

Abstract
BACKGROUND & AIMS:
Colorectal cancer is a leading cause of cancer death, and a major risk factor is chronic inflammation. Despite the link between colitis and cancer, the mechanism by which inflammation leads to colorectal cancer is not well understood.
METHODS:
To investigate whether different forms of inflammation pose the same risk of cancer, we compared several murine models of colitis (dextran sodium sulfate [DSS], 2,4,6-trinitrobenzene sulfonic acid, 4-ethoxylmethylene-2-phenyloxazol-5-one, Citrobacter rodentium, Fusobacterium nucleatum, and doxorubicin) with respect to their ability to lead to colonic tumorigenesis. We attempted to correlate the severity of colitis and inflammatory profile with the risk of tumorigenesis in both azoxymethane-dependent and Dclk1/APCfl/fl murine models of colitis-associated cancer.
RESULTS:
DSS colitis reproducibly led to colonic tumors in both mouse models of colitis-associated cancer. In contrast, all other forms of colitis did not lead to cancer. When compared with the colitis not associated with tumorigenesis, DSS colitis was characterized by significantly increased CD11b+F4/80+Ly6Chigh macrophages and CD11b+Ly6G+ neutrophils. Interestingly, depletion of the CD11b+F4/80+Ly6Chigh macrophages inhibited tumorigenesis, whereas depletion of CD11b+Ly6G+ neutrophils had no effect on tumorigenesis. Furthermore, the macrophage-derived cytokines interleukin-1β, tumor necrosis factor–α, and interleukin-6 were significantly increased in DSS colitis and promoted stemness of Dclk1+ tuft cells that serve as the cellular origin of cancer.
CONCLUSIONS:
We have identified CD11b+F4/80+Ly6Chigh macrophages as key mediators of cancer initiation in colitis-associated cancer. Development of new therapies that target these cells may provide an effective preventative strategy for colitis-associated cancer.
Keywords: Colorectal Cancer, Colitis, AOM/DSS, Macrophages, Dclk1, Tuft Cells, Colitis-Associated Cancer
Graphical Abstract
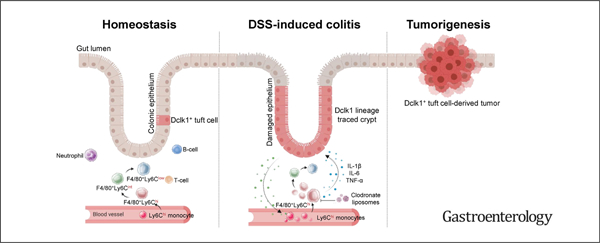
According to GLOBOCAN 2020 statistics, colorectal cancer (CRC) is the second leading cause of cancer deaths worldwide.1 A major risk factor for CRC is chronic inflammation, such as that observed in patients with inflammatory bowel disease (IBD).2–4 The link between inflammation and cancer is further supported by experimental evidence that mice treated with a single injection of the carcinogen azoxymethane (AOM) develop colon cancer only after the induction of colitis by means of the polysaccharide dextran sodium sulfate (DSS).5,6 The mechanism by which colitis leads to cancer, however, is not well understood.
To better define the factors in colitis that contribute to cancer, we examined the inflammatory responses among various models of colitis and compared their propensity to lead to tumorigenesis. Several animal models are currently used to recapitulate IBD, including Crohn’s disease (CD) and ulcerative colitis (UC). The mouse models used most include genetically engineered, adoptive T-cell transfer, infectious, chemical-induced, and spontaneous models of colitis in genetically susceptible animals. The chemical-induced models, such as DSS; 2,4,6-trinitrobenzene sulfonic acid (TNBS); and 4-ethoxylmethylene-2-phenyloxazol-5-one (oxazolone), have been the most readily studied because of their rapid onset of inflammation, methodological ease, and ability to be used in immunocompetent mice.7–10 Citrobacter rodentium, a gram-negative murine-specific bacterial pathogen, has been commonly used as an infectious model of colitis due to its alteration of epithelial barrier integrity, mucosal repair, inflammation, and the composition of commensal microbiota.11,12 Interestingly, despite differences among the models, a detailed comparison of how inflammatory responses differ, particularly with respect to their predisposition to tumorigenesis, has not been performed.
The tumor microenvironment, including inflammatory cells and surrounding stroma, form a key component of the tumor and have been associated with both pro- and antitumorigenic properties in CRC.13 T-cell infiltration within the tumor, for instance, has been proposed to predict patient prognosis in those with CRC.14–16 Furthermore, tumorassociated neutrophils are often polarized to either an antitumorigenic (N1) or pro-tumorigenic (N2) phenotype.17 N2 neutrophils have been shown to suppress T cells via transforming growth factor–β and promote tumor growth in a transgenic murine model of CRC.18 Tumor-associated macrophages, however, secrete vascular endothelial growth factor and promote angiogenesis to exert a pro-tumorigenic effect,19–24 further highlighting the role of inflammatory cells in cancer progression. The role of macrophages in tumor initiation, however, remains poorly understood.
In this study, we found that DSS-induced colitis reproducibly leads to colonic tumors, whereas other forms of colonic inflammation do not. Our data revealed that cancer initiation is highly dependent on the type of injury rather than the degree of inflammation. We further found that a specific subset of macrophages was key to colonic tumor formation and that depletion of these cells can inhibit tumorigenesis substantially. Our data showed that macrophage-derived factors interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 contribute to the stemness of epithelial tuft cells to induce colonic tumorigenesis associated with inflammation.
Methods
Animal Studies
Animal studies were approved by the Western University Animal Care Committee according to guidelines established by the Canadian Council on Animal Care. Mice were housed in the London Regional Cancer Program vivarium. Mice were exposed to a 12-hour light-dark cycle, with water and regular chow ad libitum. Equal numbers of male and female mice were used for each experiment. All mice were inbred on a C57BL/6J background. For practical reasons, the strains were maintained as homozygous when possible.
Induction of Colitis
For induction of colitis with DSS, mice were treated with 2.5% DSS (Gojira) in the drinking water (ad libitum) for 5 days. For induction of colitis with TNBS or oxazolone, mice were presensitized 7 days before injection. The mice were anesthetized with 1.5% isoflurane-mixed gas, and 2 × 2 cm area of the skin on the back of the mouse between the shoulders was shaved. Two hundred microliters of 1% (wt/vol) TNBS (Sigma) or 3% (wt/vol) of oxazolone (Sigma) presensitization solution was applied to the shaved skin. On the day of colitis induction, 3.5F catheter was inserted into the colon such that the tip was 4 cm proximal to the anus. One hundred microliters of 2.5 mg TNBS (Sigma) in 50% ethanol or 100 μL of 2% oxazolone in 50% ethanol was administered via catheter into the lumen using a 1-mL syringe. Control mice received 100 μL of 50% ethanol. For infection studies, C rodentium (kindly provided by Philip M. Sherman, Department of Laboratory Medicine and Pathobiology, University of Toronto, Ontario, Canada) was cultured in static nonaerated Luria-Bertani broth (MP Biomedicals) for 16–24 hours at 37°C to yield a final concentration of 2 × 109 colony-forming units/mL. Mice were then inoculated with 300 μL of C rodentium by oral gavage. Control mice received Luria-Bertani broth of the same volume. Fusobacterium nucleatum 12230 were grown in Columbia broth supplemented with hemin (5 μg/mL) and menadione (1 μg/mL) and incubated at 37°C in an anaerobic chamber with 5% CO2, 5% H2 and 90% N2.25 The cultures were pelleted at 5000g for 5 minutes and resuspended to an estimated titer of 2 × 1010 colony-forming units/mL. For acute macrophage depletion studies, 200 μL of 5 mg/mL clodronate (CLD) liposome (Liposoma BV) dissolved in an aqueous phosphate-buffered saline (Fisher Bioreagents) was injected intraperitoneally every other day for 6 days during DSS administration. After induction of colitis, animals were monitored and scored daily for weight loss, stool consistency, movement, and survival. Murine colitis-associated cancer models are described in the Supplementary Materials.
In Vitro Culture System
For details of organoid cultures, refer to the Supplementary Methods. In small intestinal organoid experiments involving intervention with cytokines, lipopolysaccharide, NONOate, or H2O2, Dclk1CreERT2;ROSA26tdTomato;APCfl/fl organoids were treated with 0.5 μM 4-OH-tamoxifen for 48 hours. The media and the Matrigel containing the organoids were transferred to a 15-mL Falcon tube. Cells were centrifuged at 800 rpm for 5 minutes, and pellets were resuspended in basic culture medium containing 200 ng/mL murine IL-1β (PeproTech), 20 ng/mL murine IL-13 (PeproTech), 100 ng/mL murine IL-6 (PeproTech), 80 ng/mL human recombinant TNF-α (Biovision Inc), 1 μg/mL lipopolysaccharide O111:B4 (Sigma-Aldrich), 800 μM H2O2 (BioShop), or 50 μM diethylenetriamine-NONOate (Acros Organics). Cells were then incubated in 37°C water bath for 30 minutes and centrifuged at 800 rpm for 5 minutes. Pellets were resuspended in 1:1 basic culture medium and Matrigel containing the same concentration of the treatments as listed above and seeded on prewarmed 48-well plates. Cells were then overlaid with 250 μL/well of basic culture medium.
Cultures and maintenance of organoids from primary mouse colon tumors have been described previously.26 All tumor organoids were derived from Dclk1CreERT2;ROSA26tdTomato;APCfl/fl mice treated with DSS.
Cytokine and Chemokine Measurements
Protein levels were measured using Mouse Cytokine/Chemokine Array 31-Plex (MD31) and Mouse Cytokine Array Th17 Discovery 11-Plex (MDTH17–11) from Eve Technologies. For details of the quantitative reverse transcription polymerase chain reaction methods, please refer to Supplementary Table 1 and the Supplementary Methods.
Flow Cytometry
Flow cytometry analyses of inflammatory cells were performed from mouse colonic and splenic tissues. To isolate inflammatory cells from the colonic tissue, tissues were harvested, cut open along the length, washed with cold phosphate-buffered saline, and cut into 0.5-cm pieces. Tissues were then incubated in 10 mL of Hank’s balanced salt solution containing 5% fetal bovine serum, 2 mM EDTA, 1 mM dithiothreitol, and 10 mM HEPES in 37°C on 200 rpm shaker with rotation for 30 minutes. After vortex for 30 seconds, supernatant was passed and collected through a 40-μm strainer (Corning). Remaining colonic tissues were incubated in 5 mL of RPMI containing 5% fetal bovine serum, 1.5 mg/mL collagenase IV, and 0.1 mg/mL DNase in 37°C on 200 rpm shaker with rotation for 30 minutes. Supernatant was passed through a 40-μm strainer and combined with previously collected sample. White blood cells were isolated using a Percoll (GE Healthcare Life Sciences) gradient collecting cells at the 40/80% Percoll interface.
To isolate inflammatory cells from the splenic tissue, cell suspensions were generated from the spleen by first dissociating tissue between frosted glass slides. Spleens were lysed for 2 minutes at 37°C to remove red blood cells with ammonium-chloride-potassium lysing buffer (Gibco).
Dead cells were identified by staining with a Fixable Viability Dye eFluor506 (eBioscience) according to the manufacturer’s protocol. All cells were then blocked with an anti–Fc-γ receptor, CD16/32 2.4G2 (BD Biosciences), in phosphate-buffered saline containing 2% fetal bovine serum for 30 minutes on ice. Cells were then stained on ice for 30 minutes with the listed combination of staining antibodies, followed by a secondary stain with streptavidin for 15 minutes on ice (Supplementary Table 2). Flow cytometry was performed on a BD Immunocytometry Systems LSRII cytometer. Analyses were then completed using FlowJo software (TreeStar).
Statistical Analysis
Statistical analyses were performed using 2-tailed Student t tests when comparing 2 groups or standard analysis of variance with Tukey’s multiple comparison test. Error bars denote the mean ± SEM. A P value <.05 was considered statistically significant (*P < .05, **P < .01, ***P < .001, ****P < .0001).
Results
Colitis-Associated Tumorigenesis Is Dependent on the Type of Inflammatory Response
To compare different forms of colitis with respect to their propensity to lead to colonic tumors, C57BL/6J wildtype mice were administered a single dose of the carcinogen AOM, followed by one of the colitis-inducing agents—DSS, TNBS, oxazolone, C rodentium, or doxorubicin (Figure 1A). Consistent with previous reports,27 all mice administered DSS developed colonic tumors by 20 weeks after colitis induction (Figure 1B–D). Surprisingly, TNBS, oxazolone, C rodentium, and doxorubicin did not lead to colonic tumorigenesis, even up to 52 weeks post colitis (Figure 1B–D). In separate experiments meant to model chronic, rather than acute, inflammation, we treated C57BL/6J wild-type mice with a single dose of AOM, followed by repeated courses of TNBS, oxazolone, C rodentium, or doxorubicin to recapitulate the bouts of remission and relapse often seen in patients with IBD (Supplementary Figure 1A). Interestingly, despite repeated injections of the colitis-inducing agents, none of these colitogens led to colonic tumor formation (Supplementary Figure 1B).
Figure 1.
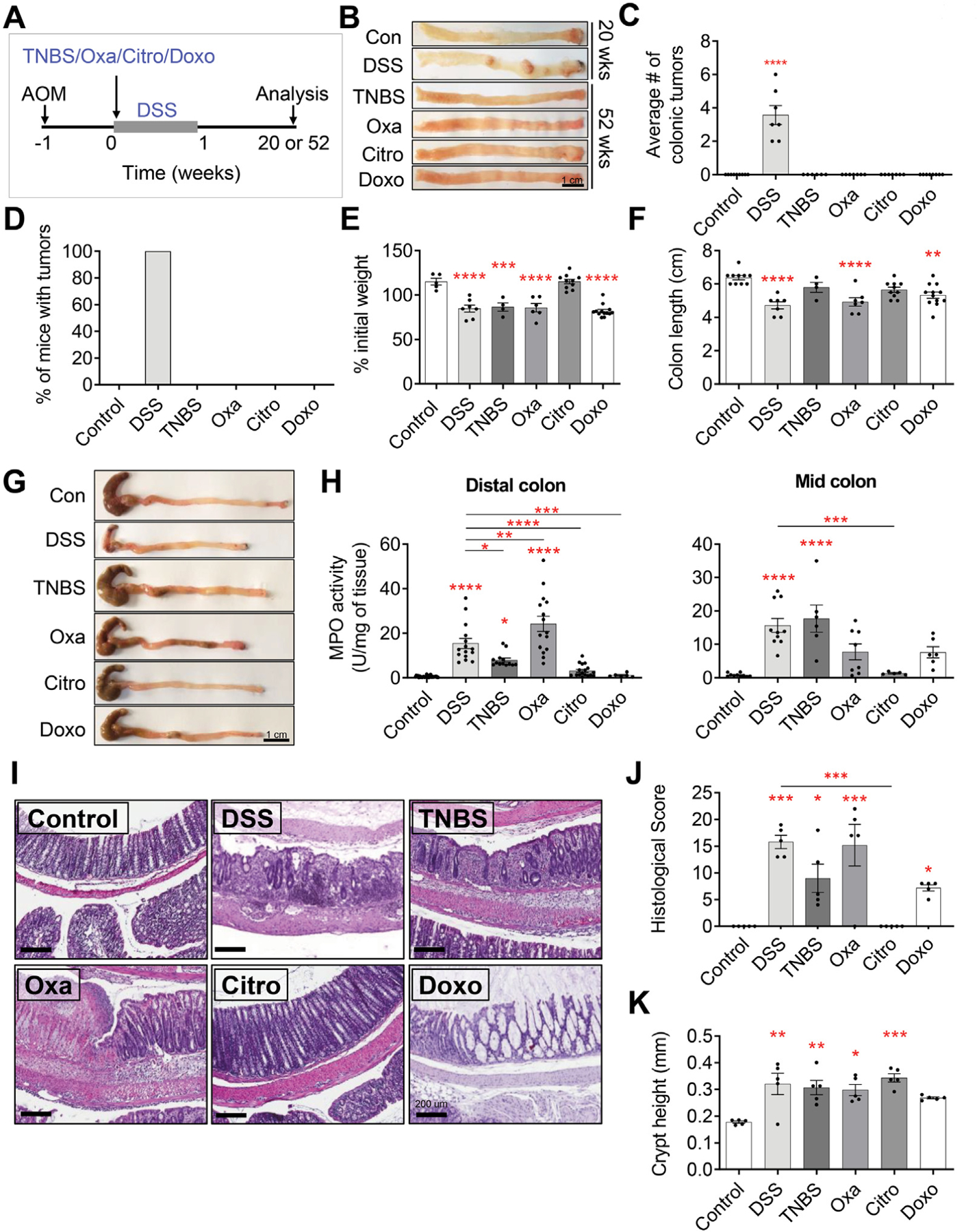
Colitis-associated tumorigenesis is dependent on the type of inflammatory response. (A) Experimental setup to assess the propensity of DSS, TNBS, oxazolone (Oxa), C rodentium (Citro), and doxorubicin (Doxo) to induce tumorigenesis in C57BL/6J wild-type mice treated with intraperitoneal injection of 10 mg/kg AOM. (B–D) Gross pathology of the colon (B), average colonic tumor number per mouse (C), and percentage of mice (D) with colonic tumors in AOM-treated C57BL/6J wildtype mice treated with vehicle or various colitis-inducing agents (n ≥ 7 per group). (E–G) Percent of initial weight (E), longitudinal colon length (F), and gross pathology of the colon (G) at peak point of inflammation from C57BL/6J wild-type mice treated with vehicle or various colitis-inducing agents. Data collected on day 8 for control, DSS, and C rodentium, day 3 for TNBS and oxazolone, and day 4 for doxorubicin (n ≥ 5 per group). (H) MPO activity measured from the distal colon (left) and mid-colon (right) at peak point of inflammation from C57BL/6J wild-type mice treated with vehicle or various colitis-inducing agents (n ≥ 6 per group). (I–J) Representative H&E-stained sections of the colon (I) and their associated histologic score (J) at peak point of inflammation from C57BL/6J wild-type mice treated with vehicle or various colitis-inducing agents (n ≥ 5 per group). (K) Measurement of the crypt height (from crypt base to the luminal side) averaged from distal to proximal colon from C57BL/6J wild-type mice treated with vehicle or various colitis-inducing agents (n ≥ 5 per group). Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
We next confirmed that the various colitis-inducing agents were effective in inducing colonic inflammation by examining the inflammatory response at the time of peak inflammation for each colitogen (ie, day 8 for DSS and C rodentium, day 3 for TNBS and oxazolone, and day 4 for doxorubicin) (Supplementary Figure 1D). The peak time point of inflammation for each colitis-inducing agent was determined on the basis of our initial examination of colitis severity (measured by weight loss, colon length, and myeloperoxidase [MPO]) at various time points (data not shown), and an extensive review of the literature.7,28,29 Mice treated with the various colitogens showed clinical and biological changes consistent with the presence of colitis. These included significant weight loss with DSS, TNBS, oxazolone, or doxorubicin colitis (Figure 1E), as well as a shortened colon in the case of DSS, oxazolone, and doxorubicin (Figure 1F and G). Mice with DSS-, TNBS-, and oxazolone-induced colitis also showed significantly increased colonic MPO activity compared with control mice, confirming the presence of increased myeloid cell infiltration (Figure 1H). Not unexpectedly, mice inoculated with C rodentium did not display any change in weight, colon length, or MPO activity (Figure 1E–H). The presence of colonic inflammation was further confirmed by means of H&E staining of histologic sections from mice in each group and scored in a blinded fashion (Figure 1I–J, Supplementary Figure 1E–G). Areas of ulceration, crypt abscesses, loss of normal crypt architecture, and infiltration of leukocytes were observed in sections taken from mice treated with DSS, TNBS, or oxazolone (Figure 1I, Supplementary Figure 1E–G). Mice treated with doxorubicin displayed copious luminal mucus, dilated crypts, and flattened epithelium with necrotic/apoptotic crypt epithelial cells (Figure 1I, Supplementary Figure 1E–G) but, interestingly, no increase in myeloid cell infiltration was observed, as measured by MPO activity (Figure 1H). In the C rodentium infectious model of colitis, mice inoculated with the bacteria showed histologic changes of transmissive murine crypt hyperplasia, a previously reported characteristic of C rodentium infection (Figure 1I and K, Supplementary Figure 1F).12,30,31 Taken together, these data suggest that the presence of colitis or colonic injury does not necessarily correlate with the risk of tumorigenesis.
Inflammatory Responses Differ Among Models of Colitis
To further characterize the different forms of colitis examined, we analyzed the messenger RNA expression of various inflammatory cytokines and chemokines in the distal colon. Our analysis revealed significantly increased messenger RNA expression of numerous inflammatory cytokines, with several differences detected among the various forms of colitis (Figure 2A, Supplementary Figure 2A). When compared with untreated controls, increased expression of TNF, IFNG, IL10, and IL17A was observed in all forms of colitis, whereas IL1B and IL6 were only increased in DSS-treated mice. Interestingly, doxorubicin-induced injury resulted in significantly increased IL4, IL5, IL10, IL13, IL28B, and IL31 within the colon (Figure 2A). We further examined colonic protein levels of 43 inflammatory cytokines and chemokines using a multiplex bead-based assay. Most of the cytokine protein levels correlated with messenger RNA expression levels (Figures 2A and B). Notably, we detected a significant increase in myeloid-derived (IL6, IL1B, and TNF) and neutrophil-derived (CSF3, CCL11, and CXCL5) cytokines in DSS- vs oxazolone-, TNBS-, or C rodentium–induced colitis (Figure 2B). RNA-sequencing analysis of DSS- and oxazolone-treated mice revealed clear differences in the expression profiles among the 2 colitis models (Supplementary Figures 2B and 3). Further interrogation of gene expression differences between DSS and oxazolone colitis using a KEGG pathway analysis revealed several distinct patterns in pathways, ranging from pathways in cancer, proteoglycans, and immune checkpoint inhibitors, to p53 and Hippo signaling pathways.
Figure 2.
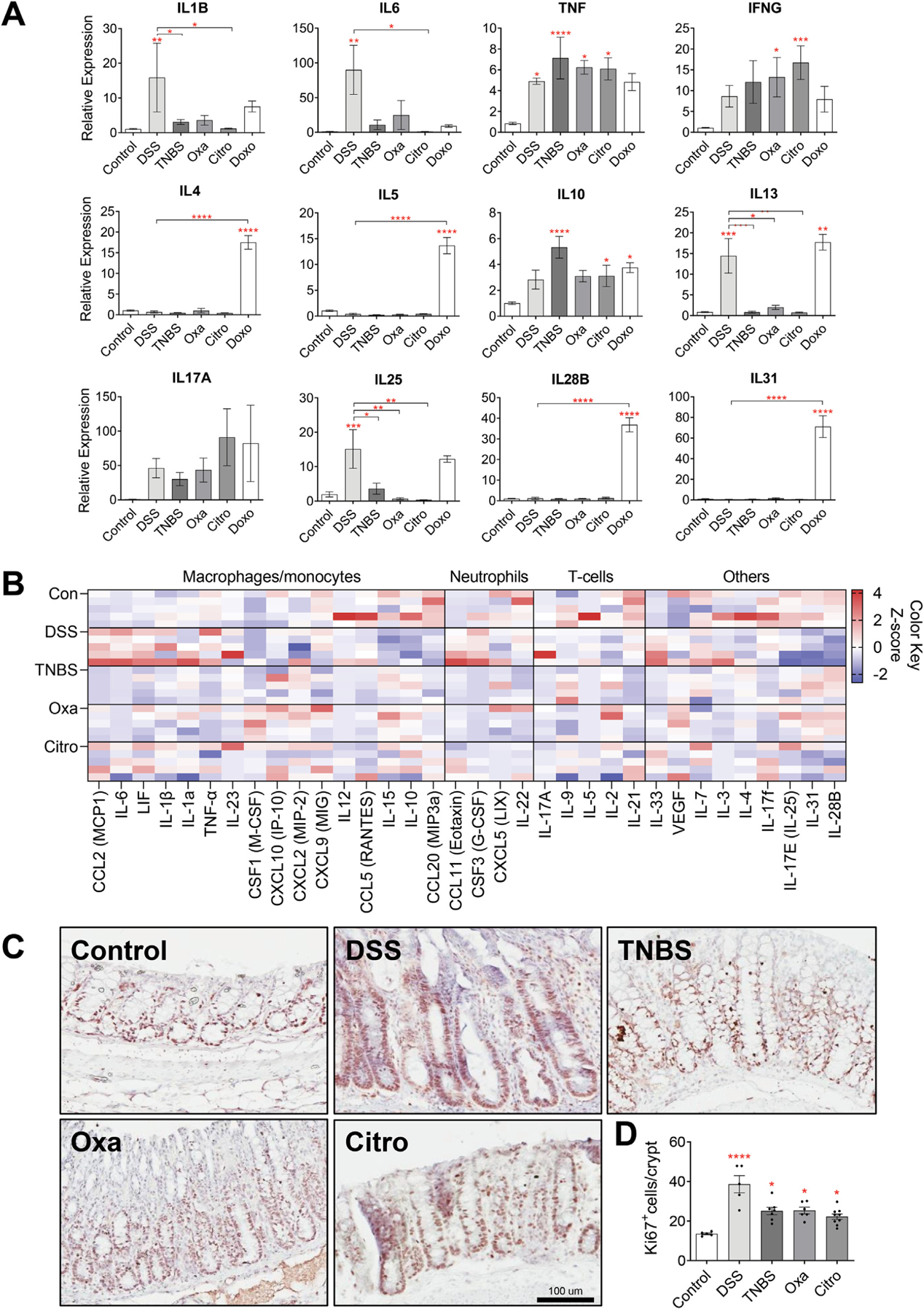
Inflammatory responses differ among models of colitis. (A) Messenger RNA levels of inflammatory cytokines in the distal colonic tissues of C57BL/6J wild-type mice treated with various colitis-inducing agents, measured by means of quantitative reverse transcription polymerase chain reaction (n ≥ 6 per group). (B) Protein levels of inflammatory cytokines in the distal colonic tissues of C57BL/6J wild-type mice treated with various colitis-inducing agents (n = 5 per group). Data are shown as a heatmap, and cytokines are organized according to the associated immune cell type. (C) Representative high-power view of Ki67+ cells in the distal colonic tissues of C57BL/6J wild-type mice treated with various colitis-inducing agents. Scale bar: 100 μm. (D) Quantification of Ki67+ cells reveal increased number of proliferating cells in all colitis models compared with the control group and an increased number of proliferating cells in DSS-treated mice compared with other models of colitis (n ≥ 5 per group). Citro, C rodentium; Doxo, doxorubicin; Oxa, oxazolone. Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Next, we compared the epithelial regenerative response across the different models of colitis by performing immunohistochemistry staining for the proliferation marker Ki67. Consistent with the presence of epithelial injury, we observed significantly increased proliferation in the colon in all of the models of colitis, with DSS resulting in the greatest proliferative response (Figure 2C and D). These data indicate that all the models of colitis (ie, DSS, TNBS, oxazolone, C rodentium, and doxorubicin) examined were effective in inducing colonic damage and inflammation. These observations additionally showed that tumorigenesis does not simply correlate with the presence or severity of colitis, but rather, the form of colitis or colonic injury.
Tuft Cells Show Stemness During Colonic Tumorigenesis Associated With Dextran Sodium Sulfate–Induced Colitis
To further explore the mechanism by which colitis leads to tumor initiation, we used a transgenic model of colitis-associated cancer in which the cellular origin of the colonic tumors is known. Using our previously described Dclk1CreERT2;ROSA26tdTomato;APCfl/fl transgenic mouse model,27 we compared how the different forms of colitis led to colonic tumorigenesis from Dclk1-expressing tuft cells (Figure 3A, Supplementary Figure 4A). Consistent with our previous observations using the AOM/DSS model, tamoxifen induction of APC loss in Dclk1+ cells followed by DSS-induced colitis resulted in 100% of mice developing colonic tumors by week 16 (Figure 3B–D, Supplementary Figure 4B). In contrast, induction of colitis by oxazolone, C rodentium, or doxorubicin did not result in any detectable tumors even up to 52 weeks post colitis (Figure 3B–D). Similarly, with TNBS- and F nucleatum–induced colitis, most of the mice did not develop any tumors. However, in both the TNBS and Fusobacterium groups, a single mouse developed 1 colonic tumor on colitis induction, confirming that the colitis induced in these models is significantly less tumorigenic than DSS-induced colitis (Figure 3B–D, Supplementary Figure 4B and C). Lineage tracing of Dclk1+ cells further revealed tdTomato+ colonic tumors in DSS-treated mice, whereas Dclk1+ cells remained quiescent as single tdTomato+ cells on treatment with other colitogens (Figure 3E, Supplementary Figure 4D and F). Interestingly, Dclk1 immunostaining showed that DSS- and oxazolone-treated mice had significantly reduced tuft cells, whereas mice inoculated with C rodentium had an increased number of tuft cells compared with untreated control mice (Figure 3F and G), suggesting that colonic tumorigenesis does not correlate with tuft cell number. Moreover, Dclk1 immunostaining showed that Dclk1+ cells were detectable only in the adjacent normal colonic tissues and not in the tumors (Supplementary Figure 4G). Analogous to the AOM colitis–associated cancer model, our Dclk1CreERT2;ROSA26tdTomato;APCfl/fl data confirmed that colonic tumorigenesis is indeed dependent on the form of colitis present. In addition, tuft cells uniquely acquire stemness only in the setting of DSS-induced colitis.
Figure 3.
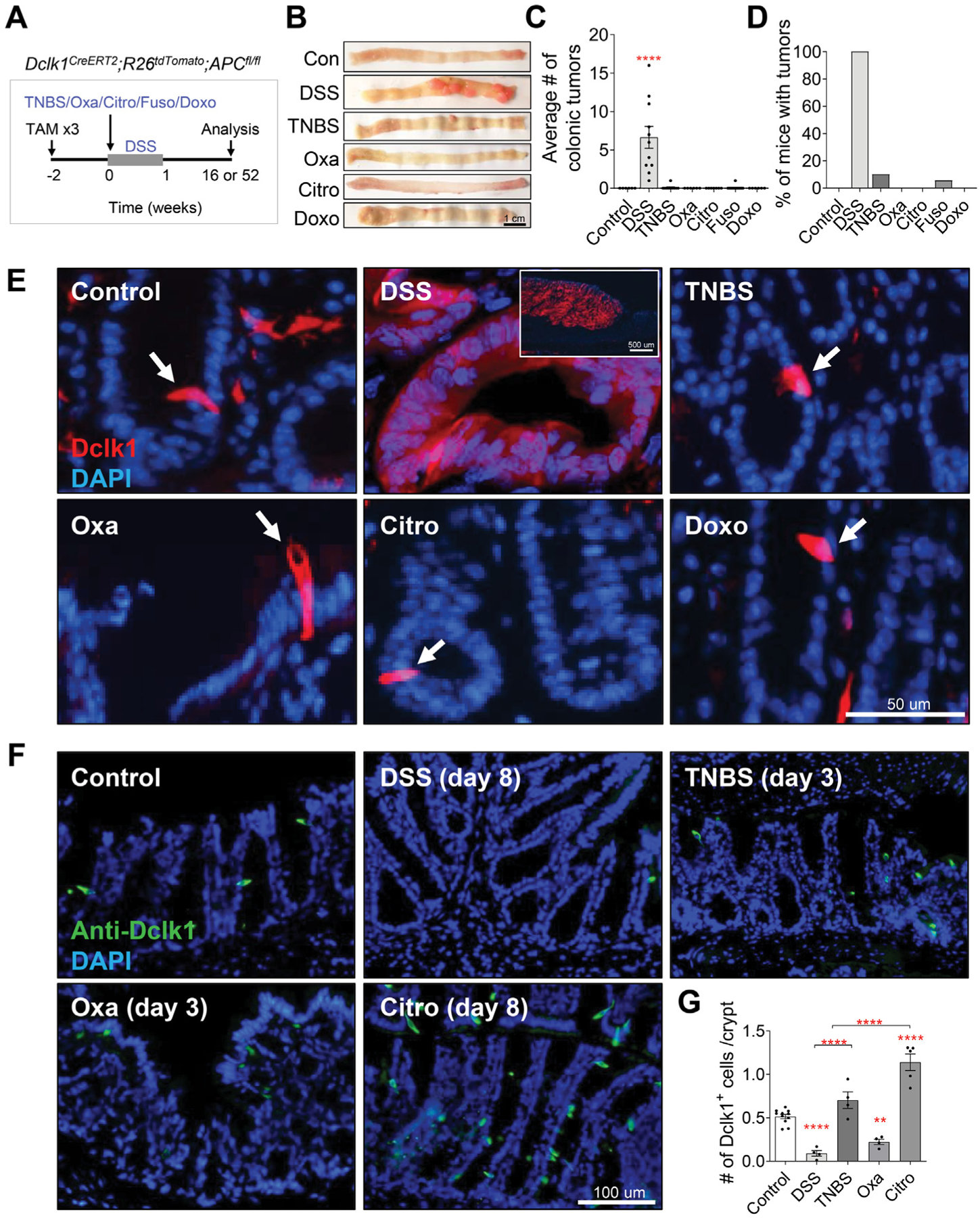
Tuft cells show stemness to give rise to colonic tumors in DSS-induced colitis. (A) Experimental setup to assess the ability of DSS, TNBS, oxazolone (Oxa), C rodentium (Citro), F nucleatum, and doxorubicin (Doxo) to induce tumorigenesis in Dclk1CreERT2;R26tdTomato;APCfl/fl mice treated with 3 injections of 6 mg/kg tamoxifen (TAM). (B–D) Gross pathology of the colon (B), average colonic tumor number per mouse (C), and percentage of mice (D) with colonic tumors in tamoxifen-treated in Dclk1CreERT2;R26tdTomato;APCfl/fl mice treated with vehicle or various colitis-inducing agents (n ≥ 6 per group). (E) Representative images of the colonic crypts from Dclk1CreERT2;R26tdTomato;APCfl/fl mice. DAPI, 4′,6-diamidino-2-phenylindole. DSS induces Dclk1 lineage-labeled tumors, whereas Dclk1+ cells remaining as single cells in mice treated with TNBS, oxazolone, C rodentium, and doxorubicin. Scale bar: 50 μm. (F–G) Representative immunofluorescence staining (F) and quantification (G) of Dclk1 in colonic tissues of C57BL/6J wild-type mice reveal reduced Dclk1+ cell number in DSS- and oxazolone-treated mice and increased Dclk1+ cell number in C rodentium–inoculated mice. Scale bar: 100 μm. Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Myeloid Cells Are Increased in Dextran Sodium Sulfate–Induced Colitis
To better characterize the inflammatory responses that promote tumor initiation, we identified differences between DSS-induced colitis that led to colonic tumorigenesis, and the other models of colitis that did not lead to tumor formation (ie, TNBS, oxazolone, C rodentium, or doxorubicin). We first performed fluorescence-activated cell sorting analysis of inflammatory cells including T cells, B cells, macrophages, and neutrophils in the colonic tissue during acute colitis (Figure 4A). Flow cytometry analysis showed a significant increase in CD11b+Ly6G+ neutrophils in DSS-treated mice and CD11b+F4/80+ macrophages in DSS-, oxazolone-, and doxorubicin-treated mice compared with mice in the vehicle-treated control group (Figure 4B, Supplementary Figure 5). We further subdivided macrophages into 3 subsets based on their levels of expression of the surface marker Ly6C, which has previously been identified as a marker of “classical monocytes” and “inflammatory macrophages” in mice32–34 (Figure 4A). We found that CD11b+F4/80+Ly6Chigh macrophages were significantly increased in DSS-treated mice compared with mice with other models of colitis that did not lead to tumors (Figure 4B, Supplementary Figure 5). In contrast, there were no differences seen in the proportion of T and B cells among mice treated with any of the different colitis-inducing agents (Figure 4B, Supplementary Figure 5). This was also validated by F4/80+ immunostaining that demonstrated an increased number of macrophages in the colon of mice treated with DSS and oxazolone compared with vehicle-, TNBS-, and C rodentium–treated mice (Figure 4C). The increased macrophages and neutrophils in DSS-induced colitis also correlated with increased macrophage-derived (IL1B, IL6, and TNF) and neutrophil-derived (CSF3, CCL11, and CXCL5) cytokines in DSS colitis vs the other models of colitis (Figure 2A and B). Collectively, these data showed that CD11b+F4/80+Ly6Chigh macrophages and CD11b+Ly6G+ neutrophils are particularly elevated in DSS colitis compared with the other models of colitis that do not lead to colonic tumor formation, suggesting that these cells may play an important role in the initiation of colitis-associated cancer.
Figure 4.
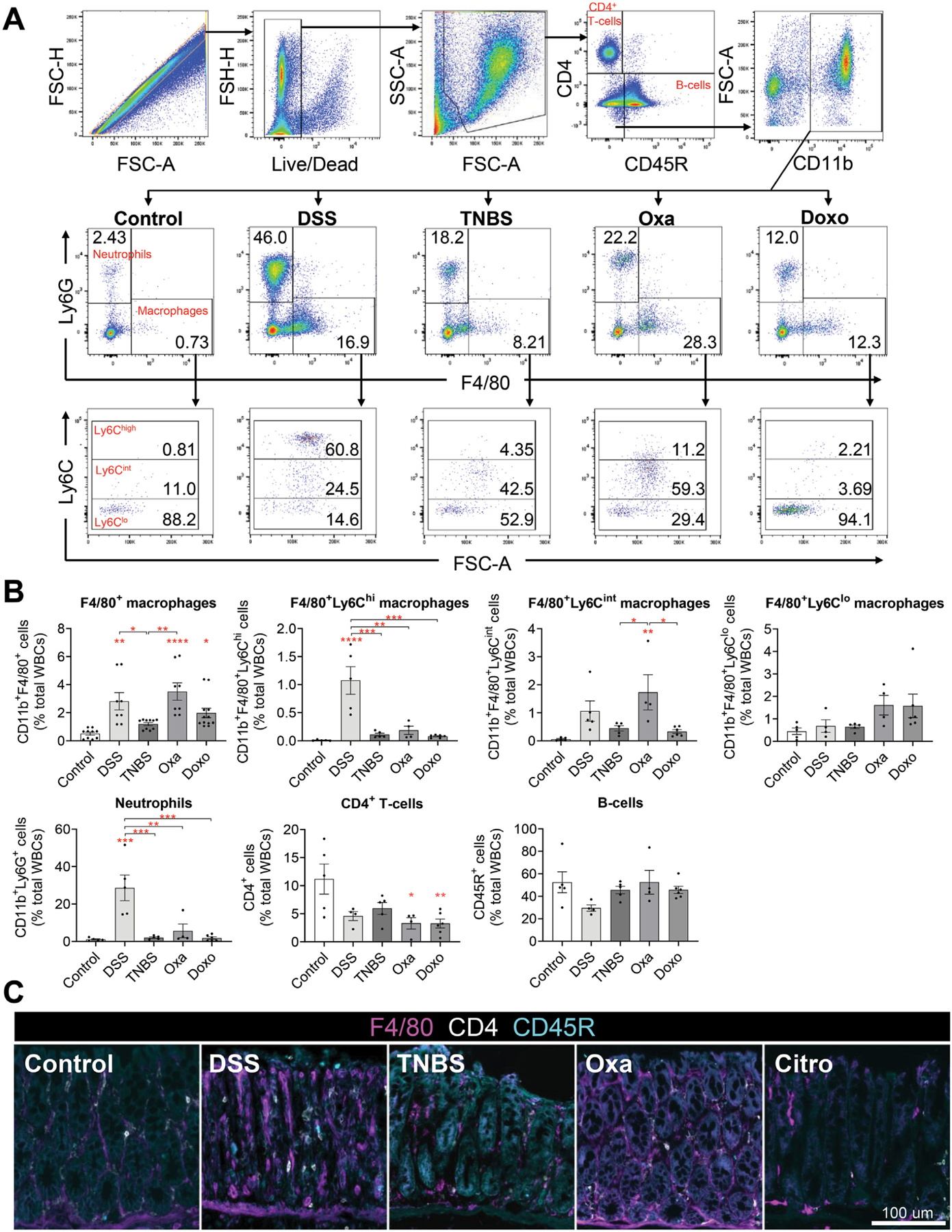
Myeloid cell infiltration is increased in DSS-induced colitis. (A) Representative gating of CD4+ T cells, B cells, Cd11b+ myeloid cells, neutrophils, macrophages, and Ly6Chigh/int/low subset of macrophages from flow cytometric analysis of C57BL/6J wild-type mice treated with various colitis-inducing agents. Citro, C rodentium; Doxo, doxorubicin; Oxa, oxazolone. One representative from each experimental group is shown. (B) Quantification of each immune cell subset as defined in panel A identifying F4/80+Ly6Chigh macrophages and Ly6G+ neutrophils as myeloid cells that are increased in DSS-treated mice compared with other models of colitis. Data are expressed as the percentage of total white blood cells (n ≥ 5 per group). (C) Representative images of immunofluorescent staining of F4/80+ macrophages, CD4+ T cells, and CD45R+ B cells in the colonic tissues of mice treated with various colitis-inducing agents. Scale bar: 100 μm. Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Dextran Sodium Sulfate–Induced Colitis Displays a Similar Inflammatory Profile to That Seen in Patients With Ulcerative Colitis
Given that DSS-induced colitis was unique in its ability to lead to colonic tumors, we next compared the inflammatory cytokine profile of DSS-induced colitis with that of patients with UC. We used a publicly available RNA-sequencing data set of DSS-induced colitis (GSE131032)35 to analyze the expression levels of inflammatory cytokines. We observed a significant increase in myeloid-derived cytokines in DSS-treated mice when compared with control mice without colitis (Figure 5A, Supplementary Table 3). We then analyzed the expression profile of inflammatory cytokines in patients with UC. Using RNA-sequencing data from rectal mucosal biopsies of 206 pediatric patients with UC and 20 healthy controls (GSE109142),36 we confirmed that myeloid cells were similarly increased in patients with UC compared with healthy controls (Figure 5B, Supplementary Table 4). Furthermore, analogous to what we observed in our DSS-treated mice, IL1B, IL6, TNF, IFNG, IL4, IL10, IL13, and IL17A were also significantly increased in patients with UC compared with healthy controls (Figure 5C, Supplementary Figure 6). To more directly compare the immune cell composition between murine colitis models and patients with UC, we used CIBERSORTx to build a signature matrix of immune cells from the published literature and estimate the abundance of myeloid cell populations using bulk RNA-sequencing data from both mouse colitis models and patients with UC.37–39 Interestingly, the changes in relative abundance of certain subsets of myeloid cells, such as macrophages and dendritic cells type 2, were consistent between mice treated with DSS and patients with UC (Figure 5D). These data suggest that DSS-induced colitis most accurately recapitulates the inflammatory cell and cytokine profile seen in patients with UC.
Figure 5.
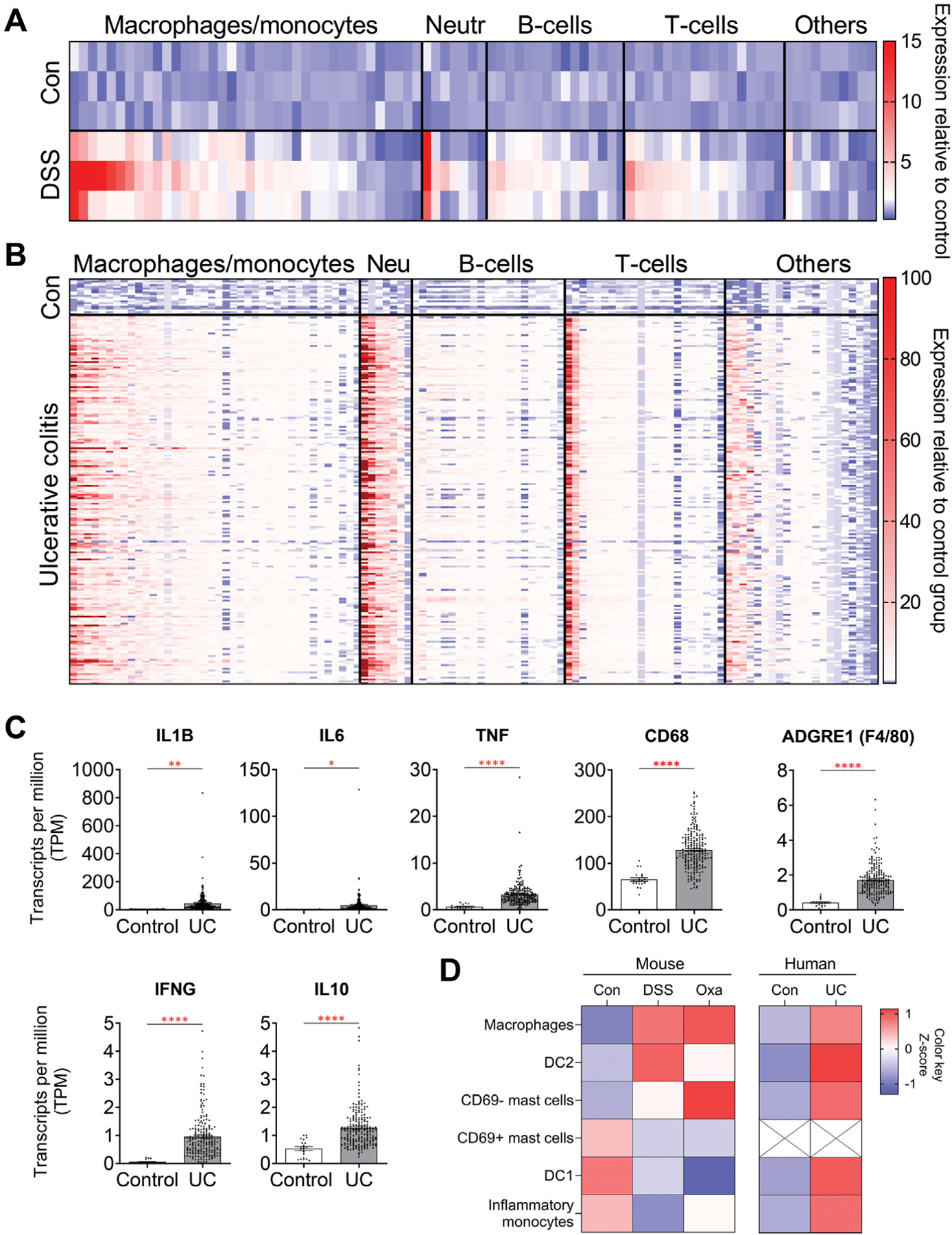
DSS-induced colitis in mice has an inflammatory profile similar to that of patients with UC. (A) Expression profiles of inflammatory cytokines and chemokines from colonic tissues of mice treated with 2.5% DSS for 5 days and analyzed on day 8. Data were derived from Gene Expression Omnibus (GEO) data set GSE131032 and normalized to control samples (n = 3 in each group). (B) Expression profiles of inflammatory cytokines and chemokines from rectal mucosal biopsies collected before treatment in pediatric patients with new-onset UC. Data were derived from GEO data set GSE109142 and normalized to control samples (n = 20 healthy controls, n = 206 patients with UC). Results in panels A and B highlight the similarities in elevated cytokines associated with myeloid cells between DSS-treated mice and patients with UC. (C) Cytokines known to be associated with colonic inflammation and that were differentially regulated in our DSS-treated mice were also confirmed to be differentially regulated in patients with UC. Data were derived from GEO data set GSE109142. (D) Abundance of myeloid cells in mouse models of colitis and patients with UC, estimated using a gene signature matrix derived from previously published single-cell RNA-sequencing data sets of patients with UC.39 Data in all bar graphs are presented as mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Macrophage-Derived Cytokines Induce Stemness of Tuft Cells In Vitro
Given that F4/80+Ly6Chigh macrophages were increased specifically in DSS-induced colitis vs the other forms of colitis examined (ie, TNBS, oxazolone, and doxorubicin), we next assessed the effects of macrophage-derived mediators on the initiation of colonic tumorigenesis. To do this, we took advantage of our Dclk1CreERT;APCfl/fl transgenic model in which the cellular origin for tumors is known to be Dclk1+ tuft cells and examined the effects of macrophage-derived mediators. We used 3-dimensional intestinal organoids derived from Dclk1CreERT2;ROSA26tdTomato;APCfl/fl mice (Figure 6A). These organoids allow visualization of Dclk1+ tuft cells and genetic lineage tracing studies to be followed in vitro on exposure to macrophage-derived cytokines IL-1β, IL-6, and TNF-α (Figure 2A and B). Incubation of the organoids with IL-1β, IL-6, or TNF-α did not change the number of Dclk1+ cells detected in vitro (Figure 6B). However, IL-1β induced spontaneous tdTomato+ lineage tracing from Dclk1+ cells that gave rise to the entire organoid (Figure 6C and D). Consistent with the labeling of a stem cell, these organoids remained tdTomato+ for at least 10 passages (Figure 6F). Interestingly, incubation of the organoids with IL-6 or TNF-α induced transient lineage tracing from Dclk1+ cells that resulted in partially traced tdTomato+ organoids that subsequently turned over and were undetectable 8 days after cytokine exposure (Figure 6C and D). When IL-1β, IL-6, and TNF-α were added to the organoids in combination, lineage tracing from Dclk1+ cells occurred at the same frequency as that observed in organoids incubated with IL-1β alone (Figure 6C and D). Treatment of the organoids with IL-13, another cytokine that was up-regulated in both DSS-induced colitis and patients with UC, resulted in an increased number of Dclk1-tdTomato+ cells but no lineage tracing of organoids was seen (Figure 6B, C, and D). Given that neutrophils were also increased in DSS-induced colitis when compared with the other models of colitis, we further tested whether any neutrophil-derived mediators affect Dclk1+ tuft cells. Specifically, we examined the effects of the nitric oxide donor, diethylenetriamine-NONOate, and the superoxide precursor H2O2, on Dclk1CreERT2;ROSA26tdTomato;APCfl/fl-derived organoids. Neither the nitric oxide donor nor hydrogen peroxide induced lineage tracing from Dclk1+ cells (Figure 6C and D). We further tested the effects of bacterial-derived lipopolysaccharide and this also did not induce lineage tracing from Dclk1+ cells (Figure 6C and D). Differences in organoid morphology and size were not observed among the different treatments compared with their respective controls (Figure 6C and E). To further assess the plasticity of Dclk1+ cells after 4-OH-tamoxifen labeling of Dclk1+ cells, Dclk1CreERT2;ROSA26tdTomato;APCfl/f-derived organoids were dissociated into single cells, replated in Matrigel, and incubated with vehicle or IL-1β. Once again, incubation with IL-1β significantly increased the proportion of tdTomato+ cells that gave rise to traced organoids, supporting our earlier findings that IL-1β promotes Dclk1+ cell lineage tracing (Figure 6G and H). Taken together, these data suggest that the macrophage-derived pro-inflammatory cytokine IL-1β, and to a lesser extent IL-6 and TNF-α, can revert quiescent Dclk1+ cells to an actively dividing stem cell state that results in lineage tracing and ultimately colonic tumors.
Figure 6.
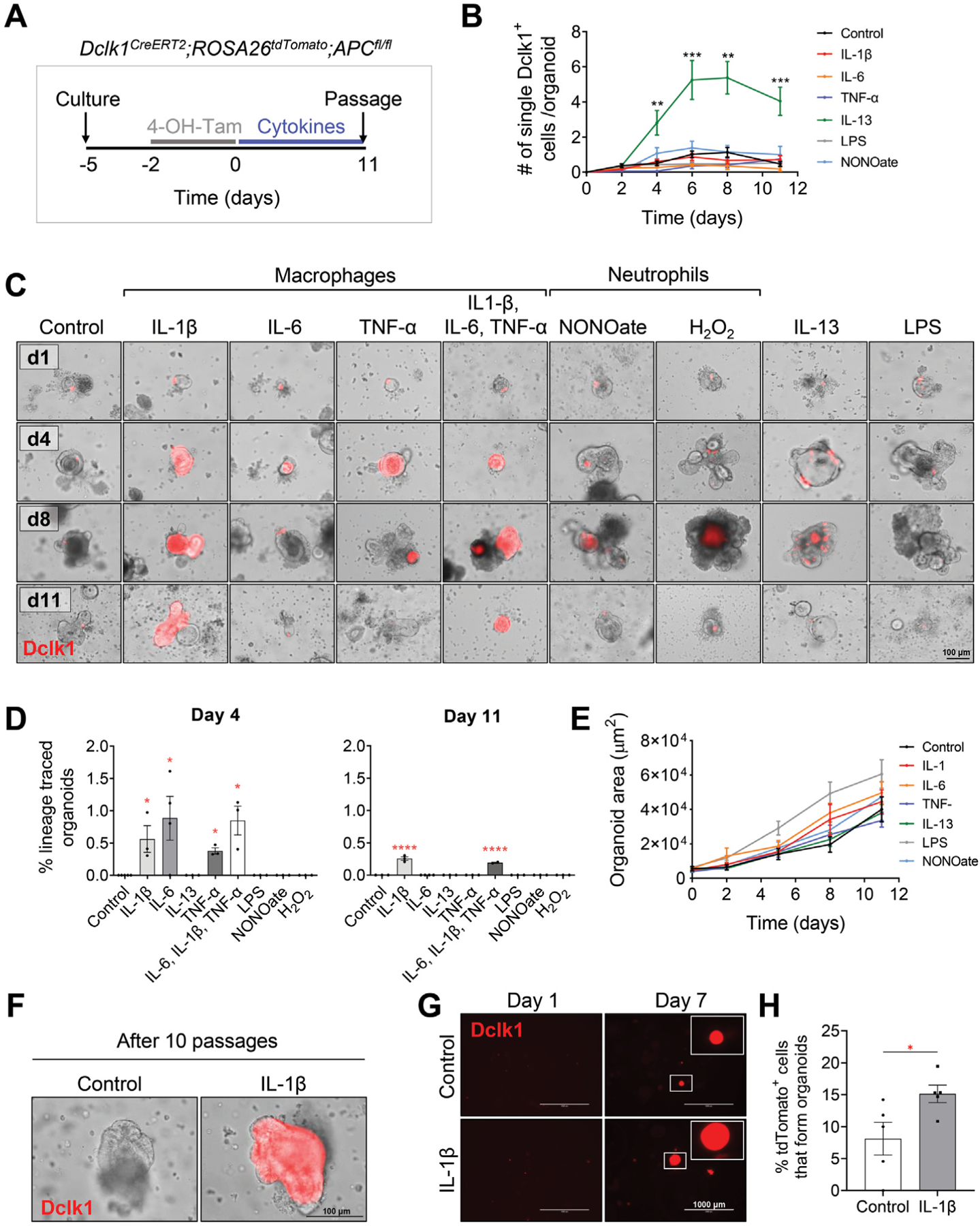
Macrophage-secreted cytokines induce lineage tracing from tuft cells in vitro. (A) Experimental setup to assess the effects of incubating inflammatory cytokines/agents with intestinal organoids cultured from proximal intestinal tissues of Dclk1CreERT2;R26tdTomato;APCfl/fl mice. (B) Quantification of Dclk1+ cells in organoids incubated with the indicated treatments. Data are collected from 3 biological replicates and are expressed as the number of single tdTomato+ cell per organoid. LPS, lipopolysaccharide. (C, D) Representative images of organoids (C) and quantification of traced organoids on days 4 and 11 (D) identify macrophage-secreted cytokines as the important factors for Dclk1+ tracing. Traced organoids were quantified as the percentage of organoids displaying entire tdTomato+ crypt tracing over the total number of organoids with detectable tdTomato+ cells. (E) Quantification of the organoid area represented as μm2 over time. Morphology and growth of organoids are not different between each group. (F) Dclk1+ tracing induced by incubation with IL-1β is maintained for over 10 passages. (G) Representative images of single cell–derived Dclk1CreERT2;R26tdTomato;APCfl/fl organoids on days 1 and 7 following culture in the presence of vehicle vs IL-1β, and (H) quantification of tdTomato+ cell–derived organoids 7 days after culture in vehicle vs IL-1β. Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Depletion of F4/80+Ly6Chigh Macrophages Reduces Colitis-Associated Tumorigenesis
Our observation that myeloid cells were increased to a greater extent in DSS-induced colitis when compared with the other models of colitis assessed led us to next examine the role of these cells in the initiation of colitis-associated cancer. To do this, we first used an anti-Ly6G antibody to deplete Ly6G+ neutrophils during acute colitis. We first confirmed that the α-Ly6G antibody was effective in depleting neutrophils in the colon. Wild-type C57BL/6J mice were treated with DSS for 5 days and administered either the isotype control or α-Ly6G antibody every other day for 6 days. Three days after the completion of DSS, colonic tissues were collected for analysis of inflammatory cells (Figure 7A). DSS-induced colitis led to a significant increase in the proportion and the number of CD11b+Ly6G+ neutrophils seen in the colon compared with mice without colitis (Figure 7B and C, Supplementary Figure 7D). Depletion of neutrophils did not affect the degree of weight loss or colon length in mice with DSS-induced colitis (Supplementary Figure 7A and B). However, as expected, mice treated with the α-Ly6G antibody during colitis showed a significant reduction in CD11b+Ly6G+ neutrophils compared with those treated with the isotype control antibody (Figure 7B and C, Supplementary Figure 7D–F). Importantly, antibody-mediated depletion did not affect other immune cells, including macrophages, T cells, and B cells (Figure 7C, Supplementary Figure 7C and D).
Figure 7.
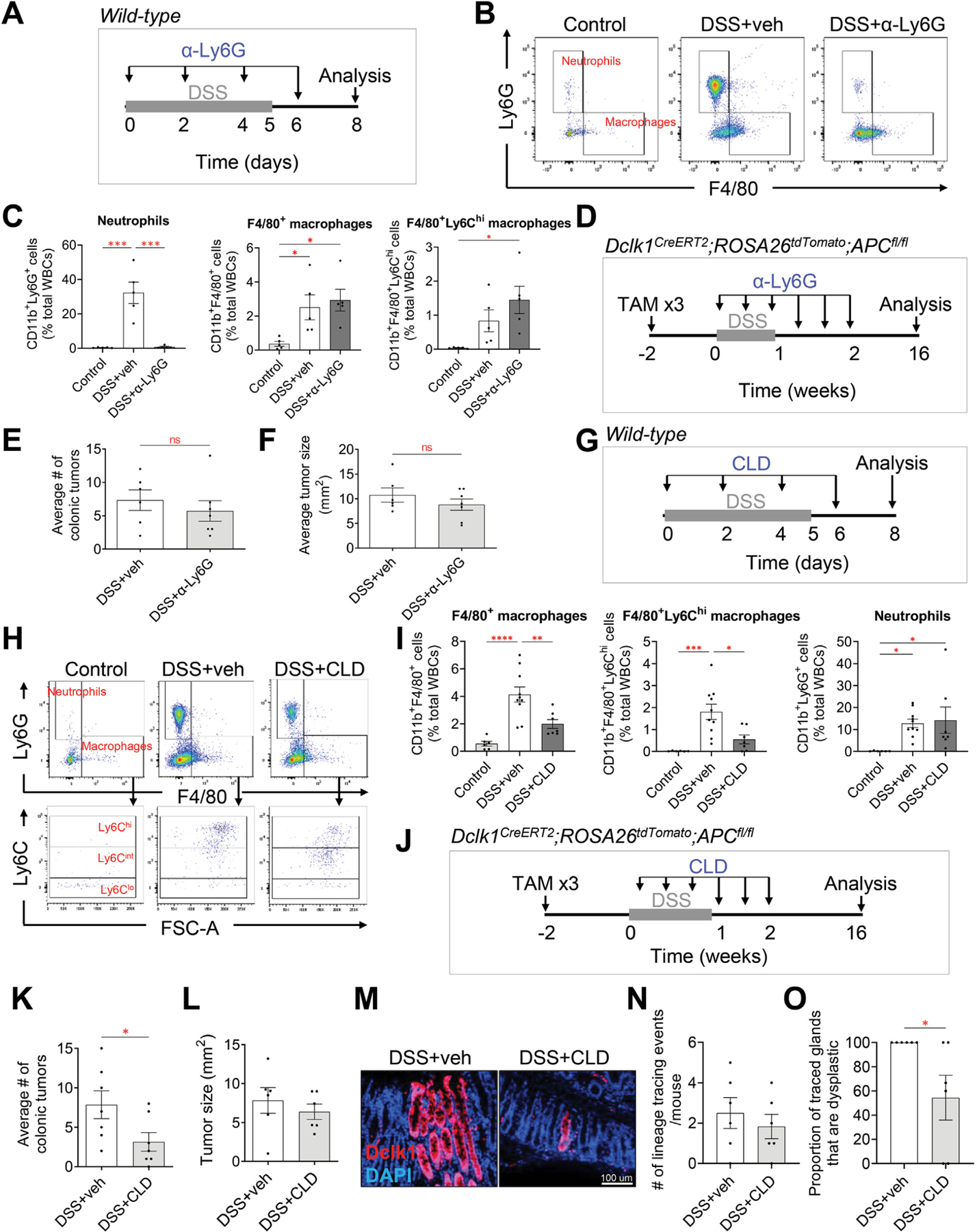
Depletion of Ly6G+ neutrophils does not affect colitis-associated tumorigenesis, whereas depletion of F4/80+Ly6Chigh cells significantly reduces colitis-associated tumorigenesis. (A) Illustration of the experimental protocol outlining α-Ly6G antibody–mediated neutrophil depletion in DSS-induced wild-type mice. (B) Representative gating of neutrophils and F4/80+ macrophages from flow cytometric analysis. One representative from each experimental group is shown. veh, vehicle. (C) Quantification of neutrophils and macrophages, as defined in Figure 4. α-Ly6G antibody is specific to neutrophils and is 98% effective. Data are expressed as the percentage of total white blood cells (n = 5 per group). (D) Illustration of the experimental protocol outlining α-Ly6G antibody–mediated neutrophil depletion in Dclk1CreERT2;R26tdTomato;APCfl/fl mice given tamoxifen (TAM) and DSS. (E, F) Neutrophil depletion does not affect colitis-associated tumorigenesis and does not affect the growth of the tumors, as indicated by the average colonic tumor number per mouse (E), and average size of the tumors (F) (n = 6 control: n = 7 α-Ly6G). (G) Illustration of the experimental protocol outlining CLD liposome–mediated macrophage depletion in DSS-induced wild-type mice. (H) Representative gating of neutrophils, F4/80+ macrophages, and Ly6Chigh/int/low subset of macrophages from flow cytometric analysis. One representative from each experimental group is shown. (I) Quantification of neutrophils and macrophages, as defined in Figure 4. CLD is specific to F4/80+Ly6Chigh subset of macrophages and is 69% effective. Data are expressed as the percentage of total white blood cells (n = 5 per group). (J) Illustration of the experimental protocol outlining CLD-mediated F4/80+Ly6Chigh macrophage depletion in Dclk1CreERT2;R26tdTomato;APCfl/fl mice given tamoxifen and DSS. (K–L) CLD prevents colitis-associated tumorigenesis and does not affect the growth of the tumors as indicated by the average colonic tumor number per mouse (K), and average size of the tumors (L) (n = 7 per group). (M–O) Representative images of Dclk1 lineage–traced glands (M), the associated quantification of tdTomato+–traced glands (N), and the proportion of traced crypts that are dysplastic (O) in the healthy colonic tissues at the 16-week time point. Data are expressed as the percentage of total number of mice. Data in all bar graphs are presented as mean ± SEM. Asterisks above each bar indicate significant differences from the control group. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Next, Dclk1CreERT2;ROSA26tdTomato;APCfl/fl mice were administered DSS in the drinking water and α-Ly6G antibody was administered every other day for 1.5 weeks to deplete neutrophils and test its effects on tumorigenesis (Figure 7D). Once again, antibody-mediated depletion of neutrophils did not result in weight loss or reduced colon length (Supplementary Figure 7G and H). Interestingly, neutrophil depletion also did not affect the number or the size of colonic tumors when compared with isotype control–treated mice (Figure 7E and F, Supplementary Figure 7I), suggesting that CD11b+Ly6G+ neutrophils are dispensable for the initiation of colitis-associated cancer.
To examine the role of macrophages in the initiation of colitis-associated cancer, we used CLD liposomes to deplete phagocytic myeloid cells during acute colitis. On administration of CLD liposomes in mice, CLD has previously been shown to accumulate intracellularly in phagocytic cells, leading to irreversible cell damage and apoptosis of cells that engulf the drug.40,41 We first confirmed that treatment with CLD liposomes was effective in depleting macrophages in the colon. Wild-type C57BL/6J mice were treated with DSS for 5 days, followed by administration of vehicle or CLD liposomes every other day for 6 days. Three days after the completion of DSS, colonic tissues were collected for analysis of inflammatory cells (Figure 7G). DSS-induced colitis led to a significant increase in the proportion of CD11b+F4/80+ cells seen in the colon when compared with mice without colitis (Figure 7H and I, Supplementary Figure 8D). As expected, mice treated with CLD liposomes during colitis had significantly fewer CD11b+F4/80+ cells in the colon when compared with those treated with vehicle (Figure 7H and I, Supplementary Figure 8D). Importantly, CLD-mediated depletion of F4/80+ macrophages resulted in preferential targeting of the Ly6Chigh subset of macrophages (Figure 7H and I, Supplementary Figure 8C and D) that were increased in DSS-induced colitis (Figure 4B). This was consistent with the findings of Schumak et al,42 who previously reported that CLD-mediated depletion mainly targets F4/80+ cells that express Ly6C.
In separate experiments, we tested the effects of macrophage depletion on colonic tumorigenesis. Dclk1CreERT2;ROSA26tdTomato;APCfl/fl mice were administered DSS in the drinking water, in addition to CLD liposomes for 1.5 weeks (Figure 7J). CLD-mediated depletion of macrophages led to a significant reduction in the number of colonic tumors when compared with vehicle-treated mice (Figure 7K, Supplementary Figure 8G). This was also true in the AOM/DSS model, where we similarly observed a significant reduction in the number of colonic tumors when compared with vehicle-treated mice (Supplementary Figure 8H and I). There was, however, no significant difference in colonic tumor size 16 weeks post colitis (Figure 7L, Supplementary Figure 8G). When we quantified tdTomato+ lineage tracing of glands within the macroscopically normal-appearing colonic tissues prepared as Swiss rolls, at 16 weeks post colitis, all Dclk1+ cell lineage–traced glands were dysplastic crypts, whereas in CLD-treated mice, 57% of the lineage tracing was non–dysplastic crypts (Figure 7M–O). Collectively, our data suggest that F4/80+Ly6Chigh macrophages promote the transformation of Dclk1+ cells to a stem cell state that leads to dysplasia and colitis-associated tumorigenesis, and this is consistent with our in vitro observations that macrophage-derived IL-1β also leads to Dclk1+ cell stemness.
Discussion
The risk of colon cancer in patients with IBD is believed to correlate with the duration and extent of colitis.43,44 In mice, recurrent bouts of colitis through repeated exposure to DSS or increased colitis severity as a result of higher DSS administration results in earlier onset of colonic tumorigenesis after AOM administration.45 The presence of colitis, however, does not always lead to cancer. Indeed, one of the major clinical challenges in caring for patients with chronic inflammation such as IBD has been identifying factors that predict tumor formation.
Our findings in this study clearly demonstrate that colonic tumor formation is not dependent on the simple presence or degree of inflammation, but rather, it is dependent on the type of inflammatory response. We have found that a myeloid cell–predominant inflammatory response, such as that seen in DSS-induced colitis, correlates with an increased cancer risk. Specifically, we have identified that a distinct F4/80+Ly6Chigh macrophage cell subset is uniquely increased in DSS-induced colitis vs other models of colitis that do not lead to tumorigenesis. Furthermore, depletion of these F4/80+Ly6Chigh macrophages is effective in inhibiting colonic tumor formation, proving that these cells play an essential role in the initiation of inflammation-associated tumorigenesis.
Our findings are consistent with previous reports that also showed that macrophages play an important role in the progression of colon cancer.46–48 Popivanova et al46 targeted the monocyte chemoattractant chemokine (C-C motif) ligand 2 by genetic and pharmacologic means and observed reduced colitis-associated tumorigenesis. Similarly, Wilson et al47 reported fewer intestinal tumors in the setting of macrophage migration inhibitory factor deficiency in a sporadic colon cancer model. More recently, macrophage depletion by CLD liposomes was found to result in fewer colonic tumors by altering the gut microbiota.48 These studies suggest that macrophages are key mediators of colonic tumor formation. Our current study further demonstrates that it is specifically the F4/80+Ly6Chigh macrophages that are most important for the initiation of colon cancer in the setting of inflammation. Importantly, these prior studies targeted macrophages throughout the duration of the experiments/tumorigenesis. Therefore, it is very likely that tumor-associated macrophages rather than macrophages that infiltrate the tissue before tumor formation were targeted to affect tumor progression. Thus, we focused on identifying cells or mediators that are key in the early steps of cancer initiation. A review by Wang et al49 described studies focused on the role of macrophages in colorectal tumor initiation. The authors highlighted the potential importance of PTGS2-expressing M1-polarized macrophages in the initial step of tumorigenesis. In addition, the authors suggested that the interactions between the immune cells and the gut microbiota may be involved in tumor initiation.49 Consistent with this, our data showed that F4/80+Ly6Chigh macrophages and the inflammatory cytokine profile detected in DSS colitis overlap, at least in part, with genes associated with M1-polarized macrophages,50 and are key to transformation of cells during the very early stages of tumor initiation.
Our findings showed that the macrophage-associated cytokines IL-1β, TNF-α, and IL-6 are increased to a greater extent in DSS-induced colitis and are key to promoting stemness of Dclk1+ tuft cells during tumorigenesis. Consistent with this, Yang et al51 previously identified 3 subsets of F4/80+ macrophages based on their expression of Ly6C and showed that these cells are the predominant source of IL-1β and TNF-α in AOM/DSS-treated mice. Here, we further showed that the same macrophage-derived cytokines are also elevated in colonic tissues from patients with UC and promote the initial steps of tumorigenesis. Importantly, when exposed to the macrophage-derived cytokines IL-1β, TNF-α, or IL-6, Dclk1+ cells, which served as the cellular origin for colonic tumors, began to lineage trace intestinal organoids. This finding suggests that these mediators promote transition of Dclk1+ cells to a stem cell state, and future work investigating how these mediators induce plasticity of Dclk1+ cells will provide better understanding for the mechanism by which colitis-induced tumorigenesis occurs. In contrast, we did not detect any effect of neutrophil-derived factors on Dclk1+ cells in vitro. This was consistent with our observation that neutrophil depletion during inflammation did not affect colonic tumorigenesis. Interestingly, several studies have proposed that neutrophils play a role in colon cancer. However, most of these studies have focused on their role in tumor progression rather than initiation.52–54 Taken together, our data indicate that macrophages are the key effectors of stemness and tumor initiation in the context of inflammation.
Thus, our study provides novel insight into the mechanism by which inflammation predisposes to cancer. Inflammation-associated tumorigenesis appears to be highly dependent on the type of inflammatory response. Specifically, F4/80+Ly6Chigh macrophages and their release of IL-1β, TNF-α, and IL-6 are critical for the stemness of cells, ultimately driving tumorigenesis. These data support the development of therapies targeting F4/80+Ly6Chigh macrophages for the prevention of inflammation-associated tumorigenesis.
Data Availability
RNA-sequencing matrix files have been deposited in the National Institutes of Health Gene Expression Omnibus database and can be accessed at PRJNA910503. All data are available in the main text or the Supplementary Materials.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Inflammation is a predisposing factor for the development and progression of cancer. However, the mechanism by which inflammation leads to colorectal cancer is not well understood.
NEW FINDINGS
The initiation of Inflammation-induced cancer is associated with a specific subset of macrophages. Cytokines produced by these macrophages promote stemness in quiescent tuft cells, and in vivo depletion of these macrophages inhibit tumorigenesis.
LIMITATIONS
Further studies are required to precisely characterize this novel subset of macrophages with respect to the cytokines, chemokines, and the pro-tumorigenic molecules they secrete.
CLINICAL RESEARCH RELEVANCE
Development of new therapies that target the subset of macrophages important for tumorigenesis may provide an effective preventative strategy for colitis-associated cancer.
BASIC RESEARCH RELEVANCE
Identification of a subset of macrophages that are key for tumorigenesis allows for further characterization of these cell types.
Funding
This work was supported by operating grants from the Canadian Institutes of Health Research and Cancer Research Society awarded to Samuel Asfaha, and by the National Institutes of Health U01-DK103155 awarded to Timothy C. Wang and Chandan Guha.
Abbreviations used in this paper:
- AOM
azoxymethane
- CD
Crohn’s disease
- CLD
clodronate
- CRC
colorectal cancer
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- IL
interleukin
- MPO
myeloperoxidase
- oxazolone
4-ethoxylmethylene-2-phenyloxazol-5-one
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- TNF
tumor necrosis factor
- UC
ulcerative colitis
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2023.01.002.
CRediT Authorship Contributions
Alice E. Shin, PhD (Conceptualization: Lead; Data curation: Lead; Formal analysis: Lead; Investigation: Lead; Methodology: Lead; Writing – original draft: Lead; Writing – review & editing: Lead).
Yodit Tesfagiorgis, PhD (Investigation: Supporting).
Frederikke Larsen, BSc (Investigation: Supporting).
Mathieu Derouet, PhD (Investigation: Supporting).
Peter Y. F. Zeng, BSc (Investigation: Supporting).
Hayley J. Good, PhD (Data curation: Supporting; Formal analysis: Supporting; Investigation: Supporting).
Liyue Zhang, MSc (Investigation: Supporting; Methodology: Supporting).
Steven M. Kerfoot, PhD (Data curation: Supporting; Formal analysis: Supporting).
Anthony C. Nichols, MD, PhD (Methodology: Supporting; Writing – review & editing: Supporting).
Yoku Hayakawa, MD, PhD (Methodology: Supporting; Writing – review & editing: Supporting).
Christopher J. Howlett, MD, PhD (Formal analysis: Supporting; Investigation: Supporting).
Timothy C. Wang, MD (Conceptualization: Supporting; Writing – review & editing: Supporting).
Samuel Asfaha, MD, PhD (Conceptualization: Lead; Methodology: Lead; Supervision: Lead; Writing – review & editing: Lead).
Conflicts of interest
The authors disclose no conflicts.
References
- 1.GLOBOCAN 2020. Colorectal Cancer. International Agency for Research on Cancer, World Health Organization. Accessed January 25, 2023. https://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf. [Google Scholar]
- 2.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology 2011;140:1807–1816. [DOI] [PubMed] [Google Scholar]
- 3.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001;48:526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: the role of inflammation, immuno-suppression, and cancer treatment. World J Gastroenterol 2016;22:4794–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc 2007;2:1998–2004. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka T, Kohno H, Suzuki R, et al. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci 2003;94:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wirtz S, Popp V, Kindermann M, et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc 2017;12:1295–1309. [DOI] [PubMed] [Google Scholar]
- 8.Zijlstra FJ, Garrelds IM, van Dijk AP, et al. Experimental colitis in mice: effects of olsalazine on eicosanoid production in colonic tissue. Agents Actions 1992. Spec No: C76–8. [PubMed]
- 9.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 1995;182:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boirivant M, Fuss IJ, Chu A, et al. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 1998;188. 1929–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins JW, Keeney KM, Crepin VF, et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 2014;12:612–623. [DOI] [PubMed] [Google Scholar]
- 12.Koroleva EP, Halperin S, Gubernatorova EO, et al. Citrobacter rodentium-induced colitis: a robust model to study mucosal immune responses in the gut. J Immunol Methods 2015;421:61–72. [DOI] [PubMed] [Google Scholar]
- 13.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008; 27:5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galon J, Pages F, Marincola FM, et al. Cancer classification using the Immunoscore: a worldwide task force. J Transl Med 2012;10:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo L, Wang C, Qiu X, et al. Colorectal cancer immune infiltrates: significance in patient prognosis and immunotherapeutic efficacy. Front Immunol 2020; 11:1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pages F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018;391:2128–2139. [DOI] [PubMed] [Google Scholar]
- 17.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009;16:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Germann M, Zangger N, Sauvain MO, et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFbeta. EMBO Mol Med 2020;12:e10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin EY, Nguyen AV, Russell RG, et al. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 2001;193:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pyonteck SM, Gadea BB, Wang HW, et al. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene 2012;31:1459–1467. [DOI] [PubMed] [Google Scholar]
- 21.Kubota Y, Takubo K, Shimizu T, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med 2009;206:1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryder M, Gild M, Hohl TM, et al. Genetic and pharmacological targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PLoS One 2013; 8:e54302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbera-Guillem E, Nyhus JK, Wolford CC, et al. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res 2002;62:7042–7049. [PubMed] [Google Scholar]
- 24.Jedinak A, Dudhgaonkar S, Sliva D. Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology 2010;215:242–249. [DOI] [PubMed] [Google Scholar]
- 25.Han YW. Laboratory maintenance of fusobacteria. Curr Protoc Microbiol 2006. Chapter 13:Unit 13A 1. [DOI] [PubMed]
- 26.Xue X, Shah YM. In vitro organoid culture of primary mouse colon tumors. J Vis Exp 2013e50210 [DOI] [PMC free article] [PubMed]
- 27.Westphalen CB, Asfaha S, Hayakawa Y, et al. Longlived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest 2014;124:1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wirtz S, Neufert C, Weigmann B, et al. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2007;2:541–546. [DOI] [PubMed] [Google Scholar]
- 29.Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol 2015;1:154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez CA, Miller BM, Rivera-Chavez F, et al. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 2016; 353:1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mundy R, MacDonald TT, Dougan G, et al. Citrobacter rodentium of mice and man. Cell Microbiol 2005; 7:1697–1706. [DOI] [PubMed] [Google Scholar]
- 32.Polletti S, Natoli G. Understanding spontaneous conversion: the case of the Ly6C(−) monocyte. Immunity 2017;46:764–766. [DOI] [PubMed] [Google Scholar]
- 33.Gross M, Salame TM, Jung S. Guardians of the gut–murine intestinal macrophages and dendritic cells. Front Immunol 2015;6:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose S, Misharin A, Perlman H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 2012;81:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czarnewski P, Parigi SM, Sorini C, et al. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat Commun 2019;10:2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 2019; 10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019; 37:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Li Z, Skrzypczynska KM, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 2020;181:442–459.e29. [DOI] [PubMed] [Google Scholar]
- 39.Smillie CS, Biton M, Ordovas-Montanes J, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019;178:714–730.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods 1994; 174:83–93. [DOI] [PubMed] [Google Scholar]
- 41.Van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods 1996;193:93–99. [DOI] [PubMed] [Google Scholar]
- 42.Schumak B, Klocke K, Kuepper JM, et al. Specific depletion of Ly6C(hi) inflammatory monocytes prevents immunopathology in experimental cerebral malaria. PLoS One 2015;10:e0124080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med 1990;323:1228–1233. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Q, Shen ZF, Wu BS, et al. Risk of colorectal cancer in ulcerative colitis patients: a systematic review and meta-analysis. Gastroenterol Res Pract 2019;2019: 5363261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seamons A, Treuting PM, Brabb T, et al. Characterization of dextran sodium sulfate-induced inflammation and colonic tumorigenesis in Smad3(−/−) mice with dysregulated TGFbeta. PLoS One 2013;8: e79182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Popivanova BK, Kostadinova FI, Furuichi K, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res 2009; 69:7884–7892. [DOI] [PubMed] [Google Scholar]
- 47.Wilson JM, Coletta PL, Cuthbert RJ, et al. Macrophage migration inhibitory factor promotes intestinal tumorigenesis. Gastroenterology 2005;129:1485–1503. [DOI] [PubMed] [Google Scholar]
- 48.Bader JE, Enos RT, Velazquez KT, et al. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am J Physiol Gastrointest Liver Physiol 2018;314:G22–G31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Undi RB, Ali N, et al. It takes a village: microbiota, parainflammation, paligenosis and bystander effects in colorectal cancer initiation. Dis Model Mech 2021:14. [DOI] [PMC free article] [PubMed]
- 50.Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS−) vs. alternatively activated macrophages. Front Immunol 2019; 10:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y, Li L, Xu C, et al. Cross-talk between the gut microbiota and monocyte-like macrophages mediates an inflammatory response to promote colitis-associated tumourigenesis. Gut 2020;70:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shang K, Bai YP, Wang C, et al. Crucial involvement of tumor-associated neutrophils in the regulation of chronic colitis-associated carcinogenesis in mice. PLoS One 2012;7:e51848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Triner D, Devenport SN, Ramakrishnan SK, et al. Neutrophils restrict tumor-associated microbiota to reduce growth and invasion of colon tumors in Mice. Gastroenterology 2019;156:1467–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka Y, Ito S, Isobe K. Vancomycin-sensitive bacteria trigger development of colitis-associated colon cancer by attracting neutrophils. Sci Rep 2016;6:23920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing matrix files have been deposited in the National Institutes of Health Gene Expression Omnibus database and can be accessed at PRJNA910503. All data are available in the main text or the Supplementary Materials.