

Abstract
Most rare diseases are caused by single-gene mutations, and as such, lend themselves to a host of new gene-targeted therapies and technologies including anti-sense oligonucleotides, phosphomorpholinos, small interfering RNAs, and a variety of gene delivery and gene editing systems. Early successes are encouraging, however, given the substantial number of distinct rare diseases, the ability to scale these successes will be unsustainable without new development efficiencies. Herein, we discuss the need for genomic newborn screening to match pace with the growing development of targeted therapeutics and ability to rapidly develop individualized therapies for rare variants. We offer approaches to move beyond conventional “one disease at a time” pre-clinical and clinical drug development and discuss planned regulatory innovations that are necessary to speed therapy delivery to individuals in need. These proposals leverage the shared properties of platform classes of therapeutics and innovative trial designs including master and platform protocols to better serve patients and accelerate drug development. Ultimately, there are risks to these novel approaches, however we believe that close partnership and transparency between health authorities, patients, researchers, and drug developers present the path forward to overcome these challenges and deliver on the promise of gene-targeted therapies for rare diseases.
Keywords: gene therapy, newborn screening, rare disease, clinical trial design, drug development, gene-targeted therapy, orphan drug, platform technologies
1. Introduction – Outlining the need
Genetic conditions account for approximately 70% of known rare diseases (Nguengang Wakap et al., 2020), which include as many as 7,000 unique conditions based on current estimates (Blencowe et al., 2018). While individually rare, collectively, rare diseases impact over 300 million individuals worldwide. Approximately 95% of rare diseases lack an existing, approved therapy. As of 2019, there are only 564 unique approved orphan drugs, with 90% of these approved only for a single indication or disease (IQVIA Institute for Human Data Science, 2020) (Figure 1).
Figure 1. Current progress in increasing available therapies, based on current approaches.
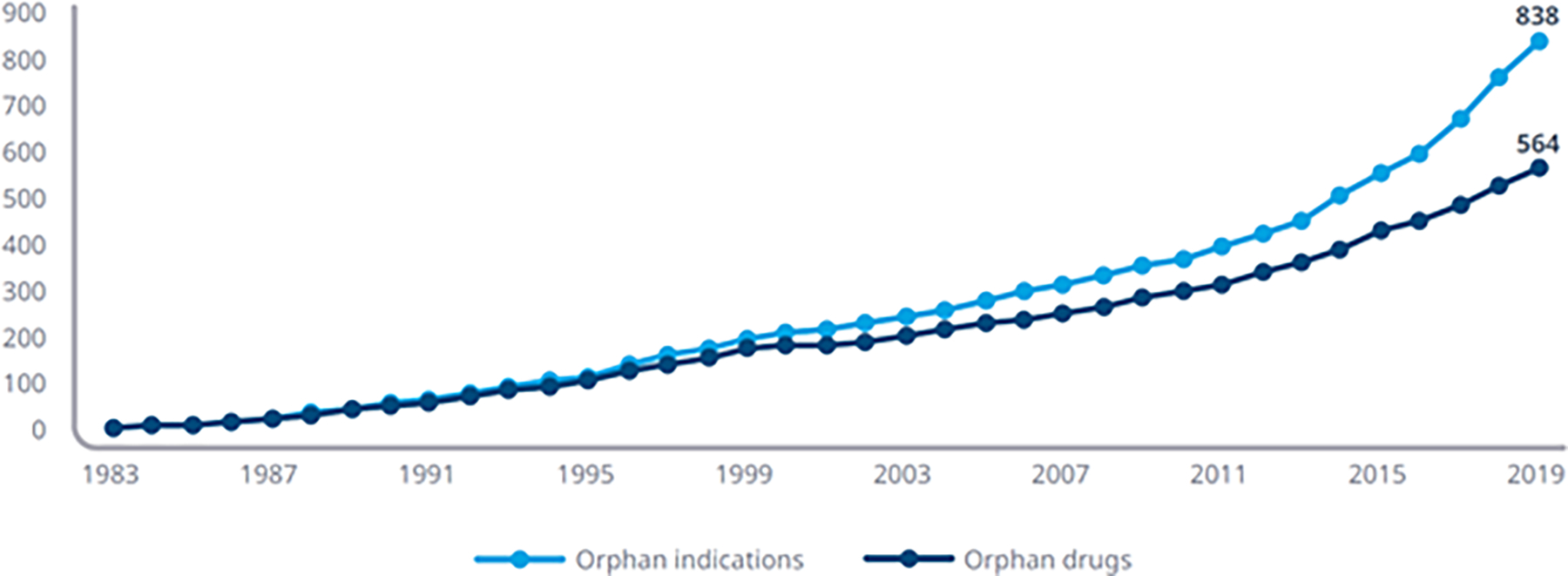
Cumulative Number of Annual Approved Orphan Indications and Orphan Drugs with at Least One Orphan Indication. Image source: IQVIA Institute (IQVIA Institute for Human Data Science, 2020)
The rapid rise of new technologies/approaches to treat monogenic diseases has sparked interest in drug development for rare genetic diseases. Financial incentives implemented in the Orphan Drug Act (“Recommendations for investigations of drugs for rare diseases or conditions,” 2011) further heightened interest. Together, these two factors have led to regulatory approvals for multiple gene-targeted therapies in recent years, including small interfering RNAs (siRNAs) (patisiran, givosiran, lumasiran, inclisiran), antisense oligonucleotides (ASOs) (nusinersen, mipomersen, inotersen), phosphomorpholinos (eteplirsen, golodirsen), gene transfer therapies using adeno-associated virus (AAV) (onasemnogene, voretigene) and other vectors, and hematopoietic stem cell approaches using lentiviral gene additions (Brown & Wobst, 2021). Genome editing and mRNA therapies will likely join this list in the near future.
These are very welcome early successes but represent just a drop in the bucket considering the vast number of distinct rare diseases. In the past 5 years, the mean number of annual Food and Drug Administration (FDA) orphan drug approvals was 87±8.3 (U.S. Department of Health and Human Services). Even at triple this approval rate, with an optimistic goal of each approval representing a distinct condition, it would not be possible to reach 7000 cumulative orphan drug approvals in a 20-year time horizon (Figure 2). Most worrisome, the costs of these development efforts remain high, and there is very real reason for concern that scaling these successes to thousands of rare diseases will be gradual in timing and societally unsustainable unless efficiencies can be gained. Further, due to low disease prevalence and global patient dispersion, the ‘gold standard’ of randomized controlled studies to demonstrate efficacy and safety sufficient for drug approvals by Health Authorities will be untenable. New solutions will be necessary to meet these challenges. Herein, we discuss approaches and challenges that must be addressed to break away from the standard one disease at a time approach.
Figure 2. Achieving 7000+ targeted rare disease therapies – modeling future FDA orphan drug approval needs.
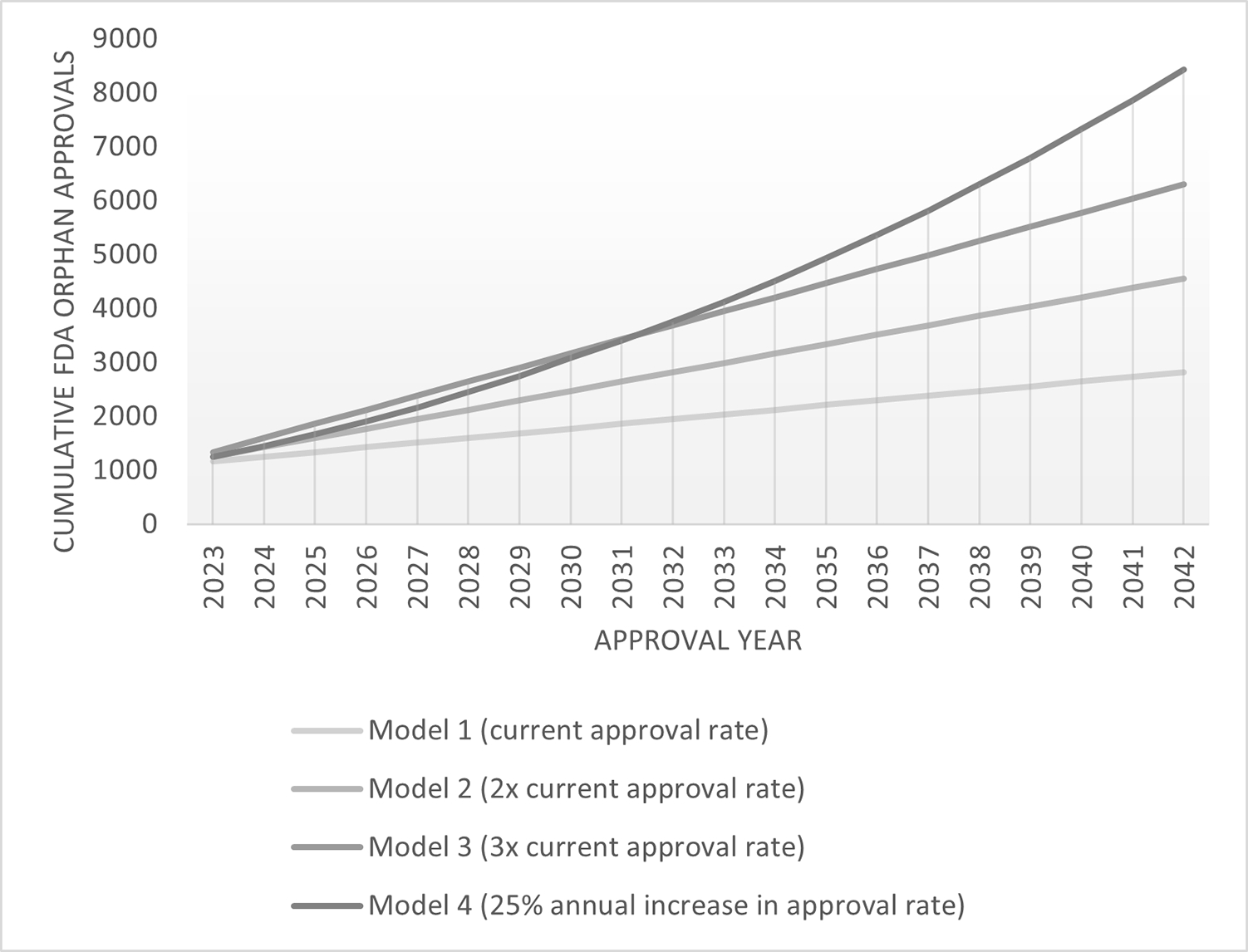
Projected cumulative FDA Orphan Drug approvals based on four annual approval rate models. Current approval rate is based on an average of 87 orphan drug approvals per year, 2017–2021. Data source, FDA (U.S. Department of Health and Human Services).
2. Moving towards modality- and mutational mechanism-centric models
One possible solution is to fundamentally rethink how we organize our drug development efforts. Conventionally, pre-clinical and clinical drug development efforts are approached “one disease at a time.” Instead, technological advances in platforms for genetically targeted therapy encourage a reframing of efforts as “one modality at a time,” leveraging shared properties of ASOs, siRNAs, or phosphomorpholinos as a class to make preclinical and clinical testing more efficient (Table 1). Furthermore, since different modalities are effective at treating different molecular classes of mutations (e.g., phosphomorpholinos can be used to modulate certain mutations that cause gene mis-splicing, siRNAs can be used to knock down genes bearing toxic gain-of-function mutations, and ASOs can be used to do either), these capabilities may even invite the reclassification of certain drug development efforts by mutational mechanism (“one mutational mechanism at a time”). We envision a future in which genetic diseases can be reclassified into more efficient therapeutic groups, combining disease, target tissue, and mutational impact: e.g., “genetic diseases of the liver addressable via siRNA-mediated knockdown” or “genetic diseases of the central nervous system addressable via ASO-mediated splice modulation,” allowing attention to be focused on reliable, and repeatable methodology for consistent delivery, dosing, safety monitoring, and efficient trial conduct (Mechler et al., 2015).
Table 1.
Platform modalities
| Status | Approach | Shared element(s) | Variable element(s) | Example/Reference | Consortia |
|---|---|---|---|---|---|
| Established | CAR-T | Cell type | T cell receptor variable region | Multiple myeloma - Idecabtagene (Gearin, 2021) | CARnation Consortium |
| Cancer vaccines | Peptide subunits and linkages | Peptide sequence | Cervical cancer - Human papillomavirus vaccine (Cheng, et al., 2020) | Translational Research Cancer Centers Consortium | |
| mRNA vaccines | RNA, modified nucleotide subunits | Sequence | COVID-19 vaccine (Thorn, et al., 2022) | K-mRNA vaccine consortium | |
| Emerging | Viral vector-mediated gene replacement | Viral vector particles (DNA or RNA, proteins) | Sequence (promoter, coding sequence) | Spinal Muscular Atrophy - Onasemnogene (Blair, 2022) | PAVE-GT, BGTC |
| Antisense oligonucleotides | Oligonucleotides, +/− lipid nanoparticles, +/− tissue-targeting conjugates | Sequence | Spinal Muscular Atrophy – Nusinersen (Hoy, S.M., 2017) | N=1 Collaborative, 1M1M | |
| Future | mRNA gene replacement | RNA, modified nucleotide subunits | Sequence | Propionic acidemia (Jiang, et al., 2020) | N/A |
| Morpholinos | DNA, modified nucleotide subunits | Sequence | Duchenne Muscular Dystrophy (Le, B.T., et al., 2022) | N/A | |
| siRNAs | RNA, modified nucleotide subunits | Sequence | Hereditary transthyretin-mediated amyloidosis - Patisiran (Adams, et al., 2018) | RNAi Consortium | |
| Genome editing | Vector, nuclease / base editor proteins | Guide RNA sequence, “donor” DNA | Amyloidosis (Gillmore, et al., 2021) | NIST Genome Editing Consortium |
BGTC – Bespoke Gene Therapy Consortium; NIST - National Institute of Standards and Technology; PAVE-GT - Platform Vector Gene Therapy; 1M1M – 1 Mutation 1 Medicine
With ten FDA approvals for rare genetic disorders, ASOs represent the most mature of current platform technologies. siRNAs and phosphomorpholinos are not far behind, with four FDA approvals, each. AAV vector-mediated gene addition may also be viewed as a platform (with the important caveats that different AAV capsids can transduce different tissue targets and possess different safety profiles), where the variable can be reduced to the cDNA contained within a vector platform. The same can be implied for gene addition through lentiviral transduction of hematopoietic stem cells. Issues related to optimal dose, delivery, and durability affect all of these technologies with some variations, because delivery to target tissues is necessary to achieve correction and efficacy. For example, any platform requiring systemic viral vector-mediated delivery will have to address the possibility of immune responses that are typically dose-related, as revealed by clinical trials with AAV vectors (Mingozzi et al., 2007, Ronzitti et al., 2020, Verdera, et al., 2020).
These types of shared issues could be more efficiently addressed for a platform technology through data sharing. For example, data sharing could allow communications regarding the toxicities of platforms across diseases by sharing preclinical data and even clinical data between programs, a long-standing goal for the field of gene therapy (O’Reilly et al., 2013). This need has been emphasized by recently emerging unexpected serious adverse events that were observed only in clinical trials, which underscores the importance of rapid, real-time reporting of serious adverse events to protect participants from harm (Buscara et al., 2020). Overall, the standardization of new technologies for gene-targeted therapies and involvement of sponsors with greater resources will continue to accelerate the development of new therapies.
Expansion of the development of therapies for monogenic disorders and leveraging the opportunities of platform therapies requires simultaneous development of novel approaches to identifying affected individuals and diagnosis, pre-clinical testing, clinical trial design and execution, and regulatory approval pathways.
3. Approaches to Early Diagnosis
With the rapid use of genomic technologies to detect, diagnose, and treat genetic disease and the promise of developing tailored therapies for specific gene variants, it is imperative to develop parallel rapid and effective methods for early diagnosis before the onset of clinical symptoms (Figure 3). For progressive conditions, early intervention may be key to optimal efficacy outcomes, as demonstrated in clinical trials of onasemnogene for spinal muscular atrophy (Mendell et al., 2021).
Figure 3.
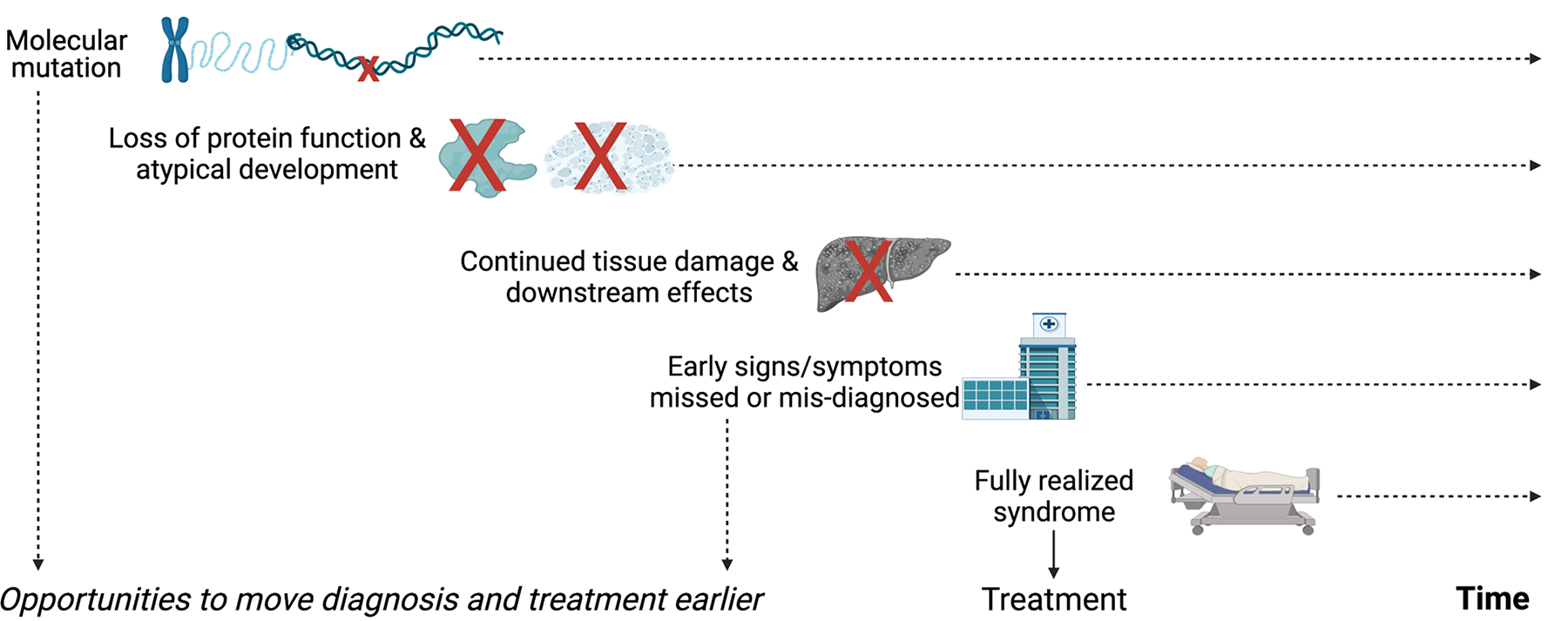
Early recognition in rare genetic diseases presents opportunities to shorten the timeline to treatment
Currently, newborn screening (NBS) is the primary method for identifying individuals pre-symptomatically for many rare diseases. Newborn screening is a long-established public health program that begins with the screening of the majority of babies born in the United States. Each state determines what conditions are screened for; most states opt for conditions that are included on the Secretary of the Department of Health and Human Services’ (HHS) recommended uniform screening panel (RUSP). Currently there are 35 core and 26 secondary conditions (those that can be detected in the differential diagnosis of a core disorder) on the RUSP. As it is the prerogative of the state to add conditions, some add conditions that are not on the RUSP (Baby’s First Test).
The established process for adding conditions to the RUSP is thoughtful, thorough and evidence based. It evaluates the sensitivity and reliability of the test, identifies a proven effective treatment or benefit of early diagnosis, as well as considers cost effectiveness. However, while scientific rigor is essential, the process as designed is only able to review one condition at a time. As such, it can take years for a condition to meet the criteria needed to be added to the RUSP, and many more for it to be adopted by all the states. With over 7,000 rare conditions identified at this time, and treatments being developed at a rate not seen in the past, our ability to identify individuals with rare conditions early—and ideally, before they are symptomatic—is falling woefully behind.
Historically, the primary methods used to screen infants in NBS programs have been biochemically based. This choice can be attributed to the available technology when NBS began in the 1960s and the large scope of the program, screening over four million newborns each year, which requires a cost-effective approach (e.g., biochemical screen for phenylketonuria). NBS was intended to be just that, a screen, and not a definitive diagnosis of a disorder. Molecular methods were used as a second tier of testing (e.g., cystic fibrosis) or as a diagnostic tool. As technology has progressed, molecular approaches have been added to the panel for Severe Combined Immune deficiency and Spinal Muscular Atrophy. There are several reasons molecular methods have not been widely implemented in public health NBS programs, with the primary reasons being high cost in a public health laboratory environment, and challenges to interpretation of the number of expected variants detected with unknown significance. Additionally, there has always been a concern that molecular tests could miss true cases. However, now with the availability of less costly Next-generation sequencing (NGS) and the expectation of more widely available gene-targeted therapies, there are many disorders that could potentially be screened for using NGS. The addition of such new approaches to standard NBS methodologies, through NGS or other molecular screening, should be considered. The wealth of genetic information generated by such an approach, and linked to phenotypic, longitudinal data, would be indispensable for the ongoing efforts to develop new gene-targeted therapies as discussed further in a companion article.
In the past, efforts to expand newborn screening (by sequencing approaches) may have been less urgent in the absence of available genetic or non-genetic therapies. This new opportunity for the rapid development of tailored therapies for specific genetic variants makes the parallel development of methods for the rapid identification of individuals through a robust screening process followed by definitive diagnosis and link to methods for treatment a necessity. Newborn screening offers an established system of population-based screening for a small group of specific diseases followed by a handover to clinicians to diagnosis and treat. However, we must move past the one-disease-at-a-time screening approach.
Since the late 1980s and early 1990s, DNA-based tests have been gradually integrated into the practice of evaluating newborns at the point of care. In a recent publication (Furnier et al., 2020), those applications were categorized into the following three groups: 1. Molecular Marker Identification as Primary Screening Methods; 2. Targeted Gene Variant Panel as Second-Tier Testing; and 3. Targeted Gene Variant or Variants as “Just-in-Time” Information. In the above applications, screening for spinal muscular atrophy using a genetic test to identify newborns with homozygous SMN1 exon 7 deletion is a prime example of how a genetic marker can accurately and expeditiously identify affected newborns in public newborn screening programs. Affected newborns can receive transformative therapies, like gene therapy, at any point while they are still in the newborn period (Kay et al., 2020) (Baker et al., 2022). The FDA predicts that there will be 10–20 such gene and cell therapy approvals per year by 2025 (Gottlieb, 2019) which will undoubtedly lead to an urgent necessity for the parallel development of newborn screening methods tailored for specific gene variant identification. Genomic newborn screening (gNBS) appears to be a cost-effective pathway to meet the demand of pre-symptomatic identification of many genetic conditions in newborns. This approach, in addition to standard NBS screening provides the advantage of assisting in the diagnosis of monogenic disorders that are not already on the RUSP.
Here we offer some points to consider in the design of a gNBS:
Expand use of high throughput and robust DNA isolation methods that reliably isolate high quality and sufficient quantity of doubled-strand DNA
Develop regulatory-compliant interpretation software that matches genetic diagnoses with available treatments in childhood. The software can be continuously updated based upon new knowledge and treatment.
Develop resources to provide full accounting of genetic findings, including accessible and available genetic counseling resources.
Create flexible genotype and phenotype combination screening protocols.
Ensure equitable access to screening and follow-up (diagnostic confirmation and treatment).
With the accelerated rise of new gene-targeted therapies both approved and those in clinical testing, implementation of genomic screening will require a new level of coordination between NBS programs and clinicians and patients and their families. NBS is a system of prenatal education, neonatal screening conducted in birthing hospitals and state departments of health, using either physiological or molecular technologies, referral to clinical care for diagnosis and treatment, and in many cases lifelong management. For the first fifty years of NBS, screen positive newborns were referred to metabolic geneticists or endocrinologist for diagnosis and treatment. Advancements in technologies to screen, diagnose, and treat genetic disease has led to a variety of clinical subspecialties now being involved in NBS. These subspecialities include cardiology, immunology, and neurology, and the number and diversity of specialists involved in the diagnosis, treatment, and management of NBS-identified newborns is expected to continue to increase. Similarly, the complexity of treatments and management approaches has expanded from diet changes to stem cell transplants, heart surgery, enzyme replacement, and gene-targeted therapies. Today, and in the future, it is possible that patient referrals will include not only health care specialists, but also information about consortia or pharmaceutical sponsors who are testing new therapies.
Surpassing the importance of adequate financial, personnel, and educational resources, are needs to address ethical and biopsychosocial considerations of genomic newborn screening, including informed consent and decision-making, issues of uncertainty, diagnostic and prognostic accuracy, and lifelong “medicalization”. We refer readers to articles in this issue for a more comprehensive exploration of these challenges. (Vockley, et al., 2022 and Vockley, et al., 2022)
4. Streamlining preclinical research that facilitates initiation of clinical trials
The rapidity of the development of these new platforms will require innovative and efficient preclinical testing approaches to assess safety and efficacy. The current situation usually requires long-term studies of pharmacology and toxicology (pharm-tox), at least in a rodent model and often in a large animal model, which is highly expensive and takes many months to complete. Such pharm-tox studies are a prerequisite to filing an Investigational New Drug application to the FDA in advance of initiating a clinical trial (Figure 4A). An abbreviated model for preclinical research could require only in vitro validation of function for the drug, as opposed to an in vivo animal study for proof of concept, followed by an abbreviated study in a rodent model (Figure 4B). This path has been used in several clinical investigations of ASOs for life-threatening orphan diseases (Kim et al, 2019, Tran et al, 2022, Korobeynikov et al, 2022), in which the time from initiation of preclinical pharm-tox study to enrollment of the first (and only) participant for the initial individualized ASO was a mere four months (Kim et al., 2019). Going forward, a streamlined model for preclinical testing could be considered based upon this precedent, under the correct circumstances (Figure 4C). In this scenario, an agent that delivers a unique nucleotide agent that does not encode a protein, such as an ASO or single guide RNA as part of a CRISPR/Cas9 nuclease, would be considered a platform technology that could be studied for its in vitro effects in human cells. This would be possible, if previously submitted preclinical studies involving an ASO or CRISPR/Cas9 that differed only with regard to the non-coding nucleotide sequence were used to review the safety of the new agent. The change that affected nucleotide sequence would be evaluated by the FDA based upon interaction with the human genome as evaluated through in vitro studies in human cells. Conceivably, with sufficient clinical experience with a given platform and supportive predictive in silico and in vitro models, the use of pharm-tox animal studies may even someday be supplanted or at least minimized (Figure 4C). This streamlined approach to preclinical research is consistent with FDA guidelines for genome editing (U.S. Department of Health and Human Services, 2022b). We anticipate that each platform category presents distinct opportunities to streamline pre-clinical research; the strategies for one category may not be appropriate for another. For viral-vector mediated gene replacement therapy platforms, initial steps towards efficiency may focus on leveraging shared knowledge related to delivery system-specific biodistribution, toxicity, and replicable manufacturing processes. The Bespoke Gene Therapy Consortium, a public-private partnership supported by the Foundation for the NIH (https://fnih.org/our-programs/AMP/BGTC), includes efforts to identify standardized sets of critical quality attributes and animal toxicology studies for assessing the safety of AAV vectors for first in human clinical trials. Only through such innovative approaches to preclinical research can novel therapies be made available to individual patients with rare, potentially lethal conditions.
Figure 4.
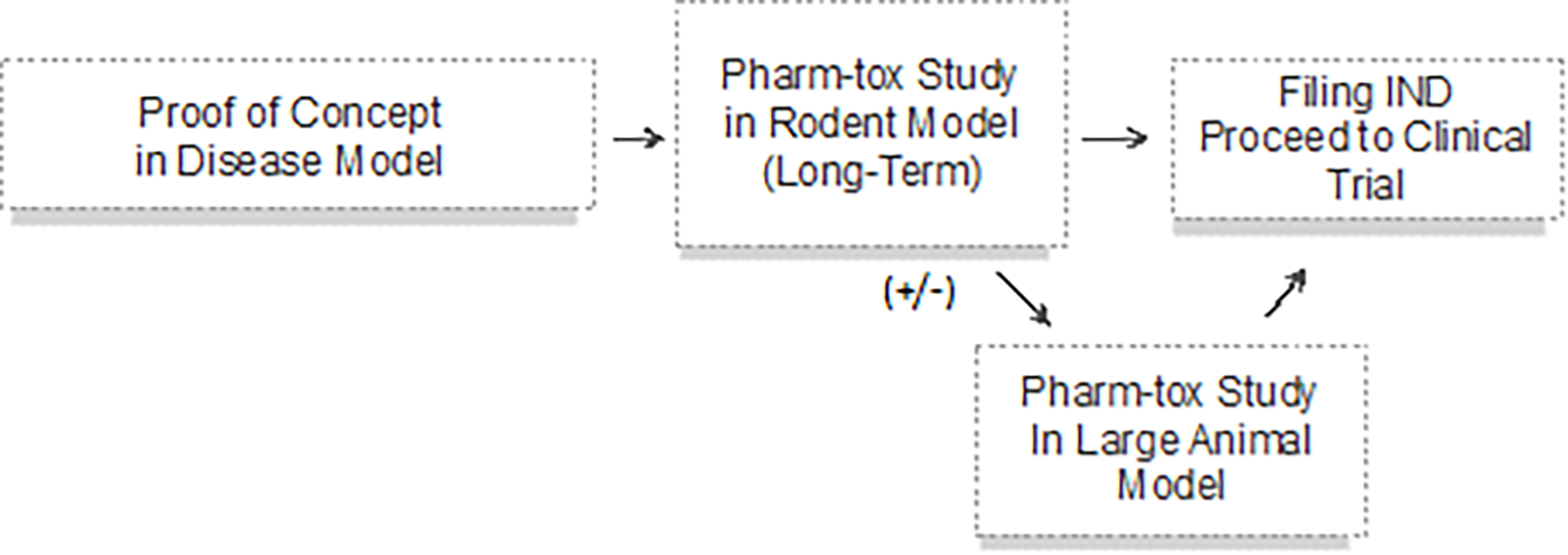

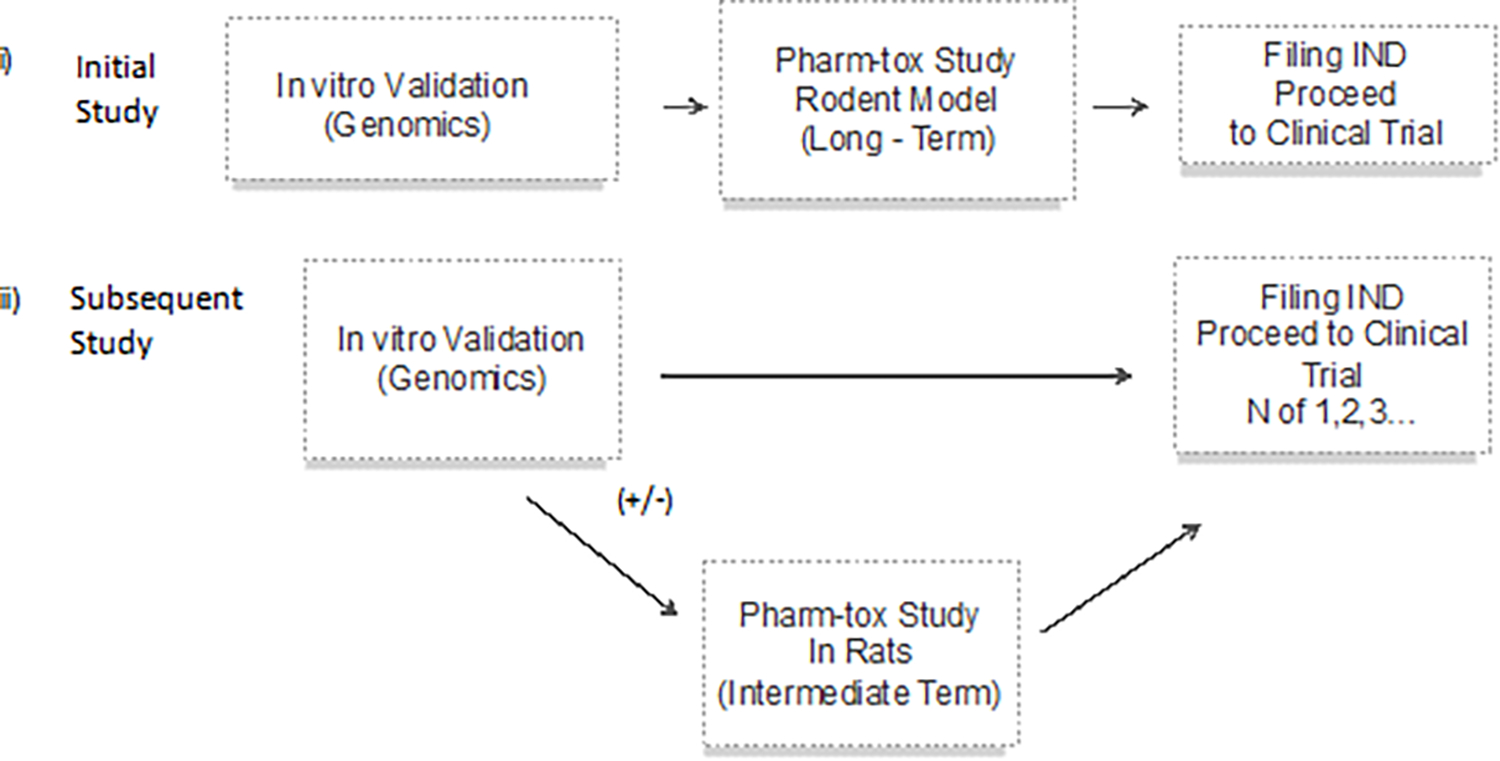
Alternative approaches to preclinical studies
5. Novel Approach to Trials/Generating Evidence
In order to bring potentially transformative gene-targeted therapies to individuals identified by NBS, particularly for those with rapidly progressive disease, efforts to expedite clinical development are paramount. Importantly, reductions in the cost and the risk of development must be paired with speedy, safe, equitable-access and high-quality clinical trials that can deliver the data needed to support health authority reviews and approvals. In tandem with these efforts, regulatory reviews of therapies for these diseases may need to embrace other approaches. Considerations for accelerated or conditional approvals based on promising surrogate endpoints, closely linked to core pathology, may need to be employed, with the stipulation for careful, rigorous, long-term follow-up of more traditional clinical endpoints, in order to address the extraordinarily high unmet need in many of these diseases. For ultra-rare or nano-rare diseases, individualized approaches to therapies are already being advanced. These efforts will require a consortium approach, with collaboration across the biomedical ecosystem including industry, researchers, physicians, regulators and government. Here, we examine novel development approaches including the use of n-of-1 studies and master protocols that can enable the study of multiple disease types and multiple therapeutics in the same trial. Further, we describe the novel programs put into place by Health Authorities, for the purpose of partnering early with sponsors and researchers in order to work together with patients and their families for the development of impactful therapies.
“N-of-1” studies are typically randomized clinical trials that apply the principles of cross-over design to an individual trial participant, where two or more treatments are given in a randomized order over multiple cross-over periods, with masking of both the participant and the researcher (Abrahamyan et al., 2016). A modification of this methodology has been used as a means of addressing the immediate needs of ultra-rare genetic disease populations and has made significant inroads into its application and the testing of gene-targeted therapies in individuals impacted by these diseases (Table 2) (Kim, et al., 2019). In 2019, Yu and colleagues reported the rational design, testing, and manufacturing (by Ionis Pharmaceuticals) of a unique investigational product, a splice-modulating ASO drug, tailored for use in a child with a novel mutation resulting in neuronal ceroid lipofuscinosis 7 (CLN7 disease). Its use was demonstrated to be safe with an objective reduction in seizure activity and demonstrated a viable approach for the efficient development of patient-customized treatments.
Table 2.
Optimal scenarios for N-of-1 Trials
| Therapeutic mutation-specificity | Applicable setting |
|---|---|
| Specific | Unique gene variant present in a very small number of individuals |
| Non-specific | Exceptional disease rarity, such that the eligible number of individuals is very small |
| Non-specific | Clinical and scientific circumstances justify accelerated trial in a very small number of individuals |
Other consortia hope to replicate this feat on a broader scale. The n-Lorem Foundation is committed to creating individual treatments for patients with nano-rare diseases, namely those affecting fewer than 30 patients in the world, using ASO technology (n-Lorem Foundation). The advantages of using ASOs lie in the relatively simplicity of manufacturing and testing, ability to tailor each ASO to patient-specific mutations, and precedence for regulatory approval and use in the treatment of rare diseases. As of Spring 2022, the n-Lorem Foundation reported more than 50 individualized drug programs in development for more than 50 patients with nano-rare diseases, anticipating the dosing of their first two patients by summer (n-Lorem Foundation, 2022).
The N=1 Collaborative comprises a group of expert physician-scientists, researchers and companies invested in the advancement of individualized therapies, beginning with ASO technologies but with plans to advance into other gene-targeted platforms (N=1 Collaborative). To date, individuals have been dosed with therapies targeting several mechanisms underpinning amyotrophic lateral sclerosis (ALS) including the Fused in sarcoma (FUS) protein (Korobeynikov et al., 2022) and the expansions of G4C2 repeats in C9orf72 (Tran et al., 2022) gene, and additionally, an individualized ASO for a specific ataxia-telangiectasia mutation (Yu et al., unpublished data) (Synofzik et al., 2022).
In 2018, the NIH Common Fund initiated the Somatic Cell Gene Editing (SCGE) consortium (National Institutes of Health), assembling a group of multidisciplinary teams designed to address the growing needs in the gene editing field, with the goal to accelerate the translation of genome-editing technologies to a wide range of tissues and diseases and provide the highest degree of transparency and sharing of discovered technologies (Saha, et al., 2021). Priorities and strategies include the discovery of new editors and improvements in existing editors, to optimize precision, the targeting and delivery of therapeutic components to specific tissue types in vivo, and the validation of approaches using both small and large animal testing. Ultimately, the goal is the creation and maintenance of an SCGE Toolkit supporting the infrastructure to promote collaborations, and provision of a platform for transparent data sharing, with SCGE investigators, the broader scientific community, and the public. In mid-2022, NIH announced the plan for Phase 2 of the SCGE, with a greater focus on moving genome editing into the clinic (National Institutes of Health, 2022). Notably, one of the FOAs will support clinical gene editing trials, with a requirement for a platform approach including at least two different diseases (Department of Health and Human Services, 2022).
In 2020, the US National Institute of Neurological Disorders and Stroke (NINDS) launched the Ultra-Rare Gene-based Therapy (URGenT) network (Schor et al., 2021). The URGenT network supports the development and delivery of gene-targeted therapies to individuals with ultra-rare neurologic diseases. Importantly, this effort will work towards standardizing and harmonizing best practices, while encouraging innovation in clinical trial design and execution. Further, NINDS offers funding and key resource support for drug product optimization, manufacturing, and Investigational New Drug-enabling studies in support of early clinical trials. The first funding opportunity was offered in late 2021 with first projects anticipated to be launched in 2022.
Launched in late 2021, the Bespoke Gene Therapy Consortium, (BGTC) (National Institutes of Health), is a public-private partnership between the NIH, the FDA, multiple pharmaceutical and life sciences companies, and non-profit organizations, with the goal to establish platforms and standards to speed the development and delivery of customized gene therapies for people affected by rare diseases. Its focus is on the therapeutic platform—developing processes for how to develop, test, and treat individuals with ultra-rare diseases, and to do so with speed and efficiency by leveraging a common gene therapy platform. To that end, another BGTC goal is to enhance the understanding of the basic biology of the adeno-associated virus (AAV), a common gene-delivery vehicle in clinical use today. BGTC hopes to streamline the approach of manufacturing, testing, and clinical trial design and execution by leveraging the commonalities that the AAV delivery platform provides, and working closely with investigators and clinical trial sites, providing manufacturing, testing, regulatory, and trial execution support and resources.
In addition to these important “n-of-1” and small sample efforts, development and regulatory innovations would also benefit the development of gene-targeted therapies for “more common” rare diseases. Development and regulatory approval of therapeutics for these diseases remain hamstrung by traditional approaches, requiring years to progress through phase 1 and 2 studies and registrational phase 3 studies. These approaches, lack efficiency and are prone to high overhead and low feasibility. Applying some of the alternative platform-based pre-clinical testing paradigms could streamline some aspects, however, additional and relatively newer innovations in clinical trial design may enable developers to test and advance therapeutics for “more than one disease at a time.”
Master protocols, classified as basket trials, umbrella trials or platform trials, are designed to investigate multiple hypotheses through concurrent studies under the same testing paradigm (Janiaud et al., 2019; Park et al., 2019; Renfro & Sargent, 2017; Woodcock & LaVange, 2017) (Figure 5). A basket trial design tests a single targeted therapy across multiple diseases for which the target is hypothesized to be relevant. Umbrella trials test multiple targeted therapies across a single disease indication. Platform trials are multi-arm, multi-stage studies that evaluate several interventions against a common control arm and can be perpetual. Some of the many benefits of platform trials is the ability to include prespecified rules for dropping non-performing investigational arms and allowing the addition of new investigational arms. Thus, novel targeted agents can continually enter and exit the trial protocol in an operationally seamless manner. Adaptive randomization schema can also be incorporated that influences the probability of newly randomized subjects to be assigned to higher performing study arms based on data from earlier subjects. Further study rules can be incorporated that pre-specify the criteria for the advancement of promising therapeutics into registrational phases of research. All master protocol approaches require endorsement and coordination by a broad consortium of stakeholders including academic researchers, industry partners, and government agencies. The goal in applying these approaches is to offer greater efficiency and feasibility to clinical testing of multiple therapies with the use of common biomarker platforms, common database modalities and consistency of study execution, adjudication and data monitoring committees, as well as a more ethical and equitable approach (Janiaud et al., 2019; Park et al., 2019; Renfro & Sargent, 2017; Woodcock & LaVange, 2017).
Figure 5.
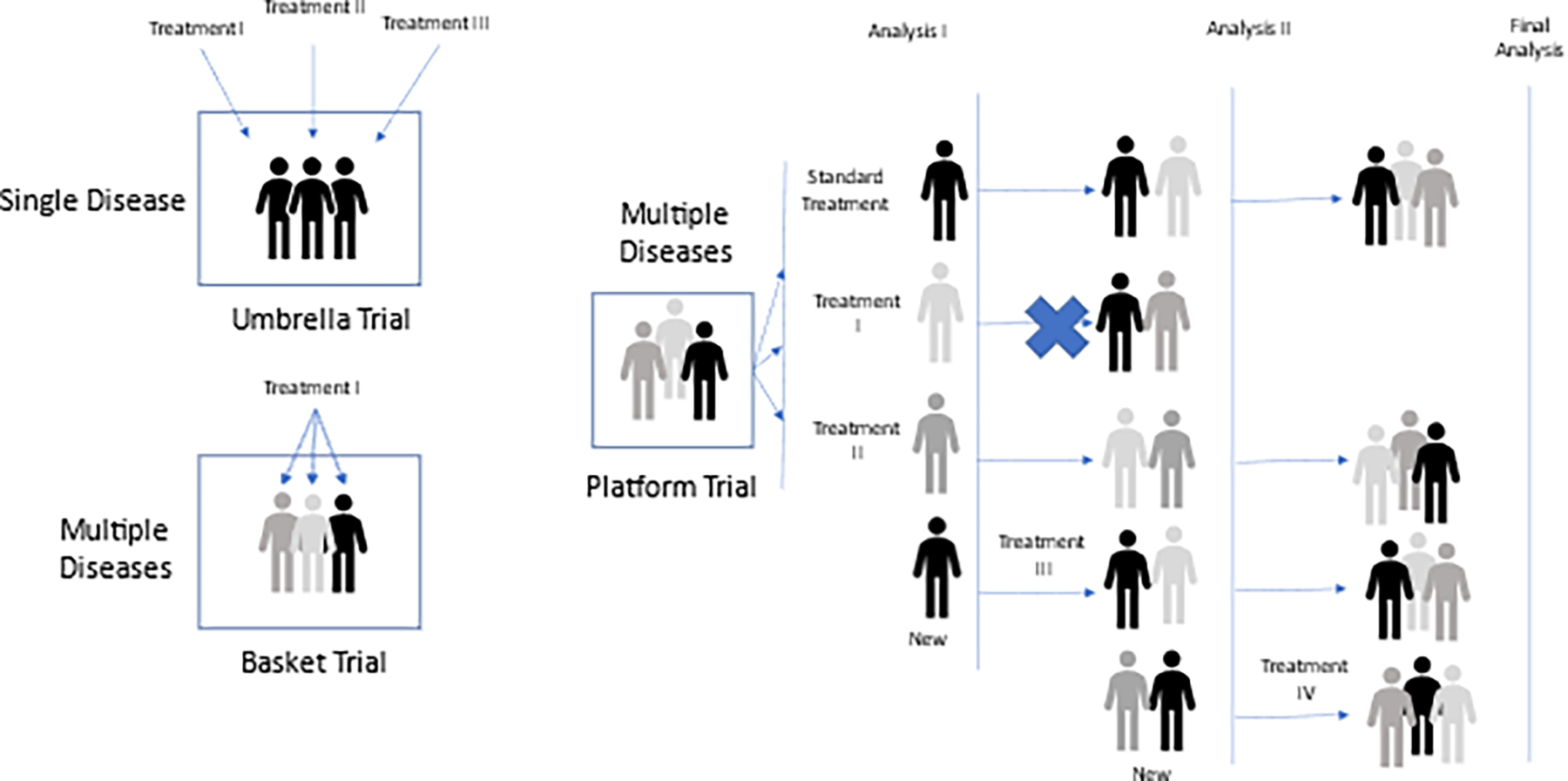
Umbrella, Basket, and Platform Trial designs
The rise of master protocols largely began in the field of oncology—driven by the principle of “precision oncology” with our ability via genetic sequencing of tumors to differentiate cancers by their genetic mutational signature, in concert with, and sometimes in lieu of, their histological elements—in essence, turning common tumor types into rare subsets, that might be better treated with targeted therapies. Today, the vast number of ongoing master protocols are being conducted for oncology indications, however a few are underway in rare disease indications (Janiaud et al., 2019; Park et al., 2019; Renfro & Sargent, 2017; Woodcock & LaVange, 2017).
The investigation of acetyl-L-leucine for the treatment of three rare neurodegenerative diseases provides an example of a master protocol that also incorporates an innovative clinical endpoint assessment based on a primary anchor test, individualized to each subject’s baseline functional abilities (Fields et al., 2021). The trial design is a basket trial, enrolling patients with Niemann-Pick type C (NPC), GM2 gangliosidosis (GM2), or ataxia telangiectasia (A-T), based on the following considerations:
Choice of investigational agent - NPC, GM2, and A-T are characterized by progressive neurodegeneration of the cerebellum and cerebrum, and as a consequence, both physical and cognitive decline, and premature death. These disorders share a number of symptoms, including cerebellar ataxia, dysarthria, and dysphagia. Owing to their common neurological manifestations, and the proposed mechanism of action of N-acetyl-L-leucine, a single master protocol was developed. In addition, pre-existing data on the use of N-acetyl-L-leucine as a compassionate use therapy, as well as in other inherited cerebellar ataxias, supported its choice as the investigation agent.
Choice of a Master Protocol - The small potential pool of participants within each disorder was anticipated to limit enrollment, hence a basket design was chosen. A basket design, testing an investigational agent across multiple rare diseases enables the consistency of study site selection, study execution and biomarker testing, study site training, including video taping of primary endpoint assessment and a centralized, blinded assessment adjudication of the study endpoints.
Choice of an open-label design - Patients and caregivers expressed legitimate ethical concerns about a placebo-controlled trial in these rapidly progressive diseases. As a consequence, use of an open-label study without masking required careful consideration of the primary endpoint and its assessment.
Choice of the primary endpoint - The broad variability of symptoms and signs within each indication, including the age at first presentation and rate of disease progression, precluded the ability to assemble well-matched cohorts. Similarly, selecting and prioritizing a single outcome measure that would be clinically meaningful across the study population would be impossible. The novel primary endpoint, the Clinical Impression of Change in Severity (CI-CS) accounts for the heterogeneity of the clinical manifestations of these different diseases as well as within the same disease. At screening, the investigator assigns each subject a primary anchor test—either the 8-m walk test or the 9-hole peg test of the dominant hand, based on the subject’s clinical symptoms. The anchor tests are videotaped in a standardized fashion at all study visits and the CI-CS assessment is conducted by an independent, sequence-blinded adjudication committee. By allowing the individual determination of the anchor test, yet maintaining a blinded and independent adjudication of performance, the criteria for efficacy can be both individualized and fairly assessed without bias.
Another example of the use of a master protocol in rare diseases is the HEALEY-ALS trial, testing multiple investigational agents in a perpetual platform design in individuals with ALS (NCT04297683) (Paganoni et al., 2022). The trial was designed in collaboration with numerous stakeholders, including the FDA, to ensure that clinically meaningful efficacy and safety data could provide substantive evidence to guide regulatory reviews of novel therapies. Additional regimens can be added as new investigational agents become available. Subjects who meet entry criteria are randomized to one of the regimens that are active at time of screening. Once randomized, they are then randomized within their cohort, in a 3:1 ratio, to either study drug or placebo. As a consequence of the design and planned statistical approach, the shared control arm allows for a reduction in the sample size per regimen of 33% and a reduction in the number of participants receiving placebo by 66%, compared with what would be required if standard trial designs were used. These elements, as well as the consistency of eligibility criteria, trial execution elements, and outcome assessments, illustrate the many advantages of testing multiple treatments in the same study. Additional considerations, well described in Paganoni et al, include the importance of a robust governance structure, including a Trial Design Committee, an Operations Committee for management of trial logistics and execution, and a Therapy Evaluation Committee for review of candidate therapies for trial inclusion. The organizers also included a Patient Advisory Committee that met initially to ensure the trial design was patient-centric and which continues to meet quarterly to provide important input into trial conduct. Further elements determined prior to study start included details of the operational structure, regulatory communication responsibilities, the candidate drug selection process and the financial structure.
Despite the attractiveness of master protocols and platform studies, it is worthwhile to highlight some of the challenges in these endeavors. Importantly, a substantial capital investment is required up-front as well as sufficient time and key-stakeholder engagement and alignment. Additionally, and particularly in rare diseases, recruitment can take time, during which standards of care may shift and render challenges in ongoing recruitment and/or data interpretation if background or control arm treatments change. Finally, the participation of pharmaceutical sponsors in umbrella or platform trials may require new ways to incentivize such as reducing the regulatory risk of drug development or novel protections of intellectual property that will not deter innovation, but rather encourage it.
6. Novel Regulatory Approaches
The FDA defines gene therapy as a technique that either a) modifies an individual’s genes to treat or cure disease, b) that seeks to modify or manipulate the expression of a gene or, c) to alter the biological properties of living cells for therapeutic use. The FDA has demonstrated strong interested in recent years in advancing cell and gene therapies, particularly in rare diseases. Correlative interest from industry is marked by dramatic increases in applications made to achieve Regenerative Medicines and Advanced Therapies (RMAT) and Breakthrough Designations (BT).
The FDA has recently added a significant number of “Guidance for Industry” position papers to address the complexity of gene-targeted therapy development. In addition to those recently issued (U.S. Department of Health and Human Services, 2020a, 2020b, 2021a, 2021b, 2021d, 2022b), additional guidance documents relevant to GTT development have been announced for 2022 including Small-entity compliance in Human Cells, Tissues, and Cellular and Tissue-Based Products, and Manufacturing Changes and Comparability of Human cells, tissues, and cellular and tissue-based products (HCT/Ps) (U.S. Department of Health and Human Services, 2022a). It is anticipated that PDUFA VII (U.S. Department of Health and Human Services, 2021c) will significantly impact gene-targeted therapy development by providing the FDA with additional resources to accelerate product development and is predicted to support the approval of 10–20 new regenerative medicines by 2025. Current proposed legislation will increase CBER staff with 228 new full-time equivalents vs 32 introduced under PDUFA VI in 2018, with most new FTEs assigned to cell and gene therapies programs. Patient-focused development of gene-targeted therapies will be an important objective including the leveraging of knowledge from public meetings and guidance. Additional pilot programs focused on rare diseases will permit early engagement with the FDA during the development process, product-specific meeting formats and other optimizations of current interactions to facilitate more efficient drug development and risk-management by sponsors.
Today, there are several regulatory designations that Sponsors can pursue in order to access regulatory authority opinions on program development:
Fast Track is intended for serious conditions with unmet medical need and requires non-clinical or clinical data demonstrating the potential to address such needs. Benefits to sponsors include increased number of FDA meetings and eligibility for priority review and rolling review,
Regenerative Medicine Advanced Therapy (RMAT) is similarly intended for serious conditions with unmet medical need. However, it is specific for regenerative therapies, meaning cell or gene therapies. It requires preliminary clinical data to show the potential to address unmet needs. The benefits include intensive guidance on the development program with the commitment of senior FDA staff, and is intended to address potential ways to support accelerated approval and satisfy post-approval requirements
Breakthrough Therapy (BT) requires a program to address a serious condition with preliminary clinical data to show substantial improvement over available therapies on clinically significant endpoints. Importantly, BTD is not limited to regenerative therapies. The benefits are the same as those for RMAT designations with intensive guidance on the proposed development program and senior FDA leadership commitment
It is anticipated that with PDUFA VII, a number of additional pathways for early regulatory authority interaction will be available, specific for the advancement of therapeutics for rare diseases.
Rare Disease Endpoint Advancement (RDEA) Pilot. One of the important challenges in drug development for rare diseases is the discovery and validation of novel surrogate and clinical endpoints. This pilot program will provide a mechanism for sponsors to collaborate with the FDA with up to four additional meetings to specifically consider endpoint development.
Advancing Real World Evidence (RWE) Pilot. This program is designed to evaluate the potential uses of real-world data (RWD) to support product labeling for effectiveness. Up to four additional meetings with sponsors specifically to address RWE utility are part of this program.
Accelerating Rare Disease Cures (ARC) Program. This program’s mission is to provide overall coordination of CDER’s rare disease activities including the development of policy, procedures and training for the review of rare disease therapies,
Offered by the European Medicines Agency (EMA), PRIME designation provides an avenue for frequent and meaningful Health Authority advice and collaboration. This early and proactive support to drug developers is intended to enable study designs that lead to the generation of robust data supportive of the benefits and risks of a new medicine and accelerated assessment of applications for products anticipated to have benefit in individuals with unmet medical needs based on early clinical data. The benefits of PRIME designation include a) the early appointment of a rapporteur from the Committee for Medicinal Products for Human Use (CHMP) or from the Committee on Advanced Therapies (CAT), b) organization of a kick-off meeting with the CHMP/CAT rapporteur and a multidisciplinary group of experts to initiate development guidance and regulatory strategy, c) assignment of a dedicated point-of-contact, and d) continued provision of scientific advice at key milestones in order speed access of patients to promising new medicines. PRIME was launched in 2016 and its first 5-year experience was recently published (European Medicines Agency, 2022). Of the 384 requests for eligibility in those 5 years, the overall acceptance rate was 25% with the majority being products indicated for oncology. Notably, whilst products targeting orphan disease represented 42% of PRIME eligibility requests, 56% were granted the designation, a likely reflection on the unmet medical need in the rare diseases. The benefits of PRIME appeared more pronounced in this first 5-year assessment for more complex products and/or those applications that relied on smaller datasets, such as those in rare diseases. Benefits included a consistent reduction on clock-stop duration, and an average overall shorter active assessment time period; further, products with the PRIME designation were more likely to be granted and to maintain accelerated assessment during their evaluation.
The Innovative Licensing and Access Pathway (ILAP) (Medicines and Healthcare products Regulatory Agency, 2022), another avenue for achieving early health authority engagement and collaboration, was put into place in January 2021 by the Medicines and Healthcare products Regulatory Agency (MHRA) of the United Kingdom. Its goal is to accelerate the time to market and facilitate patient access to medicines, including new chemical and biological entities, new indications and repurposed medicines. Achieving this designation, known as the “Innovation Passport,” provides opportunities for enhanced regulatory and other stakeholder input and triggers a portfolio of activities to create the product-specific Target Development Profile. Permanent partners in the ILAP process including the All Wales Therapeutics and Toxicology Center, the National Institute for Health and Care Excellence (NICE) and the Scottish Medicines Consortium (SMC.) ILAP does not replace the Promising Innovative Medicine (PIM) designation of the Early Access to Medicines Scheme (EAMS), which provides flexibility for earlier patient access toward the end of the development phase in indications of unmet need and where a major benefit over existing therapies can be demonstrated, and applicants can apply to both initiatives.
Conclusions
Despite the global extent and impact of rare diseases, their individual rarity and heterogeneity remain significant challenges to the efficient development of effective treatments using traditional methods. Recent platform-based technologies including ASOs, siRNAs, AAV and lentiviral gene-targeted therapies may offer much more than new hope for many of these devastating diseases; they may lend themselves to a new framework for drug development--and the enabling of wholesale efficiencies in both pre-clinical and clinical testing is envisioned. Academia, consortia, and public-private partnerships are already spearheading efforts to bring greater transparency and sharing of innovations across the biomedical ecosystem. The rapid development of tailored therapies for specific variants underscores the need for parallel development and execution of gNBS to rapidly identify individuals who would benefit. Traditional, one-disease-, or one-drug- at a time approaches continue to dominate the development landscape. However, early efforts to employ master protocols that allow the testing of multiple therapies and/or multiple indications in the same study, can demonstrate a whole host of executional efficiencies. Novel regulatory approaches, designed to enhance early collaboration with patients, researchers, and pharmaceutical partners, are in place and growing, and will foster closer communication and more robust innovation between health authorities and drug developers. These new methodologies are not without challenge—early engagement and collaboration across multiple stakeholders, including patients, researchers, pharmaceutical sponsors and regulators will be required, as well as consideration for pre-competitive interactions, the potential need for incentivization and protections of innovation, and augmentation of data sharing and transparency between all engaged parties. As a consequence, to date, no drug approvals have occurred in the context of these discussed efficiencies. Within the biomedical ecosystem, we as key stakeholders: researchers, pharma sponsors, patients, and health authorities, must enable the better and more frequent use of these modalities and de-risk their application, in terms of cost, regulatory acceptability, and time to approval, in order to fully realize the hope that gene target therapies bring to rare diseases.
Acknowledgments
The authors thank Tiina Urv and Joanne Lumsden for their respective roles in facilitating the development of this manuscript.
Footnotes
Conflicts of Interest
Erika Augustine is a member the organizing committee for N=1 Collaborative. She has served as a DSMB member for PTC Therapeutics. She has received funding from Taysha Gene Therapies and Neurogene Inc. Dwight Koeberl has served as a consultant for Sangamo Therapeutics, Genzyme Sanofi, Amicus, Takeda, AskBio, Moderna, and Vertex. He has also received grant support from Sangamo Therapeutics, Pharming, Viking Therapeutics, Genzyme Sanofi, Roivant Rare Diseases, and Amicus. Dr. Koeberl also has held equity in AskBio, which is developing gene therapy for Pompe disease. Tim Yu serves as a consultant to BioMarin Pharmaceuticals and an advisor to the N=1 collaborative, the Oligo Therapeutics Society, 1M1M Consortium, n-Lorem, Dutch Center for RNA Therapeutics, ACURARE, Wolverine Foundation, and Mila’s Miracle Foundation. Dr. Green has received compensation for advising the following companies: AIA, Allelica, Fabric, Genome Web, Genomic Life, Grail, OptumLabs, Verily, VinBigData; and is co-founder of Genome Medical and Nurture Genomics. Julie Himes is an employee of and holds stock at Takeda. PJ Brooks, Amy Brower, Aaron Goldberg, Jill Morris, and Joe Orsini have no conflicts to report.
Disclaimer
The content of this publication reflects discussions from a June 2021, 3-day workshop sponsored by the National Institutes of Health (NIH) entitled, “Gene-Targeted Therapies: Early Diagnosis and Equitable Delivery” (National Institutes of Health, 2021). This material should not be interpreted as representing the viewpoint of the U.S. Department of Health and Human Services, the National Institutes of Health, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the National Institute of Neurological Disorders and Stroke or the National Center for Advancing Translational Sciences.
Data Availability Statement
The data that support the findings reported herein are openly available at Drugs@FDA https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
REFERENCES
- Abrahamyan L, Feldman BM, Tomlinson G, Faughnan ME, Johnson SR, Diamond IR, & Gupta S (2016). Alternative designs for clinical trials in rare diseases. Am J Med Genet C Semin Med Genet, 172(4), 313–331. 10.1002/ajmg.c.31533 [DOI] [PubMed] [Google Scholar]
- Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Planté-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB (2018). Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 379(1):11–21. 10.1056/NEJMoa1716153 [DOI] [PubMed] [Google Scholar]
- Baby’s First Test. RUSP Conditions By State. Retrieved August 11, 2022 from https://www.babysfirsttest.org/newborn-screening/states
- Baker MW, Mochal ST, Dawe SJ, Wiberley-Bradford AE, Cogley MF, Zeitler BR, Piro ZD, Harmelink MM, & Kwon JM (2022). Newborn screening for spinal muscular atrophy: The Wisconsin first year experience. Neuromuscul Disord, 32(2), 135–141. 10.1016/j.nmd.2021.07.398 [DOI] [PubMed] [Google Scholar]
- Blair HA (2022). Onasemnogene Abeparvovec: A Review in Spinal Muscular Atrophy. CNS Drugs. 36(9):995–1005. 10.1007/s40263-022-00941-1 [DOI] [PubMed] [Google Scholar]
- Blencowe H, Moorthie S, Petrou M, Hamamy H, Povey S, Bittles A, Gibbons S, Darlison M, Modell B, & Congenital Disorders Expert G (2018). Rare single gene disorders: estimating baseline prevalence and outcomes worldwide. J Community Genet, 9(4), 397–406. 10.1007/s12687-018-0376-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DG, & Wobst HJ (2021). A Decade of FDA-Approved Drugs (2010–2019): Trends and Future Directions. J Med Chem, 64(5), 2312–2338. 10.1021/acs.jmedchem.0c01516 [DOI] [PubMed] [Google Scholar]
- Buscara L, Gross DA, & Daniele N (2020). Of rAAV and Men: From Genetic Neuromuscular Disorder Efficacy and Toxicity Preclinical Studies to Clinical Trials and Back. J Pers Med, 10(4). 10.3390/jpm10040258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Wang Y, Du J (2020) Human Papillomavirus Vaccines: An Updated Review. Vaccines (Basel). 16;8(3):391. 10.3390/vaccines8030391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Department of Health and Human Services. (2022). Platform Clinical Trials of Genome Editors in Multiple Diseases (UG3/UH3, Clinical Trial Required). Retrieved August 8, 2022 from https://grants.nih.gov/grants/guide/rfa-files/RFA-RM-22-016.html
- European Medicines Agency. (2022). PRIME: Analysis of the first 5 years’ experience. In.
- Fields T, Patterson M, Bremova-Ertl T, Belcher G, Billington I, Churchill GC, Davis W, Evans W, Flint S, Galione A, Granzer U, Greenfield J, Karl R, Kay R, Lewi D, Mathieson T, Meyer T, Pangonis D, Platt FM, . . . Strupp M (2021). A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia. Trials, 22(1), 84. 10.1186/s13063-020-05009-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnier SM, Durkin MS, & Baker MW (2020). Translating Molecular Technologies into Routine Newborn Screening Practice. Int J Neonatal Screen, 6(4). 10.3390/ijns6040080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearin C (2021). Approvals Expand Multiple Myeloma Treatment Options. Cancer Discov. 11(6):OF5. 10.1158/2159-8290.CD-NB2021-0338 [DOI] [PubMed] [Google Scholar]
- Gillmore JD, Maitland ML, Lebwohl D (2021). CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. Reply. N Engl J Med. 385(18):1722–1723. 10.1056/NEJMc2114592. [DOI] [PubMed] [Google Scholar]
- Gottlieb S (2019). Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics
- Hoy SM (2017). Nusinersen: First Global Approval. Drugs. 77(4):473–479. 10.1007/s40265-017-0711-7 [DOI] [PubMed] [Google Scholar]
- IQVIA Institute for Human Data Science. (2020). Orphan Drugs in the United States: Rare Disease Innovation and Cost Trends Through 2019. https://www.iqvia.com/insights/the-iqvia-institute/reports/orphan-drugs-in-the-united-states-rare-disease-innovation-and-cost-trends-through-2019
- Janiaud P, Serghiou S, & Ioannidis JPA (2019). New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat Rev, 73, 20–30. 10.1016/j.ctrv.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Jiang L, Park JS, Yin L, Laureano R, Jacquinet E, Yang J, Liang S, Frassetto A, Zhuo J, Yan X, Zhu X, Fortucci S, Hoar K, Mihai C, Tunkey C, Presnyak V, Benenato KE, Lukacs CM, Martini PGV, Guey LT (2020). Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat Commun. 11(1):5339. 10.1038/s41467-020-19156-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay DM, Stevens CF, Parker A, Saavedra-Matiz CA, Sack V, Chung WK, Chiriboga CA, Engelstad K, Laureta E, Farooq O, Ciafaloni E, Lee BH, Malek S, Treidler S, Anziska Y, Delfiner L, Sakonju A, & Caggana M (2020). Implementation of population-based newborn screening reveals low incidence of spinal muscular atrophy. Genet Med, 22(8), 1296–1302. 10.1038/s41436-020-0824-3 [DOI] [PubMed] [Google Scholar]
- Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, Pendergast MK, Goldkind SF, Lee EA, Kuniholm A, Soucy A, Vaze J, Belur NR, Fredriksen K, Stojkovska I, Tsytsykova A, Armant M, DiDonato RL, Choi J, . . . Yu TW (2019). Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N Engl J Med, 381(17), 1644–1652. 10.1056/NEJMoa1813279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, & Shneider NA (2022). Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med, 28(1), 104–116. 10.1038/s41591-021-01615-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le BT, Paul S, Jastrzebska K, Langer H, Caruthers MH, Veedu RN (2022). Thiomorpholino oligonucleotides as a robust class of next generation platforms for alternate mRNA splicing. Proc Natl Acad Sci U S A. 119(36):e2207956119. 10.1073/pnas.2207956119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechler K, Mountford WK, Hoffmann GF, & Ries M (2015). Pressure for drug development in lysosomal storage disorders - a quantitative analysis thirty years beyond the US orphan drug act. Orphanet J Rare Dis, 10, 46. 10.1186/s13023-015-0262-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency. (2022). Guidance: Innovative Licensing and Access Pathway. Retrieved from https://www.gov.uk/guidance/innovative-licensing-and-access-pathway#eligibility-through-the-innovation-passport
- Mendell JR, Al-Zaidy SA, Lehman KJ, McColly M, Lowes LP, Alfano LN, Reash NF, Iammarino MA, Church KR, Kleyn A, Meriggioli MN, & Shell R (2021). Five-Year Extension Results of the Phase 1 START Trial of Onasemnogene Abeparvovec in Spinal Muscular Atrophy. JAMA Neurol, 78(7), 834–841. 10.1001/jamaneurol.2021.1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, Ragni MV, Manno CS, Sommer J, Jiang H, Pierce GF, Ertl HC, & High KA (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med, 13(4), 419–422. 10.1038/nm1549 [DOI] [PubMed] [Google Scholar]
- n-Lorem Foundation. Retrieved August 10, 2022 from https://www.nlorem.org/
- n-Lorem Foundation. (2022). Second Anniversary for n-Lorem Foundation Marks Extraordinary Progress Toward Treating Nano-Rare Patients https://www.nlorem.org/second-anniversary-for-n-lorem-foundation-marks-extraordinary-progress-toward-treating-nano-rare-patients/
- N=1 Collaborative. Retrieved July 26, 2022 from https://www.n1collaborative.org/
- National Institutes of Health. Bespoke Gene Therapy Consortium. Retrieved August 8, 2022 from https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/bespoke-gene-therapy-consortium
- National Institutes of Health. Somatic Cell Genome Editing. Retrieved Augist 8, 2022 from https://commonfund.nih.gov/editing
- National Institutes of Health. (2022). Somatic Cell Genome Editing Funding Opportunities. Retrieved August 8, 2022 from https://commonfund.nih.gov/editing/fundingopportunities
- Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, Murphy D, Le Cam Y, & Rath A (2020). Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet, 28(2), 165–173. 10.1038/s41431-019-0508-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M, Kohn DB, Bartlett J, Benson J, Brooks PJ, Byrne BJ, Camozzi C, Cornetta K, Crystal RG, Fong Y, Gargiulo L, Gopal-Srivastava R, High KA, Jacobson SG, Jambou RC, Montgomery M, Rosenthal E, Samulski RJ, Skarlatos SI, . . . Corrigan-Curay J (2013). Gene therapy for rare diseases: summary of a National Institutes of Health workshop, September 13, 2012. Hum Gene Ther, 24(4), 355–362. 10.1089/hum.2013.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganoni S, Berry JD, Quintana M, Macklin E, Saville BR, Detry MA, Chase M, Sherman AV, Yu H, Drake K, Andrews J, Shefner J, Chibnik LB, Vestrucci M, Cudkowicz ME, & Healey ALSPTSG (2022). Adaptive Platform Trials to Transform Amyotrophic Lateral Sclerosis Therapy Development. Ann Neurol, 91(2), 165–175. 10.1002/ana.26285 [DOI] [PubMed] [Google Scholar]
- Park JJH, Siden E, Zoratti MJ, Dron L, Harari O, Singer J, Lester RT, Thorlund K, & Mills EJ (2019). Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials, 20(1), 572. 10.1186/s13063-019-3664-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recommendations for investigations of drugs for rare diseases or conditions, C.F.R. § 360aa (2011). https://www.govinfo.gov/app/details/USCODE-2011-title21/USCODE-2011-title21-chap9-subchapV-partB-sec360aa
- Renfro LA, & Sargent DJ (2017). Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol, 28(1), 34–43. 10.1093/annonc/mdw413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzitti R, Gross DA, Mingozzi F (2020). Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front Immunol, 11:670. 10.3389/fimmu.2020.00670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha K, Sontheimer EJ, Brooks PJ, Dwinell MR, Gersbach CA, Liu DR, Murray SA, Tsai SQ, Wilson RC, Anderson DG, Asokan A, Banfield JF, Bankiewicz KS, Bao G, Bulte JWM, Bursac N, Campbell JM, Carlson DF, Chaikof EL, Chen ZY, Cheng RH, Clark KJ, Curiel DT, Dahlman JE, Deverman BE, Dickinson ME, Doudna JA, Ekker SC, Emborg ME, Feng G, Freedman BS, Gamm DM, Gao G, Ghiran IC, Glazer PM, Gong S, Heaney JD, Hennebold JD, Hinson JT, Khvorova A, Kiani S, Lagor WR, Lam KS, Leong KW, Levine JE, Lewis JA, Lutz CM, Ly DH, Maragh S, McCray PB Jr., McDevitt TC, Mirochnitchenko O, Morizane R, Murthy N, Prather RS, Ronald JA, Roy S, Roy S, Sabbisetti V, Saltzman WM, Santangelo PJ, Segal DJ, Shimoyama M, Skala MC, Tarantal AF, Tilton JC, Truskey GA, Vandsburger M, Watts JK, Wells KD, Wolfe SA, Xu Q, Xue W, Yi G, Zhou J; SCGE Consortium. (2021). The NIH Somatic Cell Genome Editing program. Nature. 592(7853):195–204. 10.1038/s41586-021-03191-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schor NF, Tamiz AP, Koroshetz WJ, Group N. U.-R. G.-b. T. W., & Broome AM (2021). NINDS launches network to develop treatments for ultra-rare neurological diseases. Nat Biotechnol, 39(12), 1497–1499. 10.1038/s41587-021-01125-w [DOI] [PubMed] [Google Scholar]
- Synofzik M, van Roon-Mom WMC, Marckmann G, van Duyvenvoorde HA, Graessner H, Schule R, & Aartsma-Rus A (2022). Preparing n-of-1 Antisense Oligonucleotide Treatments for Rare Neurological Diseases in Europe: Genetic, Regulatory, and Ethical Perspectives. Nucleic Acid Ther, 32(2), 83–94. 10.1089/nat.2021.0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn CR, Sharma D, Combs R, Bhujbal S, Romine J, Zheng X, Sunasara K, Badkar A (2022). The journey of a lifetime - development of Pfizer’s COVID-19 vaccine. Curr Opin Biotechnol. 78:102803. 10.1016/j.copbio.2022.102803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, Metterville J, Shin M, Sanil N, Dooley C, Puri A, Weiss A, Wightman N, Gray-Edwards H, Marosfoi M, King RM, Kenderdine T, Fabris D, Bowser R, . . . Brown RH Jr. (2022). Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med, 28(1), 117–124. 10.1038/s41591-021-01557-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services. Orphan Drug Designations and Approvals. Retrieved August 10, 2022 from https://www.accessdata.fda.gov/scripts/opdlisting/oopd/
- U.S. Department of Health and Human Services. PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2023 Though 2027. Retrieved from https://www.fda.gov/media/151712/download
- U.S. Department of Health and Human Services. (2020a). Human Gene Therapy for Rare Diseases: Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-rare-diseases
- U.S. Department of Health and Human Services. (2020b). Long Term Follow-up After Administration of Human Gene Therapy Products: Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products
- U.S. Department of Health and Human Services. (2021a). Human Gene Therapy for Neurodegenerative Diseases: Draft Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-neurodegenerative-diseases
- U.S. Department of Health and Human Services. (2021b). Interpreting Sameness of Gene Therapy Products Under the Orphan Drug Regulations: Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/interpreting-sameness-gene-therapy-products-under-orphan-drug-regulations
- U.S. Department of Health and Human Services. (2021c). PDUFA VII: Fiscal Years 2023 – 2027. Retrieved August 8, 2022 from https://www.fda.gov/industry/prescription-drug-user-fee-amendments/pdufa-vii-fiscal-years-2023-2027
- U.S. Department of Health and Human Services. (2021d). Studying Multiple Versions of a Cellular or Gene Therapy Product in an Early-Phase Clinical Trial: Draft Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/studying-multiple-versions-cellular-or-gene-therapy-product-early-phase-clinical-trial
- U.S. Department of Health and Human Services. (2022a). Guidance Agenda: Guidance Documents CBER is Planning to Publish During Calendar Year 2022. Retrieved from https://www.fda.gov/vaccines-blood-biologics/biologics-guidances/guidance-agenda-guidance-documents-cber-planning-publish-during-calendar-year-2022
- U.S. Department of Health and Human Services. (2022b). Human Gene Therapy Products Incorporating Human Genome Editing: Draft Guidance for Industry. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-products-incorporating-human-genome-editing
- Verdera HC, Kuranda K, Mingozzi F (2020). AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol Ther, 28(3):723–746. 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley J, Defay T, Goldenberg AJ, Gaviglio AM (2022). Scaling genetic resources: New paradigms for diagnosis and treatment of rare genetic disease. Am J Med Genet C Semin Med Genet. 10.1002/ajmg.c.32016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley J, Aartsma-Rus A, Cohen JL, Cowsert LM, Howell RR, Yu TW, Wasserstein MP, Defay T (2022). Whole-genome sequencing holds the key to the success of gene-targeted therapies. Am J Med Genet C Semin Med Genet. 10.1002/ajmg.c.32017 [DOI] [PubMed] [Google Scholar]
- Woodcock J, & LaVange LM (2017). Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med, 377(1), 62–70. 10.1056/NEJMra1510062 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings reported herein are openly available at Drugs@FDA https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.