

Abstract
Studies have shown that Nrf2E79Q/+ is one of the most common mutations found in human tumors. To elucidate how this genetic change contributes to lung cancer, we compared lung tumor development in a genetically-engineered mouse model (GEMM) with dual Trp53/p16 loss, the most common mutations found in human lung tumors, in the presence or absence of Nrf2E79Q/+. Trp53/p16-deficient mice developed combined-small cell lung cancer (C-SCLC), a mixture of pure-SCLC (P-SCLC) and large cell neuroendocrine carcinoma. Mice possessing the LSL-Nrf2E79Q mutation showed no difference in the incidence or latency of C-SCLC compared with Nrf2+/+ mice. However, these tumors did not express NRF2 despite Cre-induced recombination of the LSL-Nrf2E79Q allele. Trp53/p16-deficient mice also developed P-SCLC, where activation of the NRF2E79Q mutation associated with a higher incidence of this tumor type. All C-SCLCs and P-SCLCs were positive for NE-markers, NKX1–2 (a lung cancer marker) and negative for P63 (a squamous cell marker), while only P-SCLC expressed NRF2 by immunohistochemistry. Analysis of a consensus NRF2 pathway signature in human NE+-lung tumors showed variable activation of NRF2 signaling. Our study characterizes the first GEMM that develops C-SCLC, a poorly-studied human cancer and implicates a role for NRF2 activation in SCLC development.
Keywords: Lung cancer, KEAP1/NRF2, neuroendocrine tumors, small cell lung carcinoma
Introduction
Lung cancer remains one of the deadliest cancers worldwide with an estimated 135,720 deaths in the United States in 2020 [1]. Lung cancer is divided into two histological subtypes, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for about 80% of all lung cancers which is further subdivided into lung adenocarcinoma (LUAD: 40%), lung squamous cell carcinoma (LUSC: 25%) and large cell carcinoma (LCC: 15%, including large cell neuroendocrine carcinoma, LCNEC). The remaining 20% of lung cancers represents SCLC that differs from LUAD and LUSC due to the expression of markers of neuroendocrine differentiation.
The 5-year survival rate in lung cancer rose from 17.2% in 2009 to 21.7% in 2019 [1–3], driven primarily by the development of therapies for LUAD targeting mutations in EGFR, ALK, and ROS1 [4–8]. Unfortunately, these therapies are not effective against the most aggressive types of lung cancer, including LUSC and SCLC, due to different genetic alterations [9–13]. Specifically, LUSC frequently results from inactivating mutations in CDKN2A (P16) and TRP53 along with activation of PIK3CA, P63 and/or SOX2 signaling [14–17]. In SCLC, RB1 and TRP53 inactivating mutations predominate [13, 18].
While dual TRP53/P16 loss is found in almost all types of lung cancer [9, 10, 13, 19–21], how the concurrent inactivation of these genes contributes to their development is not fully understood. Additionally, other frequent mutations, such as those recently reported to activate the NRF2 pathway, remain understudied [22]. The NRF2 (also known as nuclear factor erythroid-derived 2-like 2; NFE2L2) signaling pathway provides an important cellular defense mechanism against oxidative and metabolic stress [23]. In addition to maintaining redox homeostasis, the NRF2 pathway also regulates many other cancer hallmarks including proliferation, differentiation, apoptosis, inflammation and metastasis [24–26]. Patients with lung tumors with activating NRF2 mutations show significantly worse survival than patients with no NRF2 alterations [22, 27]. Most NRF2 mutations localize to hotspots in the DLG and ETGE motifs [22], which impair KEAP1 binding, ubiquitylation and subsequent proteasomal degradation [23], yielding constitutively-active NRF2-dependent transcription [28].
Constitutive NRF2-driven transcription in human lung cancer presumably drives tumor growth and progression under conditions of oxidative, metabolic and genotoxic stress. Thus, mutations in the KEAP1/NRF2/CUL3 complex that constitutively activate this pathway appear in most lung tumor types, including LUAD, LUSC, and LCNEC [22, 29, 30] However, only limited data exist on the frequency of KEAP1-NRF2 alterations in SCLC [13, 31]. How activating mutations, such as Nrf2E79Q/+, drive the development of various histological tumor types of lung cancer remains unclear. Therefore, we sought to determine how NRF2 signaling pathway contributes to development of NSCLC and SCLC tumors.
To understand the impact of activating mutations in NRF2 on lung tumor development driven by biallelic inactivation of the Trp53 and p16 genes, we generated and characterized lung tumors in a novel genetically-engineered mouse model (GEMM) with Trp53fl/fl;p16fl/fl with or without a common NRF2 mutation in human tumors, Nrf2E79Q [32]. We found that the dual loss in Trp53/p16 resulted in the appearance of combined-small cell lung cancer (C-SCLC), a mixture of pure-SCLC (P-SCLC) and large cell neuroendocrine carcinoma (LCNEC). C-SCLC incidence and latency were similar in mice bearing the LSL-Nrf2E79Q allele. However, the C-SCLCs developed in LSL-Nrf2E79Q mice did not express NRF2 protein, despite Cre-mediated recombination of the mutant allele. These mice also developed P-SCLC where activation of NRF2 signaling increased the incidence and decreased the survival of the mice. Thus, our study shows that the dual loss of Trp53 and p16 in mice drives the development of small P-SCLC and large, aggressive C-SCLC. Furthermore, persistent activation of NRF2 increased the incidence of P-SCLC but appeared to inhibit the appearance of large, invasive SCLC. Thus, the role of activated NRF2 signaling in lung tumor development appears dependent upon the cellular context.
Results
To dissect the role of activated NRF2 signaling in lung tumor development, we used a Trp53fl/fl;p16fl/fl genetically engineered mouse model (GEMM), as these tumor suppressor genes are inactivated in >80% of human LUAD and LUSC tumors [13, 20, 21]. We focused on dissecting the effect on tumor phenotype of a common activating mutation in the NRF2 gene, E79Q, in our Trp53fl/fl;p16fl/fl GEMM. We bred these mice with our recently reported lox-stop-lox (LSL)-Nrf2E79Q GEMM possessing a Cre-inducible Nrf2 gene harboring an activating E79Q mutation within the endogenous locus [33]. We bred two groups of mice for our study: Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;LSL-Nrf2E79Q/+. Intranasal instillation of Adenoviral-Cre (Ad5-CMV-Cre) was used to inactivate the Trp53/p16 mutant alleles in both groups and activate the Nrf2E79Q allele in the Trp53fl/fl;p16fl/fl;LSL-Nrf2E79Q/+ mice. We then monitored mice for tumor development for up to 60 weeks after treatment. We did not include an experimental group with homozygous Nrf2E79Q/E79Q alleles because these mice did not express NRF2.
Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;LSL-Nrf2E79Q/+ mice develop combined-small cell lung cancer (C-SCLC)
A previous report showed that Trp53fl/fl;p16fl/fl GEMMs developed lung adenocarcinomas at a low frequency after intranasal Ad-CMV-Cre instillation but did not further characterize these tumors [34]. In our GEMMs, both Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice and Trp53fl/fl;p16fl/fl;Nrf2+/+ mice developed large tumors, most frequently in the lung and less commonly in the brain. We also found metastatic disease in several tissues (Table 1, Supplementary Table 1a, b). We did not observe a significant change in survival after treatment with Ad-CMV-Cre between the Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ and Trp53fl/fl;p16fl/fl;Nrf2+/+ groups, with most mice developing health issues by 45 weeks (Supplementary Fig. 1a). Unexpectedly, the histological analysis of invasive tumors in the lungs of both genotypes revealed combined-small cell lung cancer (C-SCLC) (Fig. 1a) [35], based on the 2015 World Health Organization (WHO) classification of human lung tumors. Specifically, we observed a mixed cell population with histological features of P-SCLC: small cells, nuclei with finely granular nuclear chromatin, inconspicuous nucleoli, high mitotic rate, and frequent necrosis, and LCC: large cell size, low nuclear to cytoplasmic ratio, vesicular, coarse or fine chromatin, and/or frequent prominent nucleoli (Fig. 1a). There was no significant difference in C-SCLC incidence between the two groups (Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ 22% versus Trp53fl/fl;p16fl/fl;Nrf2+/+ 18%; Table 1).
Table 1.
Tumors developed by the Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice
| Genotype | Site of tumor | Tumor type | Percent from total, n (%)* |
|---|---|---|---|
| Trp53fl/fl;p16fl/fl;Nrf2+/+ | Lung | C-SCLC | 7 (18) |
| (n=38) | P-SCLC | 5 (13) | |
| Other | 3 (8) | ||
| Brain | ONB | 11 (29) | |
| Liver | Mets from lung | 1 (3) | |
| Other | 2 (5) | ||
| Other ** | SCC, lymph node tumors | 7 (18) | |
| Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ | Lung | C-SCLC | 8 (22) |
| (n=37) | P-SCLC | 12 (32) | |
| Other | 2 (5) | ||
| Brain | ONB | 11 (30) | |
| Liver | Mets from lung | 4 (11) | |
| Lymphoma | 1 (3) | ||
| Other | 1 (3) | ||
| Other ** | SCC, lymph node tumors | 8 (22) |
Some mice had more than one tumor
histological type of other tumors detailed in the supporting information Table 1; C-SCLC: Combined Small Cell Lung Cancer; P-SCLC: Pure Small Cell Lung Cancer; ONB: Olfactory Bulb Neuroblastoma; SCC: Squamous cell carcinoma
Fig. 1. Invasive tumors developed by Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice.
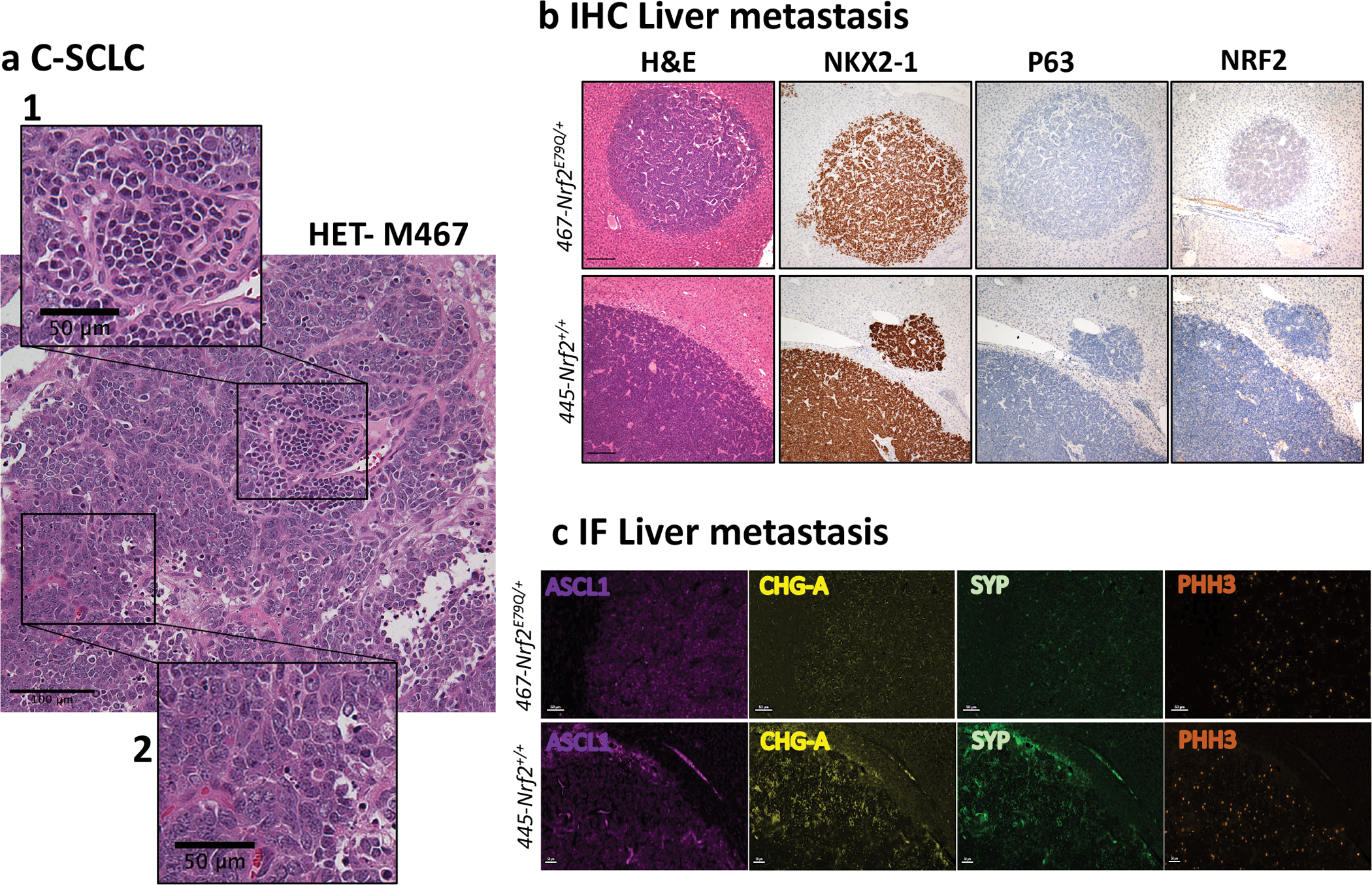
a An example of combined small cell lung cancer (C-SCLC) from a Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mouse, showing C-SCLC, scalebar=100μm with insets of 1) P-SCLC, scalebar=50μm, and 2) Large Cell Neuroendocrine Carcinoma (LCNEC), scalebar=50μm; b H&E and representative IHC for NKX2–1, P63 and NRF2 of a lung tumor metastasis in liver, scalebar=200μm. Images in a&b were taken using a BX61-Neville microscope; c Representative IF for ASCL1, CHG-A, SYP and PHH3 of a lung tumor metastasis in liver. Slides were imaged on the Vectra® Polaris Automated Quantitative Pathology Imaging System, scalebar=50μm.
Beyond the lung, we found lung tumor metastases in the parathyroid gland and liver, as well as poorly differentiated tumors of unknown origin (Table 1, Supplementary Table 1a, b and Fig. 1b,c). We also identified olfactory bulb neuroblastomas (esthesioneuroblastoma) (ONB) in the brain, a documented phenomenon in mice administered agents through the nasal cavity [36, 37] (Supplementary Fig. 1c), and squamous cell carcinoma (SCC) on the ears (Table 1, Supplementary Fig. 1d) [38–40]. The SCCs on the ears presumably developed from irritation caused by the metal ear tags used to identify individual mice [41]. No significant differences in the incidences of any of these tumors were observed between the two groups (Supplementary Fig.1b).
Expression of Nrf2E79Q/+ in the Trp53fl/fl;p16fl/fl mice increased frequency of pure-small cell lung cancer (P-SCLC)
In addition to C-SCLC, the lungs from these mice also contained discrete tumors arising mostly at the bronchial epithelium and projecting into the lumen in both Trp53fl/fl;p16fl/fl;Nrf2E79Q/+and Trp53fl/fl;p16fl/fl;Nrf2+/+ mice (Fig. 2a). Histological analysis showed that almost all tumors contained small, round to elongated cells with dark nuclei composed of coarse, condensed chromatin and no distinct nucleoli, resembling P-SCLC [42]. The incidence of these P-SCLC-like tumors, as identified by H&E section, was significantly higher in Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice (32%; n=9) compared to Trp53fl/fl;p16fl/fl;Nrf2+/+ mice (13%; n=9) (p<0.01; Table 1, Fig. 2b). To better determine the frequency of these tumors, we serially sectioned through the entire lung from an equal number of Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice (n=9) using lungs without previously detected lesions. We observed that eight out of nine (8/9) lungs of Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice contained P-SCLC compared to four out of nine (4/9) lungs of Trp53fl/fl;p16fl/fl;Nrf2+/+ mice (Fig. 2c, Supplementary Table 2). The total number of P-SCLCs observed in Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice, as well as the average number of tumors per lung, was significantly (p<0.01) higher than in the Trp53fl/fl;p16fl/fl;Nrf2+/+ mice (Fig 2b,c, Supplementary Table 2). Importantly, the Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice with P-SCLCs had significantly reduced survival compared to Trp53fl/fl;p16fl/fl;Nrf2+/+ mice with P-SCLCs, 24 vs. 33 weeks (p<0.05), eliminating the possibility that Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice developed more P-SCLCs due to longer survival times.
Fig. 2. Characterization of P-SCLC developed by Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice.
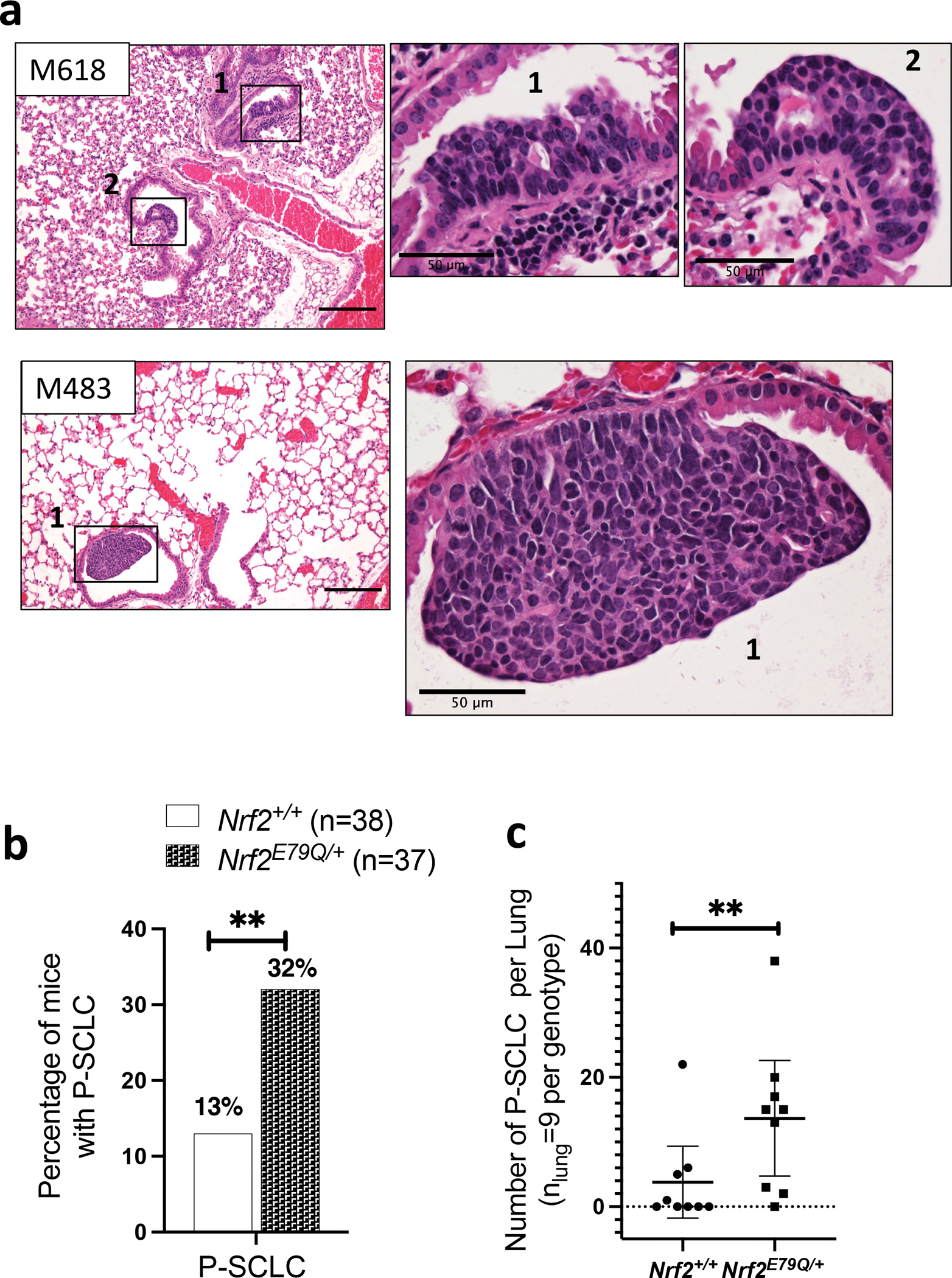
a H&E of multiple P-SCLCs in Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice, scalebar=200μm with insets showing P-SCLC lesions, scalebar=50μm. Images were taken using a BX61-Neville microscope; b Percentage of mice with P-SCLC in Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice; c Total number of P-SCLC observed in each lung from Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice (n = 9 per genotype) (See Supplementary Table 2 for more details). **P<0.01
C-SCLCs and P-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice express NKX2–1 and neuroendocrine markers
To characterize the histology of C-SCLCs we labeled them for NKX2–1 (also known as TTF1), a marker for lung tumors, and P63 (TRP63), an important factor for the development of epithelial tissues and generally considered a positive marker for human LUSC [43–45]. All C-SCLCs stained strongly positive for NKX2–1 and negative for P63 by IHC, using the scoring criteria presented in Supplementary Table 3 (Fig. 3a,b, Supplementary Tables 3 and 4a). We observed similar labeling for the lung tumor metastases in the liver from both genotypes (Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice), consistent with an origin as lung C-SCLC metastases (Fig. 1b). We also assessed expression of these markers in the SCC from the ears of Nrf2+/+ and Nrf2E79Q/+ mice. While the Nrf2E79Q/+ SCC labeled positive for P63 and NRF2, the Nrf2+/+ SCC labeled positive for P63 only; both ear tumors lacked expression of NKX2–1, as expected for a squamous tumor (Supplementary Fig. 1d). We also labeled the C-SCLCs for TRP53, and for phospho-RB1 to examine the effect of P16 loss on RB1 expression [46]. All C-SCLCs were negative for TRP53 by IHC, confirming inactivation of the Trp53 gene and positive for phospho-RB1 by IHC, consistent with p16 inactivation [46] (Fig. 3a,b, Supplementary Table 4a).
Fig. 3. Characterization and NRF2 genotyping of C-SCLC.
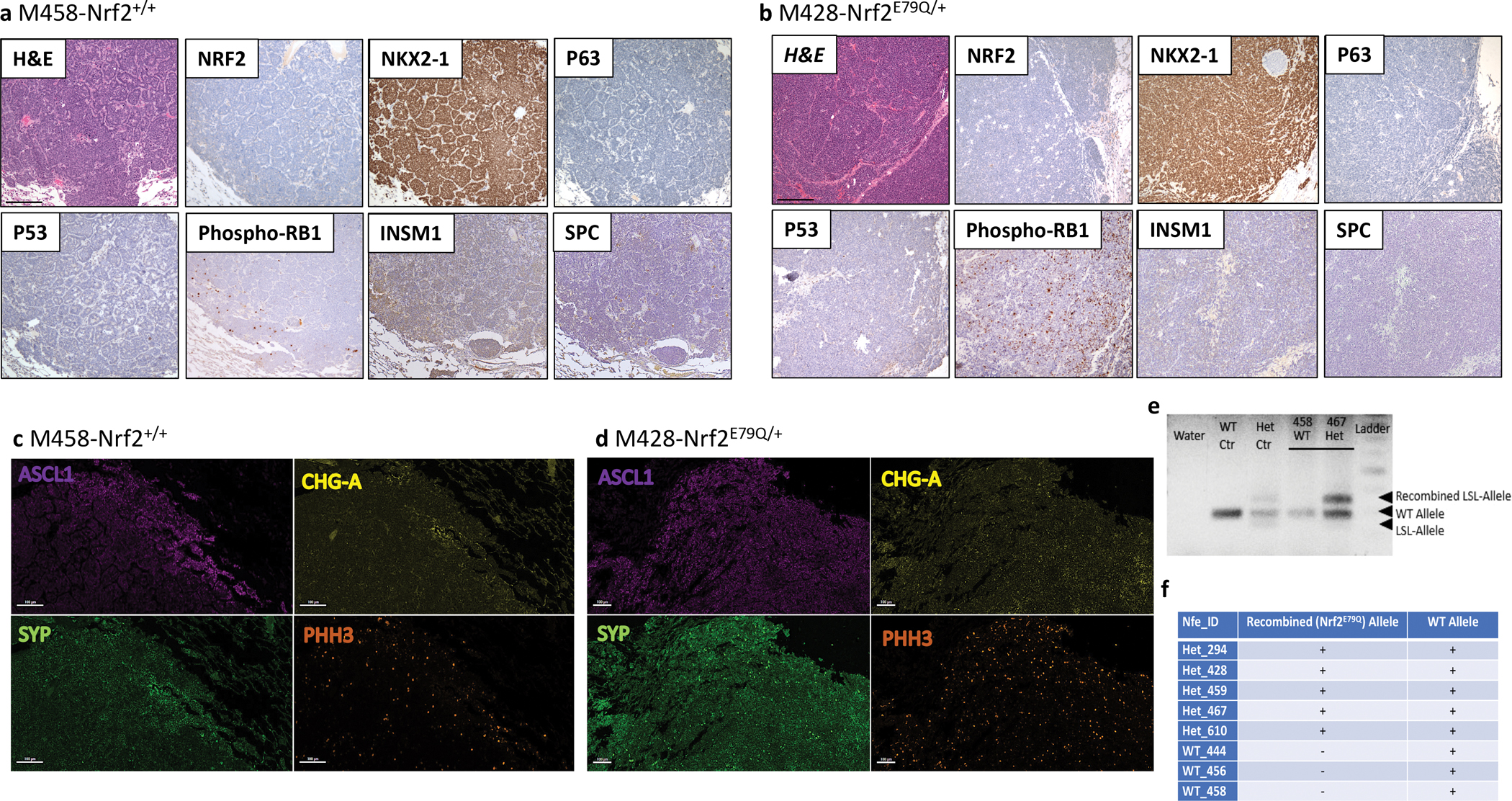
IHC for markers of lung differentiation and for tumor suppressor and NRF2 expression in C-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2+/+ (a) and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+mice (b), scalebar=200μm; IF staining for NE markers of the same tumors (c & d), scalebar=100μm; e Genotyping results of C-SCLC from (WT) Trp53fl/fl;p16fl/fl;Nrf2+/+ and (Het) Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice, showing the presence of the recombined allele in tumors from Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice; f Table summarizing the results of genotyping C-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice; a & b Images were taken using a BX61-Neville microscope; c & d Slides were imaged on the Vectra® Polaris Automated Quantitative Pathology Imaging System.
To further characterize the histology of the C-SCLCs, we labeled them for SPC (alveolar type II cell marker) and the neuroendocrine marker INSM1 by IHC. All C-SCLCs from both genotypes labeled negative for SPC and weakly to strongly positive for INSM1 (Fig. 3a,b, Supplementary Table 4a). These results were consistent with these tumors possessing both SCLC and/or LCNEC components but not LUAD. To further confirm the neuroendocrine feature of C-SCLCs, we labeled these tumors for SCLC-neuroendocrine markers including ASCL1, chromogranin-A (CHG-A) and synaptophysin (SYP) using immunofluorescence (IF). All C-SCLCs labeled positive for these markers (Fig. 3c,d, Supplementary Table 4b). Additionally, the C-SCLCs from both genotypes showed strong labeling for phospho-Histone-H3 (PHH3), a marker of mitotic cells (Fig. 3c,d, Supplementary Table 4b). Tumors in the liver from both genotypes (Nrf2+/+ and Nrf2E79Q/+ mice) had the same pattern of IF-staining, supporting their origin from metastatic C-SCLC (Fig. 1c). Similar to C-SCLCs, all P-SCLCs from both genotypes showed positive staining for NKX2–1 and phospho-RB1 and were negative for P63 by IHC (Fig. 4a, Supplementary Table 5a). These tumors labeled positive for the SCLC NE-marker, CGRP by IHC (Fig. 4a, Supplementary Table 5a). Further, the IF labeling of P-SCLC for ASCL1, SYP, and PHH3 was also positive in both genotypes (Fig. 4b and Supplementary Table 5b).
Fig. 4. Characterization of P-SCLC developed by Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice.
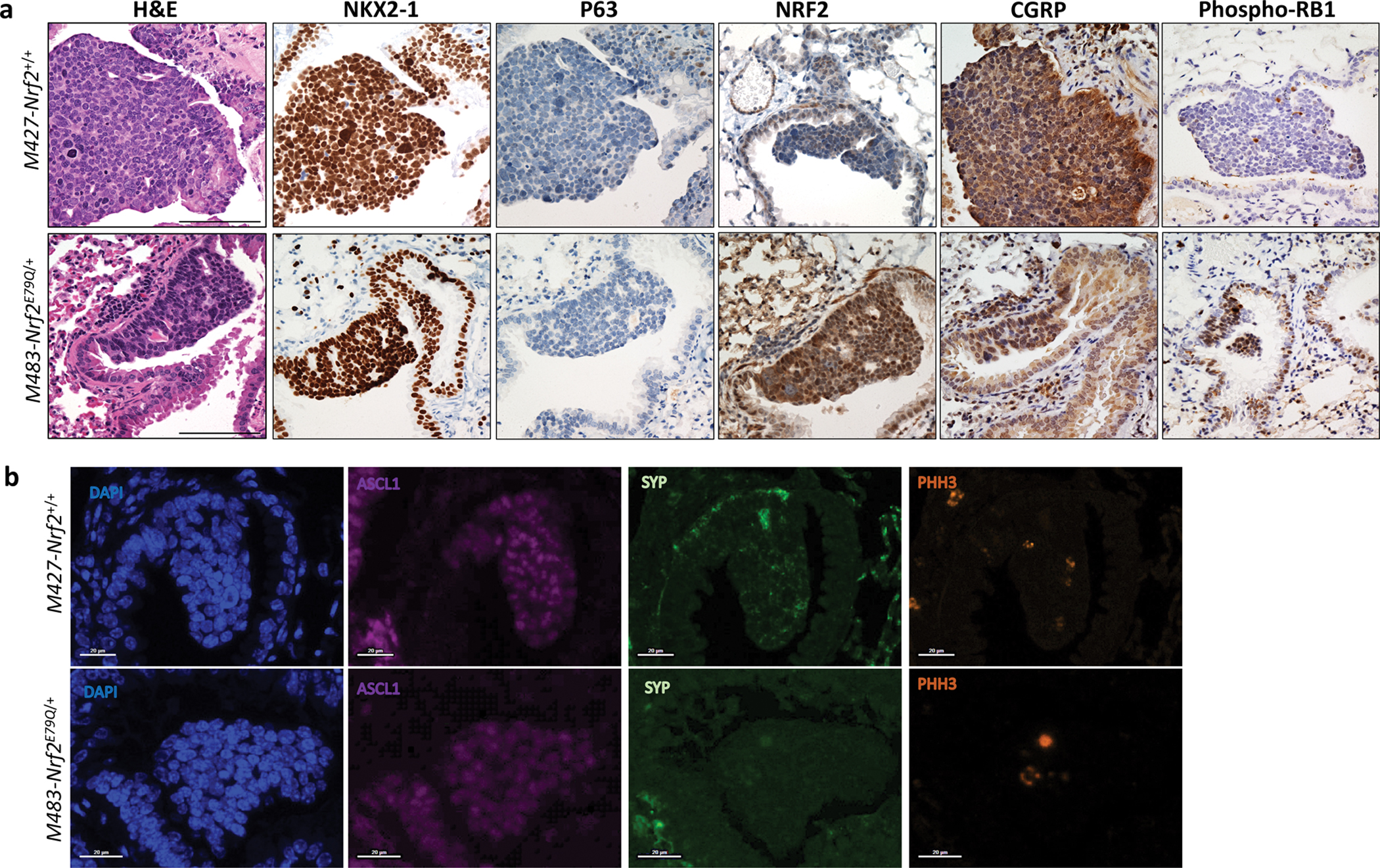
a IHC for markers of lung differentiation and for tumor suppressor and NRF2 expression in P-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+mice,. Images were taken using a BX61-Neville microscope, scalebar=200μm; b IF staining for NE markers of the same tumors. Slides were imaged on the Vectra® Polaris Automated Quantitative Pathology Imaging System, scalebar=100μm.
P-SCLCs but not C-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice express NRF2
We used immunohistochemistry (IHC) to assess NRF2 expression in the C-SCLCs and P-SCLCs from the Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice. All tissues were scored for NRF2 expression based on the criteria presented in Supplementary Table 3. Five Nrf2E79Q/+ P-SCLCs labeled positive for NRF2 while two other P-SCLCs showed weak staining in <5% of the cells (Fig. 4a, Supplementary Table 5a). The P-SCLC from the lung of a Trp53fl/fl;p16fl/fl;Nrf2+/+ mouse did not label positive for NRF2 by IHC (Fig. 4a, Supplementary Table 5a). Surprisingly, all C-SCLCs, liver metastases, and brain olfactory neuroblastomas from Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice were negative for NRF2 (Fig. 1b, Fig. 3a,b, Supplementary Fig. 1c and Supplementary Table 4a). In contrast, the SCC from the ear of Nrf2+/+ mouse was negative for NRF2, while the SCC from the ear of Nrf2E79Q/+ mouse was NRF2 positive (Supplementary Fig. 1d).
The absence of NRF2 expression in the Nrf2E79Q/+ C-SCLCs could result from a lack of activation of the mutant Nrf2 allele. To test this possibility, we performed genotyping of the tumors using DNA extracted from the paraffin embedded tissue using a pinpoint DNA isolation system [47]. We had sufficient tumor material to isolate DNA from three invasive lung tumors from Trp53fl/fl;p16fl/fl;Nrf2+/+ mice and five from Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice. As expected, the Nrf2+/+ tumors showed only the Nrf2+ wild type (WT) allele (Fig. 3e,f). Surprisingly, the Nrf2E79Q/+ tumors displayed the WT allele and the recombined mutant (Het) Nrf2E79Q allele, suggesting that the tumor cells should express elevated levels of the mutant NRF2 protein (Fig. 3e,f).
Activated NRF2 signaling in human lung neuroendocrine tumors
In order to determine the relevance of our findings to human neuroendocrine-positive lung tumors, we analyzed previously published gene expression, histopathology, and tumor mutation data in human carcinoids, SCLC, and LCNEC to assess NRF2 signaling. Neither carcinoids nor SCLCs harbored frequent NRF2 activating mutations or KEAP1 inactivating mutations, whereas LCNECs fell into two subclasses, one with frequent KEAP1/NRF2 mutations and one without (Fig. 5). Using a signature for activated NRF2 signaling based upon well-characterized NRF2 target genes [48, 49], we observed a subset of LCNEC tumors (22/69=32%) that had elevated target gene expression suggestive of NRF2 signaling activation. These NRF2-active tumors were enriched for mutations in KEAP1/NRF2 (13/22=59%). The expression of NRF2 target genes in carcinoid and SCLC was variable and independent of histopathology and molecular class.
Fig. 5. Heatmap of NRF2 target gene expression in human neuroendocrine lung tumors.
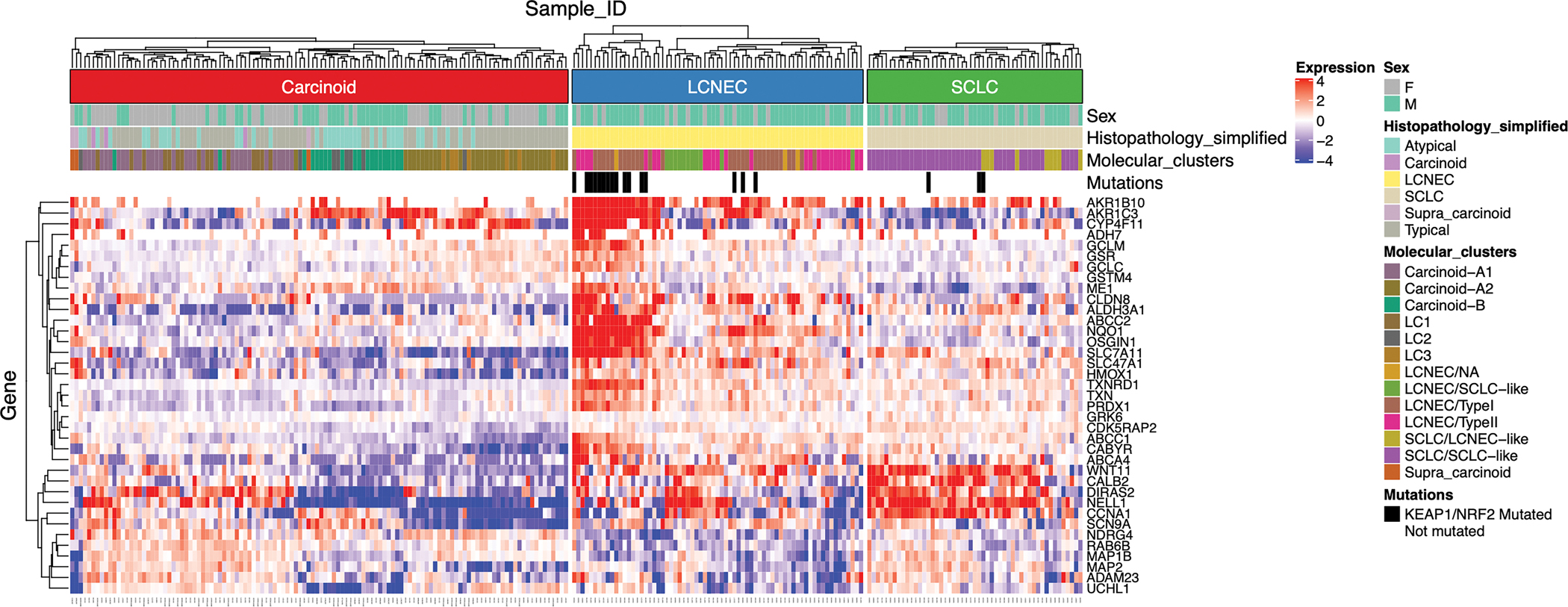
Gene expression, histopathology, and tumor mutation data were mined from a previous meta-analysis of lung tumors. Gene expression data for NRF2 target genes were VST-normalized and median-centered, and hierarchically clustered separately within each tumor super-class. Sex, histopathological classifications, molecular clusters, and mutations in KEAP1/NRF2 are designated in colors at the top based on previous annotations. Higher expression values are designated in red, and lower expression values are designated in blue. Tumors carrying KEAP1/NRF2 tumors are designated in black.
Discussion
Numerous studies have relied on GEMMs to interrogate lung cancer initiation, progression, metastasis and therapeutic response, e.g., LUAD driven by activation of KrasG12D and SCLC with predominant loss of Trp53/Rb1 [18, 45, 50]. In these well-studied models, different and mixed histological phenotypes can arise, depending on the specific genetic alterations used [51–53]. Here, we found that a novel GEMM with biallelic inactivation of the Trp53 and Cdkn2a (p16) genes developed invasive tumors in the lung that were a mixture of invasive SCLC and LCNEC, designated as C-SCLC. This is the first report showing that biallelic inactivation of the genes drives development of this poorly-characterized tumor (Fig. 6) [35]. The mice also developed small lesions that consisted of SCLC cells alone, designated as P-SCLC. The addition of an activating Nrf2E79Q/+ mutation in our model did not change the incidence of C-SCLCs (Fig. 6). However, it enhanced the development of P-SCLCs, connecting NRF2 signaling to P-SCLC development in our GEMM. We also observed a increased tumor incidence in the second study, likely reflecting the fixed genetic background of the mice after >10 generations of inbreeding.
Fig 6. Schematic representation of P-SCLC and C-SCLC development in Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ GEMM.
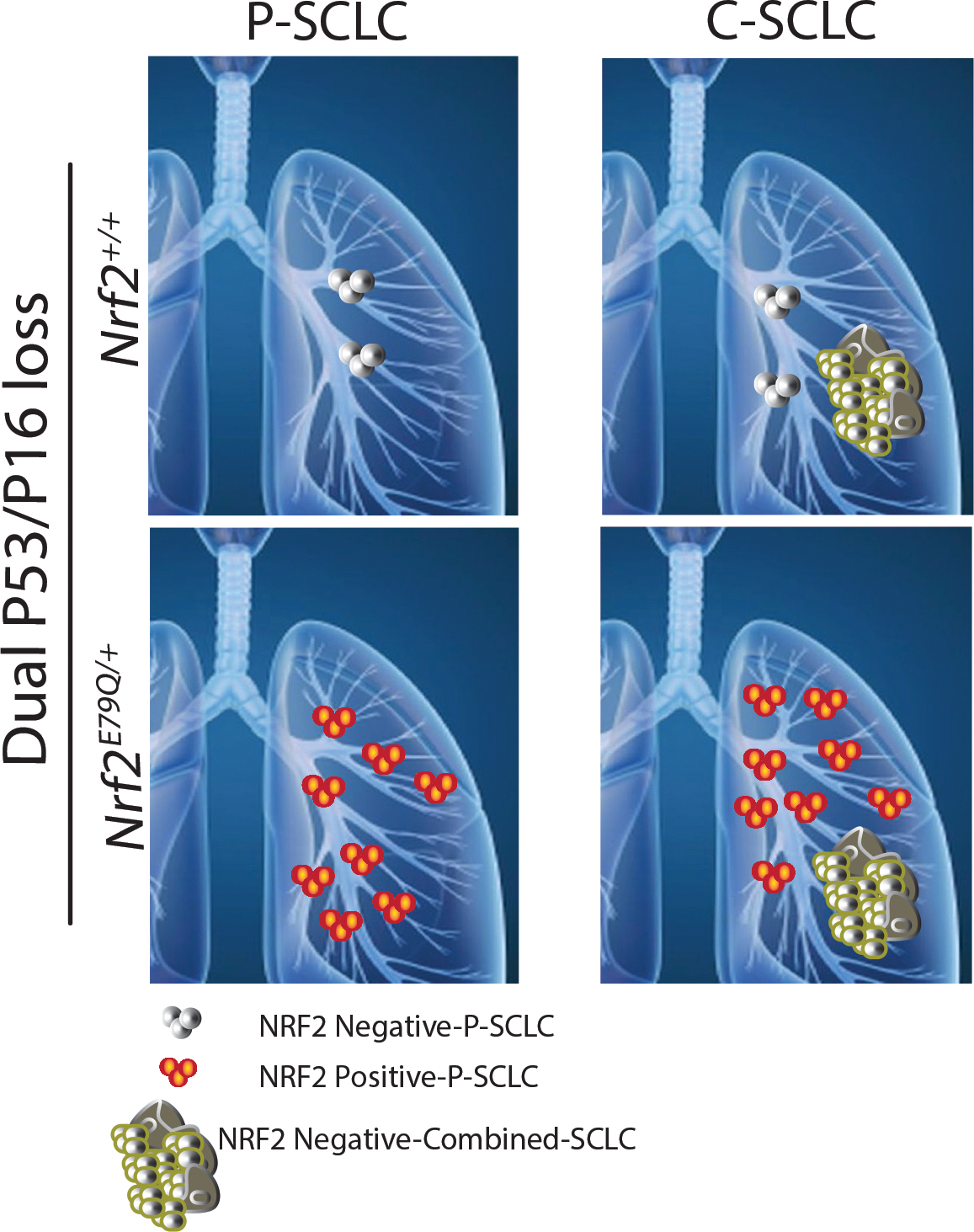
The frequency of NRF2-negative P-SCLC lesions in Trp53fl/fl;p16fl/fl;Nrf2+/+ mice was ~ten-fold lower than the frequency of NRF2-positive P-SCLC observed in Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice. Both genotypes developed NRF2-negative C-SCLC at similar frequencies. Whether the P-SCLC and C-SCLC develop independently of each other or sequentially remains an open question.
Biallelic inactivation of Trp53 and Cdkn2a (p16) tumor suppressor genes are two of the most common mutations found in human LUSC and LUAD [9, 10, 13, 19–21]. However, the appearance of P-SCLC is almost always linked with inactivating mutations in the RB1 and TRP53 tumor suppressor genes [13, 18]. Indeed, multiple groups including our own studies have shown development of P-SCLCs in Trp53fl/fl;Rb1fl/fl GEMMs [45, 54, 55]. The TRP53 loss in all tumors in our GEMM was confirmed by IHC (Figs. 3a,b, 4a). The loss of p16 deregulates the activity of cyclin dependent kinases 4/6 (CDK4/6), which promotes the phosphorylation and inactivation of RB and related proteins [56]. We confirmed this mechanism by demonstrating RB1 phosphorylation in actively dividing cells by IHC (Figs. 3a,b, 4a). Thus, the appearance of P-SCLC in the Trp53fl/fl;p16fl/fl GEMM may result from an alternative mechanism of RB1 inactivation. In contrast, Trp53fl/fl;Rb1fl/fl GEMMs generally do not develop LCNEC. The appearance of this tumor may reflect the need for p16 loss that inactivates the RB family of proteins as opposed to loss of RB1 alone. To support this notion, a recent mutational analysis of human LCNECs showed that 8/10 C-SCLC possessed dual TP53 and RB1 loss while TP53 and CDKN2A loss appeared only in three LCNECs [29]. Thus, the mixture of tumor histologies in our GEMM may reflect the high incidence of TRP53 mutations found in a wide spectrum of NSCLCs and SCLCs along with P16 and RB1 mutations, respectively [19, 57].
P-SCLCs and C-SCLCs showed similar patterns of expression of markers of lung subtypes and NE differentiation. All C-SCLCs labeled positive for the neuroendocrine marker, INSM1, and negative for SPC (alveolar Type II marker) and TRP63 (squamous differentiation marker), lowering the possibility of having LUAD or LUSC with neuroendocrine features within the C-SCLC (Fig. 3a,b) [58]. Further, these invasive tumors labeled positive for specific SCLC-markers including ASCL1, SYP and CHG-A by IF, with a high mitotic rate demonstrated by PHH3 labeling in both Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice (Fig. 3c,d, Supplementary Table 4b). All P-SCLCs also labeled positive for NE markers; however, the incidence of PHH3-positive cells appeared lower in the P-SCLCs, perhaps reflecting a slower rate of growth (Fig. 4a,b, Supplementary Table 5a,b).
Based upon previous studies using lung cancer GEMMs, we expected to observe a higher number of tumors in the Trp53fl/fl;p16fl/fl GEMM after activation of the mutant Nrf2 allele [32, 34, 59, 60]. However, expression of the activating Nrf2E79Q mutation did not change the incidence of C-SCLCs in the Trp53fl/fl;p16fl/fl GEMM. Importantly, all C-SCLCs from the Nrf2E79Q/+ mice did not express NRF2 by IHC, despite recombination of the Nrf2E79Q/+ allele (Fig. 3a,b,e,f). These results imply that the Nrf2E79Q mutation was silenced during the development of these invasive tumors, possibly through an epigenetic mechanism, implying its expression inhibits tumorigenesis [61, 62]. We also have not defined the relationship between the development of P-SCLC and C-SCLC in our GEMM i.e., does NRF2E79Q expression inhibit the progression of P-SCLC to the more aggressive C-SCLC or do they develop by independent mechanisms? We are currently investigating when and how silencing of NRF2 activation occurs during tumorigenesis using our GEMM along with cell culture models.
In contrast to the similar incidences of C-SCLC in the Trp53fl/fl;p16fl/fl;Nrf2+/+ and Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ mice, we observed a higher incidence of P-SCLCs in Nrf2E79Q/+ mice (Fig. 6) [63, 64]. All P-SCLCs developed by the Nrf2E79Q/+ mice expressed NRF2 by IHC, while the limited number of lesions from the Nrf2+/+ mice were negative for NRF2. This result implicates a role for activated NRF2 signaling in the initiation of P-SCLC (Fig. 2b,c, Fig. 4a and Supplementary Fig. 5a). How continued activation of this pathway may impact subsequent progression of these tumors remains an open question. Therefore, our ongoing studies are dissecting the mechanisms by which NRF2 activation contributes to the development of P-SCLC.
Additionally, our analysis of human neuroendocrine tumors showed high variability in NRF2 signaling (Fig. 5). While SCLC showed infrequent KEAP1/NRF2 mutations or NRF2 activation signatures, LCNEC showed two subclasses, one with frequent KEAP1/NRF2 alterations and/or activated NRF2 pathway expression and one that did not. How the silencing expression of the Nrf2E79Q mutation in the invasive C-SCLCs observed in our GEMM may mirror human SCLC and LCNEC development remains an open question.
Taken together, our studies show that activation of NRF2 signaling increases the incidence and shortens the latency of P-SCLC development in mice. In contrast, activated NRF2 signaling may inhibit tumors with a combination of P-SCLC and LCNEC. We are currently generating Nrf2E79Q/+;Trp53fl/fl;Rb1fl/fl mice to better address the role of activated NRF2 signaling in the etiology of P-SCLC. We are also characterizing the Trp53fl/fl;p16fl/fl mice at earlier time points after Ad-CMV-Cre instillation to determine when the silencing of the mutant Nrf2 allele occurs and the silencing mechanism. The availability of GEMMs with conditional Nrf2-activating mutations or Keap1 loss of function mutations will continue to provide important reagents for discovery and validation of innovative approaches for treatment approaches for SCLC that target this key signaling pathway.
Conclusions:
SCLC is more aggressive than NSCLC and occurs exclusively in smokers. Like NSCLC, SCLC is rarely detected at its early stages when the prognosis is favorable [65]. This study shows that mice with the dual loss of the Trp53 and p16 genes primarily develop invasive C-SCLC and P-SCLC, with early activation of the NRF2 pathway significantly increasing the incidence of P-SCLC only. This issue may be due, in part, to the difficulty of detecting the small, early lesions of this cancer. Our studies suggest that NRF2-activating mutations may appear early in the development of SCLC. Thus, the detection of these activating mutations or their downstream targets might provide an important tool for early detection of this cancer. Furthermore, the presence of NRF2-activating mutations in these tumors might yield a new target for therapeutic intervention.
Materials and Methods
Mouse Strains
We previously generated the LSL-Nrf2E79Q/+ mice that allow conditional expression of an endogenous Nrf2E79Q allele upon exposure to Cre recombinase [33]. The Trp53fl/fl and p16fl/fl mice were provided by the Mouse Phase 1 Unit at the University of North Carolina at Chapel Hill. The Krt14-CreERT2 mice were a kind gift from Dr. Antonio L. Amelio at the University of North Carolina at Chapel Hill. The genotype of each mouse strain as well as the GEMMs used in this study were confirmed by genotyping using the primers listed in Supplementary Table 5. Genotyping was performed by Celplor, LLC (Raleigh, NC). While the LSL-Nrf2E79Q/+ mice were generated on a pure C57Bl/6 background, the three other mouse strains were on a mixed C57Bl/6J;129SVJ background. We confirmed the genetic background of the experimental mice in both experiments by miniMUGA analysis through the UNC Mammalian Genotyping Core while backcrossing them to generate a fixed genetic background.
Generation of genetically engineered mouse model (GEMM)
In order to generate a novel genetically engineered mouse model (GEMM) for lung cancer development, we crossed LSL-Nrf2E79Q/+ mice to Trp53fl/fl and p16fl/fl, to generate two experimental groups, WT (n=38) mice (Trp53fl/fl;p16fl/fl;Nrf2+/+), and heterozygous (n=37) mice (Trp53fl/fl;p16fl/fl;LSL-Nrf2E79Q/+). We selected this number of mice/group based on a calculated P value where for a one sided Alpha of less than 0.05, Fisher’s Exact Test had 88% power with N = 20 mice per group. These mice also possess a Krt14-CreERT2 allele for use in future studies. Intranasal instillation of adenovirus expressing Cre recombinase (Ad-CMV-Cre: 2.5 X107 PFU per inoculation) at 6–8 weeks of age resulted in inactivation of the Trp53 and p16 alleles and activation of the heterozygous Nrf2E79Q/+ allele. After treatment with Ad-CMV-Cre, the mice were monitored by the University of North Carolina-Lineberger Comprehensive Cancer Center (UNC-LCCC) Animal Studies Core for weight and health status. Mice were sacrificed when they showed signs of distress (i.e., labored breathing and/or weight loss or other body conditions such as fur ruffling, difficulty in walking and hunched posture) or at the end time point (60 weeks). Upon sacrifice, we harvested the lungs, brain, liver, spleen and any other tissues that showed abnormal growth. We performed two independent studies for this report (using 18–19 mice per group).
Tissue processing, Hematoxylin & Eosin (H&E), and immunohistochemistry (IHC) labeling
Harvested tissues were fixed via submersion in 10% neutral buffered formalin at room temperature. Tissues were sent to the UNC-LCCC Animal Histopathology Core for processing, embedding into paraffin wax, and sectioning. Each tissue was stained for Hematoxylin and Eosin (H&E) and additional sections were prepared for immunohistochemistry (IHC) labeling as previously described [66]. Briefly, for H&E staining, the paraffin embedded tissue was sectioned (4 μm thickness), de-paraffinized, and rehydrated using Xylene, ethanol and dH2O. For Hematoxylin staining, slides were rinsed in hematoxylin, water, then ethanol; slides were then eosin stained, then rinsed in ethanol and xylene. IHC labeling was performed on the Ventana’s Discovery Ultra Automated IHC platform using the following primary antibodies: NRF2, ab137550, Abcam (1:500); TTF1/NKX2–1, ab133737 Abcam (1:1000); TRP63, 12143–1, Protein Tech (1:25); TRP53, VP-P956, Vector Laboratories, Inc (1:500); Phospho-RB, D20B12, Cell Signaling Technology (1:100). Discovery OmniMap anti Rabbit HRP (760–4311, ready to use) was incubated for 32 minutes at room temperature, specimens were treated with 3,3’-diaminobenzidine (DAB) and Hematoxylin II for 12 minutes, then with Bluing Reagent for 4 minutes.
Labeling with neuroendocrine markers and SPC (alveolar Type II marker) was performed with sequenza cover plate technology at the University of Utah; the primary antibodies used in this study were: CGRP, C8198 Sigma (1:250); INSM1, SC-271408 (1:1000); and SPC, Millipore AB3786 [45]. IHC slides were examined using an Olympus BX43 Light Microscope. Cells that labeled yellow to brown was considered IHC positive. All tumors were scored using both 100X and 400X magnifications for the intensity and percentage of the labeled cells. Two variable factors were considered in scoring (1) the number of positively labeled cells; and (2) the intensity of the staining (Supplementary Table 3).
Multispectral Immunofluorescent (IF) labeling
Formalin-fixed Paraffin-embedded (FFPE) 4 μm sections were cut and placed on charged slides. Slides were dried at 65°C for 2 hours. After drying, the slides were placed on the BOND RXm Research Stainer (Leica Biosystems) and deparaffinized with BOND Dewax solution (AR9222, Lecia Biosystems). The multispectral immunofluorescent (mIF) staining process involved serial repetitions of the following for each biomarker: epitope retrieval/stripping with ER1 (citrate buffer pH 6, AR996, Leica Biosystems) or ER2 (Tris-EDTA buffer pH9, AR9640, Leica Biosystems), blocking buffer (AKOYA Biosciences), primary antibody, Opal Polymer HRP secondary antibody (AKOYA Biosciences), Opal Fluorophore (AKOYA Biosciences). All AKOYA reagents used for mIF staining come as a kit (NEL821001KT). Spectral DAPI (AKOYA Biosciences) was applied once slides were removed from the BOND. They were cover slipped using an aqueous method and Diamond antifade mounting medium (Invitrogen ThermoFisher). The mIF panel consisted of the following antibodies (clone, company, and opal fluorophores): Anti-Chromogranin A (Abcam, EPR22537–248, Opal 520, 1:1000), Synaptophysin (Cell Signaling Technology, D8F6H, Opal 570, 1:200), Anti-Phospho-Histone H3 (polyclonal, Millipore Sigma, Opal 690, 1:300), Anti-ASCL1 (BD Biosciences, 24B72D11.1, Opal 780, 1:100). Slides were imaged on the Vectra® Polaris Automated Quantitative Pathology Imaging System (AKOYA Biosciences).
Quantification of in situ P-SCLC
To determine the number of P-SCLC lesions in each lung, we selected N=9 samples from each group (WT: Trp53fl/fl;p16fl/fl;Nrf2+/+ and Het: Trp53fl/fl;p16fl/fl;Nrf2E79Q/+) with no detectable lesions in the initial H&E section. We then serially sectioned through the Formalin Fixed Paraffin Embedded (FFPE) lung tissue and either stained every section or every third section for H&E. The number of P-SCLCs in each H&E section was then counted. While assessing the number of in P-SCLC, Dr. Montgomery, the veterinary pathologist, was blinded from the mice genotypes. Unstained samples/slides of the lung sections (between the two H&E slides) were stored at −80 °C until used for IHC labeling.
DNA isolation and genotyping of C-SCLC from FFPE
To determine the recombination status of the Nrf2E79Q/+ allele in C-SCLCs from Trp53fl/fl;p16fl/fl;Nrf2E79Q/+ and Trp53fl/fl;p16fl/fl;Nrf2+/+ mice, we first extracted DNA from FFPE tumor tissue. Details about the procedure are published elsewhere [47]. Briefly, 10 μm tissue were deparaffinized and pinpoint solution was used to designate the invasive tumor. After drying, the tissue was removed, DNA extracted, purified through a Zymo-spin I column (Pinpoint Solution, Zymo Research Corp, Inc) and eluted into nuclease-free water. After determining DNA concentrations, we used the following primers: Nfe2l2-ScF3: 5’ GATGCCTTCTTCTTGCCTGTAG 3’; AdSA-R: 5’ AAAGGGACAGGATAAGTATGACATCATC 3’; and Nfe2l2-ScR3: 5’ TCCACACGGGTTAGTTCACTACA 3’, to amplify the Nrf2E79Q/+ allele by PCR, separate the products by agarose electrophoresis, and image by Biorad Gel Doc XR system.
Human RNA-seq data analysis
Gene expression, histopathology, and tumor mutation data from human carcinoids, SCLC, and LCNEC tumors were accessed from https://github.com/IARCbioinfo/DRMetrics, analyzed and VST-normalized as previously described [67]. Mutation information was mined from the underlying original publications [13, 31, 68–72]. Gene expression data were focused on well-characterized NRF2 target genes [48, 49], and median-centered prior to plotting in R using tidyHeatmap.
Code availability
The code used to generate the heatmap of target gene expression in human neuroendocrine lung tumors is available at https://github.com/jeremymsimon.
Statistical analysis
We used GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA) to present the data. Fisher Exact Test was used to identify the differences between the Trp53fl/fl;p16fl/fl; Nrf2E79Q/+ and Trp53fl/fl;p16fl/fl;Nrf2+/+ mice. A P value less than 0.05 (typically ≤ 0.05) was considered statistically significant.
Supplementary Material
Acknowledgments
The authors thank Charlene Santos and her staff at the UNC-LCCC Animal Studies Core for the breeding, monitoring, weighing, and isolation of tumors of the mice; Dawud Hilliard and his staff at the UNC-Lineberger Histopathology core for tissue processing and staining for H&E and IHC; and Dr. Pablo Ariel and his staff at the UNC Microscopy Services Laboratory for their help with the microscopy studies. This study was supported by a grant from the National Institutes of Environmental and Health Sciences (T32ES007126) (S.H.H), a Golberg Postdoctoral Fellow (S.H.H.), a grant from the National Cancer Institute (CA216051) (B.E.W, M.B.M.) and by grants 5R01CA244841–02, 5U01CA231844–04, and 5U24CA213274–05 (T.G.O.). These studies were also supported in part by a UNC Lineberger Clinical/Translational Research Award
Footnotes
Conflict of interest
No conflict of interest to report
Compliance with ethical standards
This study was conducted after approval by Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill (Protocol # 19–242.0)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Society AC. Cancer Facts and Figures 2019. American Cancer Society Inc., 2019. [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69: 7–34. [DOI] [PubMed] [Google Scholar]
- 4.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–1500. [DOI] [PubMed] [Google Scholar]
- 5.Kodama T, Tsukaguchi T, Satoh Y, Yoshida M, Watanabe Y, Kondoh O et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014; 13: 2910–2918. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Choi YL, Gong Z, Liu X, Lira M, Kan Z et al. Comprehensive Characterization of Oncogenic Drivers in Asian Lung Adenocarcinoma. J Thorac Oncol 2016; 11: 2129–2140. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non-small-cell lung cancer treated with immune checkpoint inhibitors: A meta-analysis and review. Oncotarget 2017; 8: 48248–48252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai A, Menon SP, Dy GK. Alterations in genes other than EGFR/ALK/ROS1 in non-small cell lung cancer: trials and treatment options. Cancer Biol Med 2016; 13: 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Moreno P, Brambilla E, Thomas R, Soria JC. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research 2012; 18: 2443–2451. [DOI] [PubMed] [Google Scholar]
- 12.Rekhtman N, Paik PK, Arcila ME, Tafe LJ, Oxnard GR, Moreira AL et al. Clarifying the spectrum of driver oncogene mutations in biomarker-verified squamous carcinoma of lung: lack of EGFR/KRAS and presence of PIK3CA/AKT1 mutations. Clinical cancer research : an official journal of the American Association for Cancer Research 2012; 18: 1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015; 524: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal saquamous cell carcinomas. Nat Genet 2009; 41: 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian J, Massion PP. Role of chromosome 3q amplification in lung cancer. J Thorac Oncol 2008; 3: 212–215. [DOI] [PubMed] [Google Scholar]
- 16.Massion PP, Taflan PM, Jamshedur Rahman SM, Yildiz P, Shyr Y, Edgerton ME et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res 2003; 63: 7113–7121. [PubMed] [Google Scholar]
- 17.McCaughan F, Pole JC, Bankier AT, Konfortov BA, Carroll B, Falzon M et al. Progressive 3q amplification consistently targets SOX2 in preinvasive squamous lung cancer. Am J Respir Crit Care Med 2010; 182: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003; 4: 181–189. [DOI] [PubMed] [Google Scholar]
- 19.Demirhan O, Tastemir D, Hasturk S, Kuleci S, Hanta I. Alterations in p16 and p53 genes and chromosomal findings in patients with lung cancer: fluorescence in situ hybridization and cytogenetic studies. Cancer Epidemiol 2010; 34: 472–477. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Cespedes M, Reed AL, Buta M, Wu L, Westra WH, Herman JG et al. Inactivation of the INK4A/ARF locus frequently coexists with TP53 mutations in non-small cell lung cancer. Oncogene 1999; 18: 5843–5849. [DOI] [PubMed] [Google Scholar]
- 21.Leversha MA, Fielding P, Watson S, Gosney JR, Field JK. Expression of p53, pRB, and p16 in lung tumours: a validation study on tissue microarrays. J Pathol 2003; 200: 610–619. [DOI] [PubMed] [Google Scholar]
- 22.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences 2008; 105: 13568–13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical pharmacology 2013; 85: 705–717. [DOI] [PubMed] [Google Scholar]
- 24.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes to cells : devoted to molecular & cellular mechanisms 2011; 16: 123–140. [DOI] [PubMed] [Google Scholar]
- 25.Cloer EW, Goldfarb D, Schrank TP, Weissman BE, Major MB. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res 2019; 79: 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer cell 2018; 34: 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res 2010; 16: 3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol 2013; 1: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nature communications 2018; 9: 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alcala N, Leblay N, Gabriel AAG, Mangiante L, Hervas D, Giffon T et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nature Communications 2019; 10: 3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peifer M, Fernández-Cuesta L, Sos ML, George J, Seidel D, Kasper LH et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012; 44: 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerins MJ, Ooi A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 2018; 8: 12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowman BM, Montgomery SA, Schrank TP, Simon JM, Ptacek TS, Tamir TY et al. A conditional mouse expressing an activating mutation in NRF2 displays hyperplasia of the upper gastrointestinal tract and decreased white adipose tissue. The Journal of Pathology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007; 448: 807–810. [DOI] [PubMed] [Google Scholar]
- 35.Travis WD. The 2015 WHO classification of lung tumors. Pathologe 2014; 35 Suppl 2: 188. [DOI] [PubMed] [Google Scholar]
- 36.Maronpot RR, Boorman GA, Gaul BW. Pathology of the Mouse: Reference and Atlas. Cache River Press, 1999. [Google Scholar]
- 37.Reznik GK, Schuller HM, Stinson SF. Tumours of the nasal cavities. IARC Sci Publ 1994: 305–324. [PubMed] [Google Scholar]
- 38.Koike K, Jay G, Hartley JW, Schrenzel MD, Higgins RJ, Hinrichs SH. Activation of retrovirus in transgenic mice: association with development of olfactory neuroblastoma. J Virol 1990; 64: 3988–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel NW, Neumann JE, Ahlfeld J, Wefers AK, Merk DJ, Ohli J et al. Canonical Wnt Signaling Drives Tumor-Like Lesions from Sox2-Positive Precursors of the Murine Olfactory Epithelium. PLoS One 2016; 11: e0166690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lubojemska A, Borejko M, Czapiewski P, Dziadziuszko R, Biernat W. Of mice and men: olfactory neuroblastoma among animals and humans. Vet Comp Oncol 2016; 14: e70–82. [DOI] [PubMed] [Google Scholar]
- 41.Waalkes MP, Rehm S, Kasprzak KS, Issaq HJ. Inflammatory, proliferative, and neoplastic lesions at the site of metallic identification ear tags in Wistar [Crl:(WI)BR] rats. Cancer Res 1987; 47: 2445–2450. [PubMed] [Google Scholar]
- 42.Zheng M. Classification and Pathology of Lung Cancer. Surg Oncol Clin N Am 2016; 25: 447–468. [DOI] [PubMed] [Google Scholar]
- 43.Wu M, Wang B, Gil J, Sabo E, Miller L, Gan L et al. p63 and TTF-1 immunostaining. A useful marker panel for distinguishing small cell carcinoma of lung from poorly differentiated squamous cell carcinoma of lung. Am J Clin Pathol 2003; 119: 696–702. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay S, Katzenstein AL. Subclassification of non-small cell lung carcinomas lacking morphologic differentiation on biopsy specimens: Utility of an immunohistochemical panel containing TTF-1, napsin A, p63, and CK5/6. Am J Surg Pathol 2011; 35: 15–25. [DOI] [PubMed] [Google Scholar]
- 45.Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020; 38: 60–78.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 2011; 17: 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snow AN, Stence AA, Pruessner JA, Bossler AD, Ma D. A simple and cost-effective method of DNA extraction from small formalin-fixed paraffin-embedded tissue for molecular oncologic testing. BMC Clin Pathol 2014; 14: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh A, Boldin-Adamsky S, Thimmulappa RK, Rath SK, Ashush H, Coulter J et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res 2008; 68: 7975–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cescon DW, She D, Sakashita S, Zhu CQ, Pintilie M, Shepherd FA et al. NRF2 Pathway Activation and Adjuvant Chemotherapy Benefit in Lung Squamous Cell Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21: 2499–2505. [DOI] [PubMed] [Google Scholar]
- 50.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nature medicine 2017; 23: 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Travis WD. Lung tumours with neuroendocrine differentiation. Eur J Cancer 2009; 45 Suppl 1: 251–266. [DOI] [PubMed] [Google Scholar]
- 52.Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res 2010; 70: 3877–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014; 156: 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011; 19: 754–764. [DOI] [PubMed] [Google Scholar]
- 55.Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017; 31: 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672–1677. [DOI] [PubMed] [Google Scholar]
- 57.Swanton C, Govindan R. Clinical Implications of Genomic Discoveries in Lung Cancer. N Engl J Med 2016; 374: 1864–1873. [DOI] [PubMed] [Google Scholar]
- 58.Zhu Y, Chen L, Wang J, Chu X, Chen Y, Zhang Q et al. [Identification and characterization of SP cells in human lung adenocarcinoma SPC-A1 cells.]. Zhongguo Fei Ai Za Zhi 2008; 11: 681–685. [DOI] [PubMed] [Google Scholar]
- 59.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–221. [DOI] [PubMed] [Google Scholar]
- 60.Lubet R, Wang Y, Zhang Z, You M. Mouse models incorporating alterations in the major tumor suppressor genes P53 and P16: their use in screening for potential carcinogens, developing further relevant mouse models, and screening for potential chemopreventive and chemotherapetutic agents. Exp Lung Res 2005; 31: 117–133. [DOI] [PubMed] [Google Scholar]
- 61.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res 2008; 58: 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kazanets A, Shorstova T, Hilmi K, Marques M, Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim Biophys Acta 2016; 1865: 275–288. [DOI] [PubMed] [Google Scholar]
- 63.Park KS, Liang MC, Raiser DM, Zamponi R, Roach RR, Curtis SJ et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle 2011; 10: 2806–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonkin D, Thomas A, Teicher BA. Are neuroendocrine negative small cell lung cancer and large cell neuroendocrine carcinoma with WT RB1 two faces of the same entity? Lung Cancer Manag 2019; 8: Lmt13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Zimmermann S, Parikh K, Mansfield AS, Adjei AA. Current Diagnosis and Management of Small-Cell Lung Cancer. Mayo Clin Proc 2019; 94: 1599–1622. [DOI] [PubMed] [Google Scholar]
- 66.Yamano S, Gi M, Tago Y, Doi K, Okada S, Hirayama Y et al. Role of deltaNp63(pos)CD44v(pos) cells in the development of N-nitroso-tris-chloroethylurea-induced peripheral-type mouse lung squamous cell carcinomas. Cancer Sci 2016; 107: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabriel AAG, Mathian E, Mangiante L, Voegele C, Cahais V, Ghantous A et al. A molecular map of lung neuroendocrine neoplasms. GigaScience 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alcala N, Leblay N, Gabriel AAG, Mangiante L, Hervas D, Giffon T et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nature communications 2019; 10: 3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laddha SV, da Silva EM, Robzyk K, Untch BR, Ke H, Rekhtman N et al. Integrative Genomic Characterization Identifies Molecular Subtypes of Lung Carcinoids. Cancer Res 2019; 79: 4339–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.George J, Walter V, Peifer M, Alexandrov LB, Seidel D, Leenders F et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nature communications 2018; doi: 10.1038/s41467-018-03099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamad SH, Johnson NM, Tefft ME, Brinkman MC, Gordon SM, Clark PI et al. Little Cigars vs 3R4F Cigarette: Physical Properties and HPHC Yields. Tob Regul Sci 2017; 3: 459–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fernandez-Cuesta L, Peifer M, Lu X, Sun R, Ozretić L, Seidal D et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat Commun 2014; 5: 3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.