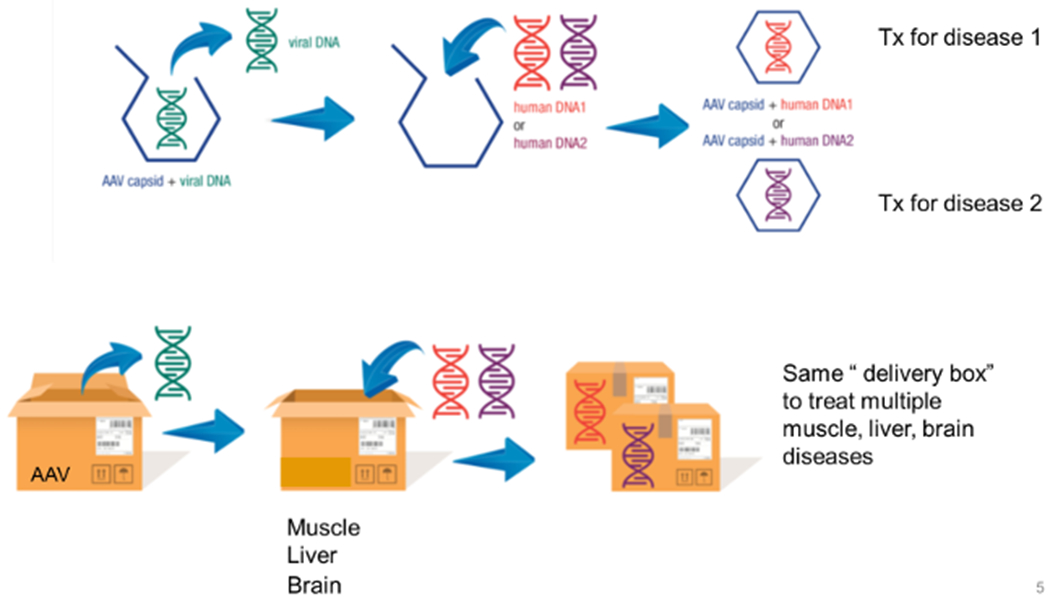
Gene therapy vectors can be considered a platform technology where the viral backbone is a near constant that can deliver multiple therapeutic genes to address multiple disorders, albeit typically one at a time. Thus, regulatory approaches for approval that view such technology as a single delivery system to treat multiple disorders will lead to faster implementation.
Equally important to approval of new medications is consideration of trial design. Double blind placebo control studies have long been viewed as the gold standard for clinical trials. However, in rare diseases, it is often challenging to truly blind a therapy and may even be unethical when the disease trajectory is one of rapid deterioration. In some situations, alternative designs, often specific to rare disease and small test populations, can even have greater power than traditional ones in proving efficacy. For example, the drug vestronidase alfa for treatment of mucopolysaccharidosis was approved on the basis of a clinical trial of 12 patients using a novel multi-domain responder index with staggered start of transition of patients randomized to treatment or placebo followed by transition to open label (Cadaoas et al., 2020; Fox et al., 2015; Tandon & Kakkis, 2021). Regardless of study design, trial design and assessment of efficacy are enormously dependent on a firm understanding of the natural history of the disease. Indeed, in a disorder with an easily predictable outcome, use of real-world data and patients as their own control can be an effective trial design, and even allows for effective N=1 trials. To reach this level of understanding in rare diseases, coordinated collection of real-world data in multi-center studies is essential. Accumulating such data optimally starts at birth, highlighting the utility of WGS-based newborn screening for disorders even in advance of the availability of a treatment.
Finally, optimization of drug development for rare diseases is the only path forward for making gene therapy financially tenable. Individual costs of $1-2 million dollars for the treatment for hundreds, if not thousands, of diseases is not sustainable in any system of health care delivery.
4. Treatment Challenges
Gene therapy, in its simplest implementation, can be viewed as the one-time process of delivery of a therapy/therapeutic vector to a patient. Common routes of delivery include intravenous, intrathecal, intramuscular, intraocular (or subretinal), or ex vivo cellular transduction with return of cells to the patient. From a medical reimbursement standpoint, such a delivery model provides very little impetus for a medical center to invest in the necessary resources to appropriately evaluate and treat such patients and would not provide even the most basic components of care to treated patients. In essence, this delivery model leads to the use of million (or multimillion) dollar drugs that generate minimal revenue for the treating center and provides no clinical safeguards for patients. To circumvent the latter, it is essential for broad delivery of gene therapy to occur in centers that include the full scope of disease diagnosis, treatment, and follow up (Pipe, 2021) (Figure 4). First, a pre-treatment protocol is necessary that encompasses confirmation of the genetic diagnosis along with ensuring that the prospective patient is in good clinical condition to undergo the therapy. Especially in inborn errors of metabolism, patients often experience variation in their biochemical control that requires optimization of metabolic care prior to any procedures. Gene therapy centers must be able to provide such service on site and in real time relative to the planned treatment. As more gene therapies become available, centers must also be able to help patients choose between vectors if more than one has been approved for individual diseases, and even confirm that gene therapy is indeed the best option to treat a specific patient. Providing such guidance requires a broad understanding by center personnel of both the molecular biology of the gene therapy vector(s) and its likely specific effects on the disease process prior to establishment of therapeutic efficacy. Understanding disease-specific risks of gene therapy is also critical to optimize therapy outcomes. For example, steroid administration, often used to reduce systemic rejection of a viral gene therapy vector, can exacerbate metabolic symptoms in some disorders, potentially requiring alterations in short-term management of the underlying disease. In light of these challenges, medical insurers in the United States will need to take a broad view of how to cover services to deliver gene therapy for any of the above suggestions to be viable. Many of these issues are also likely to be relevant in other countries, including those with national health insurance. Viewing gene therapy as a process that includes all necessary steps for evaluation of patients, delivery of the treatment, and follow up for a clinically relevant post-treatment period with bundled billing with previously agreed upon coverage by insurers offers one possible route to circumvent this problem. It is not unreasonable to consider payment policies tied to the cost of a drug with the expectation that some of that cost could, in fact, be borne by the companies that stand to profit from these extraordinarily expensive medications.
Figure 4. Models for integration of services for nucleic acid-based therapies of rare genetic diseases.
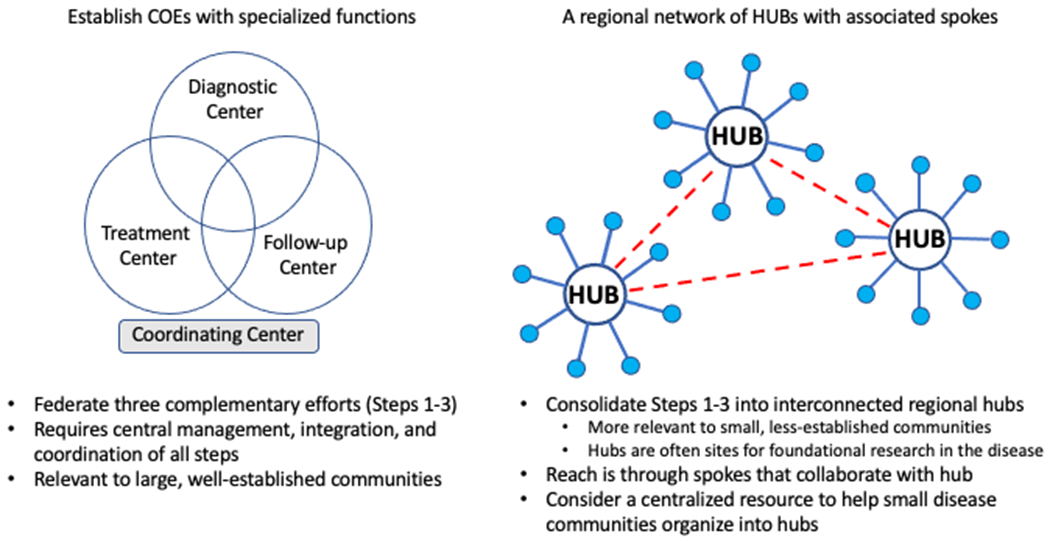
Nucleic acid-based therapies must encompass the full range of diagnostic, treatment, and long term follow up of such technologies to guarantee their appropriate utilization in a safe and effective manner. Since not all clinics and institutions will be able to or interested in participating in all aspects of such a system, a system of coordinated centers with overlapping capabilities (left) or hub and spoke model (right) with coordinating centers with full capabilities that interact with regional ones that are able to participate in some of the needed activities, typically diagnosis and long term follow up.
In many ways, the next requirement of a gene therapy center, delivering the vector, appears to be the most trivial. Most tertiary care medical centers are appropriately equipped with infusion facilities, interventional radiology suites, and day surgery centers that provide most of the resources necessary to deliver treatment. However, the physical act of gene therapy represents only a small part of the required institutional infrastructure. Specialized pharmacy facility needs include ultracold freezers for vector storage, dedicated biosafety hoods for preparation for use, and expedient delivery to the treatment center, as well as pharmacists experienced in the process (Stoner, 2018). On the delivery end, nurses and physicians must be familiar with possible infusion reactions and short-term risks such as post-treatment hyperpyrexia and thrombotic microangiopathic syndrome. In subsequent days and weeks, apparent inflammatory reactions consistent with viral induced tissue damage come into play and require careful titration of immunosuppression to avoid loss of the gene therapy vector. Longer-term management issues center on modification and titration of the prior treatment regimen as the expression of the new transgene takes effect. Often, metabolic disorders offer the most significant challenges as too rapid reduction of pre-gene therapy treatment modalities put a patient at risk for metabolic sequelae. Ideally, discontinuation of prior therapy will be possible assuming sufficient efficacy of the gene therapy. However, given the potential for loss over time of vector from transfected tissue, it might become necessary to reimplement prior metabolic therapy, thus making long-term follow up of these patients mandatory.
Long-term follow up is an essential component of any center or program offering gene therapy (Hampson et al., 2018). Here, the focus must not just be on monitoring persistence or recurrence of disease. Rather, emphasis must expand to examining the process of gene therapy itself. It is critical that data on safety and efficacy be collected systematically and submitted to a central source for storage. Access to those data should be open source, including across industry, to allow broad examination of outcomes and trends that might cross disease-specific therapies and focus instead on other commonalities such as vector backbone, mode of immunosuppression, and optimum routes of vector delivery.
Given the breadth of genetic disease and the large number of gene therapies that will ultimately become available, it is reasonable to assume that not all centers administering gene therapy will be interested and/or able to deliver all therapies for all disorders. In addition, some centers may not have sufficient resources to have a “full-service” gene therapy program, but still be able to participate collaboratively in some aspects of care of gene therapy patients. Similarly, large, tertiary medical centers likely to be able to deliver gene therapy, might not have capacity (or want) to perform all the activates related to long-term follow up. Thus, a distributive and collaborative model is likely to be necessary to meet all the requirements for safe and effective delivery of gene therapy. Many such models are likely to be viable, but one that has been successful for coordinated care of rare disease is the spoke and hub model. Here, a full-service gene therapy center, likely to grow from existing large, academic, tertiary care medical centers, would act as a hub that offers all aspects of disease assessment, gene therapy delivery, and long-term follow up for its own patients, while providing only the gene therapy for patients from institutions that are qualified to deliver the other necessary components of care. Such a model replicates the process for transplant in many regards, where a limited number of academic based transplant centers serve as referral centers for other institutitions, and it may serve as a viable starting point to build gene therapy programs. Of note, such a model could exacerbate stresses on the current genetic work force and will require explicit efforts to increase physician and genetic counselor training.
5. Public Health and Ethical Issues
Expanding or scaling newborn screening programs and related resources will also require a reexamination of its ethical, legal, and social implications. Recent debates have questioned whether adding certain disorders or new technologies may move us away from the criteria that have traditionally guided decisions about population-based screening (Figure 1). A 1982 article in the American Journal of Public Health articulated the ethical justification for compulsory PKU screening (Faden et al., 1982). Framing newborn screening within a child welfare model, the authors argued that the potential harm for an unscreened newborn with PKU should override parental choice. However, they also warned that the ethical justification for newborn screening (NBS) must be re-evaluated as new disorders are added to state panels. However, as NBS programs have evolved, this ethical re-evaluation largely has not happened. If NBS is to preserve the benefits that programs provide to newborns and their families, it will be crucial that the ethics of NBS are also “scaled” to address the potential harms of expanded screening. Economic considerations are also crucial to address such that the increased costs of screening and ultimately therapies are not barriers to broad implementation.
First, it is crucial to address how new disorders being added to panels that may weaken child welfare arguments supporting NBS, including disorders with large phenotypic variability, differential ages of onset, increased uncertainty or false positive rates, and exceedingly burdensome interventions. For example, many of the disorders being considered for expanded screening may identify variants associated with later-onset disease. Most genetic professional organizations have largely discouraged giving parents genetic information regarding adult-onset conditions, citing the child’s “right to an open future,” indicating that returning such information may violate that child’s future ability to choose whether to know genetic information about themselves (Davis, 1997). Nevertheless, current arguments regarding the “right to an open future” are evolving, and some bioethicists and genetic professionals are challenging current concerns regarding the implications of giving later-onset information to parents. For example, disclosure of adult-onset information to parents may promote the best interest of the child and could result in earlier prevention measures, increased screening, or personal life planning, and therefore outweigh the possible harms of infringing on a child’s right not to know (Garrett et al., 2019; Wasserstein et al., 2021). An increased uptake of newborn genomic sequencing would certainly necessitate further dialogue about these positions. Current pilot NBS-WGS programs like Genomic England and BeginNGS are taking a cautious approach and limiting themselves to severe early onset disorders that have effective treatments available.
Programs will also need to examine how new screening technologies might impact the justification for current NBS policies, especially when, as in genomic sequencing, these technologies exponentially expand the scope and complexity of screening results. Early data from research programs that have examined the possible psychological and emotional implications of genome sequencing in newborns have been promising, showing minimal harms to parents receiving sequencing results regarding their newborns, and little negative psychosocial impact over time (Robinson et al., 2019). Nevertheless, as the use of sequencing technology grows in the context of public health screening programs, programs must continually reevaluate the potential implications of this information on families. This is especially true for families receiving uncertain results. A number of studies have shown the possible negative implications that receiving uncertain NBS results can have on parents, including the worry and anxiety associated with becoming a “patient in waiting” where the possible onset of symptoms, disease severity, or prognosis is unknown or unclear (Timmermans & Buchbinder, 2010).
These challenges all lead to the possible need to re-examine the ethical and legal justifications for current NBS policies, including mandatory screening. With a move into a new era of NBS, it is crucial to address the need to balance parental choice with the compulsory nature of screening, in order to maintain public trust in NBS and assure that all newborns and families can continue to benefit from screening programs. It is probable, that for some new directions in NBS programs, consent will be necessary to give parents more choices about what kinds of information they would want to know about their newborns and make decisions about when to obtain those results (Ross, 2010; Tarini & Goldenberg, 2012).
Finally, the newborn screening community must also be diligent to examine the equity and justice implications of expanded or scaled NBS programs and resources. At its core, NBS programs are meant to represent a universal public health program that allows every infant born in the US to access screening. For the most part, this has been true as nearly all infants in the US currently receive newborn screening (Brosco et al., 2015). However, as NBS expands to include wider ranges of disorders and new screening technologies, new disparities might emerge. For example, given the complexity and costs of genomic technology, there is a need to address the potential that the application of genomics in newborn screening may perpetuate or even exacerbate existing NBS disparities. Specifically, as newborn screening adds disorders for which treatment is extremely expensive or harder to access, the medical system must confront the social justice implications of universal screening without equitable follow up and treatments. Additionally, programs must also address the limitations associated with genomic screening targets or screening modalities developed using data from non-diverse samples (Hindorff et al., 2018; Popejoy & Fullerton, 2016). The lack of participation of under-represented populations, including many racial and ethnic groups, has led to increase recognition that the relevance of screening results for newborns and families from these communities may be less accurate, lead to increase uncertain results, or increased false positive or negative results (Goldenberg et al., 2016; Martin et al., 2017; Pique et al., 2017). It is necessary to significantly expand research that includes under-represented populations to address the potential harms of ancestry-related test limitations with misclassification of variants as pathogenic based on limited ethnic group data, and the potential health and social implications they may have on families (Manrai et al., 2016). The goal in scaling NBS resources must be framed within the context of large equity considerations in rare diseases and gene targeted therapies, promoting the development of expanded NBS in ways that preserve the universality of newborn screening and its benefits for all newborns and families.
Acknowledgments
The authors thank Joanne Lumsden and Tiina Urv for their assistance with the manuscript.
Disclaimer
The content of this publication reflects discussions from a June 2021, 3-day workshop sponsored by the National Institutes of Health (NIH) entitled, “Gene-Targeted Therapies: Early Diagnosis and Equitable Delivery” (National Institutes of Health, 2021). This material should not be interpreted as representing the viewpoint of the U.S. Department of Health and Human Services, the National Institutes of Health, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institute of Neurological Disorders and Stroke or the National Center for Advancing Translational Sciences.
Conflicts of Interest
Thomas Defay is an employee and shareholder of Alexion Pharmaceuticals. Amy Gaviglio has received speaker fees from Worldwide Clinical Trials. Jerry Vockley and Aaron Goldberg have no conflicts to report.
References
- Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, Chen F, Kundu K, Kvale M, Mooney SD, Nussbaum RL, Randi SS, Sanford J, Shieh JT, Srinivasan R, Sunderam U, Tang H, Vaka D, Zou Y, … Brenner SE (2020). The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med, 26(9), 1392–1397. 10.1038/s41591-020-0966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Bachoud-Levi AC, Blesch A, Brenner MK, Cattaneo F, Chiocca EA, Gao G, High KA, Leen AM, Lemoine NR, McNeish IA, Meneguzzi G, Peschanski M, Roncarolo MG, Strayer DS, Tuszynski MH, Waxman DJ, & Wilson JM (2007). Progress and prospects: gene therapy clinical trials (part 2). Gene Ther, 14(22), 1555–1563. 10.1038/sj.gt.3303033 [DOI] [PubMed] [Google Scholar]
- Bailey DB Jr., Porter KA, Andrews SM, Raspa M, Gwaltney AY, & Peay HL (2021). Expert Evaluation of Strategies to Modernize Newborn Screening in the United States. JAMA Netw Open, 4(12), e2140998. 10.1001/jamanetworkopen.2021.40998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JS, Agrawal PB, Bailey DB Jr., Beggs AH, Brenner SE, Brower AM, Cakici JA, Ceyhan-Birsoy O, Chan K, Chen F, Currier RJ, Dukhovny D, Green RC, Harris-Wai J, Holm IA, Iglesias B, Joseph G, Kingsmore SF, Koenig BA, … Wise AL (2017). Newborn Sequencing in Genomic Medicine and Public Health. Pediatrics, 139(2). 10.1542/peds.2016-2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosco JP, Grosse SD, & Ross LF (2015). Universal state newborn screening programs can reduce health disparities. JAMA Pediatr, 169(1), 7–8. 10.1001/jamapediatrics.2014.2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield RJ (2021). Spinal Muscular Atrophy Treatments, Newborn Screening, and the Creation of a Neurogenetics Urgency. Semin Pediatr Neurol, 38, 100899. 10.1016/j.spen.2021.100899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadaoas J, Boyle G, Jungles S, Cullen S, Vellard M, Grubb JH, Jurecka A, Sly W, & Kakkis E (2020). Vestronidase alfa: Recombinant human beta-glucuronidase as an enzyme replacement therapy for MPS VII. Mol Genet Metab, 130(1), 65–76. 10.1016/j.ymgme.2020.02.009 [DOI] [PubMed] [Google Scholar]
- Centers for Disease, C., & Prevention. (2011). Ten great public health achievements--worldwide, 2001-2010. MMWR Morb Mortal Wkly Rep, 60(24), 814–818. https://www.ncbi.nlm.nih.gov/pubmed/21697806 [PubMed] [Google Scholar]
- Chunn LM, Nefcy DC, Scouten RW, Tarpey RP, Chauhan G, Lim MS, Elenitoba-Johnson KSJ, Schwartz SA, & Kiel MJ (2020). Mastermind: A Comprehensive Genomic Association Search Engine for Empirical Evidence Curation and Genetic Variant Interpretation. Front Genet, 11, 577152. 10.3389/fgene.2020.577152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MM, Hildreth A, Batalov S, Ding Y, Chowdhury S, Watkins K, Ellsworth K, Camp B, Kint CI, Yacoubian C, Farnaes L, Bainbridge MN, Beebe C, Braun JJA, Bray M, Carroll J, Cakici JA, Caylor SA, Clarke C, … Kingsmore SF (2019). Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci Transl Med, 11(489). 10.1126/scitranslmed.aat6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DS (1997). Genetic dilemmas and the child’s right to an open future. Hastings Cent Rep, 27(2), 7–15. https://www.ncbi.nlm.nih.gov/pubmed/9131346 [PubMed] [Google Scholar]
- De Vivo DC, Bertini E, Swoboda KJ, Hwu WL, Crawford TO, Finkel RS, Kirschner J, Kuntz NL, Parsons JA, Ryan MM, Butterfield RJ, Topaloglu H, Ben-Omran T, Sansone VA, Jong YJ, Shu F, Staropoli JF, Kerr D, Sandrock AW, … Group, N. S. (2019). Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul Disord, 29(11), 842–856. 10.1016/j.nmd.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock D, Caylor S, Waldman B, Benson W, Ashburner C, Carmichael JL, Carroll J, Cham E, Chowdhury S, Cleary J, D’Harlingue A, Doshi A, Ellsworth K, Galarreta CI, Hobbs C, Houtchens K, Hunt J, Joe P, Joseph M, … Farnaes L (2021). Project Baby Bear: Rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet, 108(7), 1231–1238. 10.1016/j.ajhg.2021.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden RR, Holtzman NA, & Chwalow AJ (1982). Parental rights, child welfare, and public health: the case of PKU screening. Am J Public Health, 72(12), 1396–1400. 10.2105/ajph.72.12.1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, Cakici JA, Benson W, Kaplan RH, Kronick R, Bainbridge MN, Friedman J, Gold JJ, Ding Y, Veeraraghavan N, Dimmock D, & Kingsmore SF (2018). Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med, 3, 10. 10.1038/s41525-018-0049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JE, Volpe L, Bullaro J, Kakkis ED, & Sly WS (2015). First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab, 114(2), 203–208. 10.1016/j.ymgme.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnier SM, Durkin MS, & Baker MW (2020). Translating Molecular Technologies into Routine Newborn Screening Practice. Int J Neonatal Screen, 6(4). 10.3390/ijns6040080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett JR, Lantos JD, Biesecker LG, Childerhose JE, Chung WK, Holm IA, Koenig BA, McEwen JE, Wilfond BS, Brothers K, & Clinical Sequencing Exploratory Research Consortium Pediatrics Working, G. (2019). Rethinking the “open future” argument against predictive genetic testing of children. Genet Med, 21(10), 2190–2198. 10.1038/s41436-019-0483-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg AJ, Comeau AM, Grosse SD, Tanksley S, Prosser LA, Ojodu J, Botkin JR, Kemper AR, & Green NS (2016). Evaluating Harms in the Assessment of Net Benefit: A Framework for Newborn Screening Condition Review. Matern Child Health J, 20(3), 693–700. 10.1007/s10995-015-1869-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson G, Towse A, Pearson SD, Dreitlein WB, & Henshall C (2018). Gene therapy: evidence, value and affordability in the US health care system. J Comp Eff Res, 7(1), 15–28. 10.2217/cer-2017-0068 [DOI] [PubMed] [Google Scholar]
- Health Resources & Services Administration. Advisory Committee on Heritable Disorders in Newborns and Children. Retrieved August 1, 2022 from https://www.hrsa.gov/advisory-committees/heritable-disorders/index.html
- Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, Manolio TA, & Green ED (2018). Prioritizing diversity in human genomics research. Nat Rev Genet, 19(3), 175–185. 10.1038/nrg.2017.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanshahi M, Gregg K, Davis G, Ndu A, Miller V, Vockley J, Ollivier C, Franolic T, & Sakai S (2021). The Use of External Controls in FDA Regulatory Decision Making. Ther Innov Regul Sci, 55(5), 1019–1035. 10.1007/s43441-021-00302-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore SF, Smith LD, Kunard CM, Bainbridge M, Batalov S, Benson W, Blincow E, Caylor S, Chambers C, Del Angel G, Dimmock DP, Ding Y, Ellsworth K, Feigenbaum A, Frise E, Green RC, Guidugli L, Hall KP, Hansen C, … Defay T (2022). A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. Am J Hum Genet, 109(9), 1605–1619. 10.1016/j.ajhg.2022.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrynski LJ (2021). Newborn Screening in the Diagnosis of Primary Immunodeficiency. Clin Rev Allergy Immunol. 10.1007/s12016-021-08876-z [DOI] [PubMed] [Google Scholar]
- Lapteva L, Purohit-Sheth T, Serabian M, & Puri RK (2020). Clinical Development of Gene Therapies: The First Three Decades and Counting. Mol Ther Methods Clin Dev, 19, 387–397. 10.1016/j.omtm.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelle TA, Feng X, Keisler M, Cohen JT, Neumann PJ, Prichard D, Schroeder BE, Salyakina D, Espinal PS, Weidner SB, & Maron JL (2022). Cost-effectiveness of exome and genome sequencing for children with rare and undiagnosed conditions. Genet Med, 24(6), 1349–1361. 10.1016/j.gim.2022.03.005 [DOI] [PubMed] [Google Scholar]
- Longo N, Sass JO, Jurecka A, & Vockley J (2022). Biomarkers for drug development in propionic and methylmalonic acidemias. J Inherit Metab Dis, 45(2), 132–143. 10.1002/jimd.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther DC, Lee YW, Nagaraj H, Scaletti F, & Rotello VM (2018). Delivery approaches for CRISPR/Cas9 therapeutics in vivo: advances and challenges. Expert Opin Drug Deliv, 15(9), 905–913. 10.1080/17425247.2018.1517746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinowski J, Miller DT, Demmer L, Gannon J, Pereira EM, Schroeder MC, Scheuner MT, Tsai AC, Hickey SE, Shen J, Practice AP, & Guidelines C (2020). Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med, 22(6), 986–1004. 10.1038/s41436-020-0771-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam K, McClain MR, Demmer LA, Biswas S, Kearney HM, Malinowski J, Massingham LJ, Miller D, Yu TW, Hisama FM, & Directors A. B. o. (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 23(11), 2029–2037. 10.1038/s41436-021-01242-6 [DOI] [PubMed] [Google Scholar]
- Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, & Kohane IS (2016). Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med, 375(7), 655–665. 10.1056/NEJMsa1507092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron JL, Kingsmore SF, Wigby K, Chowdhury S, Dimmock D, Poindexter B, Suhrie K, Vockley J, Diacovo T, Gelb BD, Stroustrup A, Powell CM, Trembath A, Gallen M, Mullen TE, Tanpaiboon P, Reed D, Kurfiss A, & Davis JM (2021). Novel Variant Findings and Challenges Associated With the Clinical Integration of Genomic Testing: An Interim Report of the Genomic Medicine for Ill Neonates and Infants (GEMINI) Study. JAMA Pediatr, 175(5), e205906. 10.1001/jamapediatrics.2020.5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD, & Kenny EE (2017). Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am J Hum Genet, 100(4), 635–649. 10.1016/j.ajhg.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandless SE, & Wright EJ (2020). Mandatory newborn screening in the United States: History, current status, and existential challenges. Birth Defects Res, 112(4), 350–366. 10.1002/bdr2.1653 [DOI] [PubMed] [Google Scholar]
- Owen MJ, Lefebvre S, Hansen C, Kunard CM, Dimmock DP, Smith LD, Scharer G, Mardach R, Willis MJ, Feigenbaum A, Niemi AK, Ding Y, Van Der Kraan L, Ellsworth K, Guidugli L, Lajoie BR, McPhail TK, Mehtalia SS, Chau KK, … Kingsmore SF (2022). An automated 13.5 hour system for scalable diagnosis and acute management guidance for genetic diseases. Nat Commun, 13(1), 4057. 10.1038/s41467-022-31446-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipe SW (2021). Delivering on the promise of gene therapy for haemophilia. Haemophilia, 27 Suppl 3, 114–121. 10.1111/hae.14027 [DOI] [PubMed] [Google Scholar]
- Pique L, Graham S, Pearl M, Kharrazi M, & Schrijver I (2017). Cystic fibrosis newborn screening programs: implications of the CFTR variant spectrum in nonwhite patients. Genet Med, 19(1), 36–44. 10.1038/gim.2016.48 [DOI] [PubMed] [Google Scholar]
- Popejoy AB, & Fullerton SM (2016). Genomics is failing on diversity. Nature, 538(7624), 161–164. 10.1038/538161a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell CM (2018). What Genomic Sequencing Can Offer Universal Newborn Screening Programs. Hastings Cent Rep, 48 Suppl 2, S18–S19. 10.1002/hast.878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, & Batshaw ML (2003). Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab, 80(1-2), 148–158. 10.1016/j.ymgme.2003.08.016 [DOI] [PubMed] [Google Scholar]
- Robinson JO, Wynn J, Biesecker B, Biesecker LG, Bernhardt B, Brothers KB, Chung WK, Christensen KD, Green RC, McGuire AL, Hart MR, Griesemer I, Patrick DL, Rini C, Veenstra D, Cronin AM, & Gray SW (2019). Psychological outcomes related to exome and genome sequencing result disclosure: a meta-analysis of seven Clinical Sequencing Exploratory Research (CSER) Consortium studies. Genet Med, 21(12), 2781–2790. 10.1038/s41436-019-0565-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman TS, Crowley SB, Roche MI, Foreman AKM, O’Daniel JM, Seifert BA, Lee K, Brandt A, Gustafson C, DeCristo DM, Strande NT, Ramkissoon L, Milko LV, Owen P, Roy S, Xiong M, Paquin RS, Butterfield RM, Lewis MA, … Berg JS (2020). Genomic Sequencing for Newborn Screening: Results of the NC NEXUS Project. Am J Hum Genet, 107(4), 596–611. 10.1016/j.ajhg.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross LF (2010). Mandatory versus voluntary consent for newborn screening? Kennedy Inst Ethics J, 20(4), 299–328. https://www.ncbi.nlm.nih.gov/pubmed/21338027 [PubMed] [Google Scholar]
- Smith LD, Willig LK, & Kingsmore SF (2015). Whole-Exome Sequencing and Whole-Genome Sequencing in Critically Ill Neonates Suspected to Have Single-Gene Disorders. Cold Spring Harb Perspect Med, 6(2), a023168. 10.1101/cshperspect.a023168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner N (2018). Are UK hospital pharmacy departments ready for the rise of gene therapy medicinal products? Expert Opin Biol Ther, 18(8), 837–840. 10.1080/14712598.2018.1495192 [DOI] [PubMed] [Google Scholar]
- Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, Chong B, Phelan D, Brett GR, Creed E, Jarmolowicz A, Yap P, Walsh M, Downie L, Amor DJ, Savarirayan R, McGillivray G, Yeung A, Peters H, … White SM (2017). Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr, 171(9), 855–862. 10.1001/jamapediatrics.2017.1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon PK, & Kakkis ED (2021). The multi-domain responder index: a novel analysis tool to capture a broader assessment of clinical benefit in heterogeneous complex rare diseases. Orphanet J Rare Dis, 16(1), 183. 10.1186/s13023-021-01805-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarini BA (2007). The current revolution in newborn screening: new technology, old controversies. Arch Pediatr Adolesc Med, 161(8), 767–772. 10.1001/archpedi.161.8.767 [DOI] [PubMed] [Google Scholar]
- Tarini BA, & Goldenberg AJ (2012). Ethical issues with newborn screening in the genomics era. Annu Rev Genomics Hum Genet, 13, 381–393. 10.1146/annurev-genom-090711-163741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans S, & Buchbinder M (2010). Patients-in-waiting: Living between sickness and health in the genomics era. J Health Soc Behav, 51(4), 408–423. 10.1177/0022146510386794 [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services. (2019). Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products: Guidance for Industry Retrieved from https://www.fda.gov/media/133660/download
- Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, Mitchell J, Smith WE, Thompson BH, Berry SA, American College of Medical, G., & Genomics Therapeutics, C. (2014). Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med, 16(2), 188–200. 10.1038/gim.2013.157 [DOI] [PubMed] [Google Scholar]
- Wang D, Tai PWL, & Gao G (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov, 18(5), 358–378. 10.1038/s41573-019-0012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein MP, Orsini JJ, Goldenberg A, Caggana M, Levy PA, Breilyn M, & Gelb MH (2021). The future of newborn screening for lysosomal disorders. Neurosci Lett, 760, 136080. 10.1016/j.neulet.2021.136080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MS (2006). Current status of newborn screening: decision-making about the conditions to include in screening programs. Ment Retard Dev Disabil Res Rev, 12(4), 230–235. 10.1002/mrdd.20127 [DOI] [PubMed] [Google Scholar]
- Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, & Howell RR (2006). Newborn screening: toward a uniform screening panel and system. Genet Med, 8 Suppl 1, 1S–252S. 10.1097/01.gim.0000223891.82390.ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner AC, Gallagher RC, Vockley J, & Adhikari AN (2021). The Use of Whole Genome and Exome Sequencing for Newborn Screening: Challenges and Opportunities for Population Health. Front Pediatr, 9, 663752. 10.3389/fped.2021.663752 [DOI] [PMC free article] [PubMed] [Google Scholar]