

Abstract
Humans and nonhuman primates (NHPs) harbor complex gut microbial communities that affect phenotypes and fitness. The gut microbiotas of wild NHPs reflect their hosts’ phylogenetic histories and are compositionally distinct from those of humans, but in captivity the endogenous gut microbial lineages of NHPs can be lost or replaced by lineages found in humans. Despite its potential contributions to gastrointestinal dysfunction, this humanization of the gut microbiota has not been investigated systematically across captive NHP species. Here, we show through comparisons of well-sampled wild and captive populations of apes and monkeys that the fraction of the gut microbiota humanized by captivity varies significantly between NHP species but is remarkably reproducible between captive populations of the same NHP species. Conspecific captive populations displayed significantly greater than expected overlap in the sets of bacterial 16S rRNA gene variants that were differentially abundant between captivity and the wild. This overlap was evident even between captive populations residing on different continents but was never observed between heterospecific captive populations. In addition, we developed an approach incorporating human gut microbiota data to rank NHPs’ gut microbial clades based on the propensity of their lineages to be lost or replaced in captivity by lineages found in humans. Relatively few microbial genera displayed reproducible degrees of humanization in different captive host species, but most microbial genera were reproducibly humanized or retained from the wild in conspecific pairs of captive populations. These results demonstrate that the gut microbiotas of captive NHPs display predictable, host-species specific responses to captivity.
Keywords: archaea, bacteria, captive populations, conservation biology, microbial transmission, microbiome
1 |. INTRODUCTION
The gut microbial communities of humans and wild living primates reflect the evolutionary histories of their hosts (Amato et al., 2019; Moeller et al., 2014; Ochman et al., 2010). Each host species’ gut microbiota contains a distinct set of microbial lineages, some of which have codiversified with primate species (Moeller, Caro-Quintero et al., 2016). However, gut microbial lineages that are confined to geographically separated wild populations of host species can be transmitted between host species when individuals come into direct contact. Similarly, shared environments can lead to the parallel gain or loss of lineages from the gut microbiota in co-occurring host populations, reducing differentiation of the gut microbiota. For example, studies of captive nonhuman primates (NHPs) have demonstrated that captivity leads to partial convergence of NHP gut microbiota with human gut microbiota (Clayton et al., 2016). In both apes (Campbell et al., 2020; Frankel et al., 2019; McKenzie et al., 2017; Narat et al., 2020; Uenishi et al., 2007) and monkeys (Clayton et al., 2016; Frankel et al., 2019; Hale et al., 2019; Lee et al., 2019; McKenzie et al., 2017; Nakamura et al., 2011; Tsukayama et al., 2018), the gut microbiotas of captive NHP populations are distinct from those of wild populations. In addition, captive the gut microbiotas of captive NHPs are often more compositionally similar to human gut microbiotas than are the gut microbiotas of wild living conspecific NHP populations (Clayton et al., 2016). The disruption and humanization of the endogenous gut microbiota in captive primates has been implicated in the gastrointestinal diseases often experienced by these populations (Amato et al., 2016; McKenna et al., 2008; Shigeno et al., 2018).
Despite the potential importance of humanization of the gut microbiota for the health of captive NHPs, this process, which can proceed through the gain of microbial lineages found in humans or the loss of gut microbiota constituents private to wild NHPs, has not been systematically evaluated across captive NHP populations. It is currently unclear whether humanization of the primate gut microbiota tends to be underlain by specific sets of microbial lineages or whether all lineages in the primate gut microbiota are equally prone to humanization. Similarly, the degree to which the probability of humanization of specific microbial lineages in captivity varies across primate populations and species is unknown.
One limitation of previous studies that has hindered the identification of specific microbial lineages that respond to captivity in NHP species is the lack of population-level host sampling. Several previous studies have examined the effects of captivity in the gut microbiota in multiple NHP species (Frankel et al., 2019; Hale et al., 2019; Lee et al., 2019; McKenzie et al., 2017; Nakamura et al., 2011; Tsukayama et al., 2018), but these have rarely examined more than 10 individuals in captivity per NHP species. Sample size is often an insurmountable constraint given the limited sizes of captive NHP populations. Previous studies have had power to detect broad differences in gut microbiota composition between captive and wild populations (e.g., differences in microbiota alpha and beta diversity), but have typically not had power to test for effects of captivity on each individual microbial lineage in the gut microbiota. Another limitation of previous studies has been the lack of replicate captive NHP populations from the same species—a requirement in order to test the reproducibility and predictability of the effects of captivity on the gut microbiota of NHP species. One exception to these limitations is a previous study of wild and captive red-shanked doucs that sampled >30 captive individuals from two replicate captive populations, observing some evidence of reproducible effects of captivity (Clayton et al., 2016). Repeating such sampling regimes, in which relatively large numbers of individuals are sampled from multiple independent conspecific captive populations, in other NHP species promises to reveal whether NHP species display reproducible, NHP-species specific responses to captivity.
Here, we sequenced the gut microbiota of captive chimpanzees retired from the New Iberia Research Center and combined our data with gut microbiota data sets from humans, wild chimpanzees, and additional captive and wild populations of chimpanzees, gorillas, and red-shanked doucs. Each captive and wild population was represented by >15 individuals, enabling tests for differentially abundant microbial lineages between wild and captive individuals as well as microbial signatures of humanization within individual gut microbial clades. Results showed that gut microbial lineages were remarkably consistent in the degree to which they were humanized in replicate captive primate populations from the same species. However, the fraction of the gut microbiota that was humanized in captivity varied significantly among primate species. These results indicate that the sets of microbial taxa that are humanized in the captive primate gut microbiota are predictable but dependent on host species identity.
2 |. MATERIALS AND METHODS
2.1 |. Sample collection and processing
Fresh faecal samples were collected from 17 captive-born captive chimpanzees during their transport from the New Iberia Research Center, Louisiana, USA to Project Chimps Sanctuary, Georgia, USA in March and September of 2018. Samples were immediately stored at −20°C then transferred to −80°C within one month. DNA was extracted using DNeasy PowerSoil PowerLyzer DNA Extractions kits according to the manufacturer’s protocol. Amplicons of the V4–V5 regions of the 16S rRNA gene were generated using 515F/926R primers based on the Earth Microbiome Project (EMP) 16S Illumina Amplicon protocol (Thompson et al., 2017). Amplicons were sequenced on an Illumina MiSeq platform using V3 chemistry and 2 × 250 bp paired-end reads at the Weill Cornell Medicine Microbiome Core Laboratory following methods of the EMP.
2.2 |. Combining NHP and human data for meta-analyses
Raw FASTQ sequence files and metadata from the captive chimpanzee samples were uploaded to the public database Qiita (Gonzalez et al., 2018) as study ID 12140 and processed using the database’s implementation of the quantitative insights into microbial ecology (QIIME2) software. We combined our data from captive chimpanzees (Pan troglodytes subspp.) retired from the New Iberia Research Center (USA2) with gut microbiota 16S rDNA data sets from wild chimpanzees (Pan troglodytes schweinfurthii) (n = 96) from Tanzania (Moeller, Foerster et al., 2016) (TZA), wild chimpanzees (P. t. troglodytes) (n = 18) and gorillas (Gorilla gorilla gorilla) (n = 28) from the Democratic Republic of the Congo (DRC), captive chimpanzees (P. t. subspp.) (n = 18) and gorillas (G. g. subspp.) (n = 15) from the United States (USA1) (Campbell et al., 2020), wild red-shanked doucs (Pygathrix nemaeus) from Vietnam (VNM) (n = 66), and captive red-shanked doucs (P. n.) from Singapore (SGP) (n = 15) and the United States (USA) (n = 12) (Clayton et al., 2016). This combined wild and captive NHP gut microbiota data set was then merged with a dataset from humans (n = 528) living in the USA (Yatsunenko et al., 2012). Forward and reverse sequences were demultiplexed and quality filtered using default settings. High-quality reads were trimmed to equal length and amplicon sequence variants (ASVs) were identified using Deblur. Data were rarefied to 10,000 sequences per sample and exported from Qiita for analyses in QIIME2 v 2018.11 (Boylen et al., 2019).
2.3 |. Taxonomy and diversity analyses
Taxonomy was assigned to ASVs by fitting a naive-Bayes classifier trained on the Greengenes v.13_8 99% OTUs reference using the sk-learn classifier. A phylogenetic tree was constructed by inserting the sequences into the Greengenes 13_8 reference tree using the QIIME2 plugin q2-fragment-insertion, which uses the SEPP insertion method. Shannon entropy and Chao1 alpha diversity indices (Chao, 1984) were calculated for each sample. The Shannon index measures richness and evenness (Shannon & Weaver, 1949), whereas Chao1 measures the abundance weighted ASV richness within a microbiota. To examine the sharing of ASVs between captive and wild NHPs and humans, the binary Sorensen-Dice dissimilarly was calculated for each pair of samples (Dice, 1945). Binary Sorensen-Dice dissimilarities were calculated as one minus the quotient of the total number of ASVs shared between two samples by the sum of the total number of ASVs in each sample.
2.4 |. Statistical analyses of alpha and beta diversity
Alpha diversity metrics including Shannon index and Chao1 were compared using Kruskal-Wallis tests. For Sorensen-Dice beta diversity, permutational multivariate analysis of variation (PERMANOVA), tests for homogeneity of multivariate dispersions (PERMDISP), and Monte-Carlo nonparametric permutation tests were conducted in QIIME to statistically partition the sources of variation in community structure using 999 permutations. Sorensen-Dice dissimilarities among all samples were visualized through ordination using principal coordinates analysis (PCoA) and plots in EMPeror. Pearson correlations and Spearman ranked correlations among log-transformed HSSs and MCSs were conducted with the “stats” package in R v 4.0.4 (R Core Team, 2013).
2.5 |. Detection of differentially abundant microbial ASVs and genera between captive and wild populations
ANCOM2 (Mandal et al., 2015) was employed in r (github.com/FrederickHuangLin/ANCOM) to test whether any individual microbial ASVs or genera were differentially abundant between the captive and wild individuals for every captive NHP population. Each analysis focused on ASVs or genera present in >10% of samples from the pair of captive and wild populations. Volcano plots displaying differentially abundant ASVs and genera were created in R with ggplot2. For these analyses, captive chimpanzees retired from the New Iberia Research Center (USA2) were paired with wild chimpanzees from Tanzania (TZA) and zoo captive chimpanzees (USA1) were paired with wild chimpanzees from the DRC (DRC). Pairings were chosen such that samples from wild and captive conspecific populations were processed and sequenced by the same laboratories, minimizing any potential study effects that could confound downstream analyses.
In addition, we tested whether microbial ASVs and genera differed repeatedly in relative abundance between the wild and captivity in multiple captive NHP populations. Specifically, we employed hypergeometric tests to assess the significance of the overlap between the sets of microbial taxa (i.e., ASVs and genera) whose abundances differ from the wild in pairs of captive NHP populations. These analyses tested whether the number of microbial taxa that shifted in relative abundance in the two captive NHP populations relative to wild conspecific NHP populations was significantly greater than the number expected by chance, given the observed number of microbial taxa shared between the multiple replicate captive NHP populations. In addition, we employed regression analyses to test for significant associations of W statistics between pairs of captive NHP populations.
2.6 |. Host specificity scores and microbiota convergence scores
We developed a statistic, the host specificity score (HSS), to assess microbial genus-specific patterns of humanization in captive NHP gut microbiotas. This statistic was defined for a microbial genus in a captive NHP population as the mean binary Sorensen-Dice dissimilarity of the ASV composition of the genus between the captive NHP population and humans divided by the mean binary Sorensen-Dice dissimilarity of the ASV composition of the genus between the captive NHP population and a conspecific wild-living population. HSSs of >1 indicate that the ASV composition of the microbial clade is more similar between captive NHPs and wild-living conspecifics than between captive NHPs and humans. In contrast, a score of <1 indicates that the ASV composition of the microbial clade is more similar between captive NHPs and humans than between captive NHPs and wild-living conspecifics.
In order to calculate HSSs, we split the ASV table containing all samples by microbial genus and calculated genus-level mean binary Sorensen-Dice (BSD) dissimilarities between captive NHPs and wild conspecifics as well as between captive NHPs and humans. ASVs without taxonomic assignments and microbial genera detected by fewer than five reads were excluded from downstream analyses. As in the ANCOM analyses described above, captive chimpanzees retired from the New Iberia Research Center (USA2) were paired with wild chimpanzees from Tanzania (TZA) and zoo captive chimpanzees (USA1) were paired with wild chimpanzees from the DRC (DRC). The HSS for each genus was calculated and log-transformed for regression analyses.
In addition to HSSs, we also calculated for each microbial genus in each captive NHP population a microbiota convergence score (MCS). This index measures the degree to which the ASV composition of the genus in a captive NHP has converged with the ASV composition of the genus in humans relative to the ASV composition of the genus in the wild conspecific NHP population. Descriptions of these analyses are presented in Appendix S1: Materials and Methods. For both HSSs and MCSs, heatmaps were calculated in R using heatmap.2 with the parameter scale = “col” and default settings. In addition to analyses based on the Yatsunenko et al. (2012) data set, we also calculated per genus HSSs and MCSs for each captive NHP population using an American Gut data set (McDonald et al., 2018; Appendix S1: Materials and Methods). All scripts used to calculate HSSs and MCSs are publicly available at github.com/CUMoellerLab/Houtz_etal_2021-NHP_microbiome.
3 |. RESULTS
3.1 |. Sequencing and comparisons of NHP and human gut microbiomes
We sequenced 16S rDNA extracted from 18 faecal samples collected from captive chimpanzees upon their arrival at Project Chimps sanctuary, generating 1,406,091 million high-quality sequences with an average of 80,704 reads per sample. These data were combined with published V4–V5 16S rDNA amplicon data sets containing matched populations of captive and wild primates from the same species, including doucs, chimpanzees, and gorillas as well as 16S rDNA data sets collected from human populations in the United States, the country in which most captive NHP individuals represented in the combined dataset resided. One faecal sample from a captive chimpanzee (chimp.17) dominated by a single ASV (>50% of reads) was removed from downstream analyses. The total data set contained 796 samples rarefied to an even depth of 10,000 reads per sample. A list of samples and their corresponding metadata are presented in Table S1. Sample metadata corresponding to published data sets are available in the supplementary materials of their respective studies (Campbell et al., 2020; Clayton et al., 2016; Moeller, Caro-Quintero et al., 2016). A map of sampling locations for wild and captive NHPs is presented in Figure S1.
3.2 |. Altered microbiota diversity in captive NHPs relative to wild conspecific NHPs
We tested for effects of host species identity and captivity state on microbiota alpha and beta diversity using 16S rDNA sequences from captive chimpanzee samples generated by the present study (Captive chimpanzees USA2) and published data from humans (Humans), wild chimpanzees (Wild chimpanzees TZA; Wild chimpanzees DRC), and captive and wild red-shanked doucs (Captive douc SGP, Captive douc USA, Wild douc VNM). Captive populations tended to display reduced Shannon entropy relative to wild conspecifics (Figure S2; Table S2). However, results from Chao1 analyses were mixed, with captive doucs displaying lower alpha diversity than wild conspecifics but captive chimpanzees and gorillas displaying higher alpha diversity than wild conspecifics (Figure S2; Table S2).
Analyses of beta diversity indicated that the gut microbiotas of captive primate populations were compositionally distinct from the gut microbiotas of wild conspecifics (PERMANOVA p-value = .001 in each comparison). In contrast, wild and captive conspecifics did not display significant differences in dispersion (PERMDISP p-value > .05 in each comparison). The distinctiveness of captive and wild NHP gut microbiotas was evident in principal coordinates analyses based on Sorensen-Dice distances (Figure 1a). Moreover, for each NHP species the gut microbiotas of captive populations were significantly more similar to those of humans than were the gut microbiotas of wild-living conspecific populations (Figure 1b) (Monte-Carlo nonparametric permutation tests p-value = .001). The relative abundances of ASVs for each sample are presented in Table S3. The relative abundances of microbial genera for each sample are presented in Table S4.
FIGURE 1.
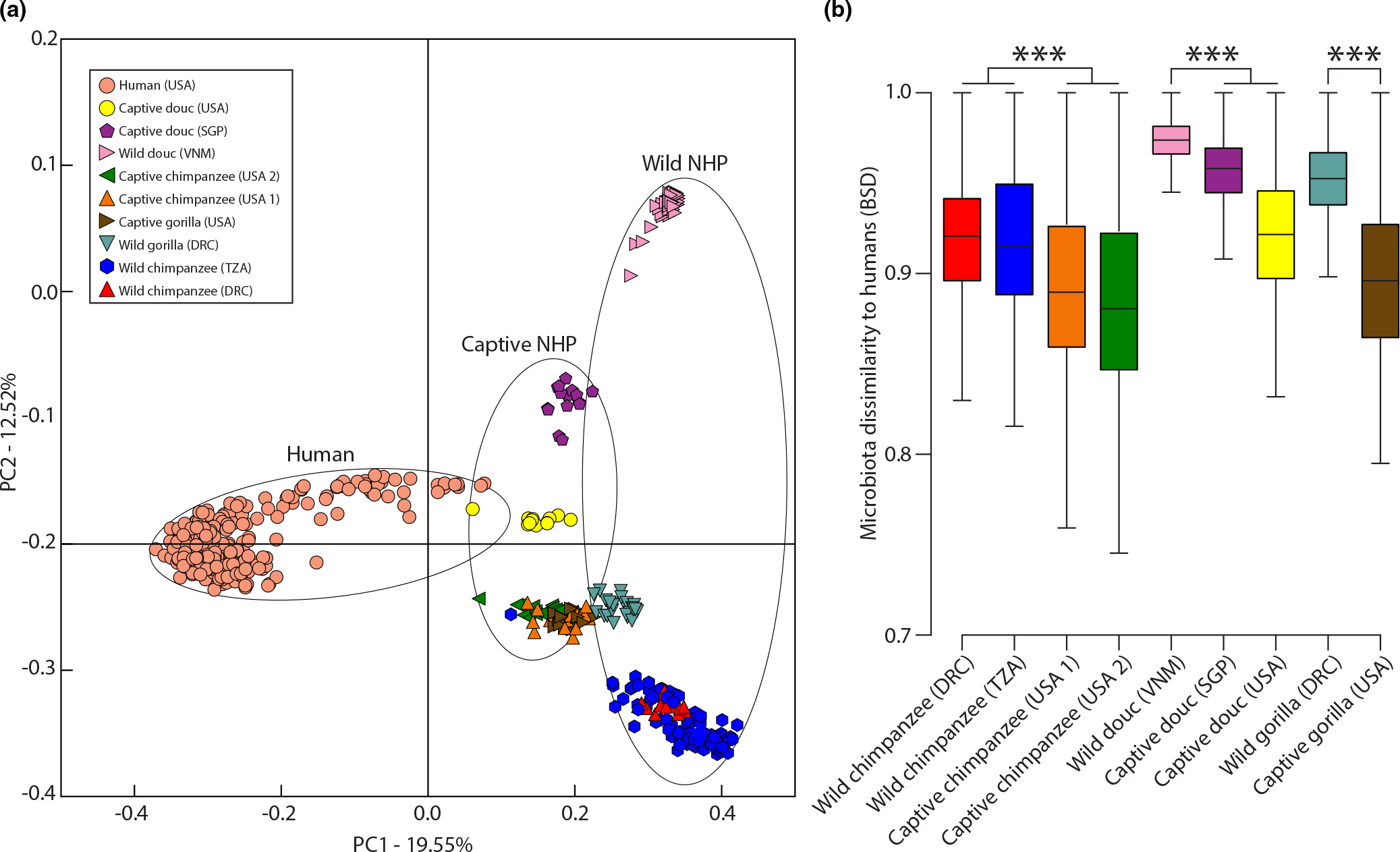
Humanization of captive NHP gut microbiotas. (a) Plot shows the first and second principal coordinates of the Binary Sorensen-Dice dissimilarities among wild primates, captive primates, and humans. Shapes represent faecal samples collected from individual primates and are coloured based on host species identity as indicated by the inset. Circles unite groups of shapes corresponding to wild, captive, or human samples. (b) Boxplots show maxima, minima, interquartile ranges, and medians of BSD dissimilarities between primates (wild or captive) and humans. Boxes are coloured by primate species as in (a). Significant differences between means based on permutation tests are indicated by asterisks; ***p = .001
3.3 |. No ASVs or genera were differentially abundant between captive and wild in all NHP species
We next employed ANCOM to test for ASVs that were differentially abundant between captive and wild individuals for each captive NHP population. These analyses indicated that hundreds of ASVs were differentially abundant between captive and wild individuals in at least one captive NHP population (Figure S3, Table S5). However, of the 262 ASVs detected in >2 of the six captive NHP populations, none were differentially abundant between captive and wild individuals in >2 of the six captive NHP populations (Table S5). Similarly, dozens of microbial genera were differentially abundant between captive and wild individuals in at least on captive NHP population (Figure 2, Table S6), but none were differentially abundant between captive and wild individuals in >3 of the six captive NHP populations (Table S6). This observation indicates that the individual ASVs and genera that were overrepresented or underrepresented in captive NHP population relative to wild conspecific populations varied across NHP species.
FIGURE 2.
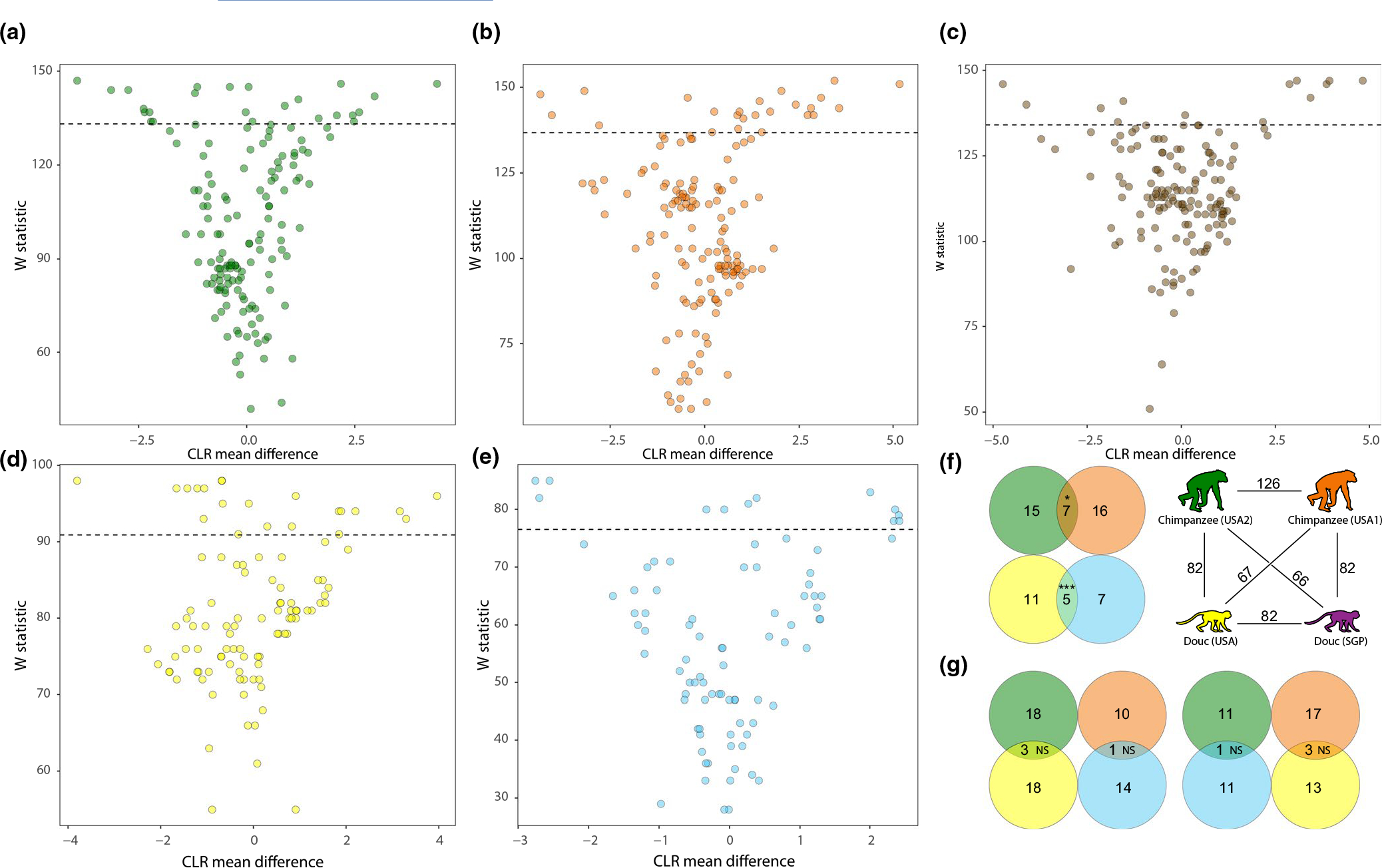
Differential abundance of microbial genera between captive and wild NHP individuals. Volcano plots show the ANCOM test statistic (y-axis) and the centered logarithmic transformed (CLR) log-fold difference in relative abundance between captive and wild individuals for each microbial genus. Dots represent microbial genera coloured by captive NHP population based on Figure 1, and each plot shows results for a single captive primate population: captive chimpanzee (USA2) (a), captive chimpanzee (USA1) (b), captive gorilla (c), captive douc (USA) (d), and captive douc (SGP) (e). Dashed line in each plot represents W statistic cutoff of 0.9. Statistics for all ASVs for each pair of captive and wild NHP populations are presented in Table S3. Venn diagrams (f and g) show overlap of the sets of differentially abundant microbial genera between pairs of captive NHP populations, represented by circles coloured by captive NHP population as in Figure 1 and in the key. In (f), the number of genera shared by each pair of captive NHP populations is shown above lines connecting cartoons coloured as in Figure 1. In (f and g), numbers in nonoverlapping portions of circles represent differentially abundant microbial genera detected in both NHP populations but differentially abundant in only one NHP population. Numbers in overlapping portions of circles represent microbial genera that were differentially abundant in both NHP populations. Significance of overlap for each comparison is shown by asterisks: ***p-value < .001; NS, not significant
3.4 |. Reproducible shifts of microbial ASVs and genera to captivity in replicate conspecific NHP populations
We observed significant overlap in the microbial ASVs and genera that displayed significant shifts in relative abundance from the wild, based on ANCOM analyses, between replicate captive NHP populations of the same species (i.e., conspecific populations; Table S5). Of the 627 ASVs detected in both replicate captive populations of chimpanzees (USA1 and USA2), 102 and 153 were significantly overrepresented or underrepresented in USA1 and USA2 chimpanzees, respectively, relative to their matched wild populations (each captive chimpanzee population was matched with a different wild chimpanzee population, Materials and Methods). Of these ASVs displaying significantly different relative abundances between captive and wild chimpanzees, 53 displayed significant differences in both captive populations of chimpanzees (USA1 and USA2) (2.1-fold more than random expectation, hypergeometric p-value = 1.429e-11). Similarly, of the 573 ASVs detected in both replicate captive populations of doucs (USA and SGP), 61 and 66 were significantly overrepresented or underrepresented in USA and SGP doucs, respectively, relative to their matched wild population. Of these ASVs displaying significantly different relative abundances between captive and wild doucs, 53 displayed significant differences in both captive populations of doucs (USA and SGP; 6.8-fold more than random expectation, hypergeometric p-value = 5.43e-43). In contrast, this degree of overlap between ASVs displaying significantly different abundances between captivity and the wild was not observed between heterospecific captive NHP populations. For example, of the 64 ASVs shared by USA1 chimpanzees and SGP doucs, 10 and 15 were significantly overrepresented or underrepresented in chimpanzees and doucs, respectively, relative to their matched wild populations. Of these ASVs, only three displayed significant differences in both USA1 chimpanzees and SGP doucs (1.3-fold more than expected, hypergeometric p-value = .215).
We also observed significant overlap between the sets of microbial genera that were differentially abundant between captivity and the wild in replicate captive conspecific NHP populations (Figure 2; Table S6). Of the 126 microbial genera detected in both replicate captive populations of chimpanzees (USA1 and USA2), 23 and 22 were significantly overrepresented or underrepresented in USA1 and USA2 chimpanzees, respectively, relative to their matched wild populations. Of these genera, seven displayed significant differences in both captive populations of chimpanzees (USA1 and USA2; 4.2-fold more than random expectation, hypergeometric p-value = .0355; Figure 2f). Similarly, of the 82 genera detected in both replicate captive populations of doucs (USA and SGP), 16 and 12 were significantly overrepresented or underrepresented in USA and SGP doucs, respectively, relative to their matched wild population. Of these genera, five displayed significant differences in both captive populations of doucs (USA and SGP; 6.8-fold more than random expectation, hypergeometric p-value = 5.95e-5; Figure 2f). In contrast, this degree of overlap between genera displaying significantly different abundances between captivity and the wild was not observed between heterospecific captive NHP populations. For example, of the 67 genera shared by USA1 chimpanzees and SGP doucs, 15 and 11 were significantly overrepresented or underrepresented in chimpanzees and doucs, respectively, relative to their matched wild populations. Of these genera, only three displayed significant differences in both USA1 chimpanzees and SGP doucs (1.2-fold more than expected, hypergeometric p-value = .234; Figure 2g).
In addition to hypergeometric tests for significant overlap of ANCOM results between pairs of captive NHP populations, we also conducted regression analyses to test whether W statistics of ASVs and genera we associated between pairs of captive NHP populations. These analyses revealed qualitatively similar results to the hypergeometric tests. Specifically, ASV and genus W statistics tended to display significant positive relationships between pairs of conspecific NHP populations, but less significant and more variable relationships between pairs of heterospecific NHP populations (Tables S5 and S6).
3.5 |. HSSs reveal reproducible and host-species specific humanization of microbial genera in captive NHPs
Identifying differentially abundant ASVs and genera between matched wild and captive populations can reveal the effects of captivity on specific constituents of the NHP gut microbiota. However, these analyses alone do not provide information about the extent to which specific microbial clades within the captive NHP gut microbiota are humanized. Here, we define humanization of a microbial clade in a captive NHP population as the replacement of endogenous ASVs in the clade (i.e., ASVs found in wild NHP populations) by ASVs found in humans or the parallel absence from captive NHP populations and humans of ASVs in the clade found in wild conspecific NHP populations.
To assess the degree of humanization for each gut microbial clade in each captive NHP population, we tested whether the ASV-compositions of individual microbial genera in captive NHPs were more similar to those of the genera in humans (i.e., humanized) or to those of the genera in wild living conspecific individuals. These analyses allowed us to identify microbial genera in each captive NHP population whose endogenous host-species specific ASVs (i.e., ASVs private to wild conspecific hosts) displayed evidence of loss or of replacement by ASVs found in humans. We developed a statistic, Host Specificity Score (HSSs) (Materials and Methods), which can be calculated for any microbial clade or taxonomic rank of arbitrary phylogenetic depth. Here, we calculated these scores at the level of microbial genus, which was the finest-scale taxonomic resolution allowed by the 16S rDNA amplicon sequences in our dataset. Log-transformed HSSs are normally distributed around 0 and provide a quantitative ranking of the degree of humanization for all microbial genera in the gut microbiotas of a captive population, with lower scores indicating a greater degree of humanization. Log-transformed HSSs >0 indicate the ASV composition of the genus is more similar between captive primates and wild-living conspecifics than between captive primates and humans. Conversely, log-transformed HSSs <0 indicate that the ASV composition of the genus is more similar between captive primates and humans than between captive primates and wild-living conspecifics. Humanization of the ASV composition of a microbial genus in a captive NHP population, as indicated by a negative HSS, could be underlain by the loss of ASVs found in wild conspecific NHPs but not found in humans or the gain of ASVs found in humans but not found in wild conspecific NHPs.
Calculating HSSs for each microbial genus for each captive NHP population indicated that some genera in the captive NHP gut microbiota were humanized, whereas others were retained from the wild. An example of a bacterial genus that was humanized in captive chimpanzees in the United States (i.e., the genus Collinsella) is shown in Figure 3a. An example of an archaeal genus that was retained from the wild in captive chimpanzees in the United States (i.e., the genus vadinCA11) is shown in Figure 3b. A list of all microbial genus-level HSSs for each captive NHP population are shown in Table S7.
FIGURE 3.
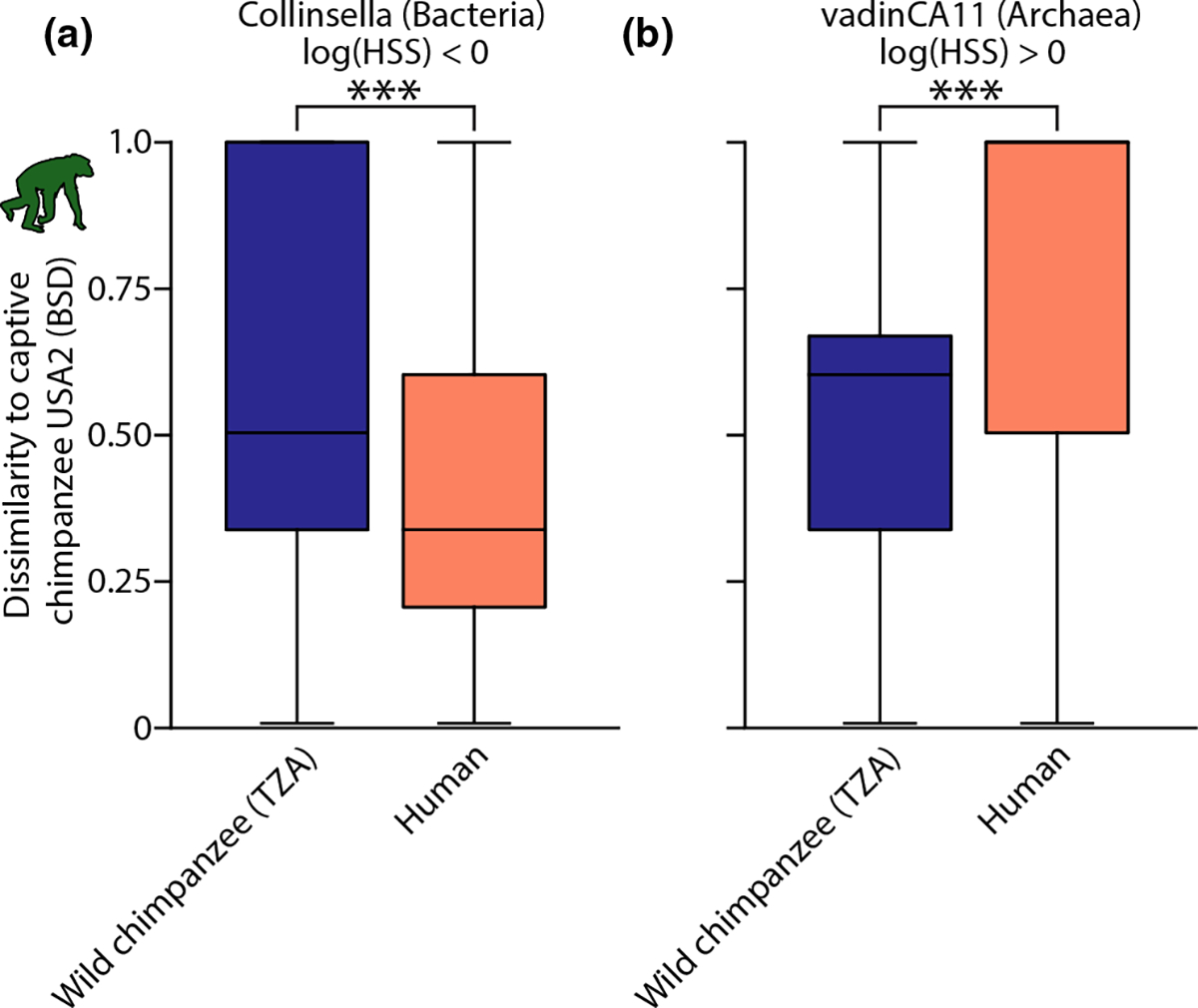
Host specificity scores reveal microbial genus-specific humanization of captive NHP gut microbiotas. Shown are examples of two microbial genera that display evidence of humanization (a) or retention from the wild (b) in captive chimpanzees. Boxplots show the maximum, minimum, interquartile range, and median of BSD dissimilarities between wild chimpanzees and captive chimpanzees or between humans and captive chimpanzees. The logarithm of the host specificity score, calculated as the ratio of the means of boxplots within each panel, was <0 for Collinsella (a) and >0 for vadinCA11 (b), indicating humanization and retention from the wild, respectively. Boxes are coloured by primate species as in Figure 1. Significant differences between means based on permutation tests are indicated with asterisks; ***p = .001
Comparing HSSs between pairs of captive primate populations indicated that no microbial genus was humanized in every NHP population (Table S7). However, replicate captive populations of the same species displayed highly consistent genus-level patterns of gut microbiota humanization. The HSSs between two independent captive populations of chimpanzees were significant positively associated (R2 = .82; p-value = 5.51e-24), as were the HSSs between two independent captive populations of doucs (R2 = .8; p-value = 8.46e-18). In addition, the HSS of chimpanzees were significantly associated with those of gorillas (R2 = .16; p-value = .000871 and R2 = .25; p-value = 1.05e-05), but not with those of doucs (Figure 4). Similar results, in which no genus displayed evidence of humanization in all captive NHP populations but replicate captive populations of the same NHP species displayed similar patterns of humanization of microbial genera, were observed in analyses of a related statistic, microbiota convergence scores (MCSs; Appendix S1: Materials and Methods, Figure S4, Table S8). Heatmaps of HSSs and MCSs of taxa found in all captive NHP populations are presented in Figure S5. Qualitatively similar results, in which HSSs and MCSs were significantly positively associated between conspecific captive NHP populations but tended to be not associated or negatively associated between heterospecific captive NHP populations, were observed in analyses based on data from the American Gut Project (McDonald et al., 2018; Appendix S1: Materials and Methods; Tables S9 and S10).
FIGURE 4.
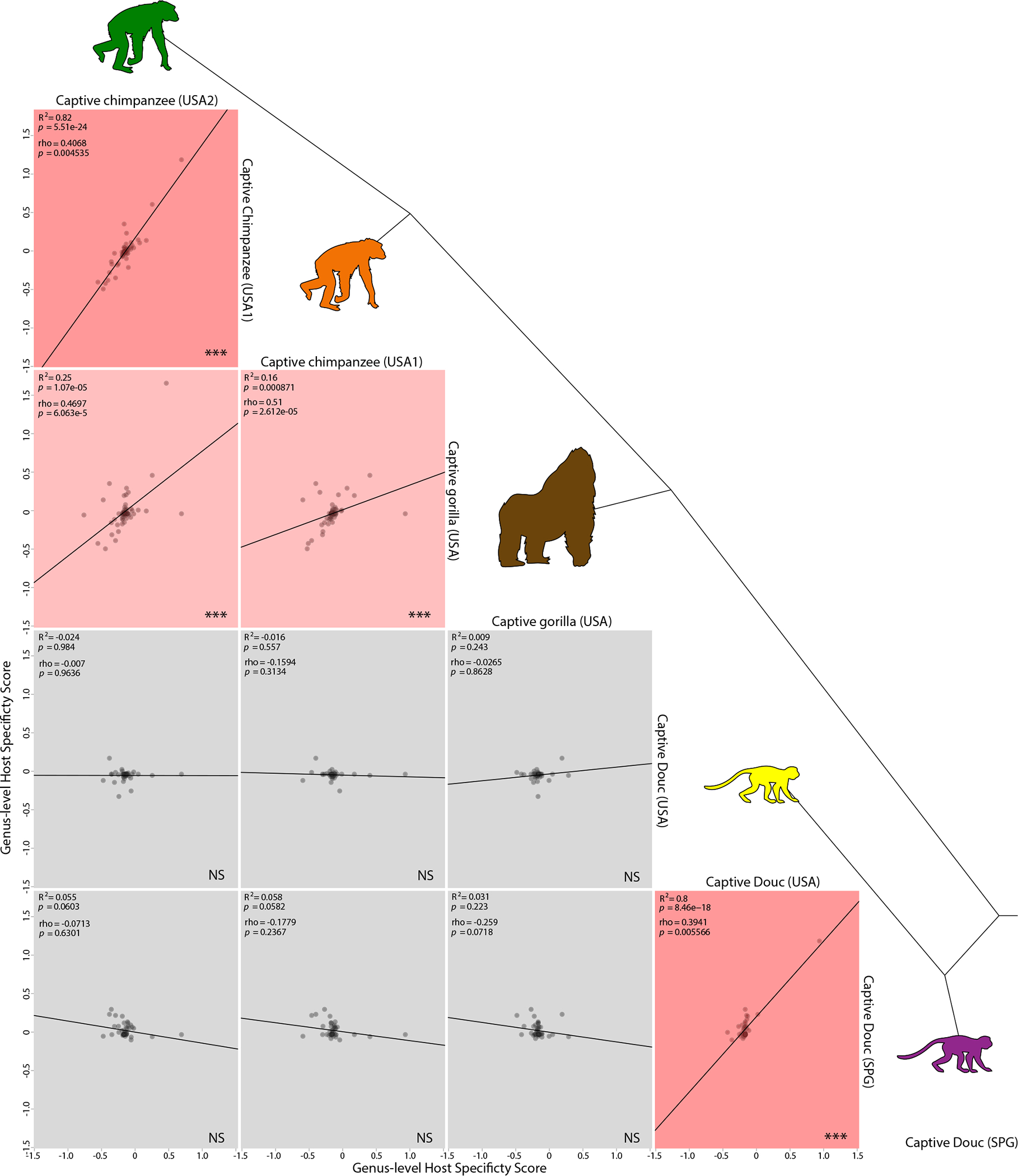
Humanization of gut microbial genera in captive NHPs is reproducible and host-species specific. Plots show relationships of microbial genus-level Host Specificity Scores (HSSs) between pairs of captive NHP populations. Each point represents the HSSs of a bacterial genus in two captive NHP populations (rows and columns), and lines indicate best-fit linear regressions. Cartoons represent NHP species and are coloured as in Figure 1. Tree connecting cartoons shows phylogenetic relationships among NHPs. Rows and columns correspond to NHP species, with each plot showing a regression between HSSs for a pair of NHP species. Boxes are coloured based on R2 of regression, with R2 > .8 coloured dark red, 0.8 > R2 > .1 coloured light red, and R2 < .1 coloured grey. R2, Spearman’s rho, and corresponding p-values are reported in the top left corner of each plot. Significance for nonzero slope of regression line (based on Pearson’s correlation) is indicated in the bottom right corner of each plot; NS, not significant; *p < 0.05; ** p < .01; ***p < .001
4 |. DISCUSSION
Comparing multiple well-sampled wild and captive populations of chimpanzees, gorillas, and doucs with humans allowed us to test for reproducible effects of captivity on specific microbial lineages in the gut microbiotas of nonhuman primate (NHP) species. Our results indicate that captivity humanizes the primate gut microbiota, but that the microbial taxa underlying this process of humanization vary substantially and consistently among NHP species. No microbial ASV or genus was significantly overrepresented or underrepresented in all captive NHP populations relative to wild conspecific populations (Tables S5 and S6). Similarly, no microbial genus displayed evidence of humanization, defined by host-specificity scores (HSSs), in every captive NHP population (Table S7). In contrast, we observed striking consistency of the effects of captivity on gut microbiota constituents between replicate captive NHP populations of the sample species. The same ASVs and genera reproducibly shifted in relative abundance in replicate captive conspecific NHP populations relative to matched wild NHP populations (Tables S5 and S6, Figure 2). Similarly, most microbial genera displayed reproducible signatures of humanization in replicate captive conspecific NHP populations: the HSSs of one captive population predicted >80% of the variation in HSSs in the other captive population in both NHP species for which replicate captive populations have been sampled (i.e., chimpanzees and doucs; Figures 4 and S4). The consistency of per-genus measures of humanization in replicate conspecific captive NHP populations was also observed in analyses based on human gut microbiota data from the American Gut Project (Appendix S1: Materials and Methods; Tables S9 and S10. Previous results have shown that the gut microbiotas of monkeys are humanized in captivity (Clayton et al., 2016); our results demonstrate that this humanization also occurs in captive ape populations. In addition, our results indicate that the humanization of the gut microbiota is underlain by distinct sets of microbial lineages in captive NHP populations of different species, but that the sets of microbial lineages that are humanized by captivity can be predicted with high accuracy by the species identity of the NHP population.
Interestingly, certain microbial taxa showed consistent patterns of humanization between replicate captive NHP populations of the same species but opposite patterns between replicate captive NHP populations of different species. For example, Oribacterium and Roseburia, genera within the Lachnospiraceae, showed evidence of humanization, based on HSSs, in both captive douc populations, but these genera were robust to humanization in both captive chimpanzee populations (Table S8, Figure S5). The observations that different NHP species display discordant but reproducible patterns of humanization of gut microbial lineages in captivity raises questions about the mechanisms underlying this pattern. One explanation is that each NHP species contains a distinct set of microbial lineages that are better adapted to the gut environment of their respective host species relative to the congeneric microbial lineages found in humans. For example, microbial genera whose ASVs were retained from the wild in captive NHP populations (i.e., genera displaying a log-transformed HSS >0) represent microbial lineages that may, in captive NHPs, outcompete microbial lineages found in humans. Discordance of per-genus HSSs between NHP species could arise if the microbial lineages that outcompete congeneric lineages derived from humans belong to different genera in different NHP species. In addition, differences in diet may affect the process of gut microbiota humanization in captivity, leading to discordance between species but reproducibility within species. For example, previous work has shown that global patterns of gut microbiota composition (i.e., alpha and beta diversity) are more affected by captivity in folivorous NHP species than in less dietary specialized NHP species (Frankel et al., 2019). Under this scenario, differences in the degree of dietary shifts experienced in captivity could lead to different effects on individual microbial lineages and genus-level patterns of humanization between NHP species.
Our results highlight the importance of population-level sampling of captive NHPs in order to identify specific gut microbial lineages most affected by captivity. Studies of fewer numbers of captive individuals per NHP species have statistical power to detect differences in alpha and beta diversity between captive and wild gut microbiotas and have yielded important insights into the effects of captivity on the gut microbiota (Campbell et al., 2020; Clayton et al., 2016; Frankel et al., 2019; Frankel et al., 2019; Hale et al., 2019; Lee et al., 2019; McKenzie et al., 2017; Nakamura et al., 2011; Narat et al., 2020; Tsukayama et al., 2018; Uenishi et al., 2007). However, identifying individual microbial lineages or clades (e.g., ASVs or genera) that display significantly different relative abundances between populations requires sampling sufficient numbers of individuals to overcome high false-discovery rates inherent in multiple testing. Our results suggest that future studies focused on identifying specific microbial lineages in the gut microbiota that are affected by captivity should when possible prioritize replication at the level of host individual.
The statistics that we developed (HSSs and MCSs) provide quantitative means to identify the specific clades of microbial lineages that display the strongest evidence of humanization in captive hosts. HSSs and MCSs can be applied to any microbial clade or taxonomic rank as well as any captive population for which microbiota data from wild conspecifics are available. In addition to captive populations, these statistics could also be applied to identify humanized gut microbial taxa in animals associated with humans in other contexts, such as urban settings or other habitat disruptions that may affect the gut microbiota (e.g., Amato et al., 2013). In the case of captive NHPs, the identification of individual microbial taxa that are repeatably humanized has important implications for managing the health of captive hosts. Humanized microbial taxa represent candidate contributors to gastrointestinal dysbiosis in captive NHP populations, in particular if they are reproducibly humanized in individuals that develop gastrointestinal dysfunction (Amato et al., 2016). For example, the genus Collinsella was repeatably humanized in replicate captive chimpanzee populations, and overrepresentation of this genus in the gut microbiota has been associated with reduced fibre intake, insulin resistance, and rheumatoid arthritis in human populations (Chen et al., 2016; Gomez-Arango et al., 2018; Mena-Vázquez et al., 2020). Similarly, Clostridium lineages were repeatably humanized in replicate captive chimpanzee populations, and lineages from this genus are the most common causes of gastrointestinal infections in humans (Smits et al., 2016). The gut microbial lineages that are reproducibly humanized in captive populations represent high priority for targeted culturing from wild NHP populations. Culturing and biobanking representatives of these microbial lineages from wild NHPs could provide opportunities to restore the gut microbiotas of captive NHP populations.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank two anonymous reviewers for their constructive comments. We gratefully acknowledge Dr Weiwei Yan for assistance in the laboratory. We thank the Project Chimps animal sanctuary for aiding in sample collection. This work was funded by a grant from the National Institutes of Health (1R35GM138284) to AHM. JLH was supported by a Cornell Presidential Life Sciences Fellowship and National Science Foundation Graduate Research Fellowship. All procedures conducted in this study were approved by the Institutional Animal Care and Use Committee at Cornell University. We declare we have no competing interests.
Funding information
This work was funded by award R35GM138284 from the National Institutes of Health to AHM. JLH was supported by a Cornell Presidential Life Sciences Fellowship and National Science Foundation Graduate Research Fellowship
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
All sequence data have been made available in the European Nucleotide Archive under accession PRJEB44013.
REFERENCES
- Amato KR, Sanders G, J., Song SJ, Nute M, Metcalf JL, Thompson LR, Morton JT, Amir A, J. McKenzie V, Humphrey G, Gogul G, Gaffney J, Baden L, A., Britton AO, G., Cuozzo P, F., Di Fiore A, Dominy J, N., Goldberg L, T., Gomez A, … R. Leigh S (2019). Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. The ISME Journal, 13(3), 576–587. 10.1038/s41396-018-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato KR, Metcalf JL, Song SJ, Hale VL, Clayton J, Ackermann G, Humphrey G, Niu K, Cui D, Zhao H, Schrenzel MD, Tan CL, Knight R, & Braun J (2016). Using the gut microbiota as a novel tool for examining colobine primate GI health. Global Ecology and Conservation, 7, 225–237. 10.1016/j.gecco.2016.06.004. [DOI] [Google Scholar]
- Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Rex Gaskins H, Stumpf RM, Yildirim S, Torralba M, Gillis M, Wilson BA, Nelson KE, White BA, & Leigh SR (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. The ISME Journal, 7(7), 1344–1353. 10.1038/ismej.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, … Caporaso JG (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8), 852–8 57. 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell TP, Sun X, Patel VH, Sanz C, Morgan D, & Dantas G (2020). The microbiome and resistome of chimpanzees, gorillas, and humans across host lifestyle and geography. The ISME Journal, 14(6), 1584–1599. 10.1038/s41396-020-0634-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A (1984). Nonparametric estimation of the number of classes in a population. Scandinavian Journal of Statistics, 11, 265–270. [Google Scholar]
- Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, & Taneja V (2016). An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Medicine, 8(1), 1–14. 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton JB, Vangay P, Huang HU, Ward T, Hillmann BM, Al-Ghalith GA, Travis DA, Long HT, Tuan BV, Minh VV, Cabana F, Nadler T, Toddes B, Murphy T, Glander KE, Johnson TJ, & Knights D (2016). Captivity humanizes the primate microbiome. Proceedings of the National Academy of Sciences, 113(37), 10376–10381. 10.1073/pnas.1521835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice LR (1945). Measures of the amount of ecologic association between species. Ecology, 26(3), 297–302. 10.2307/1932409. [DOI] [Google Scholar]
- Frankel JS, Mallott EK, Hopper LM, Ross SR, & Amato KR (2019). The effect of captivity on the primate gut microbiome varies with host dietary niche. American Journal of Primatology, 81(12), e23061. 10.1002/ajp.23061. [DOI] [PubMed] [Google Scholar]
- Gomez-Arango LF, Barrett HL, Wilkinson SA, Callaway LK, McIntyre HD, Morrison M, & Nietert MD (2018). Low dietary fiber intake increases Collinsella abundance in the gut microbiota of overweight and obese pregnant women. Gut Microbes, 9(3), 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Navas-Molina JA, Kosciolek T, McDonald D, Vázquez-Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, Sanders JG, Shorenstein J, Holste H, Petrus S, Robbins-Pianka A, Brislawn CJ, Wang M, Rideout JR, Bolyen E, … Knight R (2018). Qiita: Rapid, web-enabled microbiome meta-analysis. Nature Methods, 15(10), 796–798. 10.1038/s41592-018-0141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale VL, Tan CL, Niu K, Yang Y, Zhang Q, Knight R, & Amato KR (2019). Gut microbiota in wild and captive Guizhou snub-nosed monkeys, Rhinopithecus brelichi. American Journal of Primatology, 81(10–11), e22989. [DOI] [PubMed] [Google Scholar]
- Lee W, Hayakawa T, Kiyono M, Yamabata N, & Hanya G (2019). Gut microbiota composition of Japanese macaques associates with extent of human encroachment. American Journal of Primatology, 81(12), e23072. 10.1002/ajp.23072. [DOI] [PubMed] [Google Scholar]
- Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, & Peddada SD (2015). Analysis of composition of microbiomes: A novel method for studying microbial composition. Microbial Ecology in Health and Disease, 26(1), 27663. 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, DeRight Goldasich L, Dorrestein PC, Dunn RR, Fahimipour AK, Gaffney J, Gilbert JA, Gogul G, Green JL, Hugenholtz P, … Gunderson B (2018). American gut: An open platform for citizen science microbiome research. mSystems, 3(3), e00031–18. 10.1128/mSystems.00031-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z, Lozupone CA, Hamady M, Knight R, & Bushman FD (2008). The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathogens, 4(2), e20. 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie VJ, Song SJ, Delsuc F, Prest TL, Oliverio AM, Korpita TM, Alexiev A, Amato KR, Metcalf JL, Kowalewski M, Avenant NL, Link A, Di Fiore A, Seguin-Orlando A, Feh C, Orlando L, Mendelson JR, Sanders J, & Knight R (2017). The effects of captivity on the mammalian gut microbiome. Integrative and Comparative Biology, 57(4), 690–704. 10.1093/icb/icx090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena-Vázquez N, Ruiz-Limón P, Moreno-Indias I, Manrique-Arija S, Tinahones FJ, & Fernández-Nebro A (2020). Expansion of rare and harmful lineages is associated with established rheumatoid arthritis. Journal of Clinical Medicine, 9(4), 1044. 10.3390/jcm9041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, & Ochman H (2016). Cospeciation of gut microbiota with hominids. Science, 353(6297), 380–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Foerster S, Wilson ML, Pusey AE, Hahn BH, & Ochman H (2016). Social behavior shapes the chimpanzee pan-microbiome. Science Advances, 2(1), e1500997. 10.1126/sciadv.1500997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Li Y, Ngole EM, Ahuka-Mundeke S, Lonsdorf EV, Pusey AE, Peeters M, Hahn BH, & Ochman H (2014). Rapid changes in the gut microbiome during human evolution. Proceedings of the National Academy of Sciences, 111(46), 16431–16435. 10.1073/pnas.1419136111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Amato KR, Garber P, Estrada A, Mackie RI, & Gaskins HR (2011). Analysis of the hydrogenotrophic microbiota of wild and captive black howler monkeys (Alouatta pigra) in palenque national park, Mexico. American Journal of Primatology, 73(9), 909–919. 10.1002/ajp.20961. [DOI] [PubMed] [Google Scholar]
- Narat V, Amato KR, Ranger N, Salmona M, Mercier-Delarue S, Rupp S, Ambata P, Njouom R, Simon F, Giles-Vernick T, & LeGoff J (2020). A multi-disciplinary comparison of great ape gut microbiota in a central African forest and European zoo. Scientific Reports, 10(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Worobey M, Kuo CH, Ndjango JBN, Peeters M, Hahn BH, & Hugenholtz P (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biology, 8(11), e1000546. 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing. http://www.R-project.org/. [Google Scholar]
- Shannon CE, & Weaver W (1949). The mathematical theory of communication. University of Illinois Press. [Google Scholar]
- Shigeno Y, Toyama M, Nakamura M, Niimi K, Takahashi E, & Benno Y (2018). Comparison of gut microbiota composition between laboratory-bred marmosets (Callithrix jacchus) with chronic diarrhea and healthy animals using terminal restriction fragment length polymorphism analysis. Microbiology and Immunology, 62(11), 702–710. [DOI] [PubMed] [Google Scholar]
- Smits WK, Lyras D, Lacy DB, Wilcox MH, & Kuijper EJ (2016). Clostridium difficile infection. Nature Reviews Disease Primers, 2(1), 1–20. 10.1038/nrdp.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, & Navas-Molina JA (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature, 551(7681), 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukayama P, Boolchandani M, Patel S, Pehrsson EC, Gibson MK, Chiou KL, Jolly CJ, Rogers J, Phillips-Conroy JE, & Dantas G (2018). Characterization of wild and captive baboon gut microbiota and their antibiotic resistomes. Msystems, 3(3), e00016–18. 10.1128/mSystems.00016-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uenishi G, Fujita S, Ohashi G, Kato A, Yamauchi S, Matsuzawa T, & Ushida K (2007). Molecular analyses of the intestinal microbiota of chimpanzees in the wild and in captivity. American Journal of Primatology, 69(4), 367–376. 10.1002/ajp.20351. [DOI] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, & Heath AC (2012). Human gut microbiome viewed across age and geography. Nature, 486(7402), 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data have been made available in the European Nucleotide Archive under accession PRJEB44013.